
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mild and quantitative route towards well-defined strong anionic/hydrophobic diblock copolymers: synthesis and aqueous self-assembly†
Anton H.
Hofman
 *ab,
Remco
Fokkink
a and
Marleen
Kamperman
b
*ab,
Remco
Fokkink
a and
Marleen
Kamperman
b
aPhysical Chemistry and Soft Matter, Wageningen University, Stippeneng 4, 6708 WE Wageningen, The Netherlands. E-mail: a.h.hofman@rug.nl
bPolymer Science, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
First published on 30th September 2019
Abstract
Block copolymers that accommodate both hydrophobic and ionic elements are interesting materials for numerous applications, such as stabilizing agents, lubricants and proton-exchange membranes. Frequently these copolymers are based on weak polyelectrolytes, but the pH-dependent charge density restricts their use to a limited pH window. Although strong polyelectrolytes do not suffer this problem, the most commonly employed post-modification approach limits the choice of the hydrophobic component, as harsh reaction conditions are usually involved. Moreover, this method often results in incomplete functionalization of the precursor copolymer. To avoid these difficulties a mild route was developed that is based on a hydrophobic protected poly(3-sulfopropyl methacrylate) intermediate that enables the preparation of well-defined strong anionic polyelectrolytes. The potential of this method was demonstrated by synthesizing hydrophobic/strong anionic diblock copolymers, and their self-assembly in aqueous solution was studied.
Amphiphilic block copolymers that contain both hydrophobic and ionic features are promising materials for various applications, such as surfactants for emulsion polymerizations,1,2 thickening agents for enhanced oil recovery,3 lubricants in aqueous media,4 the preparation of photonic fluids,5 fabrication of proton-exchange membrane fuel cells,6 and the preparation of adhesives for use in wet environments.7,8 The ionic block is frequently based on a weak polyelectrolyte, i.e., the charge density is dependent on the pH, making the hydrophobic/ionic block copolymer soluble in polar organic solvents. Examples of the most commonly employed weak electrolyte monomers include anionic (meth)acrylic acid9–11 and cationic N,N-dimethylaminoethyl methacrylate,12–14 with poly(acrylic acid) generally being synthesized from a tert-butyl-protected hydrophobic precursor.15,16 While the weak ionic nature allows straightforward synthesis and characterization of amphiphilic block copolymers, it limits the applicable pH range if a high charge density is desired.
Strong polyelectrolytes do not suffer this problem, but their permanently charged structure is accompanied by a significantly reduced solubility.17,18 Therefore, the strong ionic functionality is usually introduced via post-modification of an analyzable, fully hydrophobic intermediate. An intriguing approach was recently reported by the Hawker group, in which thiol–ene click chemistry enabled the transformation of an uncharged precursor into either a strong cationic or anionic polyelectrolyte, depending on the character of the thiol.19,20 For polycations, a more commonly employed method involves quaternization of ortho- or para-substituted vinylpyridines.21–25 These quaternization reactions are, however, often incomplete and thus require thorough analysis in order to determine the exact charge density, which is a crucial parameter when polyelectrolyte complexes are concerned.26,27
Polystyrene sulfonate is among the easiest accessible strong polyanions, and while it can be synthesized directly from commercially available styrene sulfonate, sulfonation of polystyrene continues to be the most frequently applied route toward this polyelectrolyte. Similar to the previously discussed quaternization reactions, sulfonation of polystyrene is often incomplete as well, and in addition, involves the use of corrosive and toxic reagents.28–30 Quantitative sulfonation can be achieved under even more aggressive reaction conditions, but may lead to meta substitution31 and other side reactions.32 Under these conditions the hydrophobic block should remain intact and thus its choice is limited.
Although direct synthesis of sulfonate-containing block copolymers remains a feasible option, either by using a water-based solvent mixture33,34 or through polymerization-induced self-assembly (PISA),35,36 the lack of a common solvent hinders proper analysis of such highly amphiphilic copolymers. A more subtle approach towards well-defined anionic/hydrophobic block copolymers involves the use of protection chemistry: first a fully hydrophobic characterizable copolymer is synthesized, and is followed by deprotection of the sulfonic ester to give a strong anionic/hydrophobic block copolymer.37 Several research groups have made use of this alternative route to obtain polystyrene sulfonate-containing diblock copolymers, with polystyrene,38 poly(n-butyl acrylate)39 or poly(3-hexylthiophene)40,41 as the hydrophobic component. Still, a few problems arise when employing this method: (1) the low reactivity of protected styrene sulfonate limits the molecular weight of the polyelectrolyte block (molecular weights above 10 kg mol−1 are rarely obtained), (2) deprotection by thermolysis is uncontrolled and requires prolonged heating (typically above 150 °C for several hours), and (3) thermolysis results in the sulfonic acid form and thus causes a local and potentially undesired pH decrease. As the copolymer should survive these conditions, the choice of the hydrophobic block is restricted by the deprotection method.
