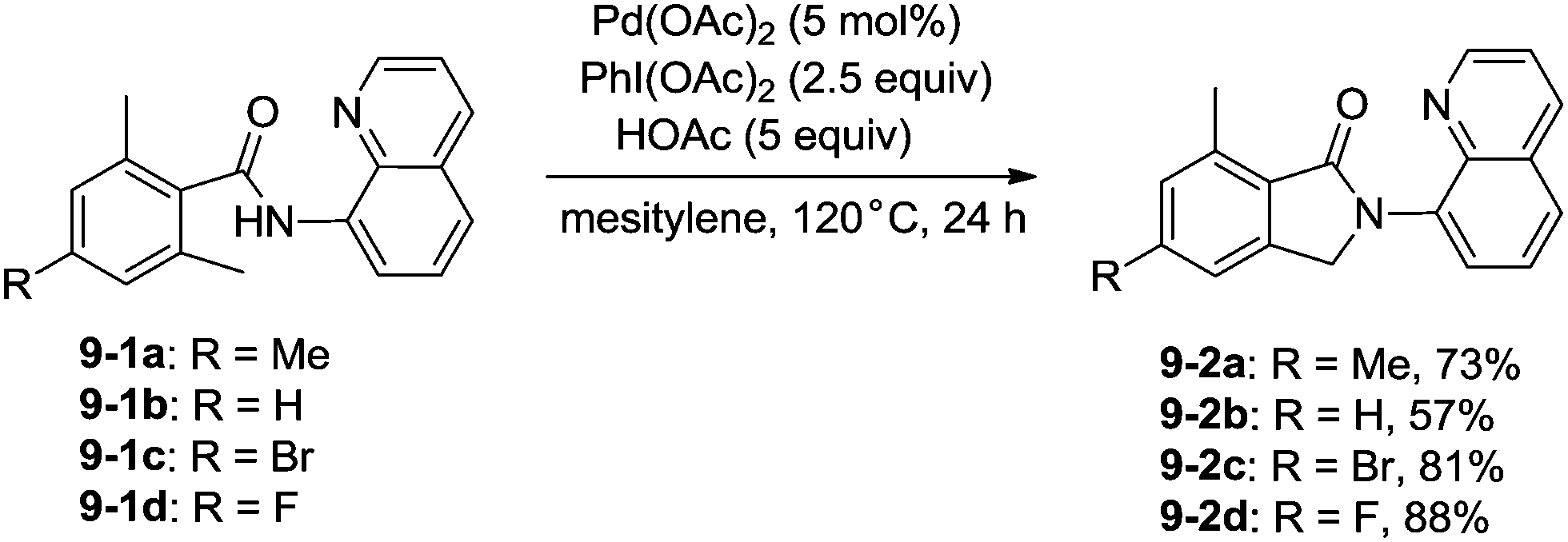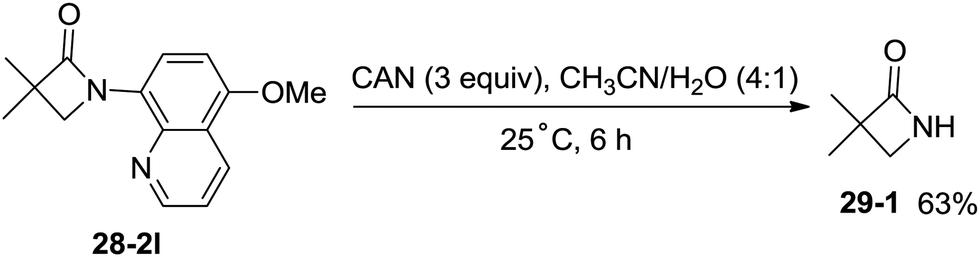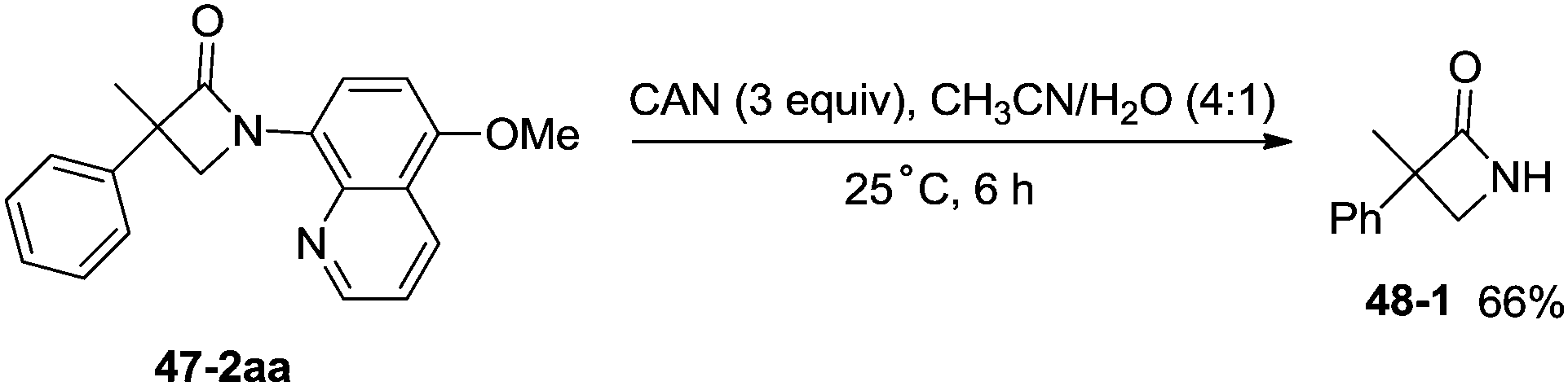DOI:
10.1039/C9CC06609H
(Feature Article)
Chem. Commun., 2019,
55, 13048-13065
Transition metal-catalyzed sp3 C–H activation and intramolecular C–N coupling to construct nitrogen heterocyclic scaffolds
Received
25th August 2019
, Accepted 5th October 2019
First published on 7th October 2019
Abstract
Nitrogen heterocycles are of great medicinal importance, and the construction of nitrogen heterocyclic scaffolds has been one of the focuses in synthetic organic chemistry. Recently, the strategy of transition metal-catalyzed sp3 C–H activation and intramolecular C–N coupling to construct nitrogen heterocyclic scaffolds has been well developed. Palladium, copper, silver, nickel, cobalt, ruthenium and rhodium catalysis were successfully used for the construction of nitrogen heterocyclic scaffolds, aziridines, azetidines, pyrrolidines, pyrrolidine-2,5-diones, indolines, isoindolines, isoindolinones, tetrahydropyridines, oxazolidinones, oxazinanones, β-lactams, γ-lactams etc., which have been synthesized by the sp3 C–H activation strategy. Here, we summarize the progress of transition metal-catalyzed sp3 C–H activation/intramolecular C–N bond formation, and introduce both the reaction development and mechanisms in numerous synthetically useful intramolecular sp3 C–H catalytic aminations/amidations.

Ming Zhang
| Ming Zhang was born in Jiangxi, China, in 1968. He studied at East China Normal University from 1985 to 1992 for bachelor's and master's degrees. After he left ECNU, he joined research works at Dalian Institute of Chemical Physics and Zhejiang University. He synthesized a series of cyclopentadienyl titanium complexes, and applied them in hydrogenation of olefins. Now he is working at Jiangxi Normal University, and his current interest is development of organic reactions catalyzed by transition metals. |

Yiyuan Peng
| Yiyuan Peng received his BS degree from the Department of Chemistry at Jiangxi Normal University in 1985. He received his MS degree from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, under the supervision of Prof. Xikui Jiang and Prof. Chengxue Zhao in 1988. He obtained his PhD degree from Nankai University under the supervision of Prof. Jinpei Cheng in 2003. Then he joined the College of Chemistry & Chemical Engineering, Jiangxi Normal University, as a professor. His research interests focus on organic synthesis and chemical biology. |

Zhiyuan Chen
| Zhiyuan Chen received his PhD from Fudan University in 2011. After a postdoctorate research period with Prof. Dr Naohiko Yoshikai at Nanyang Technological University from 2012 to 2013, he then moved to Jiangxi Normal University to carry out his independent research. He was appointed as an associate professor in 2013, and a full professor of organic chemistry in 2018. His research interest is focused on the diversity-oriented synthesis (DOS) of natural-product like molecules, and the development of synthetic methodologies for the preparation of biologically active heterocyclic compounds. |

Junmin Chen
| Junmin Chen obtained his MS degree in 2005 from Jiangxi Normal University (China). He earned his doctoral degree at the Chengdu Institute of Organic Chemistry of the Chinese Academy of Science in 2010 under the supervision of Prof. Jian Liao. He became an associate professor at Jiangxi Normal University in 2012. His current research is focused on transition metal-catalyzed C–H bond functionalization and new methods for heterocycle synthesis. |

Ai Qin Zhang
| Ai Qin Zhang was born in Jiangxi, China, in 1971. She studied at Nanchang University from 2007 to 2010 for her PhD. She is currently an associate professor at Nanchang Hangkong University. Her research interest focuses on the development of organic reactions catalyzed by transition metals. |
1. Introduction
Nitrogen heterocycles are important structural units in many biologically active natural products and synthetic pharmaceuticals, and thus the construction of nitrogen heterocyclic scaffolds has attracted the attention of many chemists.1–4 Intramolecular direct amination/amidation of sp3 C–H bonds is an effective method to construct nitrogen heterocyclic scaffolds, and there are thus far three main approaches to this problem: (1) rhodium-,5 iron-,6 cobalt-,7 or silver-8 catalyzed nitrene insertion reaction of sulfamides/carbamates or aryl/alkyl azides, (2) metal-free intramolecular oxidative coupling of sp3 C–H and N–H,9–17 and (3) transition metal-catalyzed intramolecular coupling of sp3 C–H and N–H via C–H activation. The first two approaches have limitations, as the products by the first approach are secondary amides or amines and tertiary C–H bonds are preferentially functionalized and benzylic or allylic C–H bonds are aminated in most cases using the second approach; primary C–H bonds are always preferentially functionalized by the third approach, and benzylic or allylic C–H bonds can also be functionalized, so the last approach complements the first two with respect to the reactivity patterns, and it has been well developed in recent years. A variety of metal catalysts have been found to be useful in such reactions, some reactions have been mentioned in recent reviews,4 but they have less content describing the types of heterocyclic scaffolds constructed and sorts of catalysts related to intramolecular sp3 C–H amination/amidation, so it is necessary to write a review to summarize the progress in this field. The following content includes construction of nitrogen heterocyclic scaffolds via sp3 C–H activation and intramolecular C–N coupling under palladium, copper, silver, nickel, cobalt, ruthenium or rhodium catalysis respectively, affording aziridines, azetidines, pyrrolidines, pyrrolidine-2,5-diones, indolines, isoindolines, isoindolinones, tetrahydropyridines, oxazolidinones, oxazinanones, β-lactams, γ-lactams etc. Two types of transformations with respect to disconnections will be introduced, one is the direct intramolecular coupling of sp3 C–H and N–H via C–H activation, and the other is carbonylative cyclization via sp3 C–H activation followed by insertion of CO and subsequent C–N coupling.
2. Palladium catalysis
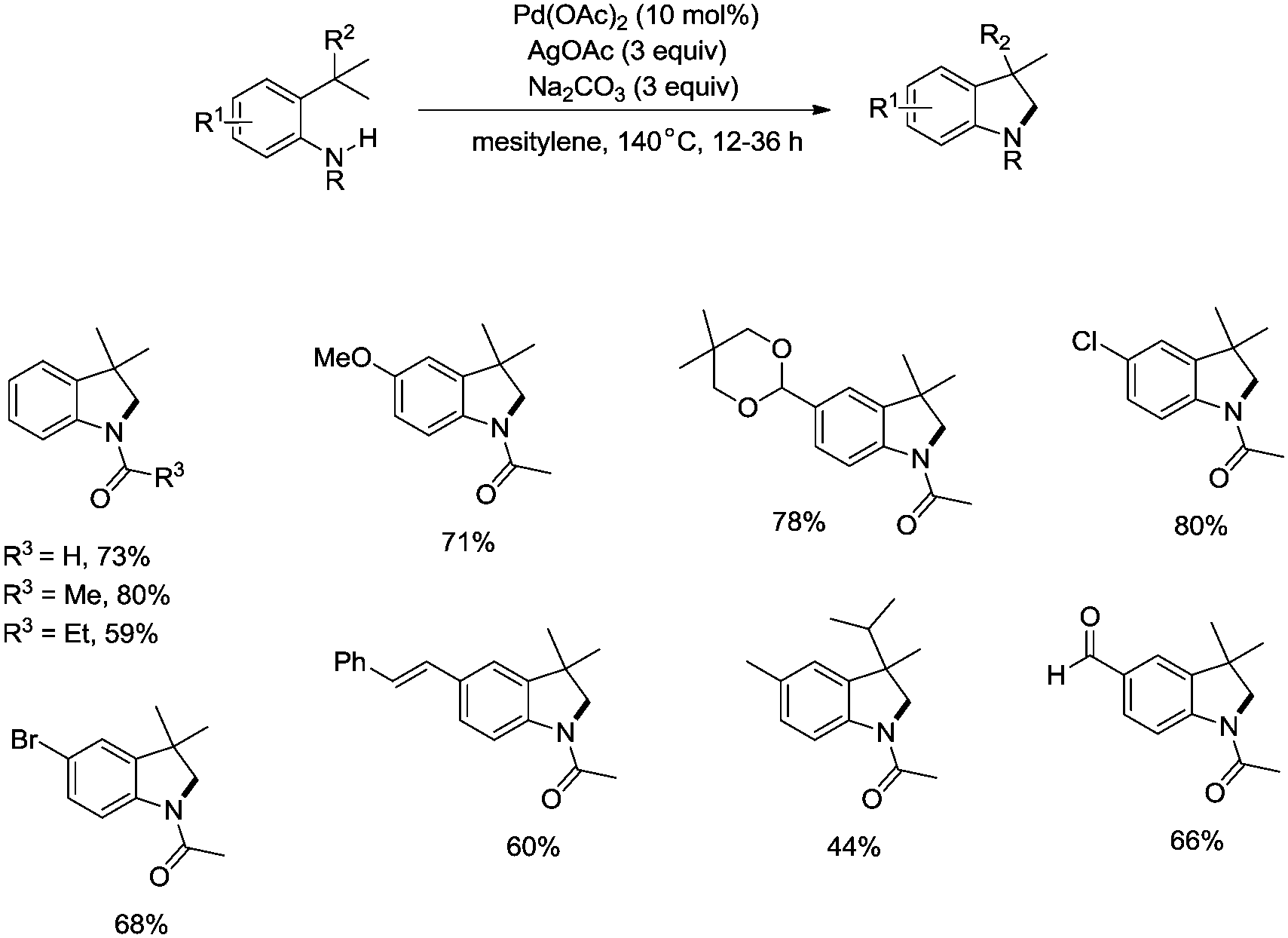
In 2009, Glorius and co-workers reported the pioneering work about the intramolecular amidation of unactivated sp3 C–H bonds, and indolines were synthesized via palladium-catalyzed cyclization of anilines (Scheme 1).18 An unactivated sp3 C–H bond is activated with the assistance of an amide directing group, and then the amide nitrogen is coupled with the sp3 carbon via reductive elimination; the transformation proceeded smoothly, affording indolines in good yields up to 80%, and the functional group tolerance is broad, halogen (Br, Cl) is also tolerated, which can be used in further manipulations. This transformation is the first example of the construction of nitrogen heterocyclic scaffolds via transition metal-catalyzed unactivated sp3 C–H bond activation and intramolecular coupling of C–H and N–H, and nitrenes are not required in this transformation.
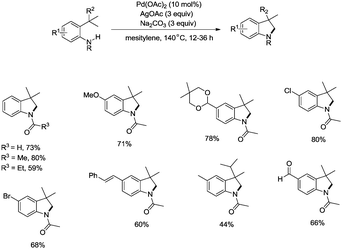 |
| | Scheme 1 Synthesis of indolines by Glorius. | |
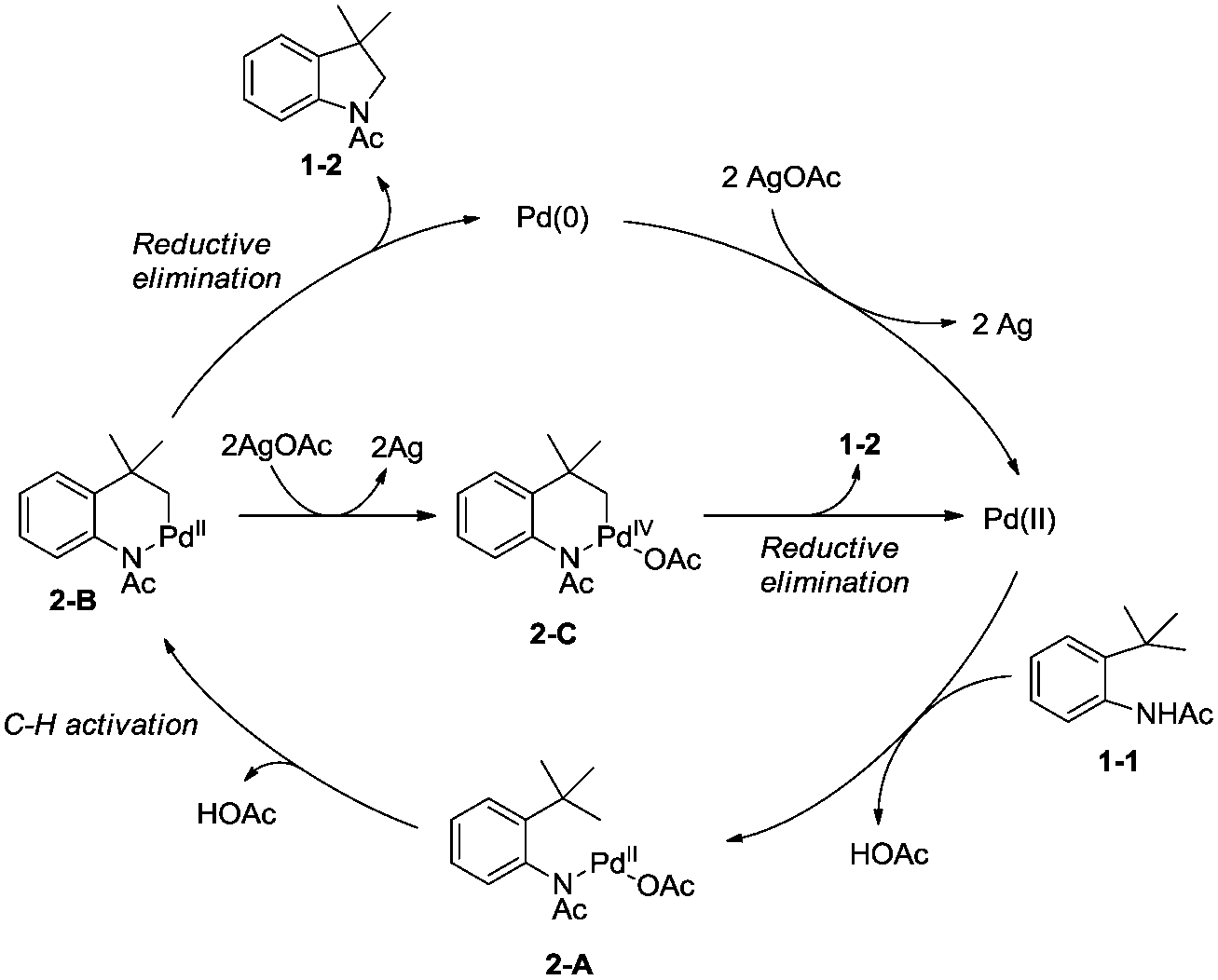
A Pd(II)/Pd(0) catalytic cycle and a Pd(II)/Pd(IV) catalytic cycle are considered as the possible mechanism for the cyclization of anilines (Scheme 2). Coordination of the aniline 1-1 with Pd(II) gives Pd(II) complex 2-A, subsequent C–H activation gives the six-membered ring Pd(II) complex 2-B, which gives product 1-2via reductive elimination, and the generated Pd(0) species is reoxidized by AgOAc to a Pd(II) species, which completes the Pd(II)/Pd(0) catalytic cycle. The Pd(IV) complex 2-C can be formed via the oxidation of 2-B, and the product 1-2 can also be obtained via a Pd(II)/Pd(IV) catalytic cycle. It is concluded that a sequential acetoxylation/amidation pathway in this reaction is impossible according to the control experiments.
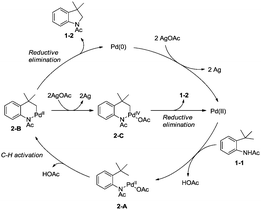 |
| | Scheme 2 The mechanism for cyclization of anilines. | |
In 2012, Daugulis and co-workers developed a palladium acetate-catalyzed method for the synthesis of pyrrolidines, indolines, and isoindolines via a C–H/N–H coupling (Scheme 3a),19a employing a picolinamide directing group. Sp2 C–H bonds, benzylic sp3 C–H bonds and C–H bonds of a methyl group are reactive. Methylene can’t undergo the amination. The directing auxiliary can be removed by LiEt3BH.
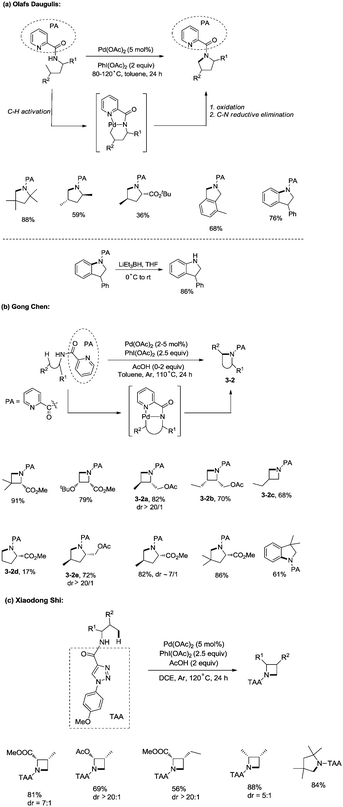 |
| | Scheme 3 Formation of cyclic amines. | |
In the same year, Chen and co-workers reported a palladium-catalyzed intramolecular amination of sp3 C–H and sp2 C–H bonds, affording azetidines, pyrrolidines, and indolines highly efficiently (Scheme 3b).19b,c The N,N-bidentate picolinamide was used as a directing group, the sp3 C–H and sp2 C–H bonds at the γ or δ positions were activated by the palladium(II) species, and intramolecular C–N bonds were formed via reductive elimination. Primary sp3 C–H bonds of methyl groups on both γ and δ positions could be preferentially functionalized, and certain geminal dimethyl substrates underwent the cyclization smoothly with high diastereoselectivity (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (3-2a, 3-2e). The order of C–H activation reactivities of different sp3 C–H bonds under the reaction conditions was as follow: secondary and tertiary γ-C–H bonds < primary δ-C–H bonds < primary γ-C–H bonds (3-2b, 3-2c, 3-2d).
:1) (3-2a, 3-2e). The order of C–H activation reactivities of different sp3 C–H bonds under the reaction conditions was as follow: secondary and tertiary γ-C–H bonds < primary δ-C–H bonds < primary γ-C–H bonds (3-2b, 3-2c, 3-2d).
When the N,N-bidentate moiety of N1-p-methyoxyphenyl-1,2,3-triazole-4-carboxylic acid derived amides was used as a directing group, sp3 C–H bonds could also undergo intramolecular amination under palladium catalysis (Scheme 3c).19d The C–H bonds at γ positions were preferentially aminated over those at δ positions, the primary C–H bonds were selectively functionalized over the secondary C–H bonds, and a series of azetidines were obtained successfully. A pyrrolidine was also synthesised in 84% yield with the directing group.
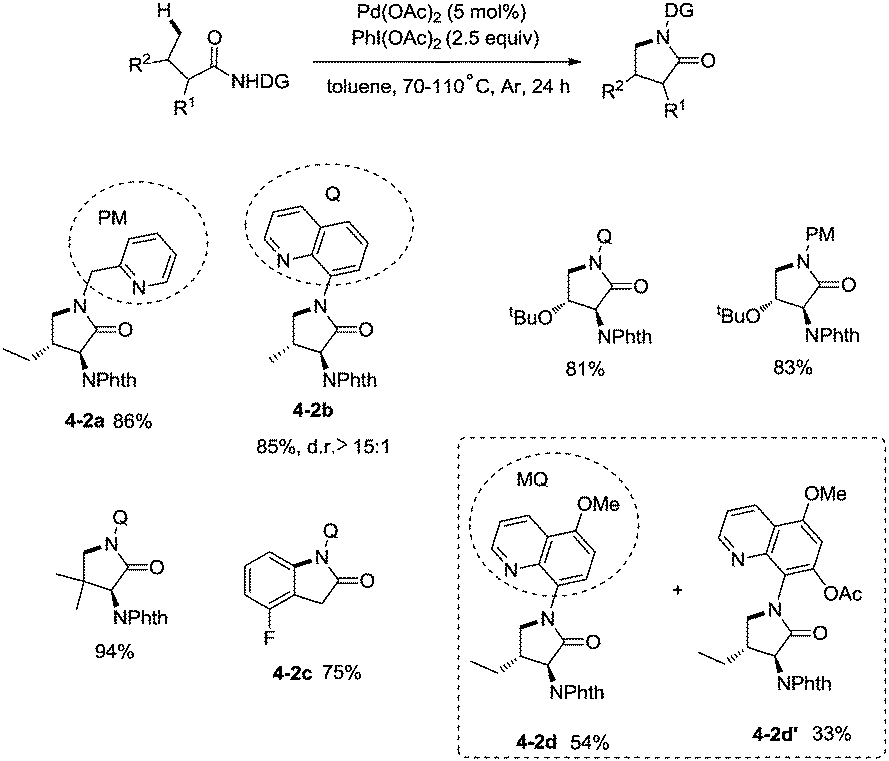
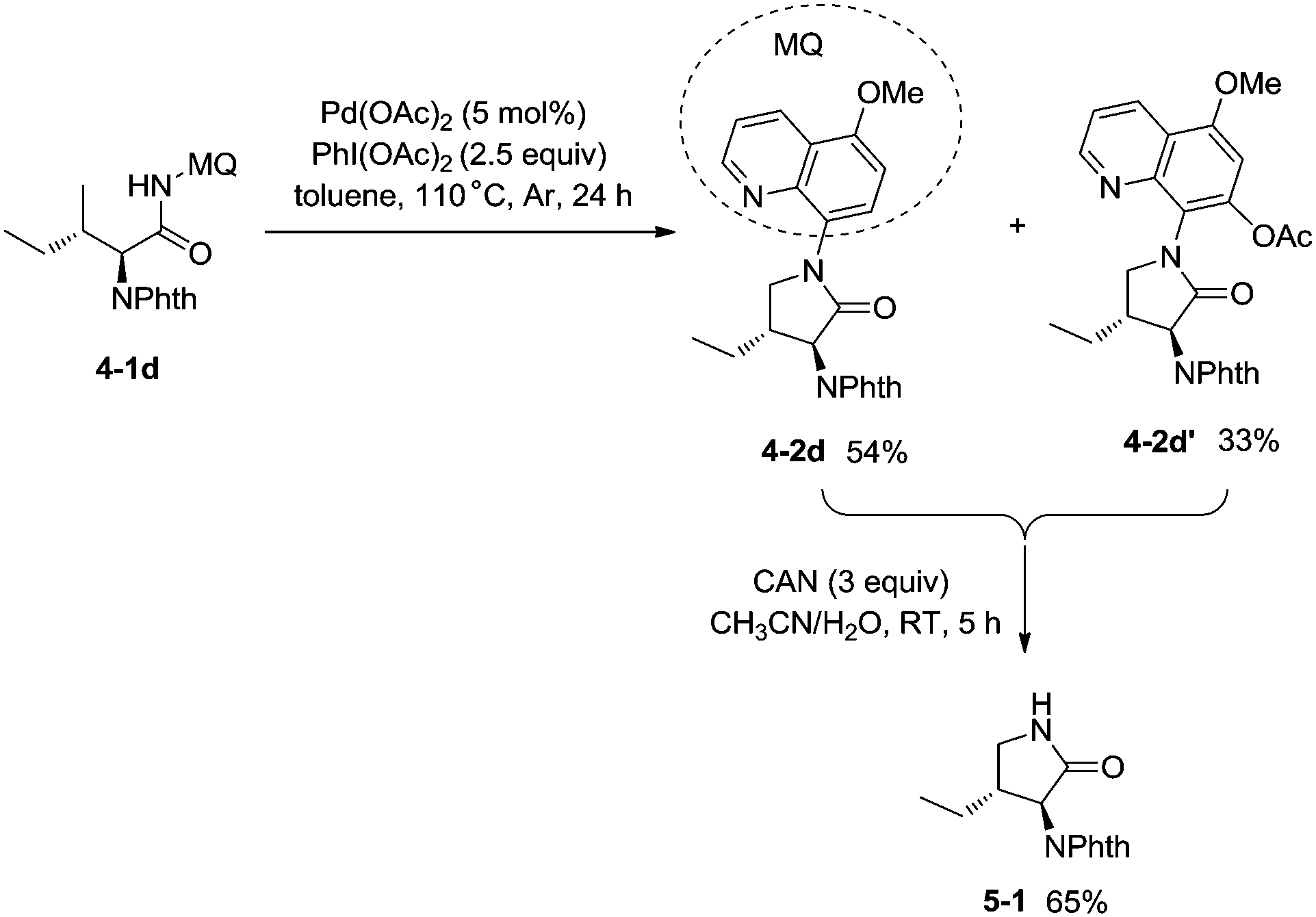
In 2013, Chen and co-workers reported another example of constructing nitrogen heterocycles via palladium-catalyzed sp3 C–H activation/intramolecular coupling of C–H/N–H, affording pyrrolidones (γ-lactams) effectively (Scheme 4).20 8-Aminoquinoline or 2-pyridylmethyl amine were used as bidentate directing groups, the γ-sp3 C–H and sp2 C–H bonds of the amides were activated, forming intramolecular C–N bonds [(4-2a)-(4-2d)], the primary sp3 C–H bonds of the methyl groups on the γ positions could be preferentially functionalized (4-2a, 4-2d), and the diastereoselectivity is excellent with certain geminal dimethyl substrates (for example, 4-2b, dr > 15:1). The 5-methoxyquinolin-8-amine derived substrate 4-1d was converted into a mixture of pyrrolidones (4-2d, 4-2d′). The 5-methoxy-quinolin-8-yl group (MQ) and its derivative of the directing group in the target molecules can be removed readily by treatment with ceric ammonium nitrate (CAN), and the NH pyrrolidone 5-1 was obtained in 65% yield, increasing the applications of the method in synthetic organic chemistry (Scheme 5).
