
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal complexes of the PPO/POP ligand: variable coordination chemistry and photo-luminescence properties†
Franziska
Flecken
 a,
Toni
Grell
b and
Schirin
Hanf
*a
a,
Toni
Grell
b and
Schirin
Hanf
*a
aInstitute for Inorganic Chemistry, Karlsruhe Institute of Technology, Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: schirin.hanf@kit.edu
bDipartimento di Chimica, Università degli Studi di Milano, Via Camillo Golgi 19, 20131 Milano, Italy
First published on 13th May 2022
Abstract
In the current work the tautomeric equilibrium between tetraphenyldiphosphoxane (Ph2P–O–PPh2, POP) and tetraphenyldiphosphine monoxide (Ph2P–P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)Ph2, PPO) in the absence and presence of transition metal precursors is investigated. Whereas with hard transition metal ions, such as Fe(II) and Y(III), PPO-type complexes, such as [FeCl2(PPO)2] (1) and [YCl3(THF)2(PPO)] (2), are formed, softer transition metals ions tend to form so-called coordination stabilised tautomers of the POP ligand form, such as [Cu2(MeCN)3(μ2-POP)2](PF6)2 (3), [Au2Cl2(μ2-POP)] (4), and [Au2(μ2-POP)2](OTf)2 (5). The photo-optical properties of the PPO- and POP-type transition metal complexes are investigated experimentally using photo-luminescence spectroscopy, whereby the presence of metallophillic interactions was found to play a crucial role. The dinuclear copper complex [Cu2(MeCN)3(μ2-POP)2](PF6)2 (3) shows a very interesting thermochromic behavior and intense photo-luminescence with remarkable phosphoresence lifetimes at 77 K, which can probably be attributed to short intramolecular Cu–Cu distances.
O)Ph2, PPO) in the absence and presence of transition metal precursors is investigated. Whereas with hard transition metal ions, such as Fe(II) and Y(III), PPO-type complexes, such as [FeCl2(PPO)2] (1) and [YCl3(THF)2(PPO)] (2), are formed, softer transition metals ions tend to form so-called coordination stabilised tautomers of the POP ligand form, such as [Cu2(MeCN)3(μ2-POP)2](PF6)2 (3), [Au2Cl2(μ2-POP)] (4), and [Au2(μ2-POP)2](OTf)2 (5). The photo-optical properties of the PPO- and POP-type transition metal complexes are investigated experimentally using photo-luminescence spectroscopy, whereby the presence of metallophillic interactions was found to play a crucial role. The dinuclear copper complex [Cu2(MeCN)3(μ2-POP)2](PF6)2 (3) shows a very interesting thermochromic behavior and intense photo-luminescence with remarkable phosphoresence lifetimes at 77 K, which can probably be attributed to short intramolecular Cu–Cu distances.
1 Introduction
Bidentate diphosphine ligands and their corresponding transition metal complexes have attracted a great deal of attention in homogeneous catalysis, such as in hydrogenation, hydroformylation or carbonylation reactions.1 In this context, also diphosphines with an additional ether linkage, so-called “POP-type ligands” have been investigated, with a few examples being shown in Fig. 1.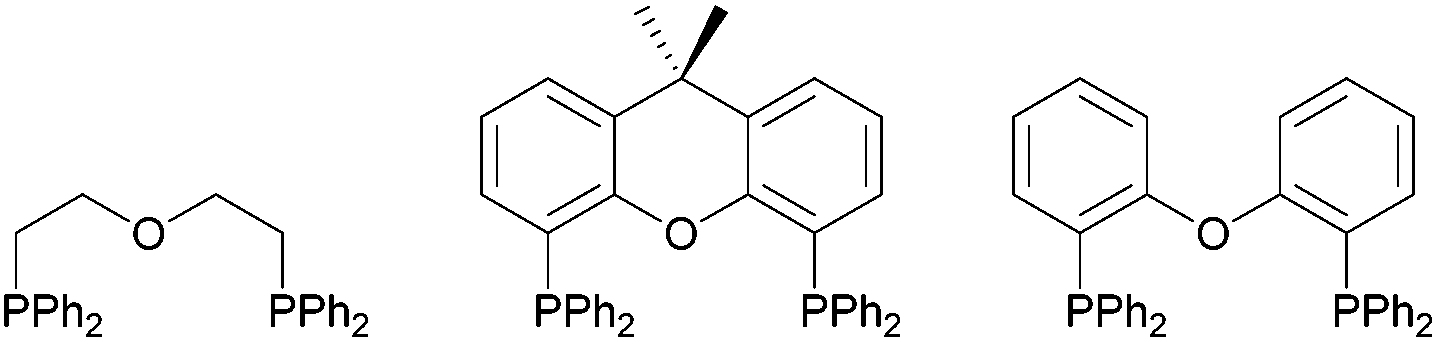 | ||
| Fig. 1 Examples of POP-type ligands. | ||
They have gained significant attention, since they can act as hemilabile ligand sets and therefore enable a reversible coordination/decoordination of the hemilabile donor atom/group. Consequently, the labile donor atom/group can offer sites for substrate binding, when removed during the catalytic reaction from the pre-catalyst and trigger the activity of the transition metal catalyst, whereas in the absence of a substrate, the chelate effect of the multidentate ligand confers stability on the metal center.2 Especially Xantphos (9,9-dimethyl-4,6-bis(diphenylphosphino)xanthene) and DPEphos (bis(2-(diphenylphosphino)phenyl) ether), originally developed for Rh-catalysed hydroformylation reactions,3 have become to the fore in this context, due to the versatile modification potential of their bite angle. Furthermore, the ability of such POP-type ligands to coordinate metal centres either in mono-, bi- or tridentate fashion,4 the latter of which is very similar to classical tridentate PNP-type pincer ligands,5 has made them suitable coordination partners for various transition metals.
In the context of POP-type ligands, a related ligand set, namely tetraalkyl- or aryl-diphosphoxane (R2P–O–PR2, POP), has been widely overlooked, despite the very interesting tautomeric equilibrium with tetraalkyl- or aryl-diphosphine monoxide (R2P–P(O)R2, PPO, Scheme 1).6 The POP tautomer is the anhydride of the phosphinous acid R2P(OH), which readily converts into the thermodynamically more stable secondary phosphine oxide R2P(O)H, unless the R substituents on the phosphorus atom are strongly electron-withdrawing (e.g. CF3).7 Secondary phosphine oxides have been widely employed as bifunctional ligands in homogeneously catalysed transformations (e.g. hydrogenation, hydroformylation, C–H arylation and cross coupling reactions)8,9 and have also found application as stabilising agents for nanoparticles.10–12 Apart from being the anhydride of the corresponding secondary phosphine oxide, the POP tautomer can be regarded as oxygen bridged analogue of well-known dppm-type (bis(diphenylphosphine)methane)13,14 or PNP-type ligands,15–18 which have found to show interesting catalytic and photo-optical properties.
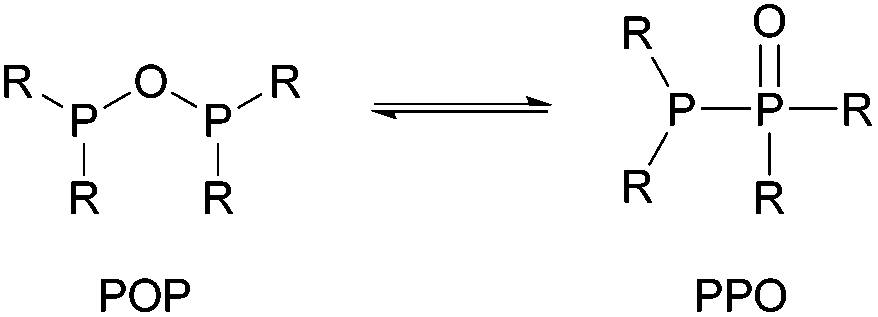 | ||
| Scheme 1 Tautomeric equilibrium of R2P–O–R2 (POP) and R2P–P(O)R2 (PPO), R = Me, Et, CF3, Ph. | ||
Metal complexes of diphosphine monoxide/diphosphoxane ligands are scarce in coordination chemistry and only a handful compounds have been structurally characterised up to now. Among those compounds, only very few were obtained from targeted synthetic approaches, which involved the direct reaction of a PPO/POP ligand set with metal precursors. In this context, Mo(II) complexes, such as [Mo(CO)3X2(μ2-R2POPR2)] (R = Ph or p-tolyl, X = I, Br),19 or related bimetallic complexes, such as [Mo(CO)5–(Ph2POPPh2)–Cr(CO)5] and [Mo(CO)5–(Ph2POPPh2)–Fe(CO)4],20 were reported. In all these cases the POP ligand acts as bidentate ligand to either one or, in bimetallic complexes, two metal centres.
In contrast, many more reactions involve metal promoted rearrangements of related ligand sets leading to the in situ formation and isolation of PPO/POP-based transition metal complexes. Often these reactions are based on the coupling/isomerisation of secondary phosphine oxide derivatives.11,21–27 For example, the attempted reactions of secondary phosphine oxides R2P(O)H (R = nBu, (CH2)4Ph) with Cu(OAc)2 in the presence of NH4Cl resulted in the formation and isolation of tetranuclear Cu complexes of the form [Cu4(μ2-Cl)4(μ2-R2POPR2)2]. The POP ligand R2P–O–PR2 was thereby formed in situ from the dehydration of two secondary phosphine oxide molecules.23 Other reactions leading to PPO/POP-based transition metal complexes via metal promoted rearrangements are based on P–N,28–35 P–O36–44 or P–H22 bond scissions and subsequent reactions (often in the presence of water) with transition metal precursors. Reviewing the literature more closely, it becomes obvious that almost all of these reported transition metal complexes comprise the POP form of the ligand with only a very few PPO complexes being described.25,32–35,45
Spurred by the lack of a systematic coordination study concerning the PPO/POP ligand set, we set out to investigate the coordination behaviour towards a variety of transition metals with different hard/soft character. The ultimate aim was hereby to rationally synthesise coordination-stabilised tautomers of either the PPO or the POP ligand. Furthermore, the investigation of the photo-optical properties of the PPO/POP-transition metal complexes was a key concern of this study, based on the similarity with PNP-type ligands, which have shown remarkable photo-luminescence properties.15–18
2 Results and discussion
2.1 PPO/POP tautomeric equilibrium
To gain a deeper understanding of the equilibrium between the POP ligand tetraphenyldiphosphoxane (Ph2P–O–PPh2) and the tautomeric PPO form tetraphenyldiphosphine monoxide (Ph2P–P(O)Ph2), DFT calculations were carried out. These show that the PPO form is indeed about 41 kJ mol−1 more stable at room temperature (Fig. 2, B3LYP, def2-TZVP, gas phase). This was also found to be true for the PPO/POP equilibrium in solution, as demonstrated by the 31P{1H} NMR spectrum of the ligand in various solvents, shown exemplarily in CDCl3 in the ESI.† Here two doublets at 35.8 ppm and −22.5 ppm (in CDCl3) with a 1JPP coupling constant of 228 Hz become obvious, belonging to the P(V) and P(III) phosphorus atoms, respectively. A singlet corresponding to the POP form could not be observed. This is accordance with the calculated energy difference between both forms which corresponds to the presence of less than 10–5% for an equilibrium at room temperature.
