
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic covalent chemistry in polymer networks: a mechanistic perspective
Johan M.
Winne
*a,
Ludwik
Leibler
*b and
Filip E.
Du Prez
 *a
*a
aPolymer Chemistry Research Group and Laboratory for Organic Synthesis, Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281 S4-bis, B-9000 Ghent, Belgium. E-mail: Johan.winne@ugent.be; Filip.duprez@ugent.be
bUMR Gulliver 7083 CNRS, ESPCI Paris, PSL Research University, 10 rue Vauquelin, 75005 Paris, France. E-mail: Ludwik.leibler@espci.fr
First published on 16th October 2019
Abstract
The incorporation of dynamic covalent linkages within and between polymer chains brings new properties to classical thermosetting polymer formulations, in particular in terms of thermal responses, processing options and intrinsic recycling abilities. Thus, in recent years, there has been a rapidly growing interest in the design and synthesis of monomers and cross-linkers that can be used as robust but at the same time reactive organic building blocks for dynamic polymer networks. In this perspective, a selection of such chemistries is highlighted, with a particular focus on the reaction mechanisms of molecular network rearrangements, and on how various mechanistic profiles can be related to the mechanical and physicochemical properties of polymer materials, in particular in relation with vitrimers, the recently defined third category of polymer materials. The recent advances in this area are not only expected to help direct promising emerging polymer applications, but also point towards the need for a better fundamental understanding of chemical reactivity within a macromolecular context.
Introduction
Classical polymer materials are expected to be chemically inert and thus to have a fixed molecular make-up. Normally, chemical reactivity within polymers is considered as a nuisance that needs to be avoided or controlled with stabilizers. This situation has changed over the last few decades by a growing interest in the area of dynamic polymer systems, designed to achieve functional characteristics such as self-healing, shape-memory, repairing, stimuli-responsiveness and adaptability.1–8 Whereas these desirable polymer dynamics can be influenced in many ways and in different contexts, such as in supramolecular systems, one particular emerging strategy in this area has been to deliberately incorporate some degree of chemical reactivity into the polymer chains. Thus, many recent studies have focused on the design of macromolecular matrices with exchangeable, reversible or adaptable covalent bonds.1,2,7–9 In light of these developments, chemical reactivity considerations can no longer be ignored in considering the physical properties of such polymer materials, and often even take on a dominant role.In this perspective paper, we would like to focus on one particular subclass of dynamic polymer materials, i.e. those made up of a (highly) cross-linked macromolecular network, wherein the covalent links between or within polymer chains can be dynamically exchanged via a chemical reaction. Such dynamic covalent polymer materials can thus undergo a net process corresponding to a molecular network rearrangement (MNR). In a broader context, with focus on polymer applications, such polymer systems have also been defined as covalent adaptable networks (CANs),7,8,10 as such materials can ‘adapt’ their macromolecular architecture in response to an external stimulus or trigger, despite their cross-linked nature. The response or triggered behavior of such polymer materials will not only rely on the structural features of the polymer matrix but will also strongly depend on the rate and nature of the chemical bond exchanges within the polymer matrix. Thus, the molecular mechanisms of these macromolecular rearrangement reactions require close attention within the study of such materials, as they can affect the mechanical material properties.
Building a fundamental understanding of the variables that govern material properties of dynamic polymer networks or rearranging polymer networks should provide further guidance for the design of new materials and for the selection and optimization of suitable building blocks. Herein, we provide the reader with a selection of exchange chemistries that have been explored in this regard, with a focus on their reactivity and mechanistic profiles, pointing towards opportunities both in material as well as chemical science. For recent overviews of such chemistries with a focus on their diversity and applications in polymers, we refer to the reviews by Zhao and Xie,11 and by Konkolewicz.12
Before discussing various types of network rearrangement reactions, we first provide an overview of how synthetic polymer materials have been categorized in terms of properties that are related to their macromolecular architecture, and then how the categories of thermoplastic and thermosetting polymers13 have been refined and even expanded with a third category, vitrimers,14–16 when taking into account dynamic covalent macromolecular architectures.
Thermoplastics, thermosets, and vitrimers
At low temperatures, polymer materials are either glassy or partially crystallized and behave like hard solids. Found on their response to temperature or solvent, three categories of polymer materials can be distinguished.Thermoplastic polymers consist of linear or branched polymer chains. When enough thermal energy is provided, thermoplastic polymers melt and the chains will be able to diffuse. Thus, when heated above a certain temperature level, thermoplastic polymers behave like a viscoelastic liquid and flow. When cooled, the material solidifies again. This fact allows for the typical ‘plastic’ continuous processing options that have made (thermo)plastics into the most popular materials for rapid production of consumer goods of various shapes and dimensions.
When exposed to good solvents (and heat), thermoplastic polymers will completely dissolve.
Because of their macromolecular character, molten thermoplastics behave like viscoelastic liquids, especially when chains are long and entangled.
Thermosetting polymers,13 on the other hand, when heated above their glass or melting temperature become softer, but they do not flow. Thermosetting polymers derive their enduring and wide-ranging resistance to deformation from their (densely) cross-linked macromolecular structure, encompassing a macroscopic covalent network which spans the whole volume of the material. Segmental motions are hindered by connectivity and chain segments cannot diffuse. Thus, well above their glass transition temperature, thermosets exhibit rubber-like elasticity and behave like viscoelastic solids. When the network does not contain many defects such as dangling ends or unattached chains, it exhibits mainly elastic responses to deformations with little viscous losses. Conventionally, rubbers (or elastomers) are defined as thermosets with a glass transition temperature below room temperature.
Even at high temperatures, when immersed into a good solvent, a thermosetting polymer material can only swell, and only a limited fraction of the material, corresponding to chains unattached to the network, can be dissolved.
Thermosets have an enhanced dimensional stability as they show resistance to polymer wear processes such as dissolution, creep or solvent stress cracking. By necessity, thermosetting materials need to be chemically synthesized (cross-linked) into their final shape. This so-called ‘curing’ process puts limitations on the facility with which various shapes and dimensions can be given to such materials. Once this final shape is chemically fixed, the material becomes unprocessable.
Vitrimer polymers are the third category of organic polymer materials, introduced by Leibler and coworkers in 2011.14–16 When heated, vitrimers behave like a viscoelastic liquid, but when exposed to solvent they will only show a limited soluble fraction, related to defects and unattached chains. Vitrimers derive these properties from a covalent molecular network that can change its topology through molecular rearrangements, while preserving the total number of bonds in the network, i.e. the network connectivity. When a vitrimer is heated above its glass (or melting) temperature, molecular rearrangements can take place and the topology of the network fluctuates. Thus, network segments can diffuse even though the network preserves its connectivity while spanning the volume of the whole sample. In terms of material properties, the permanent nature of the network enhances the resistance to polymer wear processes such as dissolution, creep or solvent stress cracking. The dynamic nature of the network facilitates processing and reprocessing and brings recycling possibilities despite the presence of permanent covalent cross-links.
In a broader context, vitrimers are a very specific type of dynamic polymer networks. It is important to stress at this point the fundamental difference between permanent dynamic networks (i.e. vitrimers) and reversible dynamic networks. Reversible dynamic polymer networks contain non-permanent covalent links or cross-links that are able to break and reform through reversible chemical bonding reactions. Such systems have been studied theoretically and experimentally for decades,17–20 and have recently received a great deal of renewed attention from both academic and industrial research groups.1–12
When heated, reversible polymer networks will undergo topology fluctuations with fluctuations in overall connectivity, whereas in vitrimers, the postulated permanent nature of the network implies that the connectivity remains unchanged at all temperatures and during the entire experimental time. This distinction is important because it will determine to some extent the macroscopic properties that can be expected for such polymer networks. In a rearranging network with fluctuations in connectivity, i.e. changes in cross-link density, the number of bonds within the polymer material is not constant, and the polymer network can be triggered (e.g. through application of heat) to undergo a gel-to-sol transition,18–20 or to evolve from a classical thermosetting macromolecular architecture to a classical thermoplastic one, and vice versa. These materials can thus indeed be expected to either behave as a thermosetting polymer or as a thermoplastic polymer, depending on the conditions that are applied. Thus, heat can trigger a loss of connectivity in a reversible dynamic network and promote thermoplastic flow, and similarly, the addition of solvent can readily trigger a loss of connectivity and even a complete dissolution after extensive loss of the network bonds. Thus, formally, reversible polymer networks are thermoplastic, as they flow when heated and can be completely dissolved in a good solvent.
In the context of dynamic polymer networks, it should also be noted that the three categories of polymer materials outlined above are idealised systems.
On the one hand, these terms refer to combinations of properties that are expected. Perfect thermosets cannot flow when heated and should resist dissolution in a good solvent. Perfect thermoplastic polymers should flow when heated and should be soluble in a good solvent. Perfect vitrimers will flow when heated but will also resist dissolution in a good solvent, even when heated.
On the other hand, and more fundamentally, the distinction in these three classes should also be understood in connection to the underlying macromolecular architecture. Perfect thermosets are made up by a permanent and static molecular network that may have some conformational freedom (or elasticity) but cannot undergo configurational changes. Perfect thermoplastics are non-covalently or reversibly linked polymer chains (or oligomers) that can diffuse by overcoming their weak intramolecular forces or bonds. Finally, vitrimers are permanent molecular networks that can change their molecular configuration through segmental diffusion but without losing network bond integrity.
Yet, in practice, distinctions between polymer categories may be less evident to make. For example, high-molar-mass highly entangled thermoplastics are characterized by very slow stress relaxations, preventing processing by conventional methods. If such a high mass is desired, such polymers are often synthesized and shaped by thermoset technologies and the shape of the object has to be defined or controlled during the synthesis. Still, such polymers can be soluble in a good solvent. For dynamic polymer networks, the situation may be even more involved. For example, although reversible dynamic networks with inherent fluctuations in cross-link density should be categorized as thermoplastic, and not as vitrimers, it might in practice be difficult to significantly shift the bonding/debonding equilibrium to a dissociated state, depending on the reversible bond forming thermodynamics and reaction conditions. We will come back to this point later.
The possibility that permanent polymer networks with exchangeable bonds could flow has been anticipated by Tobolsky already in 1956.21 However, the swelling behavior of such dynamic polymer networks is less obvious. Indeed, unlike for thermosets, the soluble fraction of vitrimers is expected not only to contain chains that were initially not attached to the network, but also chains or segments that were detached through the network rearrangements. Theory and simulations, reported by Smallenburg, Leibler and Sciortino in 2013,22a seem to indicate that, at least for vitrimers obtained through AfB2 condensation, at swelling equilibrium, the connectivity of the swollen vitrimer network is changed with respect to the initial vitrimer network, showing less defects and becoming more perfect. Here, theory predicts that the soluble fraction and swollen network structure and connectivity will depend on solvent concentration but are independent of temperature. In contrast to thermoplastics, the equilibrium cannot be shifted to the solution state by heating. Thus, the features found in vitrimers defy the classification criteria of both thermosets as well as thermoplastics, justifying their position as the third category of polymer materials that needs to be defined.
Rearrangement reaction pathways
Before discussing detailed examples of dynamic polymer networks, it is worthwhile to first distinguish a few distinct mechanistic scenarios. The exact architecture of macromolecular networks, including how cross-links and exchangeable bonds are distributed over the polymer backbone, and the relative proximity of exchanging groups besides the availability and mobility of catalytic species, are all factors that need to be taken into careful consideration. For clarity, however, it is good to first describe a generalized picture that makes abstraction of these finer details and focuses on the nature of the bond exchange reaction itself.Within a given macromolecular network, a rearrangement reaction can proceed via any of many possible mechanistic pathways, and the nature of the preferred or kinetically dominant molecular network rearrangement (MNR) pathway will be important in considering the mechanical properties of a network, in particular with respect to distinction between the vitrimer (permanent) and reversible dynamic polymer networks described above. For our discussion, it is convenient to consider network segments as if they were single molecules (Scheme 1), that undergo elementary rearrangement reactions just like small molecules would, making abstraction of specific polymer effects such as long-range correlations or accessibility of exchangeable bonds.17–20,22b Mechanistically, MNRs can then be divided into two principal groups:
(i) Concerted (or single step) MNRs
(ii) Stepwise (or multistep) MNRs
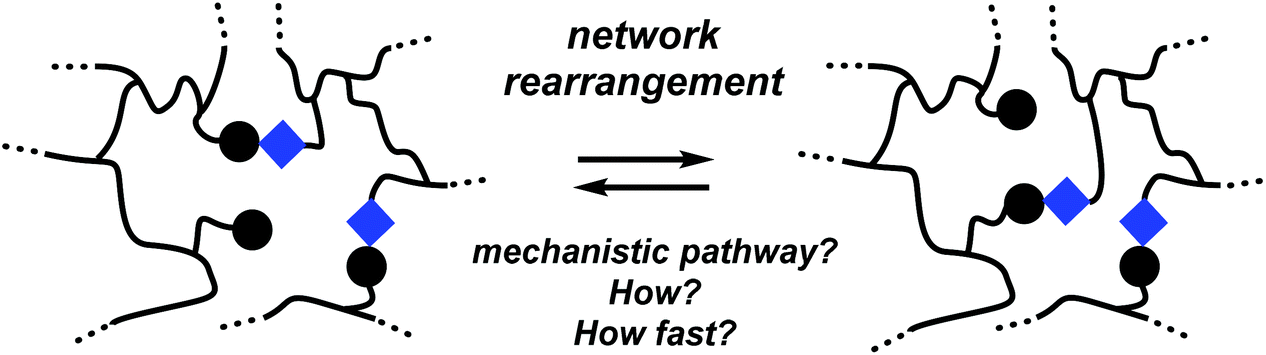 | ||
| Scheme 1 Molecular network rearrangements (MNR). | ||
In a concerted MNR (Scheme 2a), which is the least common reaction type, the new network bond can be formed at the same time as the old network bond is broken, without any kind of intermediate state, going through an ordered transition state. Polymer networks that exclusively undergo concerted rearrangements will thus always show the exact same level of connectivity or cross-link density, as all possible network topologies will have the same number of cross-links. A concerted MNR can thus be considered as an ‘ideal’ scenario to arrive at the formulated requirements for vitrimer polymers (vide supra).