To overcome these problems, we report a new route to prepare well-defined strong polyanions through a protected intermediate by starting from commercially available 3-sulfopropyl methacrylate. This methacrylate enables the synthesis of higher molecular weight polyelectrolytes, while for its deprotection we used a significantly milder route that only involves treatment with sodium iodide.42–44 Additionally, instead of the acid it immediately affords the sodium salt form of the polyelectrolyte (Scheme 1). To demonstrate the potential of our method, poly(methyl methacrylate)-containing hydrophobic/strong anionic diblock copolymers were synthesized, and their self-assembly in aqueous solution was studied.
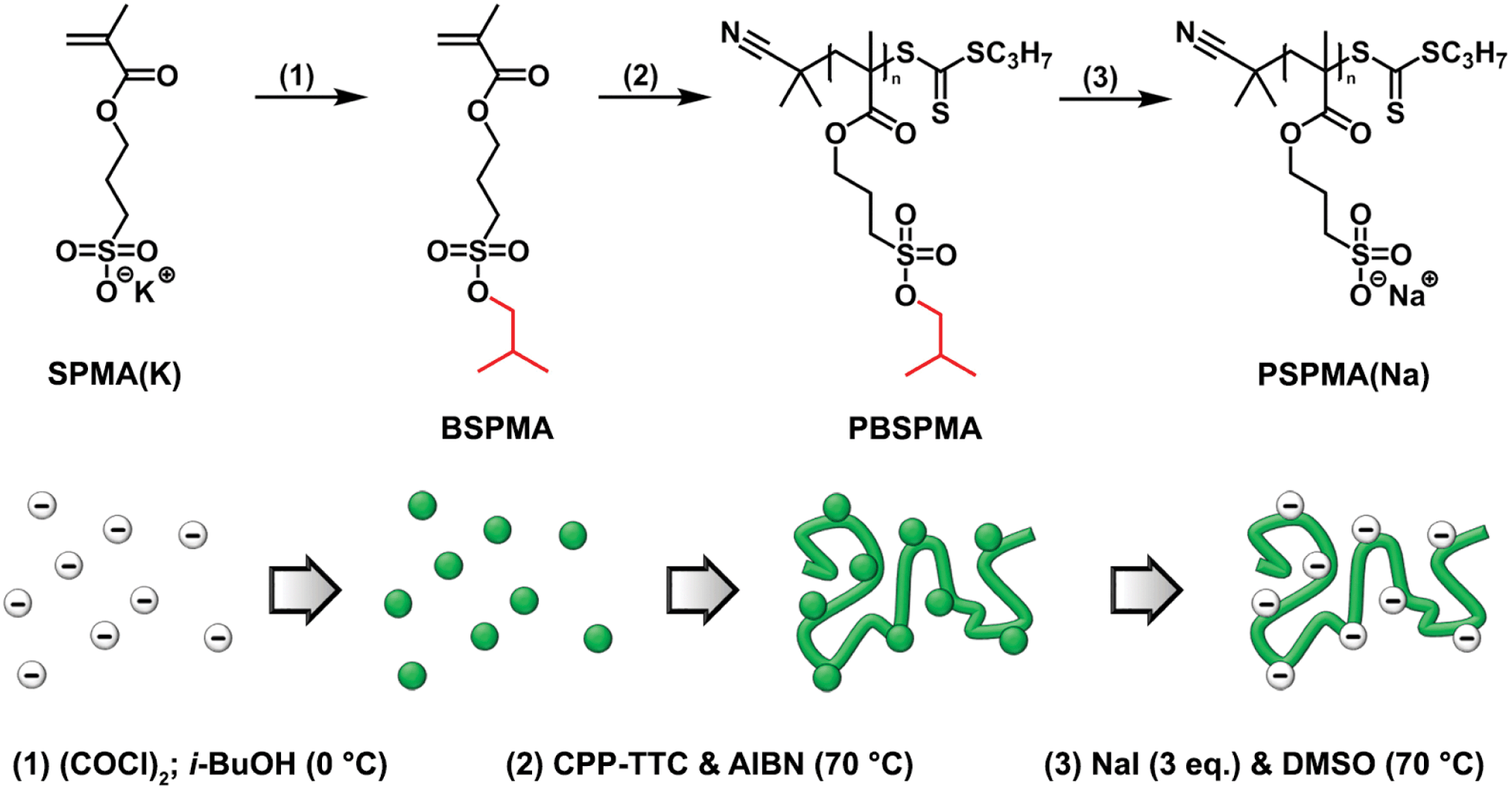 | ||
| Scheme 1 Schematic illustration and general reaction scheme describing the strategy for the preparation of protected PBSPMA sulfonate-containing homopolymers and their subsequent deprotection. | ||
The potassium salt of 3-sulfopropyl methacrylate (SPMA) was protected via a two-step, one-pot reaction. First the sulfonate was converted into 3-(chlorosulfonyl)propyl methacrylate by treating this compound with oxalyl chloride. Since only a small excess of oxalyl chloride was necessary, the in situ prepared sulfonyl chloride could be immediately used in the second step without working-up. Subsequent esterification with isobutanol in the presence of base (triethyl amine) gave 3-(isobutoxysulfonyl)propyl methacrylate (BSPMA) after purification by flash chromatography. In contrast to styrene sulfonyl chloride, it should be pointed out that the sulfonyl chloride of SPMA cannot be isolated due to its high reactivity towards moisture; still, progress of the reaction can be monitored by 1H-NMR (Fig. S1†). The optimized procedure is easy to scale up, and affords BSPMA in high yields (80+%) (Fig. 1a and S2†).