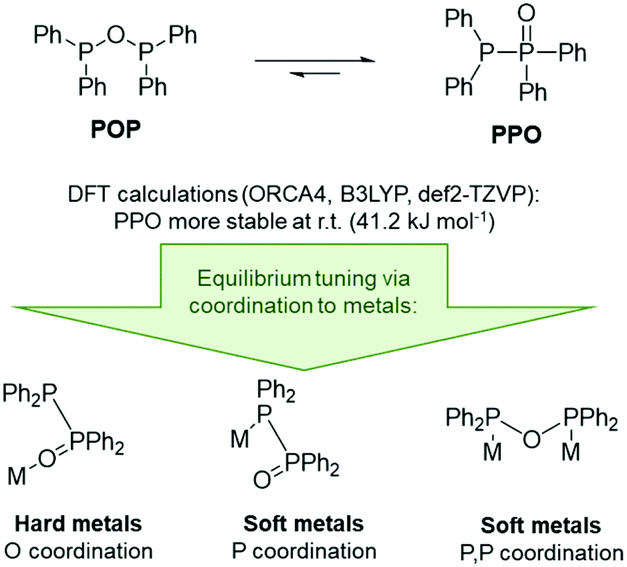 | ||
| Fig. 2 Tautomeric equilibrium and equilibrium tuning of the PPO/POP ligand. | ||
In general, the R2P(O)–PR2/R2P–O–PR2 equilibrium can be influenced by the electronic character of the R groups, whereby the anhydride POP form is stabilised through the introduction of strongly electron-withdrawing substituents, such as CF346 or 2,4-CF3-C6H3,6 or the implementation of sterically demanding substituents, such as tBu groups.47 However, the PPO/POP equilibrium can also be easily tuned via the coordination to transition metals and the synthesis of so-called coordination-stabilised tautomers. According to this, we expected the PPO form of the ligand to be present, when hard metal ions are involved, and the formation of POP-type complexes in the presence of softer transition metal ions.
2.2 Synthesis of transition metal complexes of the PPO/POP ligand
To investigate the formation of coordination-stabilised tautomers of the PPO/POP ligand framework, the PPO/PPO (Ph2P–P(O)Ph2/Ph2P–O–PPh2) ligand set was reacted with various hard and soft transition metal precursors (Scheme 2).
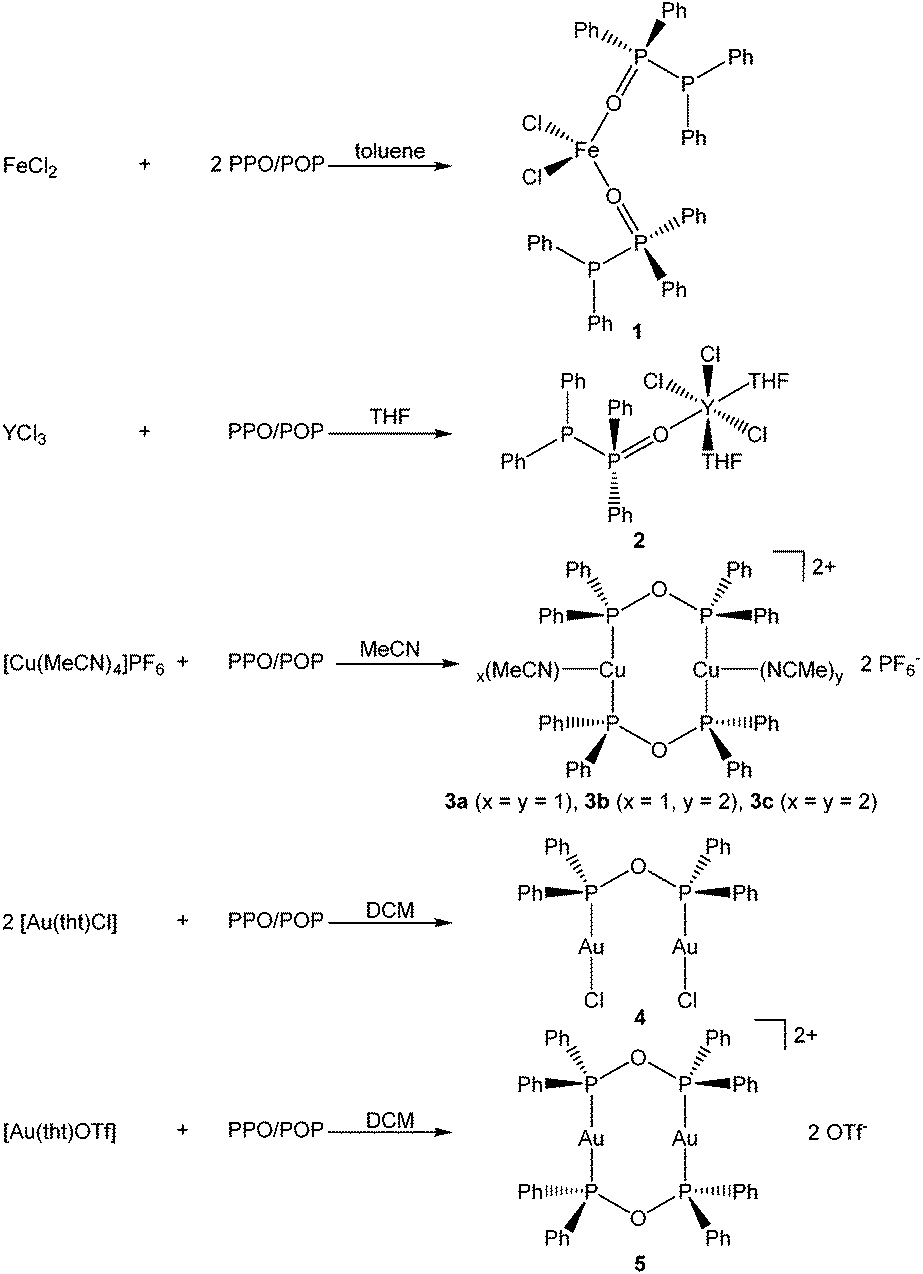 | ||
| Scheme 2 Reaction of the PPO/POP ligand with various transition metal salts. | ||
The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometric reaction of FeCl2 with the PPO/POP ligand afforded pale yellow crystals of a [FeCl2(PPO)2] complex (1), in which the iron centre is coordinated by two PPO ligands via their oxygen atoms. The pale yellow colour of the isolated crystals as well as the observation of very broad resonances in the NMR spectra indicate the paramagnetic character of the iron(II) centre and consequently a high spin configuration.
:2 stoichiometric reaction of FeCl2 with the PPO/POP ligand afforded pale yellow crystals of a [FeCl2(PPO)2] complex (1), in which the iron centre is coordinated by two PPO ligands via their oxygen atoms. The pale yellow colour of the isolated crystals as well as the observation of very broad resonances in the NMR spectra indicate the paramagnetic character of the iron(II) centre and consequently a high spin configuration.
A similar type of complex, namely [YCl3(THF)2(PPO)] (2) was isolated from the 1:1 stoichiometric reaction of YCl3 with the PPO/POP ligand in THF. Also, in this case, the PPO form is present with the phosphorus oxide moiety coordinating the yttrium centre via the oxygen atom. This coordination mode of the PPO ligand also becomes obvious from the 31P{1H} NMR resonances. Hereby a significant downfield shift of the doublet belonging to the P(V) atom to a multiplet in the range of 44.1–51.8 ppm (D8-THF) in the yttrium complex 2 compared to 35.8 ppm in the free PPO ligand (CDCl3) has been observed. The appearance of a multiplet can be attributed to the coupling with the NMR active 89Y nuclei (I = ½, natural abundance = 100%) and the related occurrence of an ABX spin system. In contrast to that, the chemical shift of the 31P{1H} NMR resonance of the P(III) atom of the free PPO ligand, appearing at −22.5 ppm (CDCl3), is almost identical with the P(III) doublet of the coordinated PPO ligand within complex 2 (−19.9 ppm, D8-THF).
When softer transition metals, such as the coinage metals copper and gold, are employed for coordination, the anhydride POP form could be stabilised within the corresponding transition metal complexes. The 1:1 stoichiometric reaction of [Cu(MeCN)4]PF6 with PPO/POP in MeCN afforded pale yellow crystals of a dinuclear copper(I) complex with the formulae [Cu2(MeCN)3(μ2-POP)2](PF6)2 (3) after the reaction workup. Single-crystal X-ray crystallography, discussed in more detail in section 2.3, revealed the 1:1:3 statistical distribution of three different copper complexes, namely [Cu2(MeCN)4(μ2-POP)2](PF6)2 (3a), [Cu2(MeCN)2(μ2-POP)2](PF6)2 (3b) and [Cu2(MeCN)3(μ2-POP)2](PF6)2 (3c). Within complex 3 two copper centres are coordinated by two bridging POP ligands. The stabilisation of the POP form is also obvious from the 31P{1H} NMR spectrum, where only one singlet at 106.1 ppm in CD2Cl2 is observed, instead of two doublets in the case of the free PPO ligand. Obviously, upon coordination to copper the signal of the P(III) atom is tremendously shifted in contrast with the P(III) doublet resonance at −22.5 ppm (CDCl3) of the free PPO ligand. Given the fact, that all 31P centres and all CH3CN proton signals of 3 are equivalent on the NMR timescale, a dynamic structure in solution can be assumed.