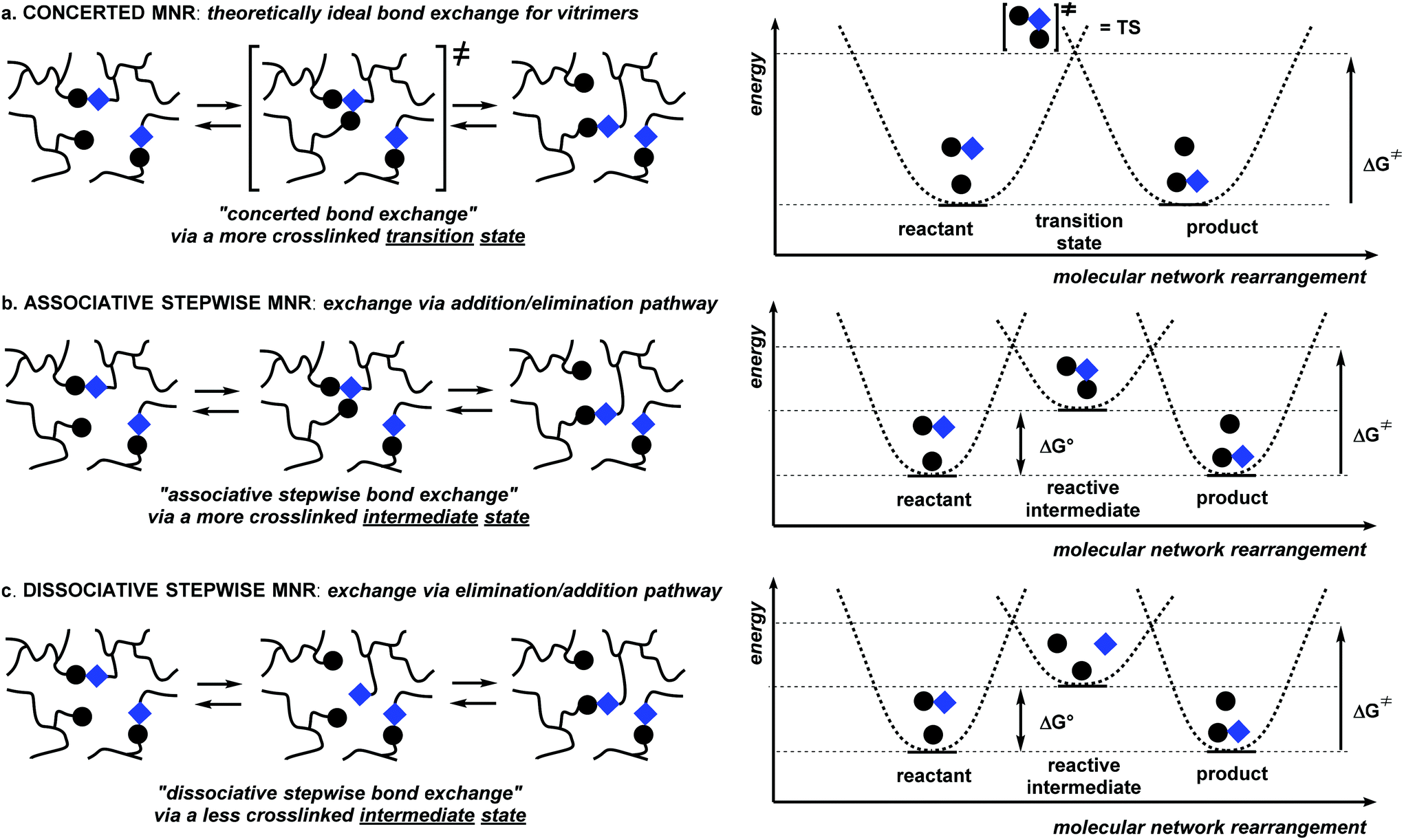 | ||
| Scheme 2 Different types of molecular network rearrangements (MNRs) within covalent networks and their energy profiles. | ||
The two most straightforward examples of non-concerted or stepwise MNRs involve just two consecutive reaction steps: a discrete bond forming and a discrete bond cleavage step. The order with which these steps occur in a two-step MNR is quite important. An associative stepwise MNR can be achieved through an addition/elimination pathway (Scheme 2b), whereas the reversed order process, or an elimination/addition pathway constitutes a dissociative stepwise MNR (Scheme 2c). The dissociative stepwise MNR has previously also been identified as a ‘reversible addition’ bond exchange process,8 whereas the associative stepwise MNR (also referred to as ‘reversible exchange’ or ‘associative exchange’) has so far been considered as an important prerequisite to arrive at vitrimer polymer network properties (i.e. flow when heated but have limited solubility).14–16,23
In a polymer network that undergoes an associative stepwise MNR (Scheme 2b), the rearrangement pathway involves the prior association of two polymer chains, resulting in a new covalent network bond, followed by an elimination step that fragments another network bond. In the reactive (higher energy) intermediate state, the cross-link density is thus temporarily increased, and the overall reaction rate will also depend on the proximity of the exchanging cross-links, as is also expected for concerted MNRs. In a strict sense, this situation does not meet the ideal requirements about the topology and connectivity fluctuations that are put forward for vitrimer polymer materials, as the overall connectivity can be increased upon heating, as the equilibrium can shift to the endothermic side. In practice, however, this effect will of course often be negligible, resulting in a practically constant cross-link density under a wide range of conditions.
In a polymer network that undergoes the bonding/debonding reactions in reverse order, i.e. the dissociative stepwise MNRs (Scheme 2c), no association of polymer chain segments is required, and a polymer chain can undergo fragmentation by itself, forming a temporarily de-cross-linked intermediate state with the formation of reactive (higher energy) chain ends. A cross-link can then reform in a second step involving another polymer chain, resulting in the overall dissociative stepwise MNR reaction. This situation also does not meet the ideal requirements about the topology and connectivity fluctuations put forward for vitrimeric polymer materials, as the overall connectivity can be decreased upon heating. Moreover, this decrease in connectivity is not limited by the conditional involvement of another reactive species and can thus become quite extensive. The decross-linked intermediate state should also be entropically favored, rendering a significant shift in the bonding equilibrium more likely here than in associative stepwise MNRs.
It should be noted that many other, more complex multistep MNR scenarios are probable and feasible. For example, a catalysed version of all three scenarios in Scheme 2 is possible, where a third species (as a catalytic additive) is involved. Association of this catalyst is then an additional prerequisite for the exchange to proceed at an appreciable rate. This can be quite important, as the ratio of this catalyst compared to the total number of dynamic cross-links will put an upper limit on the extent with which the equilibrium can be shifted to states with a different degree of connectivity (cf.Scheme 2b and c).
One particular alternative pathway we would like to finally point out here, is a dissociative stepwise MNR pathway that takes place through the mediation of a reactive small molecule as a mediator (formally speaking acting as a rearrangement catalyst). In this fourth scenario (Scheme 3), polymer chains are reversibly linked through a condensation reaction, uniting two chain fragments while eliminating a small molecule. As long as the small molecule that is formed during condensation (most typically a molecule of water) remains present in the polymer matrix, the cross-links will remain reversible via a dissociated intermediate state. In classical polymer synthesis terms, the dynamic process shown in Scheme 3 corresponds to a ‘reversible condensation’ polymerisation.8
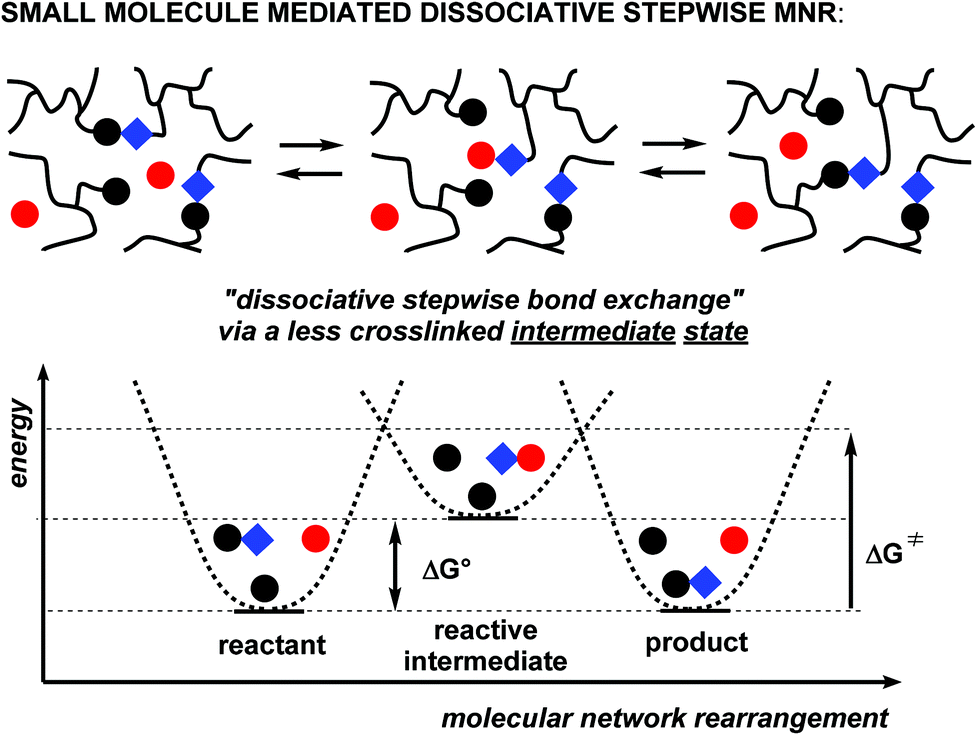 | ||
| Scheme 3 Small molecule mediated dissociative exchange. | ||
The mechanistic categories outlined above will allow a more systematic discussion of dynamic polymer networks that undergo rearrangement reactions. Examples of all four of the presented generalized mechanistic types have been reported in the literature, and below, we have selected illustrative cases. This selection is not an exhaustive listing of dynamic covalent chemistries for each mechanistic type but has rather been made based on the amount of available studies and characterisations of the chemistry underlying the rearrangement process.
An important question we would like to address in this perspective is the relationship between mechanistic profiles of MNRs and how this will influence the emergence of vitrimer properties (as opposed to thermoplastic or thermosetting ones). Previous descriptions, including those offered by our research groups in the literature,22a,23 have not fully taken into account the complete reactivity context. In fact, an ideal vitrimer should only have MNRs via a concerted pathway (Scheme 2a), where the network exists in a single well-defined potential energy well, with the exact same degree of cross-linking, and dynamics are only possible through a fleeting associated transition state that will have no lifetime. However, as we shall discuss below, this situation is very rare.
The much more common stepwise MNRs, on the other hand, will by definition show energy profiles for polymer segments with a higher energy (endothermic) state (as a separated potential energy well), where the degree of connectivity can be markedly different. The expectation then, within this conceptual framework, is that none of these materials can be ‘true’ vitrimers, as the overall degree of connectivity will always change upon heating, by shifting of the equilibrium to the endothermic side. However, it should be clear that many situations will be possible where highly dynamic MNRs can be achieved in a wide temperature window, without necessitating significant changes in the overall connectivity of the network. Thus, many stepwise MNRs, and in particular the associative ones, will result in clear vitrimer properties. This point will be addressed more explicitly later on.