 | ||
| Fig. 1 (a) 1H-NMR spectra of purified BSPMA monomer and PBSPMA homopolymer (PBSPMA-1) recorded in CDCl3. (b) 1H-NMR spectra of protected and quantitatively deprotected PBSPMA-1 (DMSO-d6). Signals corresponding to the isobutyl group [F, G, H] disappeared completely and a significant shift of protons [E] can be observed. | ||
PBSPMA homopolymers were prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization. While dithiobenzoate RAFT agents are known to offer better control over the polymerization of methacrylates, in this work a trithiocarbonate (2-cyanopropan-2-yl propyl trithiocarbonate; CPP-TTC; Fig. S3†) was selected, since these chain transfer agents (CTAs) are less prone to hydrolysis45 and are more stable towards strong nucleophiles which may be relevant in the deprotection step. Despite reaching full monomer conversion, PBSPMA homopolymers (Fig. 1a) with very decent polydispersities and molecular weights close to the theoretical values were obtained (Tables 1 and S1†). Test reactions with a dithiobenzoate CTA indeed resulted in even narrower molecular weight distributions, typically around 1.1 (not shown).
| Polymer | M n | Đ | m (MMA) | n (BSPMA) | f PMMA | R h | PDI | ζ |
|---|---|---|---|---|---|---|---|---|
| PBSPMA-1 | 31.2 | 1.28 | — | 118 | — | — | — | — |
| PBSPMA-2 | 13.5 | 1.41 | — | 51 | — | — | — | — |
| PMMA-1 | 24.5 | 1.24 | 243 | — | — | — | — | — |
| PMMA-2 | 11.4 | 1.27 | 114 | — | — | — | — | — |
| BCP-1 | 52.4 | 1.16 | 243 | 105 | 0.47 | 52.1 | 0.265 | −40.3 |
| BCP-2 | 83.3 | 1.20 | 243 | 222 | 0.29 | 70.0 | 0.330 | −45.0 |
| BCP-3 | 44.8 | 1.18 | 114 | 126 | 0.25 | 28.6 | 0.163 | −41.0 |
| BCP-4 | 74.3 | 1.27 | 114 | 238 | 0.15 | 44.8 | 0.245 | −38.2 |
| BCP-5 | 31.0 | 1.29 | 175 | 51 | 0.57 | 21.9 | 0.062 | −34.6 |
| BCP-6 | 71.7 | 1.21 | 582 | 51 | 0.81 | 37.4 | 0.142 | −33.0 |
PBSPMA degrades rapidly above 200 °C, probably due to the thermally deprotected sulfonic ester37 that in turn causes acid-catalyzed hydrolysis of the methacrylate ester, but is long-term stable up to 130 °C (Fig. S4†). Its glass transition was measured to be around 15 °C, making it to behave as a solid to slightly rubbery material at room temperature, depending on the molecular weight (Fig. S5†).
For the deprotection of PBSPMA we were inspired by a strategy that is more common in organic chemistry: the protected hydrophobic sulfonate is dissolved in an organic solvent in the presence of a nucleophile (sodium iodide).42–44 Deprotection causes precipitation of the sodium sulfonate salt; insolubility of the product drives the reaction to completion. This precipitation-driven procedure already worked remarkably well for the deprotection of PBSPMA: by using an acetone-based solvent/non-solvent system PBSPMA was converted quantitatively into the strong polyanion. As this approach may not be suitable for amphiphilic copolymers, we sought for a homogeneous system. Protected PBSPMA is soluble in a wide range of organic solvents (chloroform, THF, acetone, DMF, DMSO, etc.), whereas the sodium salt of poly(3-sulfopropyl methacrylate) [PSPMA(Na)] is only soluble in water and DMSO. Therefore, the reaction was performed in DMSO (Scheme 1), but this led to incomplete deprotection of PBSPMA when the reaction was performed under conditions comparable to the precipitation-driven method (70%; Fig. S6†). Quantitative deprotection was achieved after increasing the concentration NaI (3 eq. compared to BSPMA) and the temperature to 70 °C (Fig. 1b and S6†). Due to the high solubility of NaI in organic solvents, the excess salt can be removed by simple precipitation of the polyelectrolyte; dialysis is not necessary. The complete process (protection/polymerization/deprotection) can also be followed qualitatively by FT-IR (Fig. S7†).
Although the trithiocarbonate end group seemed to survive the NaI treatment (1H-NMR in D2O; Fig. S8†) and calculation of the degree of polymerization resulted in very similar values for PSPMA-2 (Pn,NMR = 55) and its PBSPMA-2 precursor (Pn,GPC = 51; 1H-NMR is not possible due to overlapping signals), analysis of polyelectrolytes by GPC remains challenging. To proof that a methacrylate backbone is unaffected by the deprotection conditions, a model system was developed based on a low molecular weight poly(methyl methacrylate) (PMMA) analogue. To this end, PMMA-2 was treated with NaI under identical circumstances as PBSPMA. End group analysis by 1H-NMR revealed equal degrees of polymerization, and GPC curves overlapped perfectly (Fig. S9†). The reported method is thus sufficiently mild and may potentially even allow further extension of the polyelectrolyte in aqueous media.