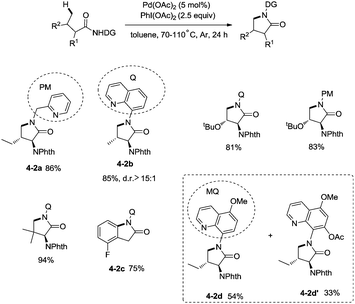 |
| | Scheme 4 Palladium-catalyzed intramolecular amination by Chen. | |
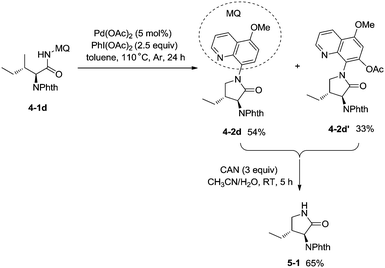 |
| | Scheme 5 Removable auxiliary group MQ. | |
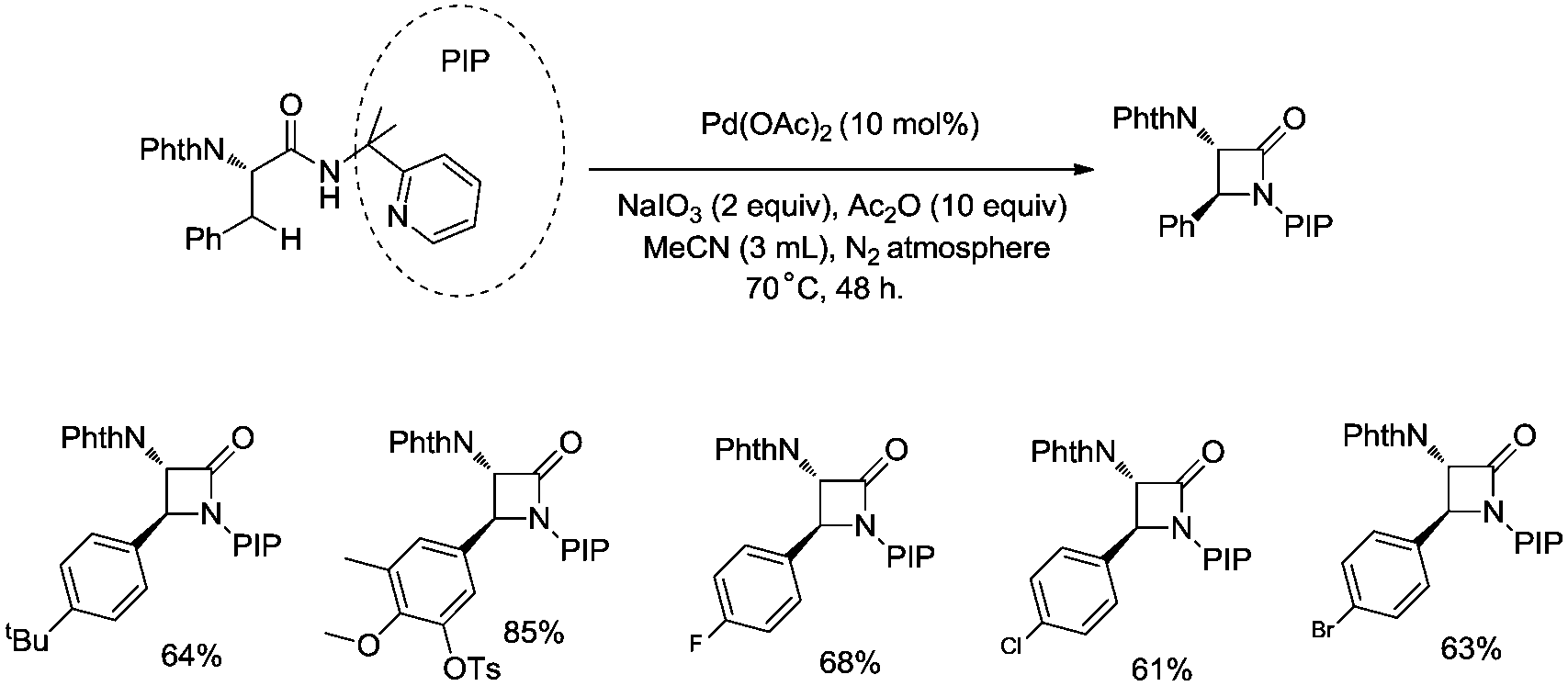
In 2013, Shi and co-workers used an N,N-bidentate directing group containing 2-(pyridin-2-yl)isopropyl (PIP) to achieve β-C(sp3)–H activation and C–N coupling cyclization, a series of α-amino-β-lactams were stereoselectively synthesized in acceptable yields, and the starting materials (substrates) can be easily prepared via palladium(II) acetate-catalyzed sp3 C–H monoarylation of alanine, undergoing the subsequent intramolecular amidation (Scheme 6).21 Secondary β-benzylic C–H bonds are amidated in this transformation. While this process is smooth and functional-group-tolerant, the PIP auxiliary cannot be removed from the β-lactams, which hinders its application in organic synthesis.
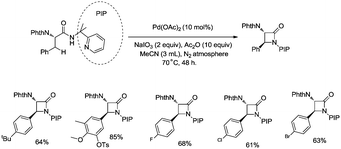 |
| | Scheme 6 The intramolecular sp3 C–H amidation with PIP as the directing group. | |
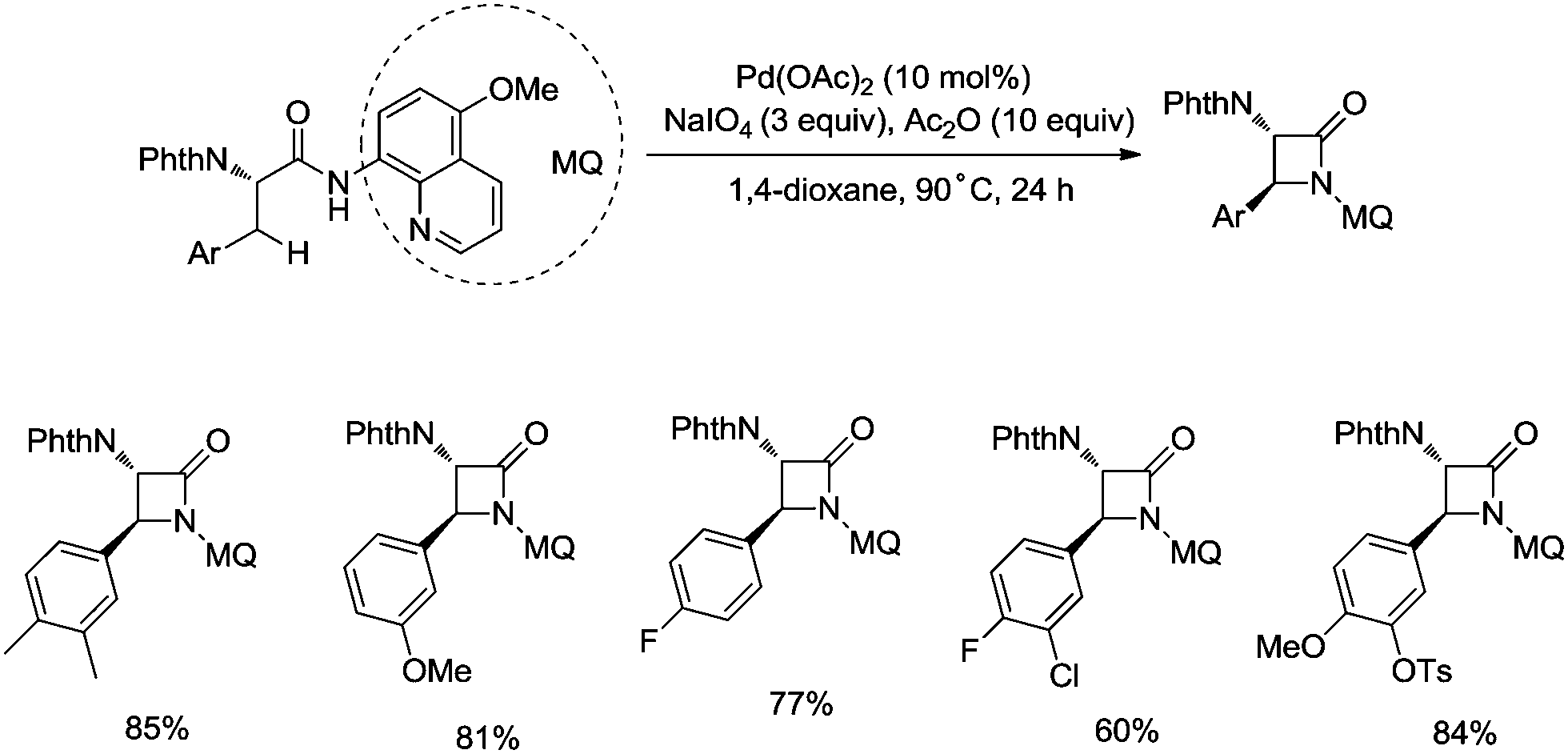
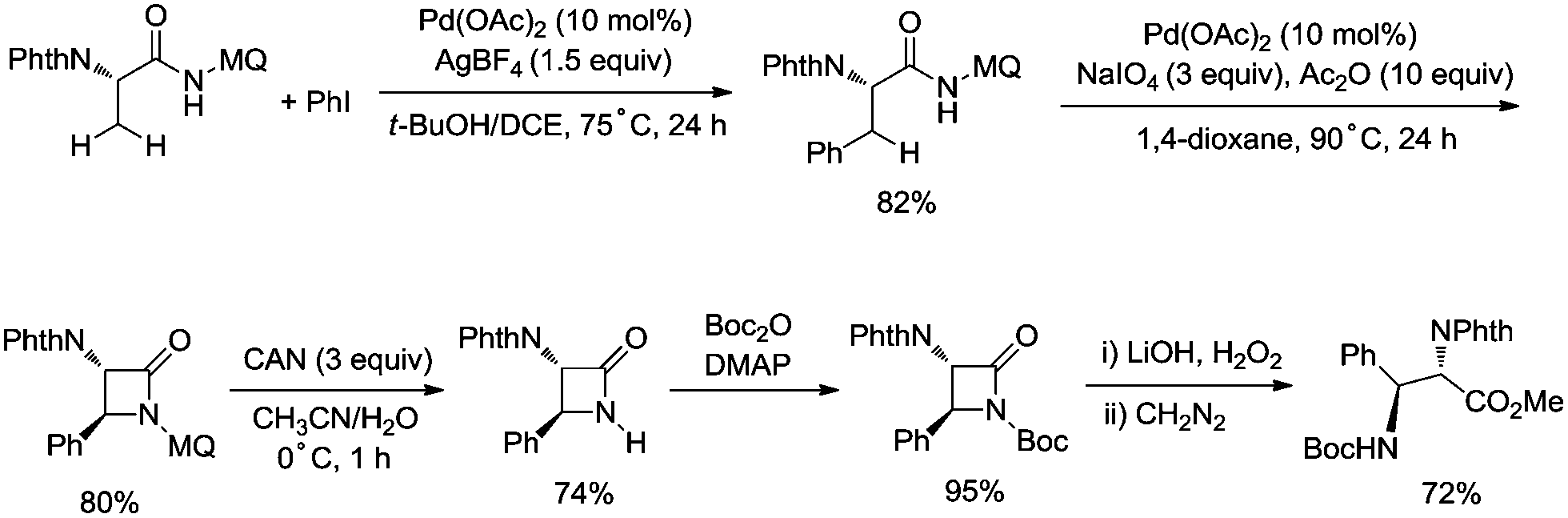
In 2017, the same group reported a revised version, where 5-methoxyquinolin-8-amine was used as a directing group, in which the auxiliary group can be removed. Palladium-catalyzed monoarylation of alanine and subsequent amidation of sp3 C–H bonds were successfully accomplished, affording various α-amino-β-lactams stereoselectively, and the reaction is highly efficient and compatible with a variety of functional groups (Scheme 7).22 The auxiliary group 5-methoxy-quinolin-8-yl (MQ) in the target molecules can be removed readily by treatment with ceric ammonium nitrate (CAN). Orthogonally protected anti-α,β-diamino acids could be synthesized stereoselectively from α-amino-β-lactams, which is useful to the total synthesis of certain natural products and biologically active compounds (Scheme 8).
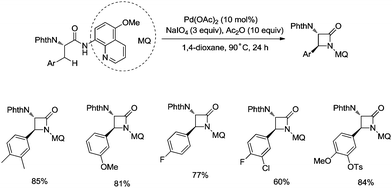 |
| | Scheme 7 Palladium-catalyzed intramolecular methylene amidation. | |
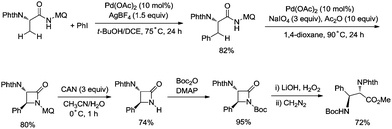 |
| | Scheme 8 Synthesis of protected anti-α,β-diamino acids. | |
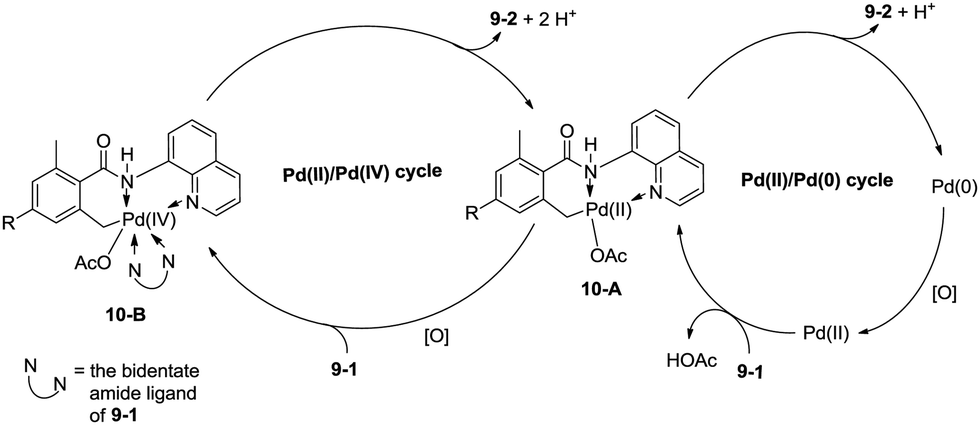
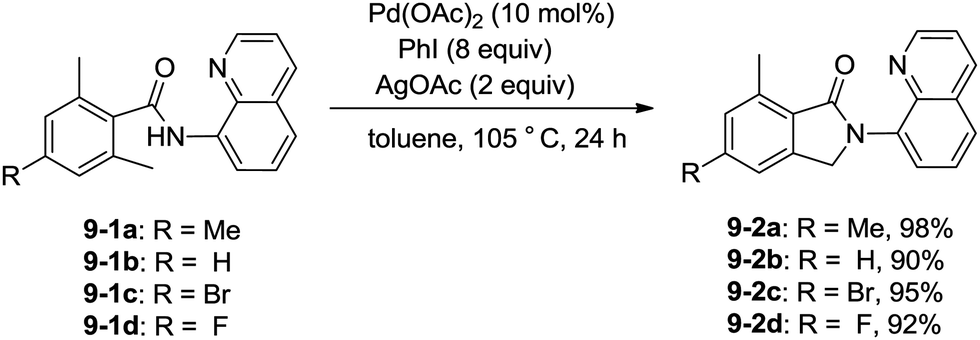
In 2013, Zhang reported isoindolinone (9-2) synthesis from 2,6-dimethyl-N-(8-quinolinyl)benzamides 9-1via palladium-catalyzed intramolecular amidation of primary benzylic C–H bonds using 8-aminoquinoline as a directing group, the N,N-bidentate amide directing group was essential to the transformation (Scheme 9).23 A possible mechanism was proposed (Scheme 10). Coordination of the bidentate 8-aminoquinoline system 9-1 to palladium and ligand exchange followed by benzylic C–H activation produce a fused five-membered ring and six-membered ring intermediate 10-A, then reductive elimination gives the product 9-2 and Pd(0), and oxidation of the Pd(0) species by PhI(OAc)2 gives the Pd(II) species, which completes the Pd(II)/Pd(0) catalytic cycle. A Pd(II)/Pd(IV) catalytic cycle is also thought to be possible. The intermediate 10-A is oxidized to the Pd(IV) species 10-B, then reductive elimination gives the product 9-2 and the Pd(II) complex 10-A.
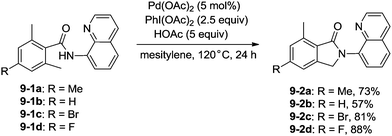 |
| | Scheme 9 Isoindolone synthesis via palladium-catalyzed intramolecular amidation. | |
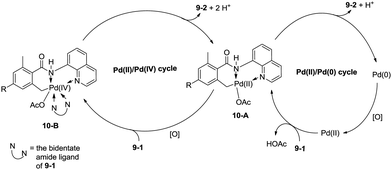 |
| | Scheme 10 The possible mechanism for isoindolone synthesis. | |
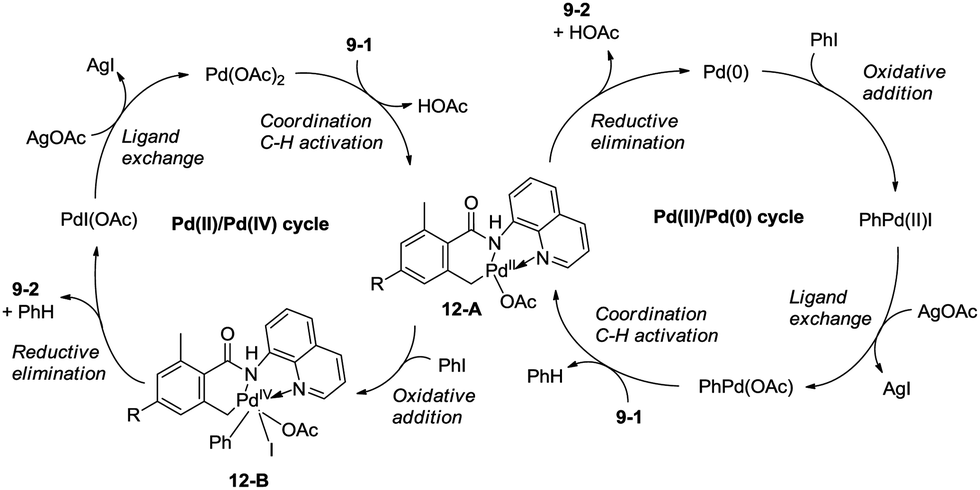
Zhang and co-workers intented to effect palladium-catalyzed arylation of benzylic C–H bonds with 2,6-dimethyl-N-(8-quinolinyl)benzamides 9-1 as substrates according to the literature methods,24 the cyclization products isoindolinones were obtained in excellent yields via intramolecular C–N coupling, and no arylation product was found (Scheme 11).25 The 8-quinolyl as a directing group and two methyl groups in the ortho positions were found to be necessary. Phenyl iodide acted as an oxidant of the catalytic cycles for intramolecular C–N coupling (Scheme 12). The reactions proceed in high yields, and the procedure is easy to perform.
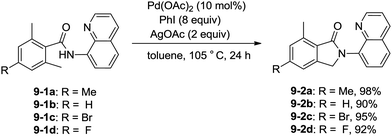 |
| | Scheme 11 Synthesis of isoindolinones with PhI. | |
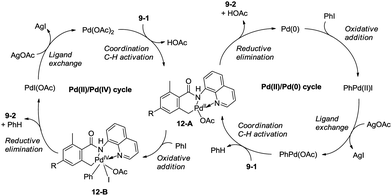 |
| | Scheme 12 The possible mechanism for isoindolone synthesis with PhI. | |
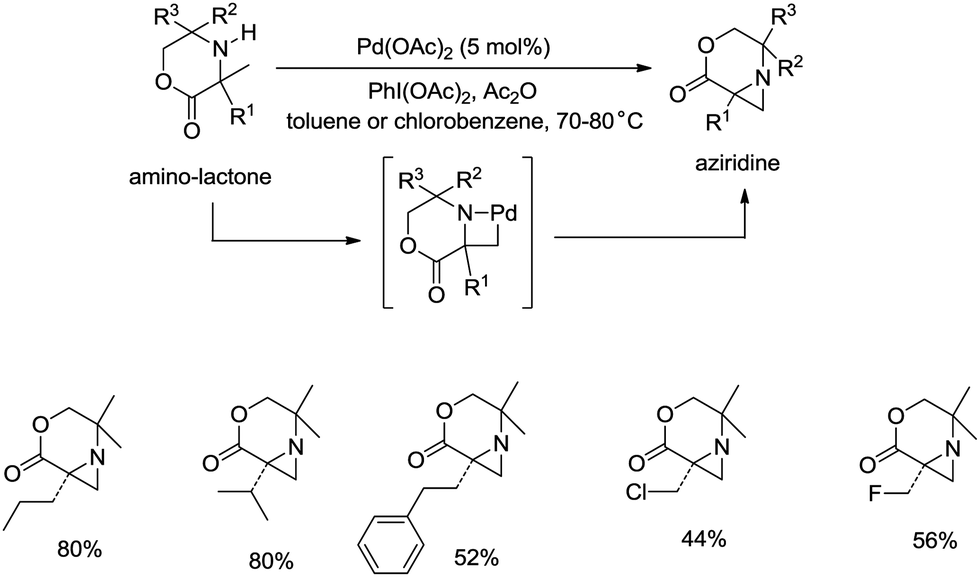
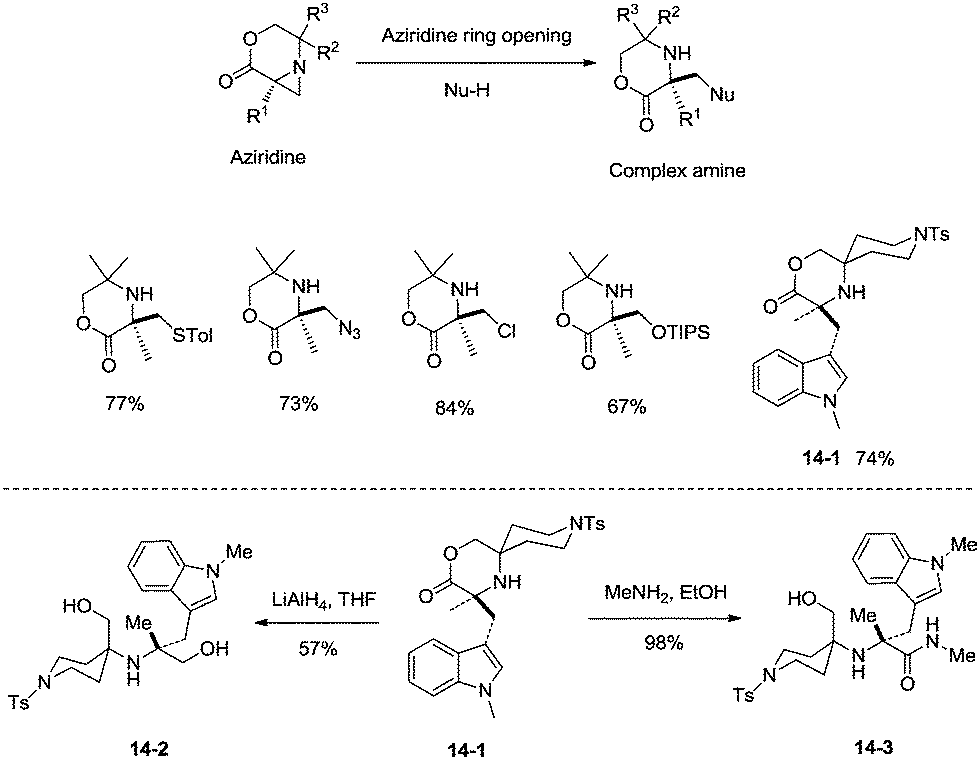
sp3 C–H bonds were activated by the assistance of N-monodentate or N,N-bidentate amide systems forming five- or six-membered cyclopalladation complexes in the above examples. In 2014, aliphatic secondary amines were found to be also useful directing groups, and strained four-membered ring cyclopalladation intermediates were formed (as confirmed by single-crystal X-ray diffraction).26a A series of amino-lactones were used as substrates, and the corresponding aziridines were obtained in moderate to good yields (up to 80%) (Scheme 13). This strategy showed that simple aliphatic amines possessing methyl groups adjacent to an unprotected amine nitrogen atom can be converted directly into strained nitrogen heterocycles. The aziridine ring opening with nucleophiles such as azide, thiol, chloride and water afforded fully substituted secondary aliphatic amines (Scheme 14). The product of an aziridine ring opening (14-1) provided a new class of secondary aliphatic amines through further conversion (14-2, 14-3), the biological properties of which are worth exploring.
