
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silicon compounds in carbon-11 radiochemistry: present use and future perspectives
Federico
Luzi
 ,
Antony D.
Gee
* and
Salvatore
Bongarzone
*
,
Antony D.
Gee
* and
Salvatore
Bongarzone
*
School of Biomedical Engineering and Imaging Sciences, 4th floor Lambeth Wing, St Thomas’ Hospital, King's College London, London SE1 7EH, UK. E-mail: antony.gee@kcl.ac.uk; salvatore.bongarzone@kcl.ac.uk
First published on 20th July 2021
Abstract
Positron emission tomography (PET) is a powerful functional imaging technique that requires the use of positron emitting nuclides. Carbon-11 (11C) radionuclide has several advantages related to the ubiquity of carbon atoms in biomolecules and the conservation of pharmacological properties of the molecule upon isotopic exchange of carbon-12 with carbon-11. However, due to the short half-life of 11C (20.4 minutes) and the low scale with which it is produced by the cyclotron (sub-nanomolar concentrations), quick, robust and chemospecific radiolabelling strategies are required to minimise activity loss during incorporation of the 11C nuclide into the final product. To address some of the constraints of working with 11C, the use of silicon-based chemistry for 11C-labelling was proposed as a rapid and effective route for radiopharmaceutical production due to the broad applicability and high efficiency showed in organic chemistry. In the past years several organic chemistry methodologies have been successfully applied to 11C-chemistry. In this short review, we examine silicon-based 11C-chemistry, with a particular emphasis on the radiotracers that have been successfully produced and potential improvements to further expand the applicability of silicon in radiochemistry.
 Federico Luzi | Federico Luzi is currently pursuing a PhD degree in Medical Imaging at King's College London under the supervision of Prof. Antony Gee. His research is focused on the development of novel methodologies for carbon-11 labelling and the application to neurodegeneration. Federico previously obtained a Master's degree in Medicinal Chemistry and Pharmaceutical Technology from Alma Mater Studiorum, University of Bologna (Italy) and a MRes degree in Medical Imaging from King's College, London. |
 Antony D. Gee | Antony Gee is Professor of PET and Radiochemistry in the School of Biomedical Engineering and Imaging Sciences at King's College London and President of the Society of Radiopharmaceutical Sciences. He obtained a BSc (Hons) in Chemistry at the University of Sussex (1985), and his PhD in Radiopharmaceutical Organic Chemistry at Uppsala University, Sweden (1991). Since then he has worked as the Director of PET Chemistry at the Guy's and St Thomas' Hospitals Clinical PET centre, UMDS, London, the Aarhus University Hospital PET Centre in Aarhus Denmark, and SmithKlineBeecham/GlaxosmithKline, before returning to academia and current position at King's College in 2010. |
 Salvatore Bongarzone | Dr. Bongarzone attained his PhD at the International School for Advanced Studies (SISSA, Trieste, Italy, 2007). Subsequent postdoctoral positions were held at the Institute for Research in Biomedicine, Barcelona (IRB) and the School of Biomedical Engineering and Imaging Sciences at King's College London (KCL). At KCL, Dr. Bongarzone conceived novel radiochemical reactions for developing PET imaging probes ([11C]Niacin, [11C]Biotin, [18F]FAMTO, and [11C]FPSZM1) and their preclinical characterization. |
1. Introduction
Positron emission tomography (PET) is a powerful functional imaging technique that allows the in vivo detection of normo- and patho-physiological changes in humans by using molecules radiolabelled with positron (β+) emitting nuclides (radiotracers).1 To achieve radiopharmaceutical targeting, radiotracers are often derived from biologically-active compounds with a known pharmacological profile, possessing high selectivity for a molecular target or physiological process.2 The inclusion of a positron-emitting nuclide in the molecule of interest enables the in vivo visualization of the molecules biodistribution and kinetics metabolism.2,3 Of all the available PET nuclides, carbon-11 (11C) is of particular interest due to the ubiquity of carbon atoms in biomolecules and because isotopic substitution of carbon-12 for carbon-11 preserves the biological properties of the non-radioactive isotope.2,4,5 However, due to the rapid radioactive decay of carbon-11 (radioactive half-life t1/2 = 20.4 minutes), the radiosynthesis, purification, formulation and quality control of carbon-11 radiopharmaceuticals must be accomplished in short times (the whole process should not exceed 60 minutes), hence quick and robust chemistry are needed to avoid substantial activity loss.2,6 The sub-nanomolar scale with which the radioisotope is produced from the cyclotron also represents a burden when performing 11C-labelling, with the non-radioactive reactants being in large stoichiometric excess. Each minor impurity in the solvents and the reagents may generate side-products or degradation of reagents resulting in unwanted intermediates, so a high degree of chemospecificity is required, as well.2To meet criteria suitable for 11C-labelling, silicon-containing compounds have received increased interest in the field. Organosilicon compounds have already demonstrated wide applicability in traditional organic chemistry enabling the development of well-known methodologies such as the Hiyama cross-coupling reactions for the formation of carbon–carbon bonds between silylated compounds and aryl halides,7,8 tosylates,9 mesylates,10 sulfinates11 and phosphates12via palladium catalysis and fluoride or base activation.7–12 Moreover, silyl compounds act as effective protecting groups due to the large number of functional groups that can be protected (e.g. alcohols, alkynes, amines, carboxylic acids…) and the ease of the protecting/deprotecting steps.13,14 Another interesting application involves the use of hydrosilicon compounds, possessing one or more Si–H bonds, for the reduction of carbon dioxide (CO2) to more reactive species.15–18 In the hydrosilylation reaction, CO2 is used as a building block for the synthesis of a variety of functional groups such as formamides,15 methylamines,15,16 aldehydes17 and aminals.18 Moreover, organosilicates have shown to be optimal substrates for the electrophilic fluorination of aryl and alkenyl substrates under mild conditions (e.g. room temperature, 18 hours) whilst having regio- and enantio-selectivity on the final product.19 Besides the large number of reactions available, silicon chemistry is cost-effective (the silylated reagents are easily synthesized or commercially available)20 and eco-friendly (organosilicon compounds are ultimately catabolised into silica gel in the environment).7,8
Given the high versatility and ease of handling of organosilane compounds, several methodologies have been successfully translated into carbon-11 and fluorine-18 chemistry in the past years. In particular, silicon-based compounds were applied in the production of a variety of fluorine-18 labelled small molecules and peptides as prosthetic groups, where the radionuclide was attached on via isotopic exchange (obtaining silicon-fluoride-acceptors – SiFAs),21 or as substrates for electrophilic fluorination.19 The application of silicon in fluorine-18 chemistry was recently reviewed by Bernard-Gauthier et al. and Tredwell et al.19,21
Herein we report the latest advances in the 11C-chemistry field based on silicon starting materials or reagents (Scheme 1). This review will initially disclose the atomic properties of silicon to then discuss the main uses of organosilane compounds in carbon-11 chemistry: (i) conversion from [11C]CO2 to [11C]CO; (ii) trapping and activating agents for [11C]CO2 and (iii) precursors of the target radiopharmaceutical. This review will also report the biologically-active molecules that were successfully radiolabelled with the discussed reactions.