The hydrophobic nature of the protected sulfonate enables facile synthesis and characterization of well-defined amphiphilic block copolymers (Scheme 2). PMMA was chosen as the hydrophobic component, although acrylates or styrenics could also have been suitable candidates. Several PMMA-b-PBSPMA diblock copolymers were prepared by starting from either a PMMA or PBSPMA macro-CTA, resulting in copolymers of varying molecular weight and composition (Tables 1 and S2†). Despite that the PBSPMA blocks were synthesized by fully converting the monomer, potentially leading to a lower end group fidelity, this approach still provided diblock copolymers with good polydispersities.46,47 Furthermore, no traces of residual homopolymer could be detected in GPC (Fig. 2a and S10†).
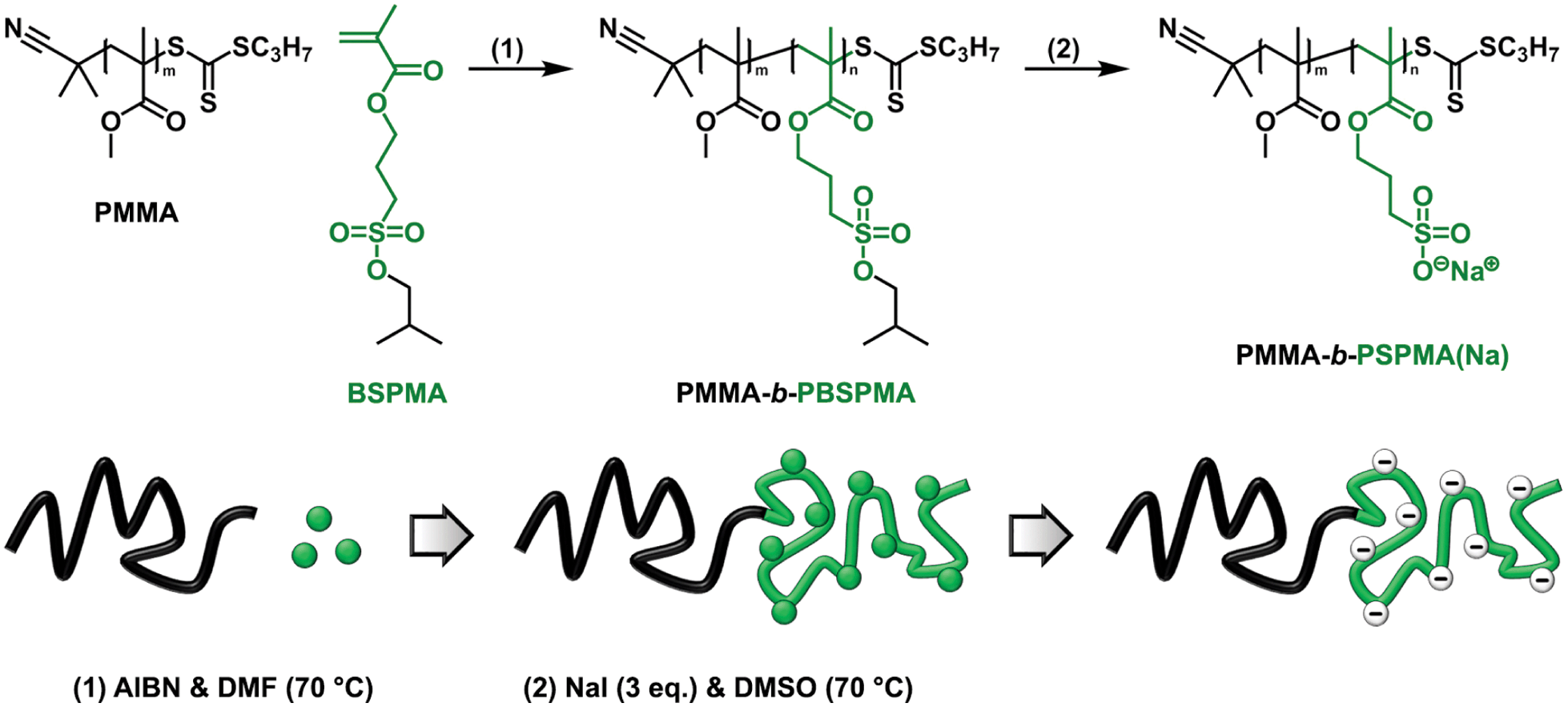 | ||
| Scheme 2 Schematic illustration and general reaction scheme demonstrating the approach towards well-defined PMMA-b-PSPMA(Na) hydrophobic/strong anionic diblock copolymers. | ||
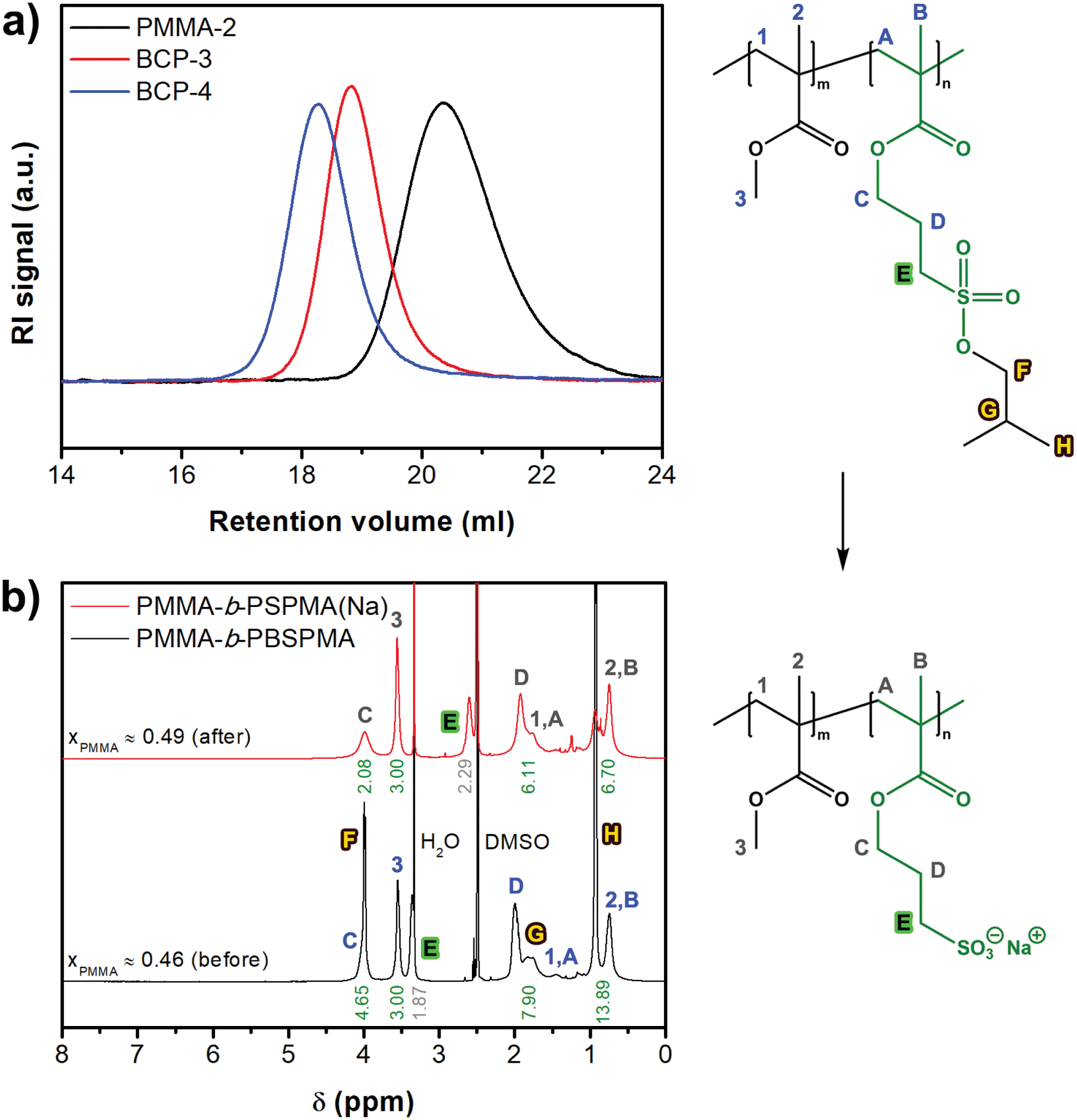 | ||
| Fig. 2 (a) Gel permeation chromatograms of a macro-CTA (PMMA-2) and protected hydrophobic diblock copolymers (BCP-3 and BCP-4). (b) 1H-NMR spectra of protected and deprotected PMMA-b-PBSPMA in DMSO-d6 (BCP-3). | ||
In order to convert the hydrophobic PMMA-b-PBSPMA precursors into PMMA-b-PSPMA(Na), the copolymers were reacted with 3 eq. NaI under the same conditions as the PBSPMA homopolymers. Block copolymers were quantitatively deprotected as evidenced by 1H-NMR, and compositions before and after the NaI treatment were almost identical (BCP-3: xPMMA = 0.46 vs. 0.49). These