Attempts to extend the coordination studies towards another copper(I) precursor, namely copper(I) chloride, resulted in the formation of the literature known tetranuclear copper(I) complex [(Cu4(μ3-Cl)2(μ2-Cl)2(μ2-POP)2], which could be identified by its 31P{1H} NMR resonance at 34.6 ppm in C6D6 as reported in the literature.32
The anhydride POP form of the PPO/POP ligand set could not only be stabilised within the copper(I) complex 3, but also in the two novel gold(I) complexes [Au2Cl2(μ2-POP)] (4) and [Au2(μ2-POP)2](OTf)2 (5). Whereas the 2:1 stoichiometric reaction of [Au(tht)Cl] with the PPO/POP ligand resulted in the formation of crystals of [Au2Cl2(μ2-POP)] (4), the use of [Au(tht)OTf] as metal precursor, including a more labile triflate anion, led to the isolation of crystals of [Au2(μ2-POP)2](OTf)2 (5). In complex 4 two gold centres are coordinated by one POP ligand, while two POP ligands are found to be involved in the coordination within complex 5. The latter structure is similar to the coordination motive found in the copper(I) analogue 3. Similar to the copper(I) analogue 3, a singlet resonance can be found in the 31P{1H} NMR spectrum of complex 5. However, the signal in 5 is shifted downfield to 132.0 ppm (CDCl3) in comparison with the 31P{1H} NMR resonance of copper complex 3 (106.1 ppm, CD2Cl2). Inspecting the 31P{1H} NMR spectrum of the gold(I) complex 4 in CDCl3, a slightly different situation can be observed (Fig. 3). Besides a singlet at 116.1 ppm, belonging to the POP phosphorus atoms within 4, two sets of doublets at 32.2 ppm and 16.7 ppm are observed. This points towards the existence of a complex in which the PPO form is present instead of the POP tautomer. In this case, coordination to the gold is probably taking place via the P(III) atom, since the 31P{1H} NMR resonance of the P(III) atom is shifted downfield in comparison with the free ligand, whereas the position of the P(V) NMR signal is almost identical. This finding can most likely be attributed to an equilibrium of the POP-complex 4 with the PPO-complex 4′ in solution (Fig. 3). Similar observations, although to a much lesser extent, were made in the solid-state, since treatment of the reaction mixture of [Au(tht)Cl] and the PPO/POP ligand with n-pentane resulted in the formation of a mixture of single-crystals of 4 and a powder, consisting of 4 and 20% 4′, calculated on the basis of elemental analysis.
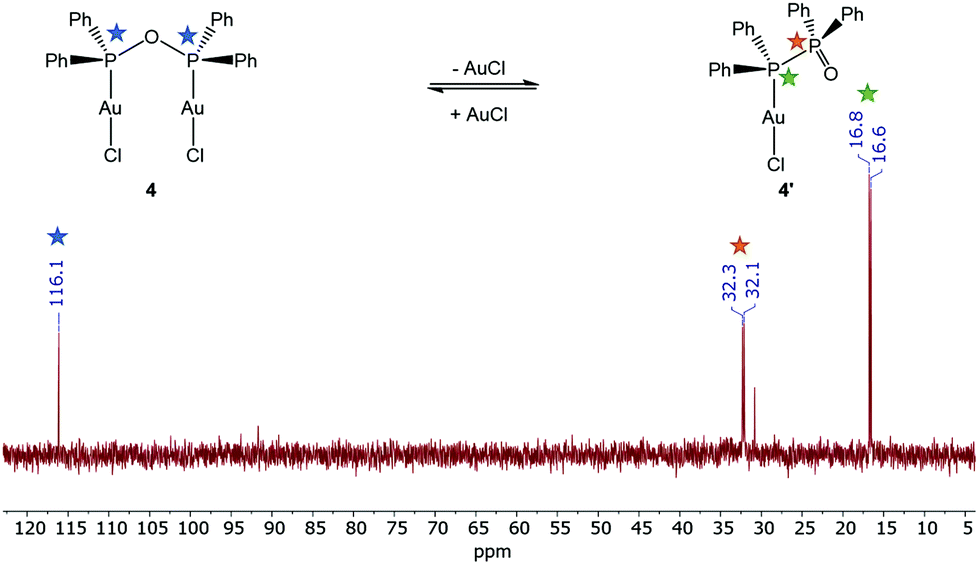 | ||
| Fig. 3 31P{1H} NMR spectrum of complex 4 in CDCl3, indicating the presence of the PPO-containing gold complex 4′. Note: A small amount of impurities (30.8 ppm) was observed, since 4 is readily hydrolysed. | ||
To verify the presence of the PPO/POP tautomers within the transition metal complexes 1–5 further, FT-IR measurements were carried out (Fig. 4). Looking at the IR spectrum of the pure ligand, the enhanced stability of the PPO over the POP form is confirmed, with a characteristic PO vibration at 1175 cm−1. This PO vibration is also found for the PPO-complexes 1 and 2 at 1139 cm−1 and 1134 cm−1, respectively. Due to the coordination to either the iron or the yttrium centres the PO vibrations are red-shifted in comparison with the free PPO ligand. In contrast, for the POP-type complexes 3 and 4 no significant PO vibrational bands in the region around 1200–1100 cm−1 are observed. This again points towards the fact, that within the isolated solid, mainly compound 4, instead of 4′, is present. Also, in the IR spectrum of the gold(I) complex 5 no PO vibrations can be assigned, since the broad band at 1150 cm−1 can be attributed to triflate vibrations instead. The POP vibrations of complexes 3, 4 and 5 can be found in the region of 830–920 cm−1, underlining the stabilisation of the anhydride POP form within the copper/gold complexes.
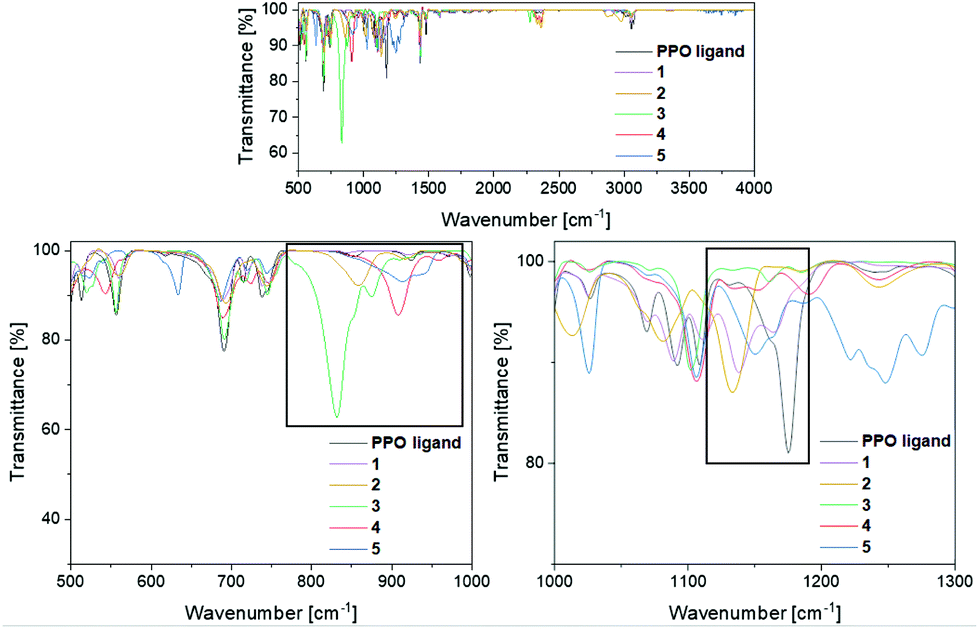 | ||
| Fig. 4 FT-IR spectra of the PPO ligand and the transition metal complexes 1–5 in the region of 500–3250 cm−1. Top: overview spectrum, bottom: of the POP (left) and PPO (right)-relevant regions. | ||
2.3 Solid-state structures of the transition metal complexes
The solid-state structures of the new complexes [FeCl2(PPO)2] (1), [YCl3(THF)2(PPO)]·0.5THF (2·0.5THF), [Cu2(MeCN)4(μ2-POP)2](PF6)2 (3a), [Cu2(MeCN)3(μ2-POP)2](PF6)2 (3b), [Cu2(MeCN)2(μ2-POP)2](PF6)2 (3c), [Au2Cl2(μ2-POP)] (4) and [Au2(μ2-POP)2](OTf)2·2.3DCM (5·2.3DCM) were determined by single-crystal X-ray crystallography. A summary of the crystallographic details can be found in the ESI.†The structure of the iron(II) complex [FeCl2(PPO)2] (1) is shown in Fig. 5. Two monodentate PPO ligands coordinate the iron(II) centre via their oxygen atoms. Additionally, two chloride ligands are attached to the metal ion, resulting in a tetrahedral coordination environment. The Fe–O bond lengths are 2.006(3) Å and 2.017(2) Å whereas Fe–Cl bond lengths of 2.264(1) Å and 2.276(1) Å were observed. Similar bond lengths were found in the related iron(II) based phosphine oxide complex [FeCl2(OPMe3)2].48 The PO bond lengths were found to be 1.493(3) Å and 1.506(3) Å, which is slightly longer than the PO bond length in the free PPO ligand (1.389(3) Å).49 The reverse trend is observed for the P–P distances of 2.188(1) Å and 2.202(1) Å within 1, which is slightly shortened compared to 2.2128(6) Å in the free PPO ligand.49 To the best of our knowledge, beside [ZrCl4(PPO)2],45 complex 1 is only the second literature-reported transition metal complex, in which O-coordination of a PPO ligand occurs. Much more common is the coordination of the P(III) atom of the PPO ligand to other transition metals, such as in the case of Mo,25 Cu,32 Co33 and Fe.34,35
 | ||
| Fig. 5 Molecular structure of [FeCl2(PPO)2] (1). Co-crystallised solvent molecules atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Fe(1)–(O1) 2.017(2), Fe(1)–(O2) 2.006(3), Fe(1)–Cl(1) 2.264(1), Fe(1)–(Cl2) 2.276(1), P(1)–(O1)1.506(3), P(1)–P(2) 2.202(1), P(3)–(O2) 1.493(3), P(3)–P(4) 2.188(1), O(1)–Fe(1)–O(2) 97.9(1), Cl(1)–Fe(1)–Cl(2) 114.90(4), O(1)–P(1)–P(2) 117.3(1), O(2)–P(3)–P(4) 117.8(1). | ||
A related structure of the yttrium(III) complex which crystallised in the form of [YCl3(THF)2(PPO)]·0.5THF (2·0.5THF) is shown in Fig. 6. The yttrium centre within this structure is coordinated by three chloride, two THF and one PPO ligand via its oxygen atom. This results in an octahedral coordination environment of the metal centre. Within the structure a very similar PO bond length of 1.502(4) Å and P–P bond lengths of 2.188(2) Å, in comparison with the iron(II) complex 1, have been observed.