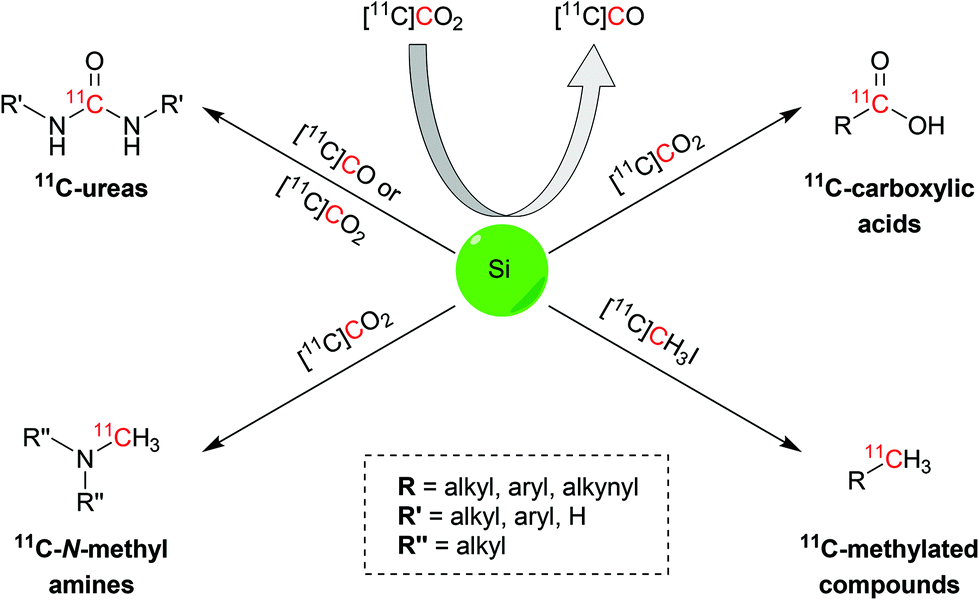 | ||
| Scheme 1 Schematic representation of the 11C functional groups and synthons obtainable via silicon chemistry. | ||
2. Characteristics and application of silicon in 11C-chemistry
2.1. Atomic properties of silicon
The versatility of silicon in organic chemistry is explainable by examining the strength of the bonds that this atom creates with other elements. The strength of Si–H and Si–C bonds (90 and 85 kcal mol−1, respectively) in organosilicates is indeed slightly lower than C–H and C–C bonds (104 and 88 kcal mol−1, respectively) in hydrocarbons, making silicon bonds more easily cleavable and silicates a favoured leaving group.22–25 The lower electronegativity of silicon would also generate a partial negative charge on carbon upon Si–C bond cleavage, favouring a higher reactivity on that carbon towards electrophiles.22–24 The higher electropositivity of silicon also favours the formation of hydride ions upon Si–H bond cleavage, making organosilicon compounds functional reducing agents.25 The high fluoro- and oxo-philicity of silicon also provokes the rearrangement or the cleavage of existing Si–C, Si–H and Si–Si bonds upon addition of fluorinated or oxygenated nucleophiles resulting in the formation of Si–F or Si–O bonds that enhances the nucleophilicity of the atom it was initially bound to, facilitating the reaction with electophiles.22–24 The involvement of d-orbitals further expands the applicability of silicon in radiochemistry, allowing the formation of penta- and hexa-coordinate compounds (e.g. the formation transition metal silylene complexes)26 which opens to more reaction pathways.26,272.2. Use of silyl compounds as [11C]CO2-to-[11C]CO converting agents
Carbon monoxide-releasing molecules (CORMs) have been exploited in the past years with either therapeutic or synthetic purposes.28–30 Within these compounds, silacarboxylic acids can produce CO upon exposure to high temperature or reaction with nucleophiles (e.g. fluoride anions, bases, water) following the 1,2-Brook rearrangement28–33 and can be readily synthesized by the reaction of the relative silyl lithium derivative with CO2.29,30 The produced CO was then employed in carbonylative coupling of aryl iodides and amines to yield amides, esters and α,β-unsaturated ketones.29,30Considering these advantageous characteristics, silacarboxylic acids were examined as potential [11C]CO2-to-[11C]CO converting agents as an alternative strategy to gas-phase reduction (reduction over thin molybdenum wires at 850 °C),34 photo-chemical (a ruthenium/cobalt solution irradiated by visible light)35 and electro-chemical methods (using transition-metal triflates as cathodes).36
Initial efforts focused on identifying the organosilyl chloride with the highest reactivity towards [11C]CO2. A variety of tri-substituted silanes were tested, with chloro(methyl)diphenylsilane (1) and chloro(tert-butyl)diphenylsilane (2) showing the overall best trapping efficiency (TE >95%) after reacting with metallic lithium to yield the corresponding lithium silane (3, Scheme 2). The reaction with [11C]CO2 forms the relative 11C-silacarboxylate ([11C]4) which could be protonated by adding HCl to obtain ([11C]5). Both [11C]4 and [11C]5 are able to release [11C]CO after tetrabutylammonium fluoride (TBAF) addition.37–39 A quantitative release of [11C]CO was achieved by adjusting reaction variables such as the amount of the starting chlorosilane, the temperature, equivalents of TBAF, the reaction time, the solvent.37–39 The [11C]CO produced with these optimised conditions was then employed in the palladium-catalysed 11C-carbonylation of several molecules of interest (Scheme 2), including the selective AMPA ligand [11C]CX546, the monoamine oxidase A (MAO-A) inhibitor [11C]moclobemide, the dopamine D2 selective antagonist [11C]raclopride, [11C]olaparib for cancer imaging and the receptor for advanced glycation end-products (RAGE) radiotracer [11C]FPS-ZM1 in radiochemical yields (RCYs) ranging between 29% and 91% (estimated by radioHPLC).37–39 Follow-up studies on the 11C-silacarboxylate production of [11C]CO and subsequent 11C-carbonylation showed that the whole process can be fully automated using a commercially available radiochemistry synthesis module.38 With an initial delivery of 10 GBq of [11C]CO2, the radiotracer [11C]FPS-ZM1 (Scheme 2) was achieved with a RCY of 34% (isolated, decay-corrected to end of [11C]CO2 delivery (EOD)) and molar activity (Am) 28 GBq per μmol within 25 minutes from end of bombardment (EOB).38