slightly deviating values are possibly caused by peak broadening in DMSO-d6 (Fig. 2b). True block copolymer compositions were therefore always calculated from the 1H-NMR spectra of the protected hydrophobic precursor in chloroform-d (xPMMA = 0.47; Fig. S11†).
To verify the amphiphilic nature of these strong anionic/hydrophobic PMMA-b-PSPMA(Na) diblock copolymers, their aqueous self-assembly was studied by dynamic light scattering (DLS) and transmission electron microscopy (TEM). A small amount of salt (10 mM KNO3) was added to prevent polyelectrolyte effects (e.g. slow modes). Aqueous solutions (1.0 mg ml−1) were prepared by directly dissolving the polymer powders in water, except for BCP-6 which required the presence of a small amount of DMSO (1/1, then diluted with water; see ESI†). All block copolymers dissolved rapidly after shortly heating the solutions. A first indication for the sizes and shapes of the copolymer aggregates was obtained by multi-angle DLS: polydispersity indices (PDIs) and hydrodynamic radii (Rh) obtained through cumulant analysis are summarized in Table 1, while size distribution plots (CONTIN algorithm; 90°) are demonstrated in Fig. 3a–c. The relatively low PDI values may indicate the formation of spherical objects for all six diblock copolymers. The measured zeta potentials confirm the charged nature of the micellar aggregates, with the order of magnitude being typical for strong polyelectrolyte nano-objects.35,36 Additionally, the corona size of the frozen aggregates, and thus their hydrodynamic radius, was found to be highly sensitive to the salt concentration (Fig. S12†).