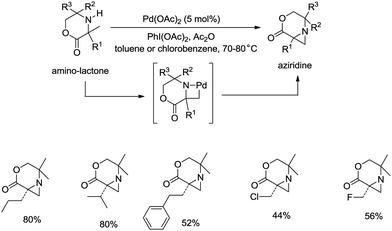 |
| | Scheme 13 Palladium-catalyzed aziridination. | |
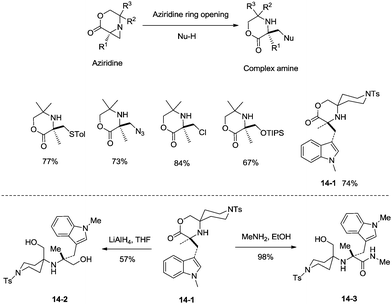 |
| | Scheme 14 Aziridine ring opening with nucleophiles. | |
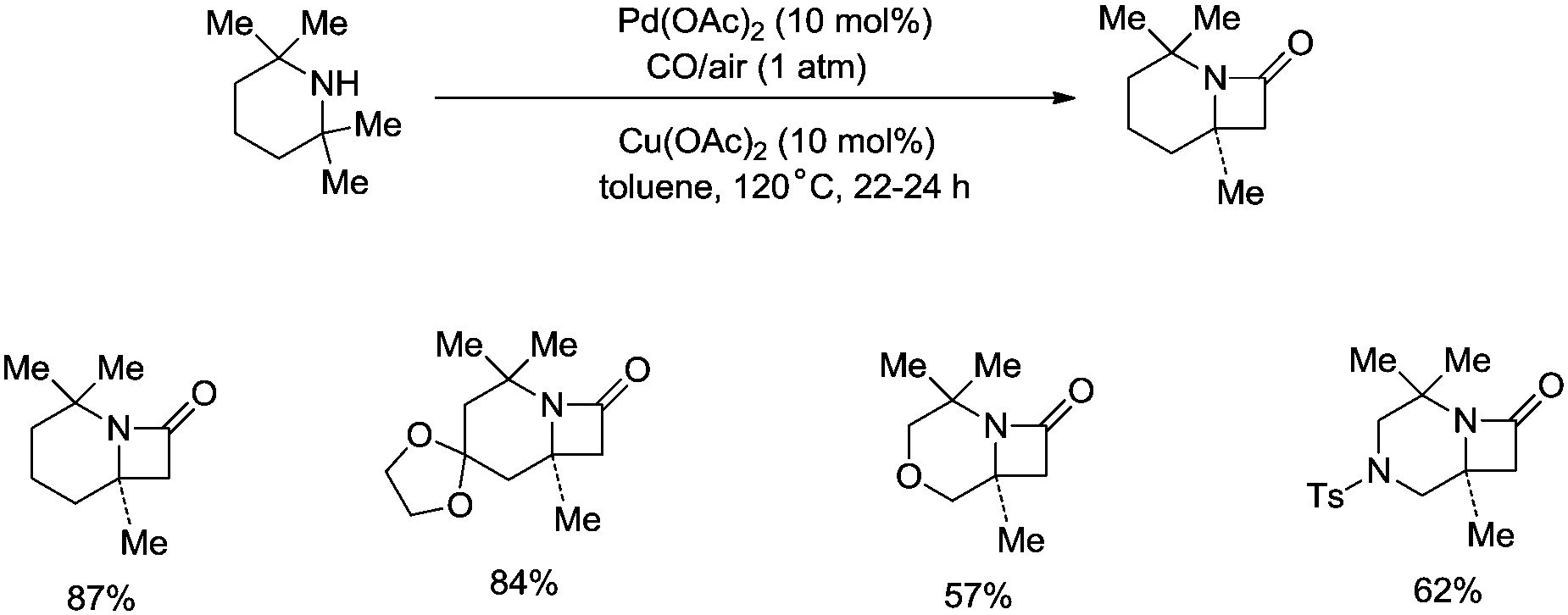
Strained nitrogen heterocycle β-lactams which are important structure units of potential pharmaceutical agents can also be produced via carbonylation of the sp3 C–H bonds of a methyl group adjacent to a secondary amine, and this further demonstrates the broad uses of the C–H activation directed by aliphatic secondary amines (Scheme 15).26a Before this report, palladium-catalyzed carbonylative cyclization of N-arylamides successfully provided succinimides via β-C(sp3)–H activation.26b
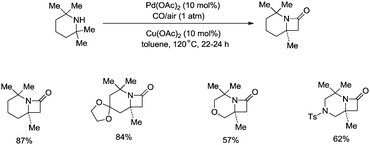 |
| | Scheme 15 Pd-catalyzed C–H carbonylation. | |
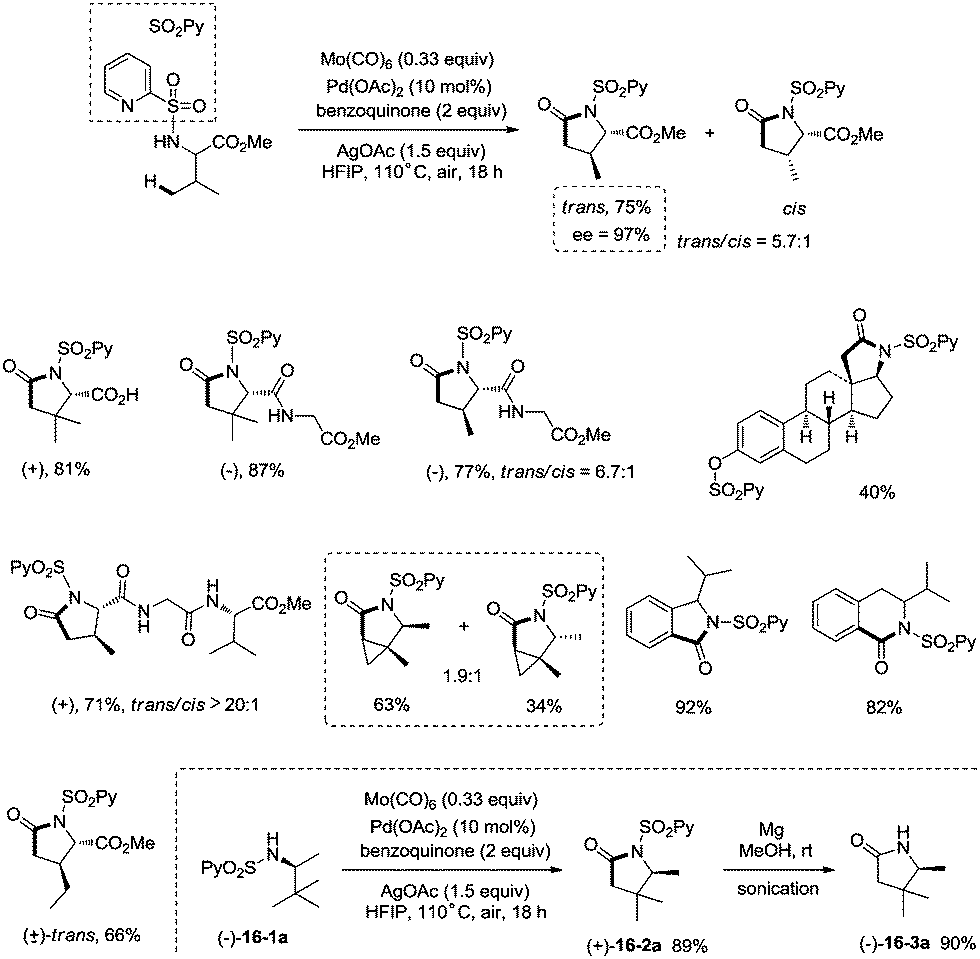
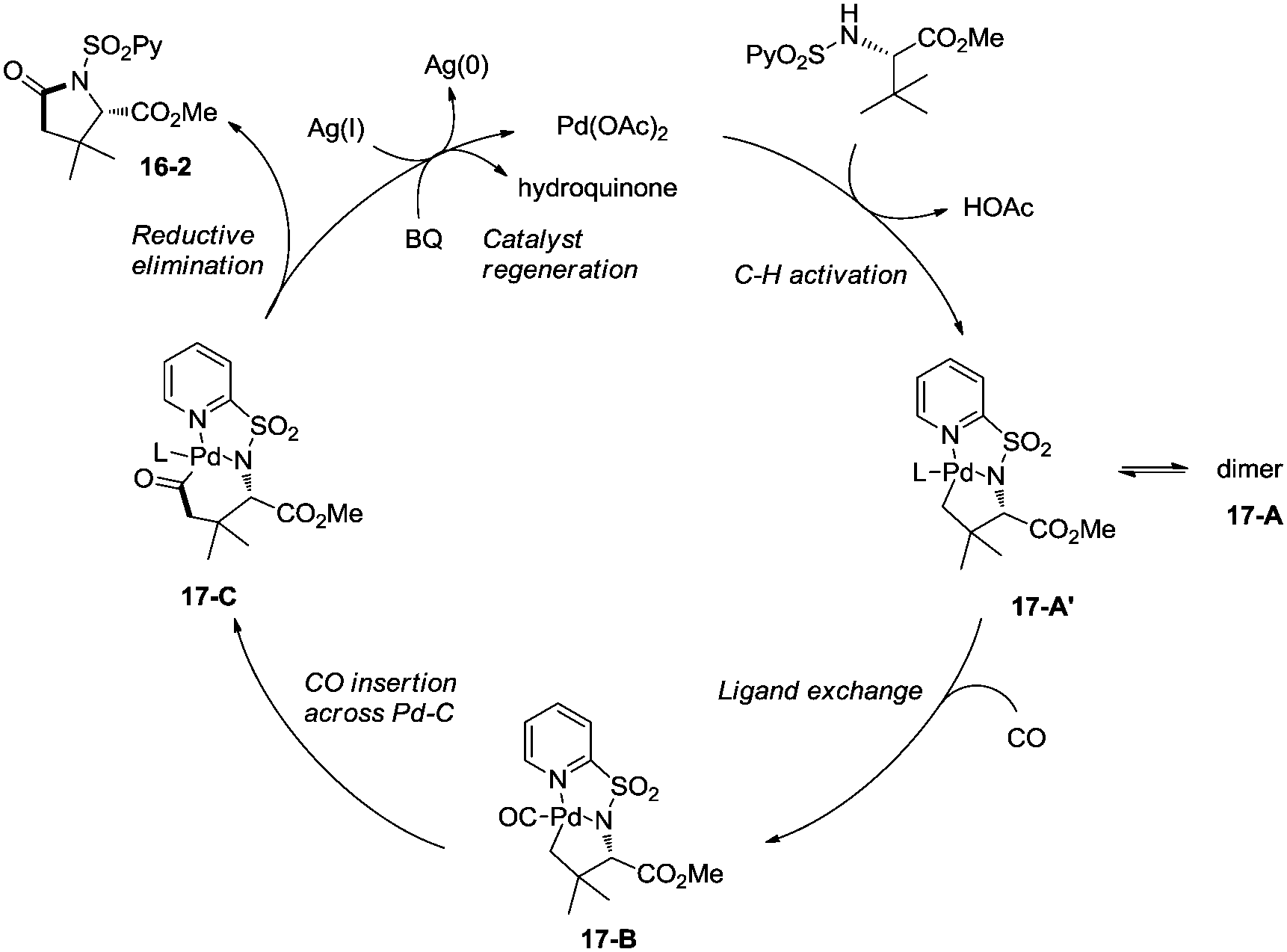
Late-stage carbonylative cyclization of an estrone derivative, amino acids and peptides via palladium-catalyzed γ-sp3 C–H bond activation was achieved, affording richly functionalized γ-lactams successfully (Scheme 16).26cN-(2-pyridyl)sulfonyl (N-SO2Py) was used as a directing group, overriding other inherent substrate coordinating elements. The directing auxiliary could be removed conveniently, and a free β-lactam (−)-16-3a was obtained in 90% yield upon treatment of (+)-16-2a with magnesium turnings at room temperature under sonication overnight. Coordination of the directing group to palladium(II) and subsequent C–H activation give the Pd(II) complex 17-A′ (Scheme 17), ligand exchange produces the intermediate 17-B, CO insertion affords the intermediate 17-C, reductive elimination affords the product 16-2, and the catalyst is regenerated by oxidants (AgOAc and benzoquinone).
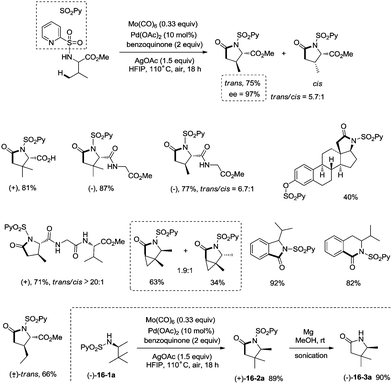 |
| | Scheme 16 Carbonylative cyclization of an estrone derivative, amino acids and peptides. | |
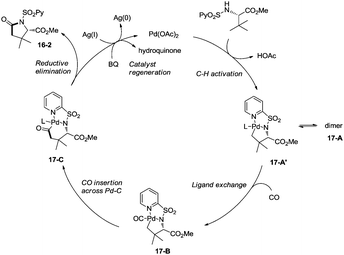 |
| | Scheme 17 The mechanism for carbonylative cyclization. | |
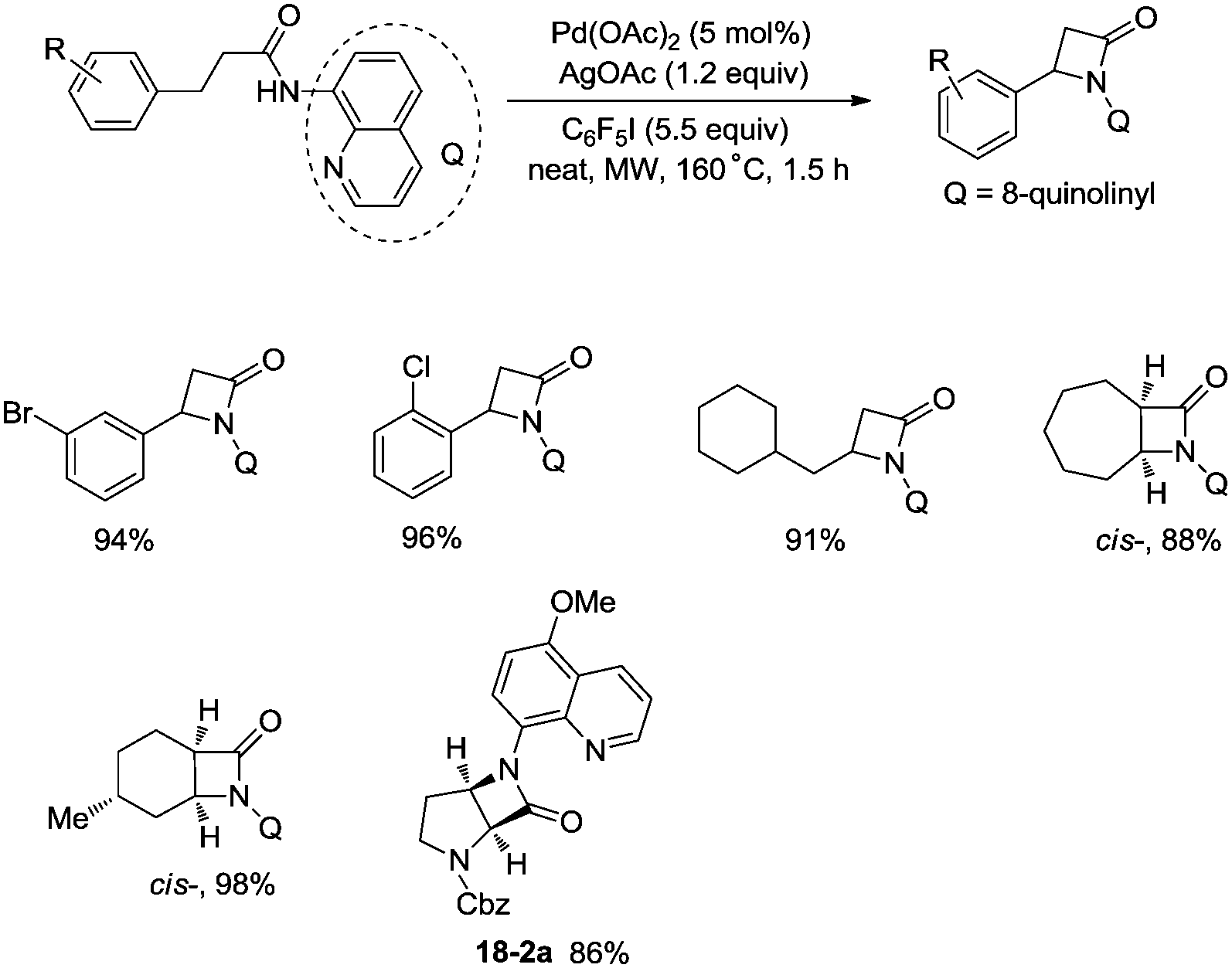
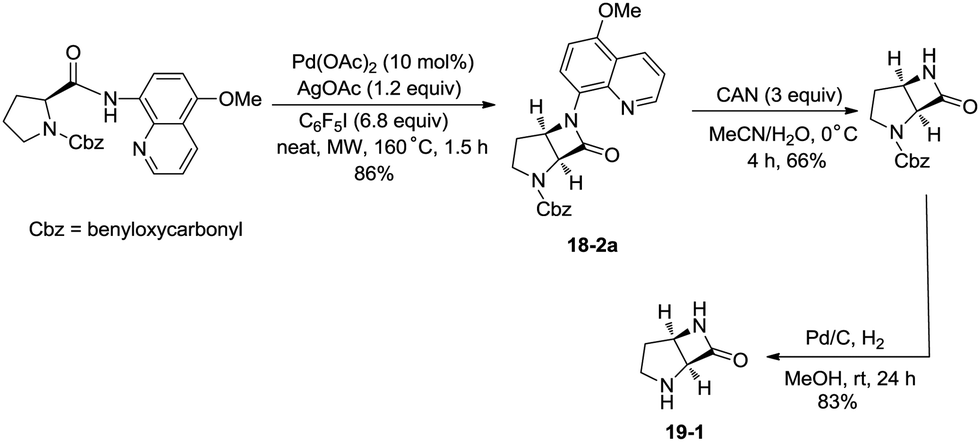
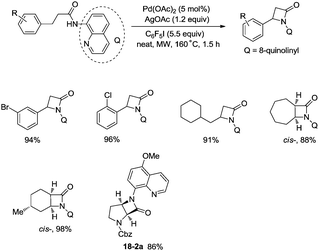
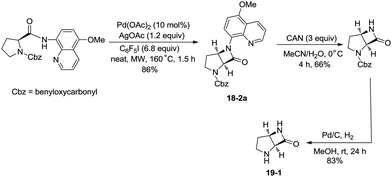
In 2016, a palladium acetate-catalyzed bidentate-ligand-directed synthesis of β-lactams via intramolecular amidation of unactivated sp3 C–H bonds was reported by Wu and co-workers (Scheme 18),27 the transformation proceeded smoothly in excellent yields up to 98%, and the bidentate amides derived from 8-amino quinoline were used as substrates in this transformation. This strategy can be applied to the stereoselective synthesis of diazabicyclic β-lactams (18-2a) with 86% yield, and 5-methoxy-quinolin-8-yl (MQ) of the directing groups and protecting group benzyloxycarbonyl (Cbz) in the target molecules can be removed readily on treatment with ceric ammonium nitrate (CAN) and hydrogen (Scheme 19). The key intermediate for the synthesis of MK-8712, cis-fused diazabicyclic β-lactams (−)-19-1 was obtained from starting material L-proline.
 |
| | Scheme 18 Synthesis of β-lactams. | |
 |
| | Scheme 19 Stereoselective synthesis of diazabicyclic β-lactams. | |
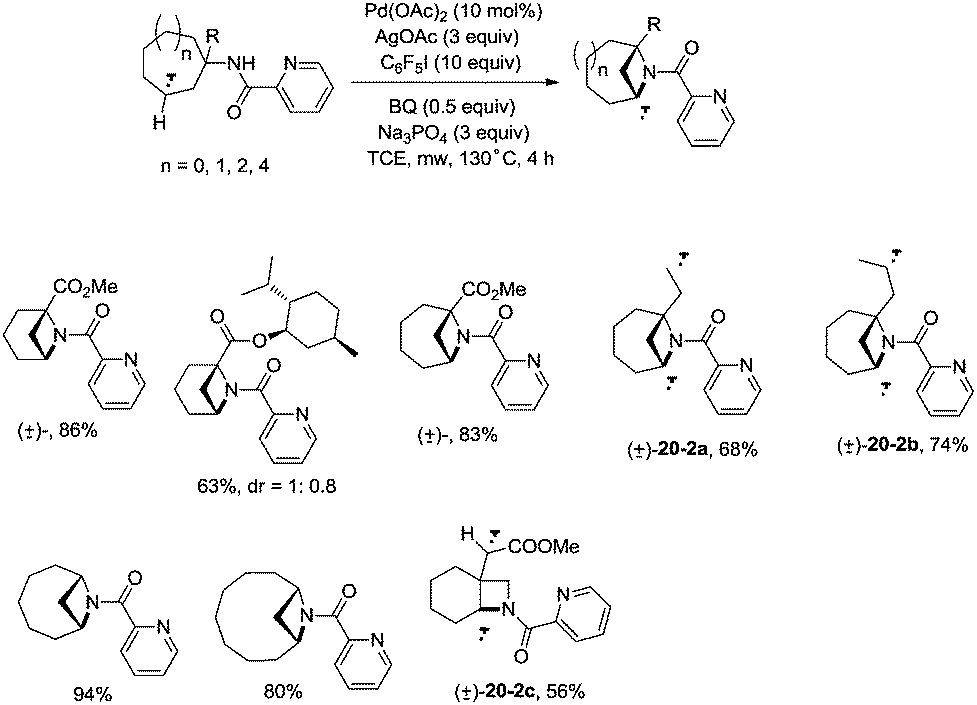
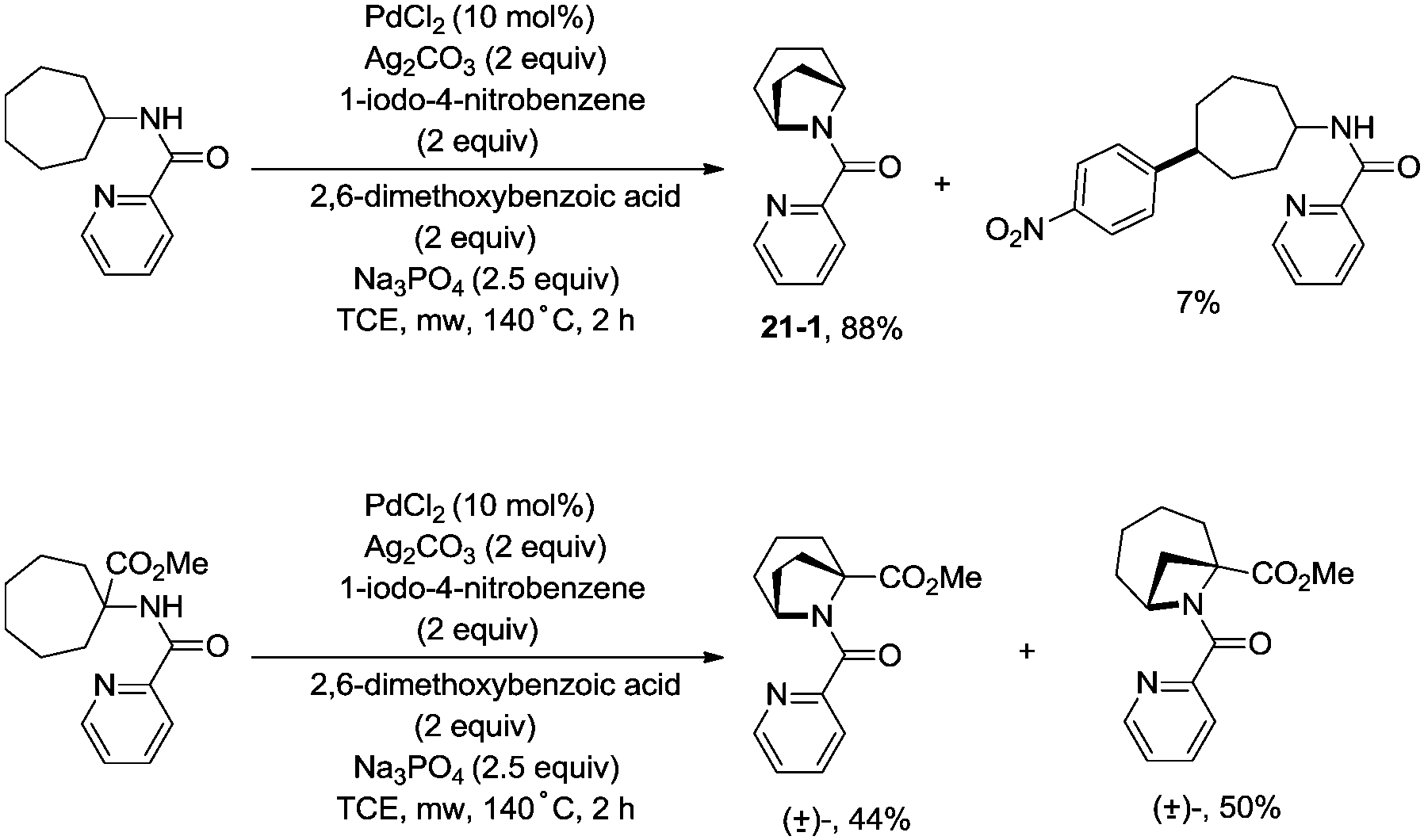
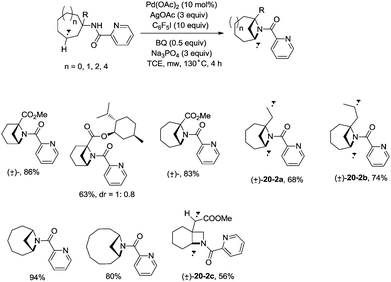
In 2017, Wu and co-workers reported the synthesis of polycyclic azetidines and pyrrolidines also using the strategy palladium-catalyzed N,N-bidentate amide-directed intramolecular coupling of sp3 C–H and N–H, but using picolinamides as the directing groups (Scheme 20).28 The secondary γ-sp3 C–H bonds on the ring were preferentially activated over the primary or secondary sp3 C–H bonds on the acyclic side chain (20-2a, 20-2b and 20-2c), affording monocyclic or bicyclic azetidines effectively. When 2,6-dimethoxybenzoic acid and 1-iodo-4-nitrobenzene were used as the key additives, the intramolecular amination also occurred at the δ position, the polycyclic pyrrolidines were obtained, the highest yield is 88% (21-1), and the other substrates afforded a mixture of polycyclic azetidines and pyrrolidines in an approximate 1:1 ratio (Scheme 21).
 |
| | Scheme 20 Azetidines synthesis. | |
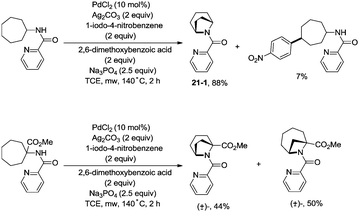 |
| | Scheme 21 Pyrrolidine synthesis. | |
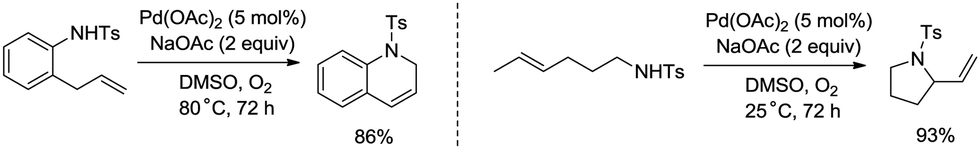
Palladium-catalyzed intramolecular allylic C–H amination is a valuable access to construct nitrogen heterocyclic scaffolds. In 1996, Pd(II)-catalyzed cyclization of olefinic tosylamides was achieved (Scheme 22).29a A π-allylpalladium intermediate was more likely involved than an aminopalladation intermediate in this transformation. Allylic C–H bonds were coupled with the nitrogen atom of amides, affording 2-vinylpyrrolidines, 1,2-dihydroquinolines, etc.