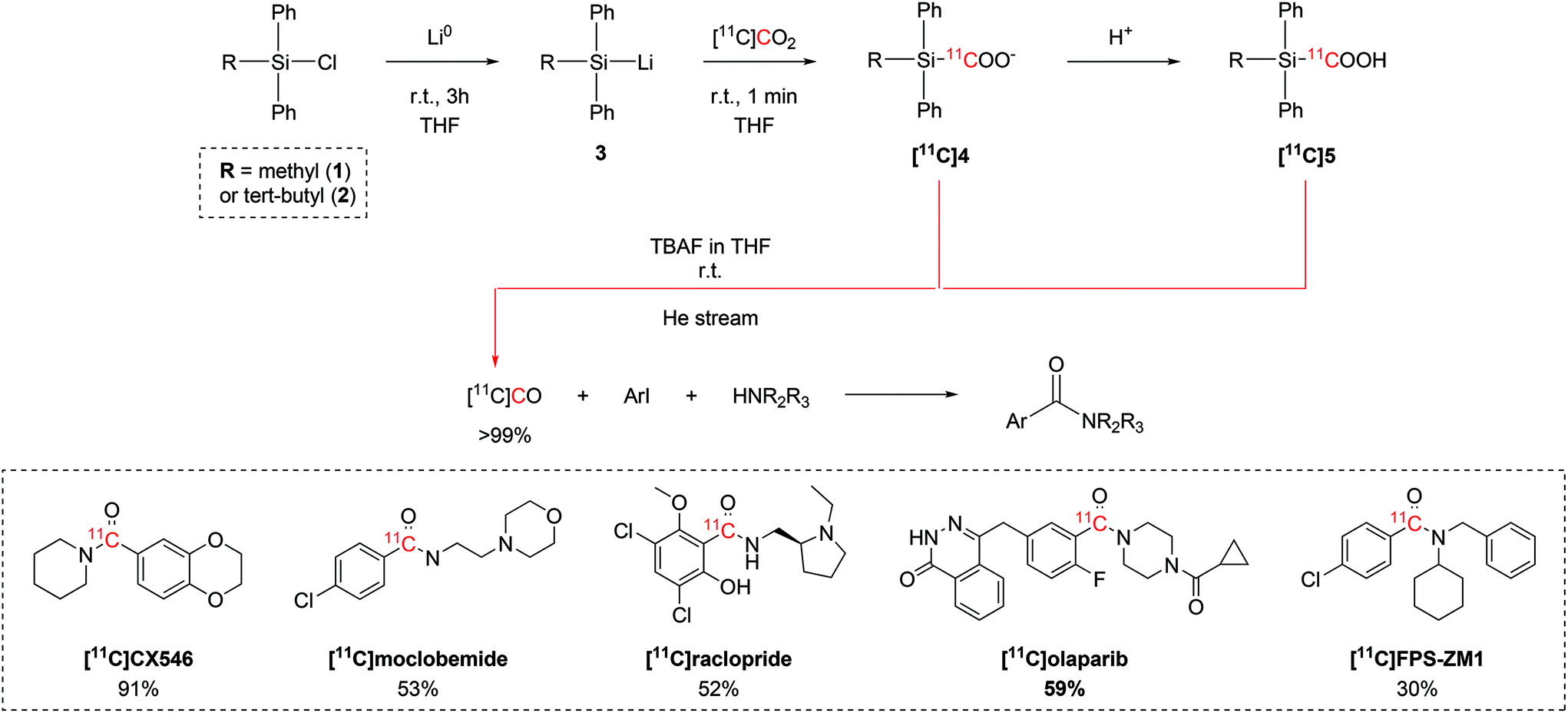 | ||
| Scheme 2 [11C]CO2-to-[11C]CO conversion via11C-silanecarboxylate derivatives, 11C-carbonylation of aryl iodides and biologically-active molecules successfully synthesized with the produced [11C]CO.37–39 | ||
Another approach to produce [11C]CO was reported by reacting disilanes with [11C]CO2. Four different disilane species were tested for their ability of trapping [11C]CO2 and converting it into [11C]CO, with 1,2-diphenyl-1,1,2,2-tetramethyldisilane ((Me2PhSi)2, 6, Scheme 3) being the most effective.40 With the aid of catalytic amounts of TBAF (0.1 equiv.) as a fluoride source, the production of [11C]CO reached 74% using mild reaction conditions and within 3 minutes from EOB.40 The reaction mechanism is believed to proceed by the initial formation of a pentavalent fluorosilyl anion which rapidly interacts with [11C]CO2 forming an unstable intermediate that spontaneously rearranges into a silylfluoride derivative (7), a silyloxide derivative (8) and [11C]CO (Scheme 3). As a proof-of-concept, the applicability of the produced [11C]CO in 11C-carbonylation reactions was tested via the radiosynthesis of [11C]benzylbenzamide which was obtained with a good RCY (74%, estimated by radioHPLC).40
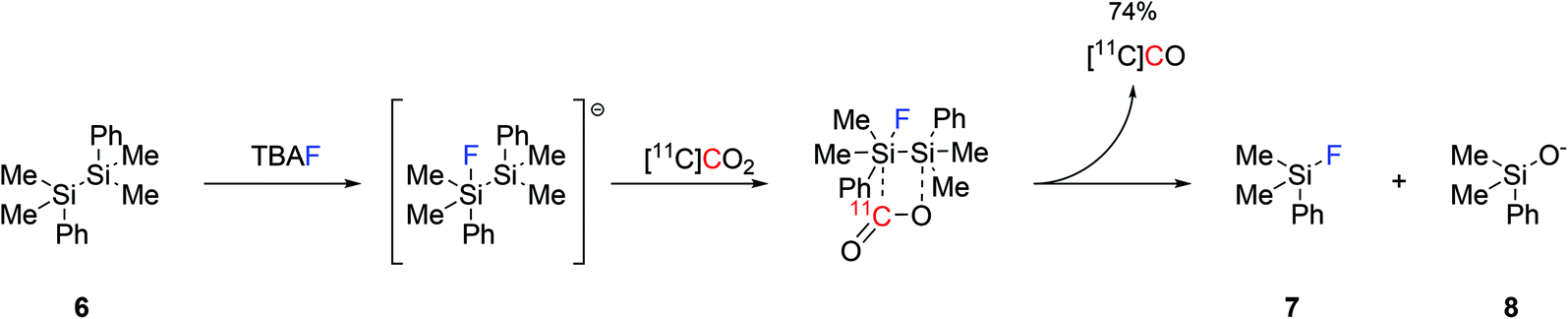 | ||
| Scheme 3 Proposed mechanism for the fluoride-promoted [11C]CO production from disilane species.40 | ||
2.3. Reductive functionalisation of [11C]CO2 using organosilanes
The high reactivity that organosilanes have towards CO2 enabled other interesting applications, such as the conversion of the cyclotron-produced [11C]CO2 to a 11C-N-methyl group without the need of using [11C]CH3I. This approach was vastly exploited in synthetic chemistry to use CO2 as a C1 building block in hydrosilylation reactions of CO2.16,41,42 The gas is initially trapped by the organosilicon compound in the form of silyl formate (Scheme 4)41 with the aid of transition-metal catalysis (e.g. Cu, Zn, Ni) and bulky ligands (e.g. σ-donor N-heterocyclic carbenes, NHCs).16,41,42 Then, the carbonyl group reacts with the target amino compound to form formamides (Scheme 4).42 The presence of an excess of silane provokes the reduction of the formamide to N-methylamine (Scheme 4).16 | ||
| Scheme 4 Hydrosilylation of CO2 forming a silyl formate. The reaction of an amine with a silyl formate produces a formamide which can be reduced by an excess of hydrosilane to yield an N-methylamine.16 | ||
The hydrosilylation of CO2 was later translated into carbon-11 chemistry by Liger et al.43 Similarly to the aforementioned non-radioactive reactions,16 this methodology exploited a NHC (1,3-bis(2,6-diisopropylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene, iPr, Scheme 5) as ligand, zinc catalysis and high temperature (150 °C) to yield 11C-N-methylamines within 20 minutes from EOD (Scheme 5). The radiolabelling of the amyloid-β plaque imaging agent [11C]PiB was achieved, as well, with an overall time of 50 minutes from EOD and in good RCY (38%, isolated, decay-corrected) but low Am (15 GBq per μmol).43
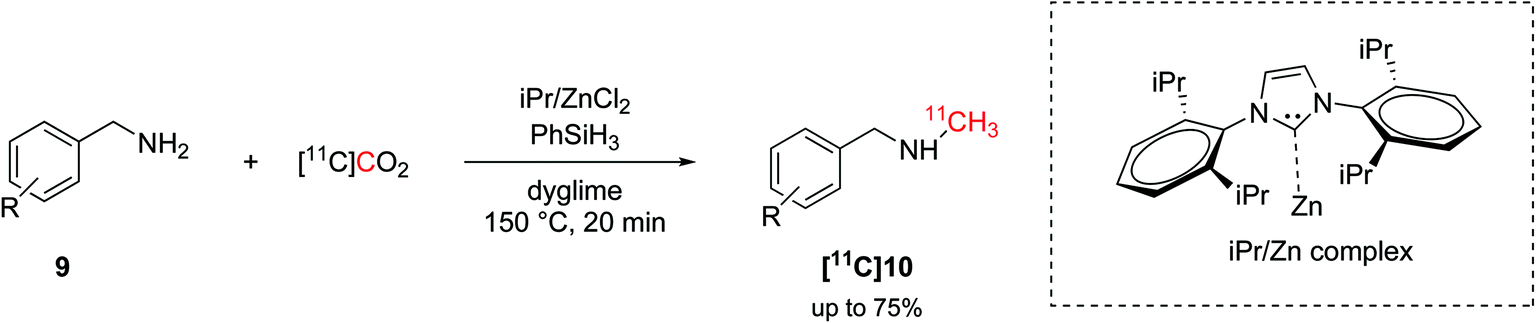 | ||
| Scheme 5 Hydrosilylation of [11C]CO2 and application on the 11C-N-methylation.43 | ||