 | ||
| Fig. 3 (a–c) DLS size distribution plots of self-assembled PMMA-b-PSPMA(Na) hydrophobic/strong anionic diblock copolymers in 10 mM aqueous KNO3 (1.0 mg ml−1). (d–i) Transmission electron micrographs of negatively stained block copolymer aggregates: BCP-1 (d), BCP-2 (e), BCP-3 (f), BCP-4 (g), BCP-5 (h), BCP-6 (i). | ||
More information on the self-assembled block copolymer structures was obtained by electron microscopy. Samples were negatively stained with a 2 wt% uranyl acetate solution (Fig. 3d–i). Indeed, only spherical micelles are observed, even for the highly asymmetric BCP-5 and BCP-6 copolymers (Fig. S13†). While cryo-TEM is required for a detailed and quantitative analysis of the objects (e.g., core sizes, aggregation numbers, etc.), still a few words can be spend on the behavior of these hydrophobic/strong anionic diblock copolymers. It seems reasonable to assume that only the PMMA cores are observed in these images. Core radii vary between roughly 10 and 20 nm, which is in all cases significantly smaller than the measured hydrodynamic radii. These differences imply that the strong anionic PSPMA corona chains are highly stretched in solution. Although being a staining effect, aggregated and hexagonally packed particles never touch, further supporting the strong repulsion between the charged coronas in the hydrated state. Also, when comparing block copolymers with a similar core size (BCP-1 vs. BCP-5), the distance between packed BCP-1 particles is indeed significantly larger due to its longer PSPMA block. BCP-3 and BCP-4 on the other hand were synthesized from the same PMMA macro-CTA, but differ in size of the PSPMA block. Stronger repulsion of BCP-1's larger corona block caused the formation of a smaller, more curved PMMA core (i.e., lower aggregation number) and simultaneously resulted in a larger inter-particle distance. Finally, BCP-6 was found to give the largest PMMA nanoparticles due to its highly asymmetric composition.
The copolymers poor in the hydrophobic component (such as BCP-2, BCP-3 and BCP-4) self-assembled into the expected star-shaped micelles.9,10,48 Interestingly though, the copolymers that contained a large hydrophobic component (BCP-5 and BCP-6 in particular) still self-assembled into spherical micelles, even in the presence of KNO3. This behavior differs significantly from the thoroughly studied polystyrene-b-poly(acrylic acid) weak polyelectrolyte system,49 since weak polyelectrolyte crew-cut micelles already transformed to other non-spherical objects (rods, plates, etc.) in the presence of very little salt (<10 mM).50 These observations may be a result of poly(acrylic acid) being only partially charged under neutral conditions, reducing the inter-polyelectrolyte chain repulsion and consequently allows the formation of a less curved interface. Moreover, DMF was often added as a cosolvent, which further lowers the charge density and induces additional electrostatic shielding. Thus, charge repulsion plays a dominant role in the self-assembly of the strong anionic/hydrophobic diblock copolymers.
It should be noted that the PMMA cores are glassy at room temperature, and therefore the observed nano-objects are expected to be kinetically trapped non-equilibrium structures. To investigate whether the formed micelles are static or dynamic, the solution of BCP-6 was annealed at 80 °C overnight. No morphological changes were observed (Fig. S14a†). This result is not very surprising though, since previously studied liquid core-containing systems based on poly(n-butyl acrylate) were even found to be static due to the high interfacial tension.10 Because the preparation method of micellar aggregates can have a dramatic effect on the final morphology,51 preliminary tests with the solvent addition method (DMSO/H2O) were performed. DLS experiments, however, indicated no significant difference between both methods as very similar hydrodynamic radii and PDIs were measured (Table S3†). Indeed, BCP-6 still formed the unexpected spherical micelles as evinced by TEM (Fig. S14b†). Furthermore, as opposed to weak polyelectrolyte systems, the presence of salt was required in order to obtain monodisperse aggregates. These findings already demonstrate the deviating solution behavior of our strong polyelectrolyte/hydrophobic copolymers, although one should be careful with drawing any hard conclusions at this point due to the glassy state of the PMMA micelle cores. For a more detailed study on such hydrophobic/strong anionic diblock copolymers, it is therefore recommended to replace the PMMA block by a more liquid-like hydrophobic block, such as polyisobutylene9 or poly(n-butyl acrylate).10 This will be the subject of future work.