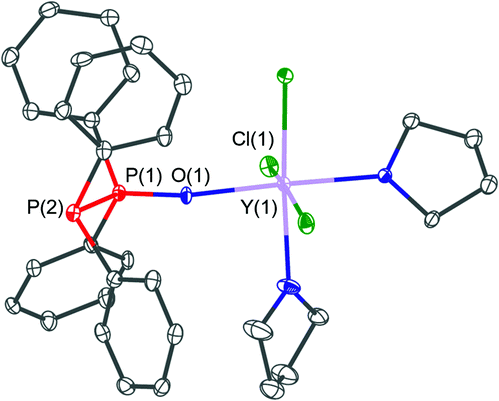 | ||
| Fig. 6 Molecular structure of [YCl3(THF)2(PPO)]·0.5THF (2·0.5THF). Selected bond lengths (Å) and angles (°): Y(1)–Cl(1) 2.591(2), Y(1)–Cl(2) 2.536(1), Y(1)–Cl(3) 2.600(2), Y(1)–O(1) 2.204(4), P(1)–O(1) 1.502(4), P(1)–P(2) 2.188(2), O(1)–P(1)–P(2) 116.3(2). | ||
As mentioned before, when softer transition metal ions were employed for the coordination studies, the anhydride POP form could be stabilised within the corresponding transition metal complexes. From the reaction of [Cu(MeCN)4]PF6 with the PPO/POP ligand, a bimetallic copper complex could be obtained, in which two bridging POP ligands coordinate two copper centres. Single-crystal X-ray crystallography revealed the 1:1:3 statistical distribution of three different copper complexes, namely [Cu2(MeCN)4(μ2-POP)2](PF6)2 (3a), [Cu2(MeCN)2(μ2-POP)2](PF6)2 (3b) and [Cu2(MeCN)3(μ2-POP)2](PF6)2 (3c), as shown in Fig. 7. The main difference lies in the number of MeCN ligands coordinated to the copper centres, varying between two and one for each Cu(I) ion. The number of coordinated MeCN molecules clearly has an impact on the distance between the two copper centres. Whereas in complex 3a and 3c the metal–metal distances are 3.371(3) Å and 2.8965(24) Å and thus lie above the sum of the van der Waals radii of 2.80 Å, a short copper–copper distance of 2.4408(23) Å is present in complex 3b. The latter could be termed as intramolecular and fully ligand-supported cuprophillic interaction. However, it has to be mentioned at this stage, that despite the fact, that d10–d10 interactions in gold(I) complexes are well-established,50 the existence of weak cuprophillic interactions is still controversial.51,52 Within the structures 3a, 3b and 3c the Cu–P bond lengths were found to be in the range of 2.1987(21) Å–2.2671(21) Å whereas P–O bond lengths of 1.6173(27) Å–1.6408(24) Å were observed. Within the core of all structures of 3, an 8-membered copper-ligand ring is formed, similar to dinuclear copper(I) complexes with bridging diphosphine ligands.53–55
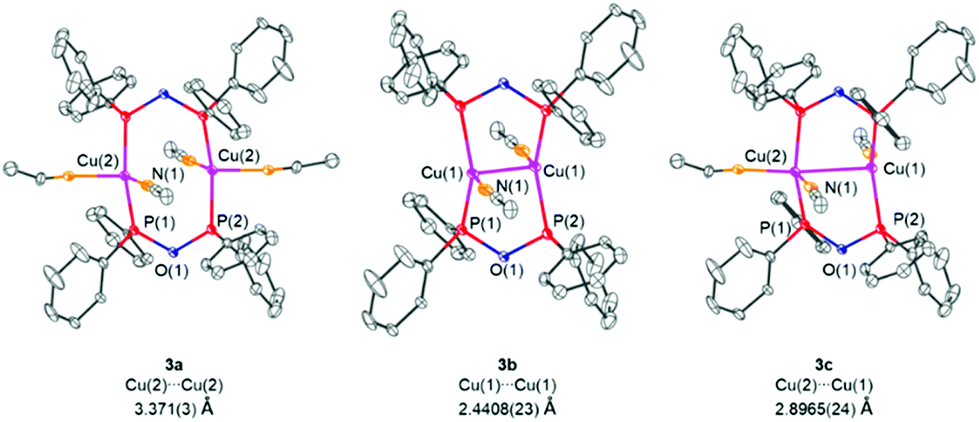 | ||
| Fig. 7 Molecular structures of the cations of [Cu2(MeCN)4(μ2-POP)2]2+ (3a), [Cu2(MeCN)2(μ2-POP)2]2+ (3b) and [Cu2(MeCN)3(μ2-POP)2]2+ (3c). The PF6− anions are omitted for clarity. Selected bond lengths (Å) and angles (°) for 3a: Cu(2)–P(1) 2.2449(21), Cu(1)–P(2) 2.1987(21), Cu(2)–Cu(2) 3.371(3), P(1)–O(1) 1.6408(24), P(2)–O(1) 1.6173(27), P(1)–O(1)–P(2) 119.85(12), P(1)–Cu(2)–P(2) 139.97(8). Selected bond lengths (Å) and angles (°) for 3b: Cu(1)–P(1) 2.2272(21), Cu(1)–P(2) 2.2671(21), Cu(1)–Cu(1)2.4408(23), P(1)–O(1) 1.6408(24), P(2)–O(1) 1.6173(27), P(1)–O(1)–P(2) 119.85(12), P(1)–Cu(1)–P(2) 136.55(7). Selected bond lengths (Å) and angles (°) for 3c: Cu(1)–P(1) 2.2271(21), Cu(1)–P(2) 2.2672(21), Cu(2)–P(1) 2.2449(21), Cu(1)–P(2) 2.1987(21), P(1)–O(1) 1.6408(24), P(2)–O(1) 1.6173(27), P(1)–Cu(1)–P(2) 136.55(7), P(1)–Cu(2)–P(2) 139.97(8). Comment about the X-ray structure: the complex crystallized on an inversion centre. The second acetonitrile molecule (N2) must therefore have an occupancy of 50% because of its vicinity to the inversion centre, which does not allow a full occupancy. An additional disorder related to the former is observed for the Cu atom, which splits to two positions corresponding to two different coordination environments (one for a coordination by three donor atoms and one for a coordination of four donor atoms). Because the Cu site with four, instead of three, donor atoms is caused by the coordination of the second acetonitrile (N2), and thus its occurrence is linked to the presence of the latter, the occupancies of this Cu position and in turn also the one of the second one were fixed to 50%. | ||
The solid-state structures of the gold(I)–POP complexes 4 and 5 are shown in Fig. 8. In both cases the POP form of the PPO/POP molecule is present, whereby the ligand acts as bridging ligand to two gold centres. When [Au(tht)Cl] was employed (complex 4), the two gold atoms are coordinated by one bridging POP ligand and two chloride ligands, therefore attaining a linear geometry, which is the most typical gold(I) coordination geometry.56 The P–O bond distances were found to be 1.615(3) Å and 1.616(3) Å, the P–O–P bond angle amounts 125.3(2) Å and the Au–P bond distances are 2.204(1) Å and 2.208(1) Å, respectively. Through the bridging mode of the POP ligand, the gold centres come in close contact of 3.2055(5) Å, which is well below the sum of two van der Waals radii (3.80 Å). In the literature such type of interaction, in a structural arrangement similar to 4 have been referred to as semi-supported aurophilic interactions.50 A similar complex with an Au–Au distance of 3.1816(3) Å, namely [Au2Cl2(μ2-Et2POPEt2)] has been synthesised unintentionally from a diethylphosphine oxide and [Au(tht)Cl] in the context of nanoparticle stabilisation approaches.11 When instead of [Au(tht)Cl], the triflate analogue [Au(tht)OTf] was employed as metal precursor, the dimeric complex [Au2(μ2-POP)2](OTf)2·2.3DCM (5·2.3DCM) could be crystallised. In this complex two POP ligands bridge two gold centres with P–O bond distances of 1.626(4) Å–1.629(4) Å and Au–P bond distances of 2.304(2) Å–2.313(2) Å. The structural motive of 5·2.3DCM is very similar to the one observed within the copper(I) complex 3. However, due to the additional MeCN ligands attached to the copper(I) centres in 3, the P–Cu–P bond angles amount 136.55(7)°–139.97(8)°, whereas P–Au–P bond angles of 177.52(6)° and 179.00(6)° were found for 5·2.3DCM. Again, as seen before in 4, short Au–Au distances of 2.9302(7) Å have been observed in complex 5·2.3DCM, which are commonly referred to as fully-supported aurophilic interactions in this case.50 The solid-state structure of [AuCl(PPO)] (4′) could not be determined via single-crystal X-ray crystallography, despite several attempts.
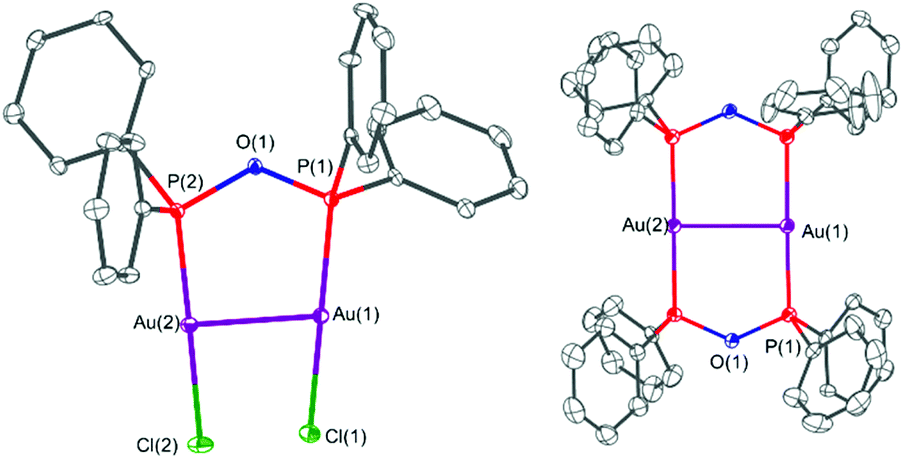 | ||
| Fig. 8 (Left) molecular tructure of [Au2Cl2(μ2-POP)] (4) and (right) molecular structure of the cation of [Au2(μ2-POP)2]2+ of 5·2.3DCM. Triflate anions and DCM solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (°) for 4: Au(1)–P(1) 2.204(1), Au(2)–P(2) 2.208(1), Au(1)–Cl(1) 2.275(1), Au(2)–Cl(2) 2.287(1), Au(1)–Au(2) 3.2055(5), P(1)–O(1) 1.615(3), P(2)–O(1) 1.616(3), P(1)–O(1)–P(2) 125.3(2). Selected bond lengths (Å) and angles (°) for 5·2.3DCM: Au(1)–P(1) 2.312(1), Au(1)–P(2) 2.313(2), Au(2)–P(3) 2.309(1), Au(2)–P(4) 2.304(2), Au(1)–Au(2) 2.9302(7), P(1)–O(1) 1.626(4), P(2)–O(2) 1.627(4), P(3)–O(1) 1.626(4), P(4)–O(2) 1.629(4), P(1)–O(1)–P(3) 131.2(2), P(2)–O(2)–P(4) 131.7(2), P(1)–Au(1)–P(2) 179.00(6), P(3)–Au(3)–P(4) 177.52(6). | ||
2.4 Photo-luminescence properties
Despite the fact, that transition metal complexes of PNP-type ligands such as Ph2PNRPPh2 (R = Et,15 C6H5,16 2,6-Me2C6H3,16,17 C6H3N2S),18 and coordination complexes of related PNNP-type ligands, e.g. N,N′-bis[(2-diphenylphosphino)phenyl] formamidinate,57–60 have been widely investigated with respect to their photo-luminescence (PL) properties, the oxygen-based POP analogue has not been considered in this context.In fact, when investigation the PPO ligand and the complexes 1–5 in the solid-state under UV light radiation (Fig. 9), it becomes obvious that all samples, beside the iron(II) complex 1, display luminescent behaviour at 77 K. Particularly interesting in this regard is the thermochromism of the dinuclear copper complex 3. Whereas at 77 K a blueish luminescence is observed, it changes to orange when the sample is warmed up to room temperature. This blueshift upon cooling can be attributed to structural changes or to lattice contractions, which have the potential to enhance the M–M interaction.61–63 Whereas a bright luminescence is observed for almost all complexes at 77 K, at room temperature only the PPO ligand and the Y complex 2 are brightly luminescent.