A simpler set up that would not require the use of unstable NHCs was lately proposed for the synthesis of 11C-N-methylamines.44 Inspired by non-radioactive experiments,45 the radiolabelling step required the use of only hydrosilane and TBAF as fluoride source.44 The hydrosilane and the fluoride source were initially mixed in order to form an activated pentavalent fluorosilyl anion (11, Scheme 6) which instantly reduced the delivered [11C]CO2 to [11C]formate, allowing the formation of a 11C-silylformate ([11C]12, Scheme 6). The 11C-formyl group was then attached to the aminic precursor, yielding a 11C-formamide that was reduced to 11C-N-methylamine by the excess of fluorosilyl anion previously formed (Scheme 6).44
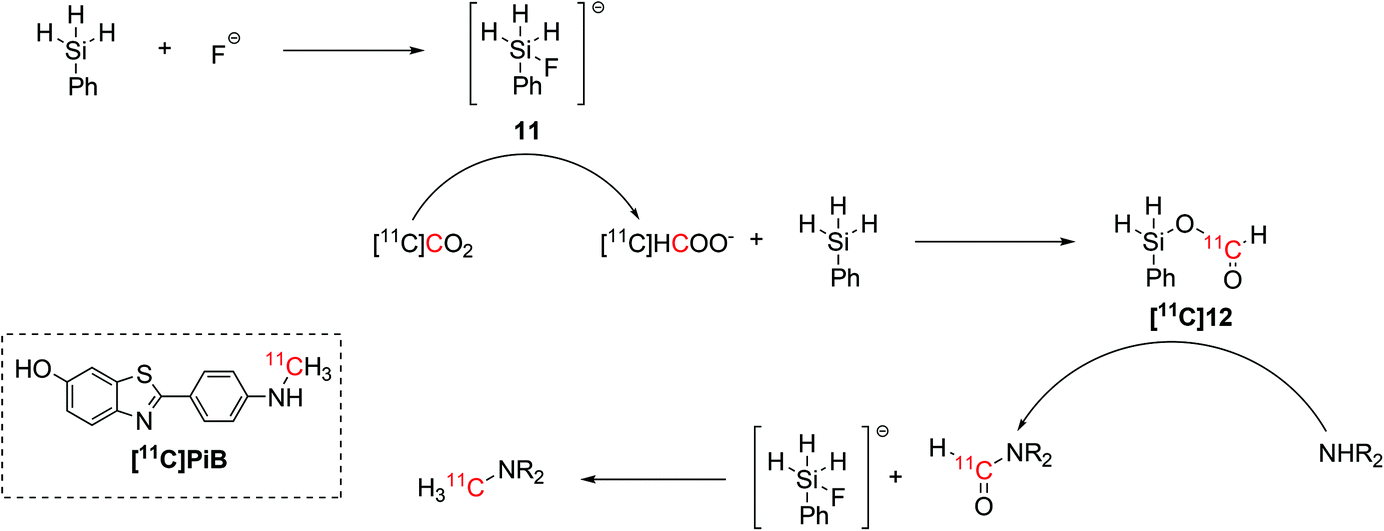 | ||
| Scheme 6 Fluoride-activated hydrosilylation of [11C]CO2 and application in the radiosynthesis of 11C-N-methylamines, including the amyloid-β plaque imaging agent [11C]PiB.44 | ||
This fluoride-activated radiosynthesis of 11C-N-methylamines was also fully automated and exploited for the synthesis of [11C]PiB with RCY of 15% (isolated, decay-corrected) and Am 61 GBq per μmol within 32 min from EOB.44
2.4. Organosilicon as precursors for carbon-11 radiolabelling
Recent findings revealed that organosilicon compounds are also viable radiolabelling precursors, especially in the form of trialkylsilyl and trialkoxysilyl arenes. The synthesis of these versatile organosilicon precursors is readily achieved via transition metal catalysis, such as Ni, Rh, Cu, allowing the silylation of a large number of aromatic compounds (e.g. Grignards, amides, cyanides, esters and acyl fluorides) with a wide functional group tolerance.20,46–49Trialkylsilyl and trialkoxysilyl arenes showed high reactivity towards copper-catalysed desilylative carboxylation reactions.50 With the aid of a fluoride source, the precursor was initially converted into a pentavalent silyl fluoride anion which then undergoes oxidative addition onto the Cu catalyst and react with the cyclotron-produced [11C]CO2. The highest reactivity was achieved when using DMF as solvent, KF/Kryptofix® 222 (crypt-222, 0.25 equiv.) as fluoride source, 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP, 0.6 equiv.) as CO2-trapping agent and a temperature of 140 °C for 5 minutes (Scheme 7). This method resulted equally efficient in the radiolabelling of alkynyl, aryl and heteroaryl precursors in short times (12 min) and with RCYs ranging between 19% and 93% (estimated by radioHPLC).50
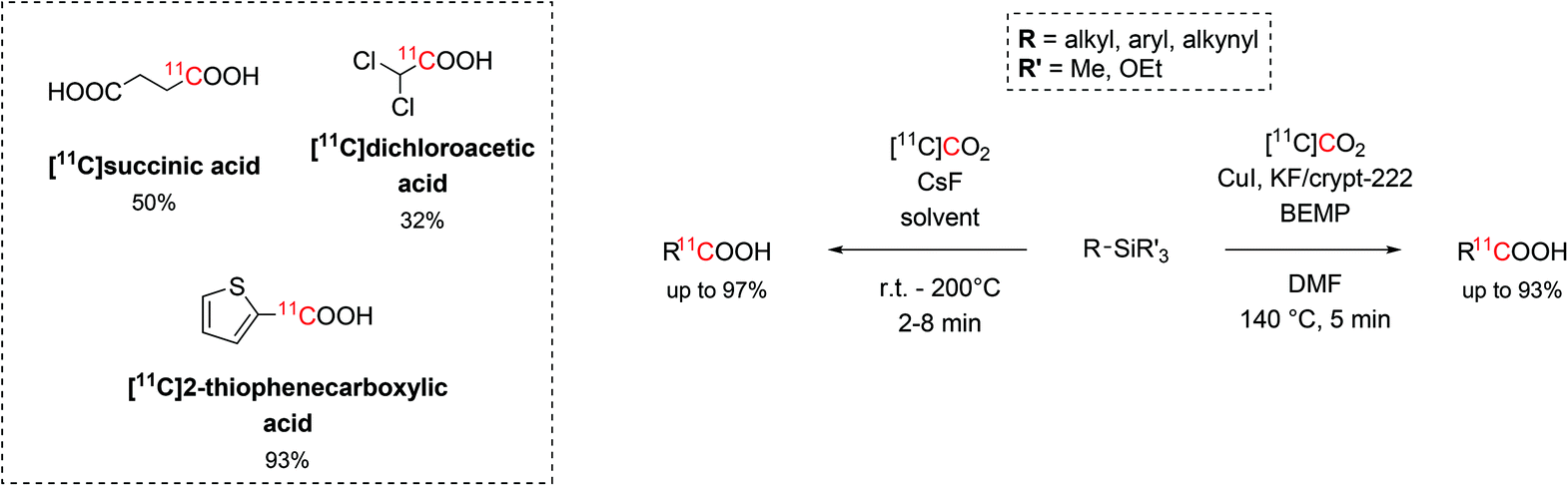 | ||
| Scheme 7 The use of trialkylsilyl and trialkoxysilyl as precursors in the 11C-carboxylation with [11C]CO2. | ||
A similar methodology that did not require copper catalysis was developed, as well. When CsF (1 equiv.) was employed as fluoride source and the reaction proceeded for 2–8 minutes at temperatures between r.t. and 200 °C, a large number of alkynyl, aryl and heteroaryl precursors (26 examples, Scheme 7) were successfully 11C-carboxylated with RCYs from 11% to 99%.51 A variety of solvents (DMF, DMA, DMSO/THF) were also tested returning similar trapping efficiency and reactivity. Using this strategy, eleven alkyl silyl precursors were successfully 11C-carboxylated (RCYs = 19%–97%), as well, including the mitochondrial kinase inhibitor dichloroacetic acid ([11C]dichloroacetic acid) and [11C]succinic acid (isolated decay-corrected RCYs = 32% and 50%, respectively, Scheme 7).51
Besides the production of 11C-carboxylic acids, the interaction between alkylsilyl precursors and [11C]CO2 showed to effectively yield other moieties, such as 11C-N-methylamines. Following a protocol that was initially developed with non-radioactive CO2,52 Ram et al. established a reliable method for 11C-N-methylation of secondary amines to methyl-11C-tertiary amines via direct use of [11C]CO2 and silyl amine precursors. The hydrochloride salt of the amine precursor (13, Scheme 8) was initially treated with hexamethyldisilazane (HMDS, Scheme 8) in the presence of either n-butyl lithium or ammonium sulphate to yield the respective silyl amine (14, Scheme 8). Upon delivery, [11C]CO2 would then be incorporated in the precursor as 11C-silyl carbamate (15, Scheme 8) when reacted for 8–10 minutes at 60–65 °C. 15 is then reduced by lithium aluminium hydride to the desired tertiary 11C-N-methylamine with RCYs ranging between 22% and 84% (isolated, decay-corrected at EOB) and Am of 1.5–15 GBq per μmol (calculated at EOB) within 36–50 minutes.52–56 This method was applied in the radiolabelling of a variety of tertiary alkylic amines, including the biologically-active molecules [11C]tamoxifen,53 [11C]imipramine,54 [11C]chlorpromazine55 and [11C]SCH 23390 (Scheme 8).56