Associative stepwise rearrangements
The most common scenario that can be expected to meet or at least approach ideal vitrimer requirements is found in polymer systems that can exclusively undergo associative stepwise MNRs (Scheme 2b). In these systems, network decross-linking is not possible, and the build-up of additional cross-links can often be suppressed, even under conditions of very rapid bond exchanges, leading to only negligible fluctuations in cross-linking density, while having very rapid fluctuations in network topology. The first truly associative bond exchanges were purposely implemented in polymer chemistry by Bowman and Cook, already in 2005, by their elegant design of cross-linked networks that undergo photoinduced radical addition–fragmentation reactions resulting in plasticity and a certain stress relaxation.24 Being chain mechanisms in nature, their overall rates critically depend on concentration of free radicals and on the relative reaction rates of many competing processes (e.g. initiation vs. termination rates). Radical addition and fragmentation reactions often proceed without barriers, operating at the diffusion limit with no real activation energy. They do not require thermal triggers, and do not adhere to the generic situations outlined in Scheme 2. Radical chemistry will thus not be explicitly considered in this perspective, and we refer readers to other dedicated reviews.7–12In 2011, Leibler and coworkers purposely introduced an associative bond exchange reaction into a polymer network.14 The underlying idea was to design a material that will flow when heated but would not be soluble owing to the permanent nature of the network. This is a combination of properties that is common for inorganic materials like silica glasses but is not found for organic polymer materials. Thus, they designed an epoxy-based polyester resin that includes free hydroxyls, judiciously placed next to ester linkages that make up the entire polymer backbone (Scheme 4a). Introduction of a suitable (Lewis) acid or base catalyst accelerated the dynamic interchange of ester linkages between hydroxyl moieties via a very feasible addition/elimination reaction inside this polymer network. This approach gave an organic material that can swell, but is completely insoluble in good solvents, even at high temperatures. Despite this insolubility, the material also displays full stress relaxation and flow, mediated by the rapid dynamic bond exchanges. These materials also showed a viscosity profile typical of strong glass formers, not showing the fragility that is classically associated with organic polymers. Without undergoing side reactions, the material also cannot show a sol-to-gel transition, as the overall cross-link density does not decrease upon heating.22a The temperature dependence of the viscosity of these materials showed a good agreement with that observed for the chemical reaction rate, showing a connection between the dynamics of viscoelastic flow and the chemical dynamics.15,23
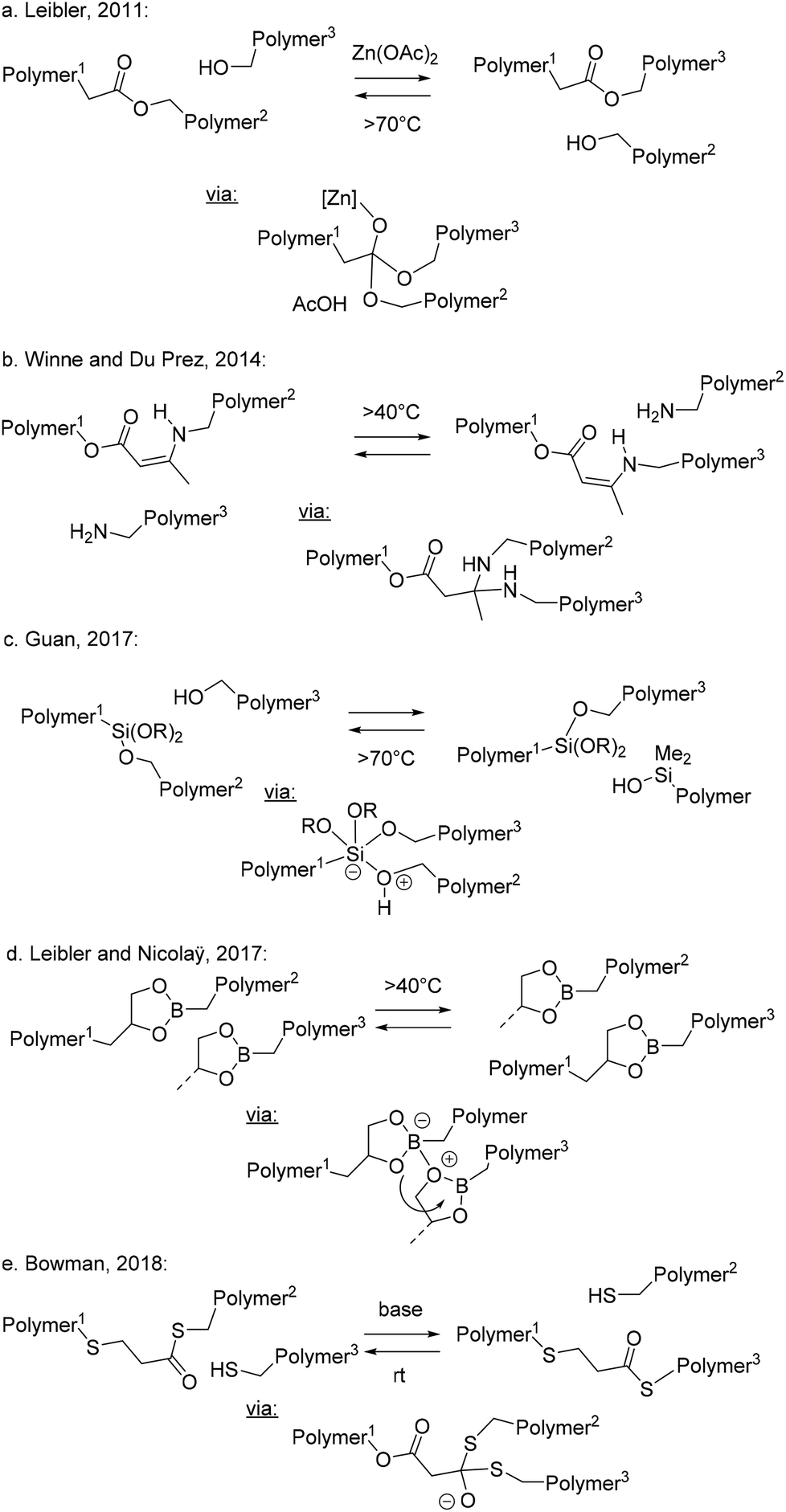 | ||
| Scheme 4 Examples of associative stepwise MNR-based materials. | ||
By changing the catalyst and its concentration, the mechanical properties of polyhydroxy-polyester resins have been shown to be directly modified and controlled.15,23 Catalyst loading shows a linear relationship to reaction rate, if the mechanism is first order in that catalyst. By keeping the catalyst to hydroxyl ratio low, side reactions and extensive additional cross-linking upon heating can be minimised. Different catalysts also result in other activation energies, which can be measured in situ by mechanical stress relaxation experiments. Thus, judicious choice of small amounts of additives that can act as a catalyst can drastically alter the mechanical properties of one and the same polymer matrix, which is a powerful design concept for polymer materials.
In 2014, Winne and Du Prez and coworkers introduced the dynamic covalent chemistry of enaminone-moieties derived from amines and β-ketoesters as a platform chemistry for the design of vitrimer materials.25 The resulting linkage can formally be considered as a vinylogous urethane, with a vinylic bond inserted in between the electron donating nitrogen and the electron-withdrawing ester moiety (Scheme 4b). This electronic effect makes vinylogous urethanes into thermodynamically stable linkages, very similar in nature to classical isocyanate-derived urethane cross-links. However, whereas normal urethanes do not readily undergo (catalyzed) addition/fragmentation reactions because of their weak electrophilic properties,26 the vinylogous urethanes are more reactive because of the Michael-type reactivity of the α,β-unsaturated carbonyl moiety. Another interesting feature of this chemistry is its resistance toward hydrolysis, as amines and β-ketoesters spontaneously undergo a condensation to form the enaminone, even in aqueous media.
Another classical associative stepwise MNR bond exchange we want to draw attention to here is that between hydroxyls and siloxanes. In 2017, Guan and coworkers implemented siloxanes as cross-linkers for hydroxy-functionalised polystyrene backbones (Scheme 4c).27 The resulting high Tg networks could undergo rapid exchange reactions upon heating via an addition/elimination reaction of the silyloxy groups to the silyl ether linkages. Interestingly, the inclusion of basic amine moieties in the cross-linkers reduced the activation energy for the exchange reaction and accelerated the reaction as a covalently embedded or internal catalyst.
Also in 2017, Nicolaÿ and Leibler reported the straightforward synthesis of cross-linked simple polyvinylic networks incorporating boron-heterocyclic cross-links (Scheme 4d).28 Dioxaborolanes are cyclic esters of boronic acids and a vicinal diol, which spontaneously undergo a condensation upon bringing the two together. This ring formation is entropically favored by the expulsion of two molecules of water. The boron–oxygen bonds are very strong because of their significant π-bonding character between the oxygen lone pair and the unoccupied p-orbital on boron, reminiscent of a classical carbonyl bond. At the same time, the boron center remains highly electrophilic and readily forms tetrahedral intermediate adducts with a wide range of nucleophiles.
The dynamic exchange with hydroxyls and diols has been used in boronic ester transesterification vitrimers developed by Guan and coworkers,29 but Nicolaÿ and Leibler found that the exchange can also happen as a metathesis reaction between two dioxaborolanes without the need for a free hydroxyl moiety, simplifying its implementation. The exchange mechanism of the metathesis is unclear, but it is likely a multistep process, initiated by the formation of a zwitterionic adduct wherein alkoxide residues can be readily exchanged between the two boron centers, ultimately enabling a complete crossover of bonding partners – and thus the attached polymer chain portions – through a series of addition/fragmentation reactions. The resulting MNR profile is thus likely more complex than that shown in Scheme 2b, as multiple intermediate states are formed here, wherein in total four bonds are broken and four new bonds are formed, and alternative neutral intermediate species may be involved. The timing of these intermediate steps will be of little importance, as all realistic reactive intermediates are expected to have a higher connectivity than that of the resting states of the polymer networks. Interestingly, this chemical platform allows the incorporation of associative dynamic exchanges without the need for a catalyst, but also without the need for any other functionality except the dioxaborolane, which undergoes a metathesis with itself and is a quite stable moiety.
As a final example, we also want to point out the recent work of Bowman and coworkers on the dynamic exchange of thioesters with free thiols (Scheme 4e).30,31 This transesterification is very rapid in the presence of base or acid catalysts, even at room temperature. However, by cleverly combining this chemistry with a photobase, Bowman and coworkers obtained a thiol–ene based polymer network that can be switched between dynamic and non-dynamic states by including a photobase, opening up a new concept for dynamic polymer systems.30
From a historical perspective, it is interesting to point out that associative exchanges within polymers have been speculated upon and studied even in the early days of polymer chemistry (1940s–1950s). In 2012,32 McCarthy and coworkers ‘rediscovered’ the classical work on chemical stress relaxations in PDMS elastomers reported by Osthoff et al. in 1954,33 and contemporaneously studied by Tobolsky.21 McCarthy realized that the reported exchange reactions of silyl ethers would have potential for modern self-healing dynamic materials (Scheme 2b). The well-established alkoxide-mediated addition–elimination that can happen at PDMS chain ends can indeed act as a dynamic rearrangement mechanism, revealing PDMS elastomers as an overlooked or forgotten class of dynamic materials (Scheme 5a). Siloxane transesterifications have since been used in the purposeful design of vitrimers (vide supra: Scheme 4c).27 However, alkoxide-mediated dynamic PDMS networks are prone to depolymerisation by very facile ‘backbiting’ and depolymerisation via a reverse ring opening polymerisation, severely limiting their potential as vitrimer materials.
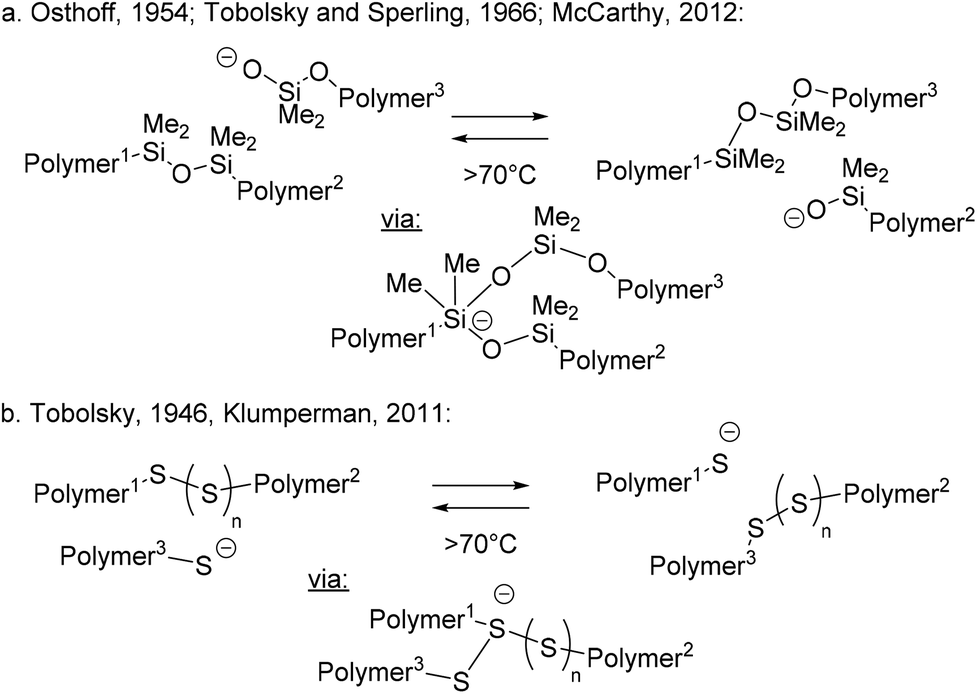 | ||
| Scheme 5 Early examples of associative exchanges in polymer networks. | ||
Likewise, classical polysulfide rubbers have been long known to incorporate dynamic S–S linkages (Scheme 5b), since the seminal studies of hydrocarbon rubber vulcanizates by Stern and Tobolsky in 1946.34,35 Although the exact exchange mechanism in vulcanized rubbers remains unclear to this day, the process is clearly associative in nature (first order kinetics in disulfide and in thiols) and does not involve disulfide bond cleavage. Alternatively, a direct metathesis exchange of disulfide cross-links via adduct formation has also been postulated by Klumperman (not shown in Scheme 5b).36 Given the prevalent associative pathway for disulfide exchange, such dynamic cross-links have also found applications in the purposeful design of vitrimer materials, as for example in the works of Odriozola.37,38 However, classical polysulfide rubber formulations rarely show complete stress relaxation,39 and do not flow when heated, possibly related to the presence of permanent (thioether) cross-links and the limited dynamics within the overall molecular architecture of these rubbers.