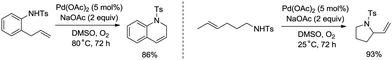 |
| | Scheme 22 C–N coupling and cyclization. | |
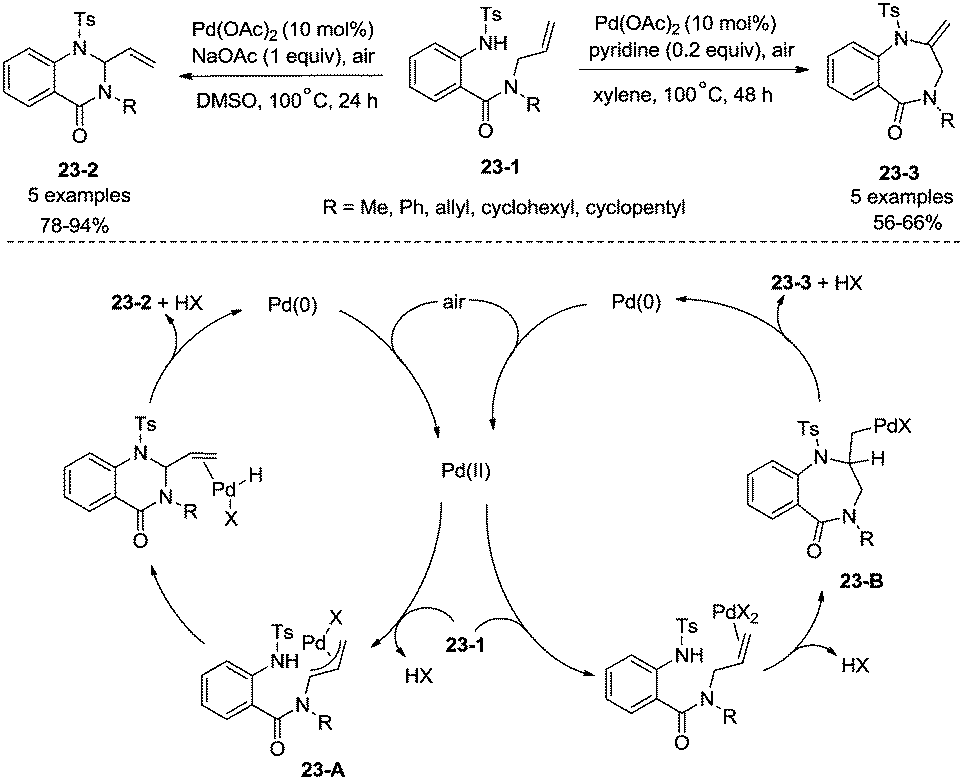
Whether a mechanism involves a η3-allylpalladium intermediate or an aminopalladation intermediate in intramolecular Pd-catalyzed amination of N-allyl-anthranilamides could be controlled by reaction conditions (Scheme 23).29b The mechanism involving a η3-allylpalladium intermediate 23-A leads to the formation of six-membered 2-vinylquinazolin-4-ones 23-2, and the mechanism involving an aminopalladation intermediate 23-B leads to the formation of seven-membered 2-methylene-1,4-benzodiazepin-5-ones 23-3.
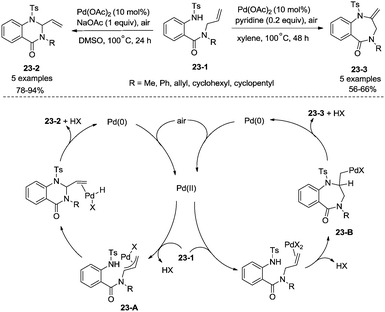 |
| | Scheme 23 Formation of a six- or seven-membered ring. | |
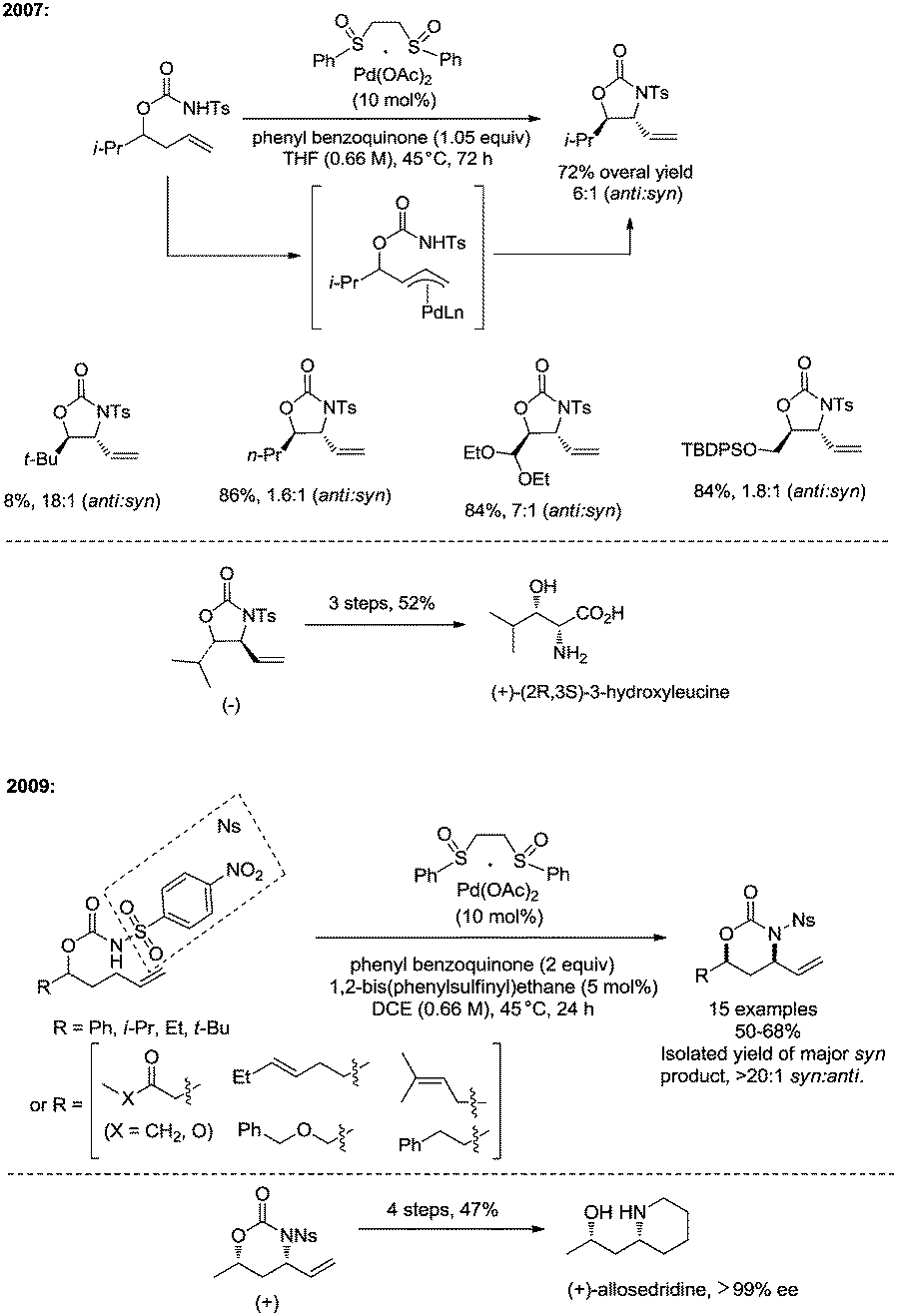
In 2007, White and co-workers developed palladium(II)/bis-sulfoxide-catalyzed intramolecular allylic C–H amination reaction of but-3-enyl N-tosyl carbamates, functionalized five-membered anti-oxazolidinones were provided with high diastereoselectivity, and a π-allyl Pd intermediate is involved in the transformation (Scheme 24).29c Useful syn-1,2-amino alcohols could be obtained via further transformation. In 2009, the same group developed a new transformation with a similar catalytic system, affording six-membered syn-oxazinanones from pent-4-enyl N-(4-nitrophenylsulfonyl) carbamates (Scheme 24), which enables the synthesis of syn-1,3-amino alcohols.29d The reaction rate of the formation of a six-membered product was increased significantly when changing the substrate from a N-tosyl carbamate to a N-(4-nitrophenylsulfonyl) carbamate. Allylic C–H bonds of terminal olefins were selectively aminated over that of internal olefins. In the same year (2009), Poli and co-workers found that using acetic acid as solvent could significantly accelerate the Pd(II)-catalyzed cyclization of but-3-enyl N-tosyl carbamates and pent-4-enyl N-tosyl carbamates,29e affording oxazolidinones and oxazinanones in high yields and with high diastereoselectivity.
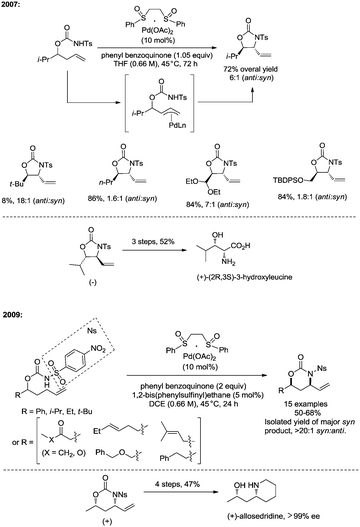 |
| | Scheme 24 Allylic C–H amination by White. | |
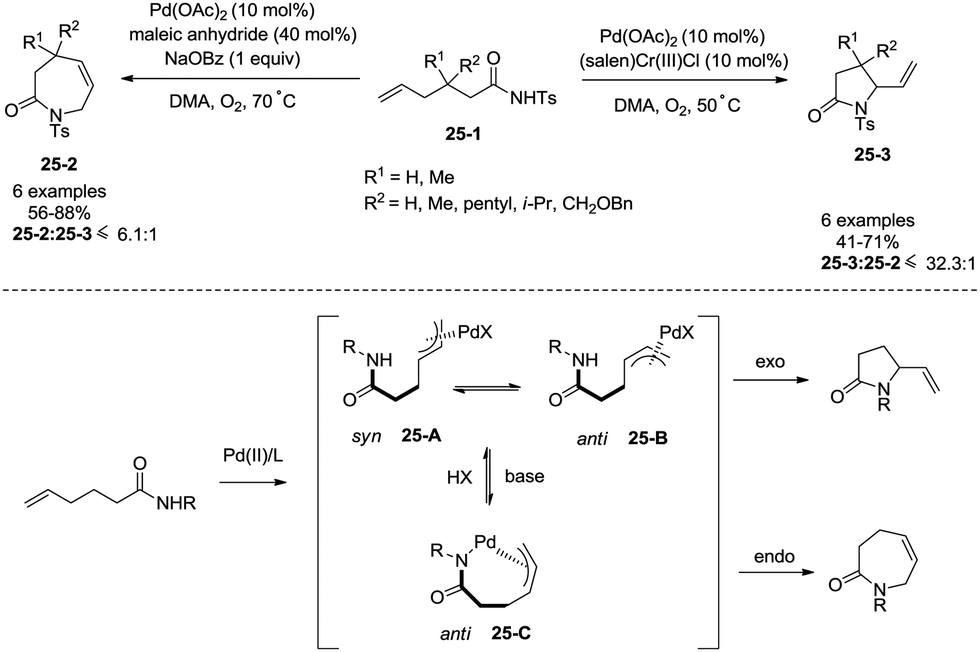
Regioselectivity of Pd(II)-catalyzed intramolecular allylic C–H amination of ω-unsaturated N-sulfonylamines could be modulated by a Brønsted base, which was reported by Liu and co-workers in 2009 (Scheme 25).29f Formation of a N–Pd bond is promoted by a Brønsted base (NaOBz), favouring the formation of intermediate 25-C, reductive elimination gives a seven-membered ring product 3,4-dihydro-1H-azepin-2(7H)-one. The intermediates 25-A and 25-B give a five-membered ring product 5-vinylpyrrolidin-2-one.
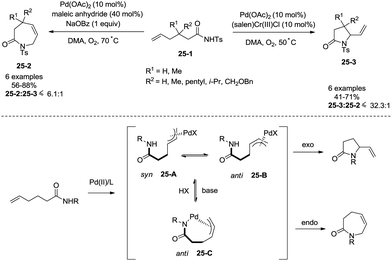 |
| | Scheme 25 Regioselectivity in intramolecular allylic amination. | |
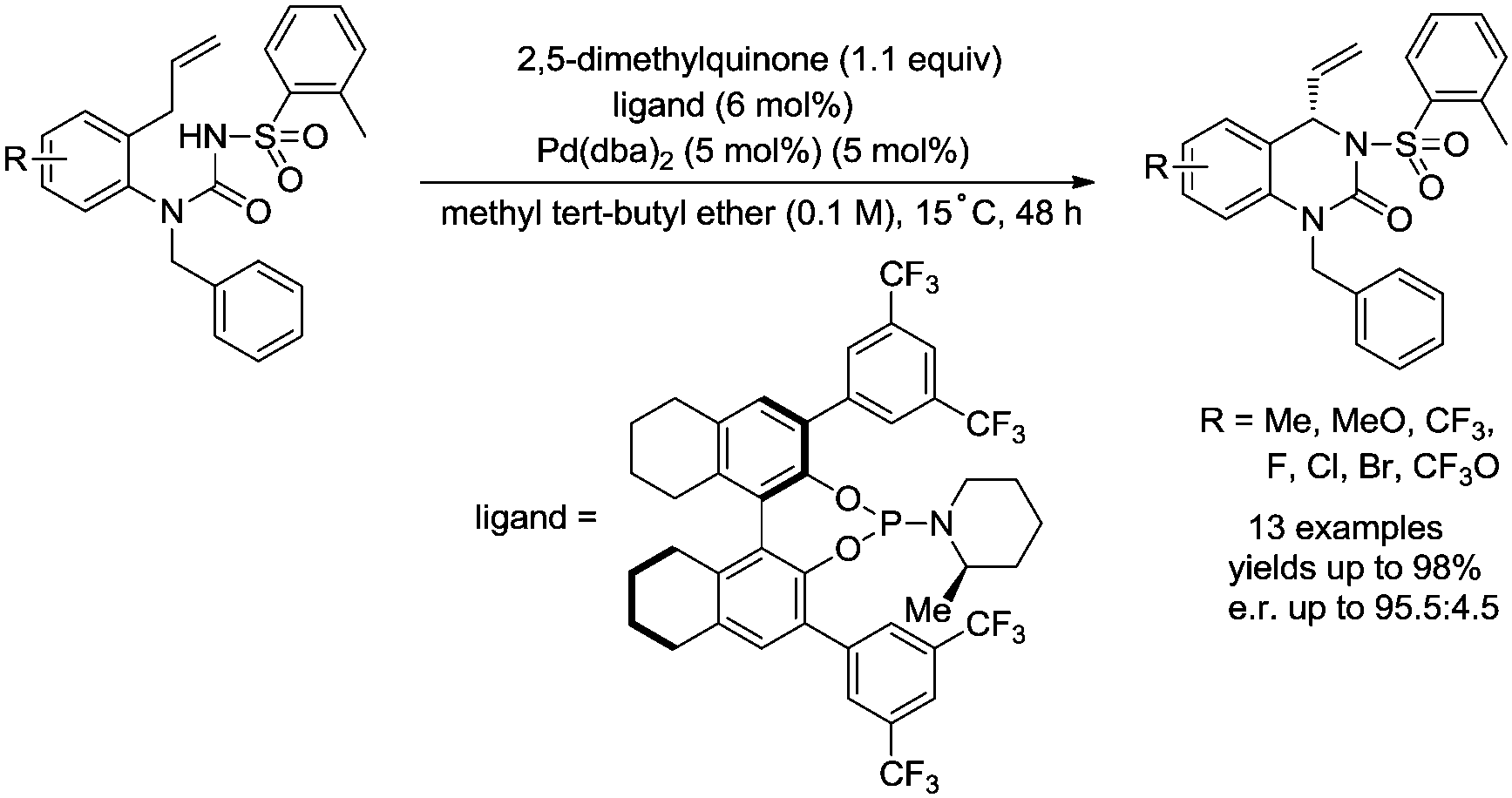
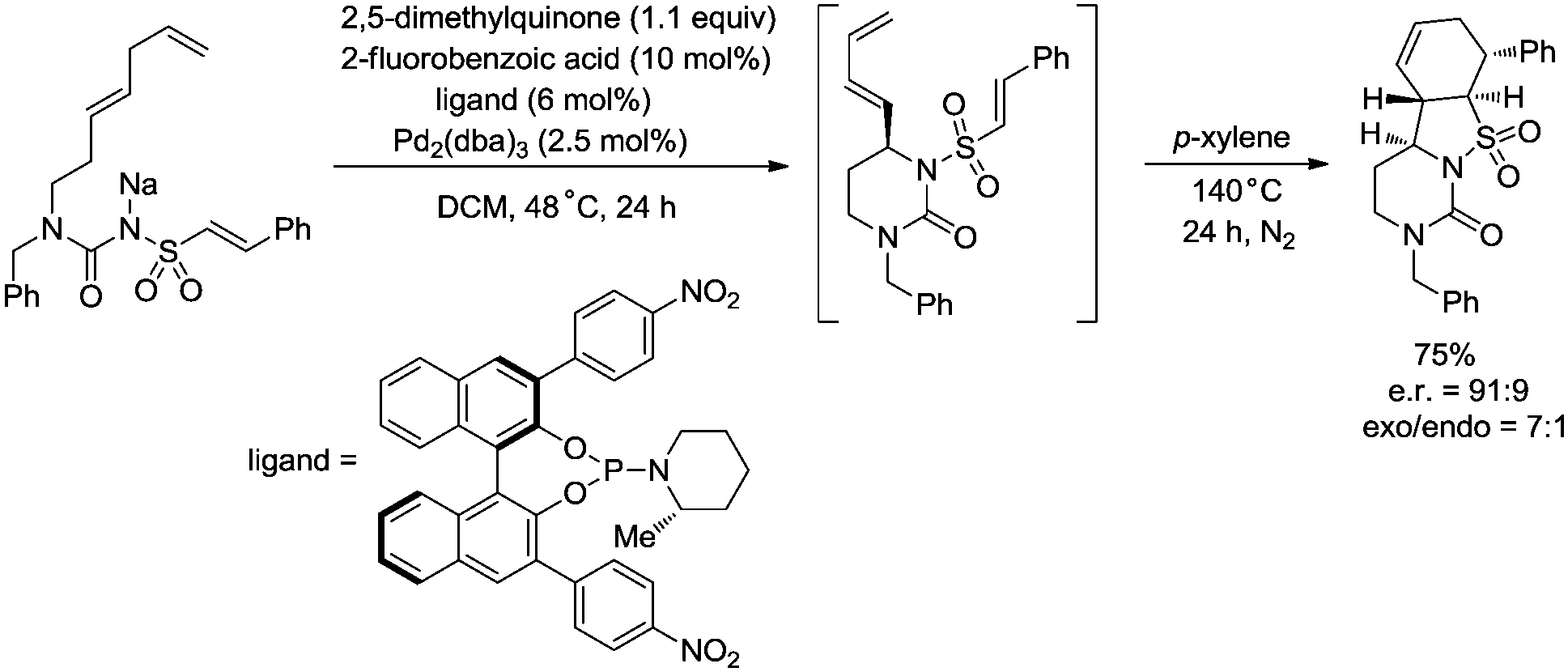
In 2017, Gong and co-workers reported an asymmetric C–N coupling to access chiral hydropyrimidinones via palladium-catalyzed allylic C–H amidation (Scheme 26),29g the cyclization proceeded smoothly with high yields (up to 98%), the enantioselectivity is high when an H8-BINOL-derived phosphoramidite ligand was used, and the enantiomeric ratio is up to 95.5:4.5. The reaction conditions of the allylic C–H amidation cyclization were slightly modified to synthesize more complex polycyclic heterocycles (Scheme 27). A dienyl sodium N-sulfonyl amide bearing an arylethene-1-sulfonyl group underwent the allylic C–H amidation cyclization, and a subsequent diastereoselective intramolecular Diels–Alder reaction afforded a chiral polycyclic heterocycle in 75% overall yield, e.r. = 91:9, exo/endo = 7:1.
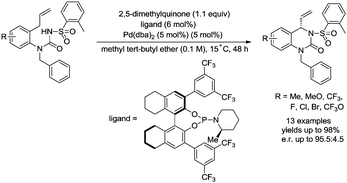 |
| | Scheme 26 Synthesis of chiral hydropyrimidinones. | |
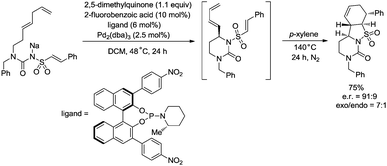 |
| | Scheme 27 Synthesis of a chiral polycyclic heterocycle. | |
3. Copper catalysis
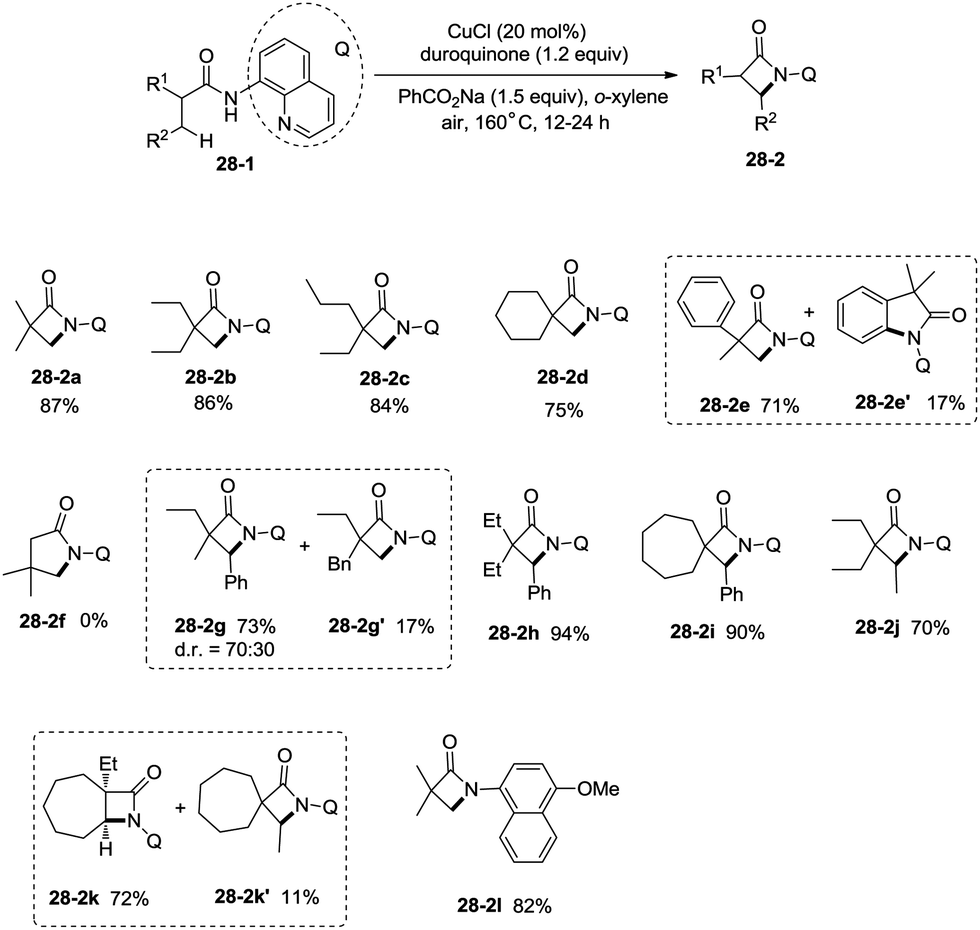
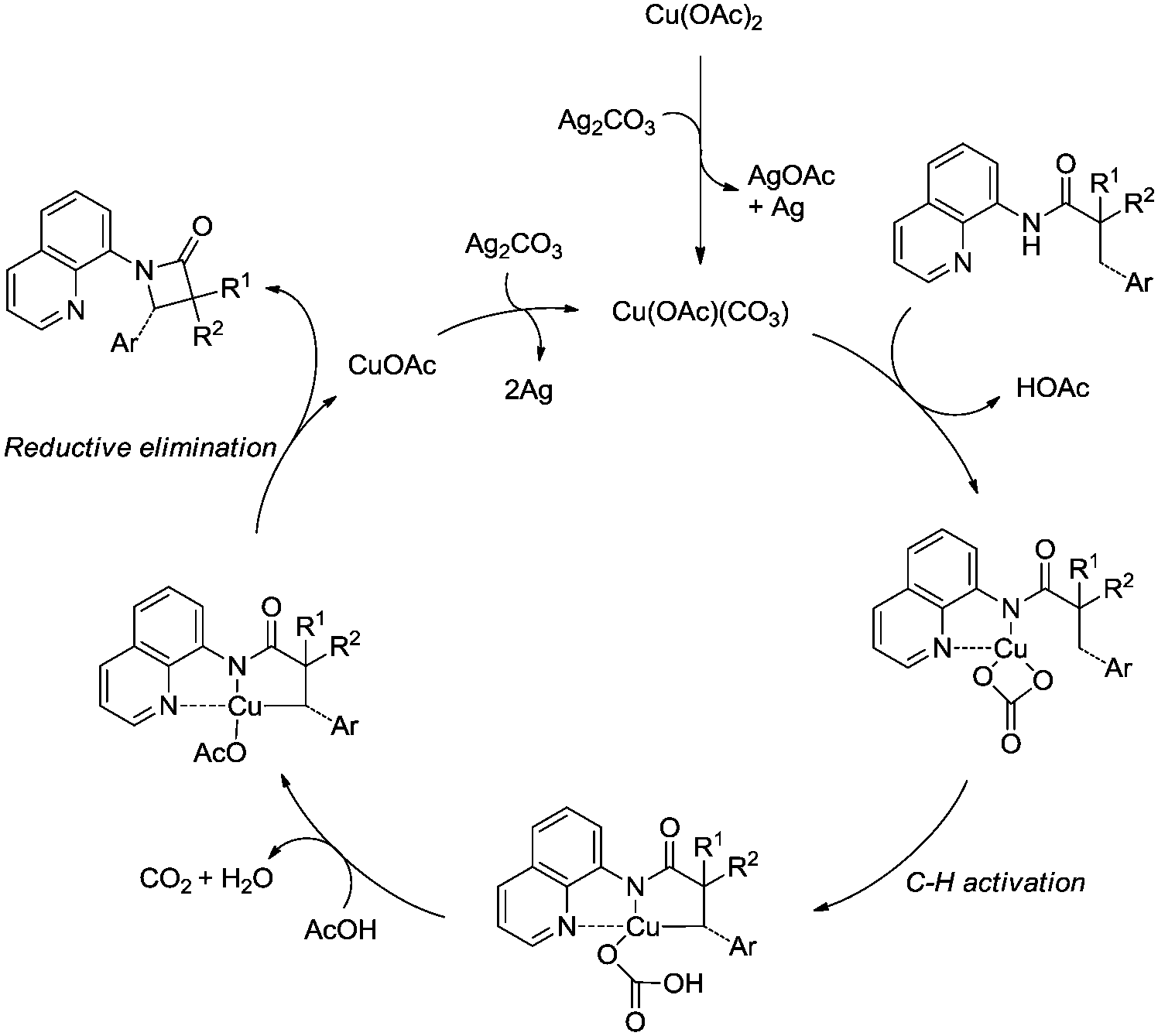
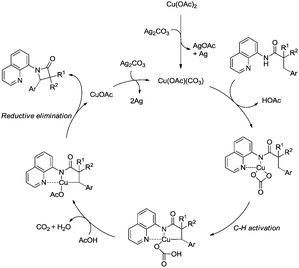
Inspired by the reports20 on the palladium-catalyzed aminoquinoline-directed intramolecular amidation of unactivated sp3 C–H bonds from Chen's group, Ge and co-workers developed copper(I) chloride-catalyzed bidentate-ligand-directed intramolecular amidation of sp3 C–H bonds in 2014 (Scheme 28).30a This strategy provides an alternative approach to afford mono-, spiro-, and bicyclic β-lactams, important structural units in many biologically active naturally-occurring products and synthetic medicines.