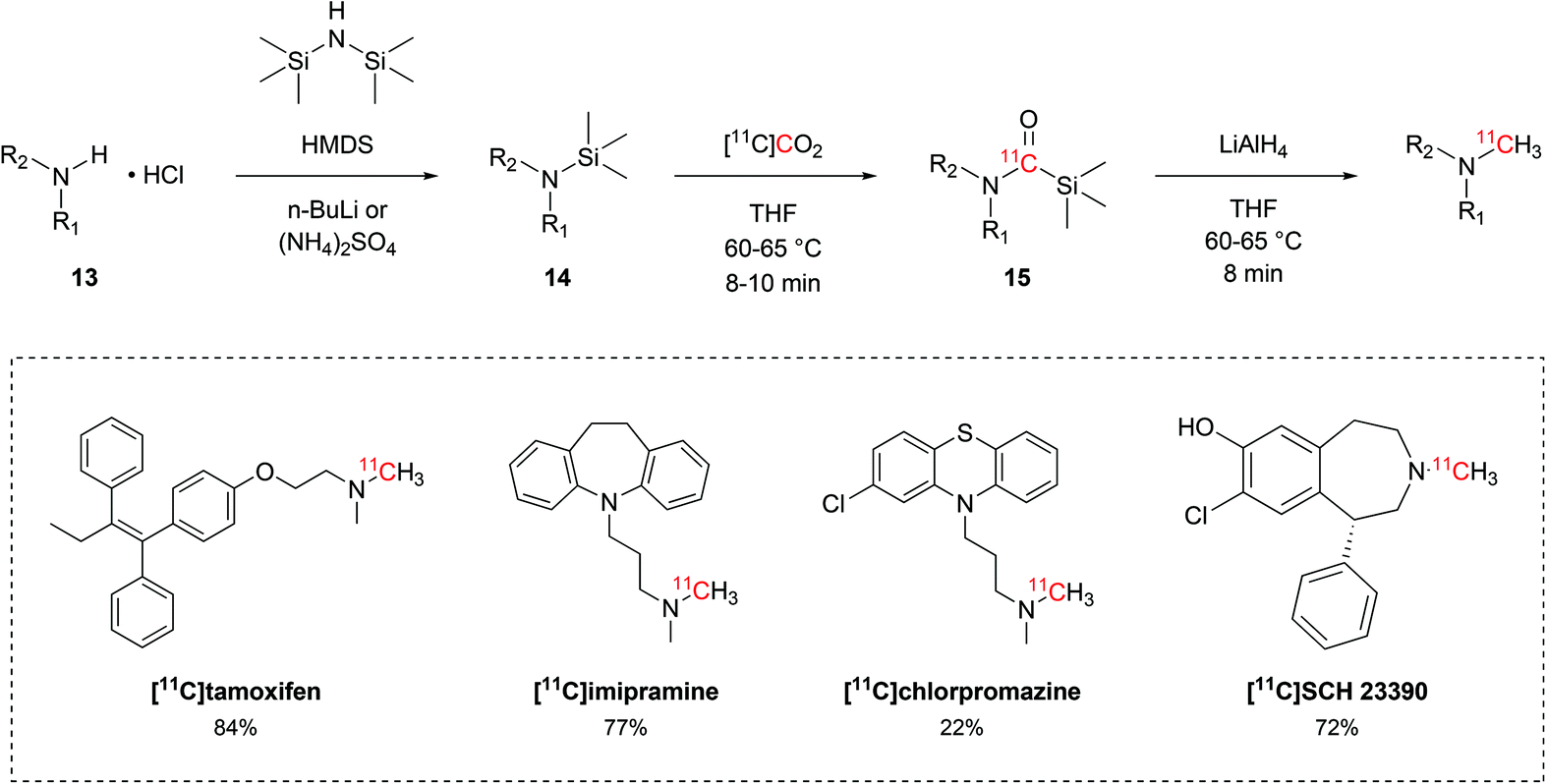 | ||
| Scheme 8 11C-methylation of secondary amines from [11C]CO2 and trimethylsilyl amines.52–56 | ||
The interaction of alkylsilyl precursors with [11C]CO2 showed to be effective in the radiosynthesis of 11C-ureas, as well.57 The reaction uses lithium bis(trimethylsilyl)amide (LBTMSA, Scheme 9) as a precursor which reacts with [11C]CO2 for 5 minutes at 65 °C to yield the corresponding 11C-carbodiimide ([11C]17) species via a 11C-isocyanate intermediate ([11C]16, Scheme 9). The hydrolysis of the 11C-carbodiimide with an aqueous solution of ammonium chloride then produced [11C]urea (Scheme 9) with RCY of 55–70% (estimated by radioHPLC) in 16 minutes from EOB. The obtained [11C]urea was then employed in the synthesis of the nucleotide [11C]uracil by condensation with diethyl malate in presence of fuming sulphuric acid (Scheme 9) at 130 °C for 5 minutes. [11C]Uracil was obtained with RCY of 40–75% (estimated by radioHPLC).57
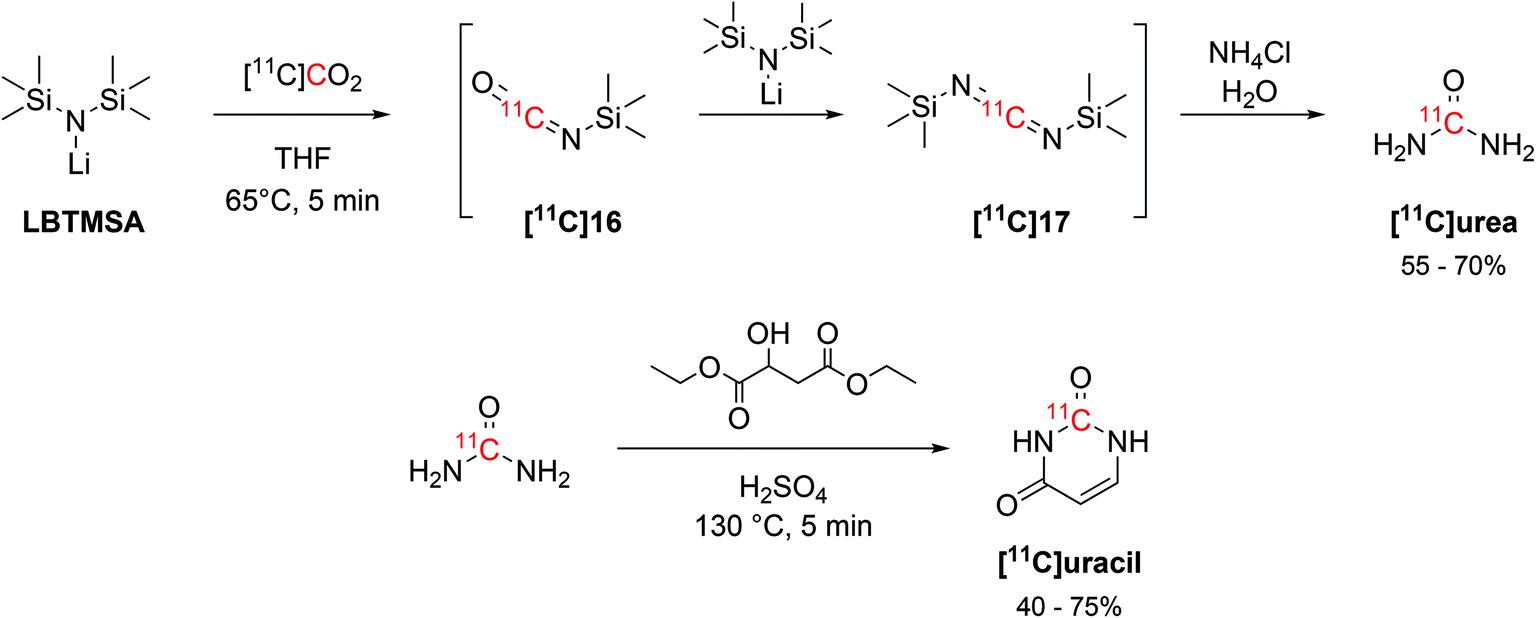 | ||
| Scheme 9 Synthesis of [11C]urea via the coupling of a silylamine precursor and [11C]CO2 and subsequent condensation with diethyl malate for [11C]uracil production.57 | ||
The radiolabelling of 11C-ureas could also be achieved by coupling organosilicon compounds with [11C]CO. This reaction requires the presence of a silylazide (18) and a (silyl)hydroxylamine (19, Scheme 10) and proceeds via transition metal catalysis in short times.58 In particular, the reaction was initially developed using trimethylsilyl azide and O-(trimethylsilyl)hydroxylamine in THF and in the presence of chloro(1,5-cyclooctadiene)rhodium(I) dimer ([RhCl(cod)]2) and 1,2-bis(diphenylphosphino)ethane (dppe) as catalyst and ligand, respectively (Scheme 10). The reaction proceeded at a temperature of 120 °C for 5 minutes and the resulting [11C]hydroxyurea was obtained with a RCY of 38% (isolated, decay-corrected, based on delivered [11C]CO).58 The suggested reaction mechanism proceeds with the formation of a Rh(I)-bound nitrene intermediate ([11C]20) which then reacts with the delivered [11C]CO to yield a Rh(III)-coordinated 11C-isocyanate ([11C]21, Scheme 10). Then, the hydroxylamine acting as nucleophile reacts with the 11C-isocyanate forming a silyl-protected 11C-urea (Scheme 10). The cleavage of the silyl groups was then easily achieved with a mixture of water and ethanol.58
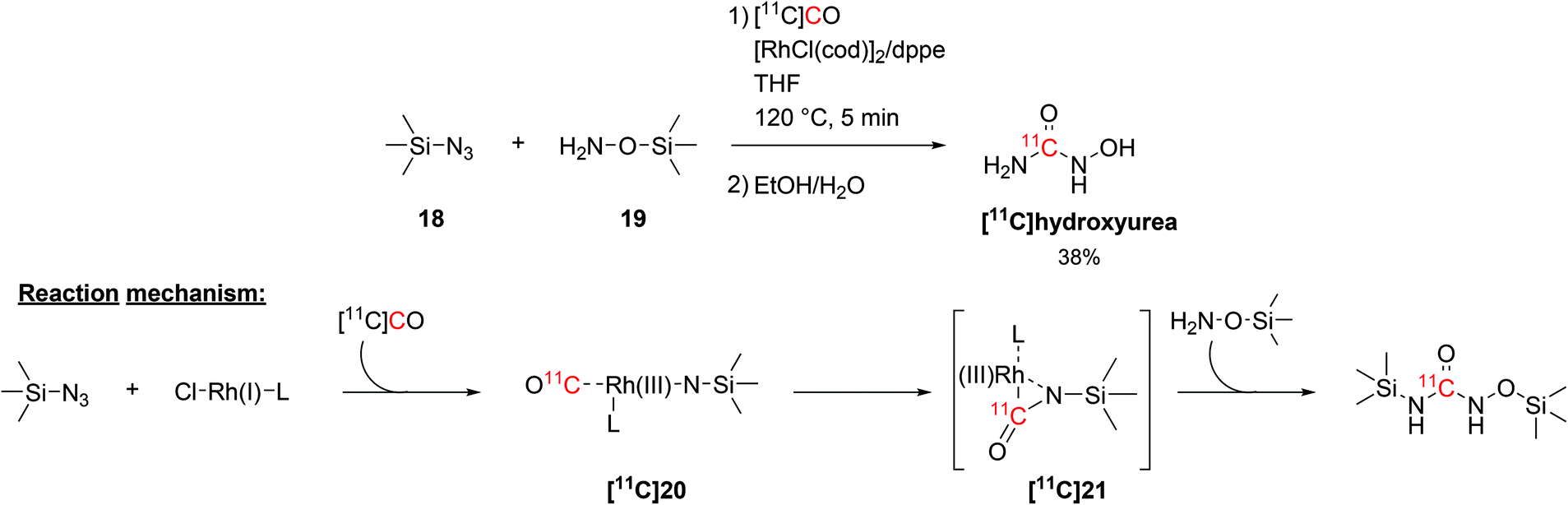 | ||
| Scheme 10 Coupling of [11C]CO with trimethylsilyl precursors to yield [11C]hydroxyurea.58 | ||
The synthesised [11C]hydroxyurea (Scheme 10), which in the body acts as a ribonucleoside reductase inhibitor, was then utilised to study its pharmacokinetics across the blood brain barrier in vivo and explore the interaction with multidrug resistance transporters.59 This was achieved by measuring the brain activity in rats with and without the simultaneous administration of multidrug resistance protein inhibitors (such as cyclosporin A and probenecid). The influx of [11C]hydroxyurea in the rat brain, however, was not significantly modified by the used intervention drugs, suggesting that hydroxyurea is not a substrate for active efflux transporters at the blood brain barrier.59
The coupling of organosilicon compounds with [11C]CO was also employed in the synthesis of 1-hydroxy-3-phenyl[11C]urea by using phenylazide (19, Scheme 11) and O-(trimethylsilyl)hydroxylamine as reagents (22, Scheme 11) whilst keeping the same conditions.58 1-Hydroxy-3-phenyl[11C]urea was successfully radiolabelled with RCY of 35% (isolated, decay-corrected, based on delivered [11C]CO) and Am of 686 GBq per μmol at 21 minutes from EOB.58