For an overview of other associative exchange chemistries, we refer to recent reports.11,12,23 Among many reactions studied in this context, a particularly promising one seems to be one reported by Hillmyer and coworkers,26 demonstrating vitrimers based on transreactions (addition/elimination) of hydroxyl moieties in polyhydroxy-polyurethanes and in polyhydroxy-polycarbonates.40 These results show that associative stepwise MNR can be designed into common and widely popular polymer matrices, using chemical functions that are widespread.
Dissociative stepwise rearrangements
In the previous section, examples of stepwise associative polymer rearrangements have been discussed. These are all relatively recent additions to the collection of dynamic covalent chemistries that have been implemented in polymer networks, striving for vitrimer polymer materials. Stepwise dissociative bond exchanges (MNRs) are in fact much more common in polymers, and the main mechanism underlying the dynamics here is a ‘reversible addition’ process, or more precisely an elimination/addition process, in which a chain segment first fragments into two new chain ends (Scheme 2c). In fact, chain fragmentation is probably one of the most common chemical reactions within a polymer matrix, and it is also a leading mechanism of polymer degradation or material failure. However, typical bond fission reactions result in high energy radical or ionic fragments. Such highly reactive chain ends will usually be very prone to undergo secondary reactions, rendering the bond cleavage effectively non-reversible, and leading to permanent damage to the material, although there are elegant examples developed by Bowman that have harnessed free radical reactivity for the development of covalent adaptable networks.24,41For a straightforward MNR, however, a fragmentation process should result in two relatively well-defined and robust chain ends, which do not readily undergo side reactions, as they should be able to recombine without permanent degradation of the polymer matrix or its material properties. Perhaps the most well-known example of a fragmentation that reliably produces closed shell, non-ionic fragments is the (retro)-Diels–Alder reaction (Scheme 6a–c). The concerted nature of the reaction implies that two Sigma bonds are broken at the same time, which immediately transform into two stable pi-bonding arrays, embedded within the diene and the dienophile.
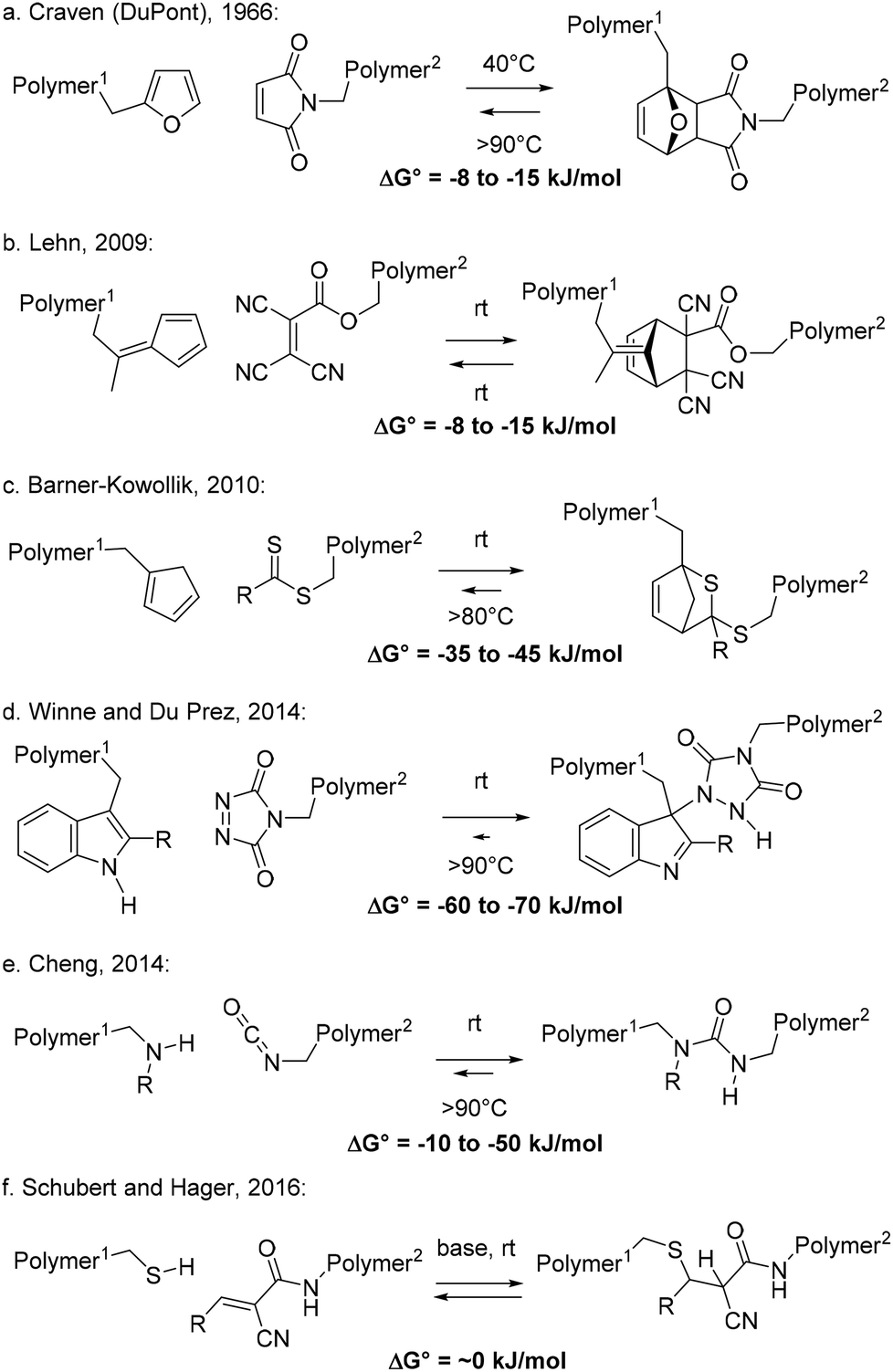 | ||
| Scheme 6 Examples of dissociative stepwise MNR-based materials. | ||
Interestingly, reversible Diels–Alder chemistry was first explored in polymers in industrial labs at DuPont,42 exploiting the thermoreversible cycloaddition reaction between furans and maleimides, as readily available bulk chemicals. Thus, already in 1966, Craven described several reversibly cross-linked polymer resins showing a unique ‘post-formability’ (Scheme 6a). This chemistry has so far not found widespread industrial applications, but interest for it has grown enormously in recent decades, since the seminal works of Hodge and Gandini,43 and Wudl.44 While the bond-forming cycloaddition of furans and maleimides is reversible, the forward reaction typically does not occur at a significant rate at room temperature and also does not readily proceed to full completion upon heating. Curing requires application of heat and is rarely complete, especially under stoichiometric conditions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 furan:maleimide). The bond forming process is characterised by a relatively high activation reaction barrier (>100 kJ mol−1) and shows only a low reaction free energy in the range of −8 to −15 kJ mol−1.45 The free energy of association between furan and maleimide is thus comparable to that of a hydrogen bond interaction, albeit one that is quite hard to break. In light of these small intrinsic thermodynamic preferences, substitution patterns of the bonding partners as well as the nature of the polymer matrix can have very pronounced effects on this equilibrium, shifting it towards one end or the other. Mild heating in a solvent can also rapidly displace the equilibrium towards the non-cross-linked state, and furan-maleimide networks are thus very fragile towards dissolution by a solvent.
:1 furan:maleimide). The bond forming process is characterised by a relatively high activation reaction barrier (>100 kJ mol−1) and shows only a low reaction free energy in the range of −8 to −15 kJ mol−1.45 The free energy of association between furan and maleimide is thus comparable to that of a hydrogen bond interaction, albeit one that is quite hard to break. In light of these small intrinsic thermodynamic preferences, substitution patterns of the bonding partners as well as the nature of the polymer matrix can have very pronounced effects on this equilibrium, shifting it towards one end or the other. Mild heating in a solvent can also rapidly displace the equilibrium towards the non-cross-linked state, and furan-maleimide networks are thus very fragile towards dissolution by a solvent.
In 2009, Lehn and coworkers developed self-healing polymers by applying a room temperature reversible Diels–Alder reaction between fulvenes and highly reactive acrylate-type dienophiles (Scheme 6b).46 The incorporation of such reactants inside a polymer network requires some synthetic effort, but network formation is straightforward, as the forward Diels–Alder reaction readily proceeds within a matter of seconds at room temperature (and takes several hours at −10 °C).47 Again, the free energy of the reaction is very small, in the range of −8 to −15 kJ mol−1. Thus, already upon moderate heating (>50 °C), the equilibrium can be quickly and significantly shifted to the endothermic, de-cross-linked side. This dynamic covalent polymer network thus actually approaches the characteristic properties of supramolecular networks that are held together by weak non-covalent interactions.
In 2010, Barner-Kowollik and coworkers used simple dithioesters, which are well known as end groups in RAFT-type controlled radical polymerisations, as reactive dienophiles that can reversibly add to cyclopentadienyl-functionalised (Cp) polymers in a swift hetero-Diels–Alder reaction (Scheme 6c).48 The authors also developed straightforward methods to incorporate reactive Cp-moieties in polymers and studied their dynamic bond formation extensively. The forward hetero-Diels–Alder reaction between Cp and thioesters proceeds within a matter of minutes at room temperature. With typical reaction free energies in the range of −35 to −45 kJ mol−1, the degree of bond formation is markedly more pronounced than in the two previously described Diels–Alder reactions, rapidly proceeding to full conversion at room temperature, but the bond formation can still be significantly reversed at higher temperatures through a retro-Diels–Alder process. Like all spontaneous bond forming processes, this reversible Diels–Alder association should involve opposing entropic and enthalpic driving forces, with the entropic factor normally favoring debonding, and the enthalpic factor favoring bonding to overcome the entropic penalty associated with linking two molecules. At high enough temperatures, the enthalpic bonding preference of all reversible cleavage reactions will diminish, revealing the entropic debonding preference according to:
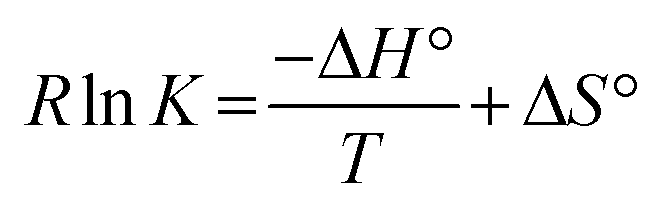
In their studies of reversible Cp-thioester Diels–Alder cross-linking,48,49 Barner-Kowollik and coworkers found that next to the effects of chemical substitution pattern on the diene and dienophile (e.g. more and less electron-rich dithioesters), the bonding-debonding equilibrium can also be strongly dependent on the exact macromolecular architecture, demonstrating significant changes in the reaction entropy as a function of the length of polymer chain fragments that separate the dynamic cross-links.49 Using a theoretical model, combined with experimental observations, highly significant differences in the bonding/debonding equilibria were found and related to the effect of chain length on the reaction of otherwise chemically identical reaction partners. These results are in line with those of previous theoretical studies on entropy effects in hydrogen bond associated polymers studied by Stadler,50 and again underline the highly significant but far from straightforward effects that the macromolecular architecture can have on the thermodynamics of a dynamic bond exchange process. In this light, it can be expected that similar effects can be found also on the reaction kinetics of MNR reactions, and such studies could be of great value for our understanding of dynamic polymer systems.