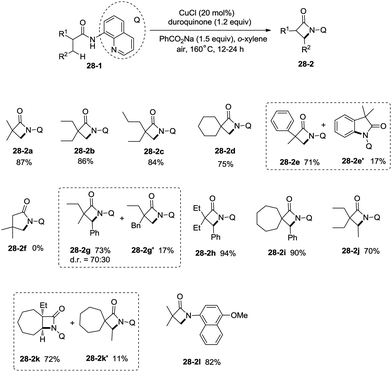 |
| | Scheme 28 Direct amidation on sp3 C–H bonds. | |
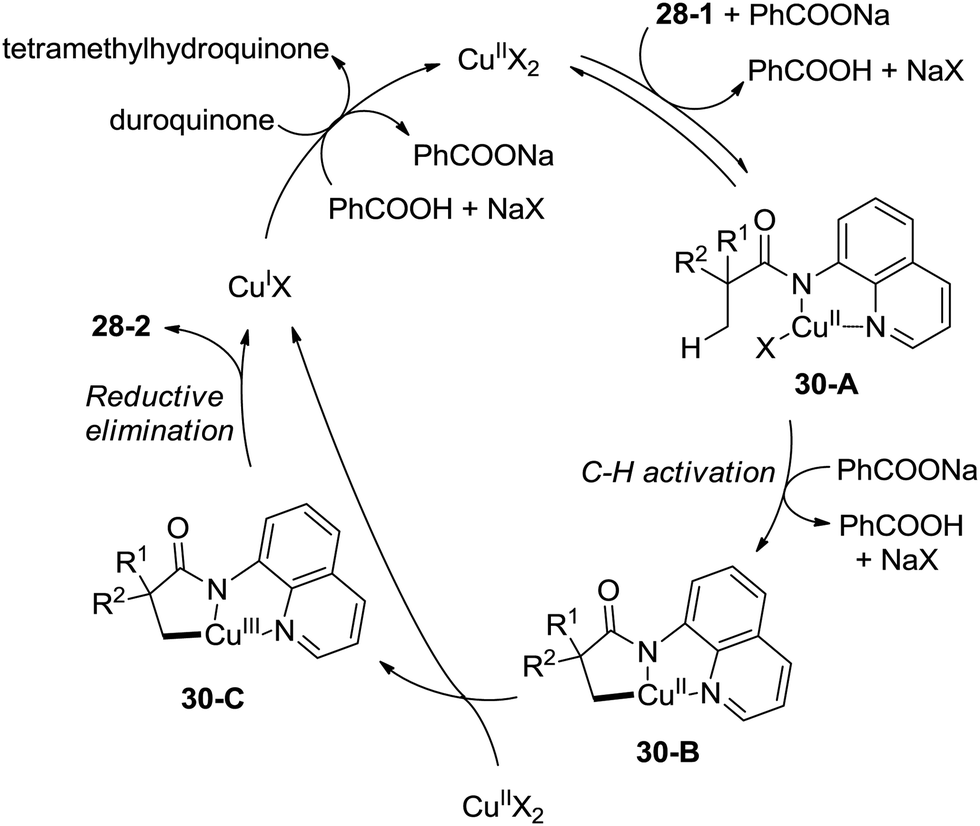
This reaction preferentially occurred at the β-methyl groups rather than the β-methylene groups [(28-2a)–(28-2d)] and the γ-positions on the phenyl groups (28-2e). The C–H bonds of the γ- or δ-methyl group did not undergo the amidation (28-2b, 28-2c, 28-2f), thus indicating that a five-membered ring cyclometalation intermediate is more easy to form than the six or seven-membered ring intermediate in the cyclometalation step under these reaction conditions. Secondary benzylic β-C–H bonds are preferentially amidated over the C–H bonds of the β′-methyl groups and unactivated β′-methylene groups [(28-2g)–(28-2i)], the unactivated secondary β-methylene groups are preferentially amidated over the γ-methyl groups because a five-membered ring cyclometalation intermediate is more easy to form than a six-membered ring cyclometalation intermediate (28-2j). Furthermore, the ring β-carbon atoms are preferentially amidated over acyclic β′-carbon atoms (28-2k), and the bicyclic β-lactam compound (28-2k) was provided with high syn diastereoselectivity. A tertiary α-carbon atom is necessary for this reaction.
The 5-methoxyquinolyl group of 1-(5-methoxyquinolin-8-yl)-3,3-dimethylazetidin-2-one (28-2l) was removed with ceric ammonium nitrate (CAN) according to the report from Chen's group,20 and the free β-lactam product 29-1 was obtained in 63% yield, which is a useful intermediate for further manipulation (Scheme 29).
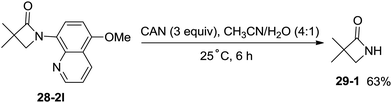 |
| | Scheme 29 Oxidative cleavage of the 5-methoxyquinolyl group. | |
A plausible mechanism for this transformation is proposed (Scheme 30). Coordination of the substrate 28-1 to a copper(II) species and ligand exchange give the five-membered ring copper(II) complex 30-A, and C–H activation at the β-position (cyclocupration) produces a fused two five-membered rings copper(II) complex 30-B. Oxidation of 30-B produces the copper(III) complex 30-C, which produces the β-lactam product 28-2via reductive elimination. The in situ formed copper(I) species is reoxidized to the copper(II) salts by duroquinone. A sequential chlorination and amidation process was also thought to be possible.
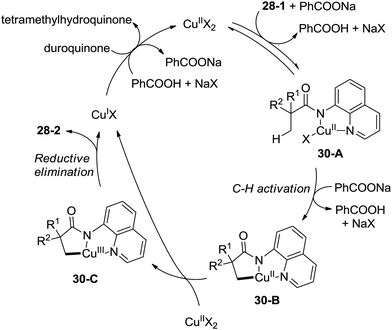 |
| | Scheme 30 Plausible reaction mechanism. | |
In the same year, Kanai and co-workers reported a copper(II) acetate-catalyzed intramolecular sp3 C–H amidation using a silver carbonate oxidant (Scheme 31).30b The reaction proceeded at the β-methyl group or benzylic position of an alkyl chain, affording β-lactams conveniently with good functional group tolerance. A Cu(III)/Cu(I) catalytic cycle was proposed for this transformation (Scheme 32).
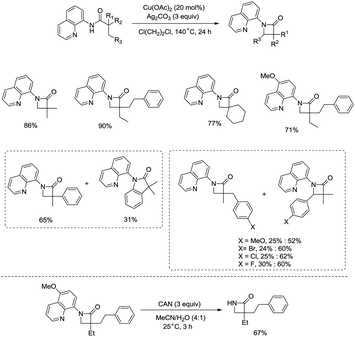 |
| | Scheme 31 Copper(II)-catalyzed cyclization by Kanai. | |
 |
| | Scheme 32 The mechanism for copper(II)-catalyzed cyclization by Kanai. | |
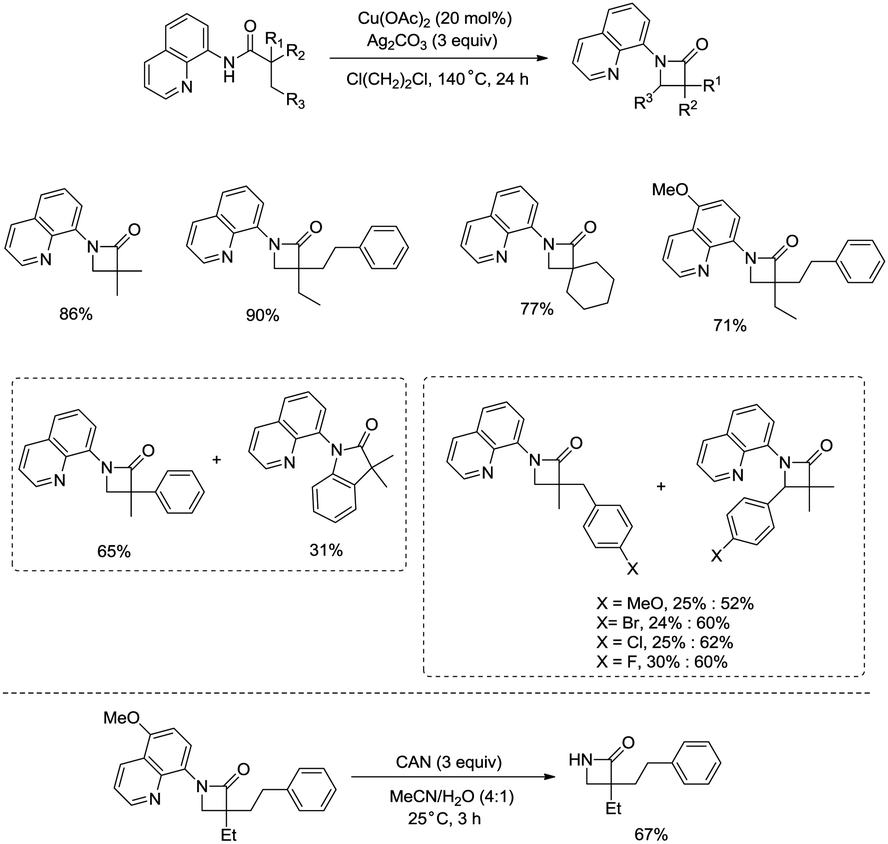
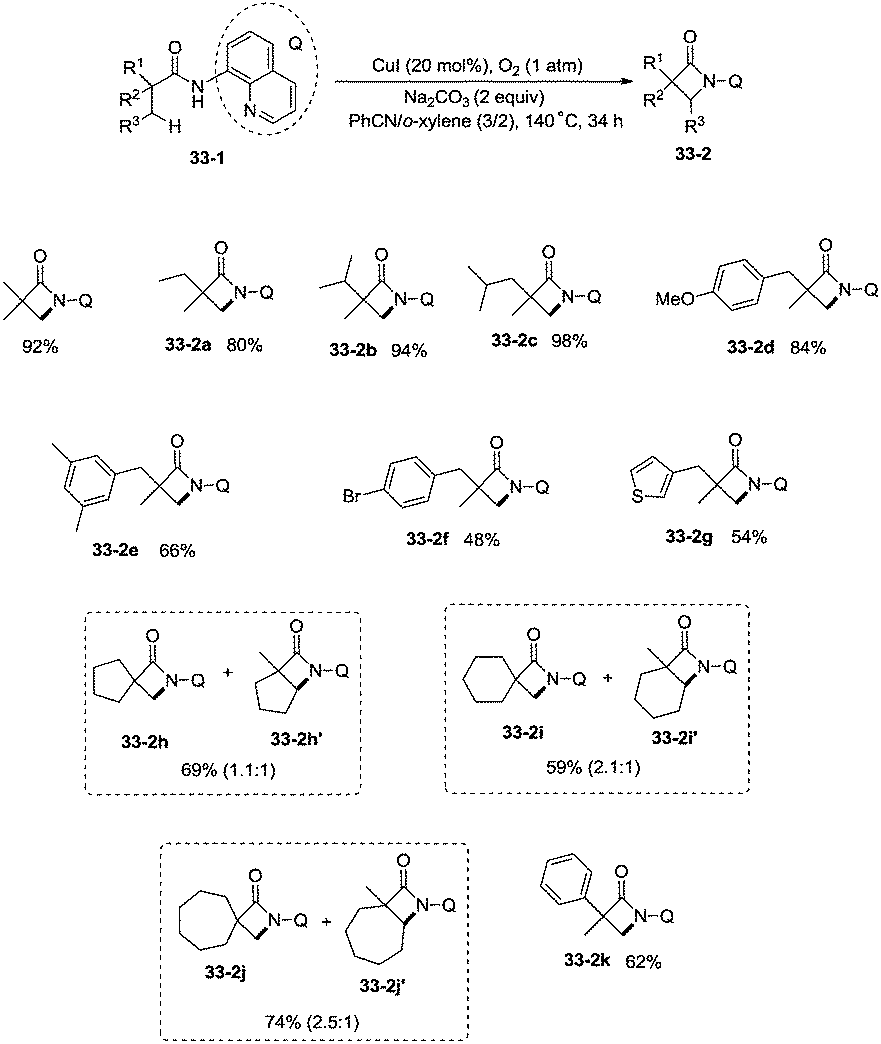
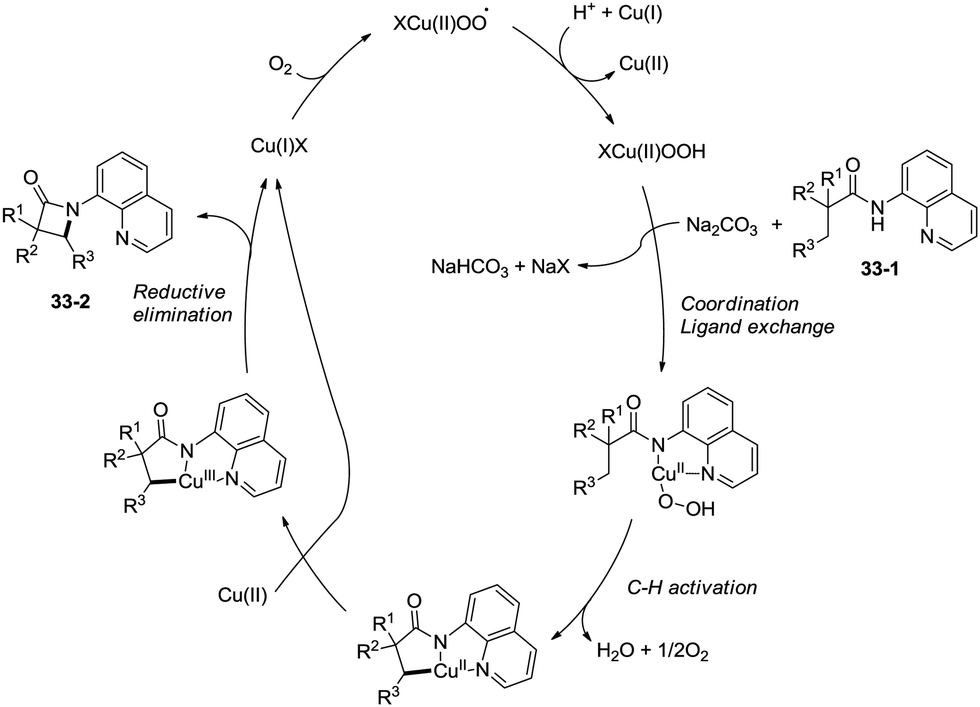
Building upon the copper-catalyzed bidentate-ligand-directed intramolecular amidation of sp3 C–H bonds,30 You et al. developed a CuI (20 mol%)-catalyzed intramolecular amidation of unactivated sp3 C–H bonds using oxygen as the sole oxidant. Oxygen is a cheap, non-poisonous and “green” oxidant, and the only byproduct of oxidation is water (Scheme 33).31a A series of aliphatic amides were transformed into the corresponding β-lactam derivatives in good to excellent yields (up to 98%). The β-methyl groups were preferentially amidated over the γ- and δ-methyl groups (33-2a, 33-2b, 33-2c), because the formation of a five-membered ring cyclocupration intermediate is more feasible than that of a six- or seven-membered ring intermediate in the cupration step. The C–H bonds of the β-methyl groups are preferentially amidated over the C–H bonds of the β-methylene groups including secondary β-benzylic C–H bonds (33-2a, 33-2c, 33-2d, 33-2e, 33-2f, 33-2g). In contrast to these results, a preference for the β-benzylic C–H bonds over the C–H bonds of the β-methyl groups was observed in the previous reports by Ge and co-workers.30 The substrates with cyclic chains produced two kinds of β-lactams from the amidation of both the β-methyl and the methylene, the yields of the spiro-products (33-2h, 33-2i, 33-2j) were slightly higher than that of the fused products (33-2h′, 33-2i′, 33-2j′). 2-Phenyl-substituted propanamide derivative afforded the β-lactam in 82% yield (33-2k) from amidation of β-methyl, and no product from amidation of phenyl was observed.
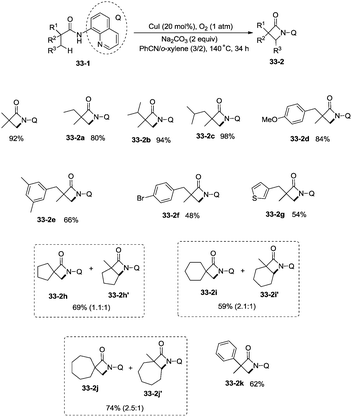 |
| | Scheme 33 Copper-catalyzed intramolecular dehydrogenative amidation. | |
The reaction systems of the amidation used by You et al. and Ge et al.30a are the same, including quinolin-8-amine derived amide substrates, Cu(I) salt catalysts, oxidants and Brønsted bases, so the plausible mechanism proposed by You et al. is similar to that proposed previously by Ge and co-workers. It is worth noting that the only byproduct is water using oxygen as the oxidant (Scheme 34).31b
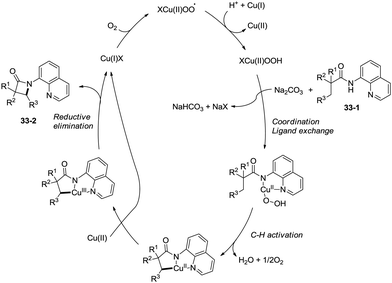 |
| | Scheme 34 The mechanism using oxygen as an oxidant. | |
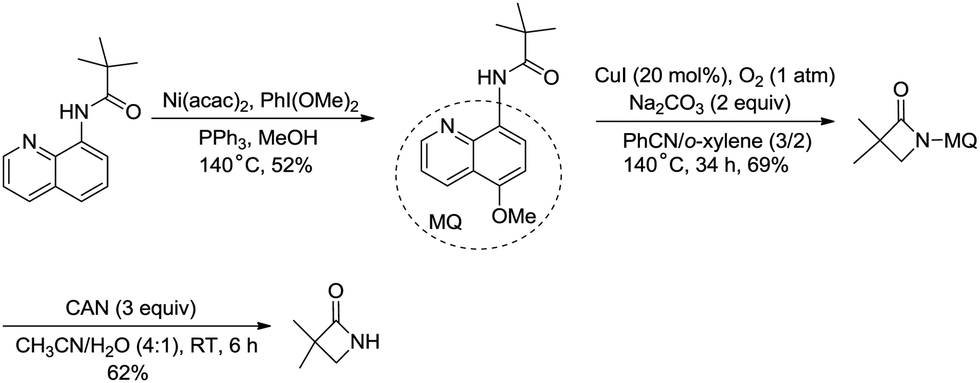
The removal of the quinolyl moiety in β-lactams needs the use of the 5-methoxy quinolyl group, and it is difficult to synthesize the 5-methoxy quinolyl derivatives due to a long synthetic route.20,30 You et al. successfully synthesized free β-lactams by sequential Ni(acac)2-catalyzed methoxylation, CuI-catalyzed amidation, and oxidative cleavage, and the synthetic route is shorter than the previous ones (Scheme 35).
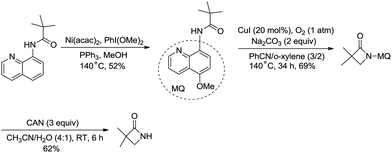 |
| | Scheme 35 Synthesis of free β-lactams. | |
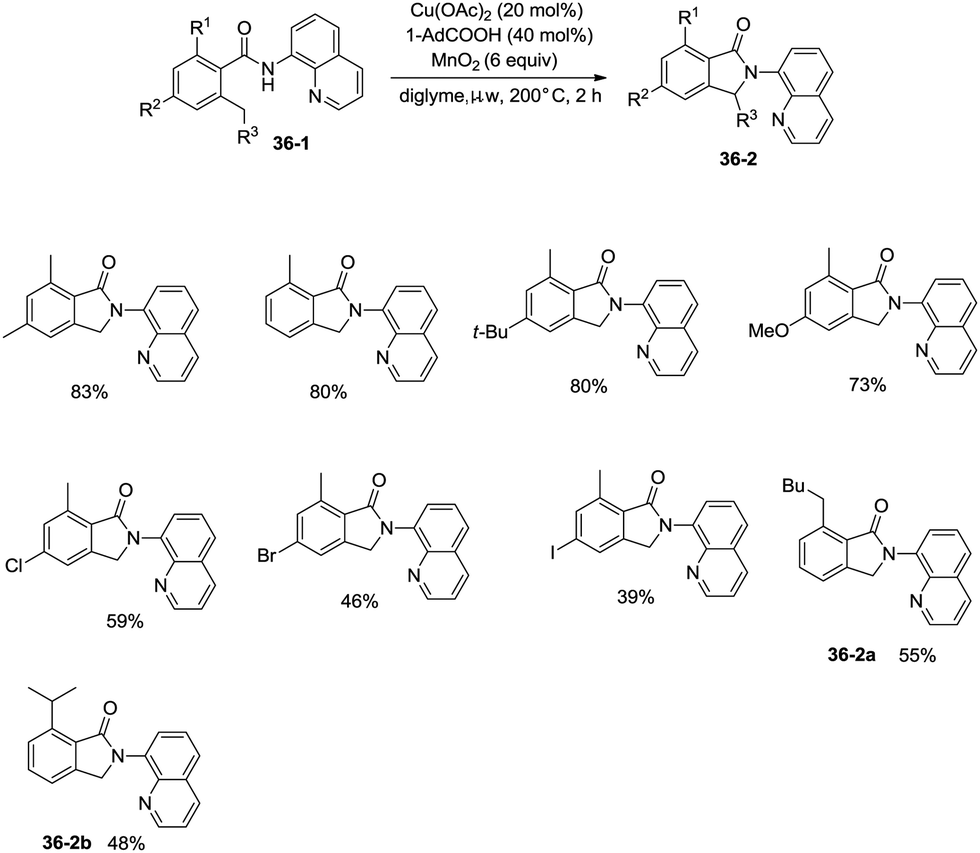
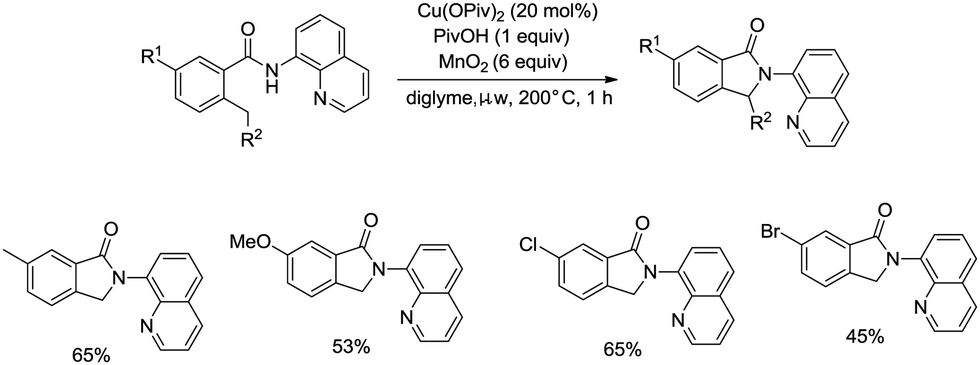
On the basis of palladium-catalyzed isoindolone synthesis from 2,6-dimethyl-N-(8-quinolinyl)benzamides as reported by Zhang,23 Miura and co-workers developed a copper acetate-catalyzed intramolecular benzylic C–H amidation for the synthesis of isoindolinones (Scheme 36).32 The reaction was compatible with electron-neutral and electron-donating substituents as well as electron-withdrawing halogen functionalities, which can be used for further coupling reactions. The amidation preferentially occurred at the ortho methyl over the ortho methylene or methyne, delivering the corresponding isoindolinones (36-2a, 36-2b). 2-Methyl-5-substituted benzamides could work well to give the corresponding isoindolinones under the revised conditions (Scheme 37).
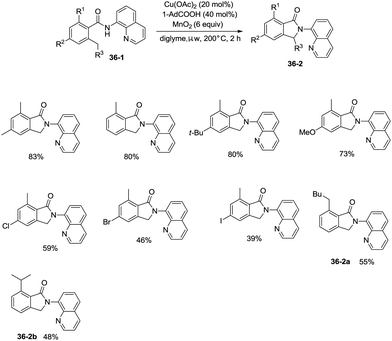 |
| | Scheme 36 Cu-catalyzed intramolecular amidation. | |
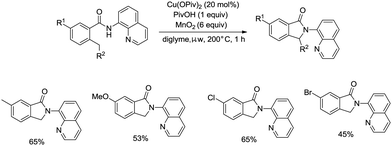 |
| | Scheme 37 Transformation of 2-methyl-5-substituted benzamides. | |
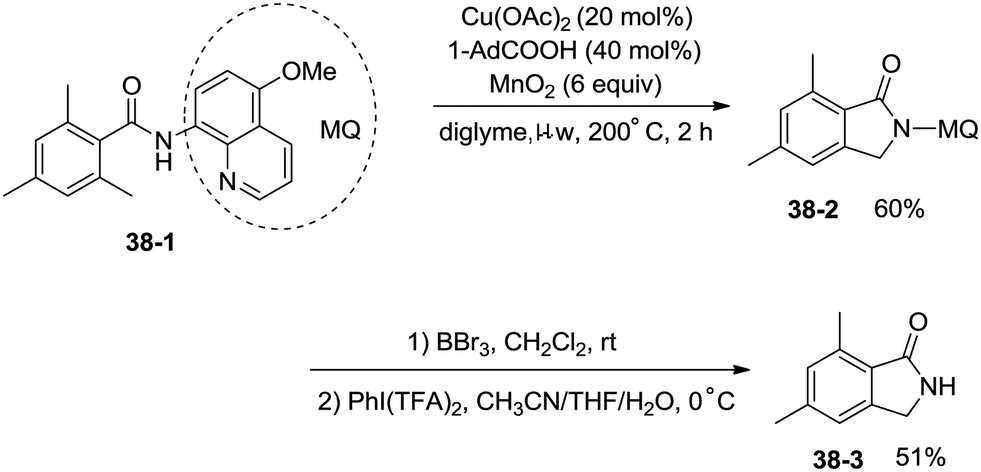
Under identical conditions, the 5-methoxyquinolin-8-amine derived benzamide 38-1 also worked well to give the desired product 38-2 in 60% yield. The NH isoindolinone 38-3 was obtained in 51% yield via demethylation with BBr3 and oxidation with PhI(TFA)2 (Scheme 38).
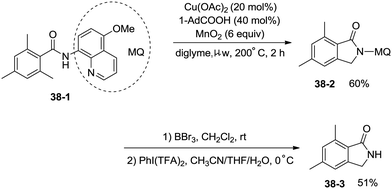 |
| | Scheme 38 Removal of MQ with BBr3etc. | |
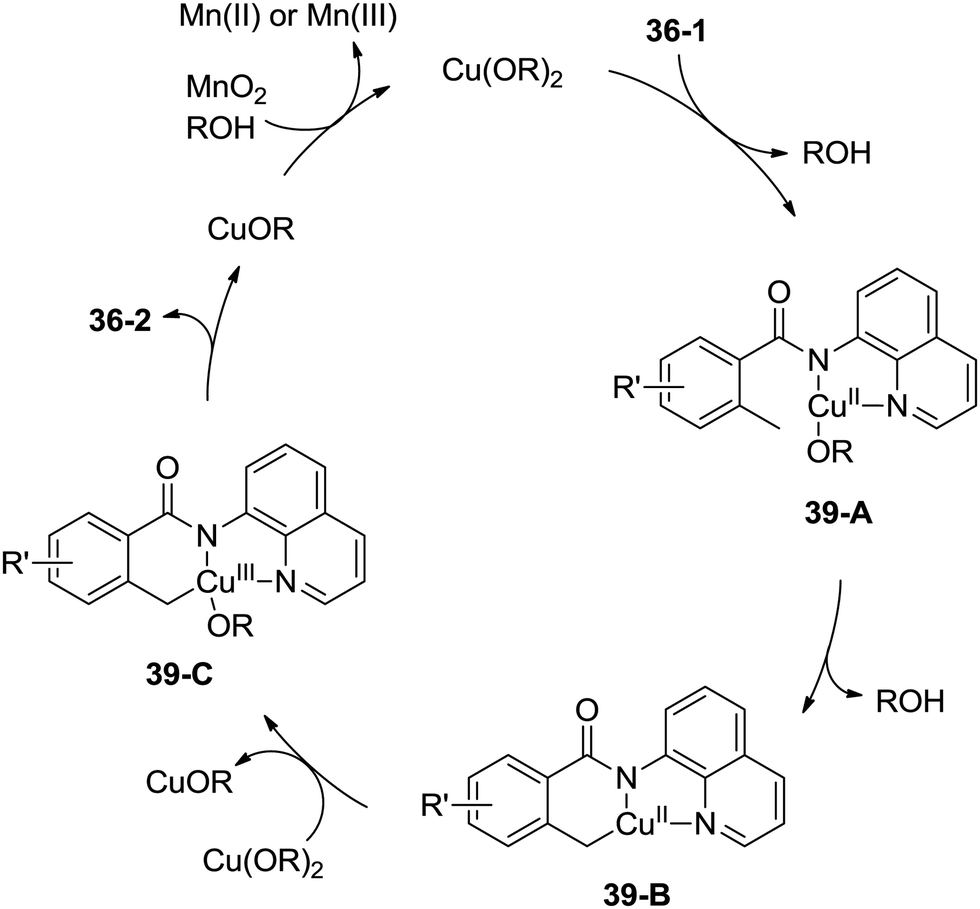
A possible mechanism was proposed (Scheme 39). Coordination of the benzamide 36-1 with Cu(OR)2 along with ligand exchange form a chelated Cu(II) species 39-A. Subsequent benzylic C–H activation (cyclometalation) generates the fused five-membered and six-membered ring Cu(II) complex 39-B, which was oxidized to the Cu(III) intermediate 39-C, and then reductive elimination gives the isoindolinone 36-2. The Cu(II) species was regenerated via oxidation of the generated Cu(I) species, which finishes the catalytic cycle.