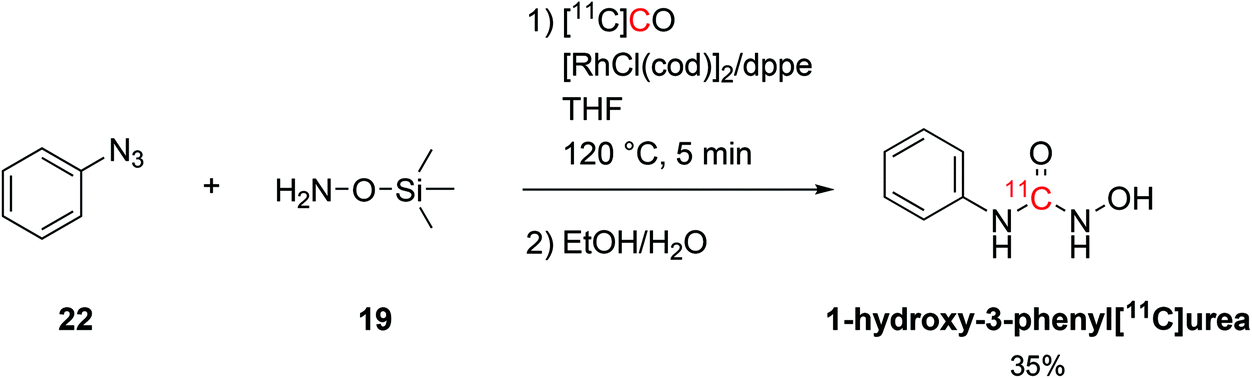 | ||
| Scheme 11 Synthesis of 1-hydroxy-3-phenyl[11C]urea.58 | ||
The use of trialkylsilyl and trialkoxysilyl precursors was then tested for 11C-methylation with [11C]CH3I (Scheme 12). The radiolabelling on heteroatoms (O, N, S), aryl and alkyl precursors was readily accomplished by either copper-catalysed or fluoride-activated desilylative reactions and two biologically-active molecules such as [11C]propiophenone and [11C]ibuprofen were also produced (isolated decay-corrected RCYs = 68% and 27%, respectively, Scheme 12).50,51
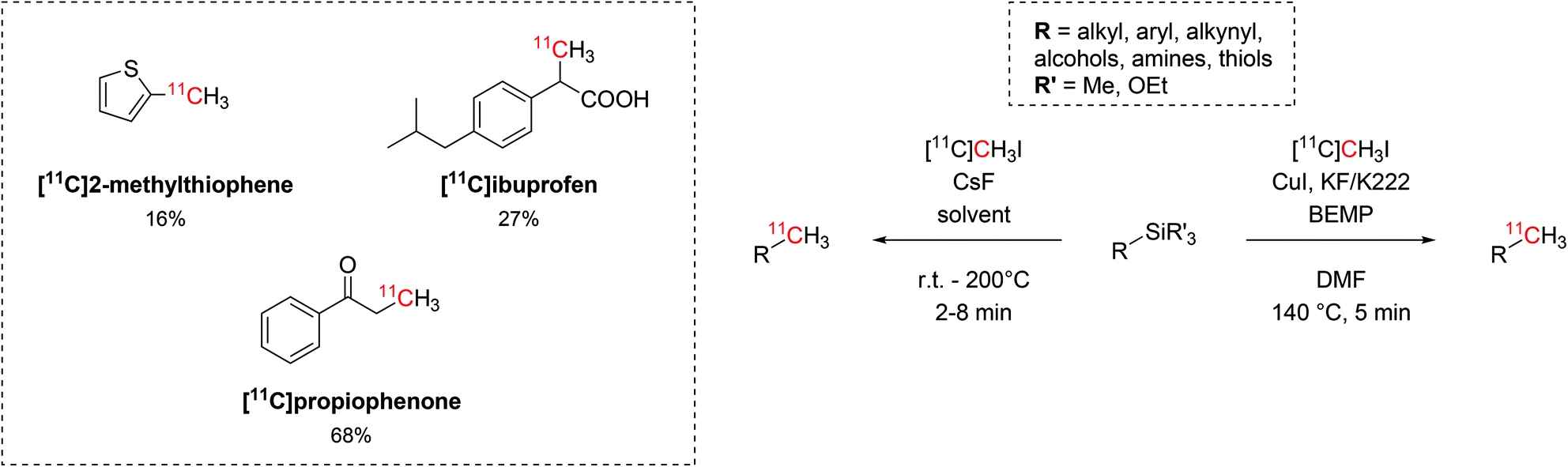 | ||
| Scheme 12 The use of trialkylsilyl and trialkoxysilyl as precursors in the 11C-methylation with [11C]CH3I. | ||
3. Conclusion
In the past years, the interesting chemistry of silicon-containing compounds has been embraced for radiolabelling purposes, meeting the need for simpler, cost-effective and more efficient radiolabelling methodologies. The versatility of the silicon atom unlocked a plethora of different applications, from [11C]CO2 converting agents to radiolabelling precursors, and enabled the production of a large number of biologically-active radiopharmaceuticals. Nonetheless, the applicability of compounds bearing silicon is yet to be fully explored. Taking inspiration from conventional organic chemistry, where silicon-based chemistry is already widely utilised, many more applications could be developed for 11C-labelling. For example, the hydrosilylation of CO2 have been used for the synthesis of a larger number of chemical structures (formamides, aldehydes, aminals)10–13 whereas in 11C-chemistry it's been used only for the production of 11C-N-methylamines.43,44Another potential route for widening the use of 11C-Si chemistry would be taking inspiration from the more established 11C-boron chemistry.60 Boron and silicon share many similarities with respect to bond energy and general chemical properties,61,62 suggesting that they may also have similar applicability in radiochemistry. Whilst some of these methodologies showed to be suitable for both boron- and silicon-containing molecules (e.g.11C-carboxylation with [11C]CO2, 11C-methylation with [11C]CH3I), organosilicates have not yet been tested in many other radiolabelling strategies, such as the 11C-carbonylation with [11C]CO or the 11C-cyanation with [11C]HCN, which instead are well-established with organoboronates.60 Regarding the potential coupling with [11C]CO, evidence from non-radioactive studies already show its feasibility: the synthesis of unsymmetrical diaryl ketones was achieved by combining a silyl arene, a iodoarene and CO in the presence of KF as activator.63 Likewise, non-radioactive experiments showed that the synthesis of thioesters is achievable by coupling CO with organosilicon precursors.64
The development of novel organosilicon radiolabelling strategies could also take inspiration from the well-established radiochemistry of organostannic compounds.65 Exploiting the Stille coupling reaction, the use of organostannanes as radiolabelling precursors found several applications and was coupled with a variety of carbon-11 synthons, such as [11C]CH3I, [11C]CO and [11C]acyl chloride, for the production of a variety of functional groups (e.g. aryl 11C-methylated compounds and 11C-ketones).65 The translation of these methodologies into silicon-based 11C-chemistry would represent a big step forward.
Besides the development of novel methodologies, organosilicon radiochemistry may also benefit from broadening the pool of radiolabel-able substrates. The previously discussed techniques are indeed still limited to certain classes of precursors, like the reductive functionalisation of [11C]CO2 (Scheme 6) which was poorly tested on alkyl precursors. The development of a “universal” radiolabelling tool, applicable to a larger number of radiotracer regardless of their chemical nature, should then also be taken into consideration.