Next to cycloadditions, other pericyclic reaction mechanisms can also establish a rapid reversible adduct formation between relatively stabilized (long living) reactants. The Alder–ene reaction is a classic example of an alternative pericyclic bond forming process, and in 2014, Winne, Du Prez and coworkers reported dynamic polymer systems based on the reversible ene reaction between an indole and a triazolinedione (TAD).51 Whereas TADs are mostly known as dienophiles in swift and mostly irreversible hetero-Diels–Alder reactions, they also act as highly reactive enophiles.52 When combined with indoles, Baran and Corey found that TADs form a very stable Alder–ene adduct (Scheme 6d), which is nonetheless reversible at high temperatures (>100 °C).53 This reversible adduct formation was investigated in a macromolecular context by Winne and Du Prez, giving a stepwise dissociative MNR pathway with an exceptionally high free energy of reaction in the range of 60–70 kJ mol−1.54 Nevertheless, the low reaction barriers that are associated with the forward TAD-indole reaction, owing to the high reactivity of TADs, results in a still feasible barrier for the backward debonding reaction. Interestingly, in light of these thermodynamic parameters, significant debonding can theoretically not be achieved in such systems, even at highly elevated temperatures. However, the small, transient fraction of debonded free TAD chain ends can kinetically still be quite relevant, and indeed significant network reorganisation, full stress relaxation and flow can be achieved in these networks, without necessitating a net depolymerisation or debonding.55
Many dissociative stepwise covalent bond exchange chemistries (reversible additions) involve pericyclic reactions, but also simple (ionic) addition reactions have been investigated in this context. For example, the reaction between isocyanates and (hindered) nucleophiles has long been known to be a thermoreversible process and has also found industrial applications in polymers through the development of so-called blocked isocyanate reagents.56 In 2014, Cheng and coworkers introduced reversible bonds between isocyanates and hindered (secondary) amines as a dissociative stepwise MNR process for polymers with dynamic urea cross-links (Scheme 6e).57 The process involves a simple proton transfer next to the urea bond cleavage event, so that neutral fragments can be formed. Although isocyanates can be prone to irreversible side reactions such as hydrolysis or allophanate formation, their availability as common monomers allows for a straightforward introduction of dynamic bonds into many polymer matrices. The reaction free energy here also strongly depends on the nature of the used hindered amine and can thus be varied or tuned over a wide range, from mildly exothermic to highly exothermic.
A final example we want to draw attention to in this section is the use of reversible Michael additions of thiols to conjugated, electrophilic alkenes. These are interesting dynamic linkages as they are in fact widely used in polymer materials as a thiol-Michael click reaction,58 and can be readily prepared from for example acrylate and thiol monomers. In this context, the formation of the thioether bond is usually considered as an irreversible ‘click’-like linkage. However, the reversible nature of this Michael reaction (in basic medium) has been known for a long time in organic chemistry.59,60 In 2016, Schubert and Hager implemented this dynamic thio-Michael addition for the design of healable polymer networks (Scheme 6f).61 They used a highly reactive, but stabilised malononitrile-derived Michael acceptor. In the presence of a mild base, the malonitrile can be readily deprotonated and a very rapid dynamic equilibrium establishes itself from which actually neither the Michael adduct or the reaction partners can be isolated in pure form, due to their unavoidable interconversion. The observed rapid exchange also prompted them to study the dynamics on a material level. One of the main issues here, was the dynamic nature of normal acrylate-based thioether linkages under the basic conditions, leading to the release of H2S over time, and thus to material degradation. Because of the widespread utility of thiol–ene chemistries, their base-promoted reversibility seems to be a promising avenue for further research, although it is clear that the design of monomers will need to take into account their longer-term stability issues under the applied conditions. In simultaneous studies by Konkolewicz and coworkers,62a the reversible thiol-Michael addition exchange has also been investigated in more common acrylate-based networks, shown to be operative at higher temperatures in the absence of a base. This work has also led this group to reinvestigate the dynamics of the thiol-Michael addition using elegant small molecules.62b
For other notable dynamic covalent chemistries that have been implemented in dissociative stepwise MNR-type polymer networks, we refer to more specialized reviews on these topics.5,7,11,12
Concerted rearrangements
By far the least common type of MNR mechanisms are single step, concerted rearrangements, where no net bonds are formed or broken under any circumstances (Scheme 2a). In fact, to our knowledge, only two dynamic covalent chemistries implemented in polymer networks have been suggested to go through a concerted bond exchange pathway (Scheme 7).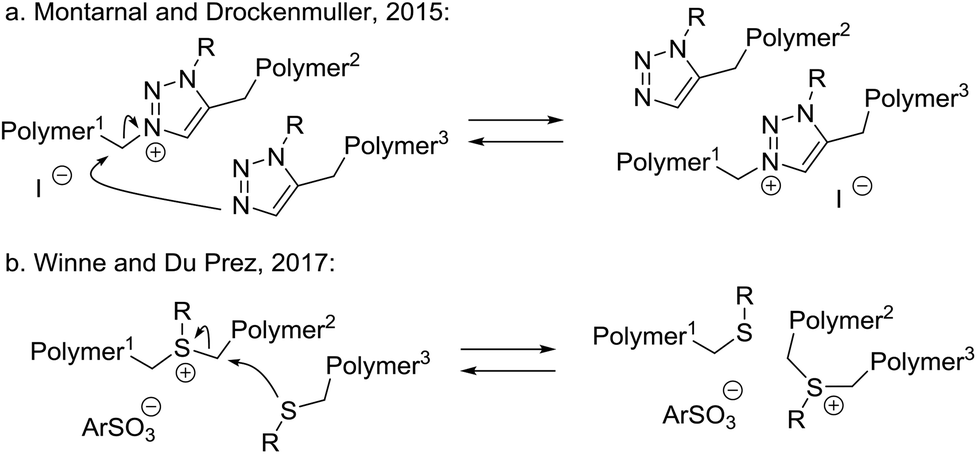 | ||
| Scheme 7 Examples of (possible) concerted MNR-based materials. | ||
In 2015, Montarnal and Drockenmuller reported the synthesis of a polyionic polymer network incorporating N-alkylated triazolium iodide moieties as cross-links (Scheme 7a), conveniently prepared using Huisgen-type azide alkyne click-like chemistry and an alkylating cross-linker for the resulting nucleophilic triazole species.63 The obtained networks could undergo stress relaxation through an associative SN2-type exchange that constitutes a transalkylating reaction of triazoles as nucleophilic species. Interestingly, however, such networks were later actually shown to be dominated by a more complex dissociative stepwise MNR pathway, wherein the iodide counterion induces a temporary decross-linking via a highly reactive alkyliodide chain end. In fact, this is a clear example of a small molecule-mediated dissociative exchange via a reversible condensation process (cf.Scheme 3). In an elaborate but elegant in-depth follow-up study, the rheology data together with data on network composition obtained from XPS measurements and statistical calculations, unambiguously confirmed an iodide-mediated stepwise dissociative MNR pathway, involving the intermediate reformation of the alkyl iodide cross-linkers, and Montarnal and Drockenmuller could actually exclude a kinetically significant SN2-type concerted MNR pathway, at least at temperatures up to 150 °C.64 This result points towards the pitfalls that can be faced when considering MNR mechanisms in dynamic covalent networks, and that care must be taken before conclusions about the nature of MNRs are drawn.
In 2017, Winne and Du Prez reported a related transalkylation chemistry, applicable to polythioether networks (Scheme 7b).65 Here, the alkylating cross-links are provided by sulfonium sulfonate ion pair. Interestingly, using a non-nucleophilic counterion for the sulfonium ion cross-links was found to be important for material properties. Alkyl halides did not give satisfactory results, likely caused by their higher nucleophilicity in SN2-type dealkylation reactions. The chemistry of the MNR bond exchange implemented here is based on the classical SN2-chemistry underlying cationic ring opening polymerisation of cyclic thioethers, as introduced to polymer chemistry by Goethals.66Via alkylation of thioether networks, a strategy for ‘vitrimerisation’ of polythioether thermosets into dynamic networks via alkylation thus seems viable. Very recently, Guo and Zhang introduced this sulfonium-sulfide transalkylation chemistry in natural rubber vulcanisates, achieving an effective recycling strategy for these classical materials via alkylation and subsequent transalkylation.67 The possibility of competing leaving group-mediated dealkylation (reversible condensation) has so far not been investigated in these systems, but this should be done in light of the recent findings of Montarnal and Drockenmuller. As it stands, this chemical platform is the only one that has a ‘theoretically ideal’ energy profile for the establishment of vitrimeric properties. As concerted MNR materials can have interesting properties, related to their strictly constant cross-link density, this certainly seems like an area within dynamic polymer systems that deserves more attention.
Small molecule-mediated dissociative stepwise rearrangements
In addition to the three above discussed generalised categories of MNR pathways (Scheme 2), the small molecule-mediated dissociative stepwise MNR pathways (Scheme 3) also deserve some discussion with specific examples. Although superficially, a very similar situation arises here as in normal stepwise dissociative MNRs, all bond cleavage events are strictly conditional on the presence of a small molecule polymer additive. Association of polymer chain segments is not required for the rearrangement reaction to occur, but rather an association between a cross-link and a (more mobile) small molecule additive must occur (a retro-polymer condensation step) before a chain can fragment into two new reactive chain ends. When only a substoichiometric amount of the decross-linking additive is present (i.e. a catalytic amount of the transfer group), the network cannot undergo extensive depolymerisation, and will remain mostly intact or permanent, while it can rearrange its structure through intermediacy of the decross-linked chain ends. In terms of approaching the dynamic network criteria for vitrimer polymers, this category can thus be also quite interesting from a practical point of view. In any case, this type of process is inherent to almost all associative MNR systems, as a low molecular weight, monofunctional additive that has the same reactivity as that of dynamic link will install such pathways at least as side reactions, or through mobile network defects.As an early example of a dynamic covalent polymer network, incorporating fast breaking and forming of bonds through a reversible hydrolysis, Leibler and coworkers pointed out already in 1990 that polysaccharide hydrogels, covalently cross-linked by borax-derived boric esters, are self-healing at room temperature (Scheme 8a).68 Although these hydrogels may be fragile towards hydrolysis at higher temperatures, the exchanges at lower temperatures are very fast and most bonds remain formed but are highly dynamic, showing an interesting compromise situation that can lead to emerging properties such as self-healing.
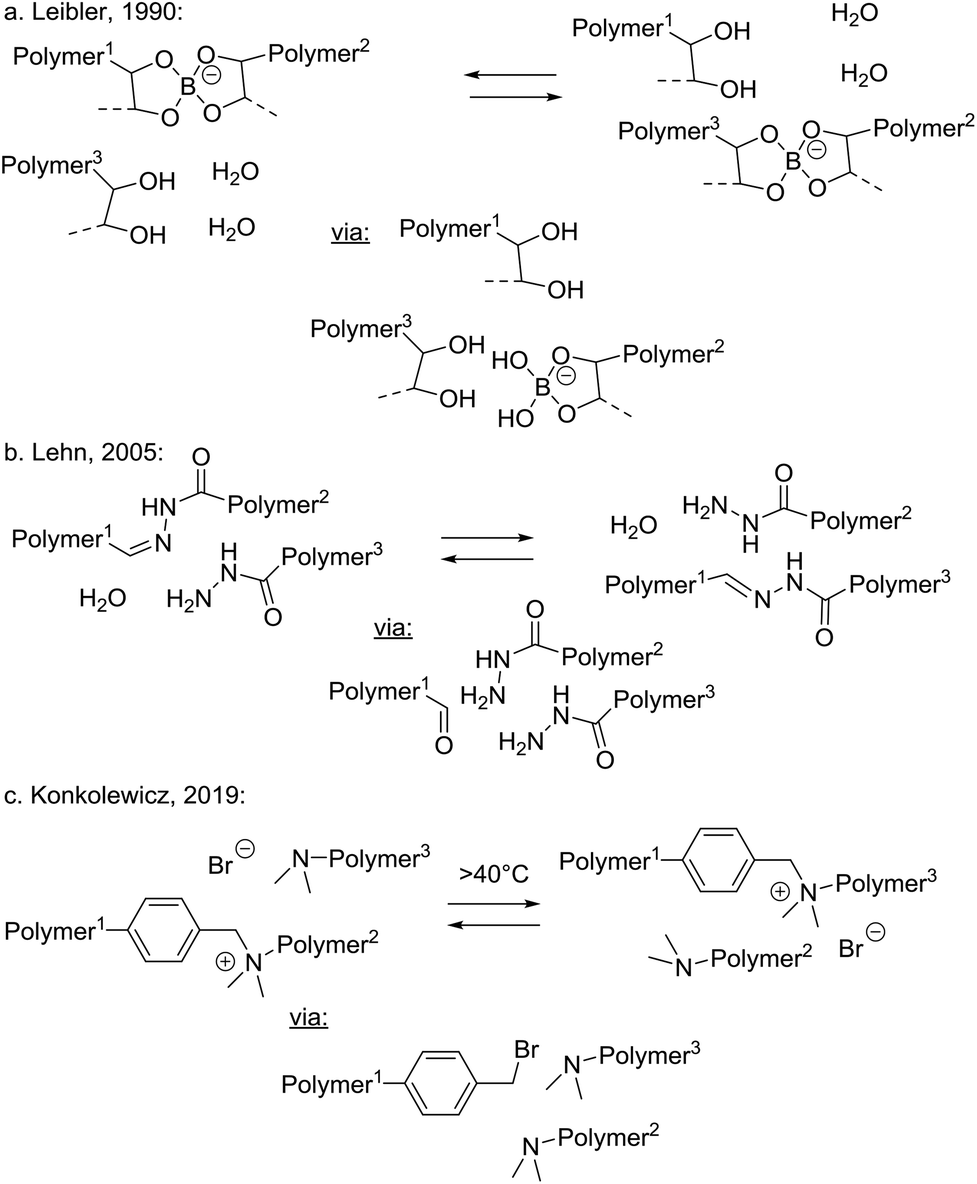 | ||
| Scheme 8 Small molecule mediated dissociative stepwise MNRs. | ||
Next to bor(on)ic esters, one of the best studied polymer systems to show a dissociative reversible polycondensation pathway, are polyimine and poly(acyl)hydrazone based networks. These networks are formally formed by condensation of aldehydes or ketones with amines or hydrazides. The carbon–nitrogen double bond spontaneously forms by mixing aldehydes with a suitable amine-type nucleophile, albeit with a limited thermodynamic driving force, which is helped by the exclusion of a molecule of water that can be used to drive the equilibrium. As this condensation reaction is inherently reversible,69 an endothermic hydrolysis can be facilitated by heating, or by increasing the concentration of water. The thermal malleability of these networks is typically found to be highly conditional on the presence of water.