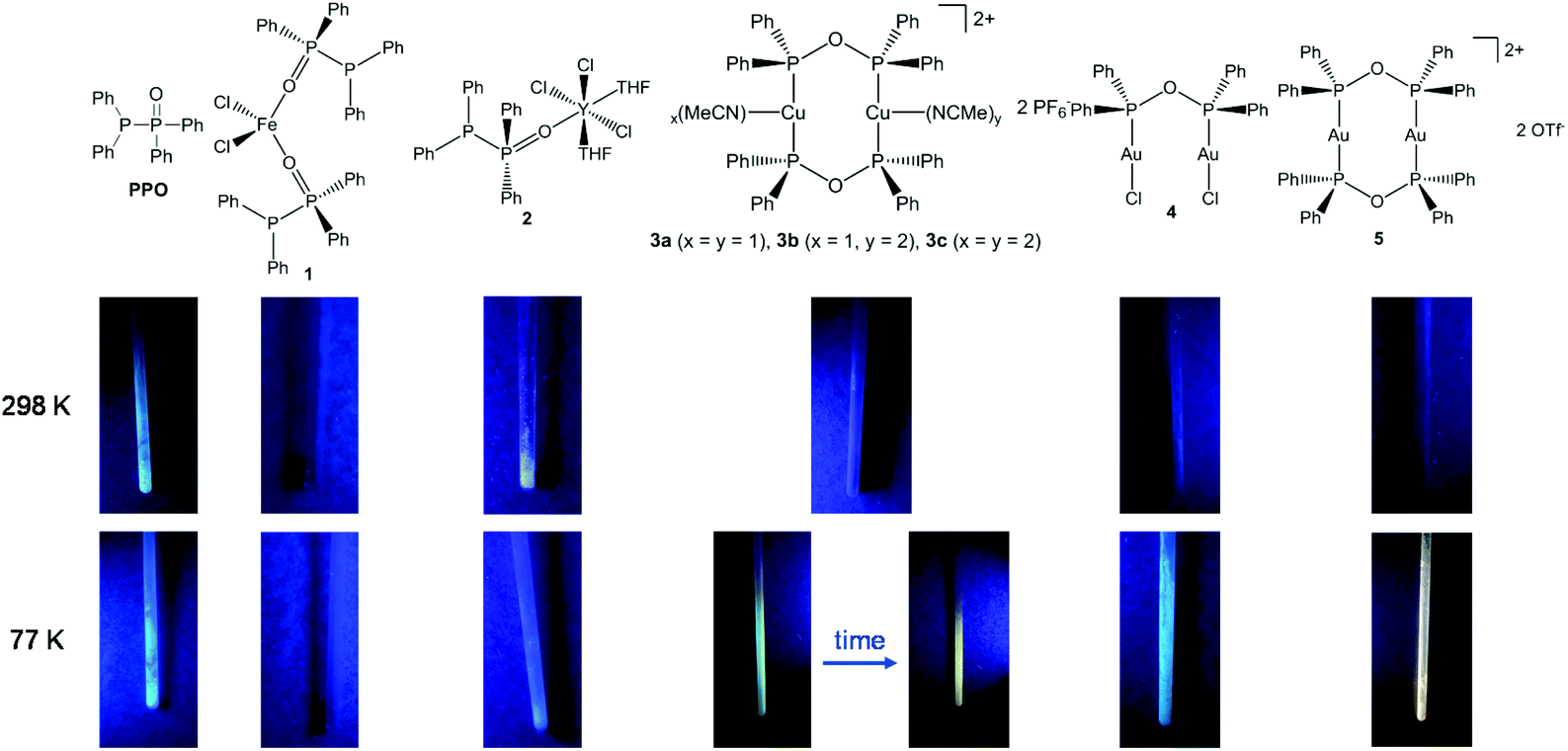 | ||
| Fig. 9 Images of the PPO ligand and the complexes 1–5 under UV light radiation (365 nm) at room temperature and under liquid N2 cooling. | ||
To examine the photo-optical properties in more detail, the PPO- and POP-type transition metal complexes 2–5 were investigated in the solid-state at RT and 77 K using photo-luminescence spectroscopy. The maximum excitation and emission wavelengths as well as the lifetimes, using a pulsed PTI XenonFlash™, were determined and are summarised in Table 1.
| T [K] | λ ex [nm] | λ em [nm] | Life-time [μs] | Metal–metal [Å] | |
|---|---|---|---|---|---|
| PPO ligand | 77 | 367 | 456 | 5.7 | — |
| 298 | 367 | 456 | 5.9 | ||
| 2 | 77 | 352 | 414 | 5.8 | — |
| 298 | 352 | 414 | 5.9 | ||
| 3 | 77 | 373 | 483 | 215.3, 66.7 | 2.4408(23)–3.371(3) |
| 298 | 400 | 440 | 4.6 | ||
| 4 | 77 | 361 | 499 | 7.2 | 3.2055(5) |
| 298 | 387 | 470 | 5.5 | ||
| 5 | 77 | 350 | 445 | 6.8 | 2.9302(7) |
| 298 | 364 | 452 | 5.9 |
The PL excitation (PLE) and emission spectra obtained at 77 K are shown in Fig. 10 (further details about measurements at room temperature can be found in the ESI†). All investigated solid compounds show broad emissions with a maximum at 456 nm (PPO ligand), 414 nm (2), 483 nm (3), 499 nm (4) and 445 nm (5), respectively. Interestingly, the binuclear coinage metal complexes 3 and 4 show similar PL spectra to each other with only slightly shifted emission maxima. This is not surprising, since d10 coordination compounds, which feature short metal–metal contacts or metallophillic interactions, have often shown similar photo-luminescence properties, often resulting from metal–ligand influenced states.53 For both complexes 3 and 4, a blue-shift of the emission maxima (3: 483 nm → 440 nm, 4: 499 nm → 470 nm) is observed when the samples are warmed from 77 K to 298 K. This can probably be explained by the contraction of the crystal lattices and consequently strengthening of the metallophillic interactions at low temperatures.64 In contrast, the position of the emission maxima of the PPO ligand and the monometallic complex 2 are temperature independent.
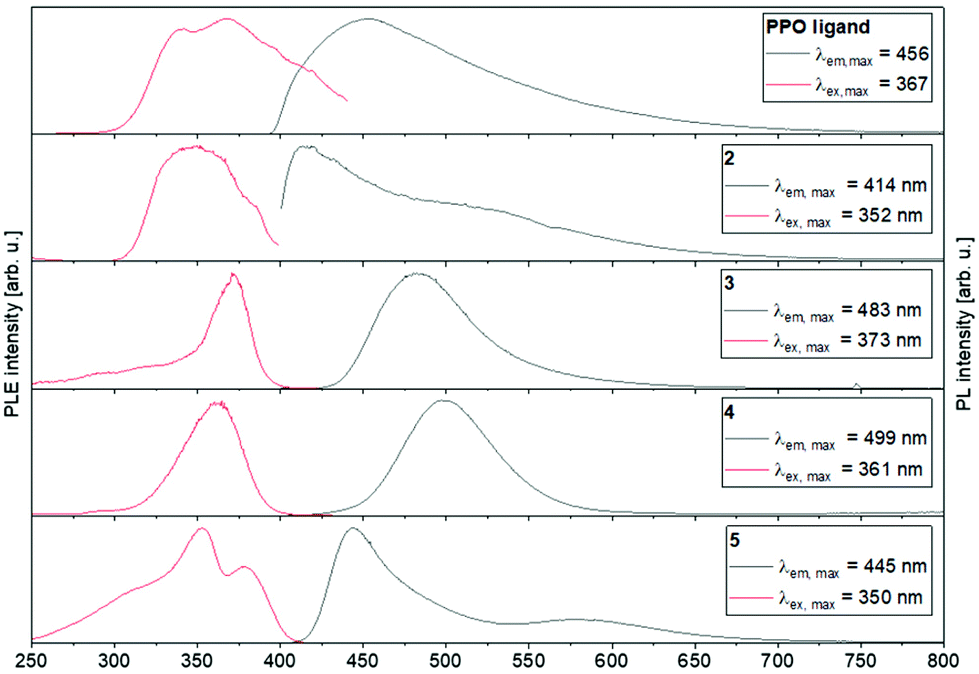 | ||
| Fig. 10 Photo-luminescence emission (PL) and excitation (PLE) spectra of the PPO ligand and complexes 2–5 at 77 K. | ||
The emission lifetimes for all investigated compounds are on the microsecond time scale, indicating that these emissions derive from triplet excited states. The longest lifetimes of 215.3 μs and 66.7 μs were determined for the biexponential decay of the copper complex 3, which is probably associated with nonradiative processes.65 Compared to other binuclear copper(I) complexes, such as bis(dicyclo-hexylphosphino)methane-,66 diphenylphosphino-pyridine-67,68 or pyridylphospholane-bridged69 copper(I) complexes, these decay times are remarkably long. In general, there is a clear decrease in phosphorescence lifetime when the temperature is increased from 77 K to ambient temperature, which also correlated with the strong decrease in PL intensity and low quantum yields through non-radiative processes70 (further information in the ESI†).