The use of the radiochemistry of organosilicon compounds should also be enhanced in regular radiopharmaceutical production. The aforementioned methods are still restricted to the research world and not have impacted the broader clinical production of radiotracers yet. Although some clinically-used radiotracers were successfully produced with the discussed methods (e.g. [11C]PiB via reductive functionalisation of [11C]CO2),43,44 traditional radiolabelling processes (e.g.11C-methylation with [11C]CH3I) are still preferred for routine clinical production in spite of the advantages that novel methods would bring to routine production such as the much shorter radiolabelling time. When considering [11C]PiB labelling, for example, the traditional production via11C-methylation with [11C]CH3I requires the prior conversion of [11C]CO2 to [11C]CH3I, a time-consuming step (around 5 minutes)66,67 that instead is not needed in [11C]CO2-based silicon chemistry. Moreover, the presence of a free hydroxyl group on [11C]PiB demands for protection/deprotection68 to avoid side reactions whilst silicon-based methods do not have these limitations as specific towards amines.44 All these factors significantly lower the radiolabelling time from 15 to 1 minute (from end of [11C]CO2 delivery to end of synthesis (EOS)),44,68 a critical improvement considering the short half-life of 11C. The use of organosilicate methodologies would also lower the costs as not requiring the purchase and maintenance of costly infrastructure such as a standalone methyl iodide production unit – potentially increasing the availability of carbon-11 labelled compounds in laboratories. The RCY of the final radiopharmaceutical would also benefit from the use of the aforementioned methods (12% with [11C]CH3I versus 26% with silicon chemistry).44,68 Hence, the translation of silicon radiochemistry into PET laboratory routine production is another important step that is required.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
King's College London and UCL Comprehensive Cancer Imaging Centre is funded by the CRUK and EPSRC in association with the MRC and DoH (England). The research was supported by the Wellcome's Multi-User Equipment ‘A multiuser radioanalytical facility for molecular imaging and radionuclide therapy research’ [212885/Z/18/Z]. It was additionally supported by the Wellcome/EPSRC Centre for Medical Engineering at King's College London [WT 203148/Z/16/Z] and the EPSRC Centre for Doctoral Training in Medical Imaging [EP/L015226/1]. The authors also acknowledge PMB Alcen for the financial support.References
- W. W. Moses, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 648(Supplement 1), S236–S240 CrossRef CAS PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- M. Conti and L. Eriksson, EJNMMI Phys., 2016, 3, 8 CrossRef PubMed.
- B. H. Rotstein, S. H. Liang, J. P. Holland, T. L. Collier, J. M. Hooker, A. A. Wilson and N. Vasdev, Chem. Commun., 2013, 49, 5621–5629 RSC.
- M. E. Phelps, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9226–9233 CrossRef CAS PubMed.
- K. Dahl, C. Halldin and M. Schou, Clin. Transl. Imaging, 2017, 5, 275–289 CrossRef PubMed.
- T. Hiyama, J. Organomet. Chem., 2002, 653, 58–61 CrossRef CAS.
- T. Hiyama and E. Shirakawa, Cross-Coupling React., 2002, 61–85 CAS.
- L. Zhang and J. Wu, J. Am. Chem. Soc., 2008, 130, 12250–12251 CrossRef CAS PubMed.
- C. M. So, H. W. Lee, C. P. Lau and F. Y. Kwong, Org. Lett., 2009, 11, 317–320 CrossRef CAS PubMed.
- K. Cheng, S. Hu, B. Zhao, X.-M. Zhang and C. Qi, J. Org. Chem., 2013, 78, 5022–5025 CrossRef CAS PubMed.
- P. Zhang, J. Xu, Y. Gao, X. Li, G. Tang and Y. Zhao, Synlett, 2014, 25, 2928–2932 CrossRef CAS.
- K. Toshima, S. Mukaiyama, M. Kinoshita and K. Tatsuta, Tetrahedron Lett., 1989, 30, 6413–6416 CrossRef CAS.