In 2005, Lehn and coworkers introduced their acylhydrazone-based dynamic covalent chemistry platform in the synthesis of dynamic polymer networks that rely on a reversible hydrolysis reaction (Scheme 8b).70 In the absence of water, imines and hydrazones can be expected to also undergo associative addition/elimination exchanges in the presence of pendant amine-functionalities. However, also under ‘wet’ conditions the extent of network dissociation can be avoided or at least controlled by limiting the availability of water. Indeed, in 2014, Zhang and coworkers reported reversible polyimine networks based on imine hydrolysis,71 that could also undergo associative exchange under dry conditions, nicely demonstrating a system that can conditionally switch between prevalent associative and dissociative pathways. Such dynamic polyimine resins are currently being developed industrially by a start-up company.72
Very recently, Konkolewicz reported another example of a reversible condensation MNR pathway operating in dynamic polymer networks, by implementing transalkylation reactions of amines, via temporary de-cross-linking by adventitious bromide ions as good nucleophiles (Scheme 8c).73 These polyionic networks undergo an SN2-type cleavage at the benzylic position, releasing a highly reactive benzyl bromide intermediate chain end. This can in turn react via an SN2-process with other free amines. This is thus in fact a very similar mechanism to that found to be operative in polyionic alkylated triazole networks developed by Montarnal and Drockenmuller (Scheme 7a).
Dissociative vitrimers?
In the recent literature, there has been some confusion over the definition, properties and conditions that define vitrimer materials.64 Several polymer networks that clearly undergo dissociative exchange reactions, have been shown to possess all the physical properties that are expected for vitrimers: they are insoluble in good solvents, but flow upon heating with a very gradual thermal viscosity profile (linear Arrhenius-like dependence of the viscosity as a function of temperature and no sol–gel transition). Thus, the question arises: “is an associative exchange reaction an absolute prerequisite for a polymer network to show vitrimer behavior?” The answer to this question, in our opinion, is quite context-dependent.One of the major characteristics of vitrimers is their gradual viscosity profile, further showing a temperature dependence in line with that of the chemical exchange reaction, with an apparent link between mechanical and chemical activation energies. Above a certain temperature, the rheological behavior of vitrimers, as assessed by mechanical stress relaxation experiments, will be dominated by the chemical exchange kinetics. In other words, the ‘rate determining step’ in chain mobility will be a chemical reaction and will show the same temperature dependence, following an Arrhenius law. It should be noted that this type of behavior is not limited to vitrimers or to polymer networks, as chemical stress relaxation can become the dominant phenomenon in the rheology of a polymer through a variety of factors under certain circumstances. Also within vitrimers, this Arrhenius-like behavior is not guaranteed under all circumstances, for example when the temperature approaches Tg and the onset of vitrification puts additional limits on chain mobility.27
In stepwise dissociative MNR pathways (Schemes 2c and 3), de-cross-linking can certainly be expected to significantly affect viscosity upon heating. However, covalent exchange reactions can become kinetically relevant at temperatures where net de-cross-linking is still very low and does not affect the viscosity. In other words, in situations where cross-links are reformed as fast as they are broken, there can be no significant build-up of free chain ends. Nevertheless, the underlying chemical exchange reactions can become the dominant stress relaxation mechanism in such dissociative networks. Chemically controlled stress relaxation or non-fluctuating degrees of connectivity is thus not an exclusive feature of associative MNR polymer systems. Dissociative exchange networks should in theory always show decross-linking, but the difference in free energy between the bonded and debonded states of the networks can prevent a sol–gel transition from happening at a reasonable temperature (e.g. <300 °C). This situation will be clarified by a hypothetical example (Fig. 1).
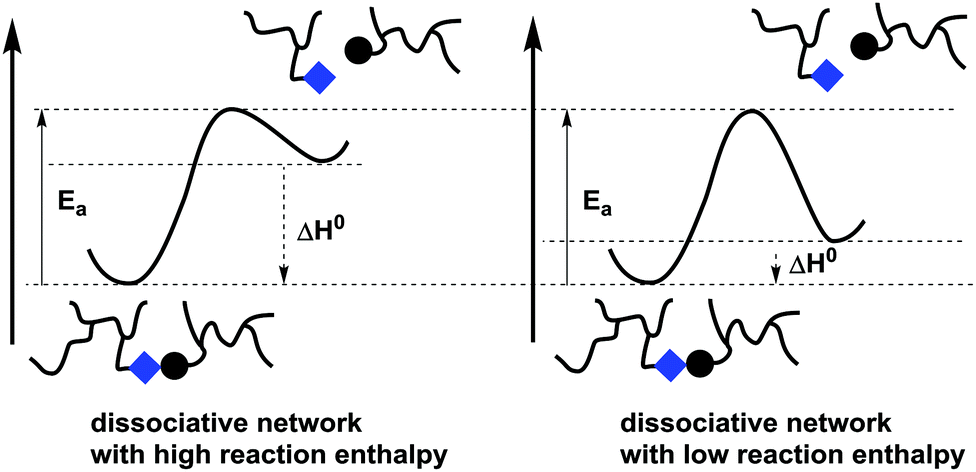 | ||
| Fig. 1 Two dissociative stepwise MNR-based networks can have the same bond exchange rate, but a different temperature response in terms of network integrity. | ||
When considering networks that undergo bond exchanges through a stepwise dissociative MNR pathway, the reaction enthalpy of the reversible bond forming step can be considered as a crucial factor. For the sake of discussion, a situation is sketched in Fig. 1 for two dissociative networks that have exactly the same activation energy barrier for the debonding step. In one case, the backward reaction barrier for rebonding is assumed to be relatively small (fast formation of bond between chain ends), while in the other case this barrier is assumed to be much higher (much slower reformation of the bond between chain ends). It should be clear that both networks will theoretically show exactly the same rate of bond exchanges at all temperatures, as the forward activation barrier is the same, while their viscosity profiles and macroscopic properties will be quite different. Network integrity can in fact be maintained in such dissociative networks if the relative abundance of the open forms, as dictated by the reaction equilibrium constant, is kept low (e.g. <5% decross-linking). A significant amount of debonding can only be expected at temperatures that shift the equilibrium to the endothermic side. This situation arises when the available thermal energy is enough to overcome the enthalpic driving force of the bond formation, in favor of the entropically more favored debonded states. When ΔH for debonding is quite large, the temperature necessary to shift the equilibrium will often be above the degradation temperature of the polymer matrix. As a result, the polymer network that is rearranging through a dissociative pathway will show a constant cross-link density or at least cross-link densities where loss of network integrity is negligible.
For many practical purposes, it can be understood from the above example that a dissociative network might show characteristics and properties of vitrimers, providing similar advantages for processing and reprocessing. This has indeed been reported in the literature in a few recent examples, where Arrhenius-like viscosity profiles have been observed, together with a constant elastic modulus, indicating a permanent network over the probed temperature range.63 However, an important difference between associative vitrimer materials and dissociative vitrimer-like materials will be their intrinsic resistance to dissolution in a good, non-reactive solvent. In a dissociative network that swells in a good solvent, the debonding equilibrium is expected to readily shift towards the debonded state by the dilution effect, and complete dissolution is in principle possible. In a permanent network, or more precisely in a vitrimer, the overall network connectivity in the sol fraction and within the remaining network will stay the same at all temperatures. As found in theoretical studies on a model AfB2 vitrimer, the sol fraction of the network will depend on solvent concentration, but not on temperature.22a
Some specific polymer backbones, however, can undergo a thermally triggered depolymerisation reaction, as discussed for the classical PDMS networks that can undergo alkoxide-mediated reversed ring opening polymerisation (cf.Scheme 5), next to network bond exchanges, leading to considerable, and thermally dependent sol fractions, even though the number of bonds stays the same. Such features are linked to the macromolecular make up and synthesis, and the likelihood of cyclative cleavages within the network. An associative mechanism thus seems to be required for all vitrimer properties but is not always sufficient. A further exception or important caveat that should be made here, is for the dissociative networks that require mediation by an additive or an associated catalyst to go to the decross-linked state (cf.Scheme 3). For such reversible condensation-type networks, the extent of decross-linking that can be attained at any temperature will be conditional on the relative amount of the reactive small molecule additive or catalyst. If this amount is low, only a small fraction of polymer segments can be in the dissociated state at any point in time, and these networks undergo dissociative MNRs, but they should nevertheless be permanent, and are also expected to be resistant to dissolution by a good solvent, as the equilibrium cannot be significantly shifted to a non-bonded state.
For demonstrating the vitrimeric properties of a given polymer network, and thus to show its non-fluctuating cross-link density, stringent solubility tests should always be performed, which is unfortunately not always reported when new ‘vitrimer’ chemistries are disclosed. In fact, we predict that many reported dissociative ‘vitrimers’ based on a stepwise dissociative MNR mechanism are likely to be readily soluble in good solvents, although it may be difficult to find a good solvent for some cases.
In summary, the distinction between vitrimer (permanent) dynamic networks and thermoplastic (reversible) dynamic networks can become a highly academic one, as also dissociative networks can show very high resistance to dissolution in solvents. Nevertheless, the interplay between macroscopic material properties and chemistry remains a fascinating area for further research and, as is expected for chemical reactivity, dynamic polymer properties can be complex and are highly context dependent. Before going to our conclusions, we would like to underline this final point in a dedicated final section, as it is something that is often overlooked or neglected in the context of materials chemistry.
Context-dependent mechanical properties
Whereas the chemical structure is an innate and absolute feature of a molecular species, its chemical reactivity is not. Care should thus be taken in considering the properties of a rearranging polymer network. Chemical reactivity is a highly relative phenomenon, and typically shows strong context dependency: reagent purity, solvents, order of mixing, and additives can all have a critical influence on reactivity. In the context of chemically reactive polymer materials, it should be realized that the precise nature and exact architecture of the polymer matrix, and possible polymer additives or even minor impurities can all have a drastic influence on material properties that can only be understood by taking into account mechanistic aspects. The findings by Barner-Kowollik and coworkers described earlier for the dynamic thioester Diels–Alder polymerisation,49 indeed point towards a significant interdependence of monomer structure and the reaction entropy for debonding, a result that should be taken as a warning against liberally comparing the situation of bond exchange reactions between small molecules (model compounds) in solutions and those taking place inside polymer network segments.Chemical reactions can be accelerated by the presence of a molecular species that acts as a catalyst, or by the specific nature of a solvent. Thus, the rheological behavior of dynamic covalent networks will certainly depend on the presence and concentration (or availability) of such catalytic species in the polymer matrix.74 This is a straightforward example of how a small change in the formulation of a polymer network can result in an important change in its mechanical properties. In the design and study of dynamic covalent polymers, such effects should be expected, scrutinized and experimentally probed.
Often, in literature dealing with vitrimers, a comparison is made between chemical and mechanical relaxation times. This can be instructive but also unwarranted. It is indeed impossible to exactly mimick the conditions of exchange reactions within a macromolecular network in a solution with low MW reagents. A material put under mechanical stress is an out-of-equilibrium situation that is not directly comparable to a freely equilibrating chemical reaction between similar species in solution. One should thus not expect a clear correlation between the reaction rates observed in chemical model experiments and in dynamic polymer networks. Indeed, many reports have found a significant difference in relaxation times, and any observed correlations are mostly qualitative. Differences have not only been observed in absolute relaxation times, but also in their temperature dependence (i.e. activation energy). Small molecule model studies are thus necessary to understand polymer dynamics, but they are also far from sufficient. Qualitative trends in reactivity observed on small molecule models are probably the most important information that can be gained from such studies. On the other hand, as there is a lack of good characterization tools for studying chemistry in networks, studying models will obviously remain critical for our understanding in this area.