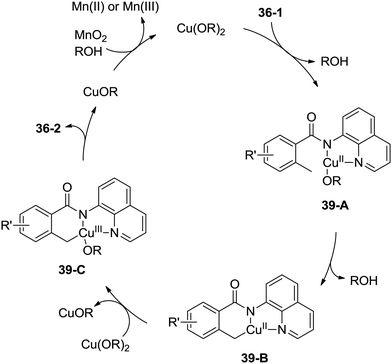 |
| | Scheme 39 Mechanism for Cu-catalyzed synthesis of isoindolinones. | |
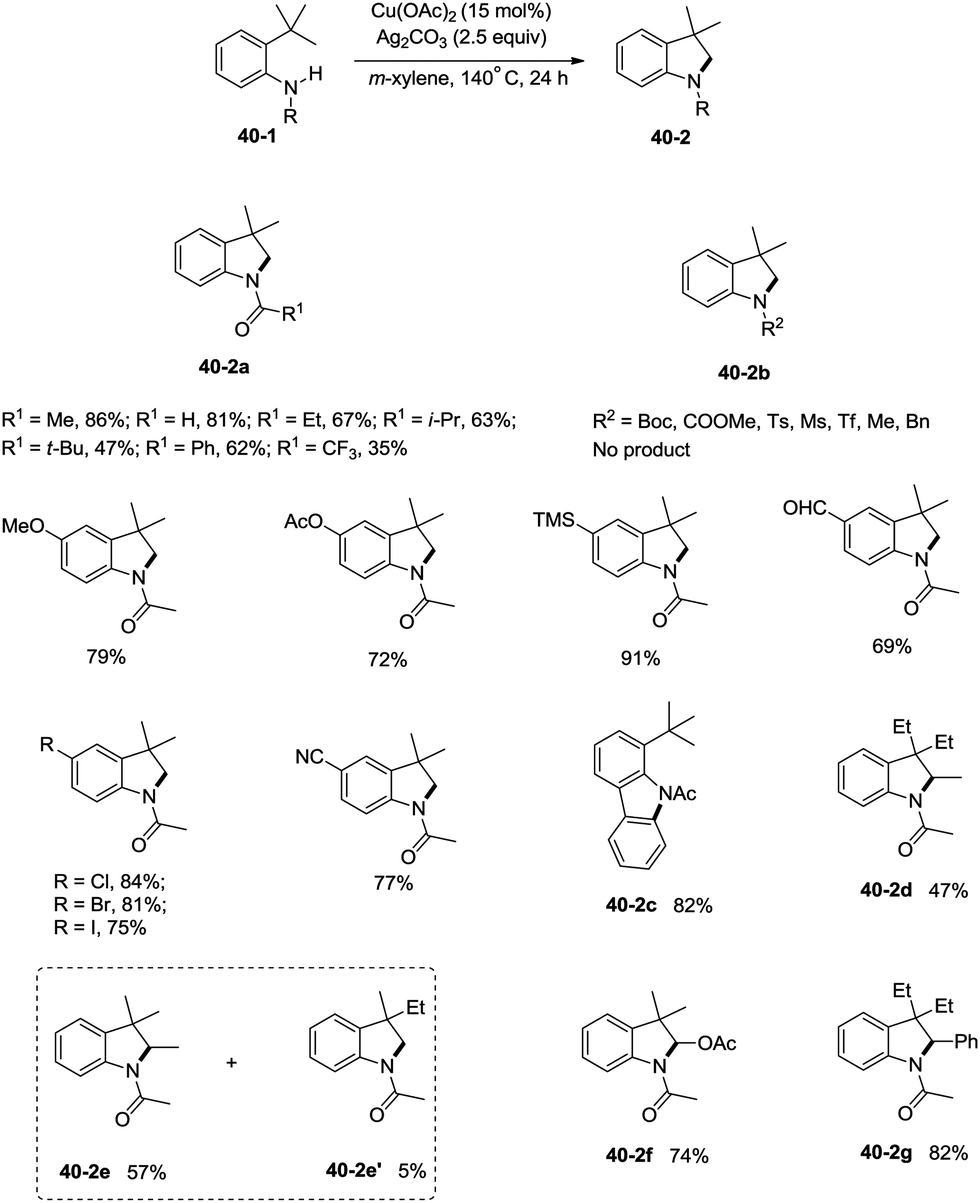
Inspired by the palladium-catalyzed synthesis of indolines from anilines as reported by Glorius and co-workers,18 Shi et al. developed a copper acetate-catalyzed intramolecular amidation of unactivated sp3 C–H bonds in 2016 (Scheme 40),33 affording N-substituted indolines efficiently. The substrates with different N-substituents could undergo the amidation (40-2a) smoothly, and the substrates with formyl, propionyl, isobutyryl, pivaloyl, and benzoyl substituents produced indolines in good to excellent yields. However, increase of the N-substituent size gradually decreased the yields. The product yield of N-trifluoroacetanilide was much lower probably due to the lower electronic density of the amide nitrogen. Moreover, substrates with carbamates, sulfonamides, and primary/secondary amines did not work, and the corresponding amidation/amination products were not found (40-2b), further implying the electronic effects of the substituents on the reaction. The reaction of acetanilides tolerated well both electron-donating and -withdrawing groups. The functionalization preference of the sp2 C–H bonds over the sp3 C–H bonds for the amidation was observed (40-2c), giving the carbazole in 82% yield. The secondary C–H bonds also worked albeit with a lower yield of 47% (40-2d). The secondary C–H bonds were found significantly preferable for the amidation over the primary C–H bonds (40-2e, 40-2e′), and the product yields ratio was 57:5. A notable preference for the activation of the activated position was discovered (40-2f, 40-2g), and the desired products were obtained in good yields (74%, 82%).
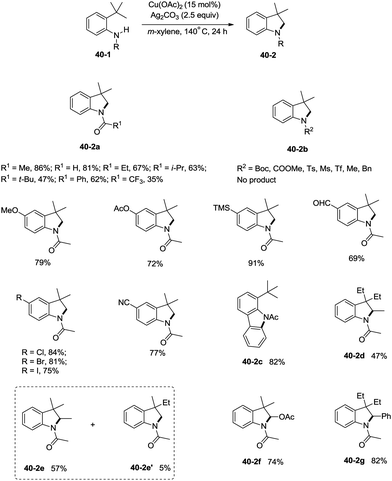 |
| | Scheme 40 Cu-catalyzed synthesis of indolines. | |
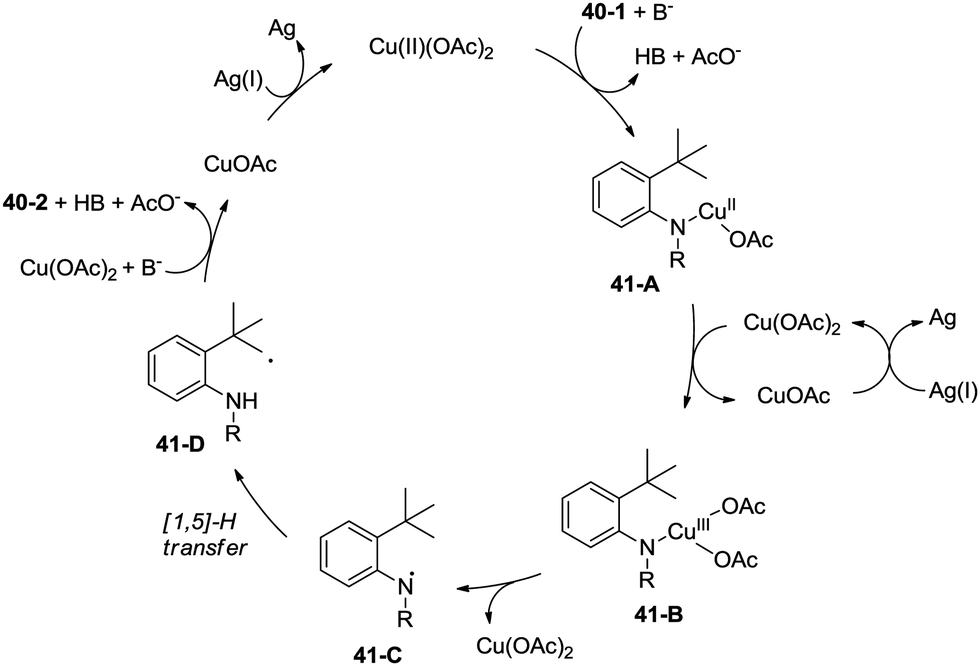
A radical mechanism was proposed for the amidation reaction (Scheme 41). Deprotonation and subsequent coordination of the amide 40-1 with Cu(OAc)2 gives Cu(II) complex 41-A, which can be oxidized to Cu(III) complex 41-B. The Cu(III) complex 38-B forms the nitrogen radical species 41-C, and then a carbon radical species 41-D was generated via a 1,5-hydrogen atom transfer (HAT), which usually occurs in the Hofmann–Loffler–Freytag (HLF) reaction.34 The carbon radical 41-D undergoes oxidation, then coupled with the amide nitrogen to form the desired product 40-2. The Cu(I) species is reoxidized to Cu(II) species by Ag2CO3, which completes the catalytic cycle.
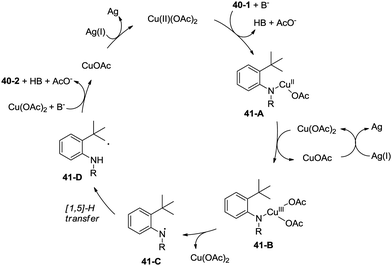 |
| | Scheme 41 Plausible reaction mechanism for the Cu-catalyzed synthesis of indolines. | |
4. Silver catalysis
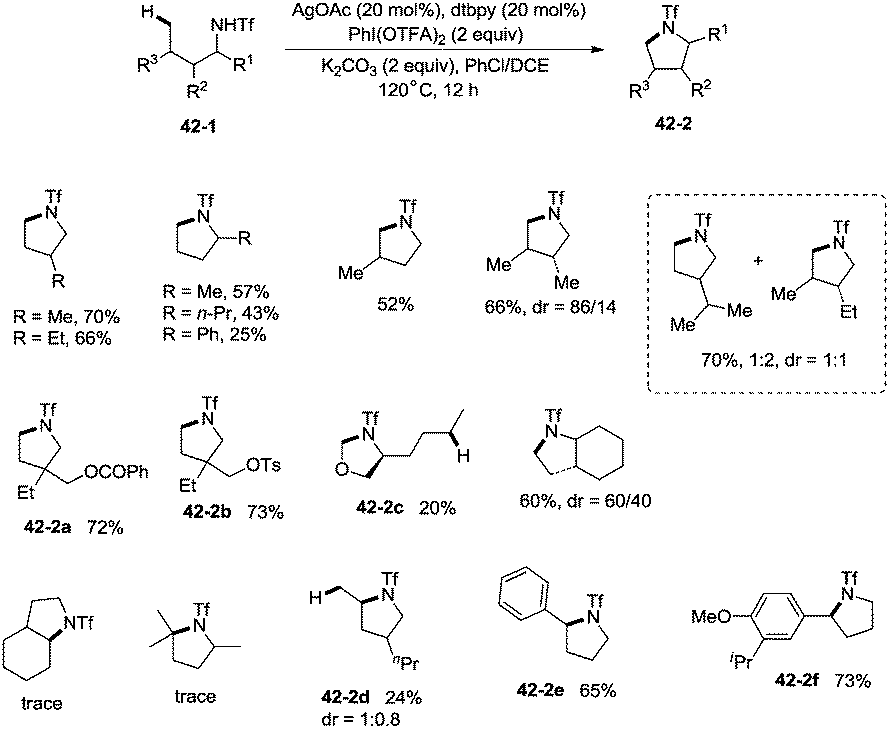
In 2014, Shi and co-workers developed the silver acetate-catalyzed direct amination of primary sp3 C–H, secondary benzylic C–H and aryl C–H bonds from linear triflamide (1,1,1-trifluoromethanesulfonamide) 42-1, providing the cyclized products 42-2 in fair to good yields (Scheme 42).35O-containing functional groups are well tolerated (42-2a, 42-2b, 42-2f), and the common leaving group -OTs (4-toluenesulfonoxyl) was also commendably compatible (42-2b). A 1,2-amino alcohol derivative afforded oxazolidine (42-2c), indicating the preference of functionalization for the C–H bonds adjacent to the heteroatom. In contrast to Hofmann–Lofler–Freytag reactions39 or nitrene insertions,5–8 primary C–H bonds are more reactive than secondary or tertiary C–H bonds. The five-membered ring product is preferentially obtained over a six-membered ring product although the reactivity of methyl is bigger than that of methylene (42-2d). Benzylic secondary C–H bonds could be functionalized well to give the cyclization products (42-2e, 42-2f).
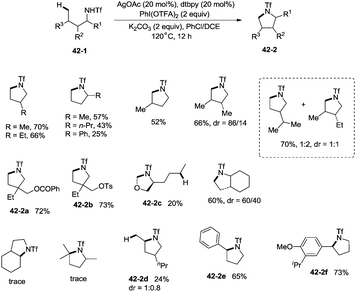 |
| | Scheme 42 Amination of primary and benzylic C–H bonds. | |
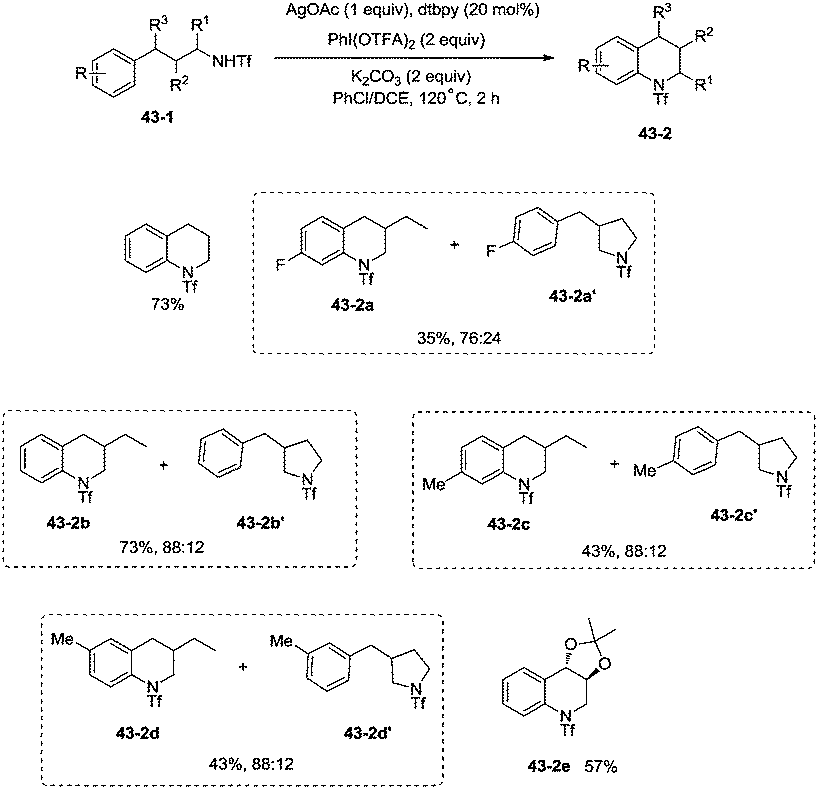
This methodology was also used to construct the structurally important tetrahydroquinoline scaffolds 43-2 from 3-arylpropyl triflamide 43-1 (Scheme 43). The sp2 C–H bonds on the benzene ring are more favorable for the amidation over the primary sp3 C–H bonds [(43-2a)–(43-2d)], and the functionalized substrate containing complex trans-2,2-dimethyl-1,3-dioxolane also gave the desired product in 57% yield (43-2e), thus indicating that the transformation tolerates well the acid-sensitive functional groups.
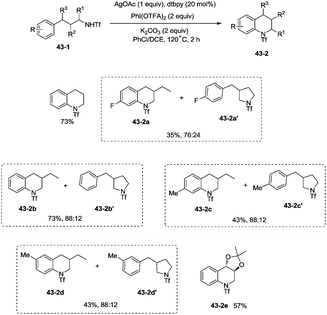 |
| | Scheme 43 Amination of aromatic C–H bonds. | |
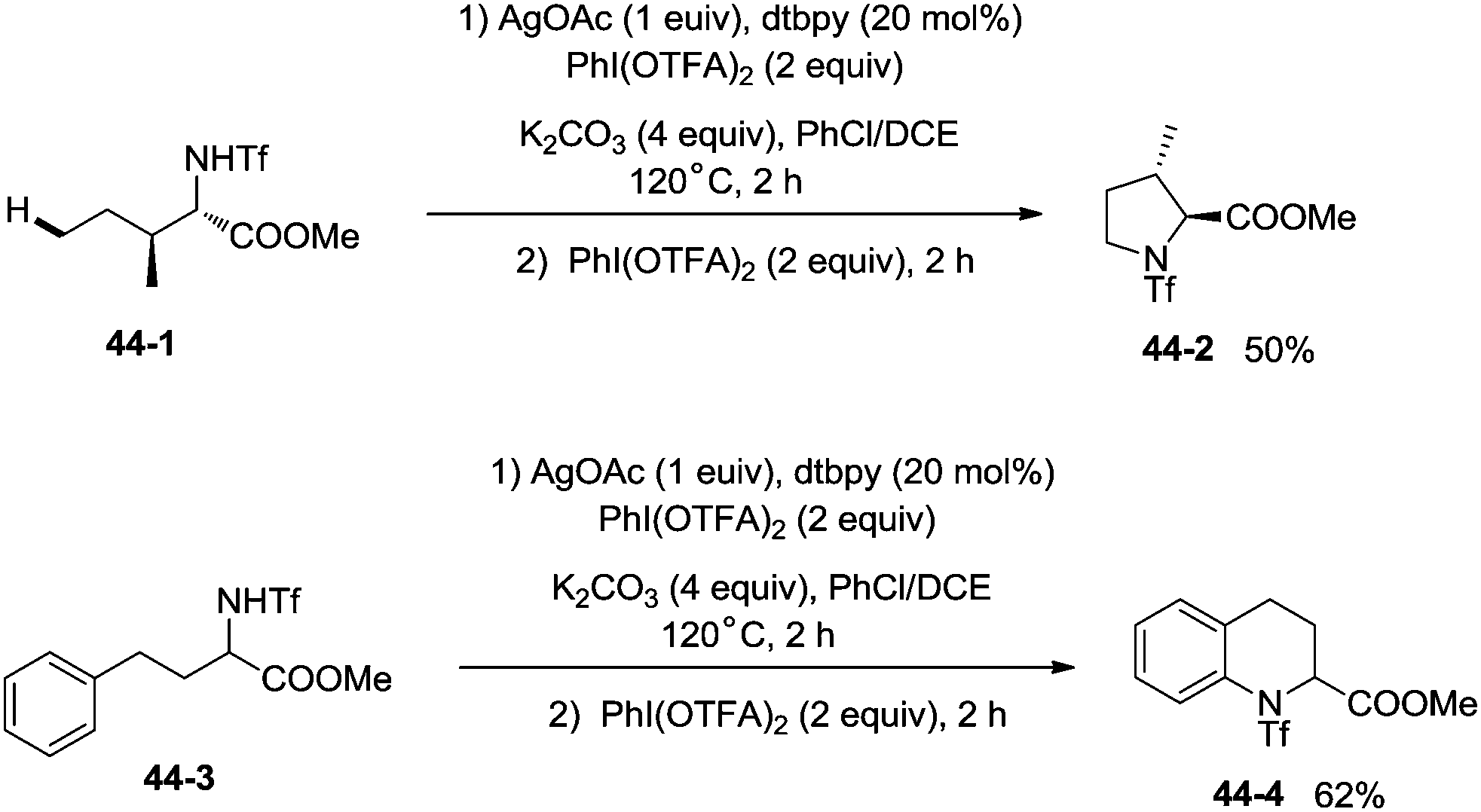
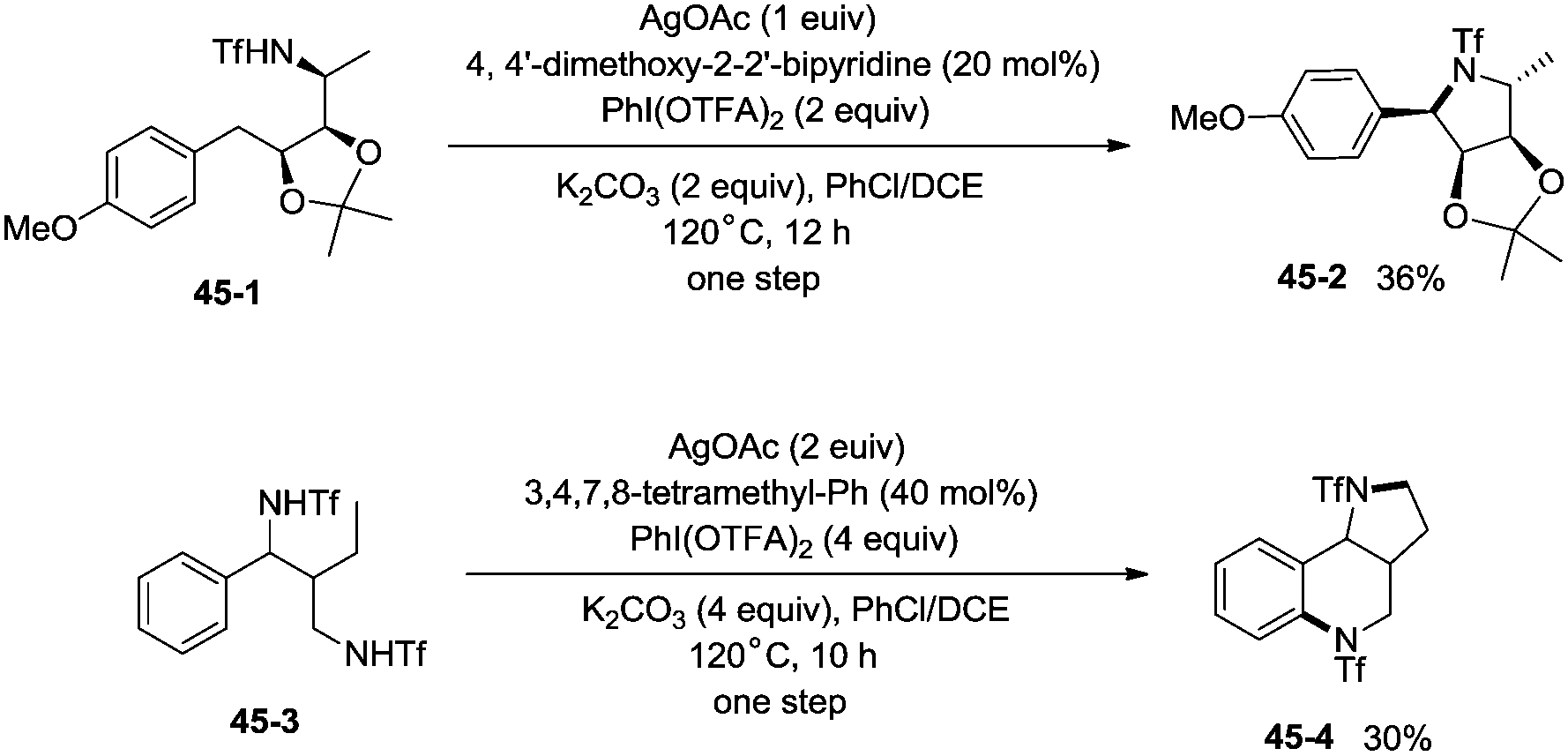
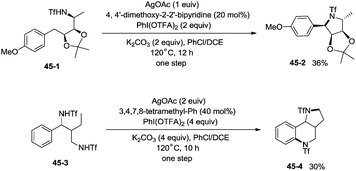
The protected linear amino acid esters 44-1 and 44-3 were transformed into 3-methylproline 44-2 and 2-tetrahydroquinonyl carboxilic ester 44-4 in acceptable yields (Scheme 44). Tetrasubstituted pyrrolidine 45-2, the epimer of natural (−)-codonopsinine could be obtained as a sole diastereoisomer in a single operation from the complex triflamide 45-1. Tricyclic scaffold 45-4, containing the core structure of the natural product (−)-martinellic acid, could also be constructed via primary C(sp3)–H and aromatic C(sp2)–H double amination in a single operation from ditriflamide 45-3, showing the power of this chemistry in organic synthesis (Scheme 45).
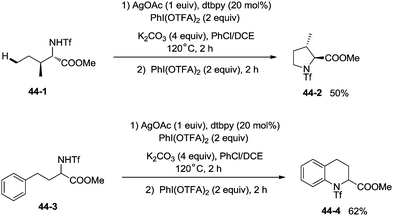 |
| | Scheme 44 Cyclization of amino acids. | |
 |
| | Scheme 45 Synthesis of natural structures. | |
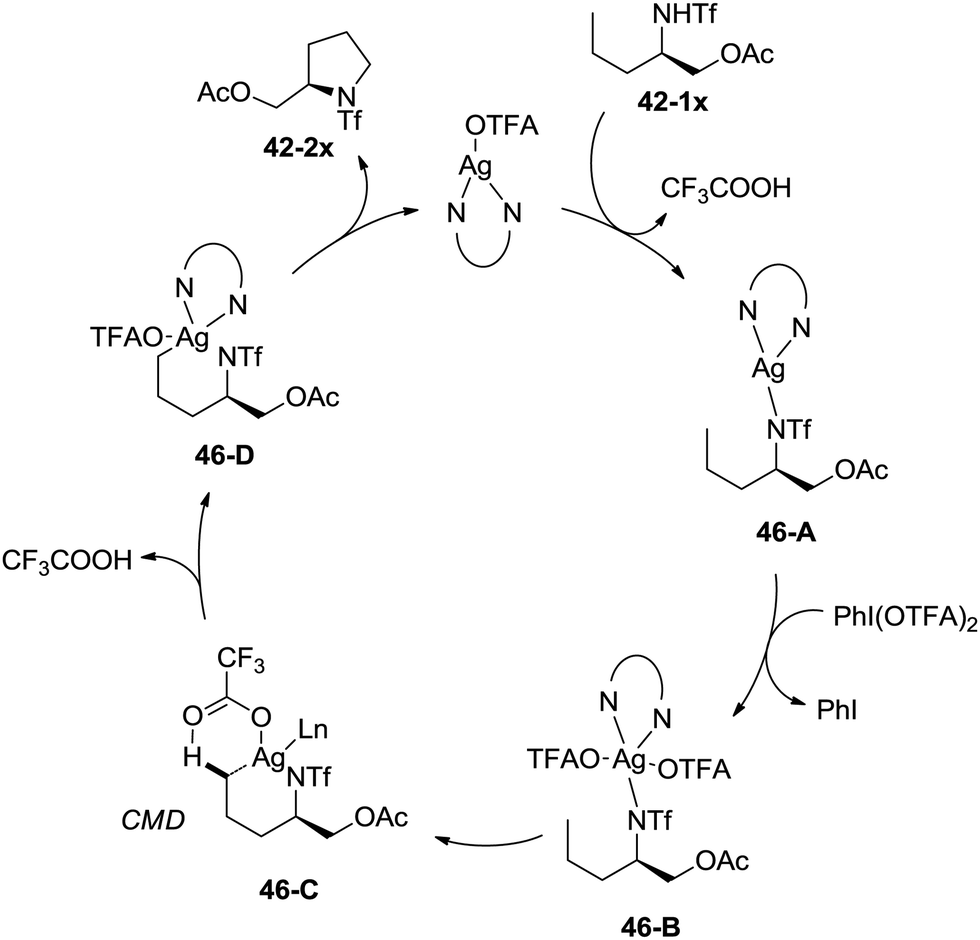
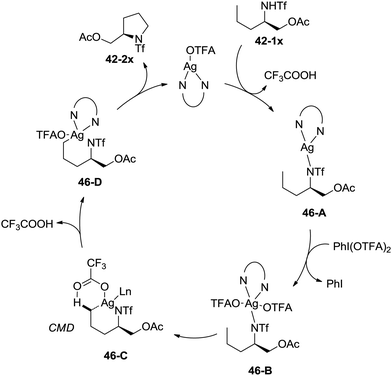
A possible mechanism involving a concerted metallation/deprotonation (CMD) process was proposed (Scheme 46). Coordination of the substrate 42-1x with silver(I) after deprotonation forms silver(I) complex 46-A, which can be oxidized to silver(III) complex 46-B. The activation of the primary C–H bonds gives a six-membered ring silver(III) complex 46-D, which probably undergoes a concerted metallation/deprotonation (CMD) process (46-C). Reductive elimination gives the cyclization product 42-2x along with the Ag(I) species, fulfilling the catalytic cycle.
 |
| | Scheme 46 Mechanism for Ag-catalyzed reactions. | |
5. Nickel catalysis
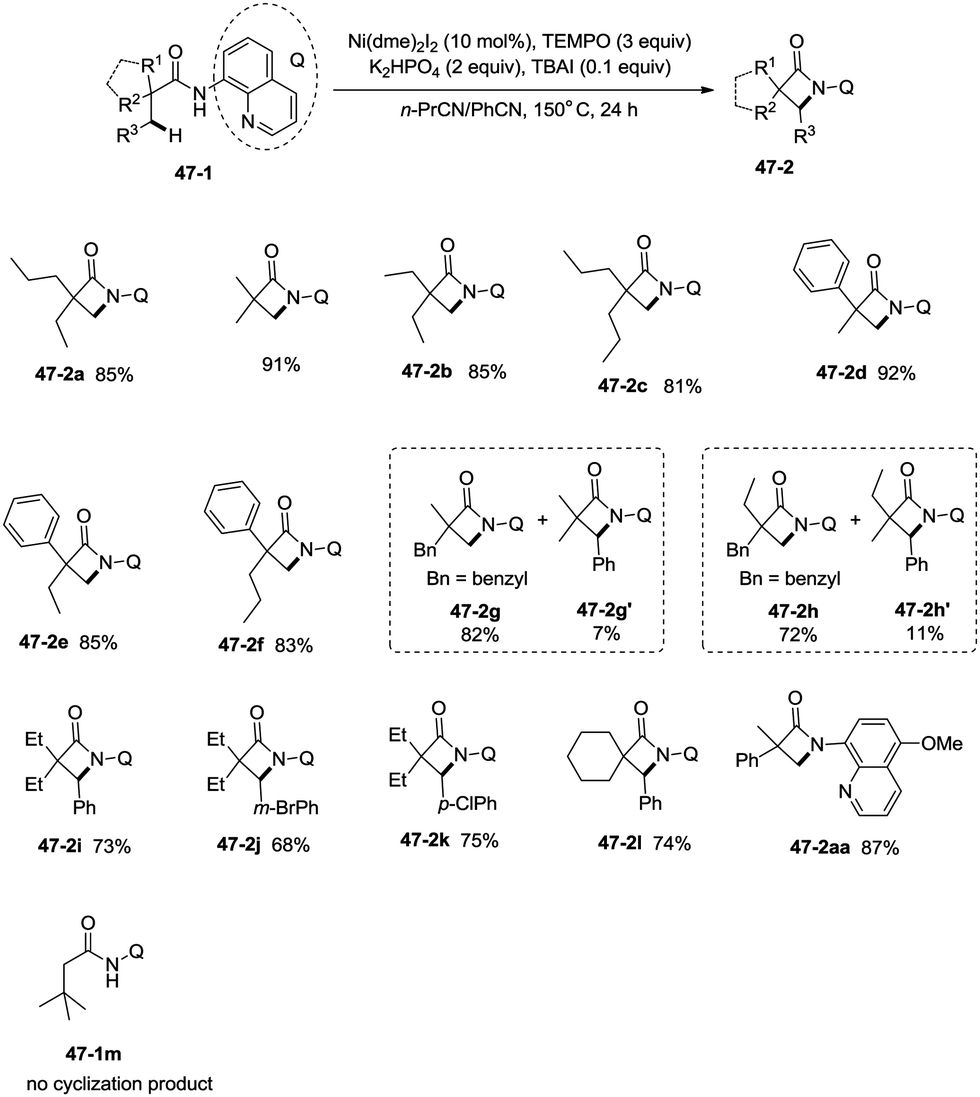
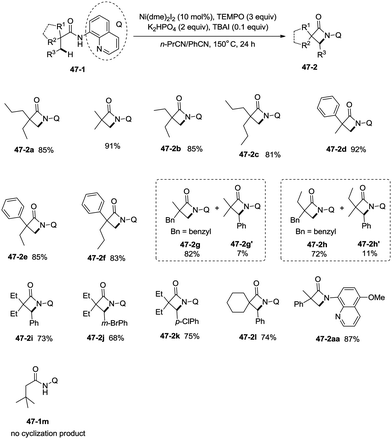
Inspired by the palladium(II)-catalyzed bidentate ligand-assisted intramolecular amidation of unactivated sp3 C–H bonds from Chen's group,20 Ge and co-workers developed nickel(II)-catalyzed intramolecular cyclization on unactivated sp3 carbon atoms in 2014 (Scheme 47).36 The monocyclic or spiro-β-lactam derivatives were produced effectively, which are important subunits in biologically active natural products and synthetic pharmaceutical compounds. Site-selectivity is high in this reaction, and the β-methyl groups are more reactive than the β-methylene and γ-methyl groups [(47-2a)–(47-2c)]. 2,2-Disubstituted propanamides 47-1 bearing both the linear and cyclic chains were converted into the desired products 47-2 in good to excellent yields. The sp3 C–H bonds of the β-methyl group were preferentially functionalized over the sp2 C–H bonds of a phenyl group with the 2-phenyl-substituted substrates [(47-2d)–(47-2f)], because the generation of a five-membered ring cyclometalation intermediate is more feasible than a six-membered ring cyclometalation intermediate in the cyclonickelation step. In the previous copper-catalyzed cyclization, sp2 C–H bonds were also amidated along with the sp3 C–H to give two kinds of amidation products (four-membered and five-membered ring nitrogen heterocycles).30 The primary C–H bonds of the β-methyl groups are more reactive than the secondary β-benzylic C–H bonds (47-2g, 47-2h). In contrast with this, the secondary β-benzylic C–H bonds are more reactive in the previous copper-catalyzed reaction.30 The functionalization of the benzylic secondary C–H bonds could also be effectively achieved [(47-2i)–(47-2l)]. The reaction conditions also tolerate well halogens (Cl and Br) (47-2j, 47-2k). The 8-amino-5-methoxyquinoline derived substrate also undergoes the cyclization smoothly, and the yield is similar to that of the cyclization of the 8-aminoquinoline derived substrate (47-2aa, 47-2d), which provides access to N-unprotected β-lactam derivatives.20,30 A tertiary α-carbon atom is necessary for this cyclization, and the C–H bonds on the γ-methyl of 3,3-dimethyl-N-(quinolin-8-yl)butanamide 47-1m were not amidated, which indicated that a six-membered ring intermediate is difficult to form in the cyclonickelation step under these reaction conditions.
 |
| | Scheme 47 Ni-catalyzed C–H amidation. | |
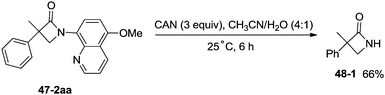
1-(5-Methoxyquinolin-8-yl)-3-methyl-3-phenylazetidin-2-one (47-2aa) was subjected to the oxidative conditions for the cleavage of the auxiliary group with ceric ammonium nitrate (CAN), and the desired NH-β-lactam product 3-methyl-3-phenylazetidin-2-one (48-1) was produced in 66% yield (Scheme 48), which benefits further structural modification.
 |
| | Scheme 48 Cleavage of the 5-methoxyquinolyl group. | |
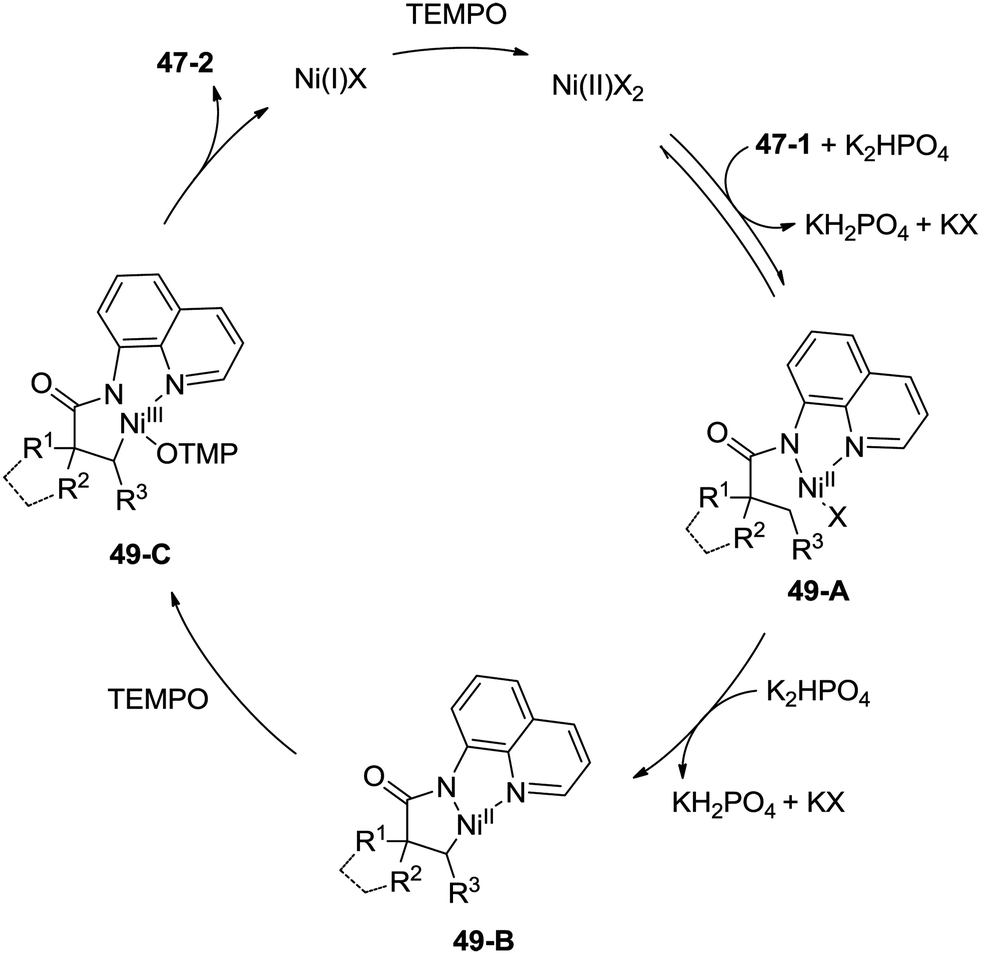
A plausible mechanism is proposed (Scheme 49). Coordination of the amide 47-1 to a Ni(II) species and subsequent ligand exchange process give a chelated nickel(II) complex 49-A, which undergoes C–H activation affording the cyclonickelation complex 49-B, the Ni(III) species 49-C was generated via the oxidation of the Ni(II) complex 49-B. Reductive elimination of the Ni(III) complex 49-C gives the cyclization product 47-2. The generated Ni(I) species is reoxidized to the Ni(II) species by TEMPO, so finishing the catalytic cycle.
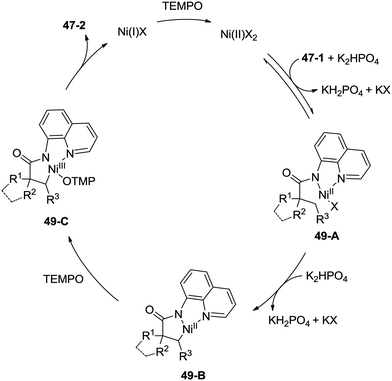 |
| | Scheme 49 Mechanism for the Ni-catalyzed amidation. | |
6. Cobalt catalysis
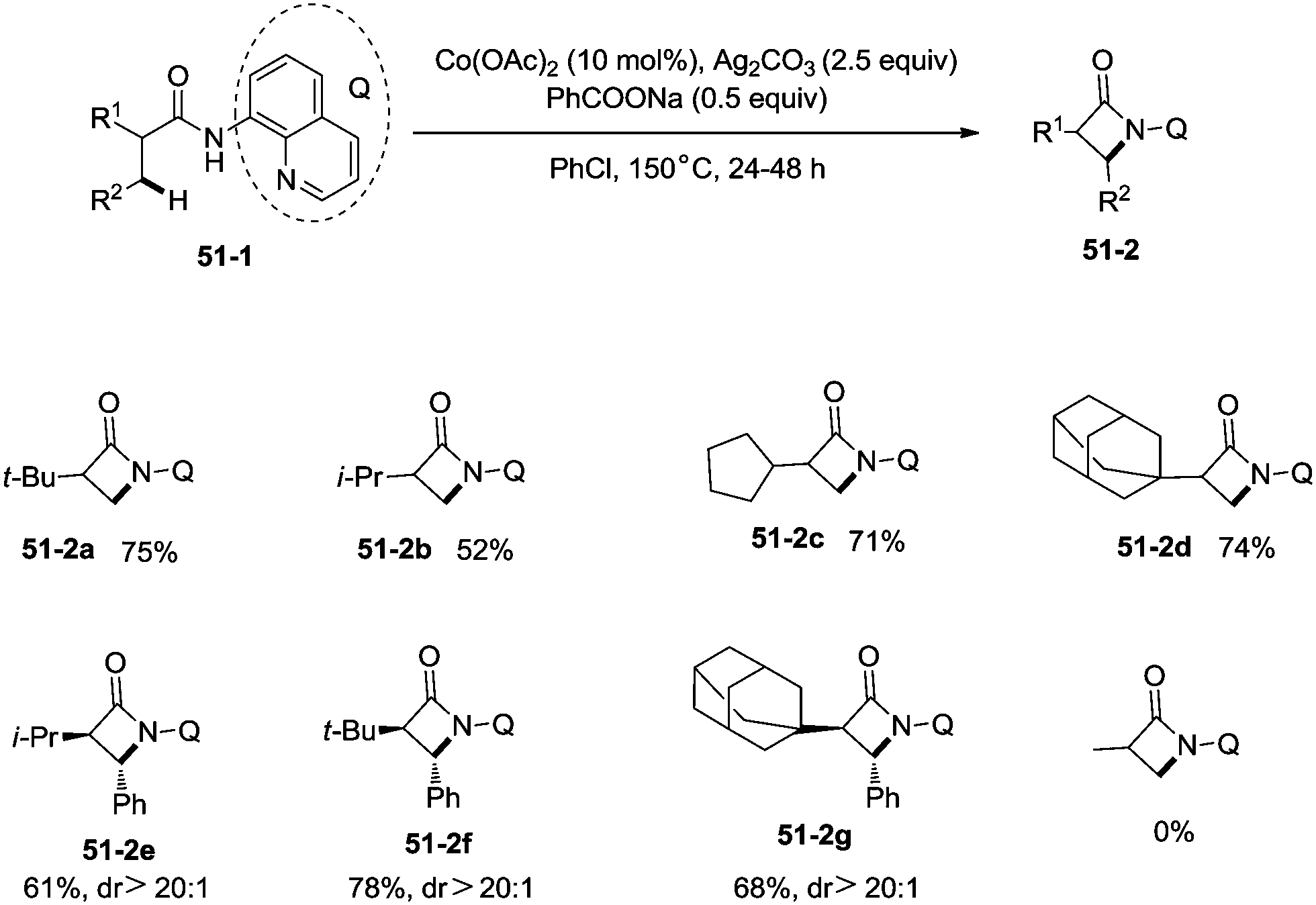
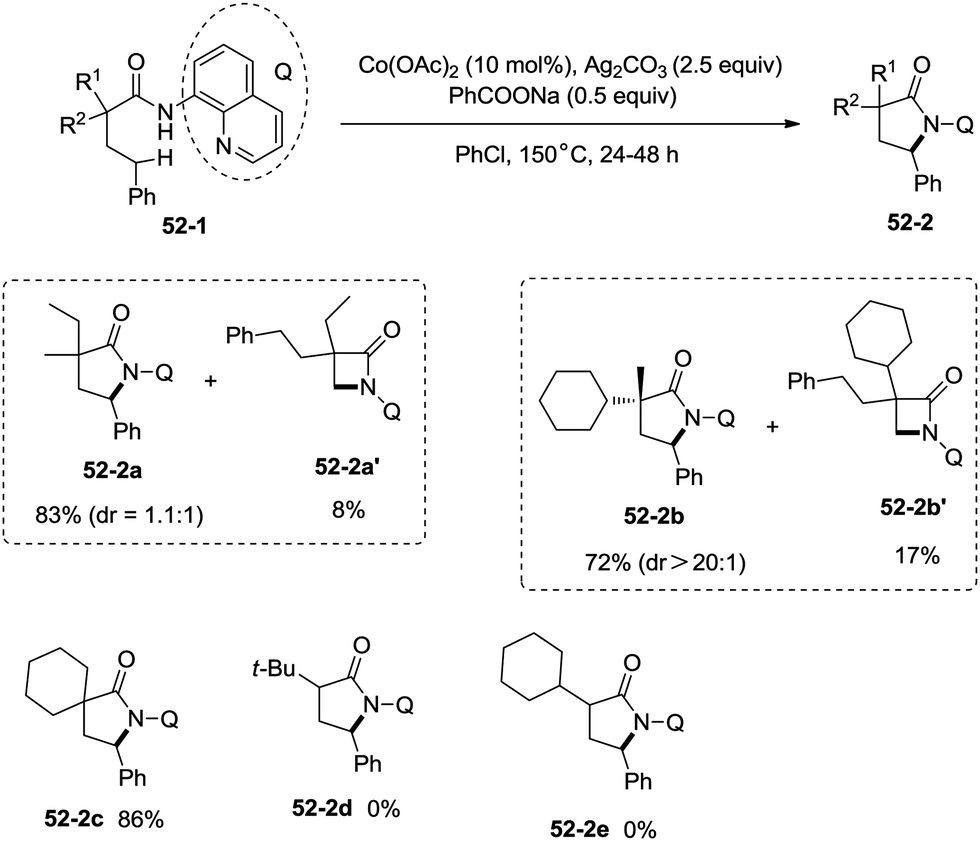
Inspired by the reports of transition metal (palladium, copper and nickel)-catalyzed bidentate ligand-directed sp3 C–H functionalization processes,20,30,36 Ge and co-workers explored cobalt acetate-catalyzed site-selective direct sp3 C–H functionalization with the assistance of a bidentate directing group in 2015.37 Cobalt-catalyzed bidentate ligand-directed intramolecular cyclization of propanamides was achieved, affording β- or γ-lactams effectively (Scheme 50). This reaction proceeded with a high site-selectivity, and the C–H bonds of a β-methyl group are more favourable for the amidation than those of β-methylene and γ-methyl groups [(50-2a)–(50-2e)]. The sp2 C–H bonds on the γ-positions of the benzene ring are more reactive than the sp3 C–H bonds of β-methyl, affording the corresponding indolin-2-one derivatives as the major products (50-2f′, 50-2g′). The quinolyl group and 5-methoxyquinolyl group derived substrates yielded similar results, and the cyclization yields were 83% and 86% respectively (50-2a, 50-2b). The method for the removal of 5-methoxyquinolyl moiety of β- or γ-lactams has been well established.20,30,35 Substrates with a trifluoromethyl, cyano, ethoxycarbonyl, sulfonyl or phthalimidyl group on the α-carbons were cyclized successfully [(50-2h)–(50-2l)], giving β-lactams in yields up to 90%. While substrates with α-methoxy and acetoxy groups could not be cyclized to provide the desired β-lactams (50-2m), β-acetoxy or phenyloxycarbonyl group substituted substrates could be cyclized to produce β-lactams 50-2n and 50-2o in good yields (78%, 74%). The C–H bonds of the β-methyl are more reactive than that of the β-benzyl group (50-2p). The β-benzylic sp3 C–H bonds could also be cyclized to give the desired β-lactams in good yields [(50-2q)–(50-2t)]. α-Bulky group substituted substrates underwent the reaction smoothly [(51-2a)-(51-2g)] (Scheme 51). β-Phenyl substituted substrates underwent the cyclization with excellent diastereoselectivity (dr > 20:1) [(51-2e)–(51-2g)]. The cyclization much more favored γ-benzylic C–H bonds over the C–H bonds of the β-methyl (Scheme 52), and the major products were γ-lactams [(52-2a)–(52-2c)]. However, α-monosubstituted substrates could not be cyclized to afford the desired γ-lactams (52-2d, 52-2e).
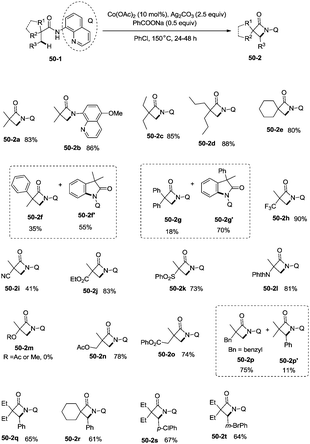 |
| | Scheme 50 Cyclization of α,α-disubstituted propanamides. | |
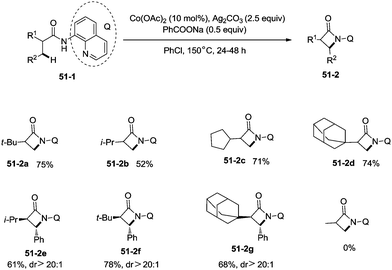 |
| | Scheme 51 Cyclization of α-monosubstituted propanamides. | |
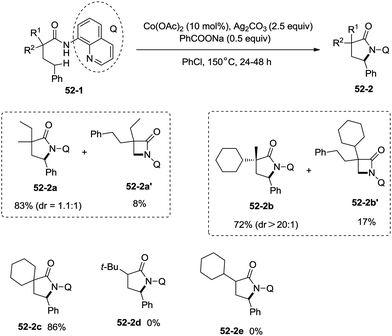 |
| | Scheme 52 Co-catalyzed synthesis of γ-lactams. | |
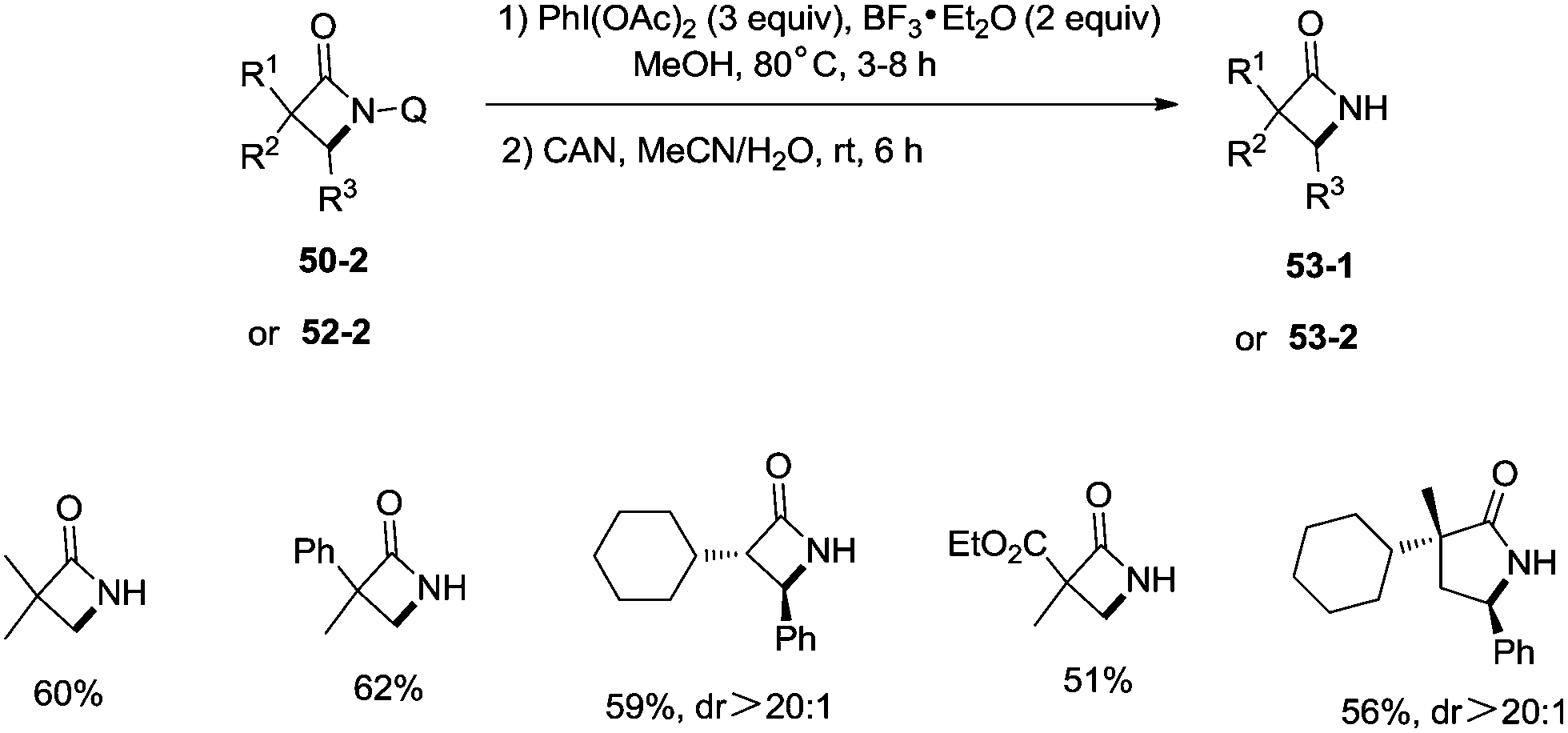
A new two-step method was developed for the removal of the quinolyl group of the lactams (Scheme 53). A methoxy group was introduced on the C5 position of the quinolyl group, and the newly generated 5-methoxyquinolyl moiety could be cleavaged with ammonium cerium(IV) nitrate (CAN). Some free α-mono and α,α-di-substituted β-lactams, and α,α-di-substituted γ-lactams (53-1, 53-2) were produced efficiently using this new method.
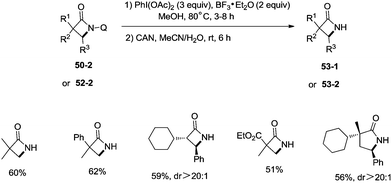 |
| | Scheme 53 The two-step removal of the quinolyl group. | |
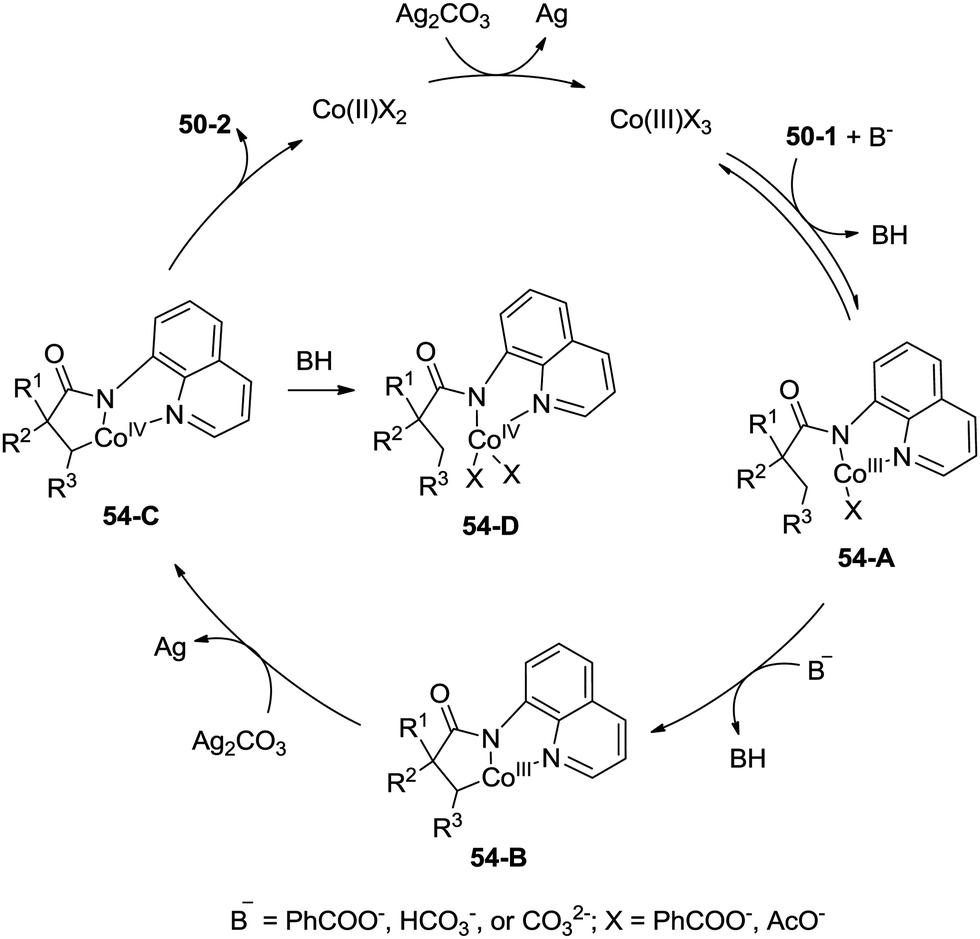
A possible mechanism for the formation of β-lactams was proposed (Scheme 54). Coordination of the amide 50-1 with a cobalt(III) species and a subsequent ligand exchange process generate the Co(III) complex 54-A. β-C–H activation (cyclometalation) gives the cyclic cobalt(III) complex 54-B, which is oxidized to the Co(IV) complex 54-C, reductive elimination affords the β-lactam 50-2. A mechanism involving the Co(II)/Co(IV) catalytic cycle is thought to be possible. The protonated Co(IV) species 54-D is possibly formed. It is thought that the mechanisms involving a radical or cationic species are possible in the formation of γ-lactam derivatives, because the γ-benzylic C–H bonds are preferentially functionalized over the β-methyl C–H bonds.
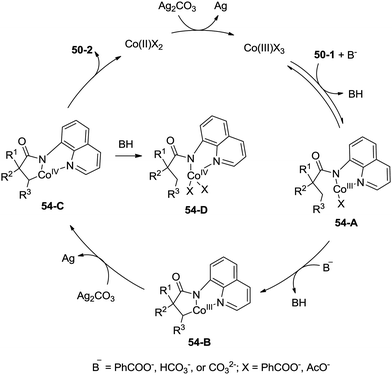 |
| | Scheme 54 Mechanism for cobalt-catalyzed reactions. | |
7. Ruthenium catalysis
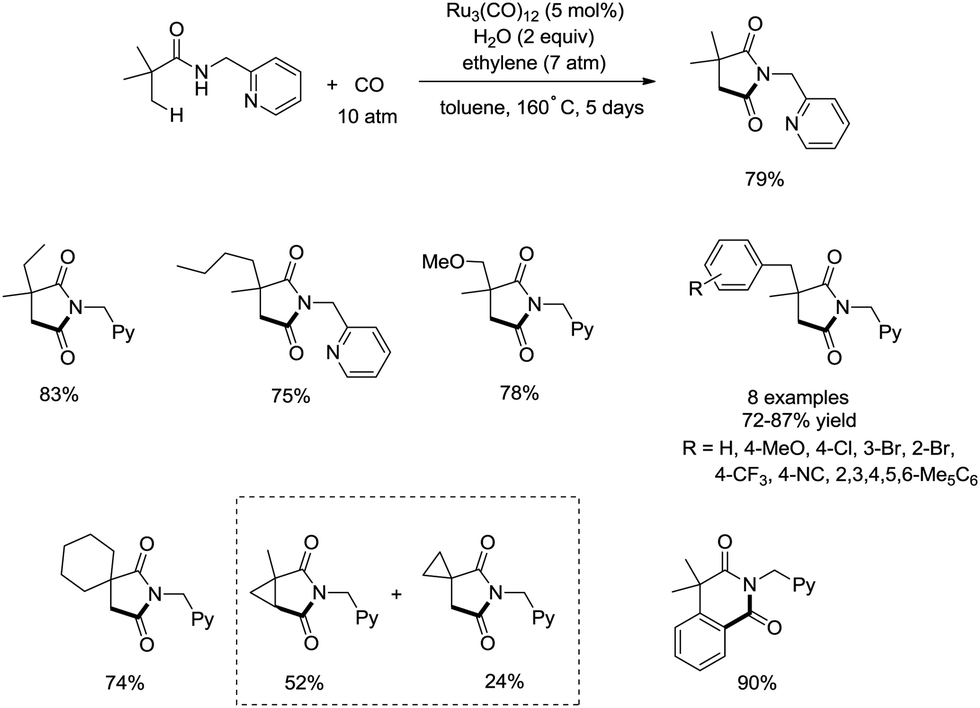
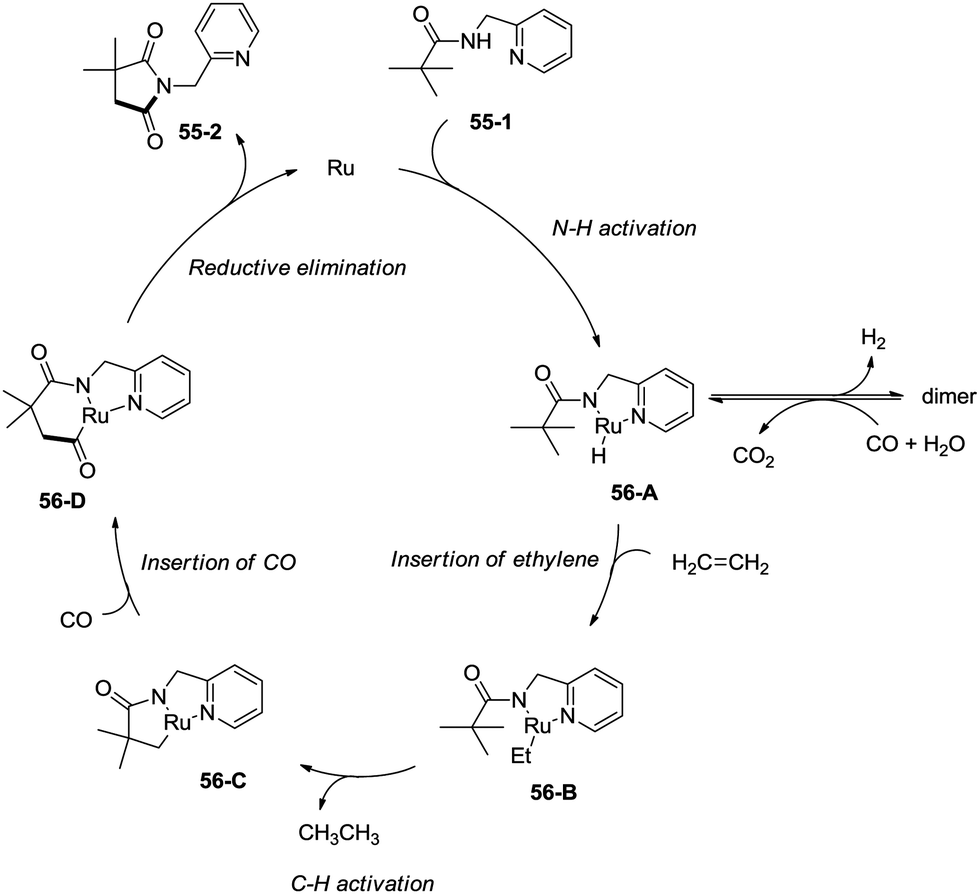
In 2011, a ruthenium-catalyzed carbonylative cyclization of sp3 C–H was reported, affording a series of 1-(pyridin-2-ylmethyl)pyrrolidine-2,5-diones successfully with good functional group tolerance (Scheme 55).38 In general, the cyclization proceeded selectively at β-methyl over methylene. A possible mechanism was proposed (Scheme 56). Coordination of the amide 55-1 to Ru and subsequent N–H activation produce the ruthenium hydride complex 56-A, insertion of ethylene gives the ruthenium complex 56-B, C–H activation forms the complex 56-C, insertion of CO produces the complex 56-D, and reductive elimination gives the product 55-2, along with the regeneration of the catalyst.
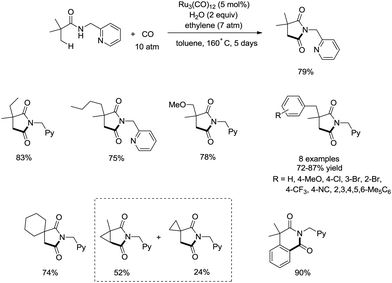 |
| | Scheme 55 Ruthenium-catalyzed carbonylative cyclization. | |
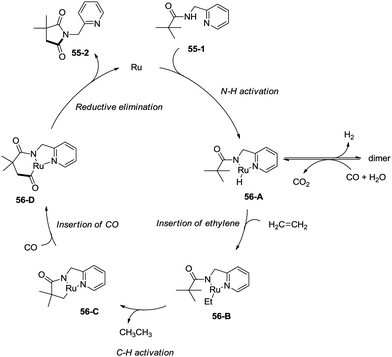 |
| | Scheme 56 The mechanism for Ru-catalyzed cyclization. | |
8. Rhodium catalysis
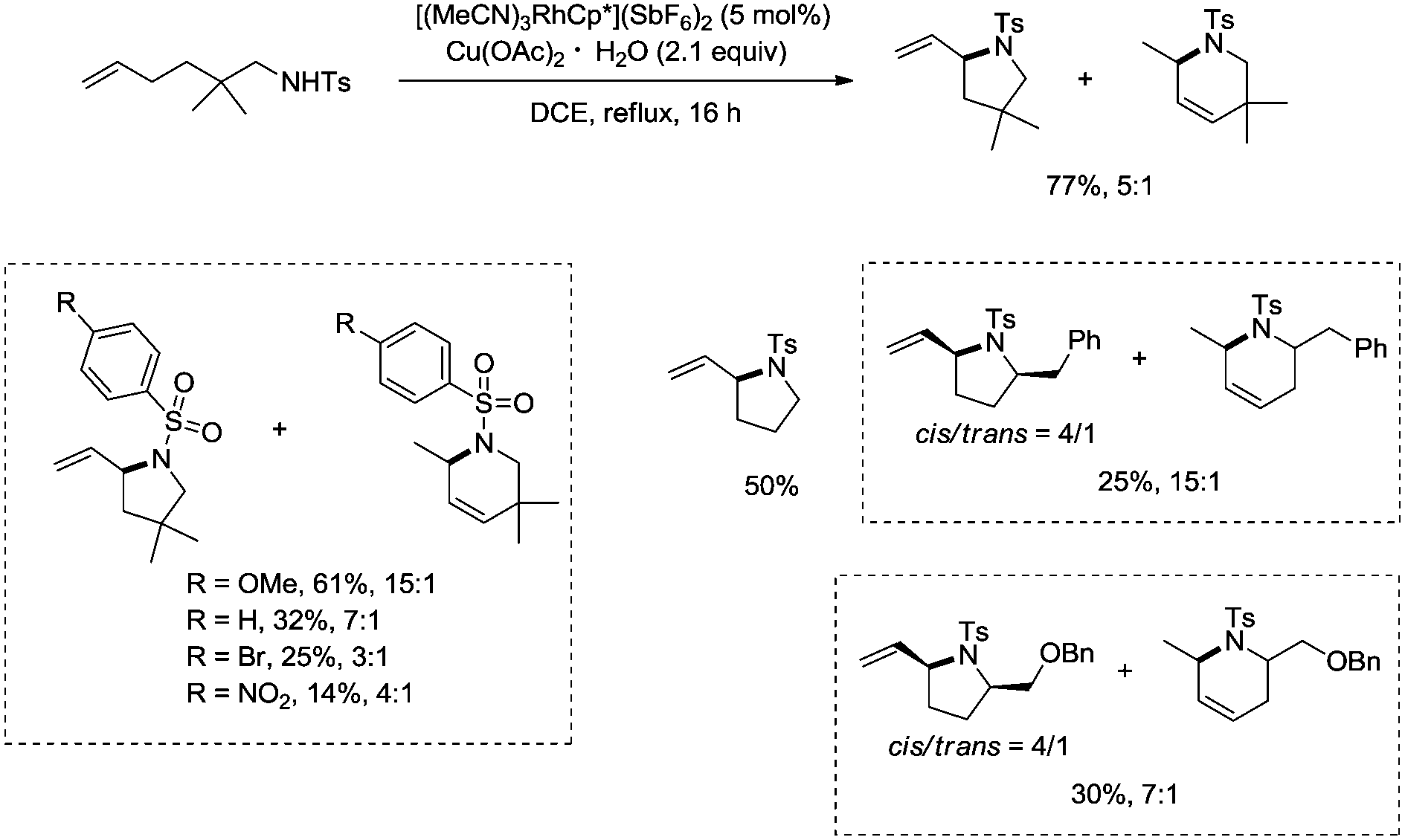
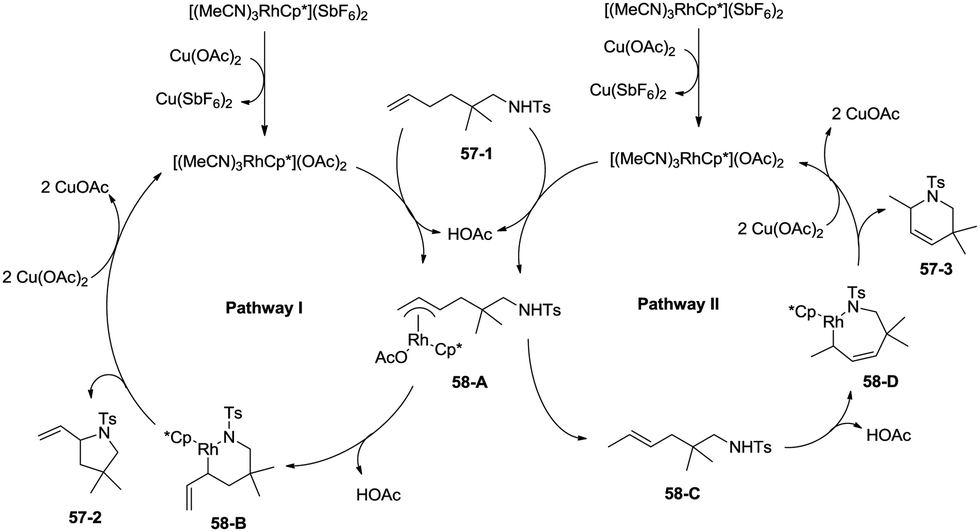
In 2012, a rhodium(III)-catalyzed intramolecular allylic C–H bond amination was developed by Cossy and co-workers, and pyrrolidines and tetrahydropyridines were synthesized in moderate to good yields from ω-unsaturated N-sulfonylamines (Scheme 57).39 Allylic C–H bonds were selectively aminated, and aromatic, benzylic and ethereal C–H bonds did not undergo the amination. The formation of tetrahydropyridines and pyrrolidines from ω-unsaturated N-tosylamides was thought to have been achieved in two pathways (Scheme 58). A π-allylic rhodium complex 58-A can be formed via C–H activation, deprotonation and coordination of the amide to Rh generate the complex 58-B (pathway I), and reductive elimination gives the pyrrolidine 57-2. The formation of the tetrahydropyridine 57-3 can be explained by pathway II. The isomeric olefin 58-C is produced after the migration of a hydrogen, a π-allylic rhodium complex can be formed from 58-C and N-metalation produces 58-D, and reductive elimination gives the tetrahydropyridine 57-3.
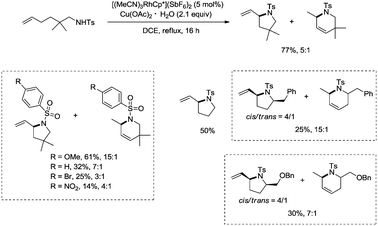 |
| | Scheme 57 Rh-catalyzed cyclization. | |
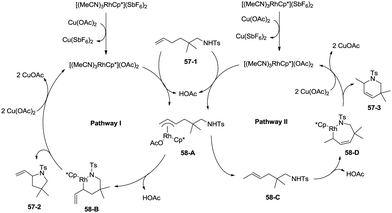 |
| | Scheme 58 The mechanism for Rh-catalyzed cyclization. | |
9. Conclusions and outlook
To date, a wide variety of heterocyclic scaffolds have been constructed by the strategy where a transition metal catalyzes sp3 C–H activation and intramolecular C–N coupling. Palladium, copper, silver, nickel, cobalt, ruthenium and rhodium catalysis have been applied successfully to these transformations, and these methods complement each other. Palladium catalysis is effective for direct intramolecular sp3 C–N coupling and carbonylative cyclization. Primary C–H bonds of a methyl group, secondary benzylic or allylic C–H bonds and secondary C–H bonds of a methylene group could be activated by palladium catalysts and undergo cyclization. Three-, four-, five-, six- and seven-membered products are all likely to be produced, and the regioselectivity depends on the reaction conditions and the substrate structure. Copper catalysis is effective for direct intramolecular sp3 C–N coupling. Primary C–H bonds of a methyl group, primary or secondary benzylic C–H bonds and sp3 C–H bonds of a methylene group could be activated and undergo cyclization under Cu catalysis. Four- and five-membered products can be formed; the four-membered products are preferentially formed over the five- or six-membered products. Silver catalysis is effective for direct intramolecular sp3 C–N coupling. Primary C–H bonds of a methyl group, and secondary C–H bonds of a methylene group could be activated and undergo cycliztion. Five-membered products are preferentially produced over six-membered products in the absence of reactive sp2 C–H bonds in substrates. Nickel catalysis is effective for direct intramolecular sp3 C–N coupling. Primary C–H bonds of a methyl group, and secondary benzylic C–H bonds could be activated and undergo cyclization. Four-membered products are preferentially produced over five- or six-membered products. Cobalt catalysis is effective for direct intramolecular sp3 C–N coupling. Primary C–H bonds of a methyl group, and secondary benzylic C–H bonds could be activated and undergo cyclization. Four- and five-membered products are likely to be produced, and the regioselectivity depends on the substrate structure. Ruthenium catalysis is effective for carbonylative cyclization, and primary C–H bonds of a methyl group and secondary C–H bonds of a methylene group could be activated and undergo cyclization. Five-membered products are preferentially produced over six-membered products in the absence of reactive sp2 C–H bonds in the substrates. Rhodium catalysis is effective for intramolecular allylic C–H bond amination, and benzylic and ethereal C–H bonds don’t undergo the amination. Five- and six-membered products could be formed, and the cyclization favours the formation of five-membered products over six-membered products.
The catalytic systems of these reactions all include catalysts and stoichiometric oxidants, and some oxidants are toxic or expensive, so improvements are needed to overcome the disadvantages existing in this area. Certain development trends of this strategy are as follows. (1) A merged photoredox and palladium-catalyzed process has been used for the synthesis of carbazoles via the intramolecular amination of sp2 C–H bonds; the terminal oxidant is oxygen, and no stoichiometric toxic oxidant is required,40 thus the combination of visible light photoredox and transition metal catalysis could be developed for intramolecular sp3 C–H amination. (2) Recently, regioselective electrochemical synthesis of N-heteroaromatics via intramolecular sp2 C–H/N–H cross-coupling was reported,41 requiring no metal catalysts, oxidizing agents, or salt additives. Electrochemical synthesis was also applied to the intramolecular amination of sp3 C–H bonds without metal catalyst and oxidant, affording pyrrolidines, oxazolines42 and isoindolinones,43 so electrochemistry could be introduced further into the sp3 C–H functionalization due to its green chemistry features, and merging electrochemistry with transition metal catalysis could provide more types of heterocyclic compounds. (3) Compared with palladium, nickel and cobalt are cheaper, but reports involving Ni and Co catalysis for intramolecular sp3 C–H amination/amidation are relatively rare, so Ni and Co catalysis could be employed more for this kind of sp3 C–H cyclization. (4) Chiral heterocyclic compounds are of great medicinal importance, so asymmetric intramolecular sp3 C–H amination/amidation could be developed further in the future. (5) The most common directing groups are amides, and free amines are rarely used as a directing group for sp3 C–H cyclization. The relationship between the basicity of directing nucleophilic nitrogen sources and C–H activation reactivity is worth investigating. It would be interesting to test other nitrogen sources (carbamates, hydrazones, oximes, etc.) as a directing group. (6) The bidentate directing groups could promote sp3 C–H activation, but some auxiliarys could not be removed, so development of better bidentate directing groups with removable auxiliarys would be desirable.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21262017 and No. 21366023).
Notes and references
- A. H. Romero, Top. Curr. Chem., 2019, 377, 21 CrossRef.
- O. V. Zatolochnaya and V. Gevorgyan, Nat. Chem., 2014, 6, 661–663 CrossRef CAS PubMed.
-
(a) M. Zhang, A.-Q. Zhang and Y. Peng, J. Organomet. Chem., 2013, 723, 224–232 CrossRef CAS;
(b) M. Zhang, Adv. Synth. Catal., 2009, 351, 2243–2270 CrossRef CAS.
-
(a) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef CAS PubMed;
(b) J. L. Jeffreya and R. Sarpong, Chem. Sci., 2013, 4, 4092–4106 RSC;
(c) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
-
(a) C. G. Espino and J. D. Bois, Angew. Chem., Int. Ed., 2001, 40, 598–600 CrossRef CAS;
(b) M. Kono, S. Harada and T. Nemoto, Chem. – Eur. J., 2017, 23, 7428–7432 CrossRef CAS PubMed;
(c) J. L. Roizen, M. E. Harvey and J. D. Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed;
(d) B. Darses, R. Rodrigues, L. Neuville, M. Mazurais and P. Dauban, Chem. Commun., 2017, 53, 493–508 RSC.
-
(a) I. T. Alt, C. Guttroff and B. Plietker, Angew. Chem., Int. Ed., 2017, 56, 10582–10586 CrossRef CAS;
(b) S. M. Paradine and M. C. White, J. Am. Chem. Soc., 2012, 134, 2036–2039 CrossRef CAS PubMed.
-
(a) P. F. Kuijpers, M. J. Tiekink, W. B. Breukelaar, D. L. J. Broere, N. P. van Leest, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2017, 23, 7945–7952 CrossRef CAS PubMed;
(b) K. Lang, S. Torker, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2019, 141, 12388–12396 CrossRef.
- J. M. Alderson, J. R. Corbin and J. M. Schomaker, Acc. Chem. Res., 2017, 50, 2147–2158 CrossRef CAS PubMed.
- P. Becker, T. Duhamel, C. J. Stein, M. Reiher and K. Muniz, Angew. Chem., Int. Ed., 2017, 56, 8004–8008 CrossRef CAS PubMed.
- D. Jing, C. Lu, Z. Chen, S. Jin, L. Xie, Z. Meng, Z. Su and K. Zheng, Angew. Chem., Int. Ed., 2019, 58, 14666–14672 CrossRef CAS PubMed.
- C. J. Evoniuk, G. dos, P. Gomes, S. P. Hill, S. Fujita, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2017, 139, 16210–16221 CrossRef CAS.
- A. Bose, S. Sau and P. Mal, Eur. J. Org. Chem., 2019, 4105–4109 CrossRef CAS.
- A. Bose, S. Maiti, S. Sau and P. Mal, Chem. Commun., 2019, 55, 2066–2069 RSC.
- K. S. Kanyiva, M. Tane and T. Shibata, J. Org. Chem., 2019 DOI:10.1021/acs.joc.9b01154.
- A. Verma, S. Patel, M. Meenakshi, A. Kumar, A. Yadav, S. Kumar, S. Jana, S. Sharma, C. D. Prasad and S. Kumar, Chem. Commun., 2015, 51, 1371–1374 RSC.
- X. Zhu and S. Chiba, Org. Biomol. Chem., 2014, 12, 4567–4570 RSC.
- M. Lingamurthy, Y. Jagadeesh, K. Ramakrishna and B. V. Rao, J. Org. Chem., 2016, 81, 1367–1377 CrossRef CAS.
- J. J. Neumann, S. Rakshit, T. Droge and F. Glorius, Angew. Chem., Int. Ed., 2009, 48, 6892–6895 CrossRef CAS.
-
(a) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7–10 CrossRef CAS;
(b) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3–6 CrossRef CAS;
(c) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2017, 139, 561 CrossRef CAS PubMed;
(d) X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Petersen and X. Shi, Chem. Sci., 2013, 4, 3712–3716 RSC.
- G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124–11128 CrossRef CAS.
- Q. Zhang, K. Chen, W. Rao, Y. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588–13592 CrossRef CAS.
- P.-X. Ling, S.-L. Fang, X.-S. Yin, Q. Zhang, K. Chen and B.-F. Shi, Chem. Commun., 2017, 53, 6351–6354 RSC.
- M. Zhang, J. Chem. Res., 2013, 606–610 CrossRef CAS.
- Y. Xie, Y. Yang, L. Huang, X. Zhang and Y. Zhang, Org. Lett., 2012, 14, 1238–1241 CrossRef CAS.
- M. Zhang, T. Chen and Q. Liu, Heterocycles, 2014, 89, 1255–1259 CrossRef CAS.
-
(a) A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS PubMed;
(b) E. J. Yoo, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 17378–17380 CrossRef CAS;
(c) E. Hernando, J. Villalva, A. M. Martinez, I. Alonso, N. Rodríguez, R. G. Arrayas and J. C. Carretero, ACS Catal., 2016, 6, 6868–6882 CrossRef CAS.
- S.-J. Zhang, W.-W. Sun, P. Cao, X.-P. Dong, J.-K. Liu and B. Wu, J. Org. Chem., 2016, 81, 956–968 CrossRef CAS PubMed.
- J. Zhao, X.-J. Zhao, P. Cao, J.-K. Liu and B. Wu, Org. Lett., 2017, 19, 4880–4883 CrossRef CAS PubMed.
-
(a) R. C. Larock, T. R. Hightower, L. A. Hasvold and K. P. Peterson, J. Org. Chem., 1996, 61, 3584–3585 CrossRef CAS PubMed;
(b) E. M. Beccalli, G. Broggini, G. Paladino, A. Penoni and C. Zoni, J. Org. Chem., 2004, 69, 5627–5630 CrossRef CAS PubMed;
(c) K. J. Fraunhoffer and M. C. White, J. Am. Chem. Soc., 2007, 129, 7274–7276 CrossRef CAS;
(d) G. T. Rice and M. C. White, J. Am. Chem. Soc., 2009, 131, 11707–11711 CrossRef CAS;
(e) F. Nahra, F. Liron, G. Prestat, C. Mealli, A. Messaoudi and G. Poli, Chem. – Eur. J., 2009, 15, 11078–11082 CrossRef CAS PubMed;
(f) L. Wu, S. Qiu and G. Liu, Org. Lett., 2009, 11, 2707–2710 CrossRef CAS PubMed;
(g) P.-S. Wang, M.-L. Shen, T.-C. Wang, H.-C. Lin and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 16032–16036 CrossRef CAS PubMed.
-
(a) X. Wu, Y. Zhao, G. Zhang and H. Ge, Angew. Chem., Int. Ed., 2014, 53, 3706–3710 CrossRef CAS PubMed;
(b) Z. Wang, J. Ni, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 3496–3499 CrossRef CAS PubMed.
-
(a) C. Wang, Y. Yang, D. Qin, Z. He and J. You, J. Org. Chem., 2015, 80, 8424–8429 CrossRef CAS PubMed;
(b) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS.
- C. Yamamoto, K. Takamatsu, K. Hirano and M. Miura, J. Org. Chem., 2016, 81, 7675–7684 CrossRef CAS.
- F. Pan, B. Wu and Z.-J. Shi, Chem. – Eur. J., 2016, 22, 6487–6490 CrossRef CAS.
- M. E. Wolff, Chem. Rev., 1963, 63, 55 CrossRef CAS.
- M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y.-F. Zhang, X.-S. Zhang, G. Chen, Y. Cheng, Z. Cao, Q.-Y. Guo, L. Wang and Z.-J. Shi, Nat. Commun., 2014, 5, 4707 CrossRef CAS PubMed.
- X. Wu, Y. Zhao and H. Ge, Chem. – Eur. J., 2014, 20, 9530–9533 CrossRef CAS.
- X. Wu, K. Yang, Y. Zhao, H. Sun, G. Li and H. Ge, Nat. Commun., 2015, 6, 6462 CrossRef CAS PubMed.
- N. Hasegawa, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 8070–8073 CrossRef CAS PubMed.
- T. Cochet, V. Bellosta, D. Roche, J.-Y. Ortholand, A. Greinerb and J. Cossy, Chem. Commun., 2012, 48, 10745–10747 RSC.
- S. Choi, T. Chatterjee, W. J. Choi, Y. You and E. J. Cho, ACS Catal., 2015, 5, 4796–4802 CrossRef CAS.
- H.-B. Zhao, Z.-J. Liu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 12732–12735 CrossRef CAS PubMed.
- F. Wang and S. S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385–6390 CrossRef CAS PubMed.
- S. Zhang, L. Li, M. Xue, R. Zhang, K. Xu and C. Zeng, Org. Lett., 2018, 20, 3443–3446 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Qiuhong
Wang
a,
Yiyuan
Peng
*a,
Qiuhong
Wang
a,
Yiyuan
Peng