3 Conclusions
In conclusion, we report a systematic study about the influence of transition metals on the PPO/POP equilibrium depending on the hard/soft character of the transition metal involved. Whereas hard metal ions, such as Fe(II) and Y(III), tend to form PPO-type complexes 1 and 2, coinage metal ions, such as Cu(I) and Au(I), have the potential to stabilise the POP form in the coordination-stabilised tautomers 3, 4 and 5. Inspired by the extensive exploration of the photo-optical properties of PNP ligand-based transition metal complexes, the PPO/POP-complexes were investigated experimentally using photo-luminescence spectroscopy. All of the complexes, beside the iron(II) compound 1, show intense luminescence in the solid-state at 77 K. Especially interesting is the thermochromic behaviour of the copper(I) complex 3, which shows a blueshift of the luminescence upon cooling. The equilibrium tuning of the PPO/POP ligand and the resulting transition metal complexes, including metallophillic interactions, open up a new path to interesting photo-optical materials, such as luminescent molecular thermometers, which we are currently investigating.4 Experimental section
4.1 Materials, methods and instruments
All experiments were carried out on a Schlenk-line under Ar atmosphere or in an Ar-filled glove box (MBraun) and all reactions involving gold(I) precursors were carried out in the absence of light. Toluene, tetrahydrofuran (THF), n-pentane and n-heptane were dried using a MBraun solvent purification system (SPS-800) and degassed. THF was additionally dried over potassium/benzophenone. Acetonitrile (MeCN), dichloromethane (DCM) and triethyl amine were distilled over calcium hydride. CDCl3 and CD2Cl2 were dried over P2O5 while C6D6, D8-toluene and CD3CN were dried over CaH2. Deuterated solvents were degassed by freeze–pump–thaw cycles prior to use. All solvents were stored over activated molecular sieves (MeCN over 3 Å, all other solvents over 4 Å). Chlorodiphenyl phosphine was purified by distillation. All other chemicals were used without further purification.NMR spectra were recorded on a Bruker AVANCE III 400 MHz spectrometer at 298 K. Chemical shifts are given in ppm and are referenced on residual solvent signals of the deuterated solvents. Unambiguous assignments were determined on the basis of chemical shifts, coupling patterns and 2D NMR experiments (1H–1H COSY, 1H–13C HMQC, and 1H–13C HMBC). The multiplicity of the NMR signals is denoted as s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, sep = septet, m = multiplet, and br = broad.
IR spectra were recorded in the region of 4000–400 cm−1 on a Bruker Tensor 37 FTIR spectrometer equipped with a room temperature DLaTGS detector, a diamond ATR (attenuated total reflection) unit and a nitrogen-flushed measurement chamber. IR signals were classified according to their intensities (vs = very strong, s = strong, m = medium, w = weak).
Elemental analyses were carried out with an Elementar Vario MICRO Cube device.
Single crystal X-ray diffraction (SC-XRD) data were collected with two different setups. Data for compound 5 were measured on a STOE IPDS II diffractometer (2-circle) equipped with a STOE Imaging Plate (34 cm diameter) at a temperature of 150 K using a sealed X-Ray tube (Mo-Kα, λ = 0.71073 Å) with a graphite monochromator as radiation source. Data for compounds 1, 2, 3 and 4 were measured on a STOE STADIVARI diffractometer equipped with an open Eulerian cradle (4-circle) and a DECTRIS PILATUS pixel detector at a temperature of 100 K using a microfocus molybdenum source (Mo-Kα, λ = 0.71073 Å) with a graphite monochromator as radiation source. The data reduction was carried out with X-Area71 version 1.73.1.0 (STOE, 2018) using the semi-empirical absorption correction by X-RED71 with scaling of the reflection intensities by LANA included in X-Area. All structures were solved by dual space methods with SHELXT 201572 and refined with SHELXL 201873 using the WinGX program suite.74 Structure refinement was done using full-matrix least-square routines against F2. All hydrogen atoms were calculated on idealised positions. Pictures were generated with Diamond.75 In all graphical representations thermal ellipsoids are shown with 30% probability and hydrogen atoms are omitted for clarity. CCDC 2163970 (1), 2163971 (2·0.5THF), 2163972 (4), 2163973 (3), and 2163974 (5·2.3DCM)† contain the supplementary crystallographic data for this paper and can be accessed under https://www.ccdc.cam.ac.uk/structures/
Photoluminescence measurements were conducted on a PTI QuantaMaster™ 8075-22 spectrometer, equipped with double monochromators (HORIBA Jobin Yvon GmbH) for emission and excitation spectra. Samples were dried and measured in solid form at 77 K by cooling with liquid nitrogen and at 298 K. For this, samples were placed into EPR Suprasil® tubes. Emission spectra were detected using a R928 photomultiplier (HORIBA Jobin Yvon GmbH) and a DSS-IGA020L/CUS detector for the IR area which was cooled with liquid nitrogen. Lifetime measurements of the luminescence were investigated with a PTI XenonFlash™. PL quantum yields at ambient temperature were determined using an integrating sphere out of optical PTFE, which was installed into the sample chamber of the spectrometer, according to the method of Friend et al.76 The uncertainty of these measurements was estimated to be ca. 10%.
Theoretical calculations were carried out with the ORCA package (version 4.0.0.1) in the gas phase.77,78 For all ORCA calculations, atom-pairwise dispersion corrections with the Becke–Johnson damping scheme (D3BJ)79,80 and the RIJCOSX81 approximation was used. Geometry optimisations and frequency analysis, to prove the absence of imaginary frequencies, were obtained employing the B3LYP82,83 functional in conjunction with the def2-TZVP84,85 basis set. Further, single point calculations on a higher level of theory, employing the B3LYP82,83 functional and the def2-QZVP84,85 basis set, were used to calculate the PPO/POP equilibrium.
4.2 Synthesis of PPO/POP
The PPO/POP ligand was synthesised according to a modified literature procedure.86 For this chlorodiphenylphosphine (0.9 mL, 1.12 g, 5 mmol) was added dropwise to a solution of diphenylphosphine oxide (1.01 g, 5 mmol) and triethyl amine (0.8 mL, 0.58 g, 5.7 mmol) in toluene (50 mL) at 0–5 °C. After 3 hours the solution was filtered to remove residual triethylammonium chloride. Colourless crystals could be grown from a saturated toluene solution at −18 °C. The crystals were washed with n-pentane and dried in vacuo. Crystalline yield: 1.20 g, 3.10 mmol, 62%. 1H NMR (298 K, CDCl3, 400 MHz), δ [ppm] = 7.83–7.72 (4H, m, P(V)–Ph–H-ortho), 7.64–7.54 (4H, m, P(III)–Ph–H-ortho), 7.47–7.40 (2H, m, P(V)–Ph–H-para), 7.41–7.34 (4H, m, P(V)–Ph–H-meta), 7.34–7.28 (2 H, m, P(III)–Ph–H-para), 7.28–7.21 (4H, m, P(III)–Ph–H-meta). 31P{1H} NMR (298 K, CDCl3, 161 MHz), δ [ppm] = 35.8 (P(V), d, 1JPP = 227.7 Hz), −22.5 (P(III), d, 1JPP = 227.7 Hz). 13C NMR (298 K, CDCl3, 101 MHz), δ [ppm] = 135.7–135.3 (m, P(III)–Ph–C-ortho), 134.6–133.3 (m, P(V)–Ph–C-ipso), 131.8–131.1 (m, P(V)–Ph–C-ortho, para), 130.3–130.0 (m, P(III)–Ph–C-ipso), 129.8 (s, P(III)–Ph–C-para), 128.8–128.4 (m, P(V)/P(III)–Ph–C-meta). Elemental analysis, calcd for PPO/POP·2Toluene C 79.98, H 6.36; found C 79.90, H 5.20. ATR-IR ṽ [cm−1] = 1958 (w), 1888 (w), 1811 (w), 1675 (w), 1583 (w), 1479 (m), 1433 (s), 1329 (w), 1305 (w), 1245 (w), 1175 (vs), 1109 (m), 1091 (m), 1071 (m), 1029 (w), 996 (w), 922 (w), 854 (w), 736 (m), 715 (m), 692 (vs), 557 (s), 513 (m), 496 (s), 460 (m), 432 (m), 375 (m).4.3 Synthesis of [FeCl2(PPO)2] (1)
FeCl2 (8.3 mg, 0.07 mmol) and PPO/POP (54.9 mg, 0.14 mmol) were reacted in toluene (10 mL) for 3 days at room temperature. The resulting yellow solution was filtered and pale-yellow crystals were obtained by slow diffusion of n-pentane into the toluene solution. Crystalline yield: 23.6 mg, 0.03 mmol, 37%. Due to the paramagnetic iron(II) centre NMR data could not be obtained. Elemental analysis, calcd for [FeCl2(PPO)2] C 64.10, H 4.48; found C 64.24, H 4.26. ATR-IR ṽ [cm−1] = 3051 (m), 2914 (w), 2849 (w), 1958 (w), 1888 (w), 1811 (w), 1675 (w), 1583 (w) 1479 (m), 1433 (vs), 1333 (w), 1305 (w), 1169 (m), 1137 (vs), 1108 (m), 1086 (s), 1068 (m), 1026 (m), 996 (m), 920 (w), 843 (w), 736 (s), 719 (m), 689 (vs), 561 (m), 500 (m), 496 (s), 460 (m), 432 (m), 389 (m).4.4 Synthesis of [YCl3(THF)2(PPO)]·0.5THF (2)
YCl3 (30.0 mg, 0.15 mmol) and PPO/POP (59.4 mg, 0.15 mmol) were dissolved in THF (10 mL) and stirred for 3 hours. The colourless solution was filtered and after layering with n-pentane colourless crystals suitable for single-crystal X-ray diffraction analysis could be grown. Crystalline yield: 20 mg, 0.03 mmol, 16%. 1H NMR (298 K, D8-THF, 400 MHz), δ [ppm] = 8.23–8.04 (4H, br, P(V)–Ph–H-ortho), 7.93–7.74 (4H, br, P(III)–Ph–H-ortho), 7.53–7.44 (2H, br, P(V)–Ph–H-para), 7.44–7.33 (4 H, br, P(V)–Ph–H-meta), 7.30–7.24 (6 H, br, P(III)–Ph–H-meta, para). 31P{1H} NMR (298 K, D8-THF, 162 MHz), δ [ppm] = 51.8–44.1 (P(V), m), −19.9 (P(III), d, 1JPP = 271.7 Hz). 13C NMR (298 K, CDCl3, 101 MHz), δ [ppm] = 136.2–135.9 (m, P(III)–Ph–C-ortho), 134.9–134.2 (br, P(V)–Ph–C-ipso), 133.3–133.0 (br, P(V)–Ph–C-para), 132.6–131.9 (br, P(V)–Ph–C-ortho), 131.1–130.7 (m, P(III)–Ph–C-para), 130.6–130.1 (br, P(III)–Ph–C-ipso), 129.7–129.2 (m, P(III)–Ph–C-meta), 129.2–128.8 (m, P(V)–Ph–C-meta). Notes: 89Y NMR could not be obtained and the solvent residual peaks of THF overlap with the signals of the coordinated THF molecules. Elemental analysis, calcd for [YCl3(THF)2(PPO)]·0.5THF C 53.60, H 5.29; found C 53.67 H 5.17. ATR-IR ṽ [cm−1] = 3051 (w), 2979 (m), 2871 (w), 1621 (vw), 1585 (vw), 1481 (m), 1432 (s), 1307 (vw), 1245 (w), 1134 (vs), 1083 (s), 1010 (s), 918 (w), 860 (s), 740 (s), 690 (vs), 561 (m), 499 (s), 438 (w), 401 (w).4.5 Synthesis of [Cu2(MeCN)3(μ2-POP)2](PF6)2 (3)
[Cu(MeCN)4]PF6 (100.0 mg, 0.27 mmol) and PPO/POP (103.7 mg, 0.27 mmol) were reacted in MeCN (10 mL) for 3 hours. The colourless solution was filtered and MeCN was removed in vacuo. The solid was re-dissolved in DCM and layered with n-heptane to form pale yellow crystals suitable for single-crystal X-ray diffraction analysis. Crystalline yield: 100.0 mg, 0.10 mmol, 38%. 1H NMR (298 K, CD2Cl2, 400 MHz), δ [ppm] = 7.58–7.52 (8H, m, Ph–H-para), 7.43–7.38 (32H, m, Ph–H-meta, ortho), 2.00 (12H, s, MeCN). 31P{1H} NMR (298 K, CD2Cl2, 162 MHz), δ [ppm] = 106.1 (s, POP), −144.5 (sep, 1JPF = 707.8 Hz, PF6). 13C NMR (298 K, CD2Cl2, 101 MHz), δ [ppm] = 135.3–134.7 (m, Ph–C-ortho, meta), 132.9–132.6 (m, Ph–C-para), 131.8–131.3 (m, Ph–C-ortho, meta), 129.8–129.5 (m, Ph–C-ortho, meta, para), 128.7–127.6 (m, Ph–C-ipso), 118.7 (s, MeCN), 2.3 (s, MeCN). 19F NMR (298 K, CD2Cl2, 377 MHz), δ [ppm] = −72.9 (d, 1JPF = 707.8 Hz, PF6−). Elemental analysis, calcd for [Cu2(MeCN)3(μ2-POP)2](PF6)2 C 49.40 H 3.76 N 3.20; found C 50.36 H 3.40 N 3.41. ATR-IR ṽ [cm−1] = 2943 (w), 1958 (vw), 1888 (vw), 1811 (vw), 1675 (vw), 1587 (w), 1585 (w), 1480 (w), 1437 (m), 1400 (w), 1366 (w), 1329 (w), 1311 (w), 1184 (w), 1157 (w), 1103 (m), 1071 (w), 1029 (w), 996 (w), 922 (w), 874 (m), 833 (vs), 745 (m), 718 (m), 692 (s), 557 (s), 520 (m), 477 (m), 420 (w).4.5 Synthesis of [Au2Cl2(μ2-POP)] (4) and [AuCl(PPO)] (4′)
[Au(tht)Cl] (40.0 mg, 0.12 mmol) and PPO/POP (24.1 mg, 0.06 mmol) in DCM (10 mL) were stirred for 3 hours. The colourless solution was filtered off and layered with n-pentane providing colourless crystals together with a white powder. Crystalline yield: 40.0 mg, 0.05 mmol, 50%. 1H NMR (298 K, CDCl3, 400 MHz), δ [ppm] = 7.96–7.87 (8H, m, POP–Ph–H-meta), 7.76–7.65 (8H, br, PPO–P(V)/P(III)–Ph–H-ortho), 7.63–7.52 (6 H, br, PPO–P(V)–Ph–H-meta, para), 7.52–7.48 (6 H, br, PPO–P(III)–Ph–H-meta, para), 7.48–7.44 (4H, m, POP–Ph–H-para), 7.44–7.38 (8H, m, POP–Ph–H-ortho). 31P{1H} NMR (298 K, CDCl3, 162 MHz), δ [ppm] = 116.1 (s, POP), 32.2 (P(V)–PPO, d, 1JPP = 33.0 Hz), 16.7 (P(III)–PPO, d, 1JPP = 33.0 Hz). 13C NMR (298 K, CDCl3, 101 MHz), δ [ppm] = 133.9 (m, Ph–C-para), 132.8–132.4 (m, Ph–C-meta), 129.8–129.5 (m, Ph–C-ortho), 129.5–129.1 (m, Ph–C-ipso). Elemental analysis calculated for 80% [Au2Cl2(μ2-POP)] and 20% [AuCl(PPO)] C 35.82 H 2.51; found C 35.45 H 2.75. ATR-IR ṽ [cm−1] = 1568 (w), 1476 (m), 1437 (s), 1404 (w), 1315 (m), 1184 (m), 1155 (m), 1130 (w), 1095 (s), 1069 (m), 1027 (w), 999 (m), 952 (w), 855 (w), 838 (w), 745 (s), 550 (w), 509 (m), 482 (m), 444 (m).4.6 Synthesis of [Au2(μ2-POP)2](OTf)2 (5)
A mixture of [Au(tht)OTf] (37.0 mg, 0.07 mmol) and PPO/POP (27.0 mg, 0.07 mmol) was stirred in DCM (10 mL) for 3 hours. The yellow solution was then filtered and layered with n-heptane resulting in colourless crystals suitable for single-crystal X-Ray diffraction analysis. Crystalline yield: 25.0 mg, 0.02 mmol, 24%. 1H NMR (298 K, CDCl3, 400 MHz), δ [ppm] = 7.81–7.73 (16H, m, Ph–H-meta), 7.61–7.55 (8H, m, Ph–H-para), 7.56–7.49 (16H, m, Ph–H-ortho). 31P{1H} NMR (298 K, CDCl3, 162 MHz), δ [ppm] = 132.0 (s). 13C NMR (298 K, CDCl3, 101 MHz), δ [ppm] = 134.4–134.3 (m, Ph–C-para), 133.3–132.9 (m, Ph–C-meta), 130.2–129.9 (m, Ph–C-ortho), 129.6–129.4 (m, Ph–C-ipso). 19F NMR (298 K, CD3CN, 377 MHz), δ [ppm] = −79.7 (s, OTf−). Elemental analysis calcd for [Au2(μ2-POP)2](OTf)2·n-heptane C 44.66 H 3.48; found C 44.96 H 2.79. ATR-IR ṽ [cm−1] = 2957 (w), 1684 (vw), 1653 (vw), 1587 (w), 1485 (w), 1480 (w), 1437 (s), 1393 (w), 1300 (m), 1278 (s), 1247 (s), 1220 (s), 1186 (m), 1148 (s), 1103 (s), 1026 (s), 998 (m), 934 (m), 911 (s), 745 (m), 718 (m), 687 (s), 633 (s), 572 (w), 524 (m), 498 (m), 426 (w).Author contributions
The manuscript was written through contributions of F.F. and S.H. T.G. solved and refined the singe-crystal X-ray data. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the Stiftung der Deutschen Wirtschaft for a doctoral scholarship for F. F. Also, we wish to thank the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster).References
-
Phosphorus(III) Ligands in Homogeneous Catalysis Design and Synthesis, ed. P. W. N. M. van Leeuwen and P. C. J. Kamer, John Wiley & Sons, 2012 Search PubMed
.
- J. C. Jeffrey and T. B. Rauchfuss, Inorg. Chem., 1979, 18, 2658–2666 CrossRef CAS
- A. M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer, P. W. N. M. Van Leeuwen, K. Goubitz and J. Fraanje, Organometallics, 1995, 14, 3081–3089 CrossRef
- C. Reviews, G. M. Adams and A. S. Weller, Coord. Chem. Rev., 2018, 355, 150–172 CrossRef
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC
- B. Hoge and B. Kurscheid, Angew. Chem., Int. Ed., 2008, 47, 6814–6816 CrossRef CAS PubMed
- B. Hoge, S. Neufeind, S. Hettel, W. Wiebe and C. Thösen, J. Organomet. Chem., 2005, 690, 2382–2387 CrossRef CAS
- P. W. N. M. van Leeuwen, I. Cano and Z. Freixa, ChemCatChem, 2020, 12, 3982–3994 CrossRef CAS
- J. Francos, D. Elorriaga, P. Crochet and V. Cadierno, Coord. Chem. Rev., 2019, 387, 199–234 CrossRef CAS
- N. Almora-Barrios, I. Cano, P. W. N. M. Van Leeuwen and N. López, ACS Catal., 2017, 7, 3949–3954 CrossRef CAS
- I. Cano, M. A. Huertos, A. M. Chapman, G. Buntkowsky, T. Gutmann, P. B. Groszewicz and P. W. N. M. Van Leeuwen, J. Am. Chem. Soc., 2015, 137, 7718–7727 CrossRef CAS PubMed
- E. Rafter, T. Gutmann, F. Löw, G. Buntkowsky, K. Philippot, B. Chaudret and P. W. N. M. Van Leeuwen, Catal. Sci. Technol., 2013, 3, 595–599 RSC
- G. R. M. Dowson, M. F. Haddow, J. Lee, R. L. Wingad and D. F. Wass, Angew. Chem., Int. Ed., 2013, 52, 9005–9008 CrossRef CAS PubMed
- A. Prades, M. Fernández, S. D. Pike, M. C. Willis and A. S. Weller, Angew. Chem., Int. Ed., 2015, 54, 8520–8524 CrossRef CAS PubMed
- J. S. Field, J. Grieve, R. J. Haines, N. May and M. M. Zulu, Polyhedron, 1998, 104, 3021–3029 CrossRef
- N. Kathewad, N. Kumar, R. Dasgupta, M. Ghosh, S. Pal and S. Khan, Dalton Trans., 2019, 48, 7274–7280 RSC
- N. Kathewad, S. Pal, R. L. Kumawat, Md. Ehesan Ali and S. Khan, Eur. J. Inorg. Chem., 2018, 2518–2523 CrossRef CAS
- T. S. Sukhikh, R. M. Khisamov, D. A. Bashirov, V. Y. Komarov, M. S. Molokeev, A. A. Ryadun, E. Benassi and S. N. Konchenko, Cryst. Growth Des., 2020, 20, 5796–5807 CrossRef CAS
- F. C. Bradley, E. H. Wong, E. J. Gabe, F. L. Lee and Y. Lepage, Polyhedron, 1987, 6, 1103–1110 CrossRef CAS
- E. H. Wong, F. C. Bradley, L. Prasad and E. J. Gabe, J. Organomet. Chem., 1984, 263, 167–177 CrossRef CAS
- G. M. Gray and C. S. Kraihanzel, J. Organomet. Chem., 1982, 238, 209–222 CrossRef CAS
- G. A. Acum, M. J. Mays, P. R. Raithby, H. R. Powell and G. A. Solan, J. Chem. Soc., Dalton Trans., 1997, 7, 3427–3434 RSC
- Y. Zhao, Y. Zhou, T. Chen, S. F. Yin and L. B. Han, Inorg. Chim. Acta, 2014, 422, 36–39 CrossRef CAS
- Y. Zhou, S. Yin, Y. Gao, Y. Zhao, M. Goto and L.-B. Han, Angew. Chem., Int. Ed., 2010, 49, 6852–6855 CrossRef CAS PubMed
- E. H. Wong, L. Prasad, E. J. Gabe and F. C. Bradley, J. Organomet. Chem., 1982, 321–331 Search PubMed
- A. Aloisi, J. C. Berthet, C. Genre, P. Thuéry and T. Cantat, Dalton Trans., 2016, 45, 14774–14788 RSC
- C. S. Kraihanzel and C. M. Bartish, J. Am. Chem. Soc., 1972, 94, 3572–3575 CrossRef CAS
- B. M. Kariuki and P. D. Newman, Inorg. Chem., 2018, 57, 9554–9563 CrossRef CAS PubMed
- A. D. Burrows, M. F. Mahon, M. T. Palmer and M. Varrone, Inorg. Chem., 2002, 41, 1695–1697 CrossRef CAS PubMed
- S. Pavlik, K. Mereiter, M. Puchberger and K. Kirchner, Organometallics, 2005, 24, 3561–3575 CrossRef CAS
- E. Piras, B. Powietzka, F. Wurst, D. Neumann-Walter, H. J. Grützmacher, T. Otto, T. Zevaco and O. Walter, Catal. Lett., 2013, 143, 673–680 CrossRef CAS
- K. Naktode, R. K. Kottalanka, H. Adimulam and T. K. Panda, J. Coord. Chem., 2014, 67, 3042–3053 CrossRef CAS
- S. G. Bott, J. C. Wang and M. G. Richmond, J. Chem. Crystallogr., 1999, 29, 603–608 CrossRef CAS
- L. C. Song, M. Cao, Z. Q. Du, Z. H. Feng, Z. Ma and H.-B. Song, Eur. J. Inorg. Chem., 2014, 1886–1895 CrossRef CAS
- X. F. Liu, Inorg. Chim. Acta, 2011, 378, 338–341 CrossRef CAS
- C. Zeiher, J. Mohyla, I.-P. Lorenz and W. Hiller, J. Organomet. Chem., 1985, 286, 159–170 CrossRef CAS
- D. J. Irvine, D. J. Cole-Hamilton, J. Barnes and P. K. Hodgson, Polyhedron, 1989, 8, 1575–1577 CrossRef CAS
- M. Wallesch, M. Nieger, D. Volz and S. Bräse, Inorg. Chem. Commun., 2017, 86, 232–240 CrossRef CAS
- T. Heurich, Z. W. Qu, G. Schnakenburg, Y. Nejatyjahromy, O. Schiemann, S. Grimme and R. Streubel, Organometallics, 2017, 36, 2877–2883 CrossRef CAS
- J. Coetzee, G. R. Eastham, A. M. Z. Slawin and D. J. Cole-Hamilton, Dalton Trans., 2014, 43, 3479–3491 RSC
- D. J. Irvine, C. Glidewell, D. J. Cole-Hamilton, J. C. Barnes and A. Howie, J. Chem. Soc., Dalton Trans., 1991, 1765–1772 RSC
- J. Bravo, J. Castro, S. García-Fontán, M. C. Rodríguez-Martínez and P. Rodríguez-Seoane, Eur. J. Inorg. Chem., 2006, 3, 3028–3040 CrossRef
- J. Simon, U. Bergsträsser, M. Regitz and K. K. Laali, Organometallics, 1999, 18, 817–819 CrossRef CAS
- A. Renz, M. Penney, R. Feazell and K. K. Klausmeyer, J. Chem. Crystallogr., 2012, 42, 1129–1137 CrossRef CAS
- T. Ogawa, Y. Kajita and H. Masuda, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1129–m1129 CrossRef CAS PubMed
- J. E. Griffiths and A. B. Burg, J. Am. Chem. Soc., 2002, 84, 3442–3450 CrossRef
- V. L. Foss, Y. A. Veits, V. A. Solodenko and I. F. Lutsenko, Zh. Obshch. Khim., 1976, 46, 46–47 Search PubMed
- F. A. Cotton, R. L. Luck and K. A. Son, Inorg. Chim. Acta, 1991, 184, 177–183 CrossRef CAS
- T. Posset, F. Rominger and J. Bümel, Chem. Mater., 2005, 17, 586–595 CrossRef CAS
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC
- N. V. S. Harisomayajula, S. Makovetskyi and Y. C. Tsai, Chem. – Eur. J., 2019, 25, 8936–8954 CrossRef CAS PubMed
- F. A. Cotton, X. Feng, M. Matusz and R. Poli, J. Am. Chem. Soc., 1988, 110, 7077–7083 CrossRef CAS
- C.-M. Che, Z. Mao, V. M. Miskowski, M.-C. Tse, C.-K. Chan, K.-K. Cheung, D. L. Phillips and K.-H. Leung, Angew. Chem., Int. Ed., 2000, 4084–4088 CrossRef CAS
- T. H. Huang, M. H. Zhang, M. L. Tao and X. J. Wang, Synth. React. Inorg. Met.-Org. Chem., 2014, 44, 986–990 CrossRef CAS
- M. I. Bruce, B. K. Nicholson, B. W. Skelton, A. H. White and N. N. Zaitseva, Inorg. Chim. Acta, 2016, 453, 647–653 CrossRef CAS
- P. Schwerdtfeger, H. L. Hermann and H. Schmidbaur, Inorg. Chem., 2003, 42, 1334–1342 CrossRef CAS PubMed
- M. Dahlen, N. Reinfandt, C. Jin, M. T. Gamer, K. Fink and P. W. Roesky, Chem. – Eur. J., 2021, 27, 15127–15135 CAS
- M. Dahlen, E. H. Hollesen, M. Kehry, M. T. Gamer, S. Lebedkin, D. Schooss, M. M. Kappes, W. Klopper and P. W. Roesky, Angew. Chem., Int. Ed., 2021, 60, 23365–23372 CrossRef CAS PubMed
- M. Dahlen, T. P. Seifert, S. Lebedkin, M. T. Gamer, M. M. Kappes and P. W. Roesky, Chem. Commun., 2021, 57, 13146–13149 RSC
- M. Dahlen, M. Kehry, S. Lebedkin, M. M. Kappes, W. Klopper and P. W. Roesky, Dalton Trans., 2021, 50, 13412–13420 RSC
- A. Burini, R. Bravi, J. P. Fackler, R. Galassi, T. A. Grant, M. A. Omary, B. R. Pietroni and R. J. Staples, Inorg. Chem., 2000, 39, 3158–3165 CrossRef CAS PubMed
- I. I. Vorontsov, A. Y. Kovalevsky, Y. S. Chen, T. Graber, M. Gembicky, I. V. Novozhilova, M. A. Omary and P. Coppens, Phys. Rev. Lett., 2005, 94, 193003–1–193003–4 CrossRef PubMed
- J. X. Zhang, J. He, Y. G. Yin, M. H. Hu, D. Li and X. C. Huang, Inorg. Chem., 2008, 47, 3471–3473 CrossRef CAS PubMed
- S. Pal, N. Kathewad, R. Pant and S. Khan, Inorg. Chem., 2015, 54, 10172–10183 CrossRef CAS PubMed
- A. V. Shamsieva, E. I. Musina, T. P. Gerasimova, R. R. Fayzullin, I. E. Kolesnikov, A. I. Samigullina, S. A. Katsyuba, A. A. Karasik and O. G. Sinyashin, Inorg. Chem., 2019, 58, 7698–7704 CrossRef PubMed
- W. F. Fu, X. Gan, C. M. Che, Q. Y. Cao, Z. Y. Zhou and N. N.-Y. Zhu, Chem. – Eur. J., 2004, 10, 2228–2236 CrossRef CAS PubMed
- D. M. Zink, M. Bächle, T. Baumann, M. Nieger, M. Kühn, C. Wang, W. Klopper, U. Monkowius, T. Hofbeck, H. Yersin and S. Bräse, Inorg. Chem., 2013, 52, 2292–2305 CrossRef CAS PubMed
- D. Volz, M. Nieger, J. Friedrichs, T. Baumann and S. Bräse, Langmuir, 2013, 29, 3034–3044 CrossRef CAS PubMed
- A. V. Shamsieva, E. I. Musina, T. P. Gerasimova, I. D. Strelnik, A. G. Strelnik, I. E. Kolesnikov, A. A. Kalinichev, D. R. Islamov, A. I. Samigullina, P. Lönnecke, S. A. Katsyuba, E. Hey-Hawkins, A. A. Karasik and O. G. Sinyashin, Dalton Trans., 2020, 49, 11997–12008 RSC
-
J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2006 Search PubMed
-
Stoe & Cie, X-RED, version 1.28b: Program for data reduction and absorption correction, Stoe &Cie. GmbH, Darmatadt, Germany, 2005 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS
-
H. Putz and K. Brandenburg, DIAMOND, Crystal Impact GbR, Bonn, Germany, 1999 Search PubMed
- J. C. de Mello, F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154104 CrossRef PubMed
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC
- S. Jin, G. C. Haug, V. T. Nguyen, C. Flores-Hansen, H. D. Arman and O. V. Larionov, ACS Catal., 2019, 9, 9764–9774 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Selected NMR spectra, details about single-crystal X-ray crystallography and photo-luminescence spectroscopy. CCDC 2163970 (1), 2163971 (2·0.5THF), 2163972 (4), 2163973 (3), 2163974 (5·2.3DCM). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt01091g |
| This journal is © The Royal Society of Chemistry 2022 |