- R. D. Crouch, M. Stieff, J. L. Frie, A. B. Cadwallader and D. C. Bevis, Tetrahedron Lett., 1999, 40, 3133–3136 CrossRef CAS.
- X. Frogneux, O. Jacquet and T. Cantat, Catal. Sci. Technol., 2014, 4, 1529–1533 RSC.
- O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- T. Murata, M. Hiyoshi, M. Ratanasak, J.-y. Hasegawa and T. Ema, Chem. Commun., 2020, 56, 5783–5786 RSC.
- X. Frogneux, E. Blondiaux, P. Thuéry and T. Cantat, ACS Catal., 2015, 5, 3983–3987 CrossRef CAS.
- M. Tredwell and V. Gouverneur, Org. Biomol. Chem., 2006, 4, 26–32 RSC.
- L. Guo, A. Chatupheeraphat and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 11810–11813 CrossRef CAS PubMed.
- V. Bernard-Gauthier, C. Wängler, E. Schirrmacher, A. Kostikov, K. Jurkschat, B. Wängler and R. Schirrmacher, BioMed Res. Int., 2014, 2014, 454503 Search PubMed.
- R. Walsh, Acc. Chem. Res., 1981, 14, 246–252 CrossRef CAS.
- J. Wagler, U. Böhme and G. Roewer, Organometallics, 2004, 23, 6066–6069 CrossRef CAS.
- E. Magnusson, J. Am. Chem. Soc., 1990, 112, 7940–7951 CrossRef CAS.
- G. L. Larson and J. L. Fry, in Organic Reactions, 2009, pp. 1–737. DOI:10.1002/0471264180.or071.01.
- K. Junold, J. A. Baus, C. Burschka, T. Vent-Schmidt, S. Riedel and R. Tacke, Inorg. Chem., 2013, 52, 11593–11599 CrossRef CAS PubMed.
- H. Kwart and K. King, d-Orbitals in the Chemistry of Silicon, Phosphorus and Sulfur, Springer Science & Business Media, 2012 Search PubMed.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, e17–e24 CrossRef CAS PubMed.
- S. D. Friis, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 18114–18117 CrossRef CAS PubMed.
- C. Lescot, D. U. Nielsen, I. S. Makarov, A. T. Lindhardt, K. Daasbjerg and T. Skrydstrup, J. Am. Chem. Soc., 2014, 136, 6142–6147 CrossRef CAS PubMed.
- A. G. Brook, Acc. Chem. Res., 1974, 7, 77–84 CrossRef CAS.
- A. G. Brook, J. Am. Chem. Soc., 1955, 77, 4827–4829 CrossRef CAS.
- A. G. Brook and H. Gilman, J. Am. Chem. Soc., 1955, 77, 2322–2325 CrossRef CAS.
- S. K. Zeisler, M. Nader, A. Theobald and F. Oberdorfer, Appl. Radiat. Isot., 1997, 48, 1091–1095 CrossRef CAS.
- J. M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701–704 CrossRef CAS PubMed.
- J. Medina-Ramos, R. C. Pupillo, T. P. Keane, J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2015, 137, 5021–5027 CrossRef CAS PubMed.
- C. Taddei, S. Bongarzone, A. K. Haji Dheere and A. D. Gee, Chem. Commun., 2015, 51, 11795–11797 RSC.
- F. Luzi, V. Savickas, C. Taddei, S. Hader, N. Singh, A. D. Gee and S. Bongarzone, Future Med. Chem., 2020, 12, 511–521 CrossRef CAS PubMed.
- P. Nordeman, S. D. Friis, T. L. Andersen, H. Audrain, M. Larhed, T. Skrydstrup and G. Antoni, Chem. – Eur. J., 2015, 21, 17601–17604 CrossRef CAS PubMed.
- C. Taddei, S. Bongarzone and A. D. Gee, Chemistry, 2017, 23, 7682–7685 CrossRef CAS PubMed.
- L. Zhang, J. Cheng and Z. Hou, Chem. Commun., 2013, 49, 4782–4784 RSC.
- L. González-Sebastián, M. Flores-Alamo and J. J. García, Organometallics, 2013, 32, 7186–7194 CrossRef.
- F. Liger, T. Eijsbouts, F. Cadarossanesaib, C. Tourvieille, D. Le Bars and T. Billard, Eur. J. Org. Chem., 2015, 2015, 6434–6438 CrossRef CAS.
- P. Buccino, E. Savio and W. Porcal, EJNMMI Radiopharm. Chem., 2019, 4, 14 CrossRef PubMed.
- X.-F. Liu, R. Ma, C. Qiao, H. Cao and L.-N. He, Chem. – Eur. J., 2016, 22, 16489–16493 CrossRef CAS PubMed.
- A. S. Manoso, C. Ahn, A. Soheili, C. J. Handy, R. Correia, W. M. Seganish and P. Deshong, J. Org. Chem., 2004, 69, 8305–8314 CrossRef CAS PubMed.
- S.-C. Lee, L. Guo, H. Yue, H.-H. Liao and M. Rueping, Synlett, 2017, 28, 2594–2598 CrossRef CAS.
- M. Tobisu, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2006, 128, 8152–8153 CrossRef CAS PubMed.
- X. Wang, Z. Wang and Y. Nishihara, Chem. Commun., 2019, 55, 10507–10510 RSC.
- S. Bongarzone, N. Raucci, I. C. Fontana, F. Luzi and A. D. Gee, Chem. Commun., 2020, 56, 4668–4671 RSC.
- W. Qu, B. Hu, J. W. Babich, N. Waterhouse, M. Dooley, S. Ponnala and J. Urgiles, Nat. Commun., 2020, 11, 1736 CrossRef CAS PubMed.
- S. Ram and R. E. Ehrenkaufer, Tetrahedron Lett., 1985, 26, 5367–5370 CrossRef CAS.
- S. Ram and L. D. Spicer, J. Labelled Compd. Radiopharm., 1989, 27, 661–668 CrossRef CAS.
- S. Ram, R. E. Ehrenkaufer and D. M. Jewett, Int. J. Radiat. Appl. Instrum., Part A, 1986, 37, 391–395 CrossRef CAS.
- S. Ram and L. D. Spicer, Int. J. Radiat. Appl. Instrum., Part A, 1989, 40, 413–416 CrossRef CAS.
- S. Ram, R. E. Ehrenkaufer and L. D. Spicer, Int. J. Radiat. Appl. Instrum., Part A, 1989, 40, 425–427 CrossRef CAS.
- P. K. Chakraborty, T. J. Mangner and H. T. Chugani, Appl. Radiat. Isot., 1997, 48, 619–621 CrossRef CAS.
- J. Barletta, F. Karimi and B. Långström, J. Labelled Compd. Radiopharm., 2006, 49, 429–436 CrossRef CAS.
- S. Syvänen, J. Barletta, G. Blomquist, B. Långström and M. Bergström, Drug Metab. Lett., 2007, 1, 189–194 CrossRef PubMed.
- T. C. Wilson, T. Cailly and V. Gouverneur, Chem. Soc. Rev., 2018, 47, 6990–7005 RSC.
- P. R. Rablen and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 4648–4653 CrossRef CAS.
- G. Rayner-Canham, Found. Chem., 2011, 13, 121–129 CrossRef CAS.
- T. Hiyama and Y. Hatanaka, Pure Appl. Chem., 1994, 66, 1471–1478 CAS.
- Z. Qiao and X. Jiang, Org. Lett., 2016, 18, 1550–1553 CrossRef CAS PubMed.
- M. Pretze, P. Grosse-Gehling and C. Mamat, Molecules, 2011, 16, 1129–1165 CrossRef CAS PubMed.
- J. M. Link, K. A. Krohn and J. C. Clark, Nucl. Med. Biol., 1997, 24, 93–97 CrossRef CAS PubMed.
- B. Långström, G. Antoni, P. Gullberg, C. Halldin, P. Malmborg, K. Någren, A. Rimland and H. Svärd, J. Nucl. Med., 1987, 28, 1037–1040 Search PubMed.
- C. A. Mathis, Y. Wang, D. P. Holt, G.-F. Huang, M. L. Debnath and W. E. Klunk, J. Med. Chem., 2003, 46, 2740–2754 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |