
Recent advances in phosphoranyl radical-mediated deoxygenative functionalisation
Xiao-Qiang
Hu
 *a,
Ye-Xing
Hou
a,
Zi-Kui
Liu
a and
Yang
Gao
*b
*a,
Ye-Xing
Hou
a,
Zi-Kui
Liu
a and
Yang
Gao
*b
aKey Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education & Hubei Key Laboratory of Catalysis and Materials Science, School of Chemistry and Materials Science, South-Central University for Nationalities, Wuhan 430074, China. E-mail: huxiaoqiang@mail.scuec.edu.cn
bSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou, 510006, China. E-mail: gaoyang@gdut.edu.cn
First published on 10th July 2020
Abstract
Alcohols and carboxylic acids have been established as versatile building blocks in the assembly of various carbon frameworks. The deoxygenative functionalisation is one of the most attractive approaches for the conversion of these abundant materials into high-value products. However, the direct deoxygenative transformation under mild conditions has long been a challenging task in organic synthesis due to the lack of efficient methods for the activation of strong C–O bonds (102 kcal mol−1). Recently, the strategy of visible light photocatalytic phosphoranyl radical fragmentation provides a novel and powerful platform for C–O, N–O and S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond activation. This highlight mainly discusses recent breakthroughs in phosphoranyl radical-mediated deoxygenative functionalisations of alcohols, carboxylic acids, oximes and sulfoxides with a particular emphasis on reaction scopes and mechanism.
O bond activation. This highlight mainly discusses recent breakthroughs in phosphoranyl radical-mediated deoxygenative functionalisations of alcohols, carboxylic acids, oximes and sulfoxides with a particular emphasis on reaction scopes and mechanism.
1. Introduction
Deoxygenative reactions are fundamental reactions in organic and biological synthesis, which have attracted considerable interest from synthetic chemists.1 In this context, the direct deoxygenative functionalisations of alcohols, carboxylic acids, oximes and sulfoxides have long been elusive transformations. Several challenges are associated with the deoxygenative process, most notably the relatively high bond strength of C–O, N–O and SO bonds.2 Representative strategies for deoxygenative transformations of these compounds mainly rely on distinct preactivated steps to generate new reactive functional groups, or the use of some strong nucleophiles and reductants.3 From a sustainable point of view, the exploration of novel and efficient strategies for the direct deoxygenative transformation of alcohols, carboxylic acids, oximes and sulfoxides is still highly desirable.
The phosphoranyl radical fragmentation strategy has been esteemed as an efficient method for deoxygenative reactions.4 Typically, the phosphoranyl radical-mediated deoxygenative reactions have been achieved under high-energy UV irradiation or other initiation conditions, such as Bz2O2 and (n-Bu)3SnH/AIBN etc.5 Recently, visible light photoredox catalysis has emerged as a preeminent handle for the formation of reactive radicals and radical ions via single electron transfer (SET) and energy transfer (ET) pathways.6 In this context, visible light-induced fragmentation of phosphoranyl radicals provides a novel and powerful platform for the direct activation of C–O, N–O and SO bonds, delivering a diversity of radical species (Scheme 1). These reactions generally obviate the need for pre-functionalisation steps and radical initiators. This concept has been successfully applied in the deoxygenative deuteration, arylation, reduction and radical cascade reactions. Distinguishing features of these reactions include mild conditions, broad scopes and high efficiency. Very recently, impressive examples on phosphoranyl radical fragmentation reactions have been summarized by Rossi-Ashton.7 This highlight mainly discusses the recent advances in phosphoranyl radical-promoted deoxygenative technologies, with particular emphasis on reaction scope and mechanism.
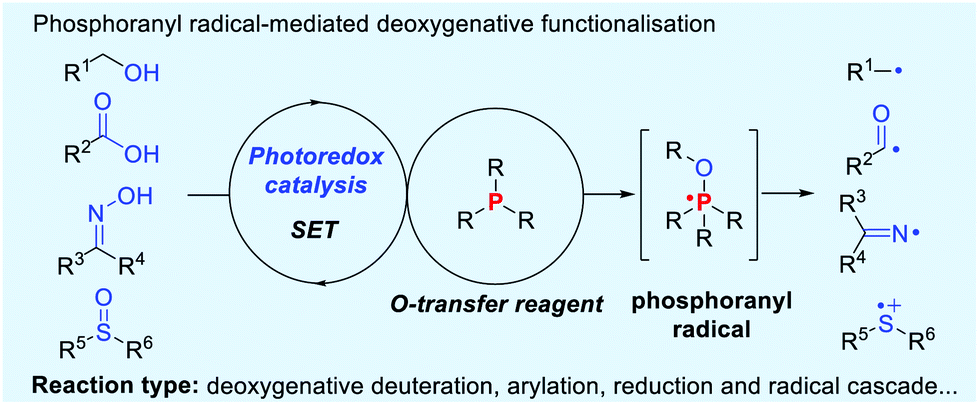 | ||
| Scheme 1 Phosphoranyl radical-mediated deoxygenative functionalisation. | ||
2. Deoxygenative functionalisation of carboxylic acids and alcohols
An important breakthrough in this field was achieved by Zhu and Xie et al.8 They developed an elegant deoxygenative addition of carboxylic acids to alkenes for the synthesis of diverse ketones. In this reaction, triphenylphosphine (Ph3P) was the optimal O-transfer reagent. A wide range of aromatic carboxylic acids and electron-deficient alkenes were compatible in this reaction, delivering the expected ketones in generally good yields. However, aliphatic carboxylic acids were not suitable in this reaction system. The synthetic potential was demonstrated by the concise synthesis of cyclophanebraced cycloketones (Scheme 2a, 6 and 7) and the drug zolpidem (Scheme 2b, 10).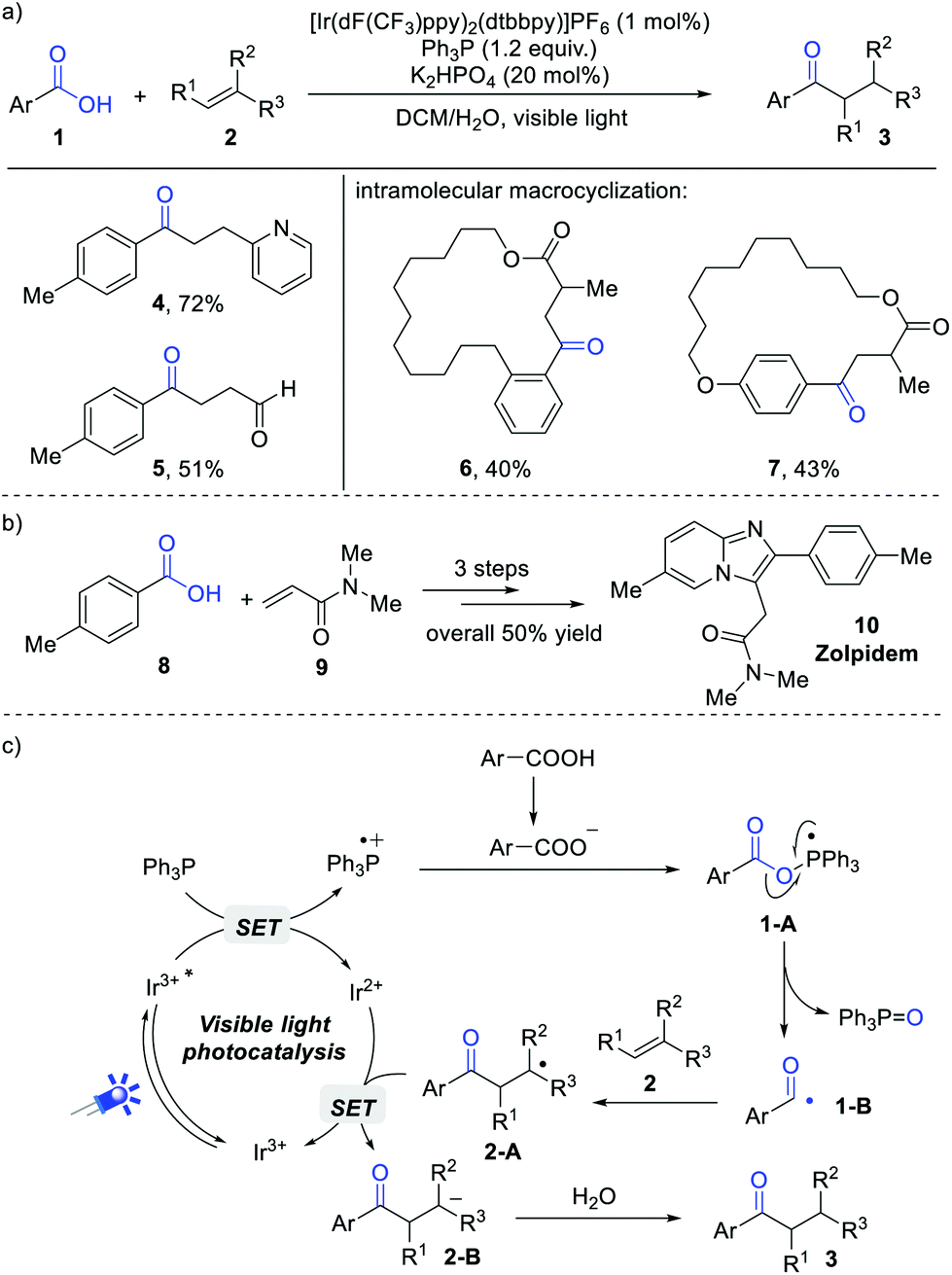 | ||
| Scheme 2 Visible light-induced deoxygenative addition of carboxylic acids to alkenes (a), (b) and proposed mechanism (c). | ||
A series of control experiments have been conducted to investigate the reaction mechanism. Stern–Volmer analysis indicated that the excited state of *Ir(III) can be efficiently quenched by Ph3P. In addition, 18O-labeling experiments revealed that the O-atom of triphenylphosphine oxide (Ph3PO) comes from the carboxylate group rather than H2O. Radical trapping reaction by 2,2,6,6-tetramethyl-1-piperidyloxy (TEMPO) demonstrated the intermediacy of an acyl radical during this transformation. Based on these results, a possible mechanism is proposed in Scheme 2c. Initially, the photoexcited *Ir(III) [Ered1/2 (*IrIII/IrII) = +1.21 V vs. SCE] was reductively quenched by Ph3P (Ered1/2 = +0.98 V vs. SCE) to generate the triphenylphosphine radical cation intermediate. Afterwards, the combination of a Ph3P radical cation with a carboxylate anion results in a radical intermediate 1-A, which undergoes a C–O bond cleavage to give the key acyl radical 1-B and Ph3PO as the byproduct. The addition of 1-B to electron-deficient alkenes gives rise to C-based radical 2-A, followed by a single electron reduction and protonation cascade to give the final products.
Almost at the same time, Doyle, Rovis et al. achieved a visible light-induced deoxygenative reduction of carboxylic acids (Scheme 3a).9 Remarkably, by rationally modifying the phosphine reagent, both aryl and alkyl carboxylic acids can be successfully reduced to the corresponding aldehydes. Ph3P was used as an efficient O-transfer reagent for the reduction of aryl carboxylic acids. However, it was ineffective for the transformation of aliphatic carboxylic acids. They proposed that a more electron-rich phosphoranyl radical intermediate was generated from Ph3P and aliphatic carboxylic acid. This electron-rich phosphoranyl radical was susceptible to oxidation, producing a phosphonium species for a rapid acyl transfer reaction. An electron-deficient phosphinite Ph2POEt proved to be suitable for the reduction of aliphatic acids. Under these reaction conditions, carboxylic acids were selectively reduced even in the presence of other reactive carbonyl groups, which demonstrated the promising potential in precise reductive reactions. Moreover, the intramolecular acyl radical-mediated cyclisations of carboxylic acids were also successful, delivering the corresponding lactones and lactams in high efficiency.
 | ||
| Scheme 3 Deoxygenative reduction of carboxylic acids (a) and benzylic alcohols (b), and proposed mechanism (c). | ||
Gratifyingly, the current catalytic system can be further applied to the direct reduction of benzylic alcohols without the requirement of preactivation steps (Scheme 3b). Alcohols bearing both electron-rich and deficient groups reacted smoothly in this transformation. In addition, secondary alcohols were also suitable for this reaction, albeit with lower yields. This reaction offers novel avenues for deoxygenative transformations of carboxylic acids and alcohols under mild conditions.
The proposed mechanism is outlined in Scheme 3c. The key phosphoranyl radical 25-A is generated from the combination of the in situ generated Ph3P radical cation with a carboxylic acid or benzylic alcohol, which proceeds through a β-scission process to give a C-centred radical 25-B with simultaneous generation of Ph2XPO. The H-atom abstraction of ArSH by radical 25-B affords the expected product.
Xie and co-workers further developed a phosphoranyl radical-mediated deoxygenative deuteration by combination of visible light photocatalysis and H-atom transfer (HAT) catalysis (Scheme 4).10 D2O was used as the deuterium reagent. It should be noted that the direct deuterium-atom abstraction from D2O by acyl radical intermediates is historically known to be difficult due to the strong bond dissociation energy (BDE) of the O–D bond. The addition of a thiol catalyst is critical for this reaction, which can bridge the energy gap between radical species and D2O. The proton exchange of a thiol catalyst (pKa = 10–17) with an excess D2O (pKa = 32) generates a D-labelled thiol, which then reacts with acyl radicals to give the deuterated products. Significantly, both aromatic and aliphatic carboxylic acids were well tolerated in this reaction. For the reaction of aromatic acids, Ph3P was employed as a good O-atom transfer reagent, while Ph2POEt proved to be the optimal transfer reagent for aliphatic acids. Various deuterated aldehydes were obtained in generally good yields with high D-incorporation.
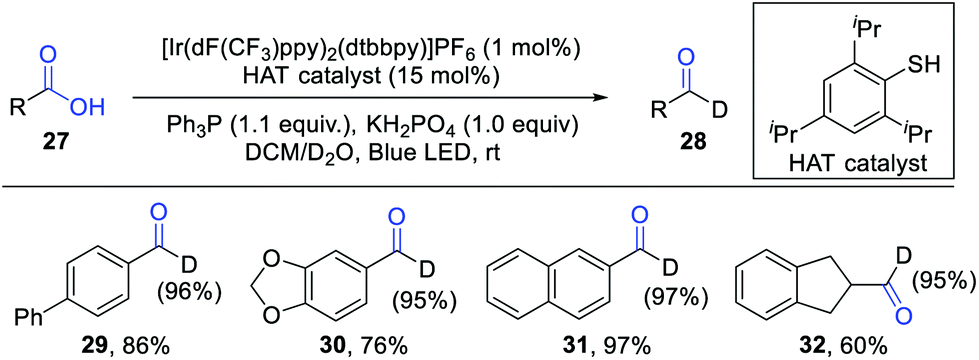 | ||
| Scheme 4 Phosphoranyl radical-mediated deoxygenative deuteration. | ||
In 2019, Doyle et al. reported an elegant intermolecular hydroacylation of olefins from aliphatic carboxylic acids by photoredox catalysis (Scheme 5).11 Key to the success of this reaction is the proper choice of an electron-rich phosphine. The previously used PPh3 and Ph2POEt delivered the desired products only in low yields. Subsequent studies demonstrated that the substrate diphenyl ethylene quenched the photoexcited *Ir(III) over two times than PPh3 or Ph2POEt, which may inhibit the formation of the key phosphoranyl radical intermediates. Therefore, a more reactive phosphine with lower oxidation potential is essential for this reaction. Dimethyl phenyl phosphine PMe2Ph turned out to be the best candidate. Under standard conditions, a wide range of aliphatic carboxylic acids reacted smoothly with styrenes, furnishing the desired dialkyl ketones in high efficiency. Importantly, this protocol was successfully performed on a 10 mmol scale under solvent-free conditions.
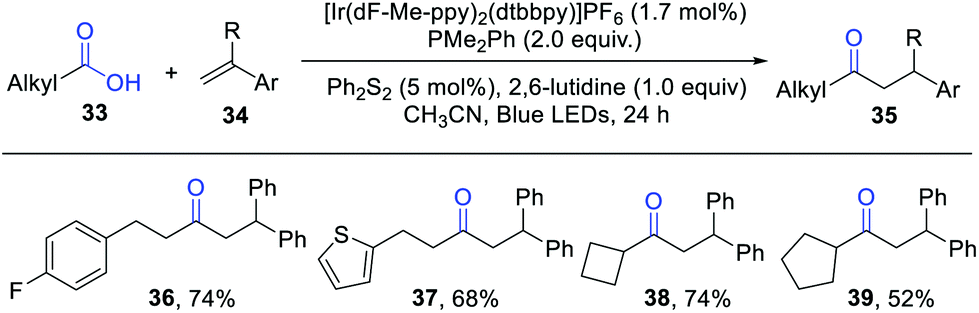 | ||
| Scheme 5 Visible light-induced intermolecular hydroacylation of olefins. | ||
Shortly after these studies, the group of Wang extended this concept to a visible light photocatalytic deoxygenation/radical addition/defluorination cascade reaction of carboxylic acids with α-trifluoromethyl alkenes for the construction of γ,γ-difluoroallylic ketones (Scheme 6).12 Consistent with the work of Zhu and Xie, the aliphatic carboxylic acids failed to give the expected products. Notably, phenylacetic acids performed smoothly in this protocol. This reaction features mild conditions, good functional group tolerance and broad substrate scope, which can be further applied in the late-stage modification of some biologically important molecules, such as adapalene and indometacin.
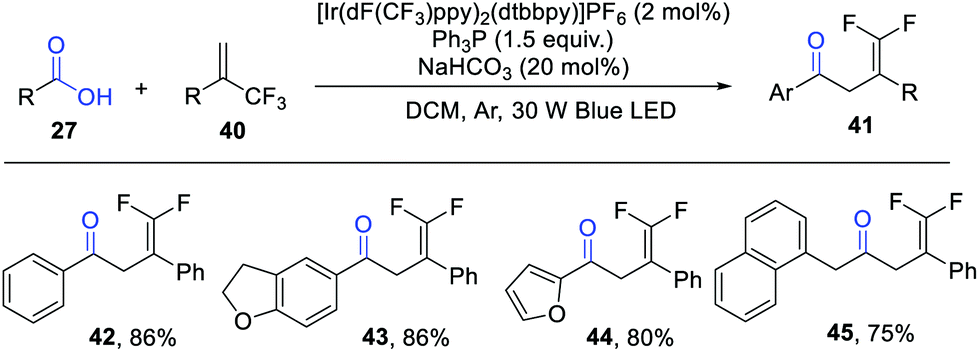 | ||
| Scheme 6 Photocatalytic deoxygenation/defluorination reaction. | ||
An intramolecular deoxygenative acyl radical cyclisation of aromatic carboxylic acids has been developed by Chu, Sun and co-workers (Scheme 7).13 They employed methylene blue (MB) as an efficient photosensitizer and O2 as a green oxidant. When Ph2POEt was used instead of PPh3, a slightly lower yield was observed. Mechanistic studies indicated the intermediacy of acyl radicals, which were generated via the β-scission of phosphoranyl radical intermediates. This protocol provides a straightforward access to diversely functionalised dibenzocycloketones.
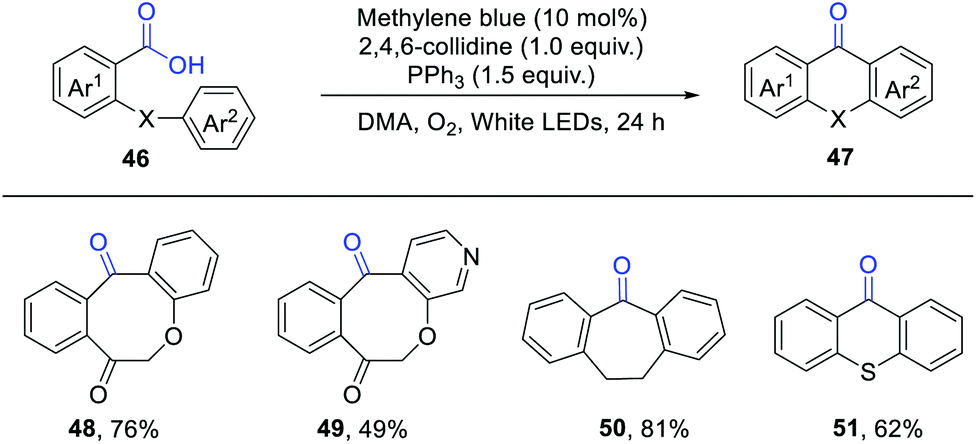 | ||
| Scheme 7 Deoxygenative acyl radical cyclisation of aromatic carboxylic acids. | ||
In 2019, Zhu and Xie et al. described an interesting deoxygenative arylation of aryl carboxylic acids via acyl radical-mediated 1,5-aryl migration (Scheme 8).14 Under the standard conditions, a wide range of o-amino and o-hydroxy diaryl ketones can be obtained in satisfying yields. Importantly, the current reaction can be successfully scaled up to 5 mmol. This reaction begins with the generation of Ph3P radical cation via a single electron oxidation of Ph3P by photoexcited *Ir(III), which reacts with the carboxylate anion to give intermediate 52-A. Subsequently, the selective C–O bond scission of 52-A forms an acyl radical 52-B, which undergoes a sequential intramolecular 1,6-ipso addition and 1,5-aryl migration process to deliver radical 52-D. Finally, the reduction of radical 52-D generates intermediate 52-E, followed by a protonation process gives the desired product. The quantum yield (Φ = 0.39) indicated that the radical chain pathway is less likely. The results of DFT calculations further demonstrated the thermodynamic and kinetic feasibility of this reaction.
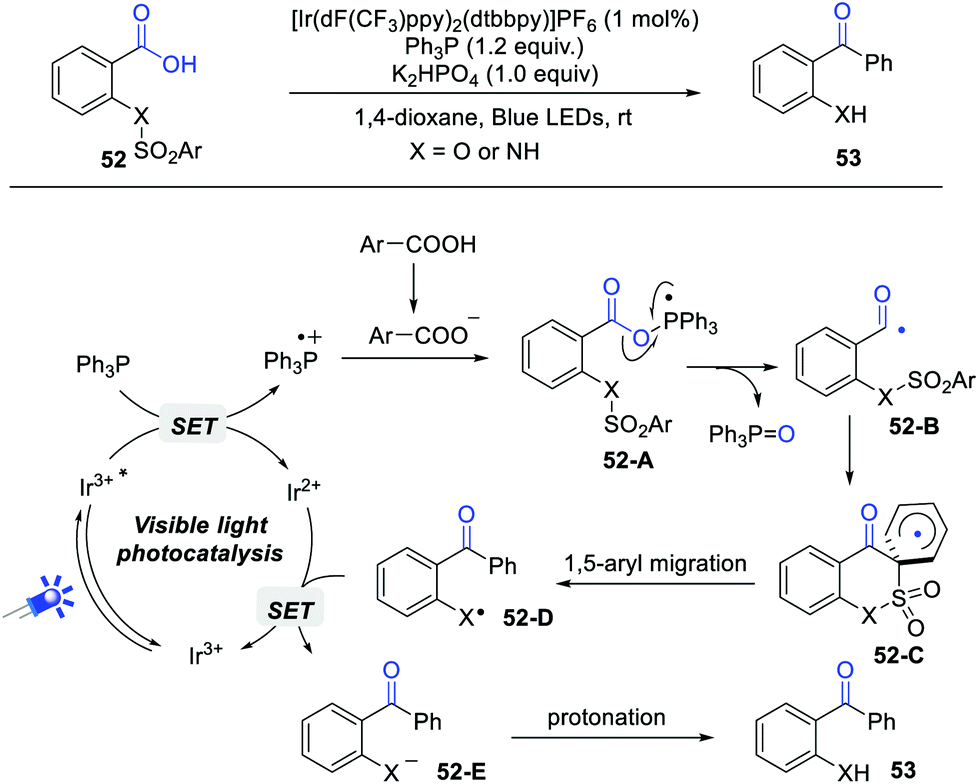 | ||
| Scheme 8 Visible light-induced deoxygenative arylation of carboxylic acids. | ||
3. Deoxygenative transformation of oximes
Iminyl radicals represent one of the most important nitrogen-centered radicals, which have been widely applied in the convenient construction of C–N bonds. Historically, these iminyl radicals are generated from the corresponding α-N-oxyacids, O-acyl and O-aryl hydroxylamines.15 However, the direct generation of iminyl radicals from simple oximes is largely unexploited. Very recently, Yang, Chen and Xiang et al. achieved an impressive iminyl radical reaction of strained cycloketone oximes via phosphoranyl radical-mediated N–O bond activation (Scheme 9).16 PPh3 was the optimal O-transfer reagent, while n-Bu3P resulted in a lower yield. Upon the irradiation by 30 W blue LEDs, various cyclobutanone oximes and alkenes or α-trifluoromethyl alkenes coupled smoothly, furnishing the elongated cyano products in good yields.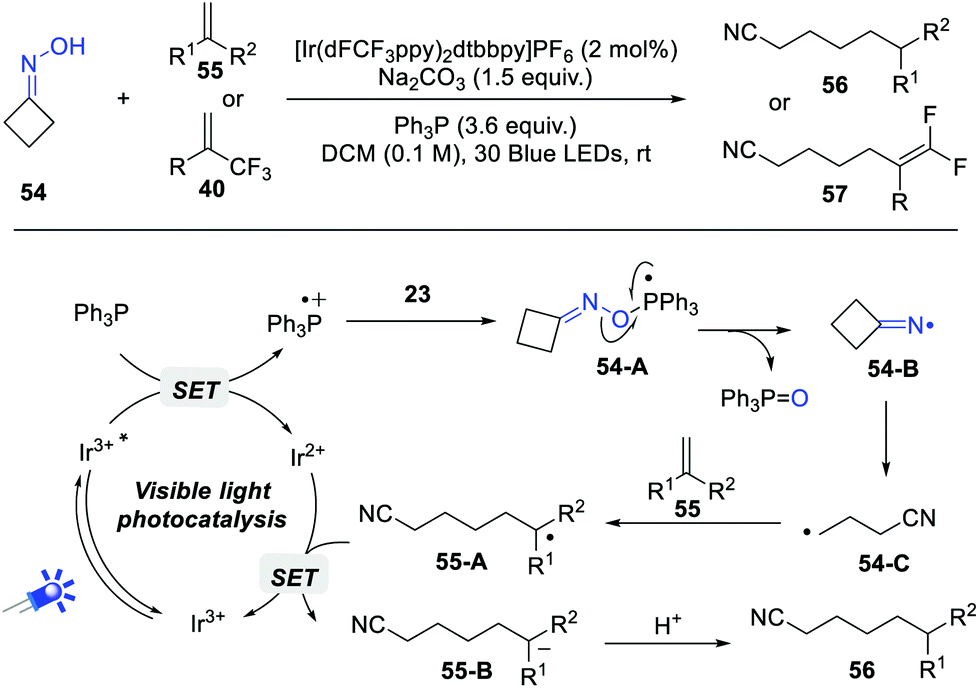 | ||
| Scheme 9 Phosphoranyl radical-mediated N–O bond activation. | ||
On the basis of control experiments, a possible reaction pathway is outlined in Scheme 9. Under photocatalytic conditions, the phosphoranyl radical 54-A is firstly formed via a polar/SET crossover process. The selective N–O bond cleavage of 54-A delivers the key iminyl radical 54-B, which undergoes a rapid β-fragmentation to give a cyanoalkyl radical 54-C. Subsequently, the radical addition of 54-C to alkenes gives a new C-based radical 55-A, which performs a sequential single electron reduction and protonation process to give the final products. The quantum yield of this reaction revealed that a radical long-chain pathway is unlikely to be involved in current protocol.
4. Deoxygenative reduction of sulfoxides
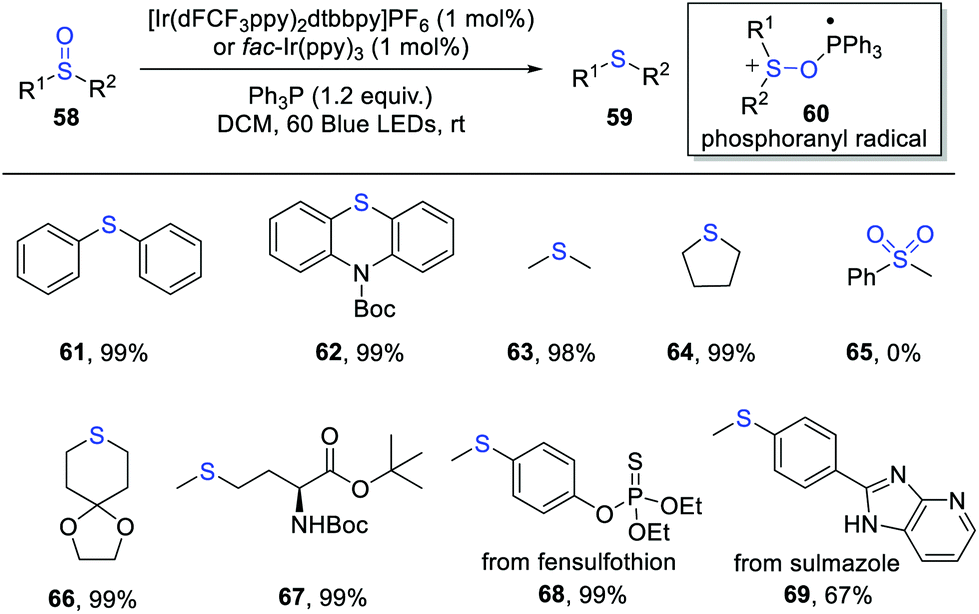
The selective reduction of sulfoxides to sulfides is an important reaction in organic synthesis. Established methods generally require the use of metal hydride reagents, phosphorus and halide ions etc., which often suffer from harsh reaction conditions and limited substrate scopes.17In 2020, Rossi-Ashton, Unsworth and Taylor et al. developed an elegant photocatalytic deoxygenation of sulfoxides for the synthesis of sulfides via phosphoranyl radical-mediated S–O bond activation (Scheme 10).18 The key phosphoranyl radical 60 is formed via a polar nucleophilic addition of sulfoxide to the phosphine radical cation. By using oxidizing and reducing photocatalysts, the authors developed several complementary reaction conditions for this deoxygenative reaction. The attractive features of this transformation are mild conditions, good functional group tolerance and simple operation. A wide range of sulfoxides worked well in this process, providing the corresponding sulfides in excellent yields. Moreover, the synthetic utility was further demonstrated by the late-stage deoxygenation of some biologically active molecules, such as fensulfothion and sulmazole. However, the sulfone substrates were not suitable for this reaction. This protocol provides an alternative and efficient means to the reduction of sulfoxides under mild conditions. Mechanistic studies suggested a radical chain pathway of this reaction.
 | ||
| Scheme 10 Visible light-induced deoxygenation of sulfoxides. | ||
5. Summary
Recently, visible light-induced phosphoranyl radical fragmentation strategies have been successfully applied in the direct activation of C–O, N–O and SO bonds under mild conditions. Phosphoranyl radicals proved to be the key intermediates in these reactions, which undergo a selective β-scission process to generate reactive radical species. By using this novel strategy, various deoxygenative deuteration, arylation, reduction and radical cascade reactions have been developed for the efficient synthesis of valuable products and late-stage functionalisations of natural products, agrochemical and drug molecules. Notable features of these reactions include mild conditions, broad scope and simple operation. We hope this highlight paper will encourage further development of new reactions in this field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (21901258, 21901045) for financial support. We also thank Florian Belitz (RUB) for revising the manuscript.Notes and references
- (a) M. M. Pichon, D. Hazelard and P. Compain, T Metal-Free Deoxygenation of α-Hydroxy Carbonyl Compounds and Beyond, Eur. J. Org. Chem., 2019, 2019, 6320–6332 CrossRef CAS; (b) X. Y. Ooi, W. Gao, H. C. Ong, H. V. Lee, J. C. Juan, W. H. Chen and K. T. Lee, Overview on Catalytic Deoxygenation for Biofuel Synthesis Using Metal Oxide Supported Catalysts, Renewable Sustainable Energy Rev., 2019, 112, 834–852 CrossRef CAS; (c) C. Li, Q. Zhang and Y. Fu, Transition Metal Catalyzed Deoxydehydration of Alcohols, Acta Chim. Sin., 2018, 76, 501 CrossRef CAS.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRS Press, Boca Raton, FL, 2002 Search PubMed.
- (a) R. A. W. Johnstone, in Comprehensive Organic Synthesis: Reduction of Carboxylic Acids to Aldehydes by Metal Hydrides, ed. B. M. Trost and I. Fleming, Oxford, Pergamon, 1991, vol. 8, pp. 259–281 Search PubMed; (b) J. M. Herrmann and B. König, Reductive Deoxygenation of Alcohols: Catalytic Methods Beyond Barton–McCombie Deoxygenation, Eur. J. Org. Chem., 2013, 2013, 7017–7027 CrossRef CAS; (c) A. P. Davis, Comprehensive Organic Synthesis: Reduction of Carboxylic Acids to Aldehydes by Other Methods, ed. B. M. Trost and I. Fleming, Oxford, Pergamon, 1991, vol. 8, pp. 283–305 Search PubMed.
- (a) D. Leca, L. Fensterbank, E. Lacote and M. Malacria, Recent Advances in The Use of Phosphorus-Centered Radicals in Organic Chemistry, Chem. Soc. Rev., 2005, 34, 858–865 RSC; (b) K. Luo, W. C. Yang and L. Wu, Photoredox Catalysis in Organophosphorus Chemistry, Asian J. Org. Chem., 2017, 6, 350–367 CrossRef CAS; (c) F. W. Hoffmann, R. J. Ess, T. C. Simmons and R. S. Hanzel, The Desulfurization of Mercaptans With Trialkyl Phosphites, J. Am. Chem. Soc., 1956, 78, 6414–6414 CrossRef CAS.
- (a) H. Maeda, T. Maki and H. Ohmori, One-Step Transformation of α-Amino Acids to α-Amino Aldehydes Effected by Electrochemical Oxidation of Ph3P, Tetrahedron Lett., 1992, 33, 1347–1350 CrossRef CAS; (b) S. Kim and D. H. Oh, Generation of 5- and 6-Membered Ring Radicals by Deoxygenation of Alkoxy Radicals, Synlett, 2014, 5, 525–527 Search PubMed; (c) L. Zhang and M. Koreeda, Radical Deoxygenation of Hydroxyl Groups via Phosphites, J. Am. Chem. Soc., 2004, 126, 13190–13191 CrossRef CAS PubMed; (d) S. W. Lardy and V. A. Schmidt, Intermolecular Radical Mediated Anti-Markovnikov Alkene Hydroamination Using N–Hydroxyphthalimide, J. Am. Chem. Soc., 2018, 140, 12318–12322 CrossRef CAS PubMed; (e) M. Bietti, A. Calcagni and M. Salamone, The Role of Structural Effects on the Reactions of Alkoxyl Radicals with Trialkyl and Triaryl Phosphites. A Time-Resolved Kinetic Study, J. Org. Chem., 2010, 75, 4514–4520 CrossRef CAS PubMed; (f) Y. Kim, C. W. Bielawski and E. Lee, Oxygen Atom Transfer: A Mild and Efficient Method for Generating Iminyl Radicals, Chem. Commun., 2019, 55, 7061–7064 RSC; (g) D. H. R. Barton, D. O. Jang and J. C. Jaszberenyi, The Invention of Radical Reactions. 32. Radical Deoxygenations, Dehalogenations, and Deaminations with Dialkyl Phosphites and Hypophosphorous Acid as Hydrogen Sources, J. Org. Chem., 1993, 58, 6838–6842 CrossRef CAS.
- (a) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Recent Advances in Radical C−H Activation/Radical Cross-Coupling, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed; (b) Q. Q. Zhou, Y. Q. Zou, L. Q. Lu and W.-J. Xiao, Visible-Light-Induced Organic Photochemical Reactions through Energy-Transfer Pathways, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS; (c) C. K. Prier, D. A. Rankic and D. W. Macmillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (d) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Exploration of Visible-Light Photocatalysis in Heterocycle Synthesis and Functionalization: Reaction Design and Beyond, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed; (e) X. L. Yang, J. D. Guo, H. Xiao, K. Feng, B. Chen, C. H. Tung and L. Z. Wu, Photoredox Catalysis of Aromatic β-Ketoester in Situ toward Transient and Persistent Radicals for Organic Transformation, Angew. Chem., 2020, 59, 5365–5370 CrossRef CAS PubMed; (f) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (g) K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Radicals in Natural Product Synthesis, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC; (h) M. Silvi and P. Enhancing, the Potential of Enantioselective Organocatalysis with Light, Melchiorre, Nature, 2018, 554, 41–49 CrossRef CAS PubMed; (i) M. C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Photocatalytic Decarboxylative Alkylations Mediated by Triphenylphosphine and Sodium Iodide, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed; (j) J. C. Tellis, D. N. Primer and G. A. Molander, Single-electron Transmetalation in Organoboron Cross-coupling by Photoredox/Nickel Dual Catalysis, Science, 2014, 345, 433–436 CrossRef CAS PubMed; (k) R. S. Shaikh, S. J. S. Düsel and B. König, Visible-Light Photo-Arbuzov Reaction of Aryl Bromides and Trialkyl Phosphites Yielding Aryl Phosphonates, ACS Catal., 2016, 6, 8410–8414 CrossRef CAS.
- J. A. Rossi-Ashton, A. K. Clarke, W. P. Unsworth and R. J. K. Taylor, Phosphoranyl Radical Fragmentation Reactions Driven by Photoredox Catalysis, ACS Catal., 2020, 10, 7250–7261 CrossRef CAS.
- M. Zhang, J. Xie and C. Zhu, A General Deoxygenation Approach for Synthesis of Ketones from Aromatic Carboxylic Acids and Alkenes, Nat. Commun., 2018, 9, 3517 CrossRef PubMed.
- E. E. Stache, A. B. Ertel, T. Rovis and A. G. Doyle, Generation of Phosphoranyl Radicals via Photoredox Catalysis Enables Voltage–Independent Activation of Strong C–O Bonds, ACS Catal., 2018, 8, 11134–11139 CrossRef CAS PubMed.
- M. Zhang, X. A. Yuan, C. Zhu and J. Xie, Deoxygenative Deuteration of Carboxylic Acids with D2O, Angew. Chem., Int. Ed., 2019, 58, 312–316 CrossRef CAS PubMed.
- J. I. Martinez Alvarado, A. B. Ertel, A. Stegner, E. E. Stache and A. G. Doyle, Direct Use of Carboxylic Acids in the Photocatalytic Hydroacylation of Styrenes To Generate Dialkyl Ketones, Org. Lett., 2019, 21, 9940–9944 CrossRef CAS PubMed.
- Y. Q. Guo, R. Wang, H. Song, Y. Liu and Q. Wang, Visible-Light-Induced Deoxygenation/Defluorination Protocol for Synthesis of γ,γ-Difluoroallylic Ketones, Org. Lett., 2020, 22, 709–713 CrossRef CAS.
- H. Jiang, G. Mao, H. Wu, Q. An, M. Zuo, W. Guo, C. Xu, Z. Sun and W. Chu, Synthesis of Dibenzocycloketones by Acyl Radical Cyclization from Aromatic Carboxylic Acids Using Methylene Blue as A Photocatalyst, Green Chem., 2019, 21, 5368–5373 RSC.
- R. Ruzi, J. Ma, X. A. Yuan, W. Wang, S. Wang, M. Zhang, J. Dai, J. Xie and C. Zhu, Deoxygenative Arylation of Carboxylic Acids via Aryl Migration, Chem. – Eur. J., 2019, 25, 12724–12729 CrossRef CAS PubMed.
- (a) X. Y. Yu, Q. Q. Zhao, J. Chen, W.-J. Xiao and J.-R. Chen, When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts, Acc. Chem. Res., 2020, 53, 1066–1083 CrossRef CAS PubMed; (b) X.-Q. Hu, J.-R. Chen, Q. Wei, F.-L. Liu, Q.-H. Deng, A. M. Beauchemin and W.-J. Xiao, Photocatalytic Generation of N-Centered Hydrazonyl Radicals: A Strategy for Hydroamination of α,β-Unsaturated Hydrazones, Angew. Chem., Int. Ed., 2014, 53, 12163–12167 CrossRef CAS PubMed; (c) X.-Q. Hu, X. Qi, J.-R. Chen, Q. Q. Zhao, Q. Wei, Y. Lan and W.-J. Xiao, Catalytic N-radical Cascade Reaction of Hydrazones by Oxidative Deprotonation Electron Transfer and TEMPO Mediation, Nat. Commun., 2016, 7, 11188 Search PubMed; (d) J.-R. Chen, X.-Q. Hu, L. Q. Lu and W.-J. Xiao, Visible Light Photoredox-Controlled Reactions of N-Radicals and Radical ions, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC; (e) T. Xiong and Q. Zhang, New Amination Strategies Based on Nitrogen-centered Radical Chemistry, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC; (f) H. Jiang, X. An, K. Tong, T. Zheng, Y. Zhang and S. Yu, Visible-Light-Promoted Iminyl-Radical Formation from Acyl Oximes: A Unified Approach to Pyridines, Quinolines, and Phenanthridines, Angew. Chem., Int. Ed., 2015, 54, 4055–4059 CAS; (g) S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Photoinduced Remote Functionalization of Amides and Amines Using Electrophilic Nitrogen-Radicals, Angew. Chem., Int. Ed., 2018, 57, 12945–12949 CrossRef CAS PubMed; (h) J. Davies, S. P. Morcillo, J. J. Douglas and D. Leonori, Hydroxylamine Derivatives as Nitrogen-Radical Precursors in Visible-Light Photochemistry, Chem. – Eur. J., 2018, 24, 12154–12163 CrossRef CAS PubMed.
- P. J. Xia, Z. P. Ye, Y. Z. Hu, D. Song, H. Y. Xiang, X. Q. Chen and H. Yang, Photocatalytic, Phosphoranyl Radical-Mediated N−O Cleavage of Strained Cycloketone Oximes, Org. Lett., 2019, 21, 2658–2662 CAS.
- (a) L. Shiri and M. Kazemi, Deoxygenation of Sulfoxides, Res. Chem. Intermed., 2017, 43, 6007–6041 CAS; (b) A. Ghosh, M. Lecomte, S. H. Kim-Lee and A. T. Radosevich, Organophosphorus-Catalyzed Deoxygenation of Sulfonyl Chlorides: Electrophilic (Fluoroalkyl)sulfenylation by PIII/PV=O Redox Cycling, Angew. Chem., Int. Ed., 2019, 58, 2864–2869 CrossRef CAS PubMed.
- A. K. Clarke, A. Parkin, R. J. K. Taylor, W. P. Unsworth and J. A. Rossi-Ashton, Photocatalytic Deoxygenation of Sulfoxides Using Visible Light: Mechanistic Investigations and Synthetic Applications, ACS Catal., 2020, 10, 5814–5820 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2020 |