Another aspect that is often overlooked in considering the mechanism of polymer rearrangements, is that a given MNR reaction can theoretically always proceed via multiple different reaction pathways, and that either of those pathways can be promoted or inhibited by the specific context of a material matrix. Care should thus be taken not to consider a particular type of chemistry as a purely associative or purely dissociative one! For the seminal example of Leibler's polyester polyhydroxyl vitrimer networks (cf.Scheme 4a),14,15,23 for example, it should be taken into account that transesterifications can actually also proceed via a dissociative pathway, for example with the intermediacy of ketenes as high energy, debonded neutral intermediates (Scheme 9, bottom pathway).75 That the chemistry of epoxy-based transesterification vitrimers is more convoluted than anticipated was also recently highlighted by Williams.76 Similar plausible alternative reaction pathways can in fact be considered for all known stepwise associative MNR chemistries. The fact that they are not observed as prevalent pathways on small molecule models in solution phase, is not a guarantee that the same will be true in a rearranging polymer network under mechanical stress.
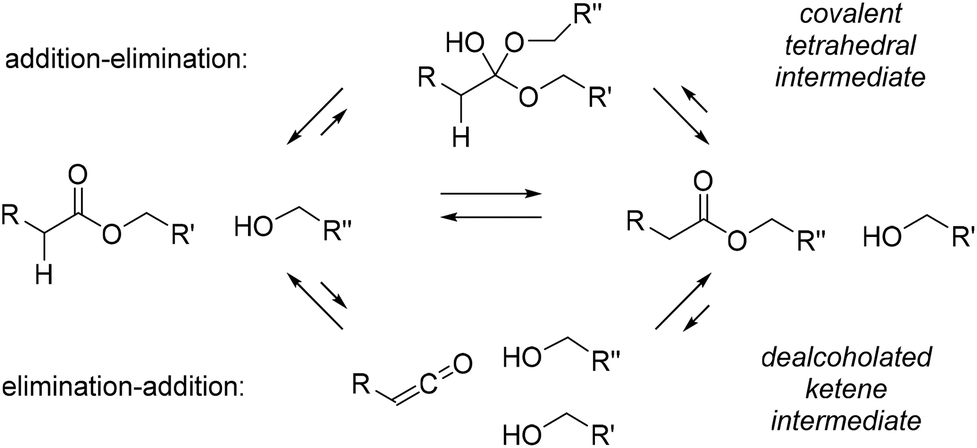 | ||
| Scheme 9 Known competing pathways for ester-hydroxyl exchange reactions. | ||
A very concrete example of two competing exchange pathways for the same exchange reaction, was observed in our own research of vinylogous urethane vitrimers, where it was found that very similar polymer networks, containing the same amount of chemically reactive moieties, can have two competing exchange pathways, as confirmed by a marked difference in temperature dependence (i.e. activation energy).77 In these vitrimers, we found that the mechanical relaxation time, as well as the dominant exchange mechanism, critically relies on small changes in formulations, additives, and also the overall polymer architecture, resulting in differences in orders of magnitude for the observed relaxation times, within chemically all but identical polymer matrices. Very recently, Guerre and Du Prez even found two distinct MNR mechanisms operating each as the dominant process within the same material, but at different temperature intervals, showing a dual temperature response.78
As another example of context dependence in dynamic covalent polymers, Winne and Du Prez have found that the dynamic bonding and debonding of TAD-indoles (cf.Scheme 6d),51 the stoichiometry of indoles and TADs in the network plays a crucial and interesting role. When there is a 1:1 equivalence between the two reactive moieties, full cross-linking or curing can be achieved owing to the high exothermicity of the reaction, and no free indole or TAD remains in the network. As there can be no significant build-up of free chain ends in this case (very fast reformation of chain ends, cf. Fig. 1a), the reaction rate for rebonding to another indole will rely mainly on the availability of such receiving indoles. Even though debonding and rebonding rates are fast in these systems, macroscopic polymer network dynamics of TAD-indole based dynamic polymer networks is not observed unless an excess of indoles, relative to the amount of TADs, is present throughout the network structure.55
As stated before, the macromolecular context can also drastically influence polymer dynamics and chemistry. For example, matrix incompatibility effects can lead to microphase separation, and completely change macroscopic properties if the exchangeable groups aggregate into domains.79
Finally, as made clear by the example poly-ionic triazolium networks (cf.Scheme 7a),63,64 the difference between concerted and stepwise pathways can be hard to determine experimentally. Accurately and effectively modeling MNR reactions is therefore predicted to be an outstanding goal for theoretical chemistry. The influence of solvatation on potential energy surfaces of small molecule reactions is already a challenging computational task. Theoretical approaches that can include the influence of a macromolecular network matrix may thus be very challenging.22a,80–82
During the last two decades, a great effort has been devoted to the design and synthesis of dynamic polymer networks. As we discussed throughout this paper, much progress has been made to introduce versatile and robust but at the same time reactive organic building blocks into polymer chemistry, enabling control over the topology and molecular dynamics of a large set of polymer matrices. Comparatively much less is known about the material and rheological properties of polymer networks that incorporate such dynamic covalent chemical linkages. Even in the absence of side reactions, the link between MNR pathways and kinetics and macroscopic properties can be quite complex and deserves closer attention. For dissociative mechanisms, some guidelines could come from the numerous studies of so-called associating polymers for which a rich spectrum of behavior has been identified both experimentally and theoretically, taking into account factors such as temperature, number of temporary bonds, their life-time, macromolecular structure and stoichiometry of the network.18,20,83 Various “sticky” reptation, Rouse or dynamic aggregate models have been developed for these systems to describe rheology or self-healing properties.84 Such models rely on the transient nature of sticky associations between chains, where the life-time of any bond can be expected to be short. Even in such systems, strong context dependence will already emerge. For example, when associating groups (stickers) are diluted, their rapid opening and closing will most of the time recombine the original partners. In this context, rapid opening and closing cannot yield a fast network topology reorganization or, from a mechanical point of view, to flow or stress relaxations.20 Thus, in the context of MNRs that involve longer lived bonds, exchanging via either dissociative or associative MNR pathways, an even more complex relationship to mechanical properties can be expected. As discussed above, in principle, for some stoichiometries, networks with dissociative MNR pathways could behave as networks with exclusive associative pathways, as net-decross-linking does not occur.85 Also, it should be stressed that in polymer matrices, reactive groups and catalysts interact with the macromolecular network and will exhibit their own dynamics, which may be different from that in ‘free’ solutions. Therefore, understanding how such interactions and motion dynamics in the network combine and influence the emergence of material properties is at this time an interesting and open research question.
Conclusions
Dynamic bond interchange between portions of macromolecular chains is a fascinating dynamic rearrangement process in polymer chemistry, which, despite its potential, has so far found limited industrial applications. However, academic and industrial interest in chemistries that allow such intrinsic reactivity within polymers has surged in the last decade, with the first applications already finding their way.72 This has been mostly brought about by academic groups interested in the design of functional polymer materials with novel useful properties. As ever more dynamic covalent chemistry platforms are being explored within a macromolecular context, it is becoming clear that studies so far have still been mostly scratching at the surface, and that a fundamental understanding of what is governing the dynamics of bond interchanges is lacking at this time.We hope that this perspective has put the various fundamental issues that need to be addressed in a logical framework, and that it can provide both inspiration and guidance for future studies in this fascinating area at the interface of polymer science and organic chemistry.
Mechanistic considerations in organic reactivity are far from trivial, and fundamental insight in even the most basic textbook organic transformations is often still lacking.86 This is especially the case for chemistry in condensed phases, for which theoretical models struggle to take adequately the effect of the reaction matrix into account. Organic reactivity within an out-of-equilibrium cross-linked macromolecular architecture can in this regard be considered as a particularly challenging setting for theoretical models. On the bright side, experimental studies of such systems, including simple mechanical measurements of macroscopic objects, allow the direct probing of such systems. Thus, opportunities and challenges are there for the theory, the synthesis, the analysis and the engineering of dynamic polymer networks. Nevertheless, we are quite optimistic when it comes to ingenuity of scientists to tackle these issues, with a bright future ahead for this clearly interdisciplinary field.
Author contributions
The perspective paper was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.References
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Using the dynamic bond to access macroscopically responsive structurally dynamic polymers, Nat. Mater., 2011, 10(1), 14 CrossRef CAS PubMed.
- N. Roy, B. Bruchmann and J. M. Lehn, DYNAMERS: dynamic polymers as self-healing materials, Chem. Soc. Rev., 2015, 44(11), 3786–3807 RSC.
- B. J. Blaiszik, S. L. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Self-healing polymers and composites, Annu. Rev. Mater. Res., 2010, 40, 179–211 CrossRef CAS.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Self-healing and self-mendable polymers, Polym. Chem., 2010, 1(7), 978–987 RSC.
- T. Maeda, H. Otsuka and A. Takahara, Dynamic covalent polymers: reorganizable polymers with dynamic covalent bonds, Prog. Polym. Sci., 2009, 34(7), 581–604 CrossRef CAS.
- M. A. C. Stuart, W. T. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Emerging applications of stimuli-responsive polymer materials, Nat. Mater., 2010, 9(2), 101 CrossRef PubMed.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Covalent adaptable networks (CANs): a unique paradigm in cross-linked polymers, Macromolecules, 2010, 43(6), 2643–2653 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Covalent adaptable networks: smart, reconfigurable and responsive network systems, Chem. Soc. Rev., 2013, 42(17), 7161–7173 RSC.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Recent advances in dynamic covalent chemistry, Chem. Soc. Rev., 2013, 42(16), 6634–6654 RSC.
- C. N. Bowman and C. J. Kloxin, Covalent adaptable networks: reversible bond structures incorporated in polymer networks, Angew. Chem., Int. Ed., 2012, 51(18), 4272–4274 CrossRef CAS PubMed.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Dynamic covalent polymer networks: from old chemistry to modern day innovations, Adv. Mater., 2017, 29(14), 1606100 CrossRef PubMed.
- P. Chakma and D. Konkolewicz, Dynamic Covalent Bonds in Polymeric Materials, Angew. Chem., Int. Ed., 2019, 58, 9682–9696 CrossRef CAS PubMed.
- J. P. Pascault, H. Sautereau, J. Verdu and R. J. Williams, Thermosetting polymers, CRC Press, 2002, vol. 64 Search PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-like malleable materials from permanent organic networks, Science, 2011, 334(6058), 965–968 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, Catalytic control of the vitrimer glass transition, ACS Macro Lett., 2012, 1(7), 789–792 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Vitrimers: permanent organic networks with glass-like fluidity, Chem. Sci., 2016, 7(1), 30–38 RSC.
- R. Stadler, Association phenomena in macromolecular systems-influence of macromolecular constraints on the complex formation, Macromolecules, 1988, 21(1), 121–126 CrossRef CAS.
- L. Leibler, M. Rubinstein and R. H. Colby, Dynamics of reversible networks, Macromolecules, 1991, 24(16), 4701–4707 CrossRef CAS.
- A. N. Semenov and M. Rubinstein, Thermoreversible gelation in solutions of associative polymers. 1. Statics, Macromolecules, 1998, 31(4), 1373–1385 CrossRef CAS.
- M. Rubinstein and A. N. Semenov, Thermoreversible gelation in solutions of associating polymers. 2. Linear dynamics, Macromolecules, 1998, 31(4), 1386–1397 CrossRef CAS.
- A. V. Tobolsky, Stress relaxation studies of the viscoelastic properties of polymers, J. Appl. Phys., 1956, 27(7), 673–685 CrossRef CAS.
- (a) F. Smallenburg, L. Leibler and F. Sciortino, Patchy particle model for vitrimers, Phys. Rev. Lett., 2013, 111(18), 188002 CrossRef PubMed; (b) E. Pezron, L. Leibler, A. Ricard and R. Audebert, Reversible gel formation induced by ion complexation. 2. Phase diagrams, Macromolecules, 1988, 21(4), 1126–1131 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, Metal-catalyzed transesterification for healing and assembling of thermosets, J. Am. Chem. Soc., 2012, 134(18), 7664–7667 CrossRef CAS PubMed.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Photoinduced plasticity in cross-linked polymers, Science, 2005, 308(5728), 1615–1617 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Vinylogous, urethane vitrimers, Adv. Funct. Mater., 2015, 25(16), 2451–2457 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, Mechanically activated, catalyst-free polyhydroxyurethane vitrimers, J. Am. Chem. Soc., 2015, 137(44), 14019–14022 CrossRef CAS PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, Silyl ether as a robust and thermally stable dynamic covalent motif for malleable polymer design, J. Am. Chem. Soc., 2017, 139(42), 14881–14884 CrossRef CAS PubMed.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis, Science, 2017, 356(6333), 62–65 CrossRef PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, Malleable and self-healing covalent polymer networks through tunable dynamic boronic ester bonds, J. Am. Chem. Soc., 2015, 137(20), 6492–6495 CrossRef CAS PubMed.
- B. T. Worrell, M. K. McBride, G. B. Lyon, L. M. Cox, C. Wang, S. Mavila, C. Lim, H. Coley, C. Musgrave, Y. Ding and C. N. Bowman, Bistable and photoswitchable states of matter, Nat. Commun., 2018, 9(1), 2804 CrossRef PubMed.