In summary, we have demonstrated a straightforward approach towards well-defined strong anionic polyelectrolytes. Our method is just as convenient as the widely employed tert-butyl acrylate/acrylic acid system. It results in a strong sulfonate polyanion, and in contrast to other reported methods, deprotection of the isobutyl sulfonic ester was found to be mild and quantitative. As a proof of concept several hydrophobic and analyzable PMMA-b-PBSPMA diblock copolymers were synthesized by controlled radical polymerization that could be converted into PMMA-b-PSPMA(Na) hydrophobic/strong anionic block copolymers. Due to their highly amphiphilic character, these copolymers readily self-assembled in aqueous solution. Although relatively simple in nature, we are convinced that this route can be easily translated to other, more advanced polymer systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Netherlands Organization for Scientific Research (NWO) via a VIDI innovational research grant. We would like to thank Wouter Teunissen and Herman de Beukelaer (Wageningen Food & Biobased Research) for their help with GPC measurements and thermal characterizations, respectively. Marc C. A. Stuart (University of Groningen) is also gratefully acknowledged for his assistance with the TEM measurements.References
- F. Li, M. Schellekens, J. de Bont, R. Peters, A. Overbeek, F. A. M. Leermakers and R. Tuinier, Macromolecules, 2015, 48, 1194–1203 CrossRef CAS.
- P. Raffa, D. A. Z. Wever, F. Picchioni and A. A. Broekhuis, Chem. Rev., 2015, 115, 8504–8563 CrossRef CAS PubMed.
- P. Raffa, P. Brandenburg, D. A. Z. Wever, A. A. Broekhuis and F. Picchioni, Macromolecules, 2013, 46, 7106–7111 CrossRef CAS.
- U. Raviv, S. Giasson, N. Kampf, J. Gohy, R. Jérôme and J. Klein, Nature, 2003, 425, 163–165 CrossRef CAS PubMed.
- M. Poutanen, G. Guidetti, T. I. Gröschel, O. V. Borisov, S. Vignolini, O. Ikkala and A. H. Gröschel, ACS Nano, 2018, 12, 3149–3158 CrossRef CAS PubMed.
- B. Date, J. Han, S. Park, E. J. Park, D. Shin, C. Y. Ryu and C. Bae, Macromolecules, 2018, 51, 1020–1030 CrossRef CAS.
- A. H. Hofman, I. A. van Hees, J. Yang and M. Kamperman, Adv. Mater., 2018, 30, 1704640 CrossRef PubMed.
- M. Dompé, F. J. Cedano-Serrano, O. Heckert, N. van den Heuvel, J. van der Gucht, Y. Tran, D. Hourdet, C. Creton and M. Kamperman, Adv. Mater., 2019, 31, 1808179 CrossRef PubMed.
- M. Burkhardt, N. Martinez-Castro, S. Tea, M. Drechsler, I. Babin, I. Grishagin, R. Schweins, D. V. Pergushov, M. Gradzielski, A. B. Zezin and A. H. E. Müller, Langmuir, 2007, 23, 12864–12874 CrossRef CAS PubMed.
- O. Colombani, M. Ruppel, F. Schubert, H. Zettl, D. V. Pergushov and A. H. E. Müller, Macromolecules, 2007, 40, 4338–4350 CrossRef CAS.
- E. Lejeune, M. Drechsler, J. Jestin, A. H. E. Müller, C. Chassenieux and O. Colombani, Macromolecules, 2010, 43, 2667–2671 CrossRef CAS.
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342–2351 RSC.
- J. E. Laaser, Y. Jiang, D. Sprouse, T. M. Reineke and T. P. Lodge, Macromolecules, 2015, 48, 2677–2685 CrossRef CAS.
- I. A. van Hees, P. J. M. Swinkels, R. G. Fokkink, A. H. Velders, I. K. Voets, J. van der Gucht and M. Kamperman, Polym. Chem., 2019, 10, 3127–3134 RSC.
- I. Dewald, J. Gensel, E. Betthausen, O. V. Borisov, A. H. E. Müller, F. H. Schacher and A. Fery, ACS Nano, 2016, 10, 5180–5188 CrossRef CAS PubMed.
- A. D. Filippov, I. A. van Hees, R. Fokkink, I. K. Voets and M. Kamperman, Macromolecules, 2018, 51, 8316–8323 CrossRef CAS PubMed.
- S. Garnier and A. Laschewsky, Macromolecules, 2005, 38, 7580–7592 CrossRef CAS.
- D. Sprouse, Y. Jiang, J. E. Laaser, T. P. Lodge and T. M. Reineke, Biomacromolecules, 2016, 17, 2849–2859 CrossRef CAS PubMed.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS PubMed.
- D. V. Krogstad, N. A. Lynd, S. Choi, J. M. Spruell, C. J. Hawker, E. J. Kramer and M. V. Tirrell, Macromolecules, 2013, 46, 1512–1518 CrossRef CAS.
- A. V. Kabanov, T. K. Bronich, V. A. Kabanov, K. Yu and A. Eisenberg, Macromolecules, 1996, 29, 6797–6802 CrossRef CAS.
- D. V. Pergushov, E. V. Remizova, M. Gradzielski, P. Lindner, J. Feldthusen, A. B. Zezin, A. H. E. Müller and V. A. Kabanov, Polymer, 2004, 45, 367–378 CrossRef CAS.
- A. Hanisch, A. H. Gröschel, M. Förtsch, M. Drechsler, H. Jinnai, T. M. Ruhland, F. H. Schacher and A. H. E. Müller, ACS Nano, 2013, 7, 4030–4041 CrossRef CAS PubMed.
- M. Billing, J. K. Elter and F. H. Schacher, Polymer, 2016, 104, 40–48 CrossRef CAS.
- Z. Zhang, M. M. Rahman, C. Abetz, B. Bajer, J. Wang and V. Abetz, Macromol. Rapid Commun., 2019, 40, 1800729 CrossRef PubMed.
- H. M. van der Kooij, E. Spruijt, I. K. Voets, R. Fokkink, M. A. Cohen Stuart and J. van der Gucht, Langmuir, 2012, 28, 14180–14191 CrossRef CAS PubMed.
- C. Facciotti, V. Saggiomo, A. Bunschoten, R. Fokkink, J. B. ten Hove, J. Wang and A. H. Velders, Soft Matter, 2018, 14, 9542–9549 RSC.
- P. L. Valint and J. Bock, Macromolecules, 1988, 21, 175–179 CrossRef CAS.
- S. Förster, N. Hermsdorf, C. Böttcher and P. Lindner, Macromolecules, 2002, 35, 4096–4105 CrossRef.
- S. Yao, A. Bethani, N. Ziane, C. Brochon, G. Fleury, G. Hadziioannou, P. Poulin, J. Salmon and E. Cloutet, Macromolecules, 2015, 48, 7473–7480 CrossRef CAS.
- J. E. Coughlin, A. Reisch, M. Z. Markarian and J. B. Schlenoff, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2416–2424 CrossRef CAS.
- S. Förster, N. Hermsdorf, W. Leube, H. Schnablegger, M. Regenbrecht, S. Akari, P. Lindner and C. Böttcher, J. Phys. Chem. B, 1999, 103, 6657–6668 CrossRef.
- G. Masci, D. Bontempo, N. Tiso, M. Diociaiuti, L. Mannina, D. Capitani and V. Crescenzi, Macromolecules, 2004, 37, 4464–4473 CrossRef CAS.
- S. Nehache, M. Semsarilar, A. Deratani, M. In, P. Dieudonné-George, J. L. K. Him, P. Bron and D. Quémener, Polym. Chem., 2018, 9, 193–202 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2012, 28, 914–922 CrossRef CAS PubMed.
- P. Gurnani, C. P. Bray, R. A. E. Richardson, R. Peltier and S. Perrier, Macromol. Rapid Commun., 2019, 40, 1800314 CrossRef PubMed.
- J. Kolomanska, P. Johnston, A. Gregori, I. F. Domínguez, H. Egelhaaf, S. Perrier, A. Rivaton, C. Dagron-Lartigau and P. D. Topham, RSC Adv., 2015, 5, 66554–66562 RSC.
- H. Okamura, Y. Takatori, M. Tsunooka and M. Shirai, Polymer, 2002, 43, 3155–3162 CrossRef CAS.
- K. Baek, H. Kim, S. Lee, K. Cho, H. T. Kim and S. S. Hwang, Macromol. Chem. Phys., 2010, 211, 613–617 CrossRef CAS.
- H. Erothu, J. Kolomanska, P. Johnston, S. Schumann, D. Deribew, D. T. W. Toolan, A. Gregori, C. Dagron-Lartigau, G. Portale, W. Bras, T. Arnold, A. Distler, R. C. Hiorns, P. Mokarian-Tabari, T. W. Collins, J. R. Howse and P. D. Topham, Macromolecules, 2015, 48, 2107–2117 CrossRef CAS.
- P. M. Reichstein, J. C. Brendel, M. Drechsler and M. Thelakkat, ACS Appl. Nano Mater., 2019, 2, 2133–2143 CrossRef CAS.
- M. Xie and T. S. Widlanski, Tetrahedron Lett., 1996, 37, 4443–4446 CrossRef CAS.
- L. S. Simpson and T. S. Widlanski, J. Am. Chem. Soc., 2006, 128, 1605–1610 CrossRef CAS PubMed.
- S. C. Miller, J. Org. Chem., 2010, 75, 4632–4635 CrossRef CAS PubMed.
- D. B. Thomas, A. J. Convertine, R. D. Hester, A. B. Lowe and C. L. McCormick, Macromolecules, 2004, 37, 1735–1741 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 3451–3460 CrossRef CAS.
- A. H. Hofman, G. O. R. Alberda van Ekenstein, A. J. J. Woortman, G. ten Brinke and K. Loos, Polym. Chem., 2015, 6, 7015–7026 RSC.
- P. Raffa, M. C. A. Stuart, A. A. Broekhuis and F. Picchioni, J. Colloid Interface Sci., 2014, 428, 152–161 CrossRef CAS PubMed.
- N. S. Cameron, M. K. Corbierre and A. Eisenberg, Can. J. Chem., 1999, 77, 1311–1326 CrossRef CAS.
- L. Zhang, K. Yu and A. Eisenberg, Science, 1996, 272, 1777–1779 CrossRef CAS PubMed.
- L. Zhang and A. Eisenberg, Macromolecules, 1999, 32, 2239–2249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, thermal analysis of PBSPMA, FT-IR spectra, GPC chromatograms, and additional TEM images recorded at a lower magnification. See DOI: 10.1039/C9PY01227C |
| This journal is © The Royal Society of Chemistry 2019 |