- B. T. Worrell, S. Mavila, C. Wang, T. M. Kontour, C. H. Lim, M. K. McBride, C. B. Musgrave, R. Shoemaker and C. N. Bowman, A user's guide to the thiol-thioester exchange in organic media: scope, limitations, and applications in material science, Polym. Chem., 2018, 9(36), 4523–4534 RSC.
- P. Zheng and T. J. McCarthy, A surprise from 1954: siloxane equilibration is a simple, robust, and obvious polymer self-healing mechanism, J. Am. Chem. Soc., 2012, 134(4), 2024–2027 CrossRef CAS PubMed.
- R. C. Osthoff, A. M. Bueche and W. T. Grubb, Chemical stress-relaxation of polydimethylsiloxane elastomers, J. Am. Chem. Soc., 1954, 76(18), 4659–4663 CrossRef CAS.
- M. D. Stern and A. V. Tobolsky, Stress-time-temperature relations in polysulfide rubbers, Rubber Chem. Technol., 1946, 19(4), 1178–1192 CrossRef.
- M. D. Stern and A. V. Tobolsky, Stress-time-temperature relations in polysulfide rubbers, J. Chem. Phys., 1946, 14(2), 93–100 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Self-healing materials based on disulfide links, Macromolecules, 2011, 44(8), 2536–2541 CrossRef CAS.
- A. Rekondo, R. Martin, A. R. de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis, Mater. Horiz., 2014, 1(2), 237–240 RSC.
- I. Azcune and I. Odriozola, (2016). Aromatic disulfide cross-links in polymer systems: Self-healing, reprocessability, recyclability and more, Eur. Polym. J., 2016, 84, 147–160 CrossRef CAS.
- L. Imbernon, E. K. Oikonomou, S. Norvez and L. Leibler, Chemically cross-linked yet reprocessable epoxidized natural rubber via thermo-activated disulfide rearrangements, Polym. Chem., 2015, 6(23), 4271–4278 RSC.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Reprocessable acid-degradable polycarbonate vitrimers, Macromolecules, 2018, 51(2), 389–397 CrossRef CAS.
- H. Y. Park, C. J. Kloxin, T. F. Scott and C. N. Bowman, (2010). Stress Relaxation by Addition – Fragmentation Chain Transfer in Highly Cross-linked Thiol–Yne Networks, Macromolecules, 2010, 43(24), 10188–10190 CrossRef CAS PubMed.
- J. M. Craven, DuPont, US Patent, 3435003, 1969 (Filed 1966) Search PubMed.
- C. Goussé, A. Gandini and P. Hodge, Application of the Diels–Alder reaction to polymers bearing furan moieties. 2. Diels–Alder and retro-Diels–Alder reactions involving furan rings in some styrene copolymers, Macromolecules, 1998, 31(2), 314–321 CrossRef.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, A thermally re-mendable cross-linked polymeric material, Science, 2002, 295(5560), 1698–1702 CrossRef CAS PubMed.
- R. C. Boutelle and B. H. Northrop, Substituent effects on the reversibility of furan–maleimide cycloadditions, J. Org. Chem., 2011, 76(19), 7994–8002 CrossRef CAS PubMed.
- P. Reutenauer, E. Buhler, P. E. Boul, S. E. Candau and J. M. Lehn, Room temperature dynamic polymers based on Diels–Alder chemistry, Chem. – Eur. J., 2009, 15(8), 1893–1900 CrossRef CAS PubMed.
- P. J. Boul, P. Reutenauer and J. M. Lehn, Reversible Diels–Alder reactions for the generation of dynamic combinatorial libraries, Org. Lett., 2005, 7(1), 15–18 CrossRef CAS PubMed.
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Rapid bonding/debonding on demand: reversibly cross-linked functional polymers via Diels–Alder chemistry, Macromolecules, 2010, 43(13), 5515–5520 CrossRef CAS.
- N. K. Guimard, J. Ho, J. Brandt, C. Y. Lin, M. Namazian, J. O. Mueller, K. K. Oehlenschlaeger, S. Hilf, A. Lederer, F. G. Schmidt, M. L. Coote and C. Barner-Kowollik, Harnessing entropy to direct the bonding/debonding of polymer systems based on reversible chemistry, Chem. Sci., 2013, 4(7), 2752–2759 RSC.
- L. de Lucca Freitas, C. Auschra, V. Abetz and R. Stadler, Hydrogen bonds in unpolar matrix—Comparison of complexation in polymeric and low molecular-weight systems, Colloid Polym. Sci., 1991, 269(6), 566–575 CrossRef CAS.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Triazolinediones enable ultrafast and reversible click chemistry for the design of dynamic polymer systems, Nat. Chem., 2014, 6(9), 815 CrossRef CAS PubMed.
- K. De Bruycker, S. Billiet, H. A. Houck, S. Chattopadhyay, J. M. Winne and F. E. Du Prez, Triazolinediones as highly enabling synthetic tools, Chem. Rev., 2016, 116(6), 3919–3974 CrossRef CAS PubMed.
- P. S. Baran, C. A. Guerrero and E. J. Corey, The first method for protection–deprotection of the indole 2, 3-π bond, Org. Lett., 2003, 5(11), 1999–2001 CrossRef CAS PubMed.
- H. A. Houck, K. De Bruycker, S. Billiet, B. Dhanis, H. Goossens, S. Catak, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Design of a thermally controlled sequence of triazolinedione-based click and transclick reactions, Chem. Sci., 2017, 8(4), 3098–3108 RSC.
- N. Van Herck and F. E. Du Prez, Fast healing of polyurethane thermosets using reversible triazolinedione chemistry and shape-memory, Macromolecules, 2018, 51(9), 3405–3414 CrossRef CAS.
- M. S. Rolph, A. L. Markowska, C. N. Warriner and R. K. O'Reilly, Blocked isocyanates: From analytical and experimental considerations to non-polyurethane applications, Polym. Chem., 2016, 7(48), 7351–7364 RSC.
- H. Ying, Y. Zhang and J. Cheng, Dynamic urea bond for the design of reversible and self-healing polymers, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, The thiol-Michael addition click reaction: a powerful and widely used tool in materials chemistry, Chem. Mater., 2013, 26(1), 724–744 CrossRef.
- C. F. H. Allen, J. O. Fournier and W. J. Humphlett, The thermal reversibility of the Michael reaction: IV. Thiol adducts, Can. J. Chem., 1964, 42(11), 2616–2620 CrossRef CAS.
- E. H. Krenske, R. C. Petter and K. N. Houk, Kinetics and thermodynamics of reversible thiol additions to mono-and diactivated Michael acceptors: implications for the design of drugs that bind covalently to cysteines, J. Org. Chem., 2016, 81(23), 11726–11733 CrossRef CAS PubMed.
- N. Kuhl, R. Geitner, R. K. Bose, S. Bode, B. Dietzek, M. Schmitt, J. Popp, S. J. Garcia, S. van der Zwaag, U. S. Schubert and M. D. Hager, Self-Healing Polymer Networks Based on Reversible Michael Addition Reactions, Macromol. Chem. Phys., 2016, 217(22), 2541–2550 CrossRef CAS.
- (a) B. Zhang, Z. A. Digby, J. A. Flum, P. Chakma, J. M. Saul, J. L. Sparks and D. Konkolewicz, Dynamic Thiol–Michael chemistry for thermoresponsive rehealable and malleable networks, Macromolecules, 2016, 49(18), 6871–6878 CrossRef CAS; (b) B. Zhang, P. Chakma, M. P. Shulman, J. Ke, Z. A. Digby and D. Konkolewicz, Probing the mechanism of thermally driven thiol-Michael dynamic covalent chemistry, Org. Biomol. Chem., 2018, 16(15), 2725–2734 RSC.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, Reprocessing and recycling of highly cross-linked ion-conducting networks through transalkylation exchanges of C–N bonds, J. Am. Chem. Soc., 2015, 137(18), 6078–6083 CrossRef CAS PubMed.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Tuning the Viscosity Profile of Ionic Vitrimers Incorporating 1, 2, 3-Triazolium Cross-Links, Adv. Funct. Mater., 2017, 27(45), 1703258 CrossRef.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, Poly, (thioether) vitrimers via transalkylation of trialkylsulfonium salts, ACS Macro Lett., 2017, 6(9), 930–934 CrossRef CAS.
- E. J. Goethals, Cationic polymerisation of cyclic sulfides, Makromol. Chem.: Macromol. Chem. Phys., 1974, 175(4), 1309–1328 CrossRef CAS.
- Z. Tang, Y. Liu, Q. Huang, J. Zhao, B. Guo and L. Zhang, A real recycling loop of sulfur-cured rubber through transalkylation exchange of C–S bonds, Green Chem., 2018, 20(24), 5454–5458 RSC.
- E. Pezron, A. Ricard and L. Leibler, Rheology of galactomannan-borax gels, J. Polym. Sci., Part B: Polym. Phys., 1990, 28(13), 2445–2461 CrossRef CAS.
- M. E. Belowich and J. F. Stoddart, Dynamic imine chemistry, Chem. Soc. Rev., 2012, 41(6), 2003–2024 RSC.
- W. G. Skene and J. M. P. Lehn, Dynamers: polyacylhydrazone reversible covalent polymers, component exchange, and constitutional diversity, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(22), 8270–8275 CrossRef CAS PubMed.
- P. Taynton, K. Yu, R. K. Shoemaker, Y. Jin, H. J. Qi and W. Zhang, Heat- or Water-Driven Malleability in a Highly Recyclable Covalent Network Polymer, Adv. Mater., 2014, 26(23), 3938–3942 CrossRef CAS PubMed.
- D. A. Kissounko, P. Taynton and C. Kaffer, New material: vitrimers promise to impact composites, Reinf. Plast., 2018, 62(3), 162–166 CrossRef.
- P. Chakma, Z. A. Digby, M. P. Shulman, L. R. Kuhn, C. N. Morley, J. L. Sparks and D. Konkolewicz, Anilinium Salts in Polymer Networks for Materials with Mechanical Stability and Mild Thermally Induced Dynamic Properties, ACS Macro Lett., 2019, 8(2), 95–100 CrossRef CAS.
- A. Demongeot, S. J. Mougnier, S. Okada, C. Soulié-Ziakovic and F. Tournilhac, Coordination and catalysis of Zn 2+ in epoxy-based vitrimers, Polym. Chem., 2016, 7(27), 4486–4493 RSC.
- J. Otera, Transesterification, Chem. Rev., 1993, 93(4), 1449–1470 CrossRef CAS.
- F. Altuna, C. Hoppe and R. Williams, Epoxy vitrimers: the effect of transesterification reactions on the network structure, Polymers, 2018, 10(1), 43 CrossRef PubMed.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Chemical, control of the viscoelastic properties of vinylogous urethane vitrimers, Nat. Commun., 2017, 8, 14857 CrossRef PubMed.
- M. Guerre, C. Taplan, R. Nicolaÿ, J. M. Winne and F. E. Du Prez, Fluorinated, vitrimer elastomers with a dual temperature response, J. Am. Chem. Soc., 2018, 140(41), 13272–13284 CrossRef CAS PubMed.
- E. Pezron, L. Leibler, A. Ricard, F. Lafuma and R. Audebert, Complex formation in polymer-ion solution. 1. Polymer concentration effects, Macromolecules, 1989, 22(3), 1169–1174 CrossRef CAS.
- E. Pezron, L. Leibler, A. Ricard and R. Audebert, Reversible gel formation induced by ion complexation. 2. Phase diagrams, Macromolecules, 1988, 21(4), 1126–1131 CrossRef CAS.
- R. G. Ricarte, F. Tournilhac and L. Leibler, Phase Separation and Self-Assembly in Vitrimers: Hierarchical Morphology of Molten and Semicrystalline Polyethylene/Dioxaborolane Maleimide Systems, Macromolecules, 2018, 52(2), 432–443 CrossRef.
- S. Ciarella, F. Sciortino and W. G. Ellenbroek, Dynamics of vitrimers: Defects as a highway to stress relaxation, Phys. Rev. Lett., 2018, 121(5), 058003 CrossRef CAS PubMed.
- Z. Zhang, Q. Chen and R. H. Colby, Dynamics of associative polymers, Soft Matters, 2018, 14(16), 2961–2977 RSC.
- E. B. Stukalin, L. H. Cai, N. A. Kumar, L. Leibler and M. Rubinstein, Self-Healing of Unentangled Polymer Networks with Reversible Bonds, Macromolecules, 2013, 46(18), 7525–7541 CrossRef CAS PubMed.
- M. Delahaye, J. M. Winne and F. E. Du Prez, Internal, catalysis in covalent adaptable networks: phthalate monoester transesterification as a versatile dynamic crosslinking chemistry, J. Am. Chem. Soc., 2019, 141(38), 15277–15287 CrossRef CAS PubMed.
- Y. Nieves-Quinones and D. A. Singleton, Dynamics and the regiochemistry of nitration of toluene, J. Am. Chem. Soc., 2016, 138(46), 15167–15176 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |