
Antileishmanial drug discovery: comprehensive review of the last 10 years
Jaiprakash N. Sangshetti
*a,
Firoz A. Kalam Khan
a,
Abhishek A. Kulkarni
a,
Rohidas Arote
b and
Rajendra H. Patil
c
aY. B. Chavan College of Pharmacy, Dr Rafiq Zakaria Campus, Aurangabad 431001, M.S., India. E-mail: jnsangshetti@rediffmail.com; Tel: +91-240-2381129
bDepartment of Molecular Genetics, School of Dentistry, Seoul National University, Seoul, Republic of Korea
cDepartment of Biotechnology, Savitribai Phule Pune University, Pune 411007, M.S., India
First published on 19th March 2015
Abstract
Leishmaniasis, a group of diseases caused by hemoflagellate obligate intracellular protozoa (trypanosomatids) from the genus Leishmania, has not received the attention it deserves and has developed into a major health problem in developing countries. No effective vaccine is available against leishmaniasis, so chemotherapy is the only effective way to treat all forms of the disease. However, the drugs currently used for treatment of human cutaneous and visceral leishmaniasis are toxic, having severe adverse reactions which limit their use. Therefore, development of novel, effective, and safe antileishmanial agents, with reduced side effects, is a major priority for health researchers, and large numbers of research reports have been published on antileishmanial agents in the last 10 years. Herein, we comprehensively review the developments of the last decade, covering all aspects of leishmaniasis including clinically used drugs, various new classes of antileishmanial agents (synthetic as well as natural), patented antileishmanial agents, and possible drug targets.
 Jaiprakash N. Sangshetti | Jaiprakash N. Sangshetti graduated from Y. B. Chavan College of Pharmacy, Aurangabad (MS), India and M. Pharmacy from Prin. K. M. Kundnani College of Pharmacy, Mumbai. He obtained his Ph.D. from Dr Babasaheb Ambedkar Marathwada University, Aurangabad (MS), India under the supervision of Dr Devanand B. Shinde in 2009 and is currently working as Associate Professor at Y. B. Chavan College of Pharmacy, Aurangabad. He has two Patents to his credit and has published more than 100 research papers in international journals. He has received research grants from different Indian government funding agencies like DST, UGC and AICTE. His major area of research includes design and synthesis of bioactive molecules, development and validation of analytical methods for drugs and pharmaceutical dosage forms. He is currently working on the different targets in antibacterial drug discovery. |
 Firoz A. Kalam Khan | Firoz A. Kalam Khan obtained his Bachelor degree in Pharmacy from Dr L. H. Hiranandani College of Pharmacy, University of Mumbai, Mumbai, India and his Master degree in Pharmaceutical Chemistry from Y. B. Chavan College of Pharmacy, Dr Babasaheb Ambedkar Marathwada University, Aurangabad (MS), India. He has been awarded as a Runner-up in the subject Pharmaceutical Chemistry for Rajnibhai V. Patel best M. Pharm. Thesis award in 2011–2012. Currently, he is working as research fellow under Dr Jaiprakash N. Sangshetti at Y. B. Chavan College of Pharmacy, Aurangabad, Maharashtra where he has applied his knowledge for the design and synthesis of novel bioactive molecules as anticonvulsant, antileishmanial, antimicrobial, and anticancer. He is currently working on Peptide deformylase as novel target for antibacterial agents. He is also working on one-pot multicomponent synthesis of various bioactive molecules using various reusable catalysts. |
 Abhishek A. Kulkarni | Abhishek A. Kulkarni graduated from Shri Bhagwan College of Pharmacy, Dr Babasaheb Ambedkar Marathwada University, Aurangabad (MS), India in 2013 with distinction. Currently, he is pursuing his master degree in Pharmaceutical Chemistry from Y. B. Chavan College of Pharmacy, Aurangabad (MS), India under the guidance of Dr Jaiprakash N. Sangshetti. He has presented various scientific posters on topics related to Pharmaceutical Chemistry at state and national level. His area of research includes design and synthesis of novel heterocyclic coupled bioactive compounds and evaluation for their antileishmanial, antimicrobial and antioxidant activities. He is also working on one-pot multicomponent synthesis of various bioactive molecules using various reusable catalysts. |
 Rohidas Arote | Rohidas Arote did his PhD in gene delivery from Seoul National University, Seoul, Republic of Korea in 2008. He continued as a postdoctoral fellow in the same University for the period of 2009–2010 and is currently working as an Assistant Professor with School of Dentistry, Seoul National University, Republic of Korea. His Ph.D. research involved development of a biodegradable polymeric gene carrier system for the treatment of various types of diseases, mainly cancer. His postdoctoral research was developing efficient gene carriers for the treatment of lung and liver cancer. Presently, he is focusing on targeted drug and gene delivery by using multifunctional nanocarriers in the field of nanomedicine. |
 Rajendra H. Patil | Rajendra H. Patil completed his master from Swami Ramanand Teerth Marathwada University, Nanded, India, in 2002. He submitted doctorate studies to the Department of Biotechnology, Savitribai Phule Pune University, Pune, India, in 2014, and later joined as an assistant professor in the same department. He is a recipient of UGC-CSIR fellowship for completing his doctorate studies. His interest has been in the area of bioorganic chemistry wherein the microbial diversity is explored for synthesis of bioactive molecules and nanoparticles. He also has published many research articles wherein the chemically synthesized nanoparticles were used to eradicate the biofilm in Pseudomonas aeruginosa. |
1. Introduction
Leishmaniasis is a vector-borne, poverty-associated disease developing in the mammalian host by protozoan parasites (obligate, hemoflagellate, intracellular in nature) belonging to the order Kinetoplastidae, family Trypanosomatidae from the genus Leishmania. These parasites reside in and are transmitted through the bites of more than 30 species of female sand flies. The World Health Organization (WHO) classified leishmaniasis as a major tropical disease, ranking second only after malaria. Ever-increasing cases worldwide are resulting in high morbidity and mortality levels with a wide spectrum of clinical syndromes. It has become a major focus of concern and a serious world problem that affects the poorer sections of the society.1–6 It is estimated that 12 million people worldwide are infected by over 20 different species of Leishmania, and about 350 million people living in the endemic areas are at risk of infection.7 This parasite exists in many tropical and temperate countries.8 Researchers have reported that more than 90% of visceral cases of leishmaniasis (visceral leishmaniasis, VL) occur in India, Nepal, South Sudan, Sudan, Bangladesh, Brazil, and Ethiopia, while about 70–75% of cutaneous cases (cutaneous leishmaniasis, CL) occur mainly in Afghanistan, Algeria, Brazil, Colombia, Costa Rica, Ethiopia, Iran, Syria, North Sudan, and Peru.9When sand flies (vectors) feed on an infected host, Leishmania parasites enter their digestive tracts and multiply therein as promastigotes, which can then be passed to a mammalian host when the sand flies bite healthy humans for blood meal. In this vertebrate host, the parasite multiplies inside the macrophages (where they survive and multiply within phagolysosomal compartment) in an amastigote form (Fig. 1). Leishmania parasites have the capability to survive in stress conditions, lyse macrophages, and are phagocytozed by new host cells.1,11–13
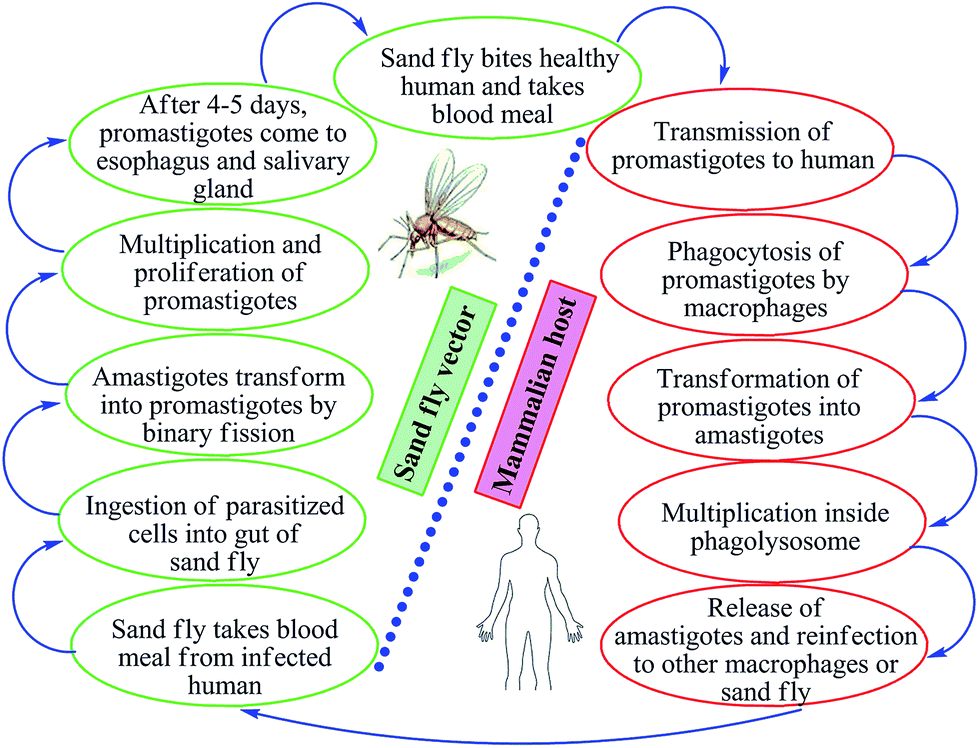 | ||
| Fig. 1 Life cycle of Leishmania. | ||
Depending on the tropism, the disease is characterized as one of four major syndromes: cutaneous leishmaniasis (CL), muco-cutaneous leishmaniasis (MCL), visceral leishmaniasis (VL), and post-kala-azar dermal leishmaniasis (PKDL). Other cutaneous manifestations such as diffuse cutaneous leishmaniasis (DCL) and recidivans leishmaniasis (RL) may also occur.8,14
Among the various causative organisms, viz. L. donovani, L. major, L. tropica, L. aethiopica, L. mexicana, L. venezuelensis, L. amazonensis, L. braziliensis, L. panamensis, L. guyanensis, L. peruviana, L. infantum (L. chagasi) etc., that infect a variety of the population, some are associated significantly with morbidity.6 If untreated, most VL caused by L. donovani is fatal. CL caused by L. major, L. mexicana, L. braziliensis, and L. panamensis is associated with morbidity, but can self-cure within 3–18 months leaving behind disfiguring scars.10,15,16 Leishmaniasis is said to be recidivans leishmaniasis, if caused by L. tropica which is difficult to treat and leaves behind extensive scars. MCL is characterized by the destruction of mucosa and cartilage of mouth and pharynx, followed by the involvement of facial tissue (several species are able to cause MCL, the most important being L. braziliensis).17
There are very few reports published regarding leishmanial disease history and parasite biology. Also, to the best of our knowledge, there are very few comprehensive reviews covering all current aspects of leishmanial disease.17 So the focus of this article is to review comprehensively developments in antileishmanial agents and their progress during the past decade. In this review, we cover disease history and parasite biology, followed by a summary of currently available treatments, and, finally, review reports of novel small molecules (synthetic as well as natural) with antileishmanial activity. We also discuss all possible drug targets reported for antileishmanial agents.
2. Current antileishmanial therapy
As there is no vaccine currently available against leishmaniasis, drugs are the only available tool for treatment and control of both VL and CL.8,17 Severity of disease is dependent on the infecting Leishmania species and the associated host-immune response. Visceral disease (may result in PKDL) caused by viscerotropic Leishmania species requires systemic treatment, whereas cutaneous disease (may further evolve into recidivans, diffuse or mucosal complication) caused by dermotropic Leishmania species is treated either systemically or locally (Fig. 2).17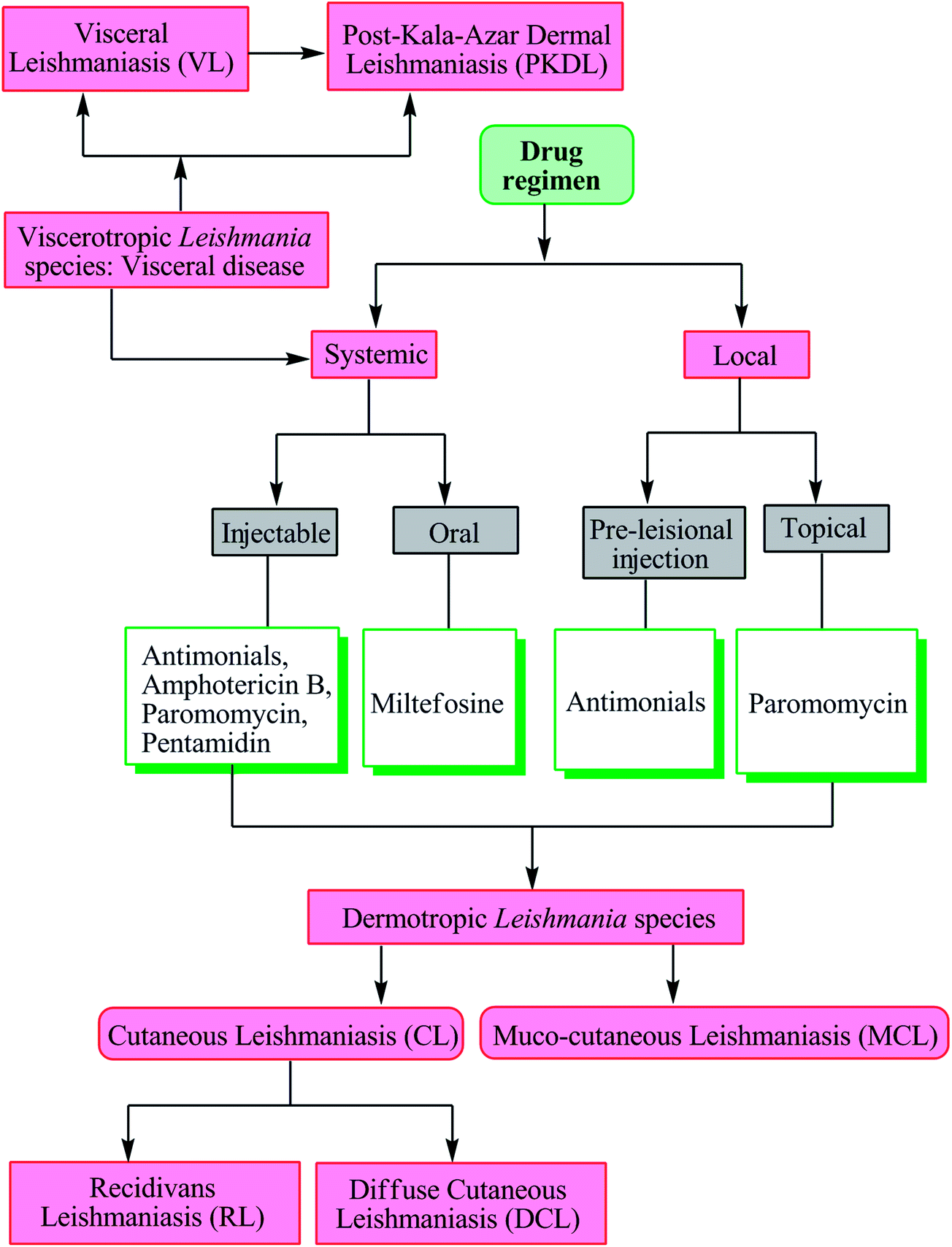 | ||
| Fig. 2 Leishmania species, associated disease, and treatment. | ||
The following details the different drugs used in current antileishmanial therapy.
2.1. Pentavalent antimonials
Prof. Brahmchari from India was nominated for a Nobel Prize (in 1929) for the first effective drug against L. donovani, the urea stibamine (discovered by him in 1912). Although it saved the lives of many poor Indians, it had some side effects. Pentavalent antimonials were developed subsequently, which showed promise in reduction of these side effects.18 The antimonials were first introduced in 1945 and remained the drug standard for about six decades. The activation mechanism of pentavalent antimonials is still not clearly known, but it has been reported that pentavalent antimonite (Sb, V) is a prodrug requiring biological reduction to its trivalent form (Sb, III) for antileishmanial activity. However, site (amastigote or macrophage) and mechanism of reduction (enzymatic or non-enzymatic) remain controversial. Studies indicate that axenic amastigotes are susceptible to Sb(V) but that promastigotes are not, suggesting that some stage-specific reduction occurs during the life cycle, but the mechanism by which amastigotes reduce Sb(V) is not clear. Both glutathione and trypanothione can non-enzymatically reduce Sb(V) to Sb(III), particularly under acidic conditions. However, promastigotes contain higher intracellular concentration of trypanothione and glutathione than amastigotes, and both stages maintain intracellular pH values close to neutral, independent of external pH. Thus, it is difficult to account for the selective action of Sb(V) against amastigotes by a non-enzymatic mechanism.19 As both stages can take up Sb(III) and Sb(V), the insensitivity of promastigotes to Sb(V) cannot be attributed to drug exclusion. Two possible candidates for the enzymatic reduction of Sb(V) to Sb(III) in amastigotes, a thiol-dependent reductase related to glutathione-S-transferase highly expressed in amastigotes and a homologue of a glutaredoxin-dependent yeast arsenate reductase, have been identified recently. However, the level of expression of arsenate reductase has not been reported and the low specific activity of the recombinant enzyme with glutaredoxin raises questions as to the physiological nature of the electron donor in Leishmania species.13,20–23These drugs are used with variable efficacy against both major types of leishmaniasis (VL and CL) and were recommended firstline treatment until observation of development of drug-resistance in the Indian state, Bihar.17 Pentavalent antimonials can be administered intramuscularly, intravenously, or even by an intralymphatic route.24 The recommended dose is 20 mg per kg of body weight for about 20–30 days, achieving more than 95% cure. In Bihar, however, 60% or more of patients did not respond, suggesting that there was development of resistance to the drugs by the parasite. However, they are still used as firstline drugs in areas in which resistance has not developed.25–27
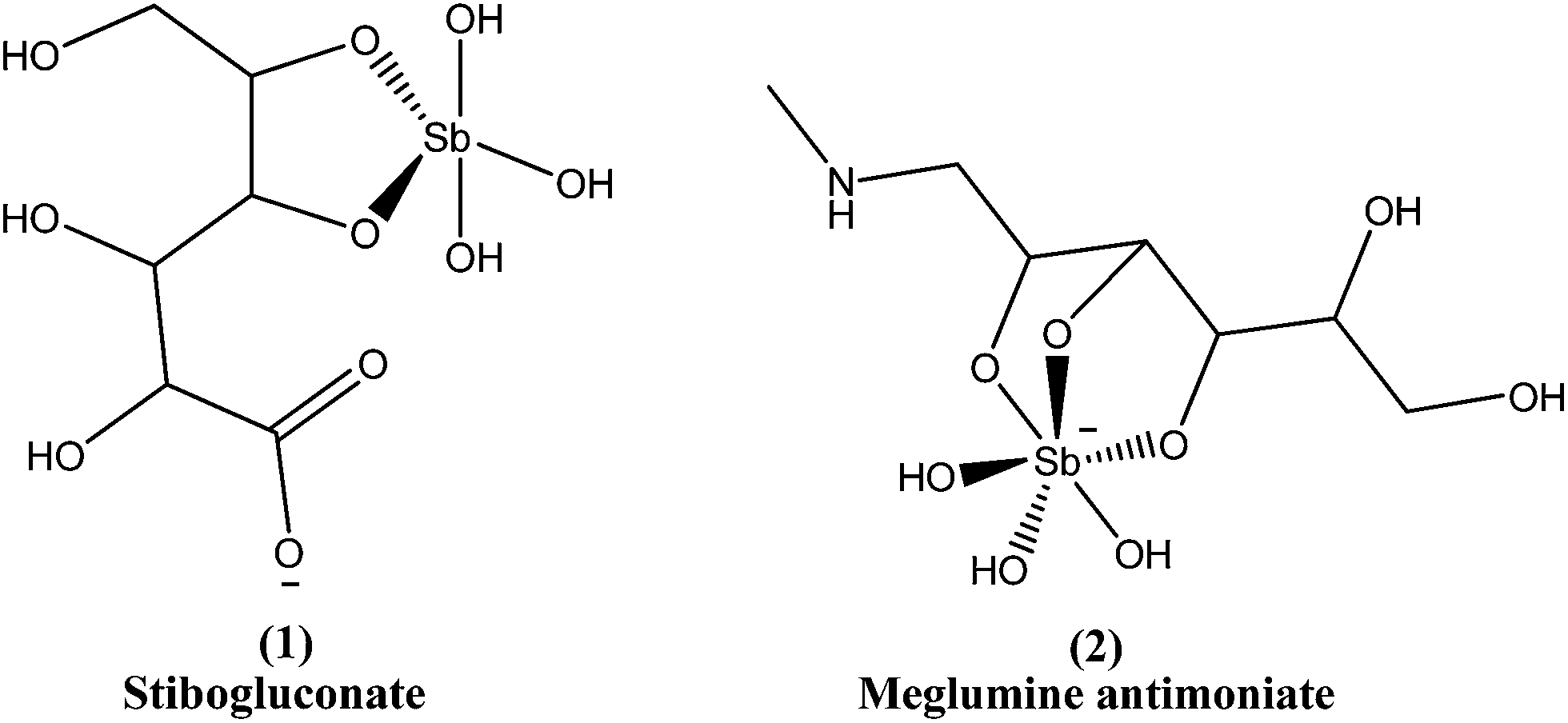
Since the mid-1940s, in English-speaking East Africa, stibogluconate (1) has been used (manufactured by GlaxoWellcome, London, UK, under the brand name Pentostam T, PSM, containing sodium stibogluconate), whereas in the former French and Italian colonies of Africa, meglumine antimoniate (2) (prepared by Rhone-Poulenc-Rohrer, Paris, under the brand name Glucantime T) has been the only drug used for treatment of kala-azar (VL) and post-kala-azar dermal leishmaniasis (PKDL). Frezard et al. proposed the structures of stibogluconate (1) and meglumine antimoniate (2), identified by ESI(−)-MS in aqueous solutions.28 However, because of increasing demand not being met by the manufacturer, the urgency of treatment, and the exceptionally high cost (approximately 200 US dollars per patient), Albert David Ltd. (Calcutta, India) began to manufacture generic sodium stibogluconate B.P. (SSG), which costs approximately 13 US dollars per patient. Since 1997, the International Dispensary Association (IDA, Amsterdam, Netherlands) also has supplied generic SSG.13,29
Several limitations have decreased the use of antimonials. They routinely cause pancreatitis during treatment, with other side effects including pancytopenia, reversible peripheral neuropathy, elevations in serum aminotransferases, pain at the site of injection, stiff joints, gastrointestinal problems, hepatic and renal insufficiency (nephrotoxicity). Cardiotoxicity may occur and this may cause sudden death. In addition, the long duration of treatment can result in accumulation of drug in the tissues in the liver and spleen.30–33
Parenteral administration, long-term treatment (up to 4 weeks), and variation in efficacy against VL and CL, along with development of resistance, are some of the factors that have led to decreased use of antimonials. Recently, Fernandes et al. reported a novel oral delivery strategy for pentavalent antimonials in treatment of visceral leishmaniasis. This was based on formation of an amphiphilic antimony(V) complex on reaction of antimony(V) with nonionic surfactants from the N-alkyl-N-methylglucamide series. Improved oral bioavailability and pharmacokinetics of Sb in mice were achieved with this strategy, compared with use of meglumine antimoniate. The resulting amphiphilic complexes were found to be active by the oral route in a murine model of VL.8
2.2. Amphotericin B
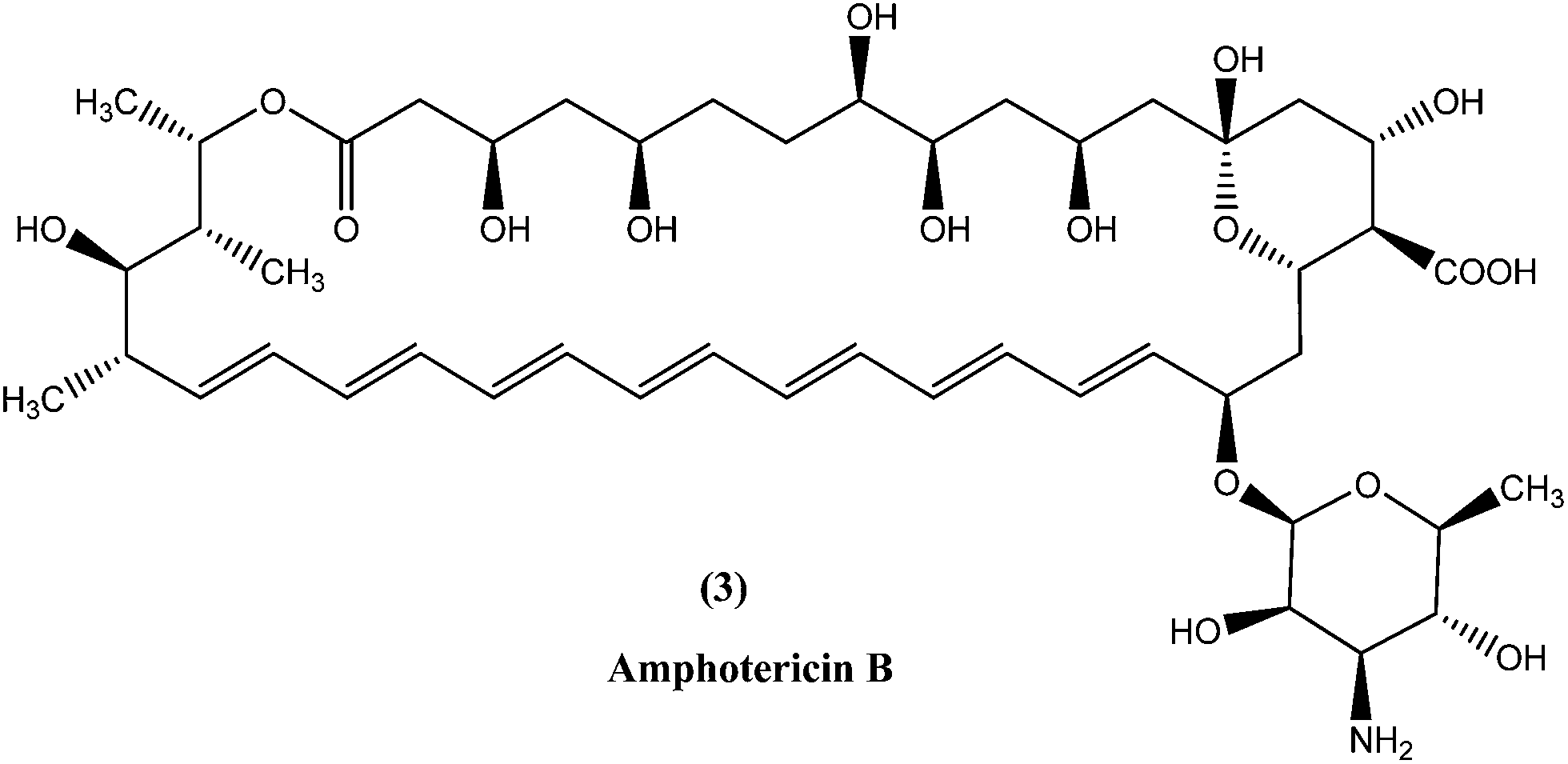
In the areas where resistance is commonly seen to antimonial antileishmanial treatment is, amphotericin B (3), a macrolide polyene antifungal antibiotic, is recommended as drug of choice.34,35 In India, amphotericin B is recommended by the National Expert Committee.36 It was discovered in 1956 in actinomycetes: Streptomyces nodosus, a bacterium collected from the soil of Orinoco River in Venezuela.14 Amphotericin B has high affinity for 24-substituted sterol, ergosterol (a major component of leishmanial cell membrane), forming a complex with it and thus interfering with the ergosterol pathway, which ultimately results in formation of aqueous pores leading to increased membrane permeability to monovalent cations, anions, and small metabolites, causing cell death (leishmanicidal action).35,37,38Currently used preparations of amphotericin B are amphotericin B desoxycholate (Fungizone®) and various liposomal formulations (e.g. AmBisome® involving unilamellar liposomes).39 Lipid formulations (liposomal) of amphotericin B are prepared to improve its bioavailability and pharmacokinetic properties, which helps in masking amphotericin B from susceptible tissues, facilitating its preferential uptake by reticuloendothelial cells and thereby reducing side effects and increasing its efficacy.40–43 Smaller liposomes reside in the bloodstream for longer duration, whereas larger lipid particles are rapidly engulfed by mononuclear phagocytes, for example hepatic macrophages (Kupffer cells, the site at which VL parasites such as L. donovani attack and accumulate, and VL develops). Thus, the mechanism of AmBisome is such that it accumulates rapidly in the liver and reaches its therapeutic concentration at a faster rate than antimonials, and with increased effect because of its longer half-life.39,41,44,45
AmBisome is registered and approved for treatment of VL in various countries such as USA and Europe, and its use is recommended by a WHO working group. It can also be used for CL and complex forms of CL (such as MCL), as well as for PKDL.34,46,47 Recently, after a single infusion therapy analysis on a particular group of patients in India, it was reported that AmBisome cures 95% of patients with minimal adverse effects.48
Despite having higher toxicity profiles and lesser efficacy than AmBisome, other formulations such as an ampB–lipid complex (Abelcets®), an ampB colloidal dispersion (Amphocil™), and a multilamellar liposomal formulation, are also in use, but these are not as common.49,50 Besides the main drawbacks of high cost, administration route, and lack of stability at high temperature (manufacturer guarantee 25 °C) which limits usefulness, liposomal amphotericin B has been proven to be an efficient drug with more than 95% efficacy over Amphocil and Abelcet.8,51,52 It can be administered intravenously in therapeutic doses of about 0.75–1.0 mg per kg body weight as 15–20 infusions either daily for about 20–30 days or 1.5–2.0 g on alternate days, or as 5–20 mg kg−1 total dose in 4–10 doses over 10–20 days.24,25,43
Although minimal, amphotericin B does bind to the cholesterol present in human cell walls and thus exhibits some toxic effects. Infusion-related adverse effects such as “nephrotoxicity, fever with rigor and chills, bone pain, hypotension, anorexia, dyspnea, thrombophlebitis, cardiac arrest (rarely), myocarditis and delayed side effects such as hypokalemia” explain fluctuations in use of amphotericin B before the 1990s, when there were no available lipid formulations.53–55 The use of amphotericin B also requires prolonged hospitalization and close monitoring.43 Although no resistance has been reported as yet, there is evidence of development of resistance in laboratory Leishmania strains.24
2.3. Miltefosine
Miltefosine (4), chemically known as hexadecylphosphocholine, was originally developed as an antineoplastic agent used in topical treatment (Miltex) of skin metastases of breast cancer. Its use as an antileishmanial agent was initiated in the mid-1980s. It is the first orally administered drug effective in treatment of leishmaniasis, and the most recent antileishmanial drug to enter in the market. In the mid-to-late 1990s, collaborative development of miltefosine by Asta Medica (now Zentaris) and a WHO/TDR partnership showed that it has oral activity in VL patients, including antimonial-unresponsive patients. On administering a dose of 2.5 mg kg−1 of miltefosine daily for 28 days in phase III clinical trials, it was observed that there was about 94% cure in VL patients. The drug was then registered in 2002 and entered the Indian market. Thereafter, a phase IV study was performed. Two years later, the drug was approved in Germany, showing usefulness in treatment of immunocompromised patients. More recently, study of miltefosine in treatment of CL was carried out in regions of Colombia, where L. panamensis is a commonly infecting parasite. A 91% cure rate was observed with the same oral dose as described above. However, in regions of Guatemala (Central America) where L. braziliensis and L. mexicana are common, only 53% cure rates were observed, much lower than the cure rate of antimonials (more than 90%).17,56 Therefore, considering these studies, miltefosine was registered in India, Germany, and Columbia (Impavido®).8In a recent study, children suffering from cutaneous leishmaniasis were given miltefosine, with results being similar to those after administration of meglumine antimoniate, and the added advantage of an oral delivery route that is more easily tolerated by pediatric patients in comparison with other routes.57 The primary effect of miltefosine is uncertain, but possible mechanisms are action by blocking proliferation of Leishmania, inhibition of phosphatidylcholine biosynthesis, alteration of phospholipid and sterol composition, activation of cellular immunity, or inhibition of signal transduction and calcium homeostasis.58,59 Also, it has been reported that the activity of miltefosine may be enhanced by intracellular accumulation, which is regulated by drug transporters. Researchers also have found that it causes apoptosis-like processes in L. donovani, but the exact mechanism is not known.60,61 Miltefosine has been shown to stimulate production of inducible nitric oxide synthetase 2 (iNOS2), causing generation of nitric oxide (NO) which further helps to kill the parasite within the macrophage.62
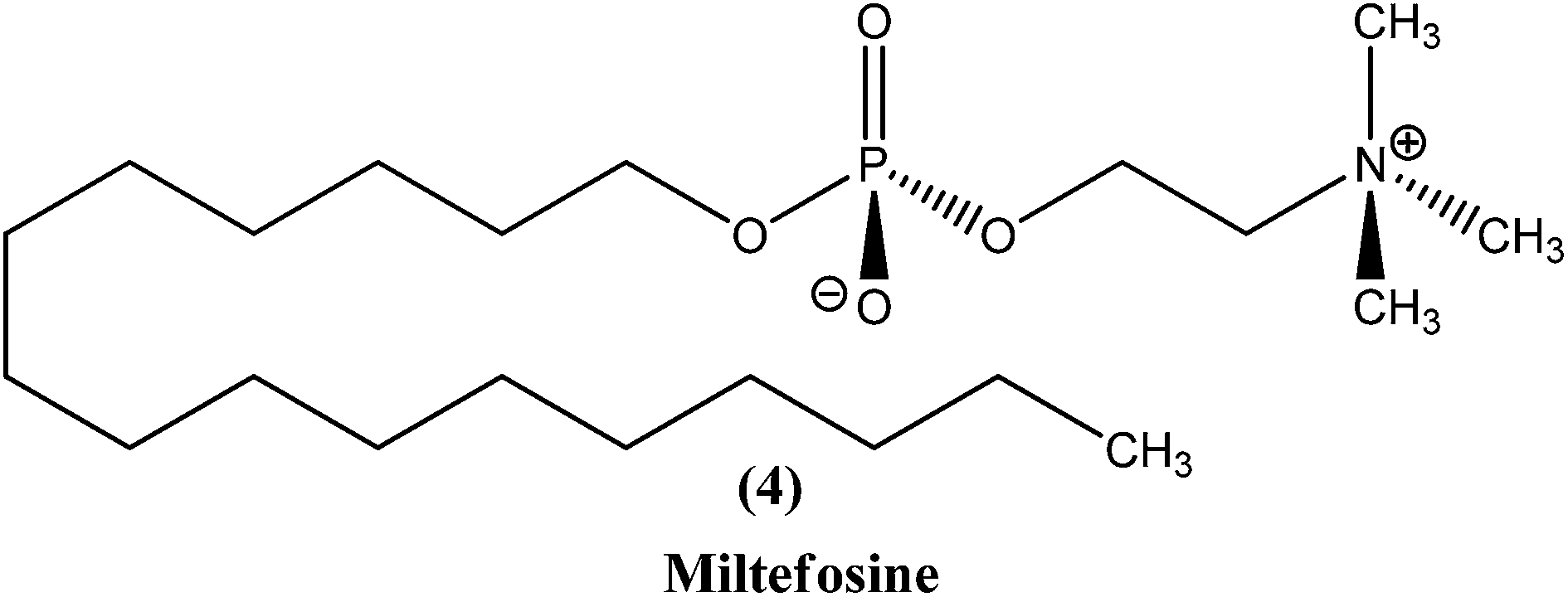
In a phase IV clinical trial of miltefosine in India, preliminary results on treatment with weekly supervision suggested that there was doubling of its relapse rate.63 It has less severe toxicity but long terminal residence time (approx. 152 h), and can cause teratogenicity, therefore its use should be avoided during pregnancy. Other adverse reactions of this drug include gastrointestinal disturbances, hepatotoxicity, and renal toxicity, but these can be reversed. Resistance for the drug was found to be established in laboratory strains.24,64 Also, some studies suggest that a few Leishmania species, such as L. braziliensis, L. guyanensis, and L. mexicana, are insensitive to miltefosine.65 There is potential for use of combination therapy of miltefosine with either paromomycin or amphotericin B, and this may even prove useful in treatment of antimonial-resistant VL patients in India.66
2.4. Paromomycin
Paromomycin (5), an aminoglycoside antibiotic, is a relatively new broad-spectrum antibiotic drug that has been used for treatment of leishmaniasis. It can be used for treatment of both types of leishmaniasis (VL as well as CL), although paromomycin is more effective for CL. Limited availability restricts its use in endemic regions.67,68 Paromomycin is available as an intramuscular injection for parenteral administration to treat systemic infections (i.e. VL), and as an ointment formulation to treat local skin infections (i.e. CL).69,70 It has been found to be effective in India, Kenya, and more recently in Tunisia, but was found to be less effective in Sudan and Colombia.71,72 It has also undergone phase IV clinical trials.73 A bacterial pathogen Streptomyces rimosus var. paromomycinus is the organism responsible for origination of paromomycin by a fermentation mechanism.74 Paromomycin is now an off-patent drug and is recognized as an Orphan Drug by the US FDA and EU EMEA.75 The drug is recommended by the WHO and was approved by the Indian government in August 2006 to treat VL-infected patients.76 It is inexpensive, but a dose of 16 mg kg−1 is required daily for about 21 days via an intramuscular route.77Although paromomycin acts specifically to treat other diseases, the mechanism specific to Leishmania requires further study and elucidation. However, it has been observed that in the cytoplasm as well as in the mitochondria of L. donovani, following low Mg2+ concentration-induced dissociation, ribosomal subunit association was promoted by paromomycin, resulting in inhibition of subunit recycling and, ultimately, inhibition of leishmanial protein synthesis.78 Other mechanisms may include alteration of membrane fluidity and lipid metabolism.79 Paromomycin can induce a local conformational change in the A-site of 16s rRNA and also respiratory dysfunction in L. donovani promastigotes.73,80 Some adverse reactions or side effects of paromomycin have been observed, including elevated hepatic transaminases, ototoxicity, pain at injection site, nausea, abdominal cramps, and diarrhea.76,81 Experimental evidence has shown that a laboratory strain of L. donovani promastigotes developed resistance to paromomycin.75

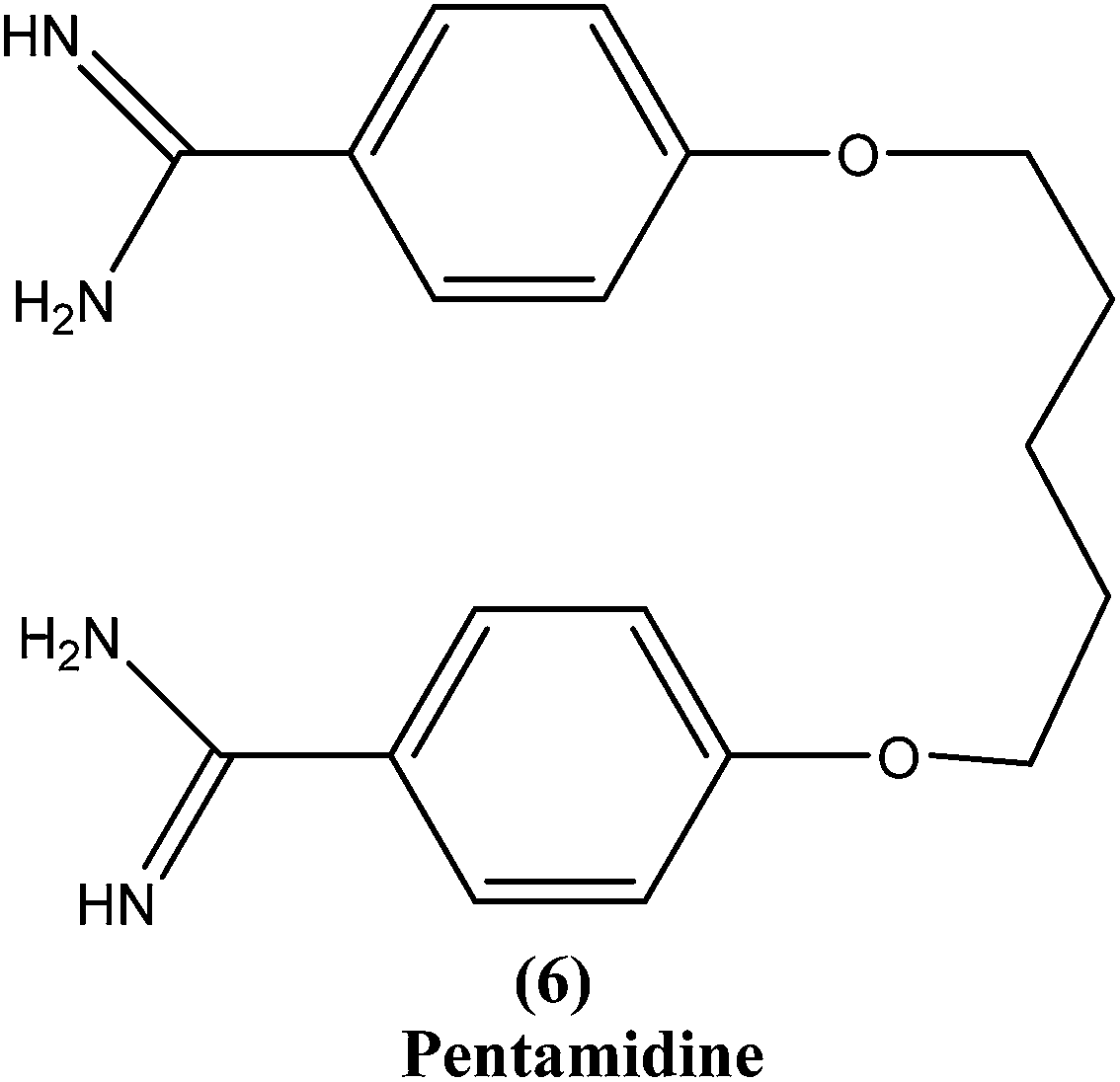
2.5. Pentamidine
Pentamidine (6) is an aromatic diamidine. This drug is used in antimonial resistance cases. It was originally used in treatment of VL via an intramuscular route, but because of increasing resistance and toxicity, its use was forfeited. It also has potential as a useful drug for maintenance treatment in immunocompromised hosts.82 Although its primary mechanism of action is unknown and yet to be explored, it is thought that the drug is accumulated in the parasite, with effects including binding to kinetoplast DNA. It also has been reported that the drug enters promastigotes through arginine and polyamine transporters and is accumulated in mitochondria, acting to inhibit mitochondrial topoisomerase II.1,83–86Treatment with pentamidine may cause myalgia, pain at the injection site, nausea, headache, and, less commonly, results in a metallic taste, a burning sensation, numbness and hypotension, irreversible insulin-dependent diabetes mellitus, and death.87 Increasing unresponsiveness in India, emergence of drug resistance, especially in HIV co-infections, and toxicity (reversible hypoglycemia and nephrotoxicity) are some other limitations to its usefulness.88
3. Drawbacks of current antileishmanial therapy
Although the range of antileishmanial drugs has expanded somewhat, currently available antileishmanial drugs do not meet the increasing requirements of managing infection in different patient populations, and drug resistance and toxicity have been reported with all the drugs (Table 1).75,89| Sr. no. | Current drugs | Toxicity | Resistance |
|---|---|---|---|
| 1 | Pentavalent antimonials | Pancreatitis, pancytopenia, reversible peripheral neuropathy, elevations in serum aminotransferases, pain at the site of injection, stiff joints, gastrointestinal problems, hepatic renal insufficiency (nephrotoxicity), cardiotoxicity, accumulate inside the tissues, such as particularly in liver and spleen, etc. | Resistance developed in Bihar state, India |
| 2 | Amphotericin B | Nephrotoxicity, fever with rigor and chills, bone pain, hypotension, anorexia, dyspnoea, thrombophlebitis, rarely cardiac arrest, myocarditis, and delayed side effects such as hypokalemia, etc. | Resistance in laboratory strains |
| 3 | Miltefosine | Gastrointestinal disturbances, hepatotoxicity, renal toxicity, etc. | Resistance in laboratory strains, L. braziliensis, L. guyanensis, and L. mexicana are insensitive towards miltefosine |
| 4 | Paromomycin | Elevated hepatic transaminases, ototoxicity, pain at injection site, nausea, abdominal cramps, diarrhoea, etc. | Laboratory strain of L. donovani promastigote developed resistance |
| 5 | Pentamidine | Myalgia, pain at the injection site, nausea, headache, and less commonly results in a metallic taste, a burning sensation, numbness and hypotension, irreversible insulin-dependent diabetes mellitus, and death | Unresponsiveness in India, emergence of drug resistance especially in HIV co-infections |
A wide range of biological assays of possible compounds have been performed with several strains and different parasite forms, but only a few have reached clinical trials.55 New antileishmanial drugs are urgently required to minimize adverse effects and overcome the increasing resistance to existing drugs, and also availability problems.
4. Recent progress in antileishmanial agents
In the last 10 years, various cores and their derivatives have been reported to possess antileishmanial activity (Fig. 3). | ||
| Fig. 3 Various cores reported as antileishmanial agents. | ||
4.1. Acridines
Acridine and its derivatives have been found to have ability for intercalation in DNA and to interfere with various metabolic processes of both eukaryotic and prokaryotic cells. In recent times, various acridine derivatives have been investigated for potential activity as antileishmanial agents.90,91 Giorgio et al. reported the synthesis and antileishmanial activity of 6-mono-substituted and 3,6-disubstituted acridines. Among the array of synthesized compounds, the most active compound, 3,6-disubstituted acridine (7), with benzoylamino groups at the third and sixth positions, demonstrated a strong affinity for both parasite forms. Its IC50 values were found to be about 1.1 ± 0.2 μM for amastigotes and 4.3 ± 1.2 μM for promastigotes, while it exhibited a lower antiproliferative activity against human monocytes (IC50 = 110.3 ± 15.2 μM) with a SI (selectivity/specificity index) value of 100.2, suggesting that acridine compounds could interact with protozoan and mammalian cells in different ways.92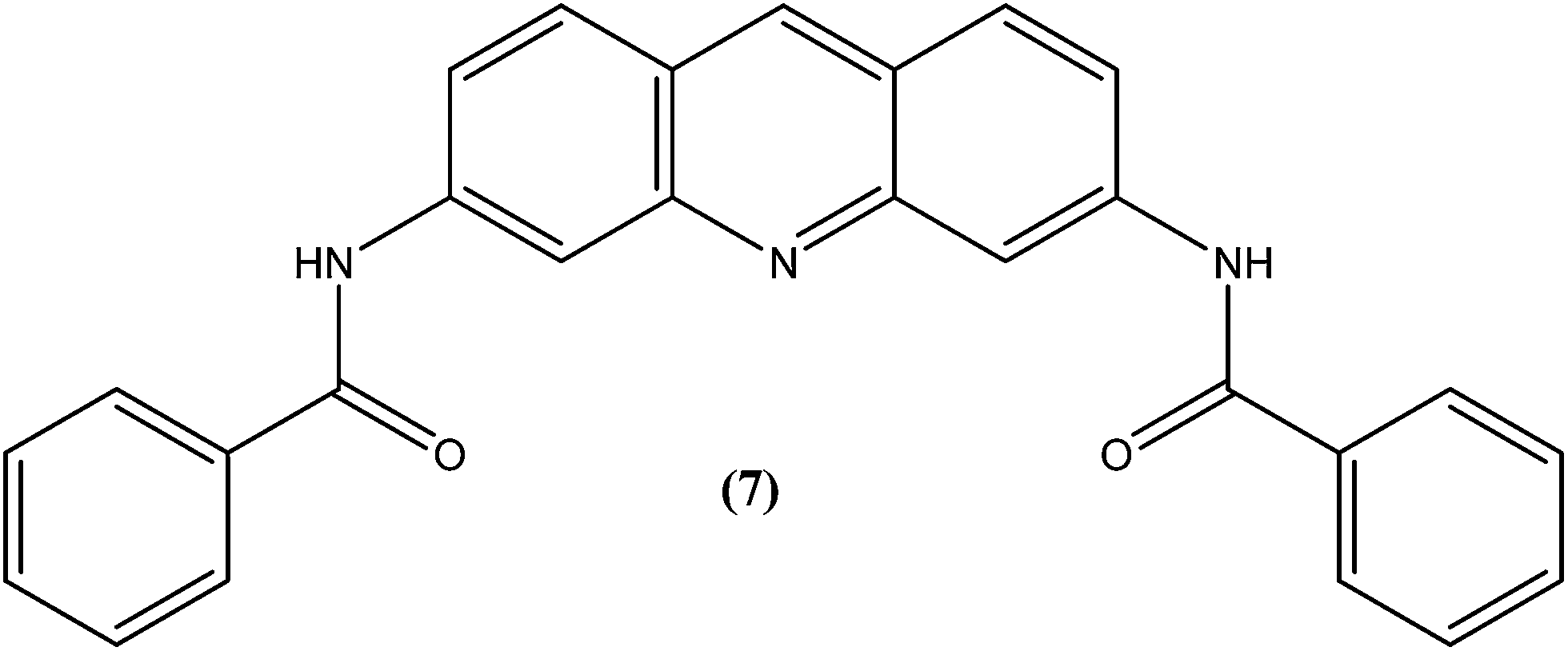
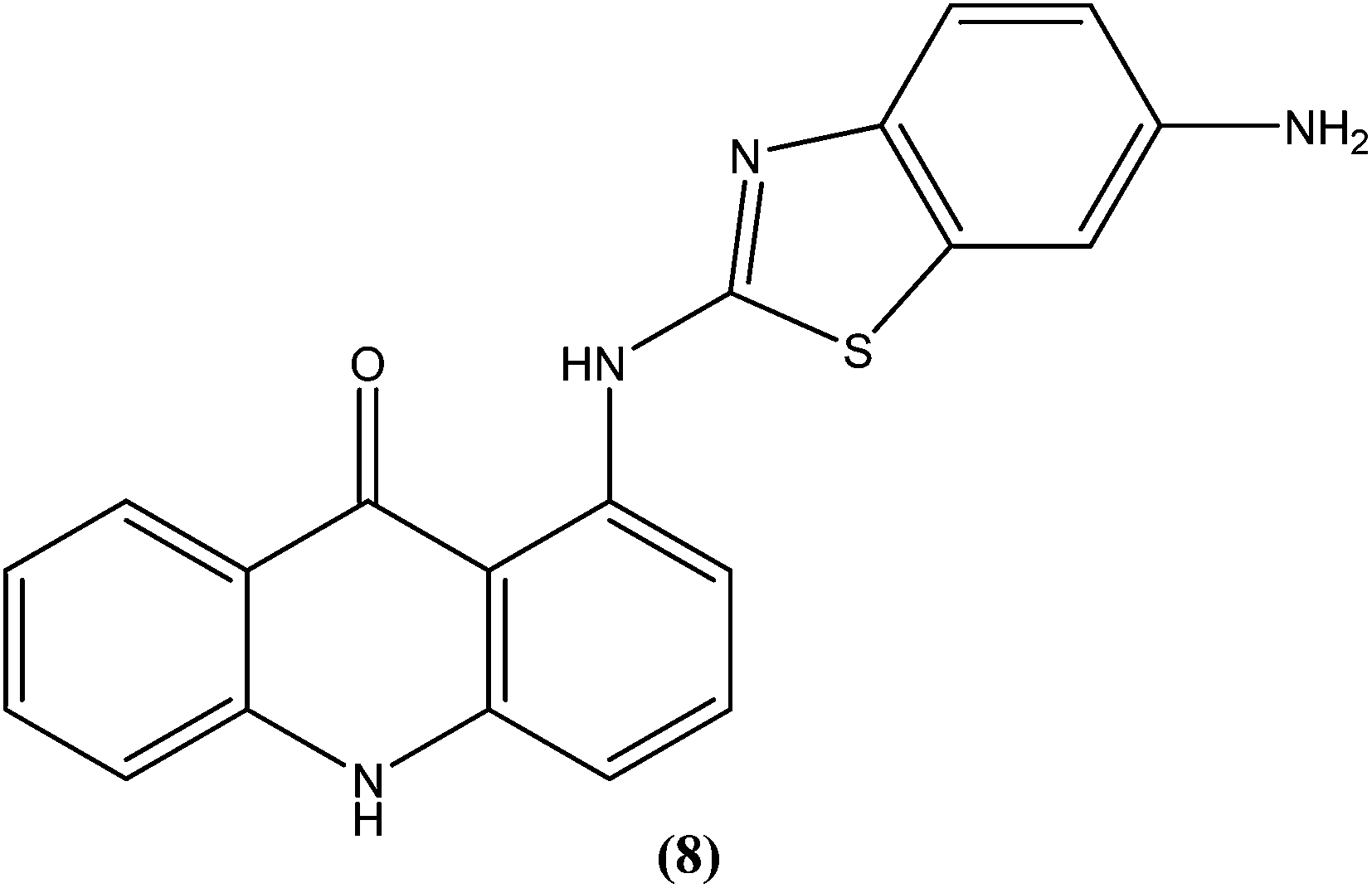
The same study group also reported synthesis of (1,3-benzothiazol-2-yl)amino-9-(10H)-acridinone derivatives as potent antileishmanial agents via a procedure based on the Ullman reaction. After evaluation for antileishmanial activity against L. infantum, compound (8) with 6-amino benzothiazole substitution (on the amino group of 1-(1,3-benzothiazol-2-yl)amino-9-(10H)-acridinone) revealed selective antileishmanial activity. It showed IC50 values of 20.1 μM against promastigote form, 4.3 μM against amastigote form, and 223.1 μM against human monocytes (lesser antiproliferative action) with a promising SI (selectivity index) of 51.9. While explaining its SAR, they reported that a benzothiazole group on a parent ‘amino-9-(10H)-acridinone’ ring could enhance antileishmanial abilities and the presence of a 6-amino-benzothiazole group at the second position of the amino chain was essential for specific anti-amastigote properties.93
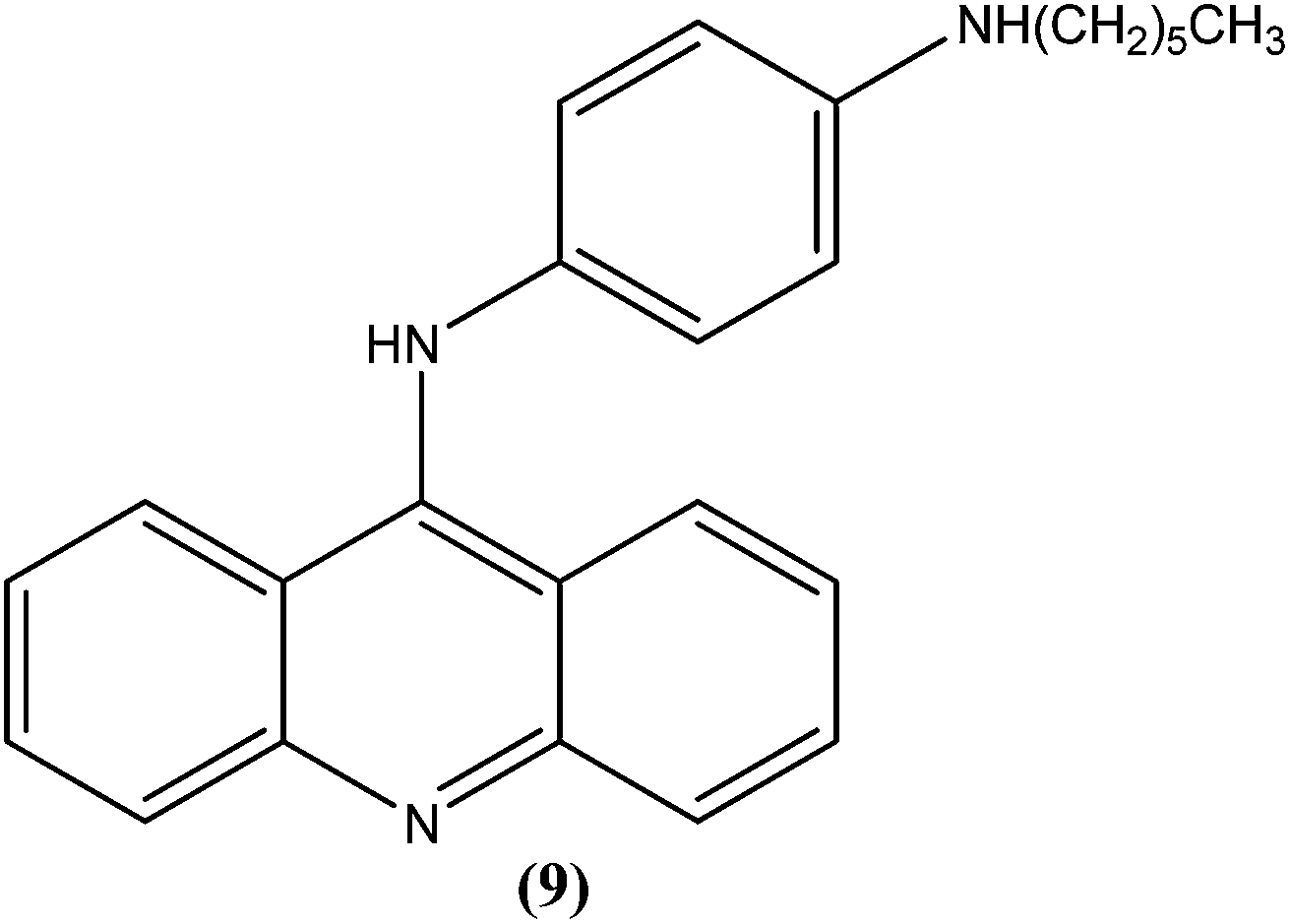
Ralph et al. synthesized several 9-anilinoacridine derivatives as antileishmanial agents. The compounds possessing 1′-NH-alkyl substituents have shown more than 80% growth inhibition of (macrophage infected) L. major amastigotes at or below a concentration of 1 μM. These compounds were also evaluated for their activities against promastigote and amastigote forms of L. major, and also for their toxicities to human Jurkat leukemia cells. Among the synthesized compounds, compound (9) was found to be one of the most active against intracellular parasites (>80% killing at 1 μM concentration). It had strong antileishmanial activity and also was found to be the least toxic compound to human Jurkat cells (IC50 = 17.0 μM). After evaluating the activity of these compounds, Ralph et al. suggested that it might be possible to modify existing anticancer drugs targeted at DNA topoisomerase II to improve their activities and specificities against other organisms, such as those causing leishmaniasis.94
4.2. Chromenes and coumarins
The 2H-chromene-2-ones, oxygenated heterocycles, are also known as natural coumarins.95 Naturally available derivatives of coumarins have been found to be effective against the promastigote form of the Leishmania parasite within a range of 17–50 μg mL−1 for IC50 determination.96–98 Therefore, coumarins have potential as antileishmanial agents. Dubey et al. synthesized a series of chromene-2-thione derivatives by molecular docking (into the active site of the trypanothione reductase (TryR) enzyme, which is required for redox balance of the parasite and the inhibition of which leads to parasite death) and evaluated their antileishmanial activity in vitro on promastigote, axenic amastigote, and intracellular amastigote forms of L. donovani. The derivatives showed high levels of antileishmanial activity together with minimal toxicity to human peripheral blood mononuclear cells compound (10) was one of the most active of the tested compounds, showing IC50 values of 36, 97, and 22 μM against axenic amastigote, promastigote, and intracellular amastigote forms, respectively, with % cytotoxicity to the human peripheral blood mononuclear cells observed at IC50 as 28.84 μM. In silico evaluation revealed that none of the synthesized ligands violated any criterion of the Lipinski rule of five, suggesting that these ligands have good potential for development as oral agents and as potentially active drug candidates.99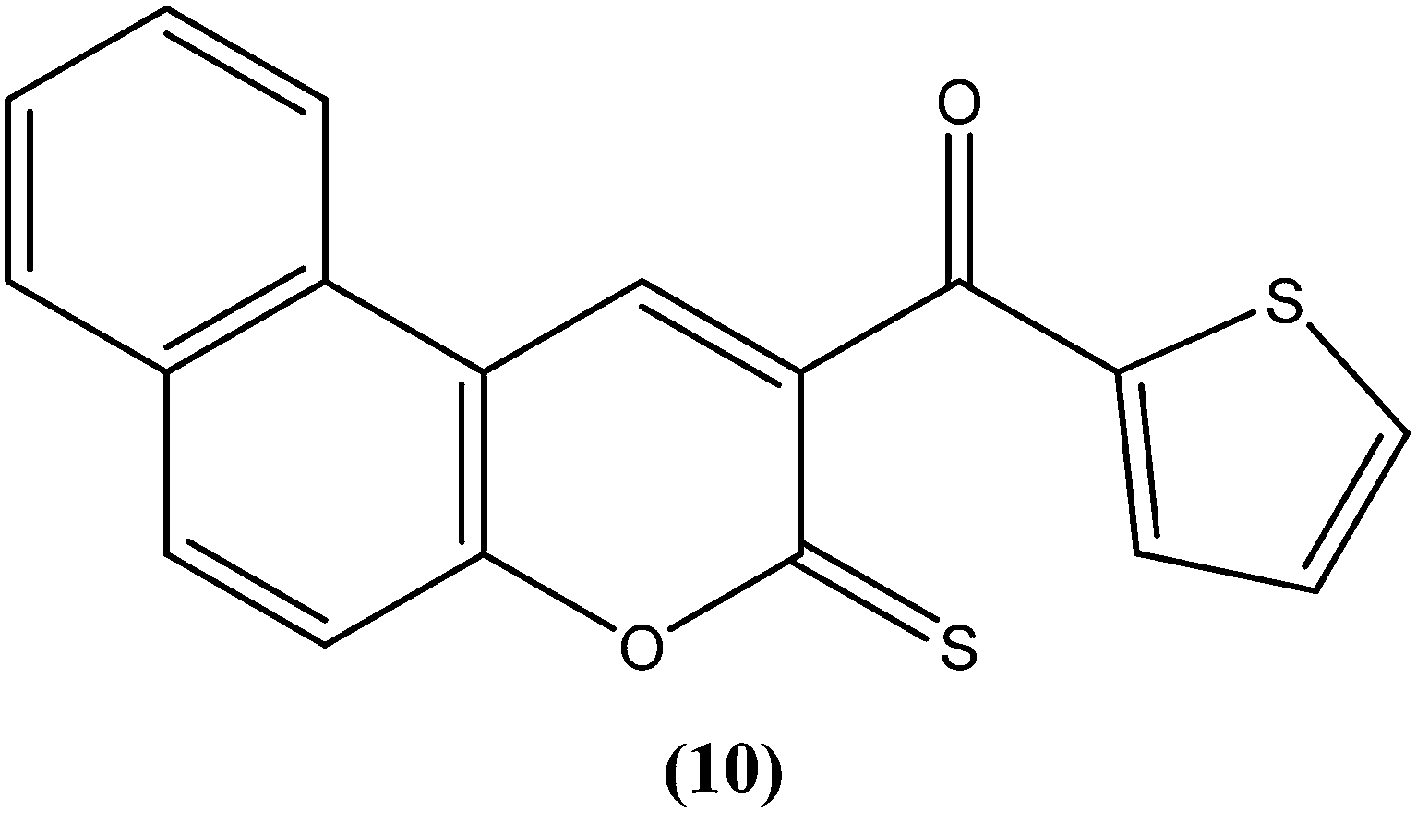
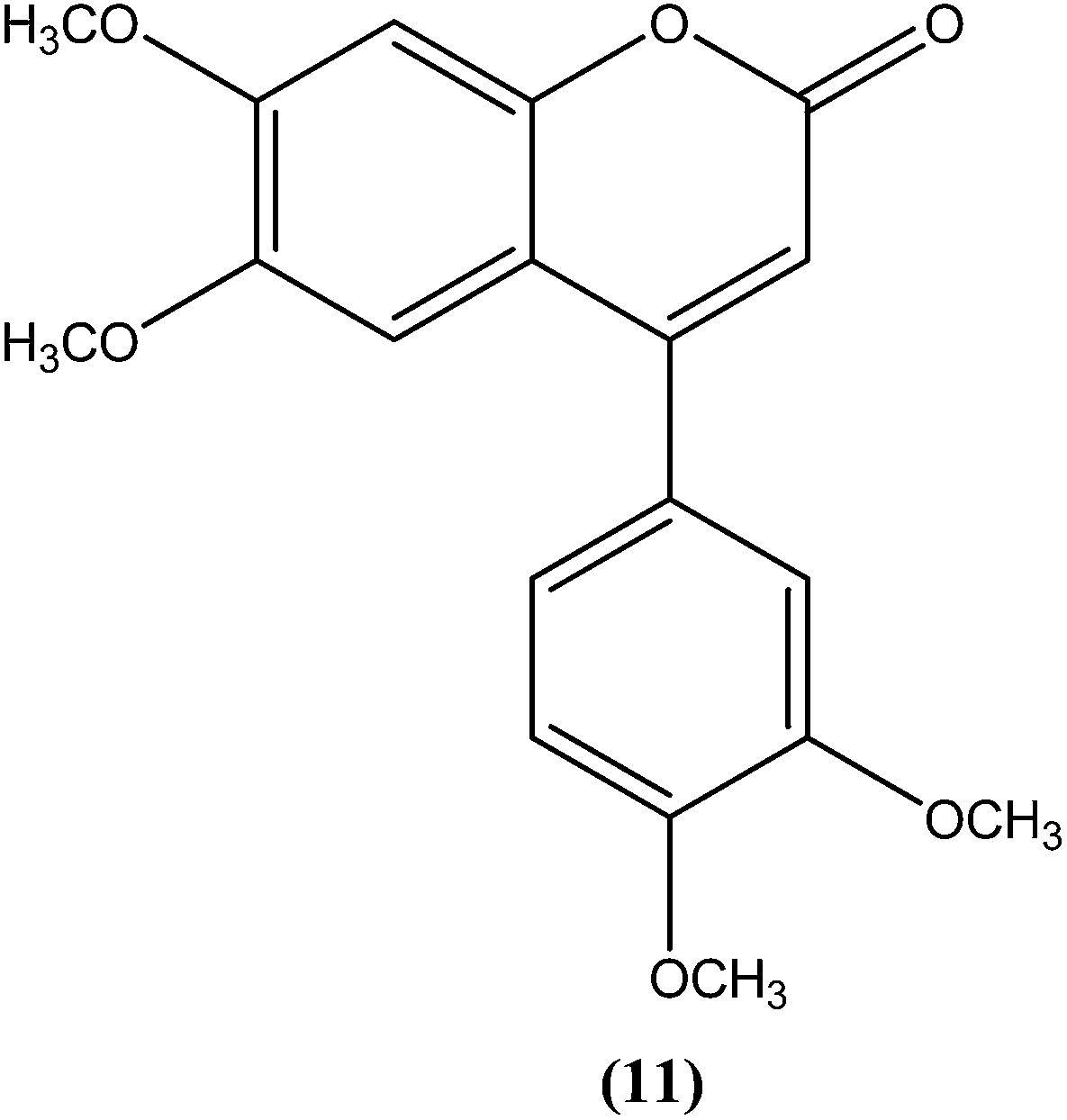
Combes et al. reported synthesis of a series of 4-arylcoumarins by a Suzuki–Miyaura cross-coupling reaction. On performing evaluation against L. donovani, 4-(3,4-dimethoxyphenyl)-6,7-dimethoxy-2H-chromene-2-one (11) exhibited potent activity against amastigote form (IC50 = 1.1 μM IC50 against human monocytes THP1: >292 μM) with a SI of 265, twice that of amphotericin B (SI = 140).100
4.3. Benzodiazepines
Mackay et al. prepared a series of synthetically amenable benzodiazepines and pyrrolobenzodiazepines structurally related to the paullone nucleus to probe for activity using a macrophage amastigote infection model. Paullones (12) can inhibit cyclin-dependent kinases, which completely inhibits growth of L. mexicana promastigotes in vitro.101 Mackay et al. reported that the tricyclic pyrrolobenzodiazepine-2,5-diones were more effective antileishmanial agents than sodium stibogluconate at the concentrations tested, with no evidence of toxicity against the host macrophage cells. Also the activity was found to be independent of chirality in the tricyclic 2,5-diones. The authors concluded that 7-chloro substituted 1,4-benzodiazepine-2,5-dione (13) had an amastigote suppression efficacy comparable with that of the clinically used sodium stibogluconate, and was non-toxic in the test model. It demonstrated efficacy at a concentration of 11.5 μM, whereas sodium stibogluconate had a plasma concentration of 1.47–2.95 μM after clinical dosing.102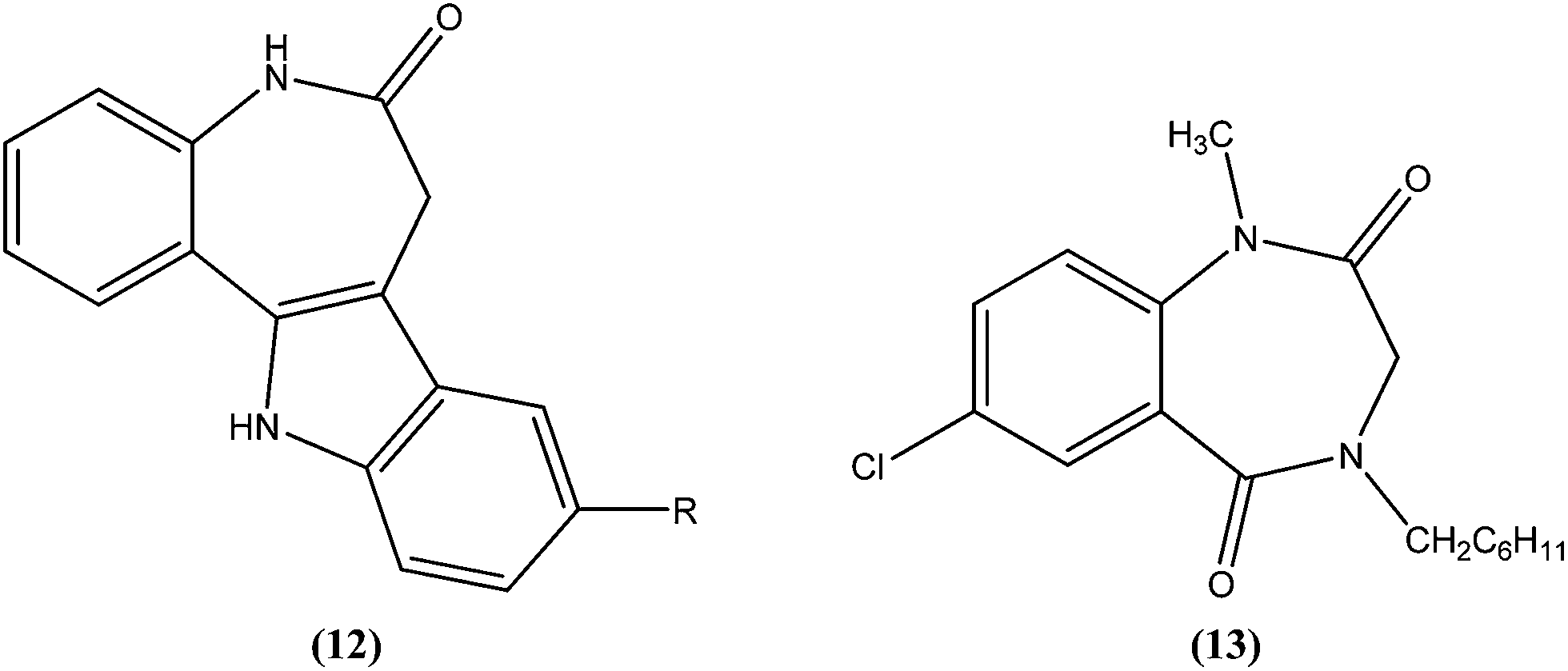
4.4. Chalcones
Chalcones are open chain flavonoids with two aromatic rings linked by a carbonyl group and two α,β-unsaturated carbon atoms.103–105 Kharazmi et al. reported that licochalcone A (14), an oxygenated chalcone, inhibits in vitro growth of both L. major and L. donovani promastigote form. Their preliminary studies showed that it destroyed the ultrastructure of the parasite's (promastigote form) mitochondria.106,107 Furthermore, while expanding their studies on the function of parasital mitochondria, they noted that licochalcone A inhibited respiration of the parasite in a concentration-dependent manner, as illustrated by inhibition of O2 consumption and CO2 production by the parasites. Moreover, licochalcone A inhibited the activity of the parasite mitochondrial dehydrogenase. These findings demonstrate that licochalcone A alters the ultrastructure and function of the mitochondria of Leishmania parasites.108 Again, further investigation on the mechanism of action of chalcones, focusing on the parasite's respiratory chain, showed that licochalcone A inhibited the activity of fumarate reductase (FRD) in a permeabilized L. major promastigote form as well as in the parasite mitochondria, and also inhibited solubilized FRD and purified FRD from L. donovani. This indicates that FRD, one of the enzymes of the parasite respiratory chain, might be the specific target for antiprotozoal chalcones. As FRD exists in the Leishmania parasite and does not exist in mammalian cells, it has potential as an excellent target for antiprotozoal drugs.109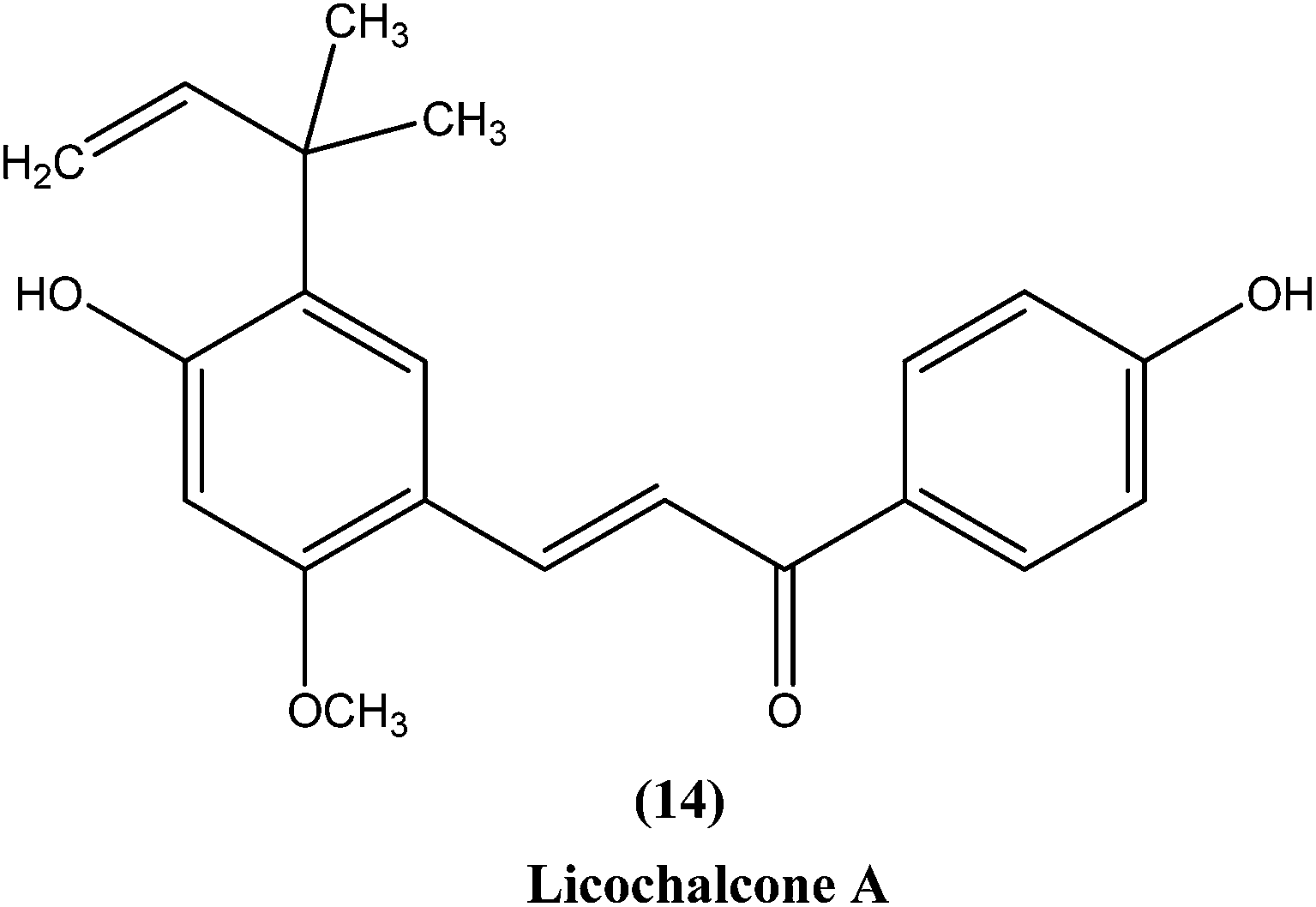
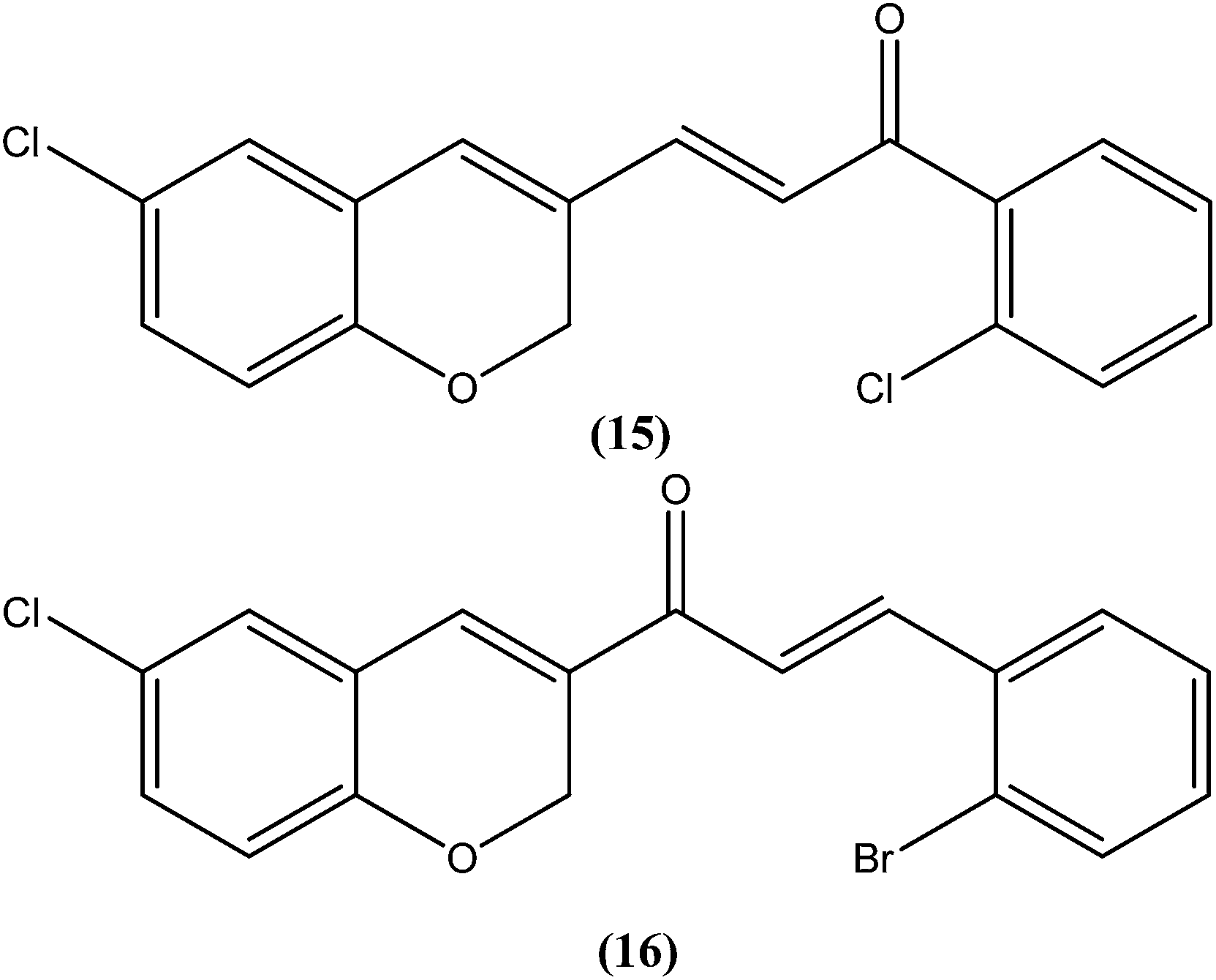
Foroumadi et al. prepared a series of novel chalconoids containing 6-chloro-2H-chromen-3-yl groups, and evaluated these against the promastigote form of L. major using MTT assay. All the compounds showed high antileishmanial activity in vitro at concentrations less than 3.0 μM. Cytotoxicity assessment against mouse peritoneal macrophage cells showed that the antileishmanial activity of these compounds was achieved at non-cytotoxic concentrations. The most potent compounds statistically were the compound containing a 2-chlorophenyl group (15) with IC50 value 1.22 ± 0.31 μM, and the compound containing a 2-bromophenyl group (16) with IC50 value 1.33 ± 0.52 μM.110
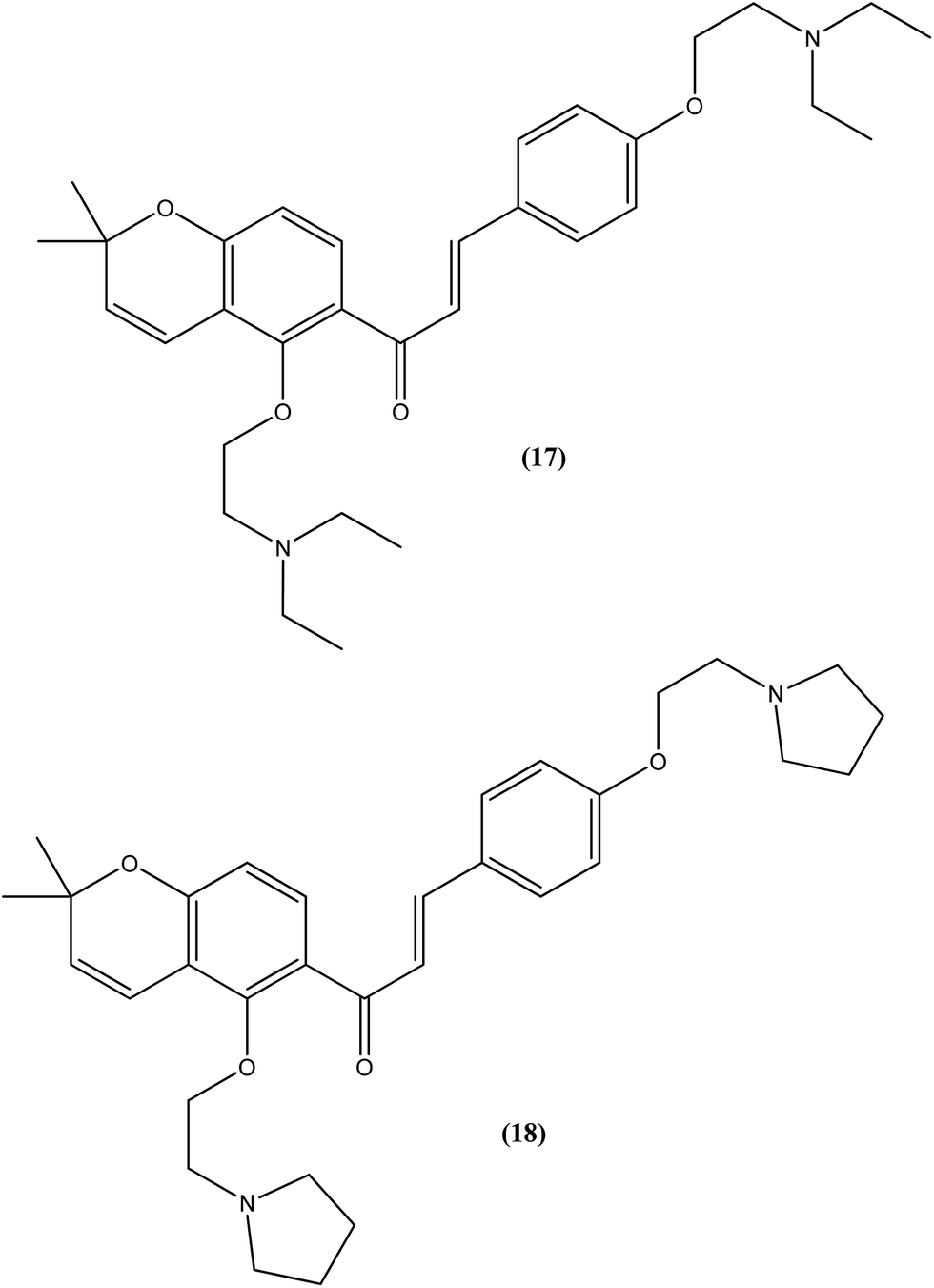
Tadigopulla et al. reported antileishmanial activities of chalcone derivatives. Compounds (17) and (18), with IC50 values of 2 and 2.5 μM, and CC50 values of 325.4 μM (SI = 162.5) and 258.1 μM (SI = 103.2), respectively, were found to be most potent against L. donovani amastigote form. These compounds also showed almost 100% inhibition of promastigote form of the same species at a concentration of 25 μM. Introduction of alkylated amino substituents on rings A and B increased the activity profile significantly from that of the parent compound (IC50 > 20 μM against amastigote form). Remarkably, these di-alkylated amino substituted analogues were also found to be more effective than standard drugs miltefosine (IC50 = 8.40 μM) and SSG (IC50 = 49.7 μM).111
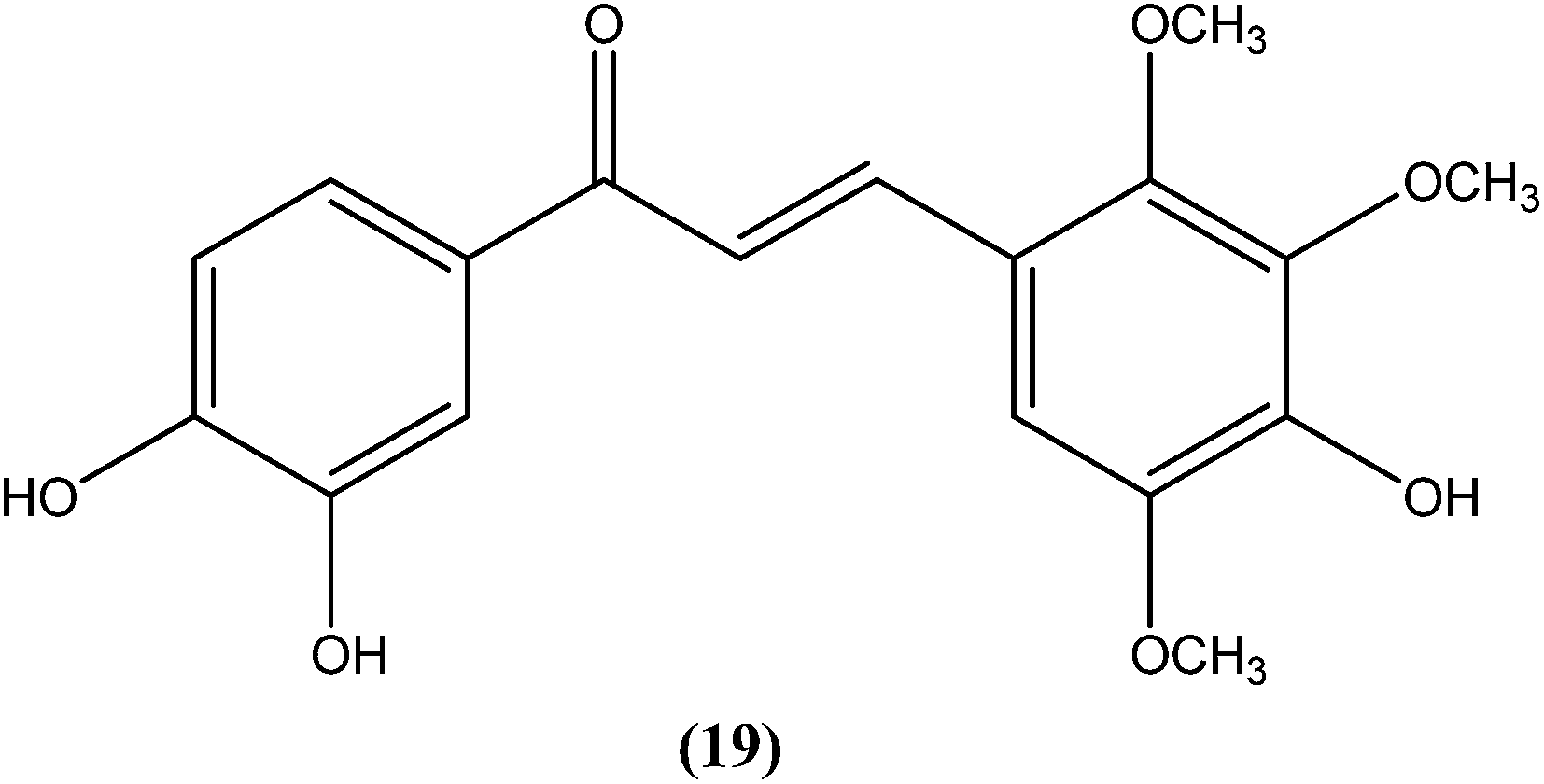
Nielsen et al. reported synthesis and evaluation of a large number of substituted chalcones with antileishmanial activities against L. donovani promastigote. Among the synthesized compounds, the IC50 value of the most potent chalcone (19) was 3.7 μM.112
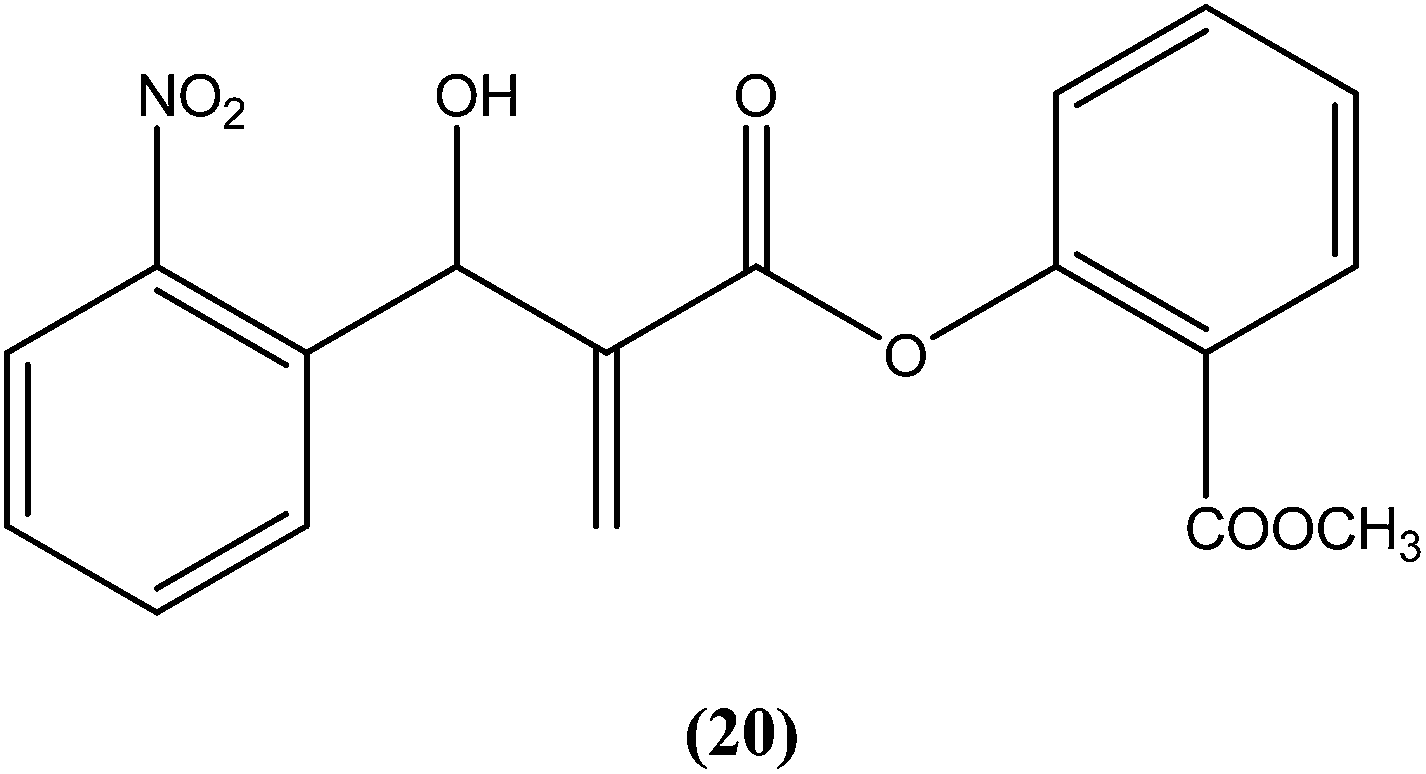
Vasconcellos et al. synthesized a new chalcone-like series using the Morita–Baylis–Hillman reaction, and reported that compound (20) bearing an o-nitro group had the most potent leishmanicidal activity, with IC50 values of 7.65 and 10.14 μM on L. amazonensis and L. chagasi, respectively.113
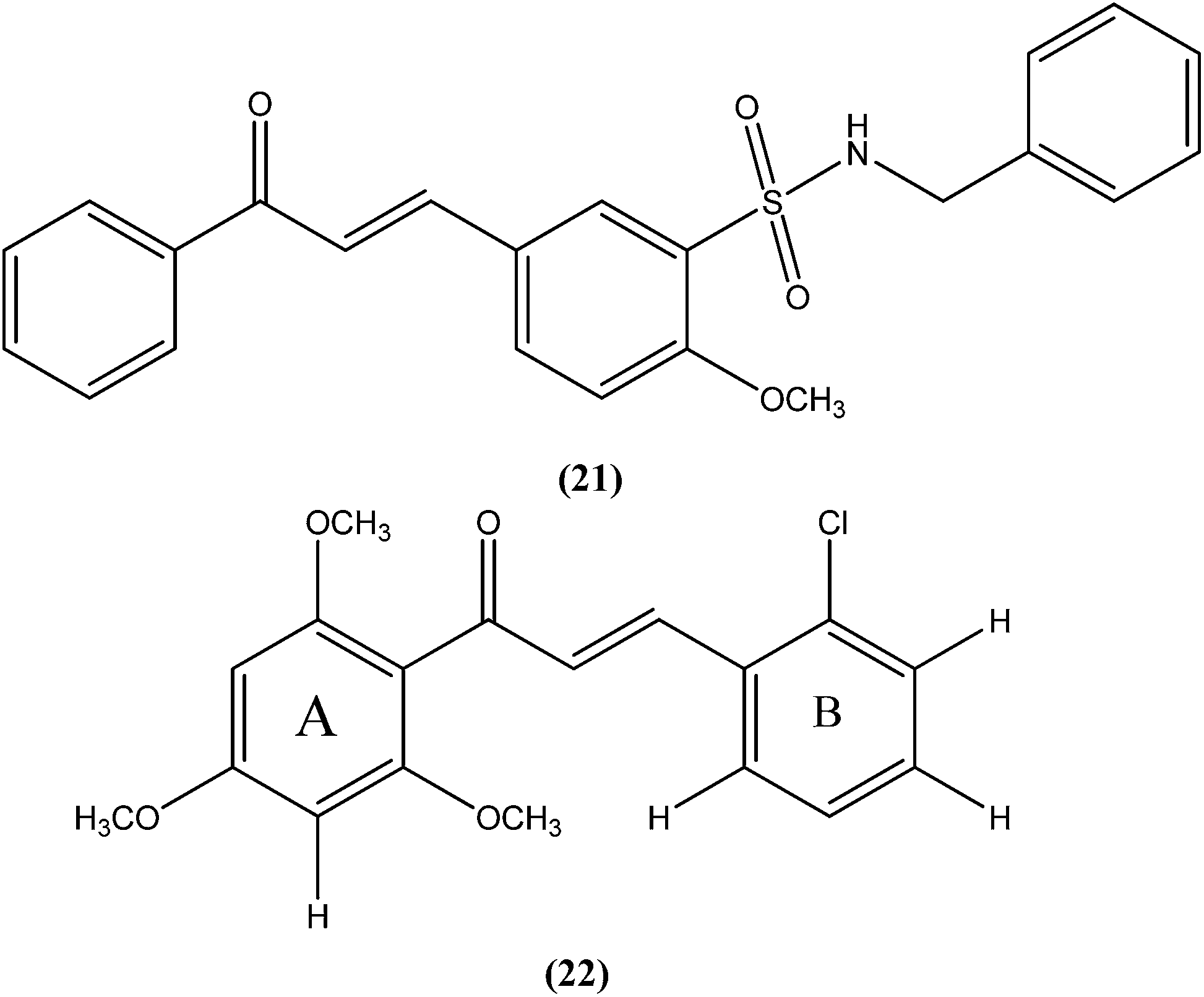
Nunes et al. reported the effects of a new set of sulfonamide 4-methoxychalcones against the promastigote form of L. braziliensis, with compound (21) presenting the best antileishmanial profile (IC50 = 3.5 ± 0.6 μM).114 The same study group also reported the synthesis, antileishmanial activity, molecular modeling, and structure–activity relationship (SAR) evaluation of a series of novel chalcone derivatives based on a 1,3-diacetyl biphenyl nucleus without sulfonamide group. After evaluation, the most active compound (22) showed reduced toxicity level in a Vero cell assay (CC50 = 216 μM) with low IC50 (3.9 μM), and the best SI of 55.4 against the promastigote form of L. braziliensis. The position of the methoxy group in phenyl ring A (especially di-ortho-substituents) and the chlorine atom in phenyl ring B (compound 22) seem to be important for antileishmanial activity. This compound fulfilled the Lipinski rule of five, identifying it as a hit compound for further exploration with potential in design of new candidates for leishmaniasis treatment.115
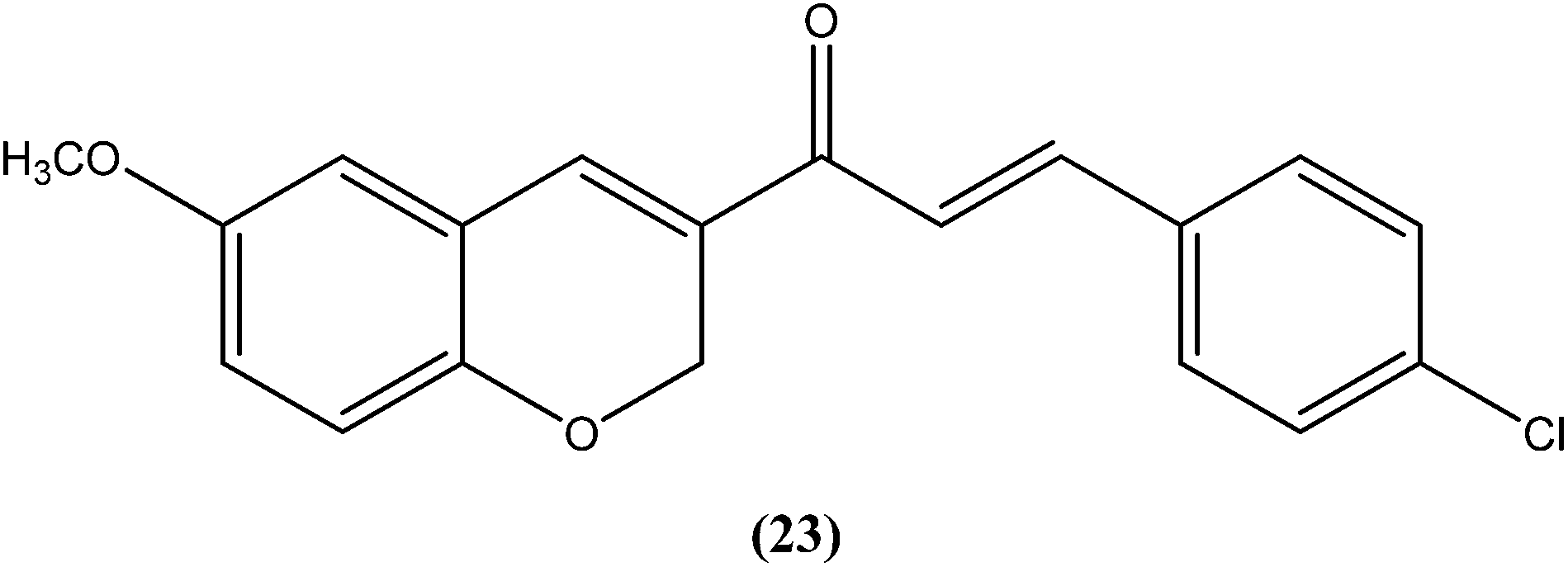
Shafiee et al. prepared two regioisomeric chromene-based chalcones and investigated these for antileishmanial activity against the promastigote form of L. major. The chloro-substituted 1-(6-methoxy-2H-chromen-3-yl)-3-phenylpropen-1-one showed excellent activity at non-cytotoxic concentrations. The compound (23) with chloro substitution at the para position of the phenyl ring was found to be most potent, having an IC50 of 0.7 ± 0.3 μM.116
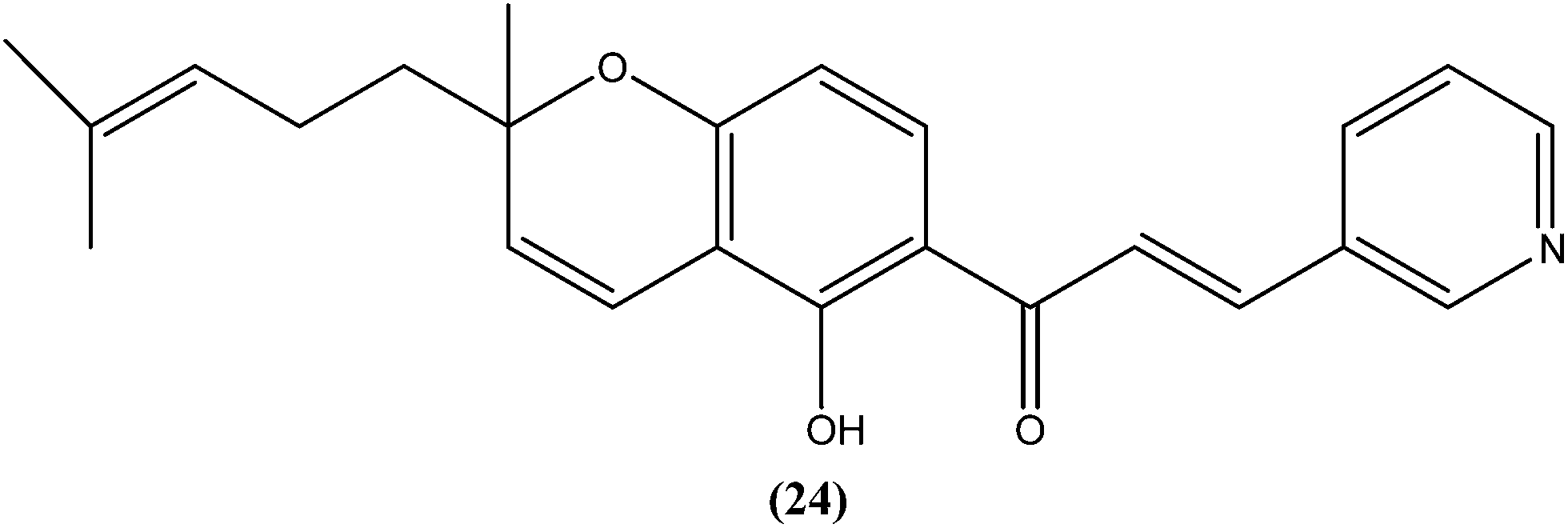
Gupta et al. reported synthesis, structure–activity relationships, and biological studies of chromenochalcones as potential antileishmanial agents. From the compounds that exhibited better activity than the marketed drug miltefosine against the intracellular amastigote form of L. donovani, a potent compound (24) showed IC50 = 0.78 and 5.4 μM against promastigote and amastigote forms, respectively, with a CC50 value of 40.07 μM on mammalian kidney fibroblast cells (Vero cell lines) and SI value of 7.5 compared with miltefosine (IC50 = 8.4 μM, CC50 = 52.5 μM with SI of 6.2). Oral administration of compound (24) in a hamster model, at a concentration of 100 mg per kg of body weight per day for 5 consecutive days, resulted in >84% parasite inhibition at day 7 post treatment and activity was retained until day 28. Molecular and immunological studies revealed that compound (24) has a dual nature to act as a direct parasite killing agent and as a host immune stimulant. Pharmacokinetics and serum albumin binding studies suggested that compound (24) has potential as a candidate for treatment of the non-healing form of leishmaniasis.117
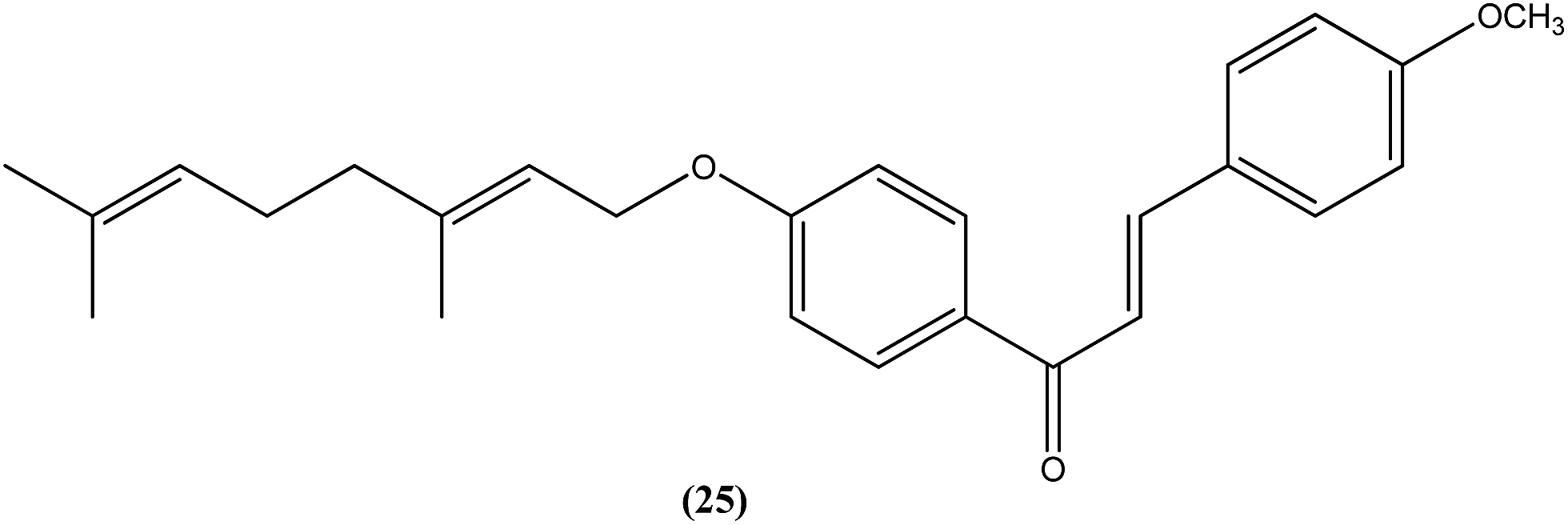
Gupta et al. also reported the synthesis and biological evaluation of chalcones as potential antileishmanial agents. The synthesized compounds exhibited potent activity in a concentration range of 1.70–8.0 μM against extracellular promastigote and intracellular amastigote forms of L. donovani. Compound (25) with IC50 = 3.1 μM against the amastigote form and CC50 = 146.5 μM on Vero cell line, showed 83.32% parasite inhibition in vivo after a dose of 50 mg kg−1 for 10 days, and 75.89% parasite inhibition in vivo after a dose of 100 mg kg−1 for 5 days by intraperitoneal route, at day 7 post treatment when tested in a hamster model.118
4.5. Indole
Vishwakarma et al. developed an efficient protocol for synthesis of 3,3′-diindolylmethane. They observed that all synthesized 3,3′-diindolylmethanes had promising antileishmanial activity against L. donovani promastigotes as well as axenic amastigotes. Of the synthesized compounds, the nitroaryl substituted diindolylmethanes showed potent antileishmanial activity, with the most potent being compound (26). Compound (26), which contains a 4-nitrophenyl moiety linked to 3,3′-diindolylmethane, showed IC50 values of 7.88 and 8.37 μM against L. donovani promastigote and amastigote forms, respectively.119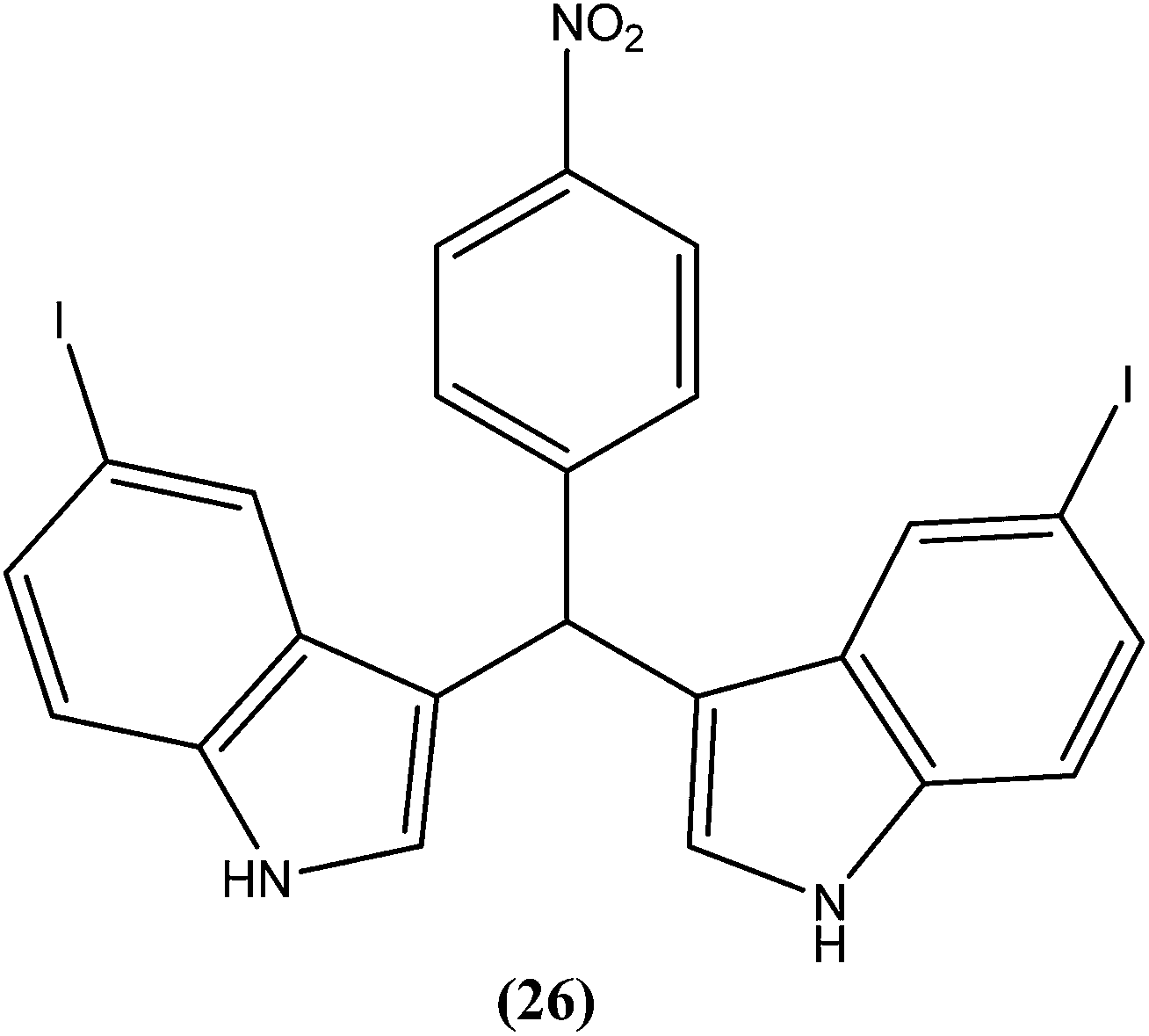
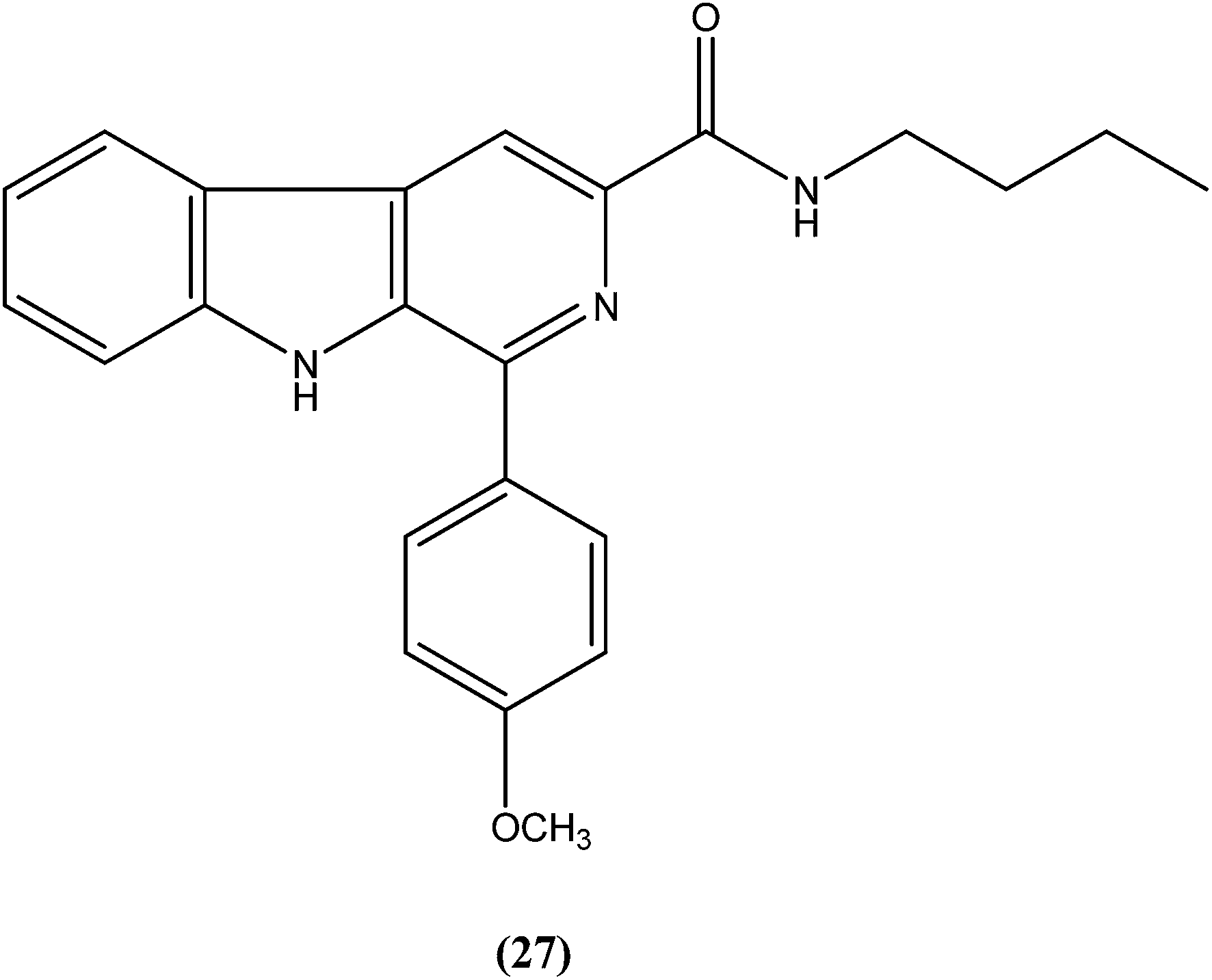
Sarragiotto et al. synthesized and evaluated a series of 1-phenylsubstituted β-carbolines containing an N-butylcarboxamide group at C-3 of the β-carboline nucleus. After in vitro evaluation of this series of compounds against the promastigote form of L. amazonensis, compound (27) with 4-methoxy phenyl as a substituent was found to be most active drug candidate with an IC50 of 0.25 ± 0.07 μM, and to have the lowest cytotoxicity to macrophages (CC50 = 521.0 ± 6.36 μM). The selectivity index ratio (SI) was 2084.120
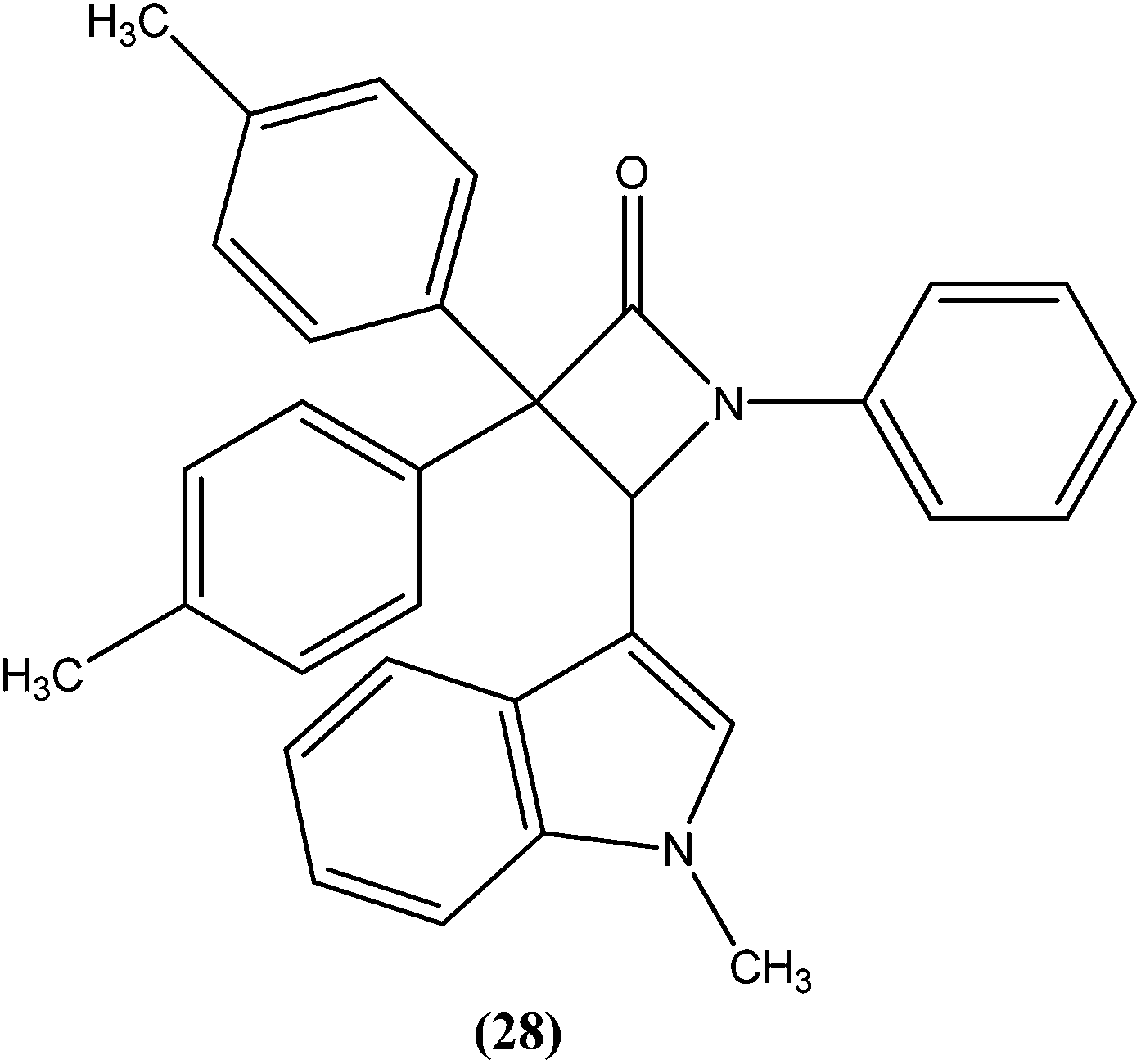
Singh et al. reported synthesis of N-(1-methyl-1H-indol-3-yl)methyleneamines and 3,3-diaryl-4-(1-methyl-1H-indol-3-yl)azetidin-2-ones as potential antileishmanial agents. After screening for antileishmanial activity against L. major, the most potent compound (28) was shown to have an IC50 value of 0.122 μM, similar to that of the standard drug amphotericin B (IC50 = 0.06 μM).121
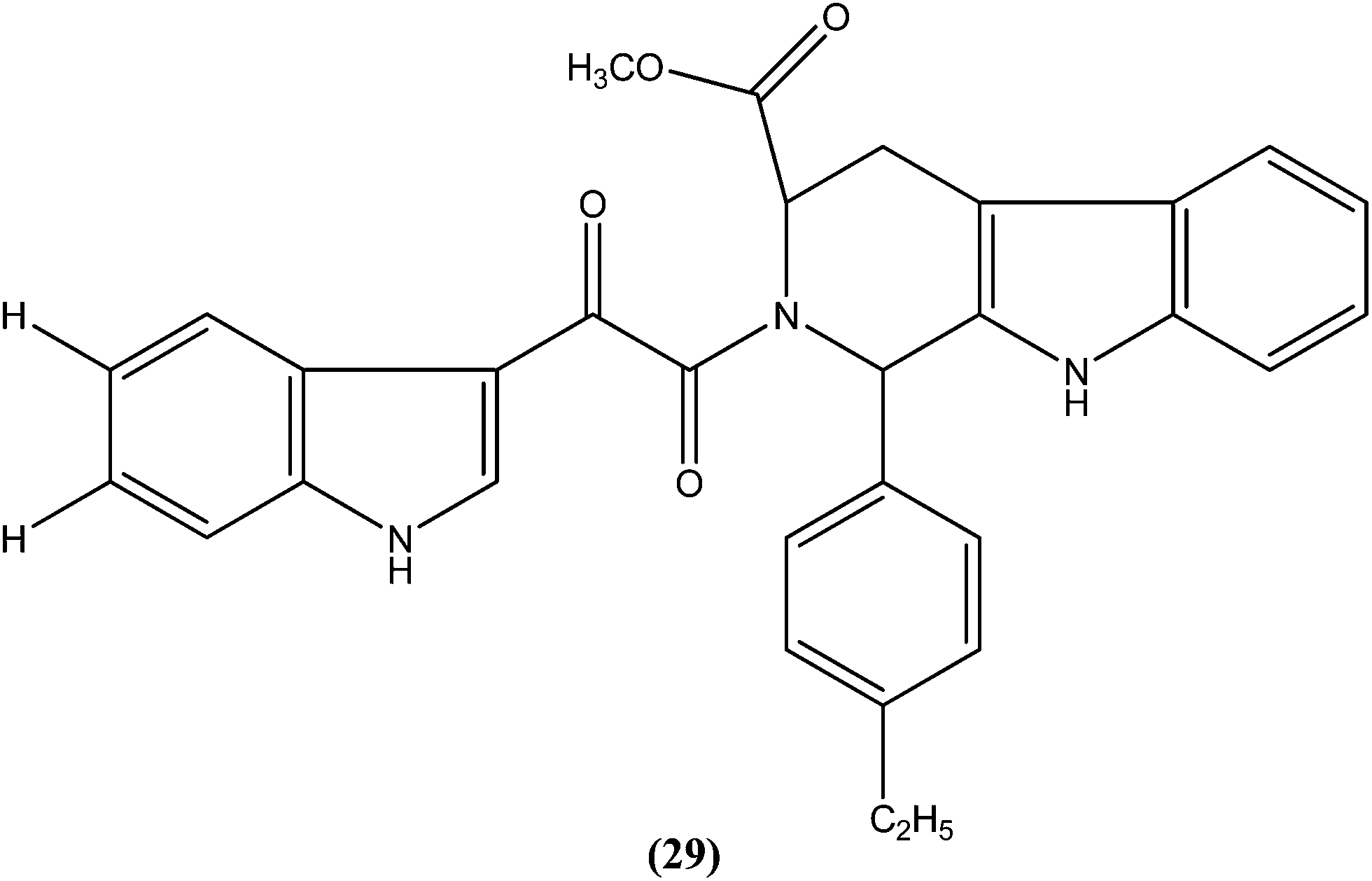
Chauhan et al. synthesized a series of indolyl glyoxylamides and evaluated their in vitro activity against the amastigote form of L. donovani. Compound (29) with a para-ethylphenyl ring on tetrahydro-β-carboline was identified as the most active analog of the series, with IC50 and CC50 values of 5.17 μM and 162.76 μM, respectively (SI = 31.48). This lead molecule was also found to be 12- and 5-fold more selective than the standard drugs pentamidine (IC50 = 20.43 μM, SI = 2.58) and sodium stibogluconate (IC50 = 71.90 μM, SI = 5.53), respectively.122
4.6. Furan
A systematic lead discovery program was employed and evaluated for in vitro and in vivo antileishmanial activities, mutagenicities, and toxicities of two novel AIAs (arylimidamides/bis-arylimidamides), DB745 and DB766 (30a). In intracellular Leishmania assays, compound (30a), which has unsymmetrical substitutions on the diphenylfuran linker, was found to be substantially more potent than miltefosine, paromomycin, and pentamidine, and similar in potency to amphotericin B (Table 2). After assessment for activity using J774 macrophages infected with clinical isolates of antimony-resistant L. donovani, IC50 values were found to range from 0.064 to 0.090 μM. An Ames screening assay was performed and it was found that compound (30a) did not exhibit any mutagenicity. Furthermore, in vivo analysis showed that compound (30a) when given orally, produced dose-dependent inhibition of liver parasitemia in two efficacy models, L. donovani-infected mice and hamsters. Most notably, compound (30a) (100 mg per kg of body weight per day for 5 days) reduced liver parasitemia in mice and hamsters by 71% and 89%, respectively. Werbovetz et al. have synthesized analogs of this antileishmanial lead (30a), along with an additional compound containing isopropoxy groups meta to the central furan. After in vitro evaluation of all the prepared compounds against intracellular amastigote forms of L. donovani and L. amazonensis, the target compounds displayed IC50 values in the nanomolar range with selectivity indices >100 compared with J774 macrophages. Compound (30b) bearing a meta-isopropoxy group was found to be the most potent (Table 2), with an IC50 of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 1000 nM against J774 macrophages and an approximate SI value of 2075. This compound was well tolerated by mice and showed activity in a murine model of visceral leishmaniasis; however, the unsymmetrical analogues were found to be toxic in nature.123
000 ± 1000 nM against J774 macrophages and an approximate SI value of 2075. This compound was well tolerated by mice and showed activity in a murine model of visceral leishmaniasis; however, the unsymmetrical analogues were found to be toxic in nature.123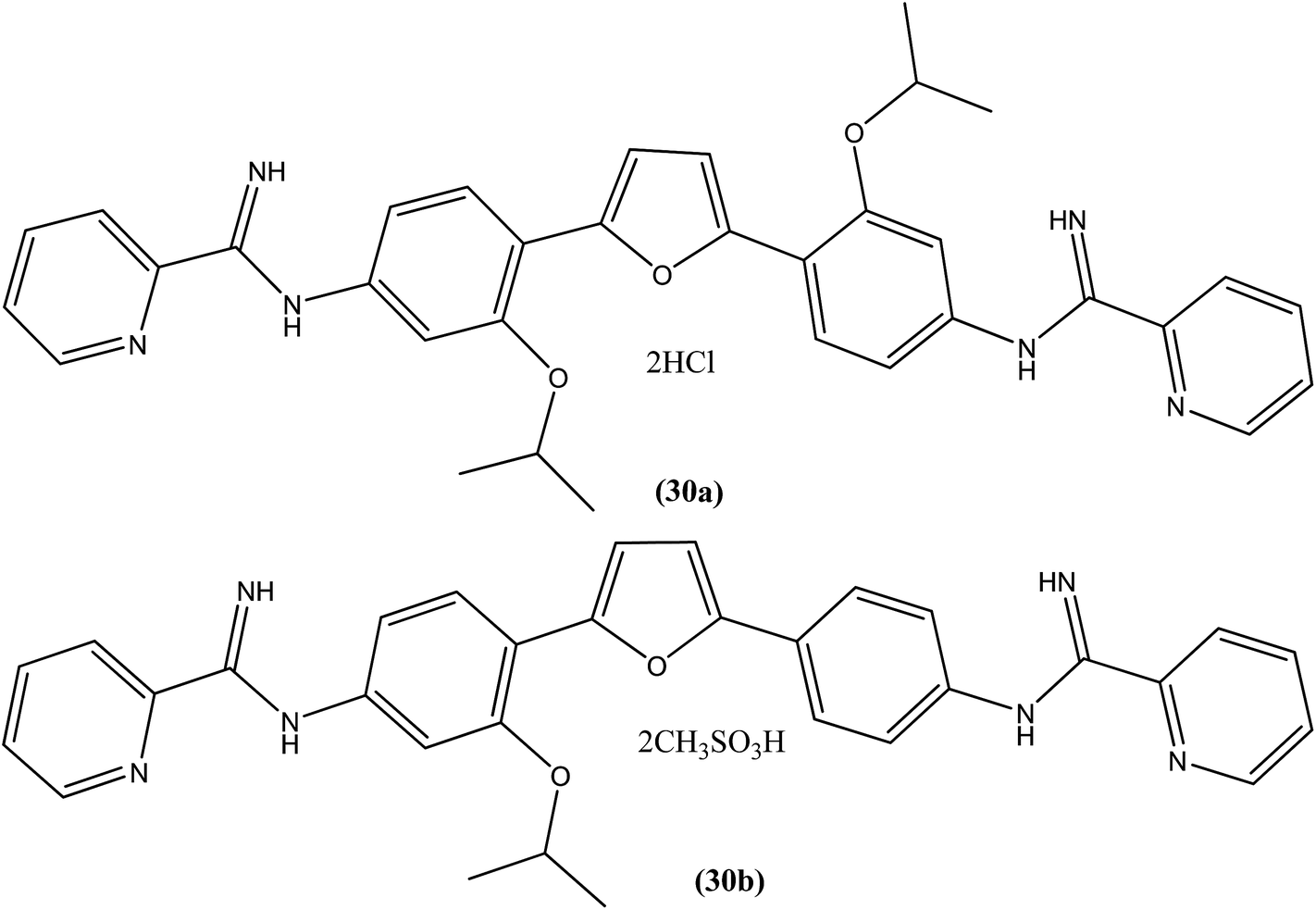
| Sr. no. | Compounds | L. donovani axenic amastigotes (IC50 in μM) | L. donovani (LV82) intracellular amastigotes (IC50 in μM) | L. amazonensis intracellular amastigotes (IC50 in μM) | L. major intracellular amastigotes (IC50 in μM) |
|---|---|---|---|---|---|
| a (—) indicates not done. | |||||
| 1 | DB766 (30a) | 0.50 ± 0.10 | 0.036 ± 0.005 | 0.087 ± 0.015 | 0.014 ± 0.004 |
| 2 | Compound (30b) | 5.3 ± 1.2 | — | 93 ± 28 | — |
| 3 | Miltefosine | 8.5 ± 1.2 | 2.7 ± 0.3 | 15 ± 3 | 25 ± 3 |
| 4 | Paromomycin | >50 | >50 | 19 ± 3 | 25 ± 2 |
| 5 | Pentamidine | 1.8 ± 0.4 | >50 | 0.83 ± 0.17 | — |
| 6 | Amphotericin B | 0.098 ± 0.013 | 0.066 ± 0.012 | 0.14 ± 0.01 | 0.21 ± 0.05 |
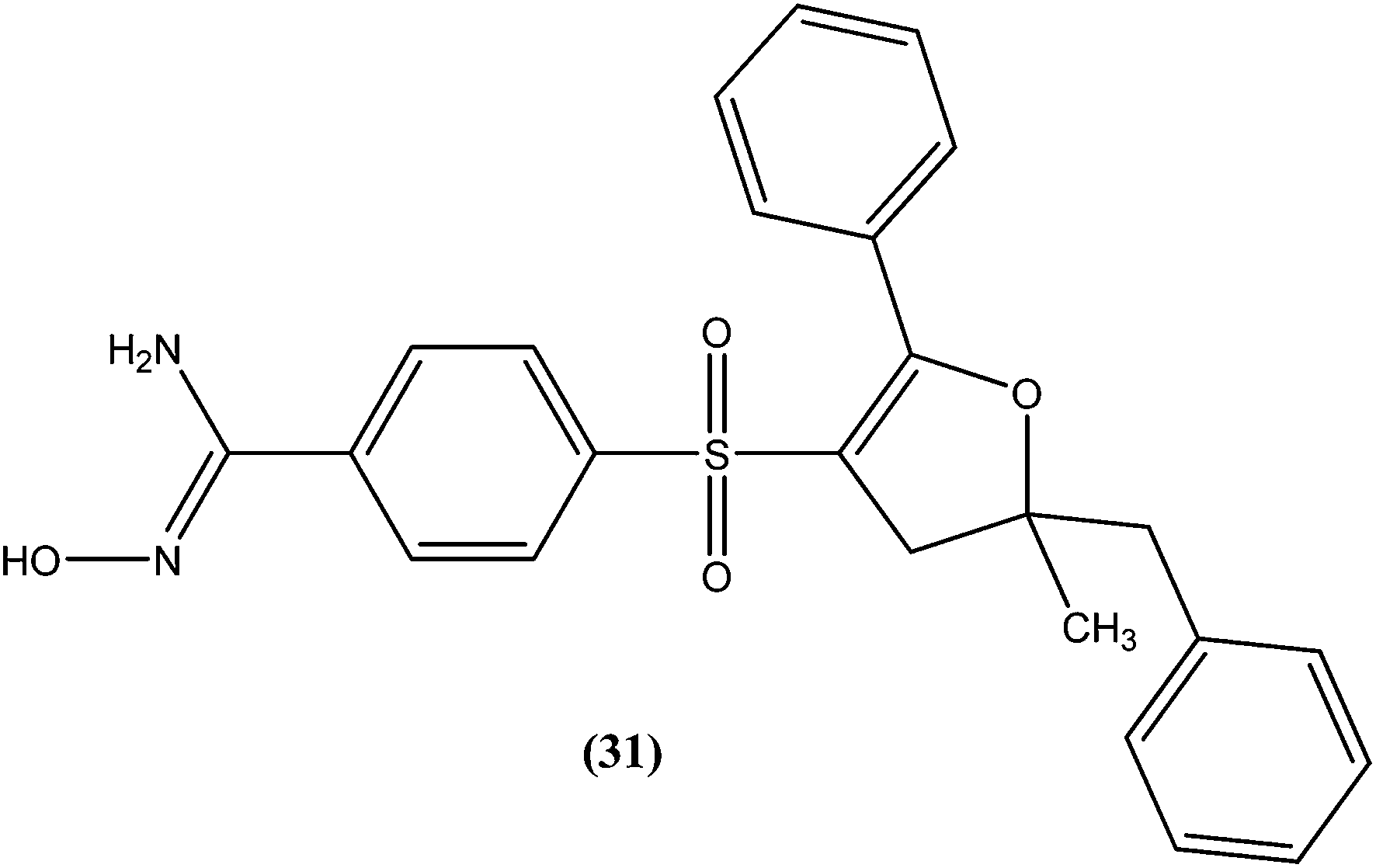
A series of amidoxime derivatives was synthesized by Vanelle et al. Among the tested compounds, compound (31), a monoamidoxime derivative with the lowest IC50 of 8.3 μM, showed better antileishmanial activity than that of pentamidine (IC50 = 11.2 μM), and had the highest SI of 6.6 (SI of pentamidine = 2.8) against L. donovani promastigote form. It was suggested that a single amidoxime group appears to be sufficient for antileishmanial activity.124
4.7. Pyridine
Tripathi et al. reported synthesis of 1-phenyl-4-glycosyl-dihydropyridines by a one-pot multicomponent reaction. The compounds were screened in vitro and in vivo for their antileishmanial activities, with most exhibiting moderate to good activity against amastigote and promastigote forms of L. donovani. From the screened compounds, the most active compound (32) exhibited in vitro IC50 values of 0.04 and 1.16 μM against promastigote and amastigote forms, respectively, with a CC50 value of 9.35 μM and SI of 8.04. In vivo administration of compound (32) showed 49.73 ± 12.0% inhibition against L. donovani in Hamster model intracellular. Molecular docking studies with these compounds revealed L. donovani PTR1 (pteridine reductase 1) as the possible target for antileishmanial activities.125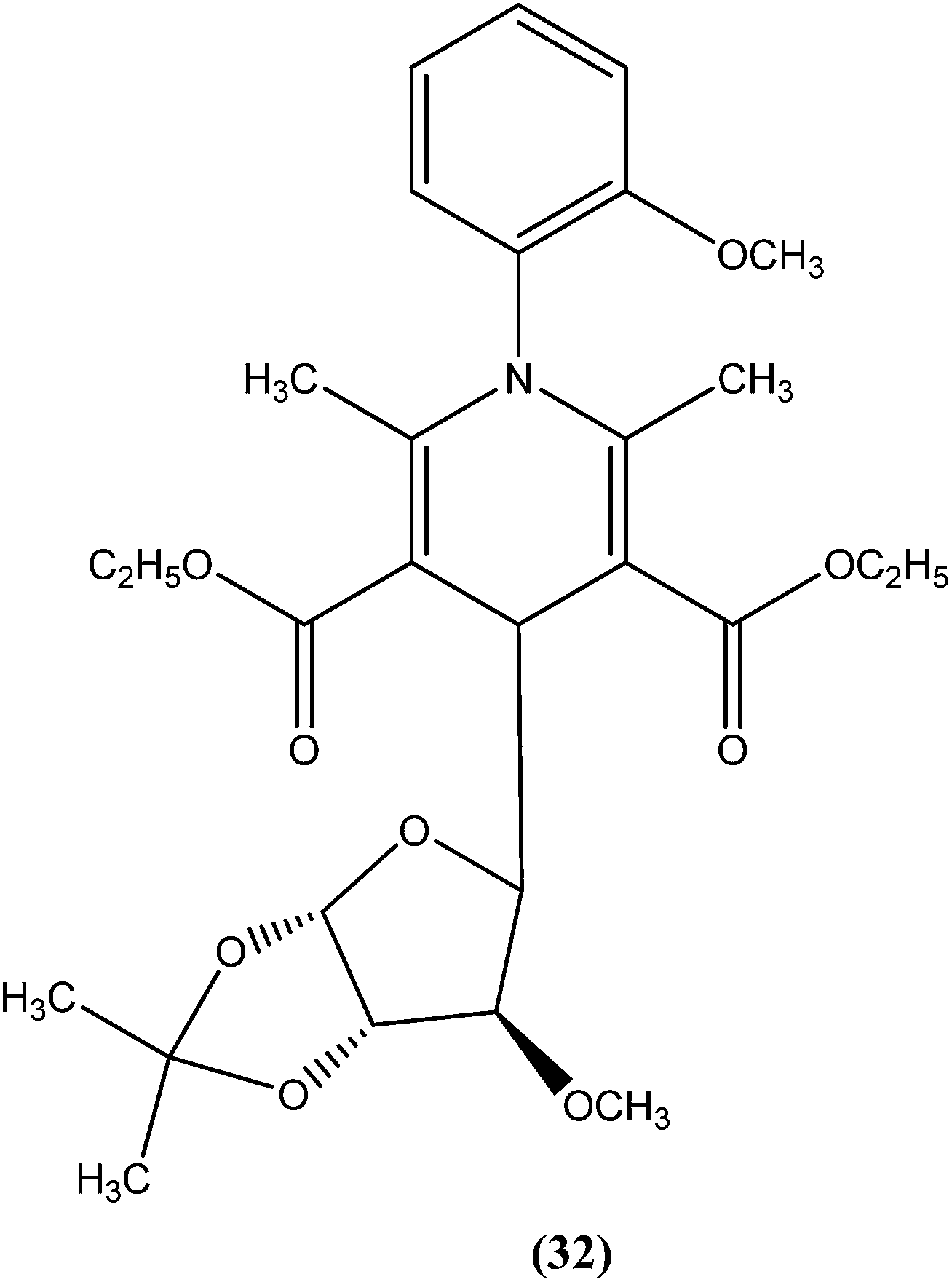
4.8. Thienopyridine
We recently reported synthesis, antileishmanial activity, and docking study of N′-substituted benzylidene-2-(6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)acetohydrazides. The synthesized series was evaluated for antileishmanial activity against L. donovani promastigotes. Among all tested compounds, 4-N,N-dimethylamino substituted phenyl ring compound (33) was found to be the most promising, with an IC50 value of 27.41 μM when compared with sodium stibogluconate (IC50 = 537.92 μM) as standard.126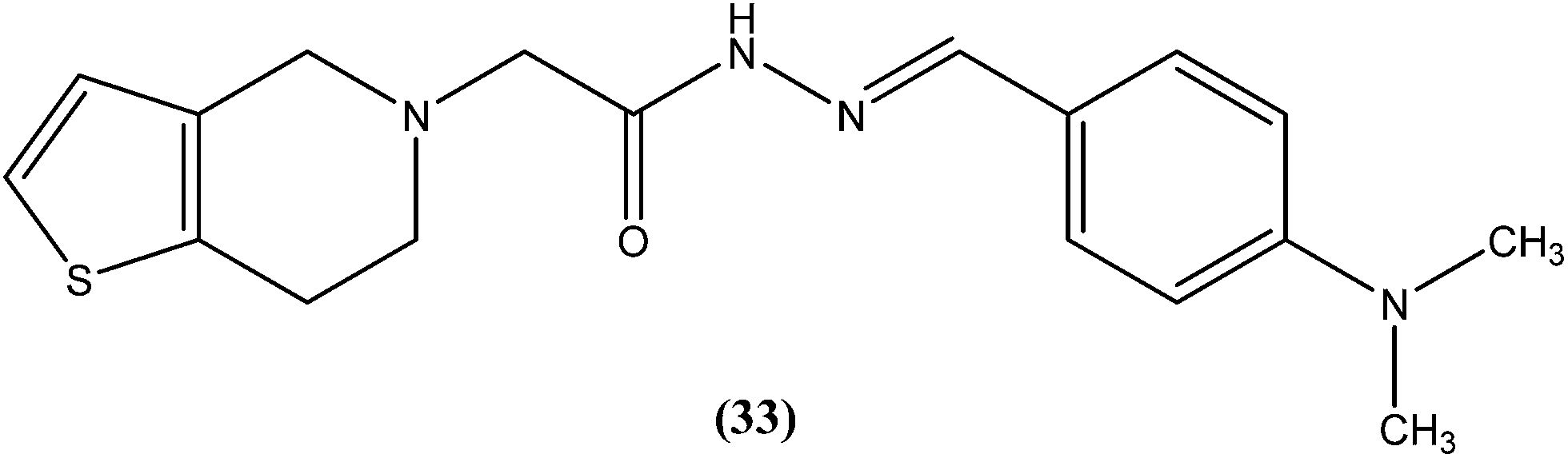
4.9. Quinoline
Quinoline based chemotherapeutic agents have attracted considerable interest as antileishmanial agents.127 The Walter Reed Army Institute (USA) discovered an 8-aminoquinoline (primaquine) analogue WR6026 (sitamaquine) (34). This was in development with GlaxoSmithKline (UK) for oral treatment against VL (caused by L. chagasi). Studies revealed that it cured 50% of patients with kala-azar in Kenya at a dose of 1 mg per kg per day for 28 days. After phase II clinical trials, WR6026 demonstrated the unusual clinical features of lack of increased efficacy against Brazilian kala-azar with increased dosing above 2 mg per kg per day and toxicity that was not present in previous investigations.128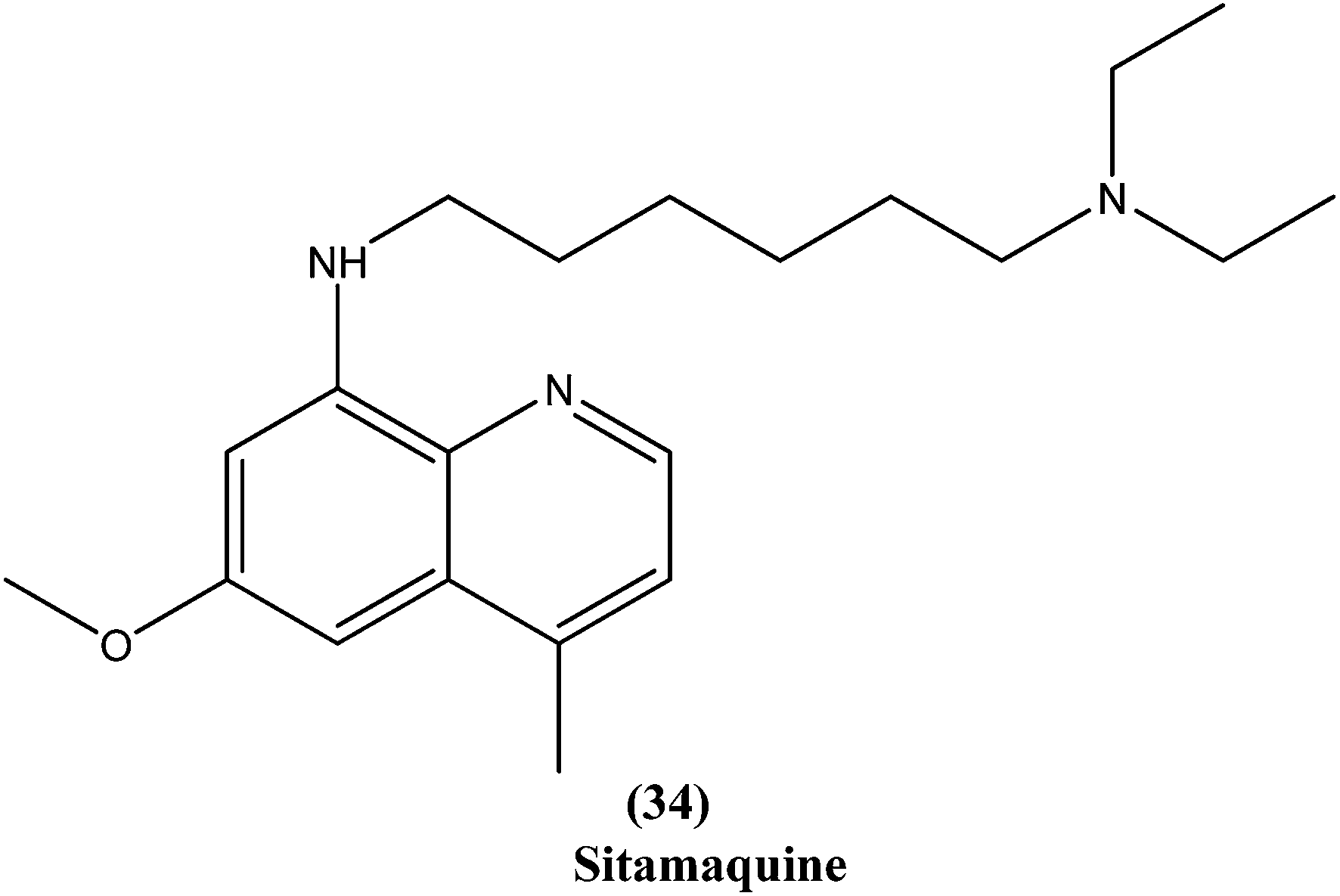
Quinolines also have been found to inhibit leishmanial GDP-mannose-pyrophosphorylase, an enzyme system producing a range of mannose-rich glycoconjugates that are essential for parasite survival and its virulence.129 Chauhan et al. synthesized a novel series of 1,2,4-triazino-[5,6b]indole-3-thiones covalently linked to 7-chloro-4-aminoquinoline. After evaluation for their in vitro activity, compound (35) was found to be the most potent, with an IC50 value of 0.36 μM and CC50 > 400 μM in Vero cell lines (SI of >1111) against amastigote form of L. donovani, which is several times more potent than the standard drugs, miltefosine (IC50 = 8.10 μM, SI = 7) and sodium stibogluconate (IC50 = 54.60 μM, SI ≥ 7).130

Cryptolepine (5-methyl-10H-indolo[3,2-b]quinoline) is an indoloquinoline alkaloid isolated from a medicinal plant Cryptolepis sanguinolenta.131 The antileishmanial properties of synthetic derivatives of cryptolepine against L. donovani parasites were evaluated for the first time by Hazara et al. From the series, compound (36), a 2,7-dibromocryptolepine, was the only drug to exhibit selective toxicity against the promastigote form of a classical L. donovani strain (AG83), with an IC50 value of 0.5 ± 0.1 μM and IC50 against mouse peritoneal macrophage cells of 9.0 ± 1.2 μM (SI ∼ 18) in comparison with cryptolepine (IC50 = 1.1 ± 0.3 μM; SI ∼ 0.7). Furthermore, compound (36) was found to inhibit substantially the intracellular amastigote forms of two clinical isolates, one of them being a Sb(V)-resistant strain of L. donovani. Compound (36) was reported to be a prospective “lead” to novel antileishmanial therapy, supported by studies on the mechanism of cytotoxicity induced by (36) in L. donovani promastigotes (AG83). This revealed a mode of cell death in L. donovani promastigotes characterized by disruption of mitochondrial membrane integrity in terms of depolarization of membrane potential, and degradation of chromosomal DNA into oligonucleosomal fragments—the characteristic event of apoptosis.132

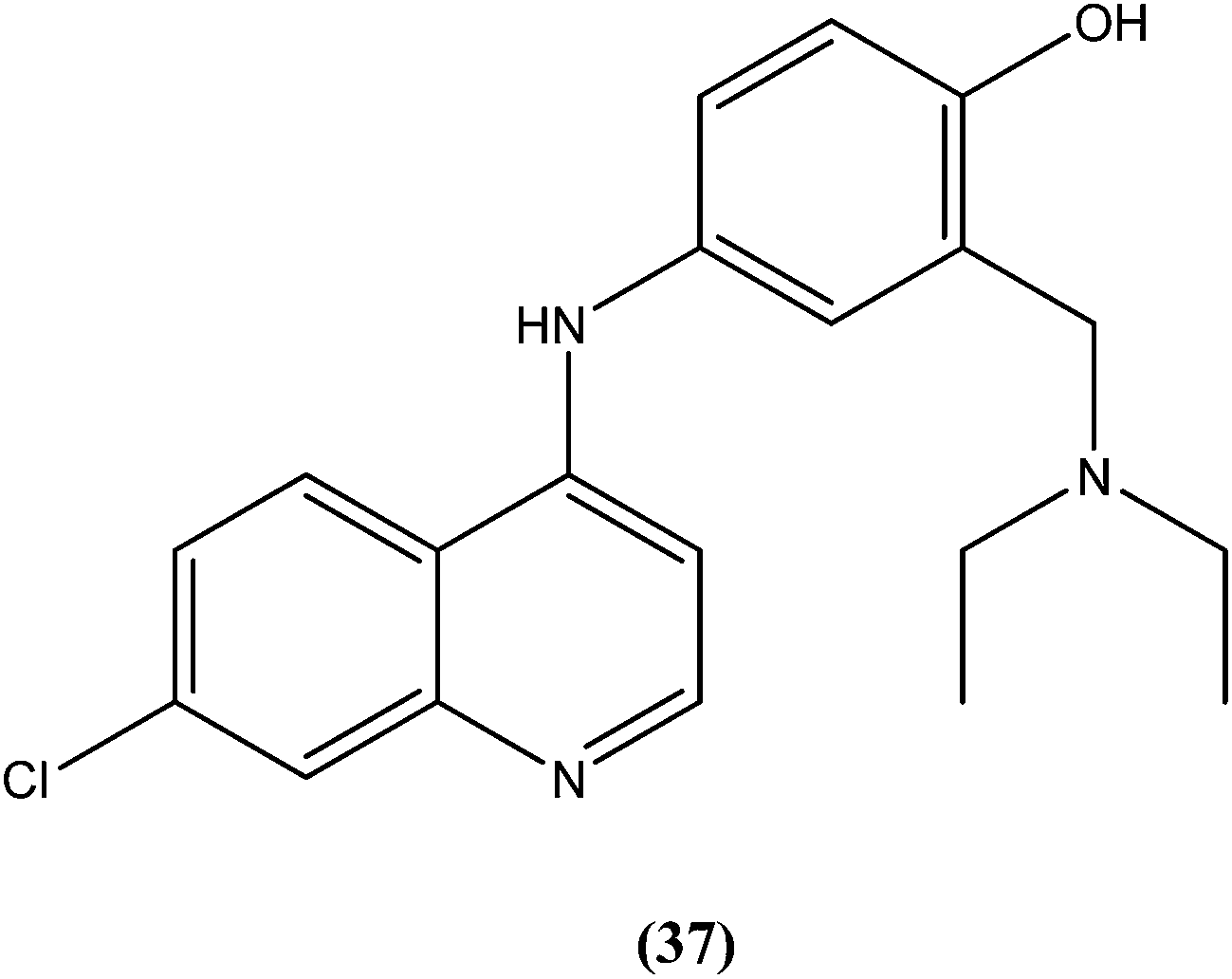
Bertinaria et al. synthesized new amodiaquine derivatives bearing modified lateral basic chains, and the compounds were tested in vitro against L. donovani (MHOM/ET/67/HU3). The authors were the first to report the antileishmanial action of amodiaquine (37) and some newly synthesized analogs against the intracellular amastigote form of L. donovani. Amodiaquine showed potent activity against leishmaniasis, with an IC50 value of 1.4 μM, but an IC50 value of 90 μM on KB cells, therefore it was found to be non-cytotoxic in nature. Derivatives of amodiaquine showed good antileishmanial activity, but were found to be cytotoxic and to have a narrow therapeutic index.133
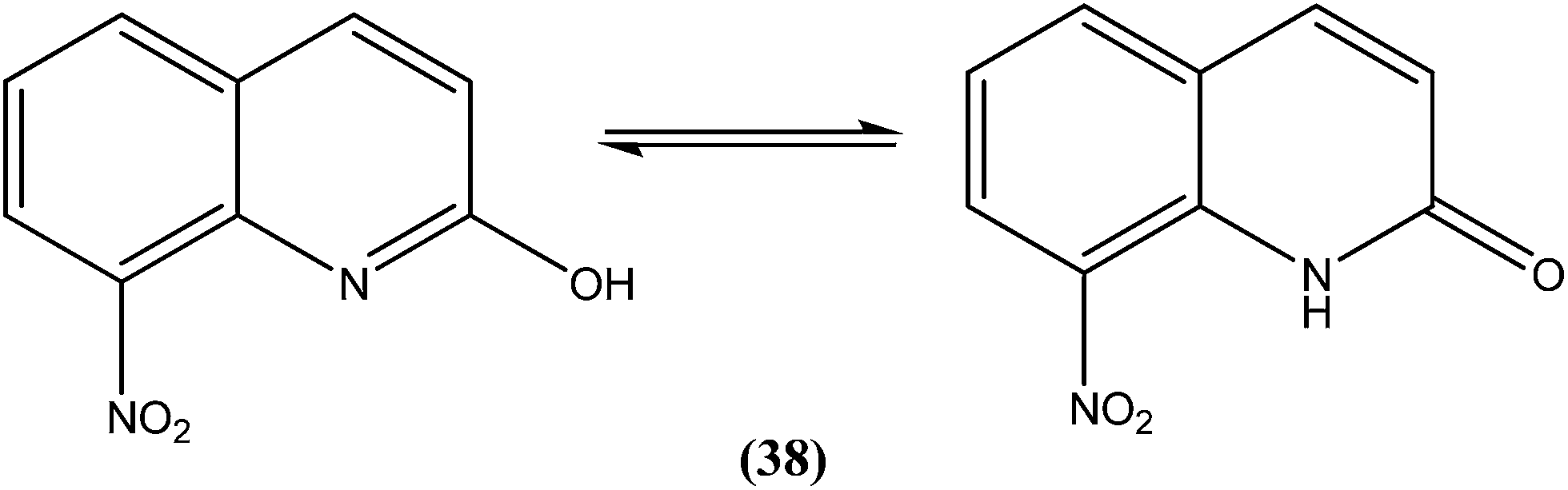
Azas et al. prepared a series of 2-substituted nitroquinolines and evaluated these for in vitro antileishmanial properties. From the series, they identified 2-hydroxy-8-nitroquinoline (38) as a hit molecule displaying IC50 values of 6.6, 6.5, and 7.6 μM against L. donovani promastigote form, amastigote form, and L. infantum promastigote form, respectively. Compound (38) was also found to possess low cytotoxicity (CC50) on human HepG2 cell lines and murine J774 cell lines, 126.3 μM and 105 μM, respectively. To explain the SAR, the authors reported that the presence of a hydroxyl group at position 2 (involved in a prototropic tautomeric equilibrium between quinoline-2-ol and its [1H]-quinolin-2-one counterpart) and a nitro group at position 8 of the quinoline ring result in reasonable activity against both promastigote (L. donovani and L. infantum) and amastigote (L. donovani) parasite stages.134
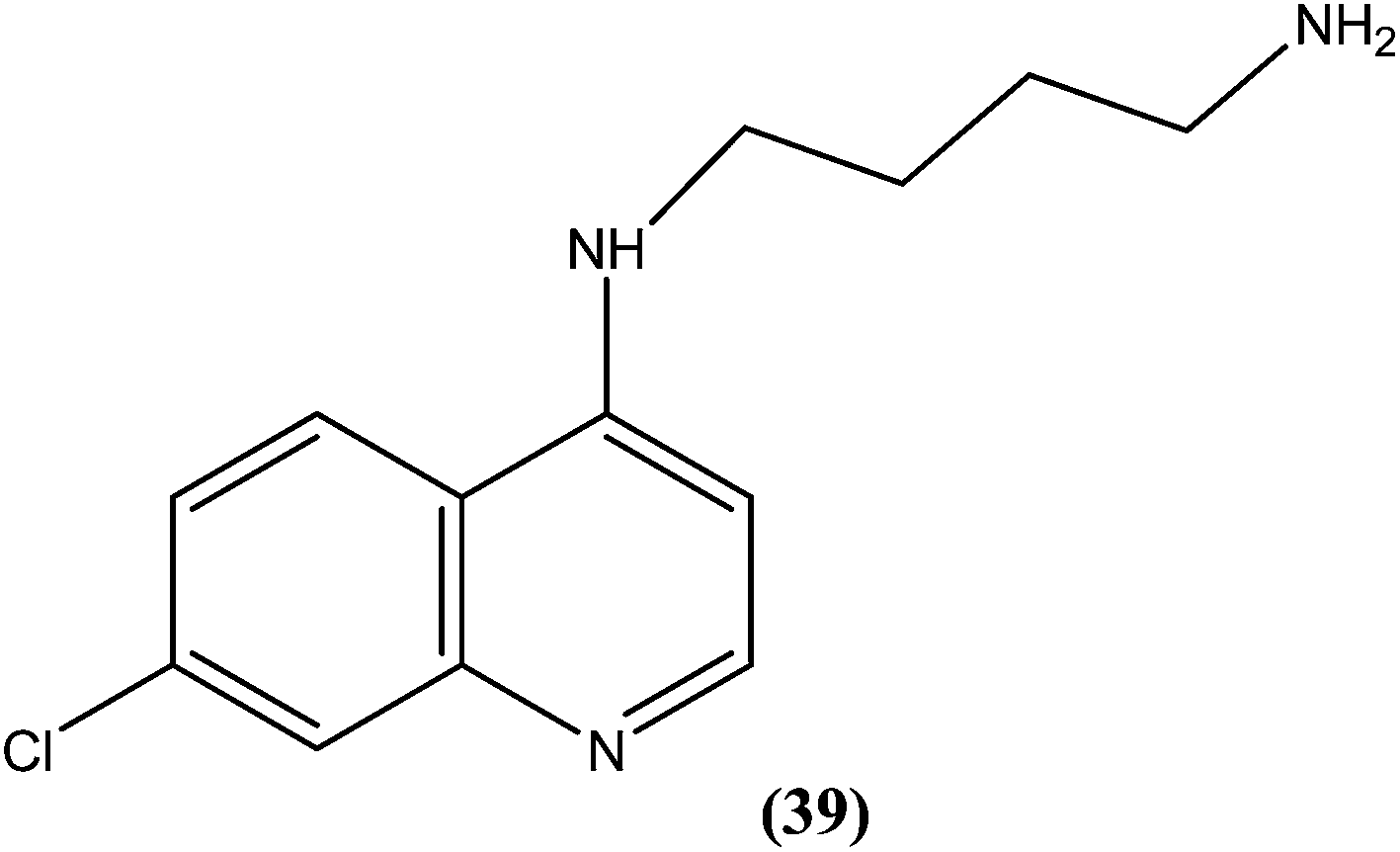
Silva et al. reported synthesis of 4-amino-7-chloroquinoline derivatives and evaluated these for antileishmanial activity against promastigotes of different Leishmania species. Among the tested compounds, compound (39) containing an amino group was found to be almost 50 times (IC50 = 0.01 μM) more active than the reference drug amphotericin B (0.004 μM) against L. chagasi. To predict the SAR, from the obtained results they stated that the presence of the amino group is essential for good activity of such compounds against Leishmania, as addition of alkyl groups, either mono or di-alkyne substituents, results in loss of antileishmanial activity.135
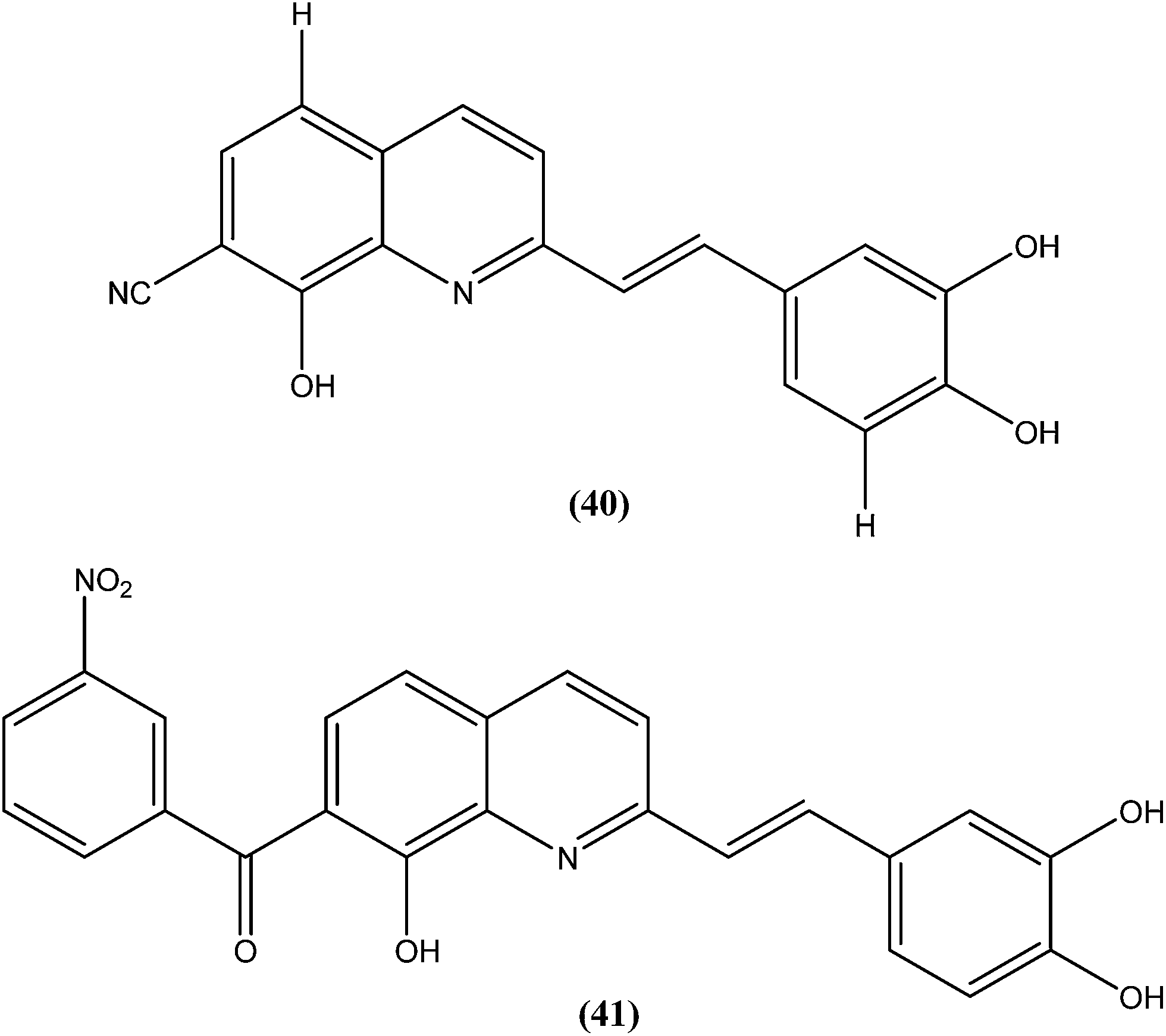
Loiseau et al. evaluated a 2-substituted series of quinolines, styrylquinolines, and 7-aroylstyrylquinolines for in vitro antileishmanial activities and cytotoxicities. Among the quinolines and the styrylquinoline derivatives, the most interesting compound was compound (40), with an IC50 of 4.1 μM for L. donovani intramacrophage amastigotes and a SI of 8.3, whereas from the7-aroylstyrylquinolines, compound (41) exhibited an IC50 of 1.2 μM and a SI of 121.5, which is 10-fold and 8-fold more active than miltefosine (IC50 = 13.4 μM; SI = 0.2) and sitamaquine (IC50 = 9.7 μM; SI = 2) with SI values 607-fold and 60-fold higher, respectively. The authors concluded that because of its high in vitro antileishmanial activity and low toxicity, compound (41) is the most interesting compound to emerge from more than 150 derivatives of 2-substituted quinolines that were synthesized and evaluated. Compound (41) has now been selected as a candidate for evaluation in vivo with L. donovani mouse or hamster models via the Drugs for Neglected Diseases initiative (DNDi) pipeline.136
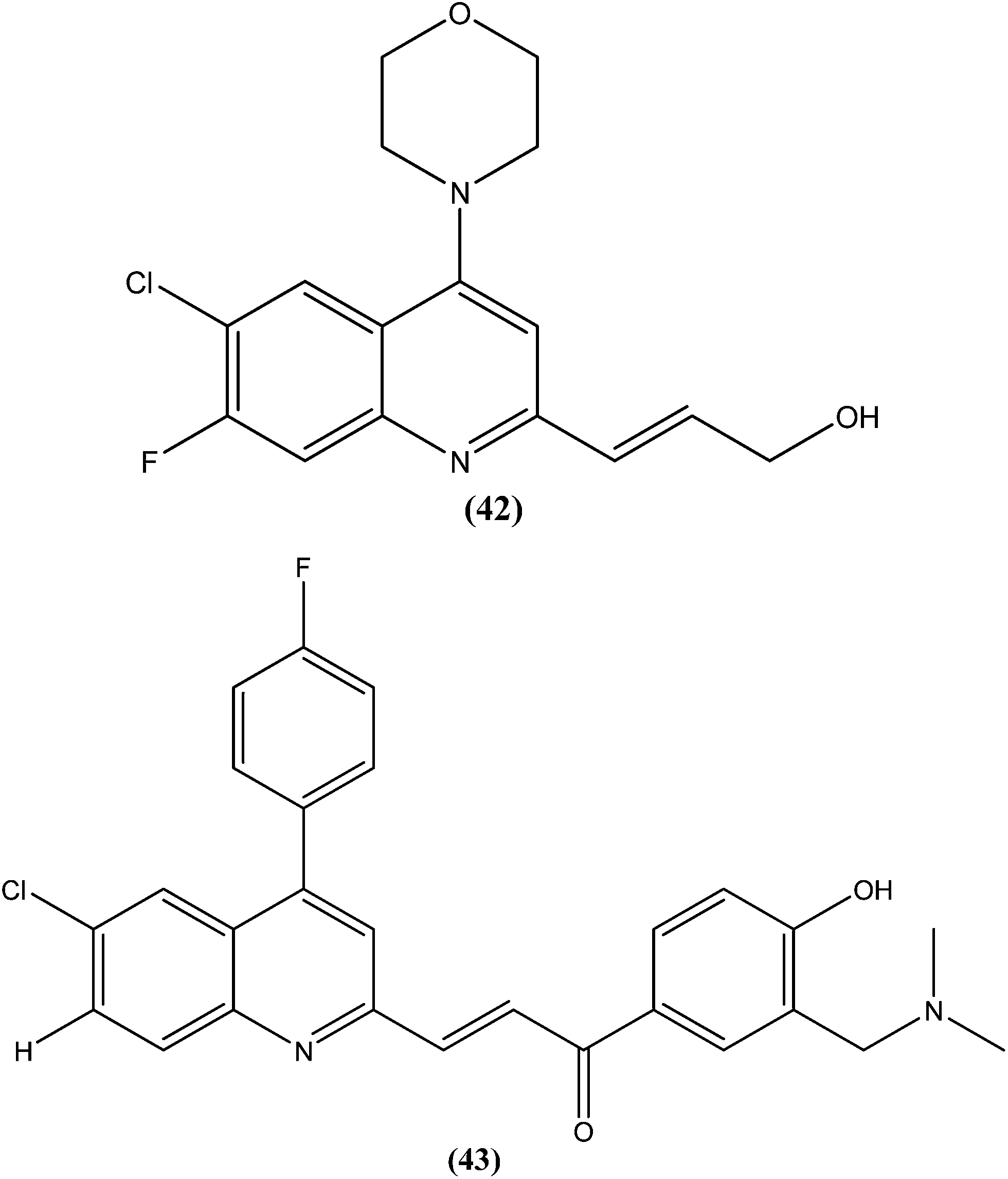
Gupta et al., researchers from Advinus Therapeutics Ltd., Bangalore, India, Division of Parasitology, CSIR-Central Drug Research Institute, Lucknow India, and Drugs for Neglected Diseases initiative (DNDi), Geneva, Switzerland, together reported that substituted quinoline, compound (42), was found to have in vitro activity (IC50 = 0.22 ± 0.06 μM) against L. donovani amastigotes. Its SI was 187.5. The compound was found to have good in vivo efficacy (84.26 ± 4.44% inhibition) and also promising ADME properties.137 The same group, in expansion of their study, reported synthesis of analogs of (42) and their subsequent characterization for in vitro activity against the intracellular form of L. donovani. The resulting quinolines were found to have similar efficacy against the parasite to that of (42). From these tested compounds, compound (43) was found to be the most active, with an IC50 value of 0.17 μM.138
4.10. Quinazoline
Sahu et al. synthesized a series of a new class of 4-(hetero)aryl-2-piperazino quinazolines and assessed these for in vitro activity against extracellular promastigotes and intracellular amastigotes of L. donovani. Among the compounds evaluated, compound (44) (a 4-methyl piperazinyl and 2,4,6-trimethoxyphenyl substituted quinoline derivative) showed the lowest toxicity, having a CC50 value above 25.38 μM. The authors reported its SI value above 8.03, which is comparable with that of sodium stibogluconate and pentamidine (SI = 6.38 and 2.07, respectively). It was also observed that compound (44) exhibited higher anti-amastigote activity against L. donovani with an IC50 value of 3.16 μM when compared with standard drugs sodium stibogluconate (IC50 = 7.92 μM) and pentamidine (IC50 = 3.56 μM). 2,3,5-Trimethoxy benzene together with an N-methyl group (44) enhanced the antileishmanial activity remarkably, and thus represent an interesting lead as antileishmanial agents.139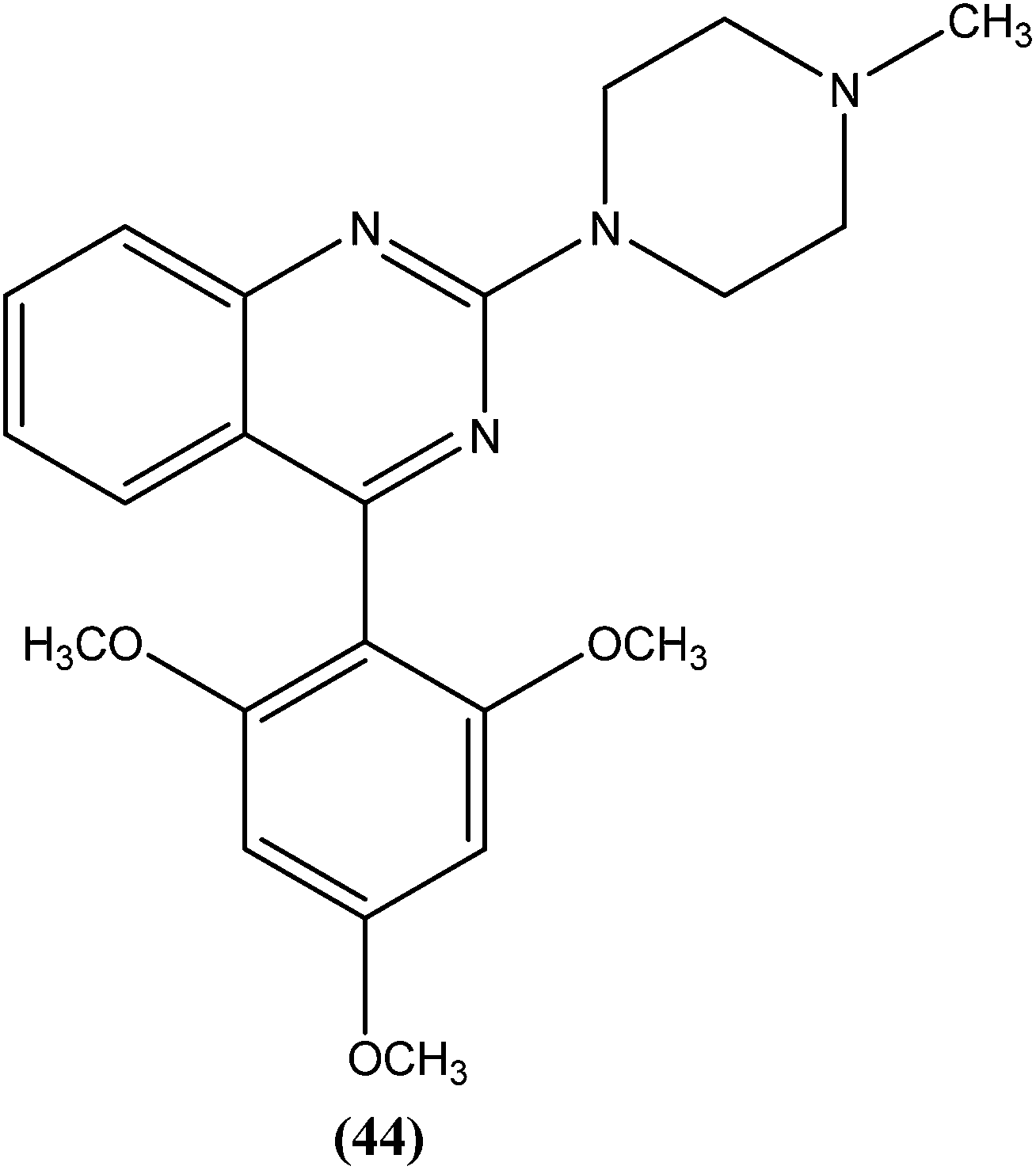
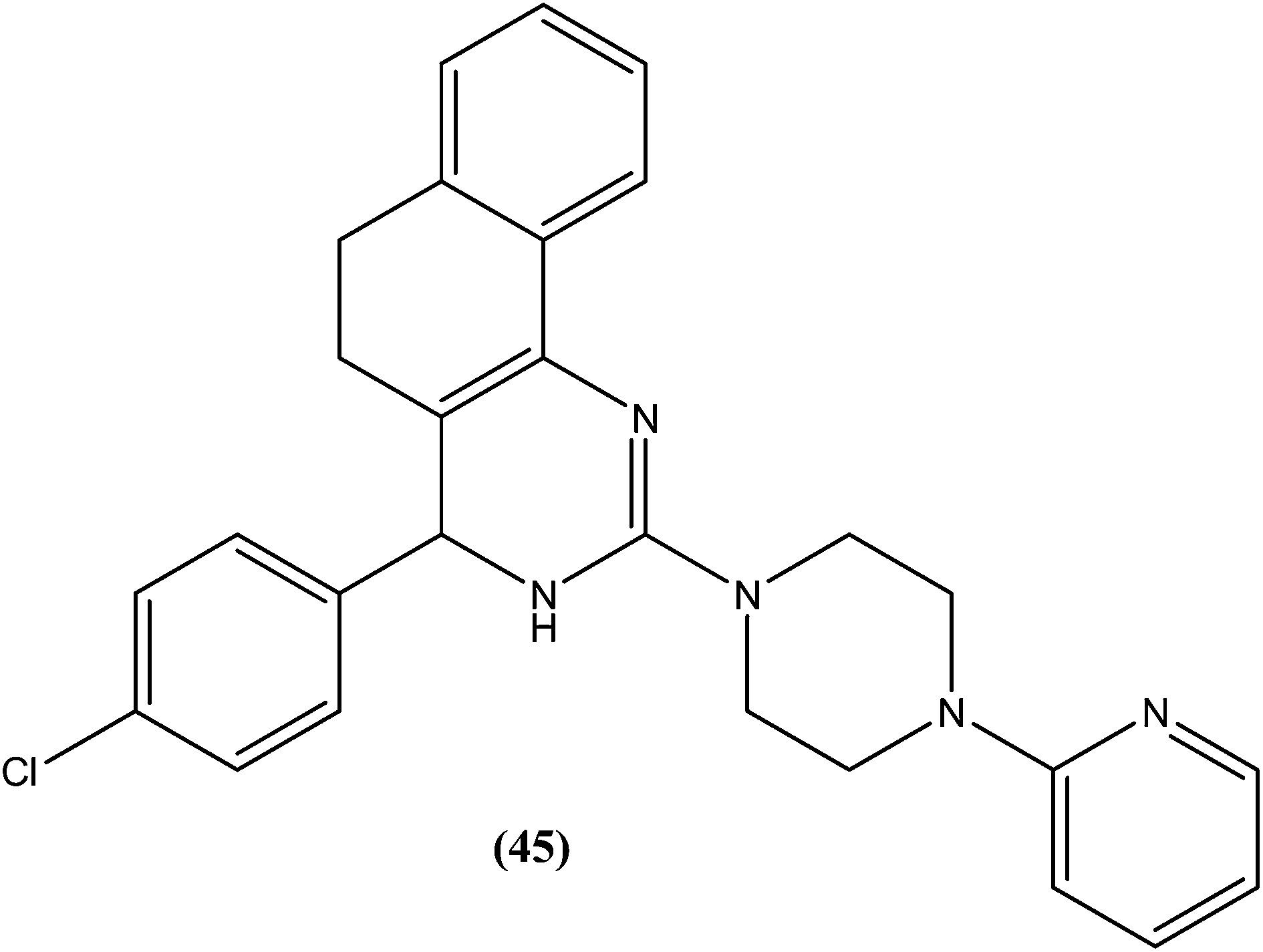
After synthesizing a series of novel substituted quinazoline derivatives and evaluating these for antileishmanial activity, Agarwal et al. reported that all the compounds exhibited higher activities against L. donovani compared with reference drugs sodium stibogluconate and pentamidine. The most active compound (45), having a 3,4,5,6-tetrahydrobenzoquinazoline ring with a pyridyl piperazinyl group and 4-chlorophenyl ring substituted at the second and fourth positions, respectively, showed 99.9 ± 0.07% inhibition against L. donovani promastigote at a concentration of 2.19 μM, whereas its antileishmanial activity in vitro (IC50) against a luciferase–amastigote system of L. donovani was found to be at its lowest at 0.58 μM when compared with the reference drugs.140
Berman et al. (Division of Experimental Therapeutics, Walter Reed Army Institute of Research, Washington, D.C.) assessed 2,4-diaminoquinazoline analogs of folate against L. major in human macrophages. After in vitro testing of these analogs, compound (46) containing a side chain with an aromatic tertiary amine, was found to be the most active, possessing an ED50 value of 0.00365 μM with a 50% macrophage toxic dose of 1.52 μM, which indicates an in vitro therapeutic index of approximately 105. It was reported that the activity of compound (46) depends on the aromatic tertiary amine being attached directly to the benzyl group of the quinazolines nucleus. Restricted amine (even if tertiary, aromatic, or bound directly to the ring) was found to be without activity. In their conclusion, they stated that “The remarkable activity of 2,4-diaminoquinazolines in vitro suggests that these or other folate analogs have strong potential to be investigated as novel antileishmanial agents”.141
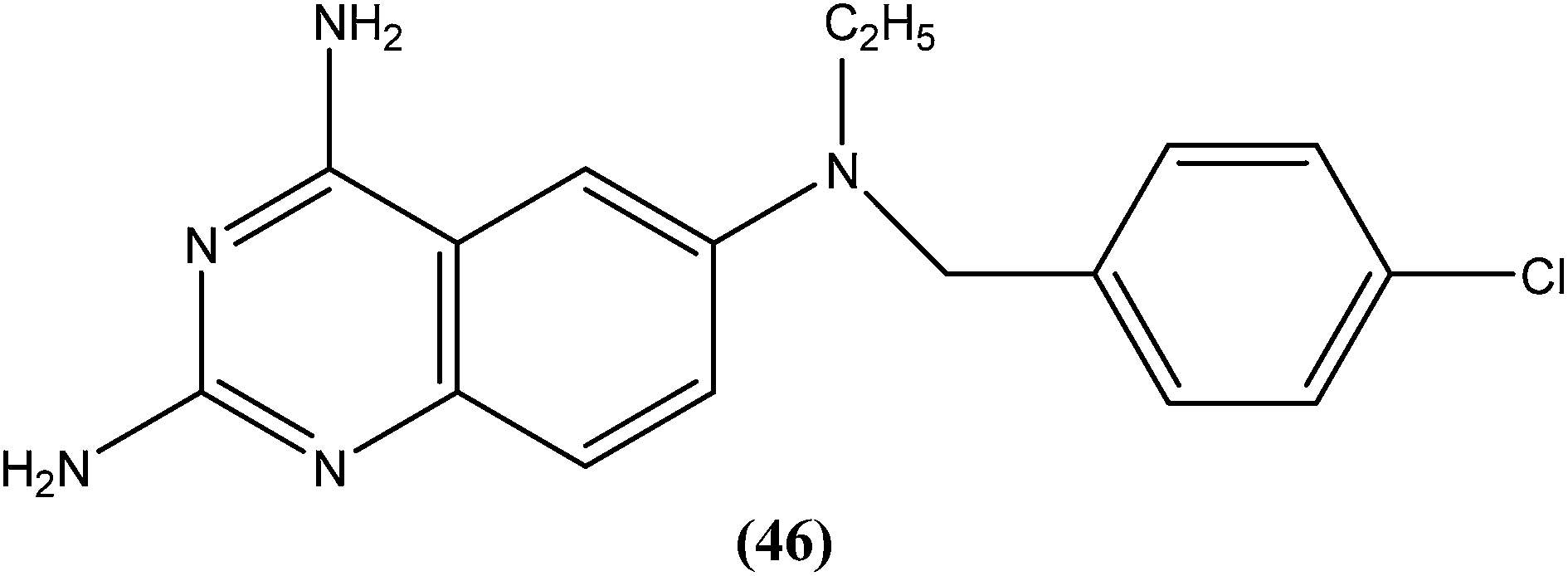
Bhattacharjee et al. reported that when tested against L. donovani amastigotes, several tryptanthrin (indolo[2,1-b]quinazoline-6,12-dione) derivatives exhibited remarkable in vitro activity at concentrations below 0.00040 μM. The parent compound can be produced by Candida lipolytica (when grown in media containing an excess of tryptophan, hence the name tryptanthrin). When tested for toxicity against murine J774 macrophages and rat neuronal NG-108-15 cells, the best selectivity was obtained with compound (47), which was found to be 69-fold more toxic to the parasites than to both mammalian cell lines (toxic at 0.00088 μM). Compound (47) was one of the most active, having an IC50 value of 0.000013 μM when compared with amphotericin B (IC50 = 0.0045 μM).142 After performing 3D-QSAR analysis, the researchers concluded that the presence of a five-membered carbonyl moiety in the molecule appears to be a structural requirement for potent activity. Stereoelectronic factors of the substituents at the third position of the D ring in indolo[2,1-b]quinazoline-6,12-dione skeleton appear to have a significant effect on potent activity. The carbonyl groups of the five- and six-membered rings in the tryptanthrin moiety, and electron transfer ability from a receptor are likely to be crucial in the mechanism of action of the compounds.143
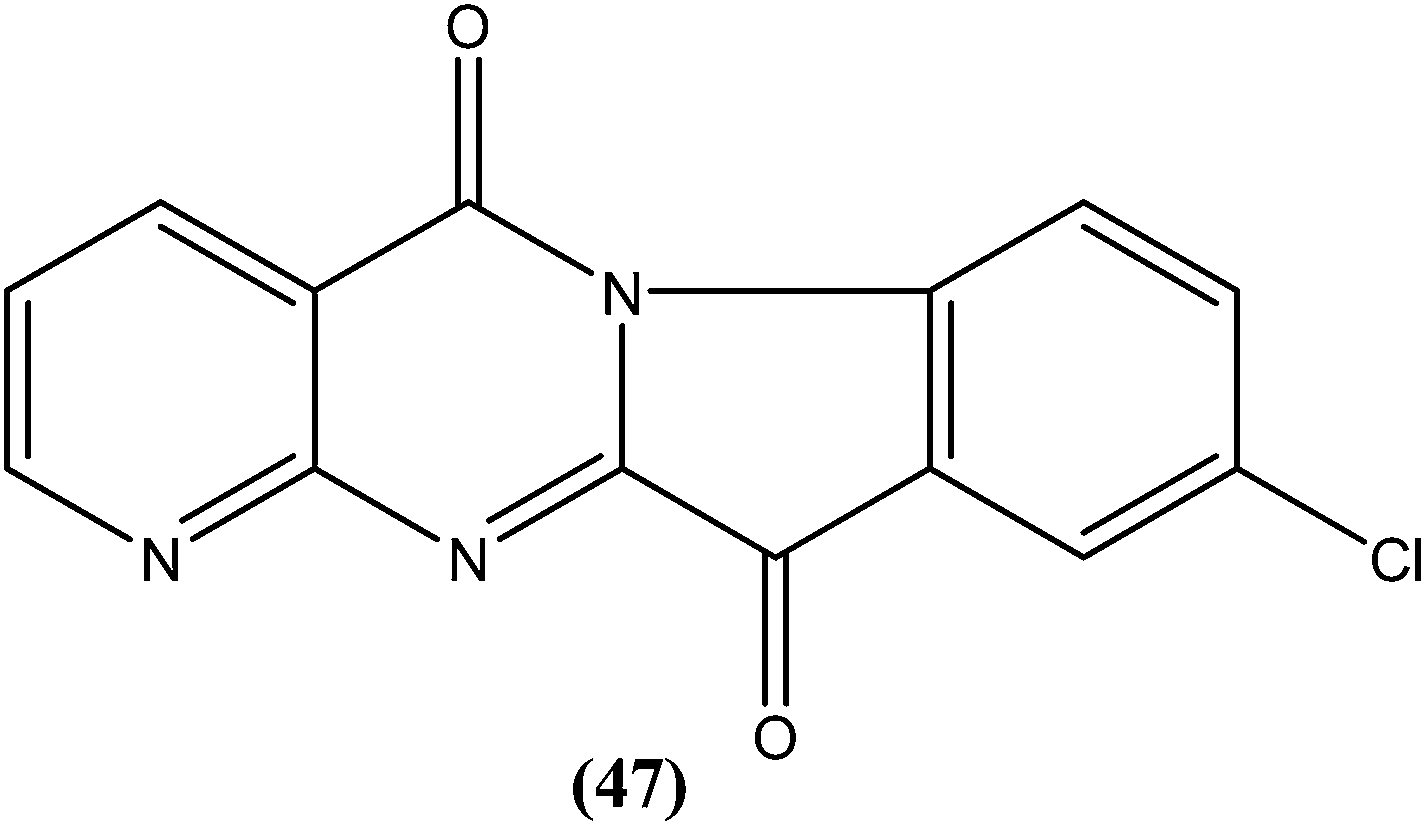
Manetsch et al. recently reported the antileishmanial activity of a series of N2,N4-disubstituted quinazoline-2,4-diamines, which were tested in vitro against intracellular amastigotes of L. donovani and L. amazonensis. Compound (48), a benzyl-substituted quinazoline, was the most potent compound (50% effective dose, EC50 = 0.15 ± 0.02 μM and 0.90 ± 0.27 μM against L. donovani and L. amazonensis, respectively), but it failed to exhibit the same actions in an in vivo murine visceral leishmaniasis model. Quinazoline (49) (EC50 of 0.83 ± 0.32 μM against L. donovani and 4.1 ± 1.2 μM against L. amazonensis) with EC50 value of >33 against J774A.1 cell line and SI value of >40, was found to reduce parasitemia by 37% when given at 15 mg per kg per day via the intraperitoneal route for 5 consecutive days. Pharmacokinetic studies of compound (49) revealed a maximum plasma concentration that was threefold higher than the EC50, and it has a terminal half-life of 5 hours after i.p. administration. Although a clear correlation among in vitro activity, in vitro physicochemical properties, and in vivo activity was not clearly observed, the potencies of front runner compounds such as (48) and (49) in conjunction with favorable physicochemical properties make N2,N4-disubstituted quinazoline-2,4-diamines a suitable platform for future development of antileishmanial agents.144
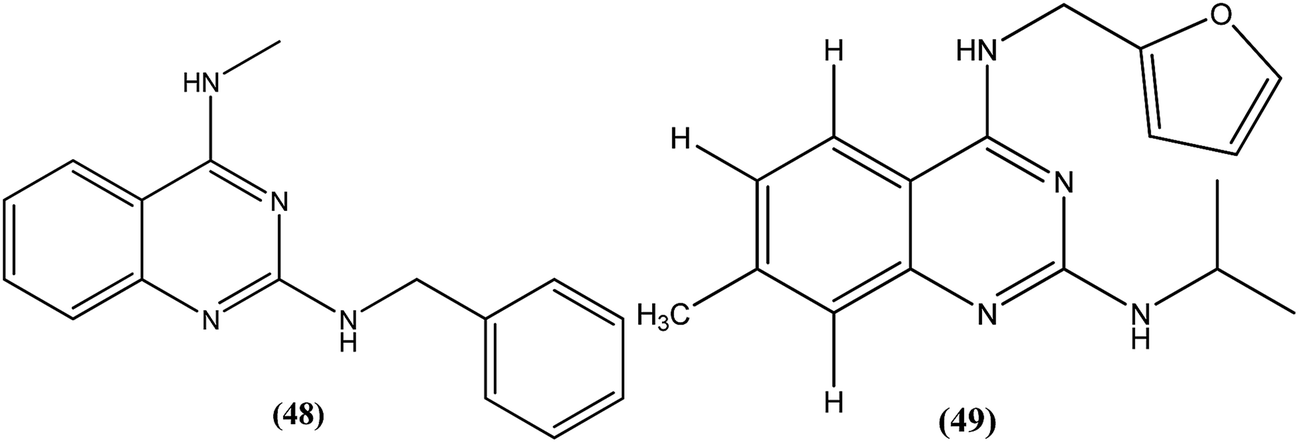
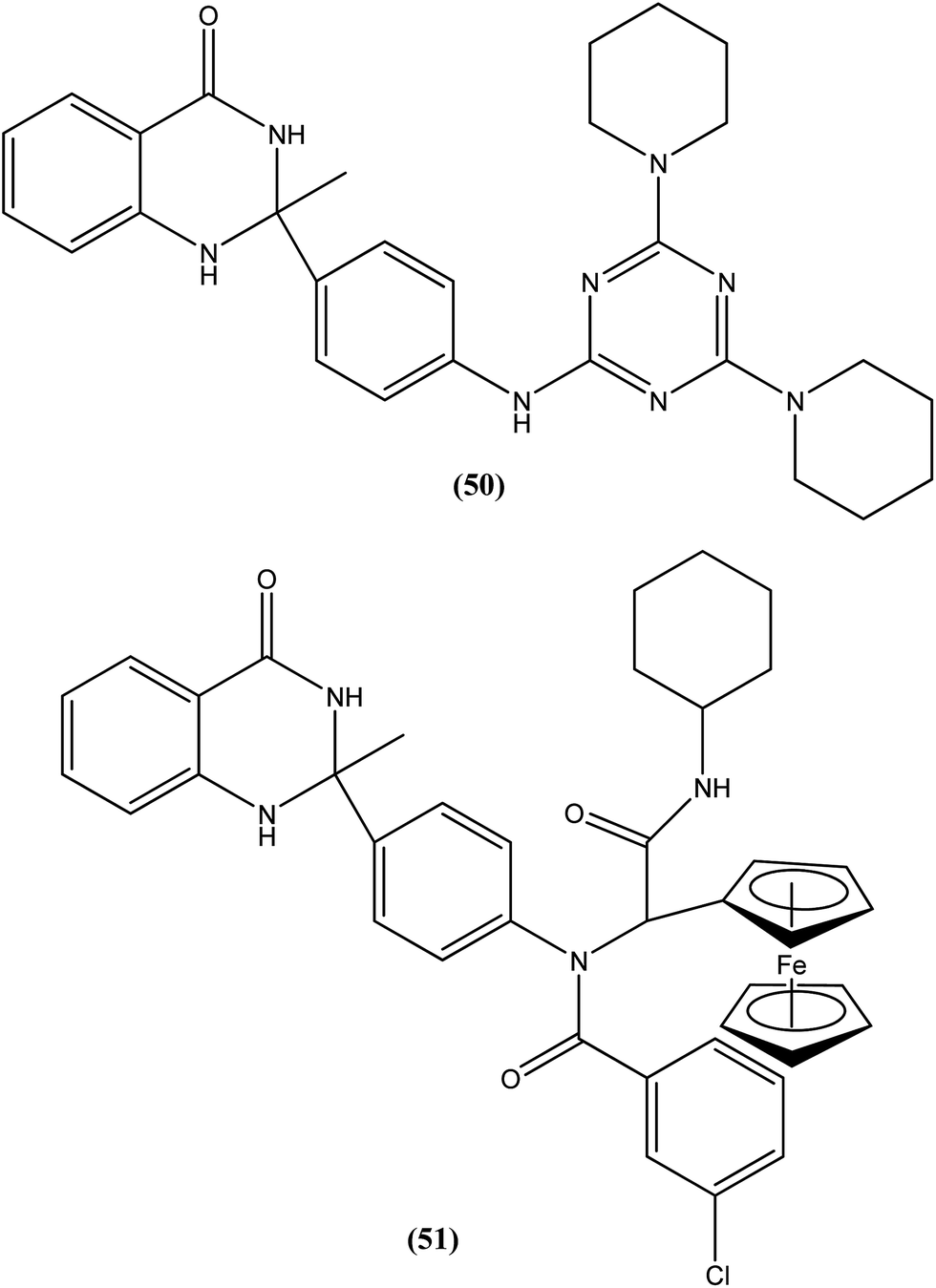
Chauhan et al. discovered four novel series of quinazolinone hybrids via introducing heterocyclic systems with different possible functionalities based on the concept of molecular hybridization: (i) among the substituted quinazolinone–triazines, compound (50), having IC50 of 7.05 ± 2.3 and 3.95 ± 0.8 μM against promastigote and amastigote forms of L. donovani, respectively (CC50 as >400 μM against both J774A.1 and Vero cell lines), was the most potent compound with SI of about 101.26; (ii) from the quinazolinone–peptide hybrids, compound (51) demonstrated potent activity, possessing IC50 of 0.73 ± 0.2 μM against amastigote form of L. donovani (CC50 as >400 μM against both J774A.1 and Vero cell lines) with the best SI value of >547.94. Both compounds were found to have higher potency compared with reference drug miltefosine (IC50 = 8.4 ± 2.1 μM and SI = 1.48). When administered in vivo in a hamster model, % inhibition of L. donovani parasite was found to be 73.15 ± 12.69 for compound (50) and 51.42 ± 15.67 for compound (51). Furthermore, it was reported that activation of T helper type 1 (Th1 type) and suppression of T helper type 2 (Th2 type) immune responses and induction in nitric oxide (NO) generation proved that compound (50) induces murine macrophages to prevent survival of parasites.145
4.11. Quinone
Costa et al. synthesized pterocarpanquinones, aza-pterocarpanquinone derivatives. Compound (52) showed the best activity against amastigote form of L. amazonensis. It was found to have IC50 values of 1.27 and 1.25 μM against promastigote and amastigote forms, respectively, with a SI (M J774 cell lines/IC50 amastigote) of 14.4.146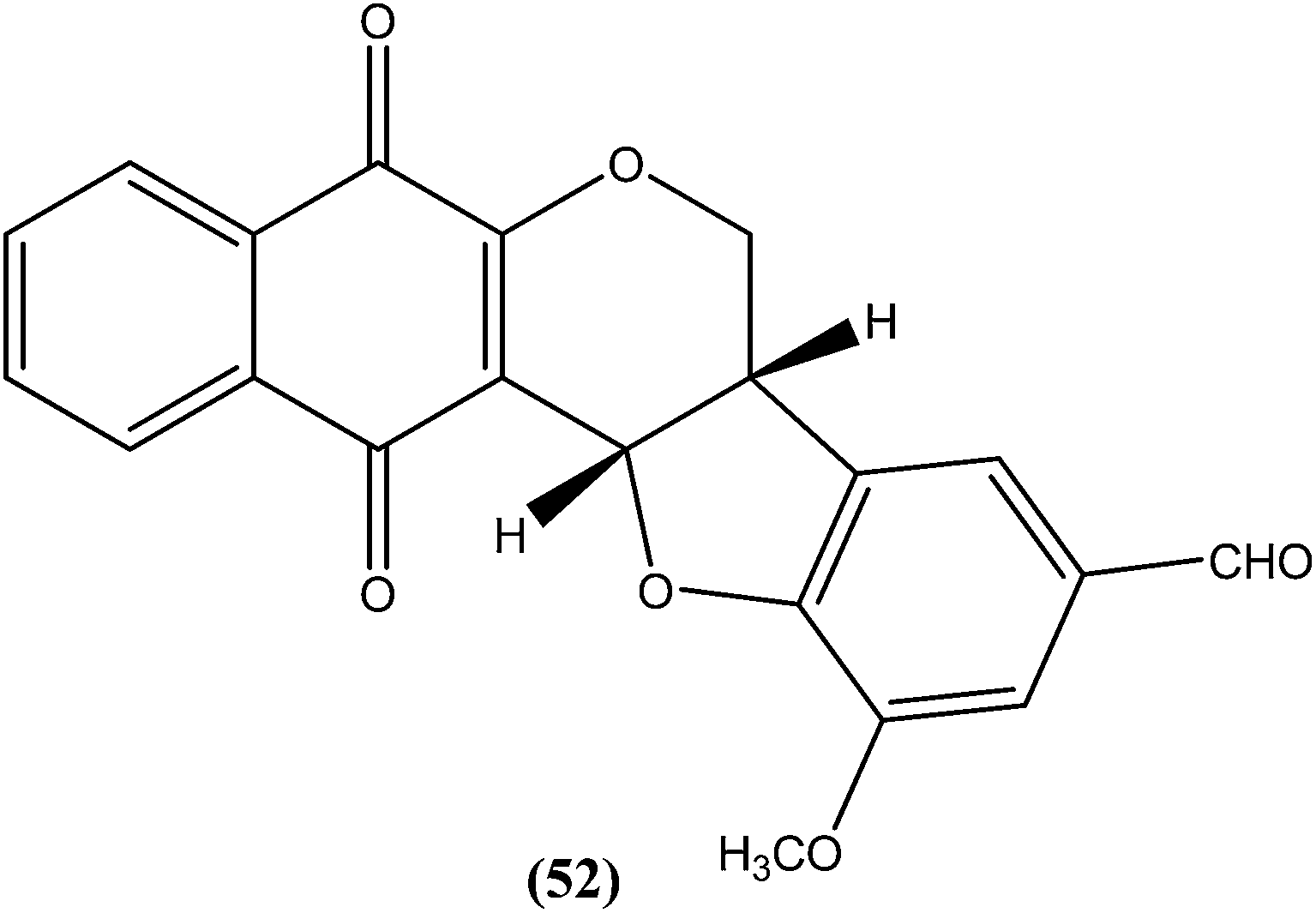
4.12. Thiophene
Robinson et al. reported potent antileishmanial activity of thiophene derivatives against L. infantum LV9.147 More than a decade later, Ram et al. reported synthesis of thiophenes and thieno[3,2-c]pyran-4-ones. On evaluating all the synthesized compounds in vitro against L. donovani promastigotes, the researchers noted that compounds (53) and (54) were the most potent, displaying 100% growth inhibition against promastigotes at a concentration of 25 μM. After performing SAR analysis, they explained that most of the highly active compounds possessed a –COOEt group except for compound (53) which has a –COOH substituent at the second position. The high order of activity may be a result of increased lipophilicity as ester groups are present. The nature of the aryl substituent also potentiates the antileishmanial activity.148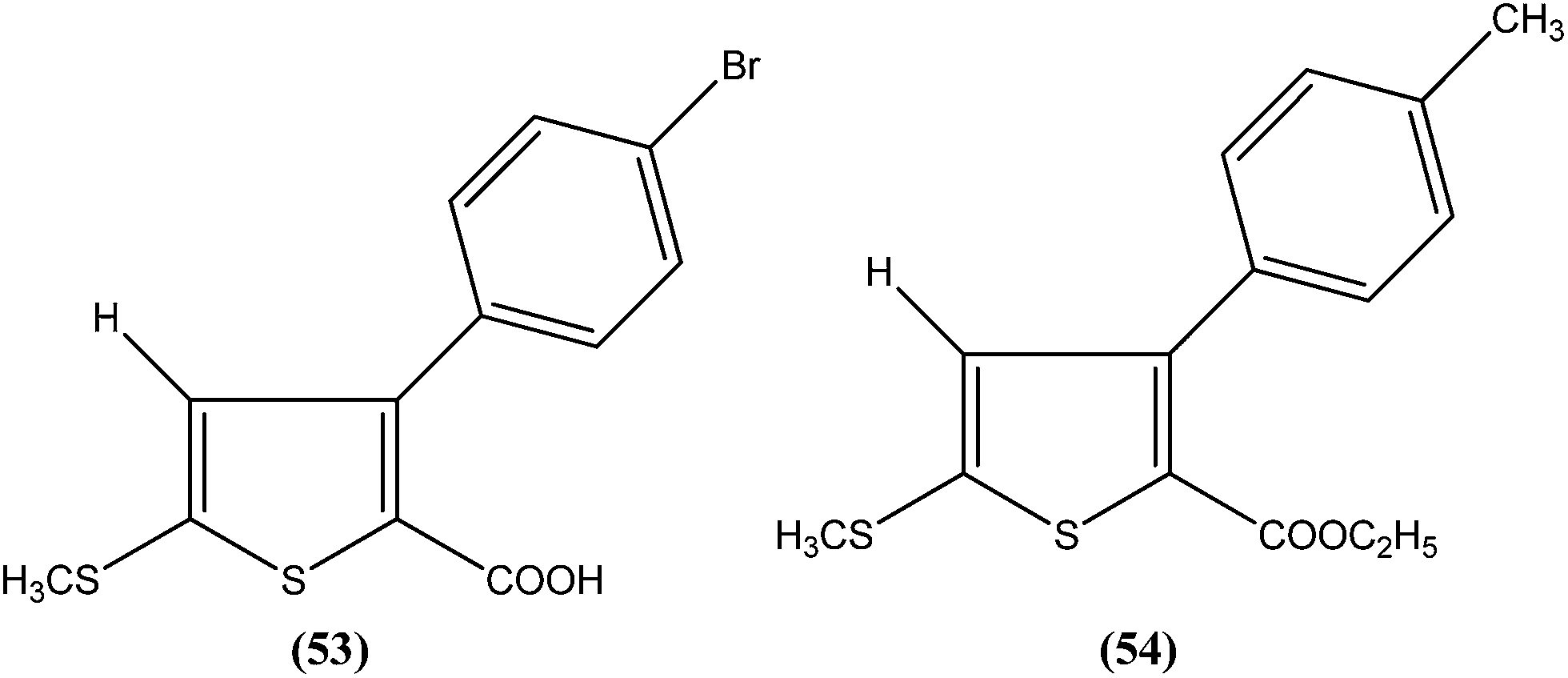
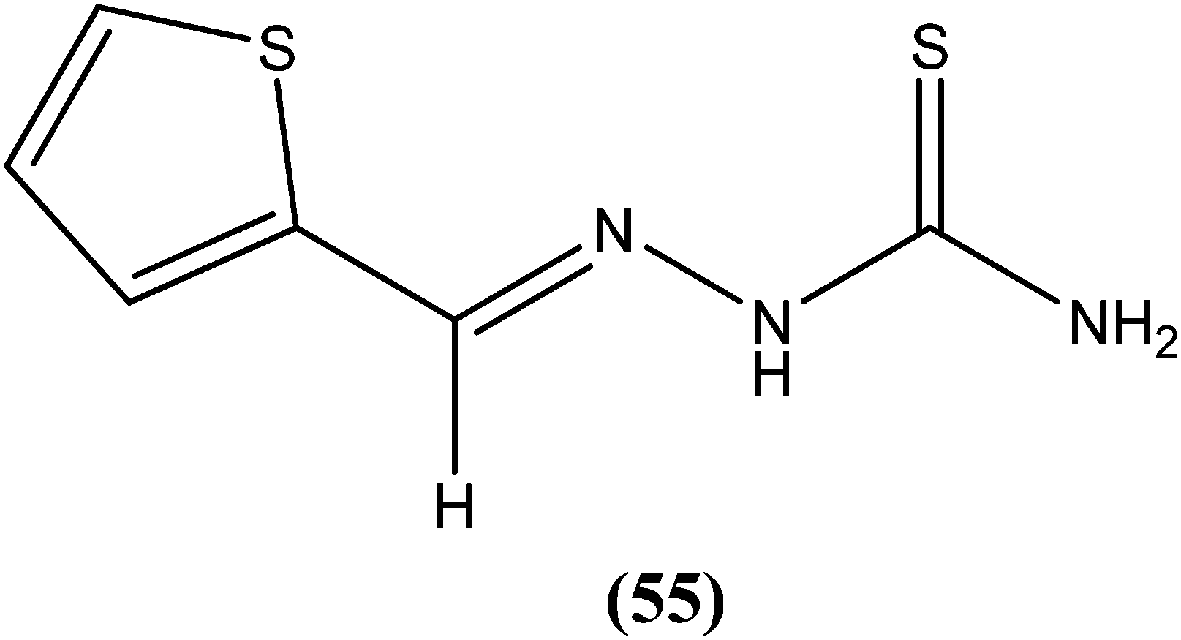
Hassan et al. prepared a series of thiosemicarbazones and reported their antileishmanial activities. In vitro assay of these compounds was performed by Zhai's method using a pre-established culture of L. major. Compound (55) showed significant antileishmanial activity (IC50 = 0.31 μM) against L. major promastigotes.149
4.13. Triazine
Chauhan et al. synthesized compounds of triazine dimers. They reported that most of the synthesized derivatives exhibited better activity against intracellular amastigotes (IC50 ranging from 0.77 to 10.32 μM) compared with standard pentamidine (IC50 = 13.68 μM), and the derivatives also were found to be non-toxic to Vero cells. Compound (56) with IC50 of 1.99 ± 0.31 μM against L. donovani intracellular amastigote form and CC50 of 216.08 ± 5.89 μM on Vero cells, possessing a SI value of 108.58, showed 74.41% inhibition in vivo in a L. donovani hamster model. Investigations of the immunostimulatory properties clearly indicated that compound (56) treated cells in Leishmania infected mouse macrophages (J-774A.1) had induced Th1 (T helper type 1) type immune responses by (i) remarkable production of interleukin (IL)-12, tumor necrosis factor (TNF)-α, and nitric oxide (NO), and (ii) effective suppression of Th2 (T helper type 2) type cytokines, IL-10, and transforming growth factor (TGF)-β. Furthermore, molecular docking studies of compound (56) revealed that it shared the same binding residues as shared by pentamidine. The docking studies also indicated that compound (56) showed H-bonding and pi-stacking with Tyr191 residue, whereas of the interaction of pentamidine with Tyr191 was limited to pi-stacking only.150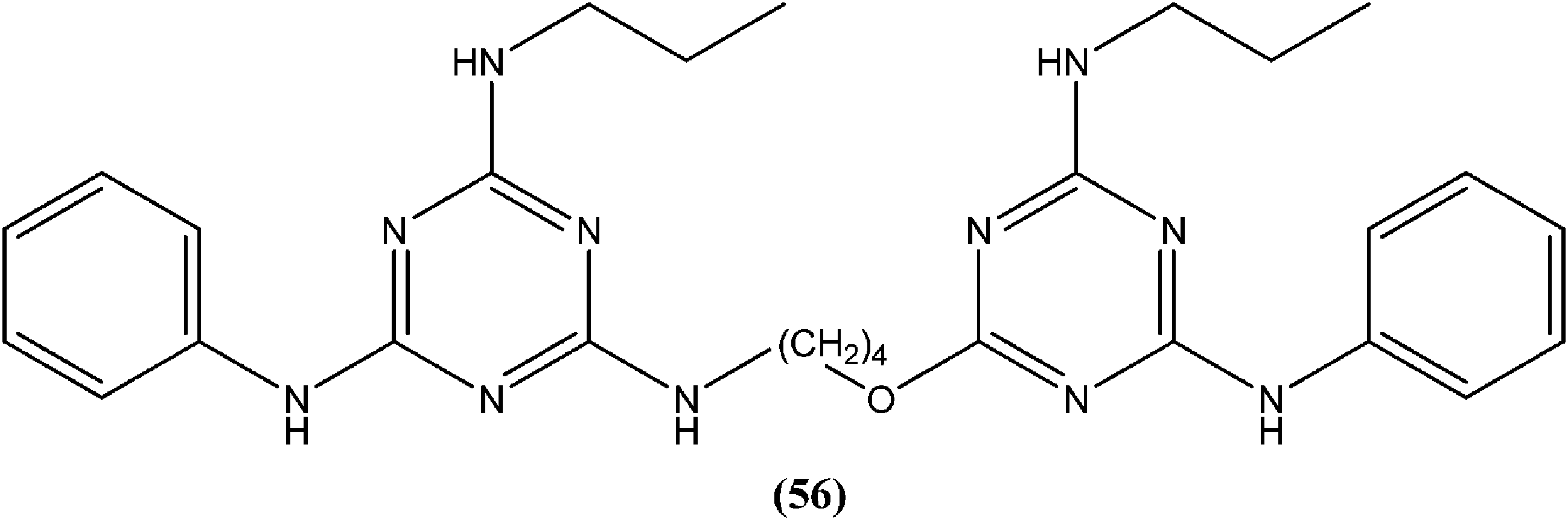
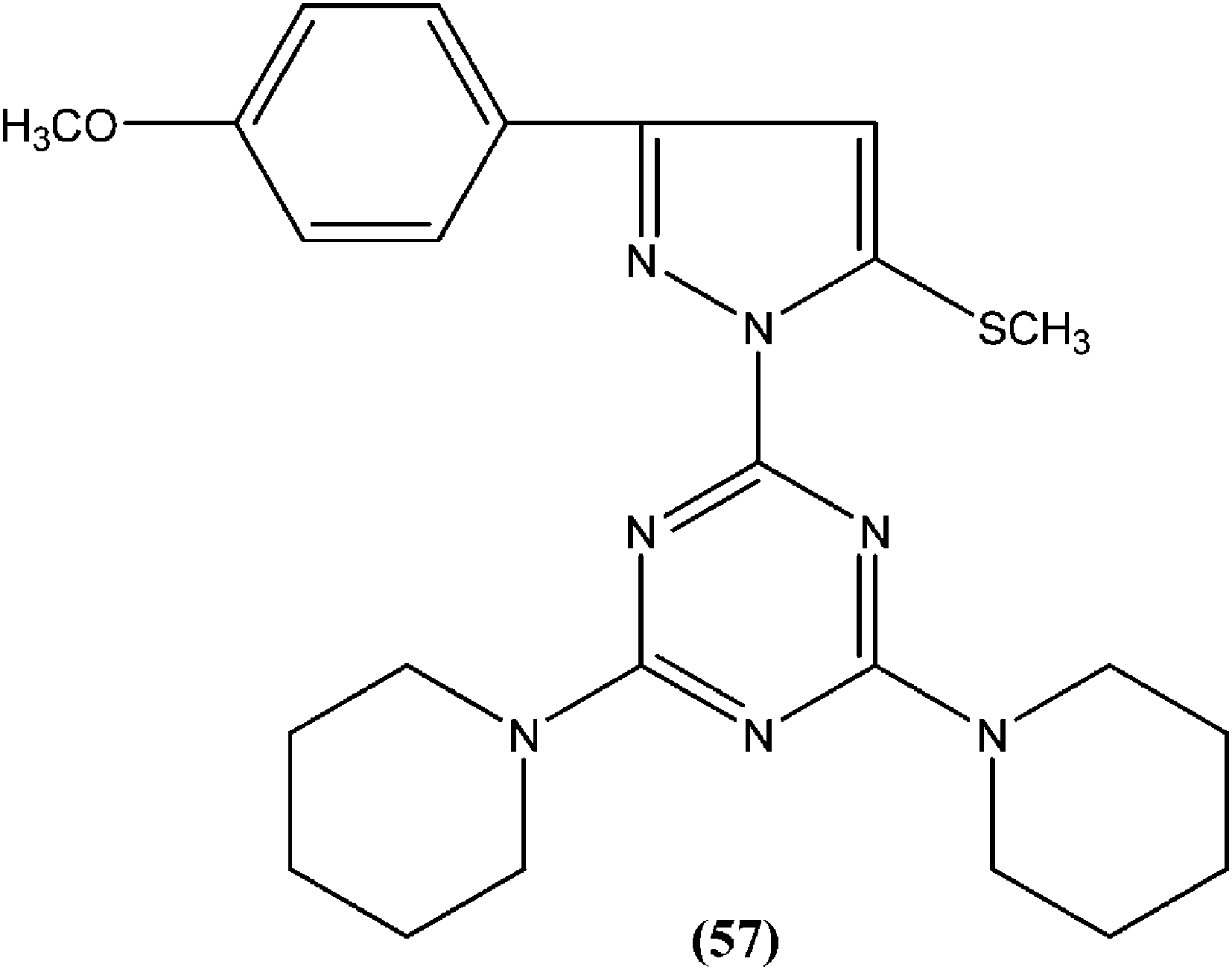
Chauhan et al. synthesized a series of 2,4,6-trisubstituted pyrimidines and triazines, and screened these for in vitro antileishmanial activity in a promastigote model of L. donovani. A 2,4,6-trisubstituted triazine compound (57) having piperidine substituted at the fourth and sixth positions showed inhibition of 98%, 94%, 78%, and 73% against the promastigote form of L. donovani at concentrations of 2.14, 1.07, 0.43, and 0.21 μM, respectively.151
4.14. Purine
Silva et al. reported synthesis and in vitro antileishmanial evaluation of a series of 6-substituted purines. Compound (58) showed the most potent activity (IC50 = 29 μM) against L. amazonensis promastigote form. Interestingly, none of the compounds were found to have significant toxicity towards mammalian cells (mouse peritoneal macrophages) at the maximal concentration used (227 mM).152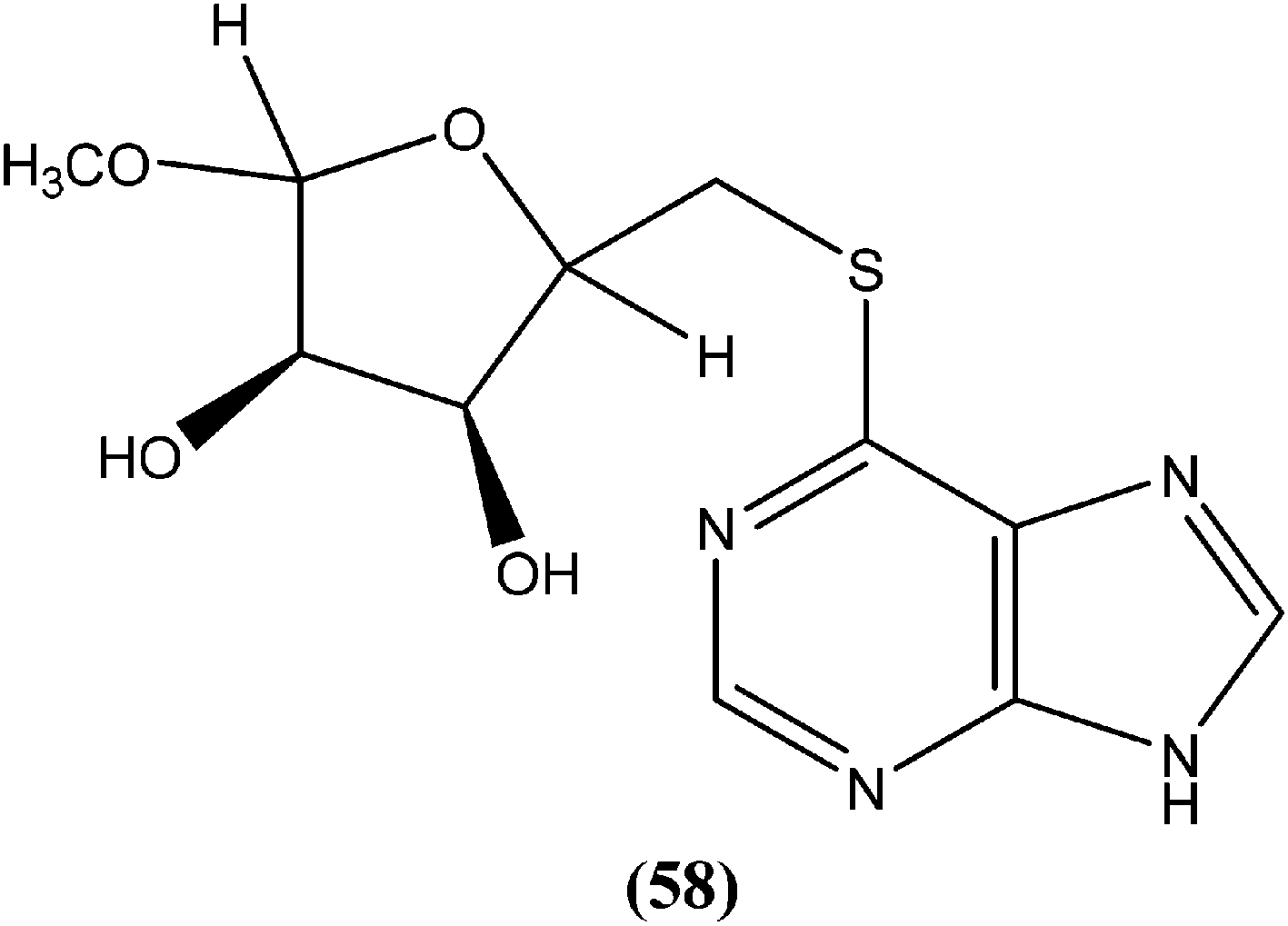
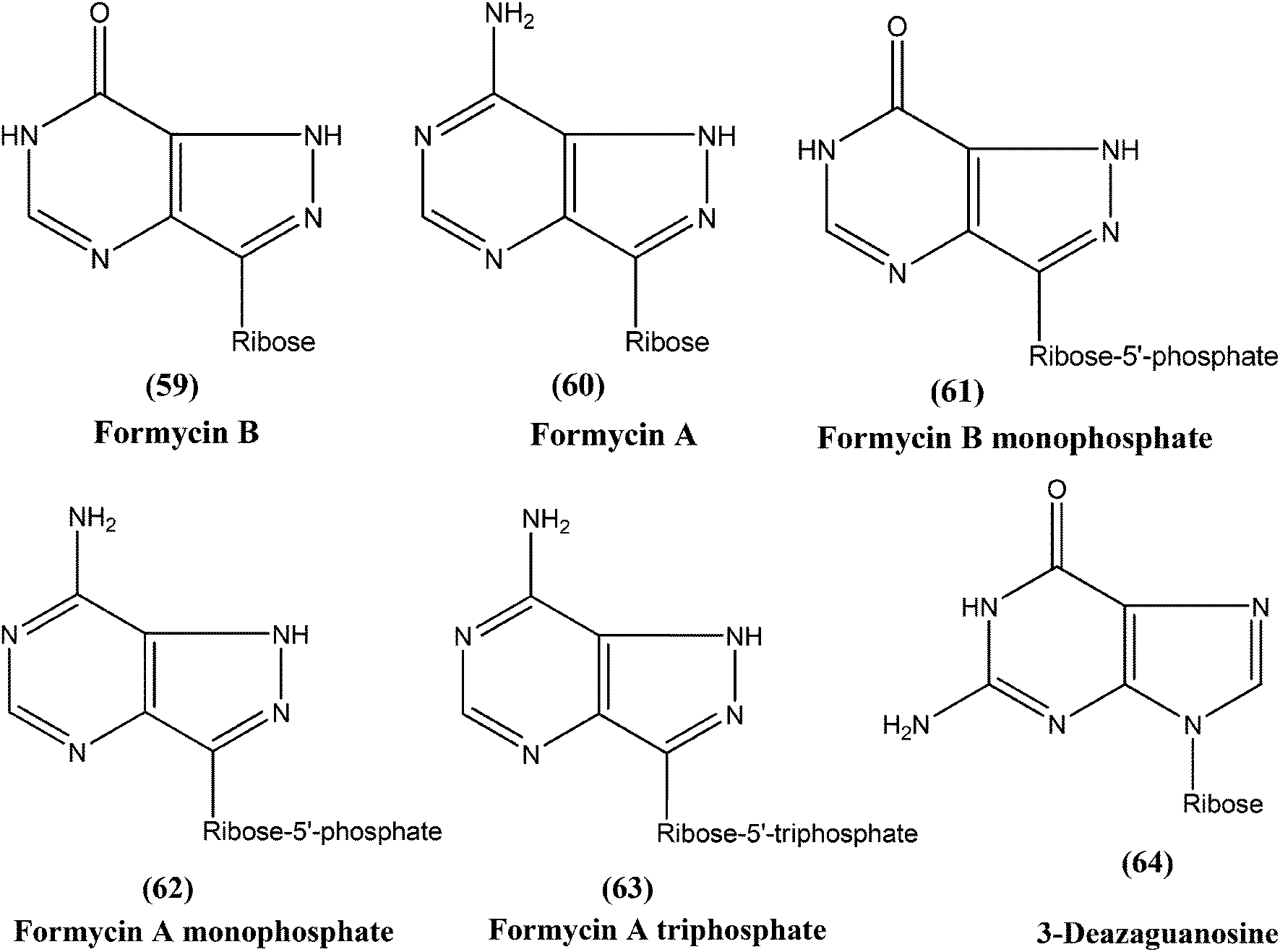
Berman et al. noted that formycin B (59), formycin A (60), formycin B and A monophosphate (61 and 62), and formycin A triphosphate (63) all had 50% effective doses of 0.02 to 0.04 μM and eliminated 90% of organisms at ≤0.5 μM, and therefore were the most active agents with favorable therapeutic–toxic ratios when tested in vitro against L. tropica infected human macrophages. They reported that the activity of 3-deazaguanosine (64) (EC50 = 3.6 μM) in the same model suggested that guanosine derivatives may have potential as antileishmanial agents. They concluded that the apparent mechanism of action of formycin B is that it gets metabolized to formycin B monophosphate, formycin A monophosphate, then formycin A triphosphate by the organisms, which then incorporate the triphosphate form into RNA.153–155
4.15. Pyrimidine
Perez-Perez et al. synthesized two series of 5′-triphenylmethyl(trityl)-substituted thymidine derivatives and tested them against L. infantum axenic promastigotes and amastigotes. Compound (65), having dipeptides coupled at the third position showed good leishmanicidal activity against intracellular parasites, similar to that observed for the control drug edelfosine (87% decrease in the number of infected macrophages), with an estimated IC50 value of 8.0 ± 0.15 μM. After performing an assay of this compound, they concluded that mitochondrial nuclease LiEndoG (L. infantum endonuclease G) was a target for the action of this family of compounds.156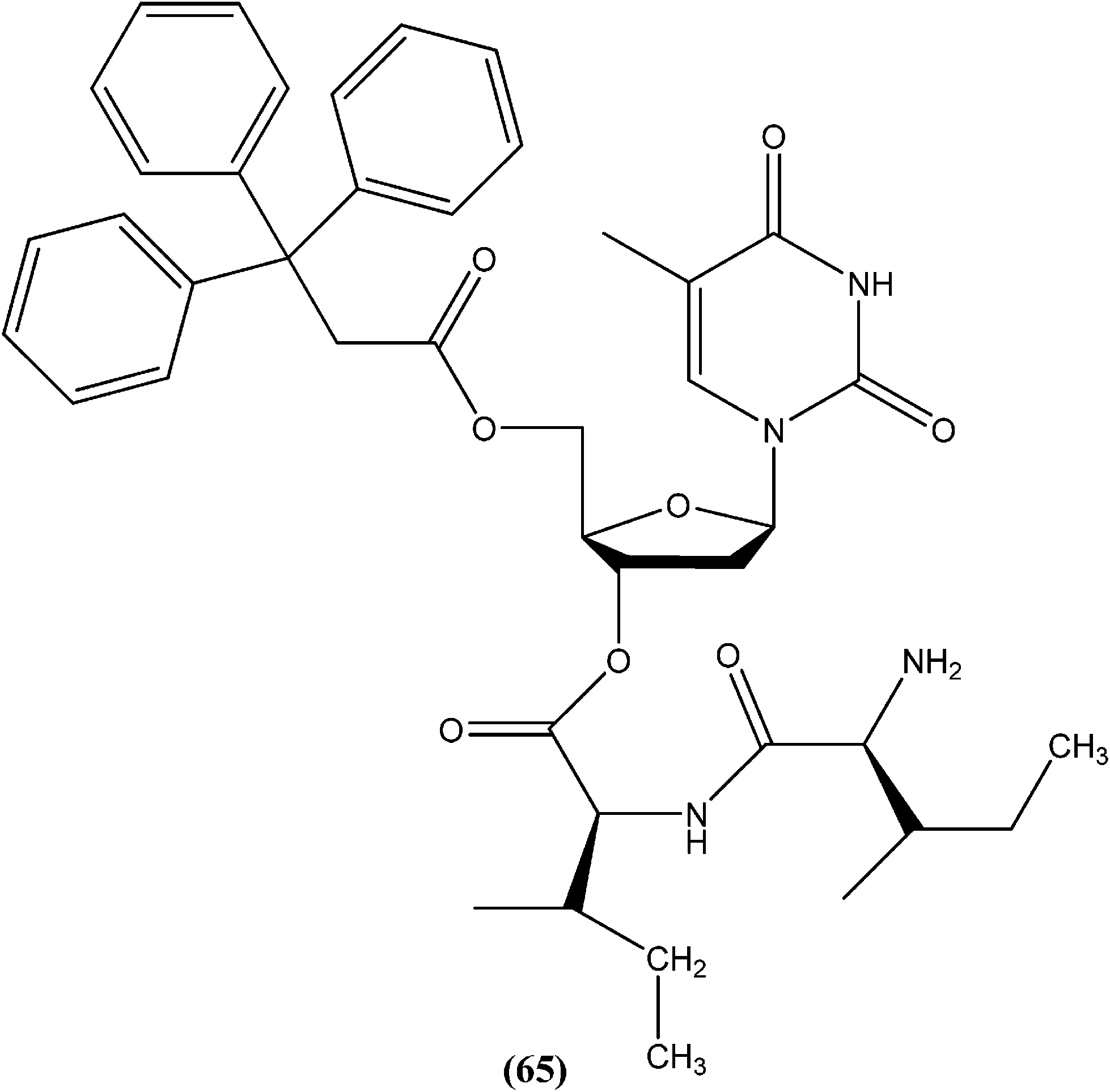
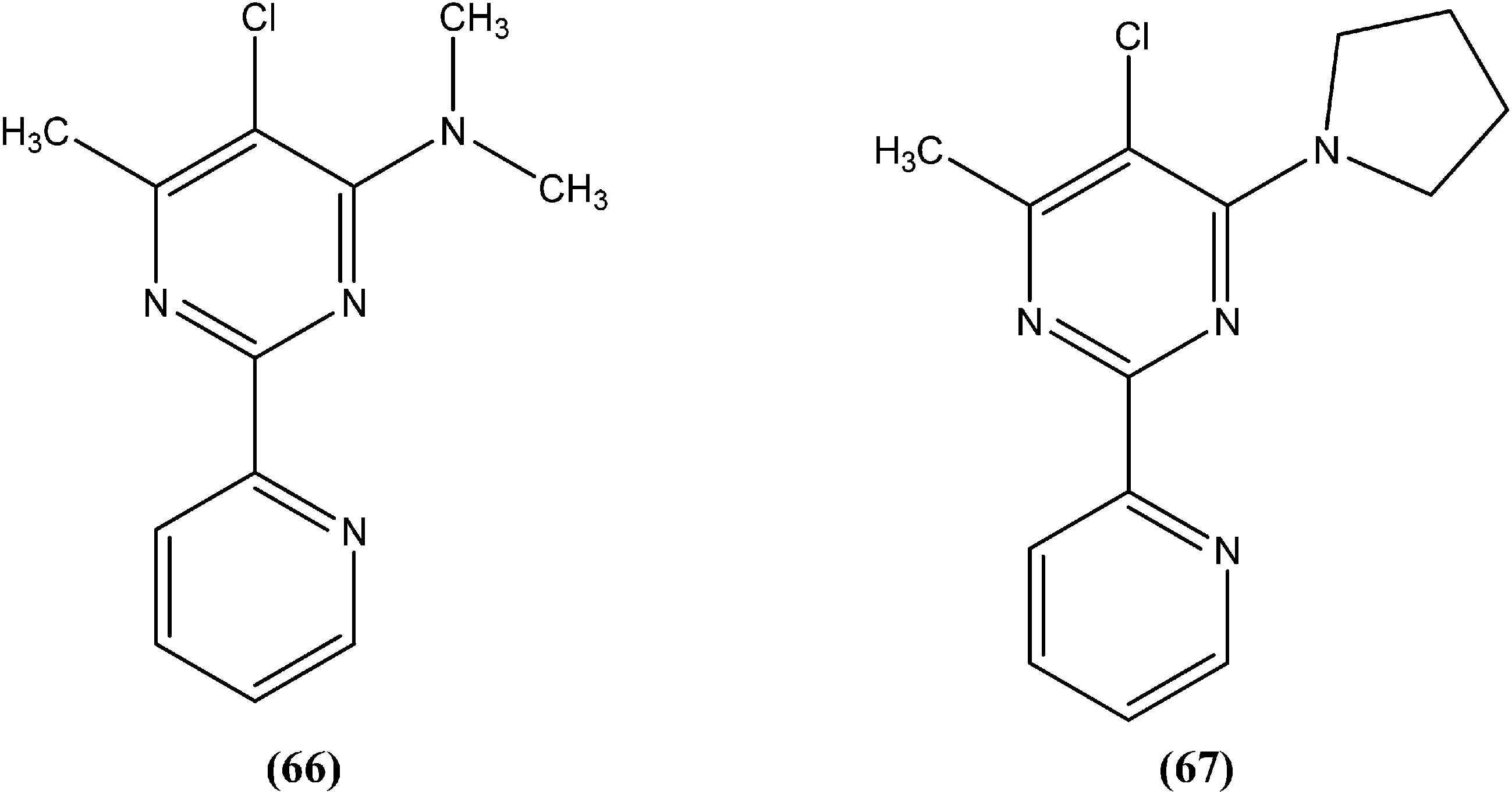
In early 2000, it was reported that methionine aminopeptidase 2 (MetAP2) inhibitors such as fumagillin and TNP-470, arrest parasite growth in L. donovani parasites.157 As a part of a collaboration between Pfizer and WHO-TDR to discover new hits and leads to treat neglected tropical diseases,158 Chen et al. reported synthesis and SAR study of the 2-(2-pyridinyl)-pyrimidine scaffold as an antileishmanial agent.159 Whitlock et al., researchers from Pfizer Global R&D, Swiss Tropical Institute, and University of Cape Town, reported that analogs (66) and (67) had the best combination of clogP (2.7 and 2.9, respectively) and L. donovani activity (IC50 values as 1.1 and 0.53 μM, whereas cytotoxic IC50 of 115 and 80 μM, respectively), therefore these could form the basis for a hit-to-lead program to identify additional compounds with increased L. donovani potency.160
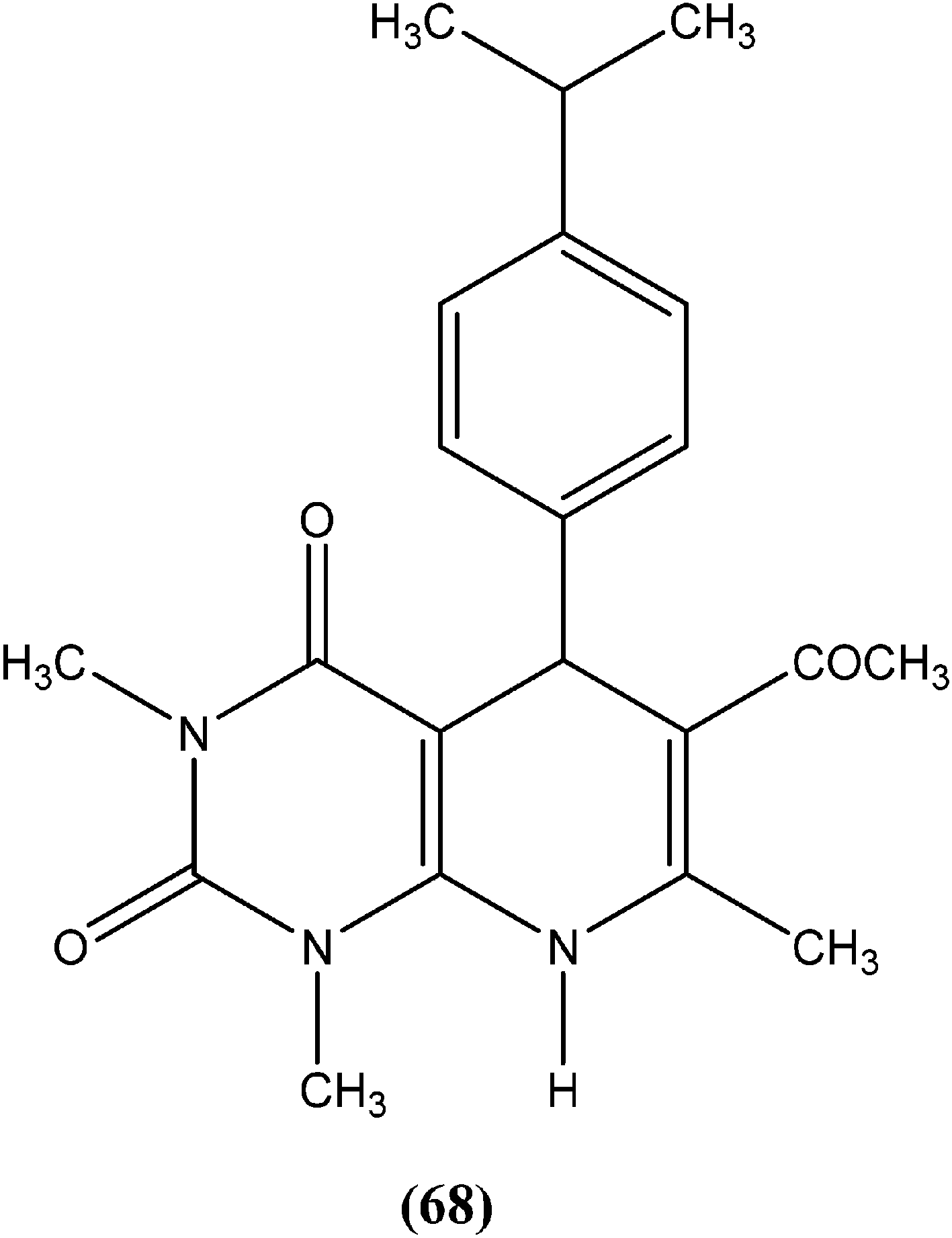
Chauhan et al. synthesized a series of dihydropyrido[2,3-d]pyrimidines and screened them for in vitro antileishmanial activity in L. donovani promastigote and amastigote models. At a concentration of 13.62 μM, compound (68), with substitution of the phenyl ring with an isopropyl group at the para position of dihydropyrido[2,3-d]pyrimidine, exhibited 100% activity in both forms of the parasite. At a concentration of 2.72 μM, it also showed 84.2% and 94.2% inhibition of promastigote and amastigote forms of L. donovani, respectively.161
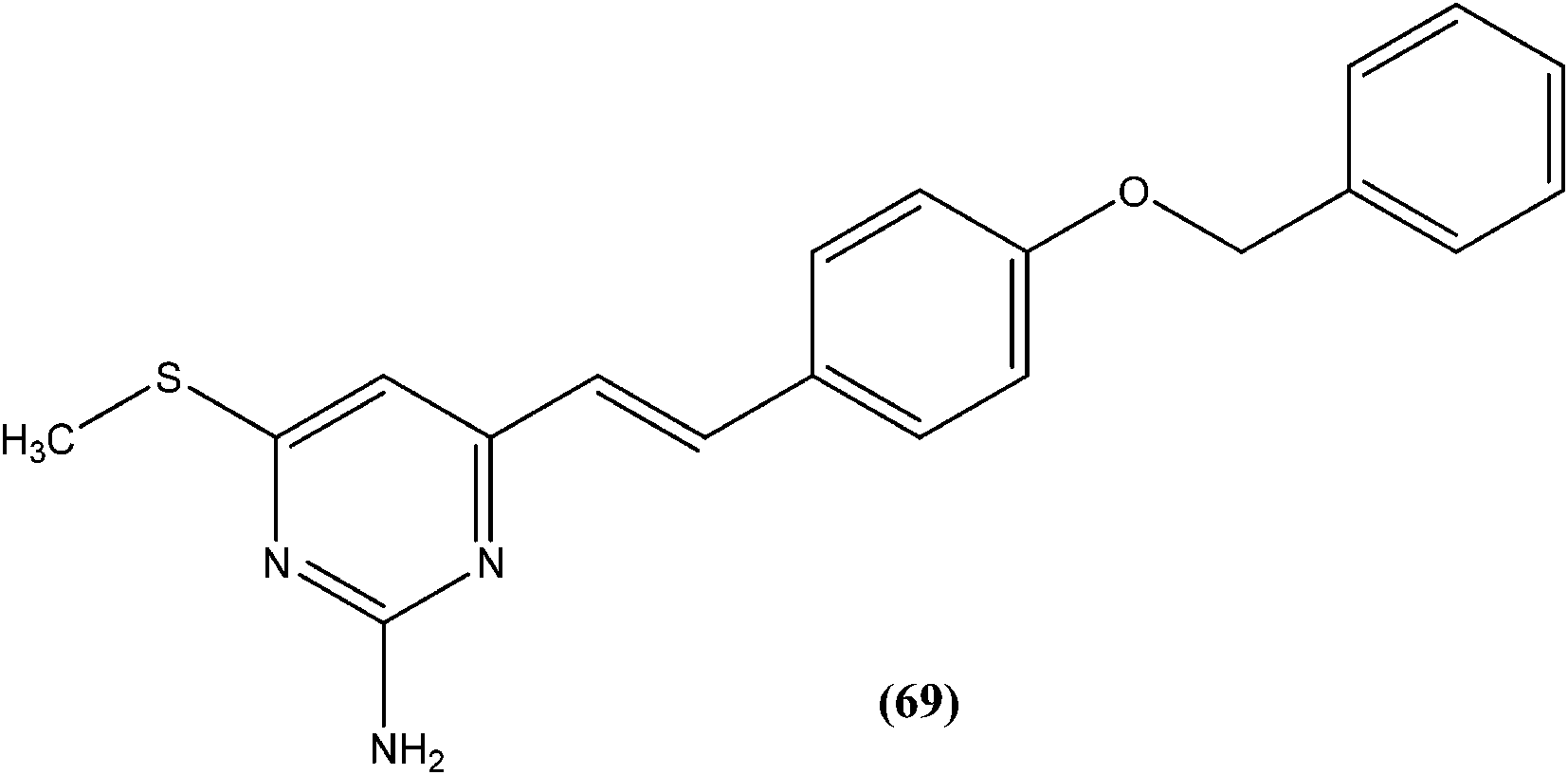
Suryawanshi et al. reported a series of substituted aryl pyrimidine derivatives evaluated in vitro for their antileishmanial potential against intracellular amastigote form of L. donovani using reporter gene luciferase assay. Among the 4-S-substituted pyrimidine derivatives and 4-N-substituted pyrimidine derivatives, they found that compound (69), a 4-S-substituted pyrimidine derivative having benzyloxy aryl substitution, was the most promising, with IC50 and CC50 (on Vero cell line) values of 2.0 ± 0.1 and 375.9 ± 5.1 μM, respectively, whereas its SI was found to be 188. On the basis of the SI, some compounds from both series were further evaluated for in vivo antileishmanial activity using a L. donovani hamster model. Again, compound (69) when administered intraperitoneally, had shown significant inhibition of parasitic multiplication (88.4%) at a daily dose of 50 mg kg−1 × 5 days. Therefore, the respective researchers concluded that compound (69) was the most promising, and may provide a new lead as an antileishmanial agent.162
4.16. Hydrazone/Schiff base
Taha et al. synthesized a library of Schiff bases of 2-methoxybenzoyl hydrazide. After evaluation of the compounds for in vitro antileishmanial activity, they found that compound (70), having 2-hydroxy-4-methoxy substituents on ring A, showed excellent activity (IC50 = 1.95 ± 0.04 μM). They suggested that 2-hydroxy substitution on ring A along with a methoxy group is vital for antileishmanial activity of this type of compound.163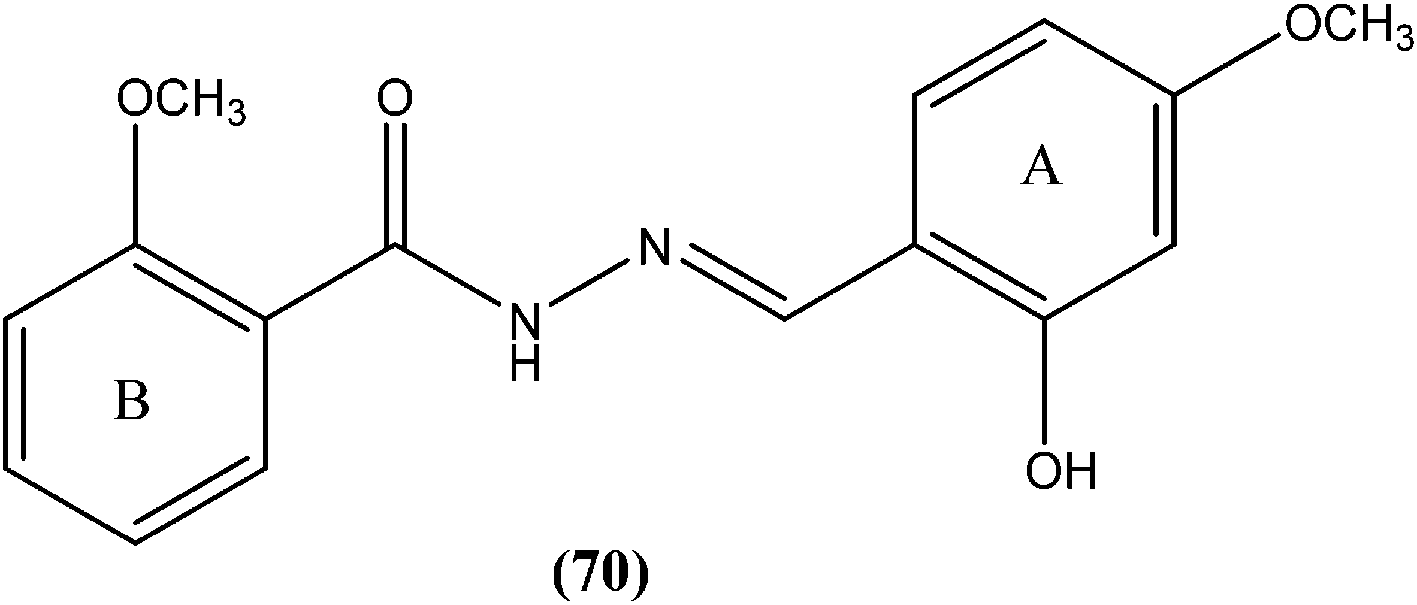
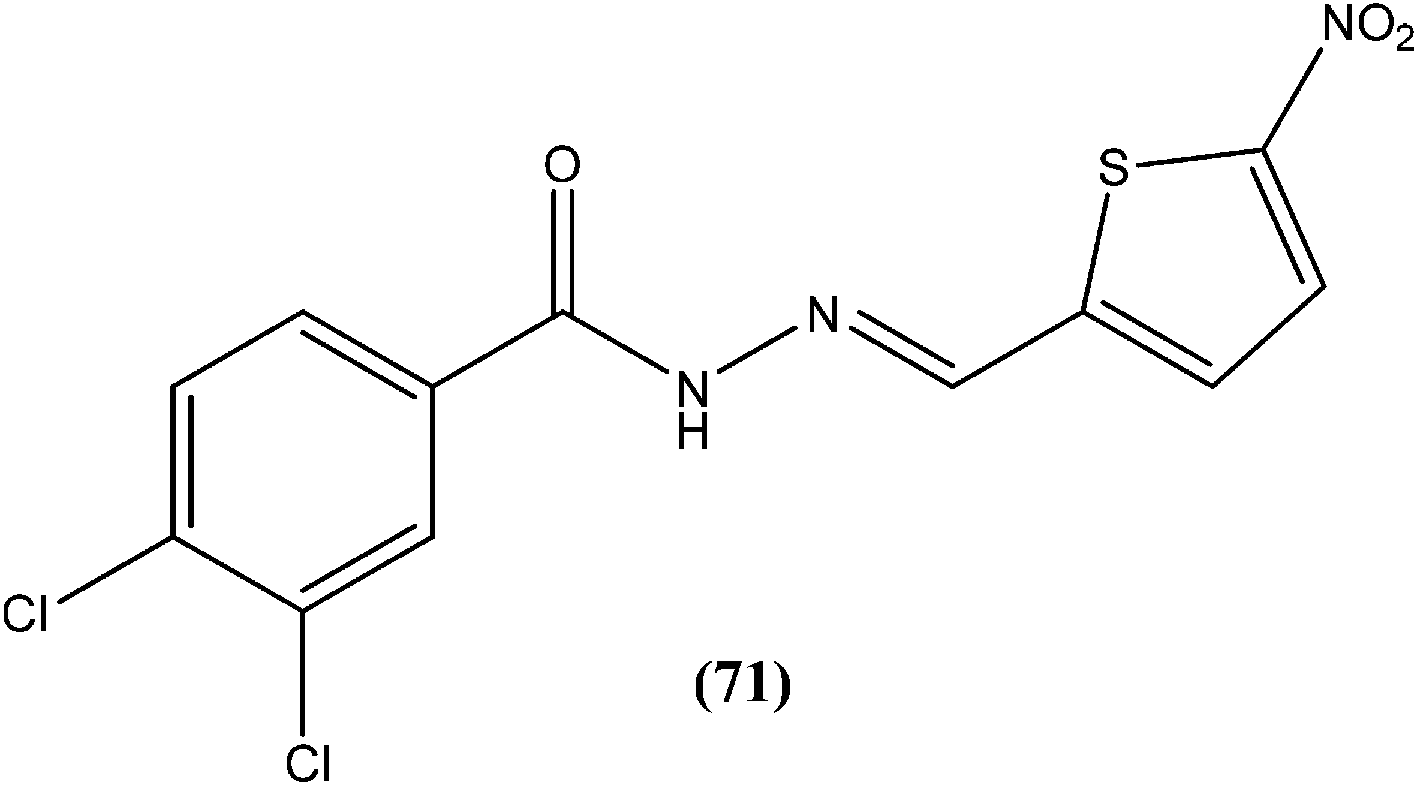
Rando et al. synthesized a series of nitro derivatives and screened these against L. donovani promastigote forms. They reported that nitrothiophene analogs were more potent than nitrofuran ones. Among the nitrothiophene analogs, compound (71) containing chloro substitutions at the meta and para positions of the phenyl ring, showed an IC50 value of 0.41 μM (with IC90 as 0.87 μM), which was lower than that of standard drugs pentamidine (IC50 = 1.06 μM) and amphotericin B (IC50 = 1.19 μM), and thus compound (71) was identified as the most potent in the series. They also mentioned that substitution was important for the activity. Further, they explained that the potency of nitrothiophene analogs was attributed to the ability of sulfur atoms to accommodate electrons from nitro groups, facilitating reduction and therefore formation of free radicals that are lethal to the parasites.164
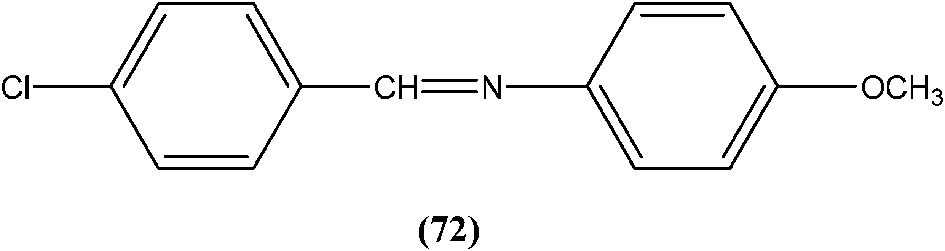
Yasinzai et al. reported that synthesized azomethines inhibited parasite growth and most showed highly potent action towards L. major promastigotes. Of these, the most potent compound (72) had an IC50 of 0.23 μM.165
4.17. Steroids
Silva et al. reported synthesis of steroids and in vitro antileishmanial screening against promastigote forms of L. amazonensis, L. braziliensis, and L. major. Compound (73), a 6-thiopurine/steroid conjugate was found to be active with IC50 values of 22.8, 13.9, and 17.3 μM for L. amazonensis, L. braziliensis, and L. major, respectively. The compound showed no toxicity on mouse peritoneal macrophages at the maximum concentration tested (100 μM).166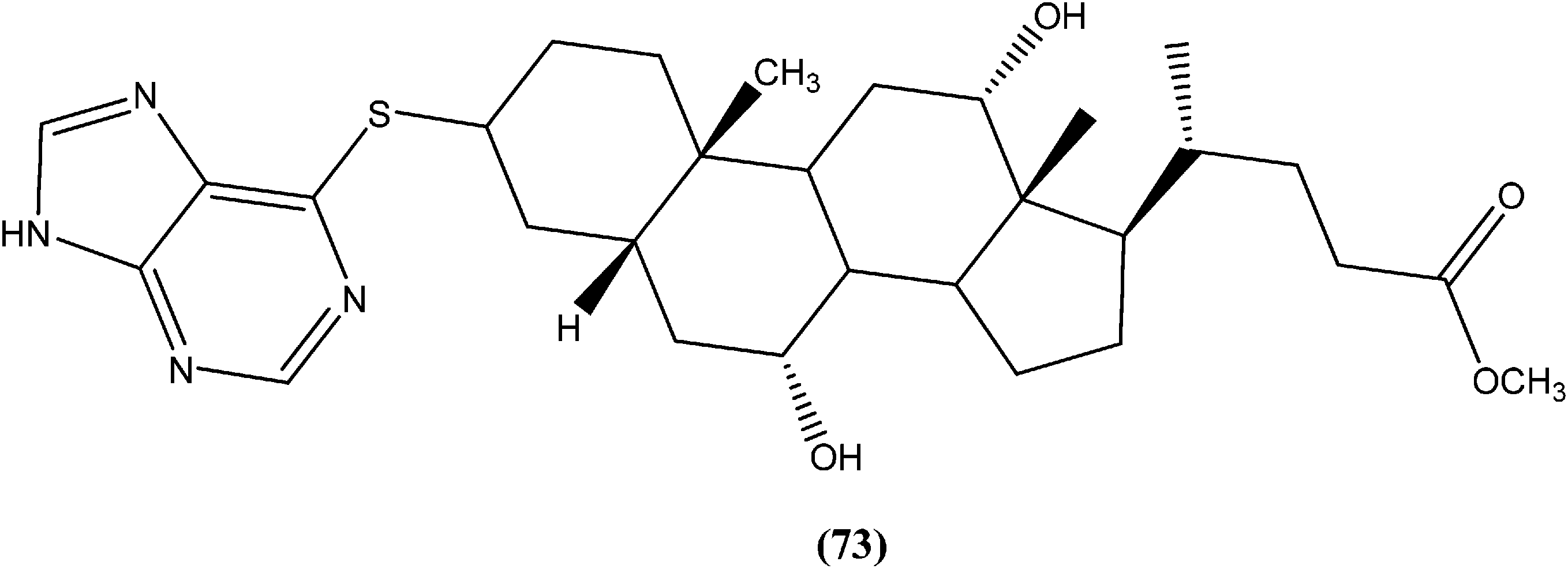
4.18. Azoles
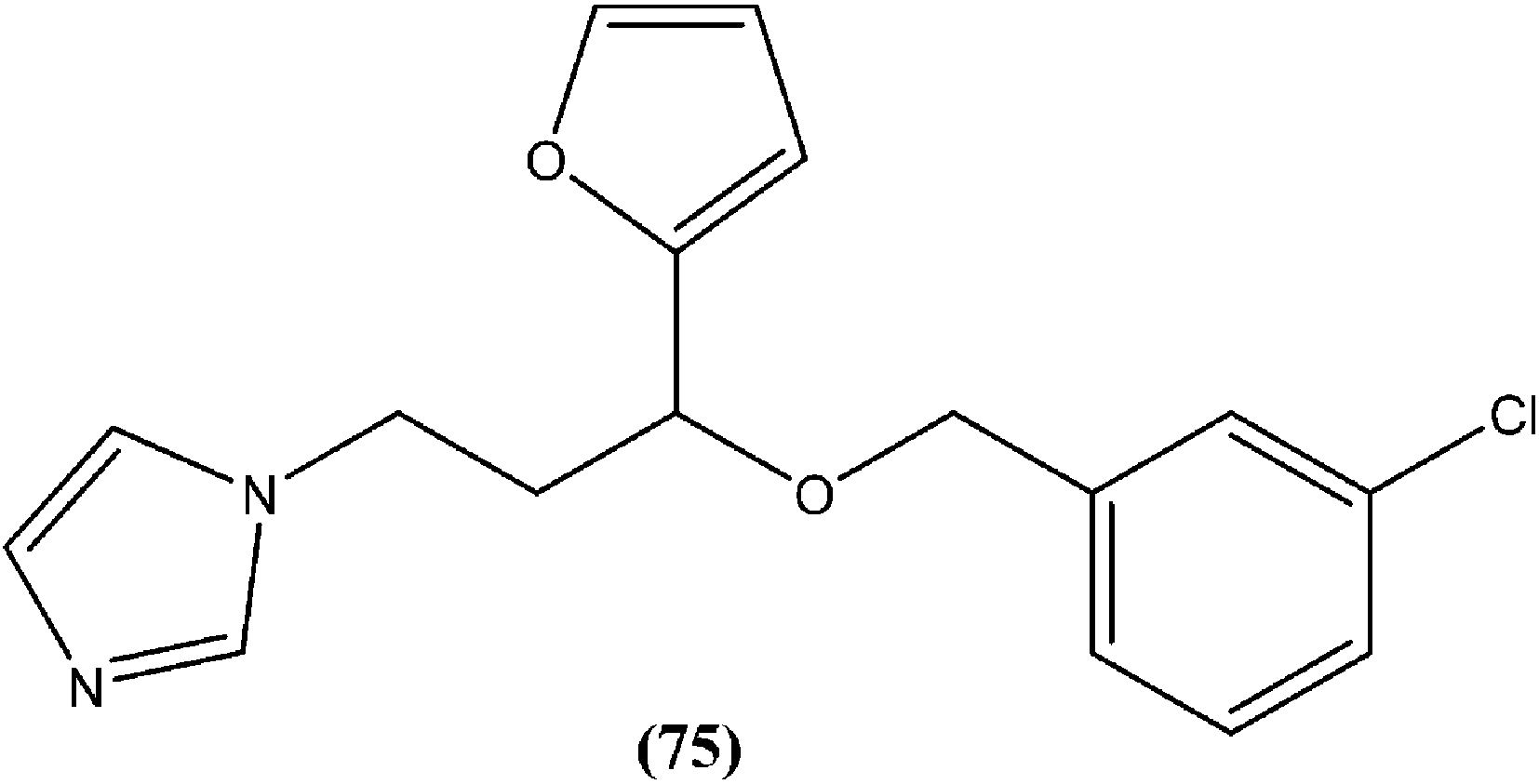
In a study on synthesis and evaluation of new furanyl and thiophenyl coupled imidazoles as antileishmanial agents, Bhandari et al. synthesized a series of benzyloxy furanyl and benzyloxy thiophenyl imidazoles and performed screening for their in vitro antileishmanial activity against both forms of L. donovani. Among the tested compounds that were found to be several times less toxic (against J774A.1 cell line) than reference drugs miltefosine (CC50 = 3.23 μM) and miconazole (CC50 = 9.93 μM), compound (75), a 3-chlorobenzyloxy furanyl imidazole, emerged as the most active, with an IC50 value of 3.04 μM (CC50 being 60.21 μM) and SI of 19.80, which was a better SI than those of miltefosine (SI = 0.24) and miconazole (SI = 1.66).168
Gupta et al. reported antileishmanial activity of synthetic oximino benzocycloalkyl imidazoles evaluated in vitro against extracellular promastigote and intracellular amastigote forms of L. donovani. Compound (76), with 5-chlorotetrahydronaphthyl and 3-chlorobenzyl moieties, showed 93.41% inhibition during anti-promastigote activity testing at a concentration of 2.5 μM. It also showed anti-amastigote activity, with an IC50 value observed at 0.23 μM. With CC50 of 5.95 μM (on mouse macrophage cell line: J-774A.1), its SI was found to be 25.59. Also, in in vivo testing in a hamster model, compound (76) showed 70.13 ± 5.23% inhibition of parasite.169

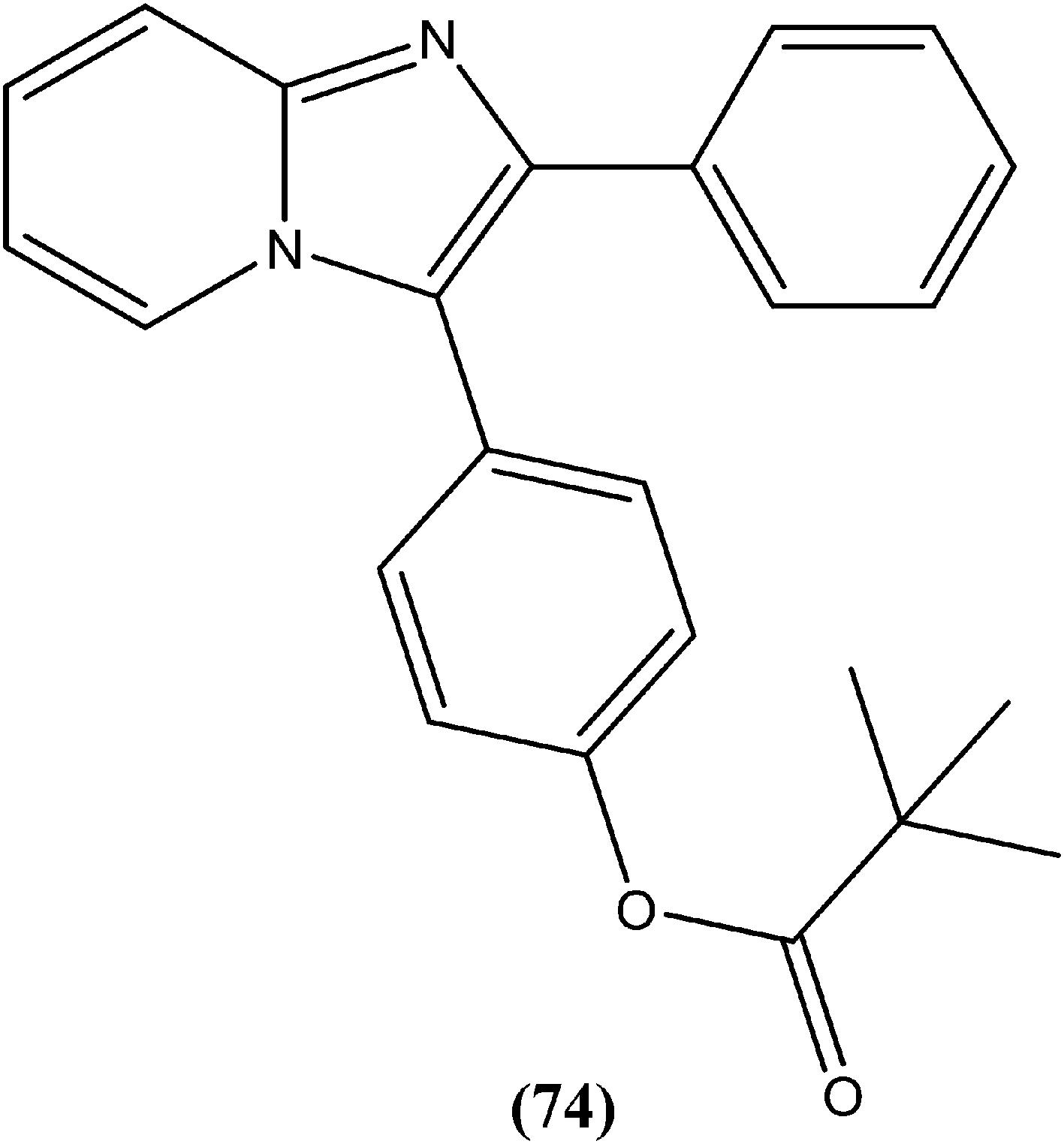
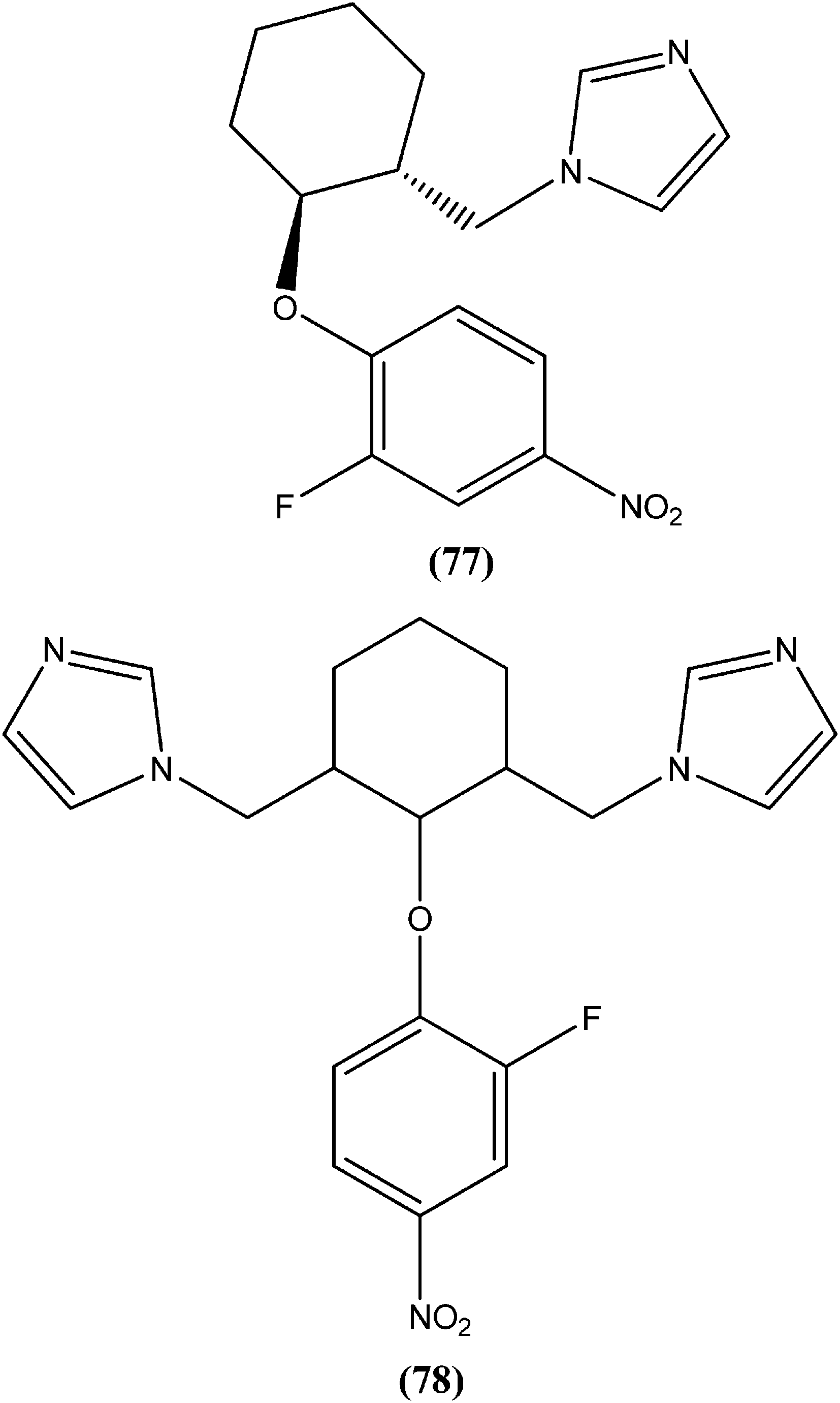
Bhandari et al. reported preparation of aryloxy cyclohexane-based mono and bis imidazoles and their in vitro antileishmanial activities against L. donovani, along with a cytotoxicity study using a mouse macrophage cell line (J-774-A.1). Their in vitro studies revealed that compound (77) was the most potent among the series, having IC50 anti-promastigote, IC50 anti-amastigote, and SI values of 0.34 μM, 0.22 μM, and 140.84, respectively. The authors reported that compound (77) was better than existing drugs sodium stibogluconate (SI = 6.38) and pentamidine (SI = 2.58). After in vivo assay along with other promising compounds in a L. donovani/hamster model, compound (77) showed 55.35% inhibition. Bis methylimidazole (78) containing a 2-fluoro, 4-nitro aryloxy group (in vitro IC50 anti-promastigote, IC50 anti-amastigote, and SI of 0.89 μM, 0.29 μM, and 33.64 respectively), exhibited significant inhibition of 77.9%. In terms of SAR of the synthesized compounds, the researchers explained that the highest activity (in vitro as well as in vivo) was shown by compounds containing 2-fluoro and 4-NO2 aryloxy moieties. They suggested that aryloxy moiety with 2-fluoro and 4-NO2 substituents should be investigated for development of highly selective antileishmanial compounds.170
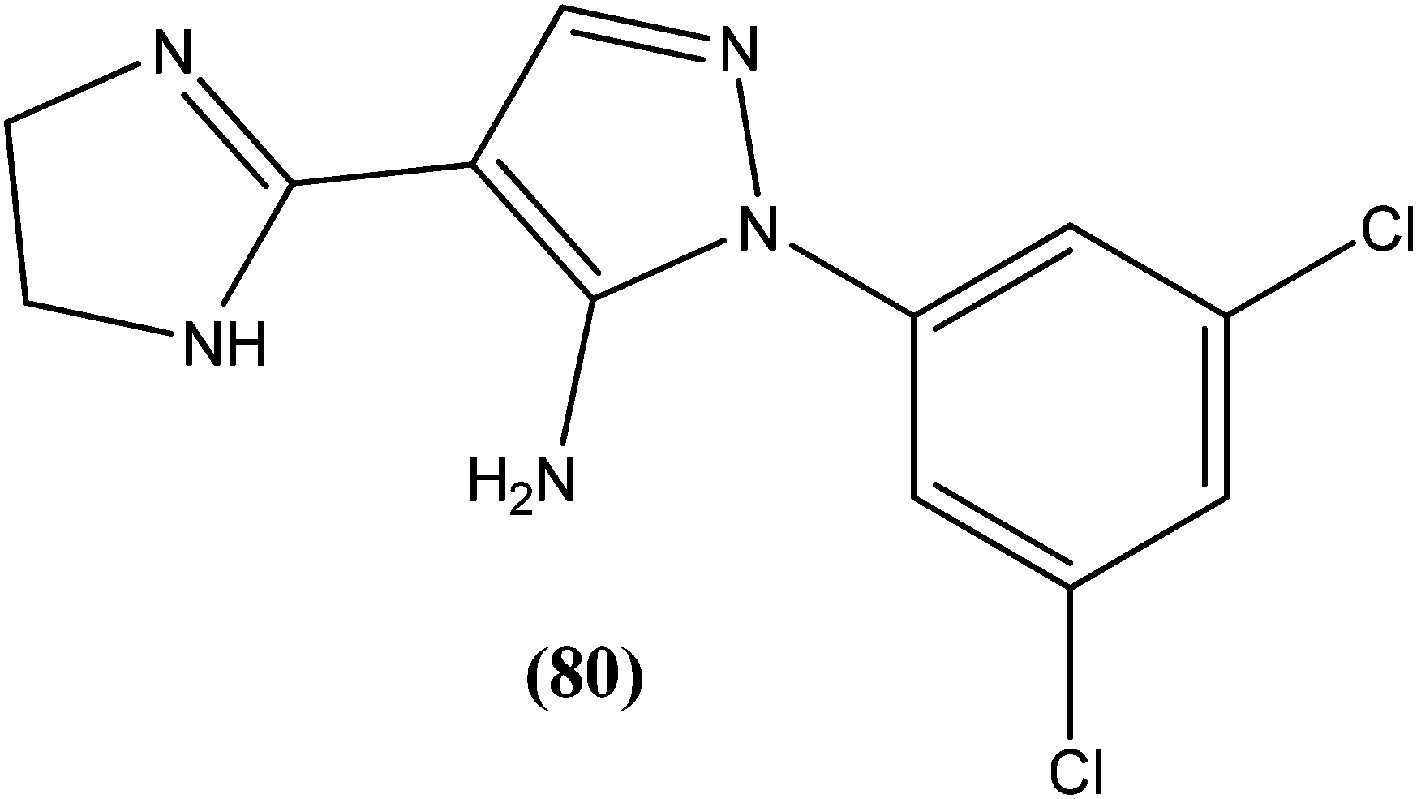
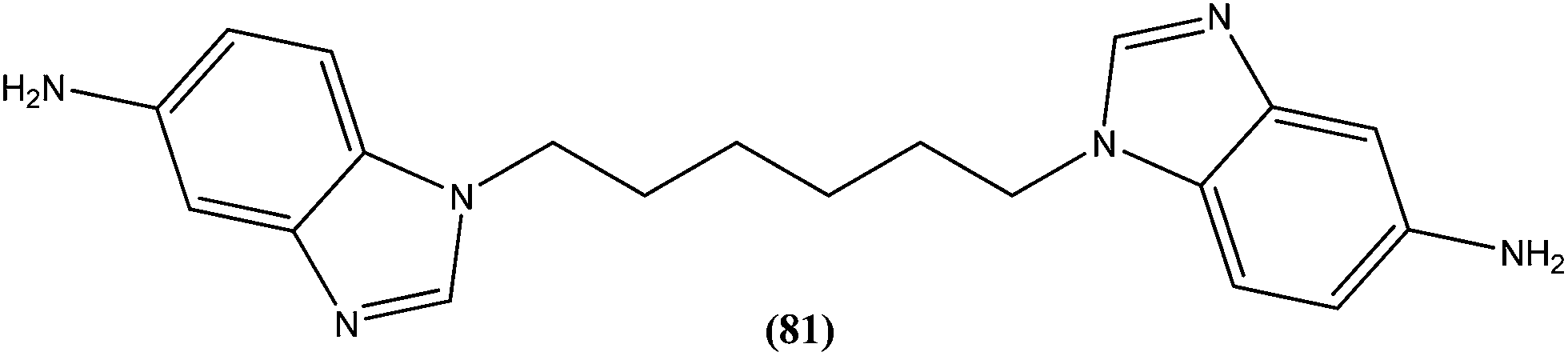
Eynde et al. prepared a small library of 2,2′-[(α,ω-alkanediylbis(oxyphenylene))]bis-1H-benzimidazoles. After in vitro screening, the synthesized derivatives emerged as promising hits characterized by IC50 values lower than that determined for pentamidine against L. donovani. Compound (82) had the lowest IC50 of 1.4 μM and IC90 of 3.1 μM, with an IC50 of 28.7 μM against Vero cells.174
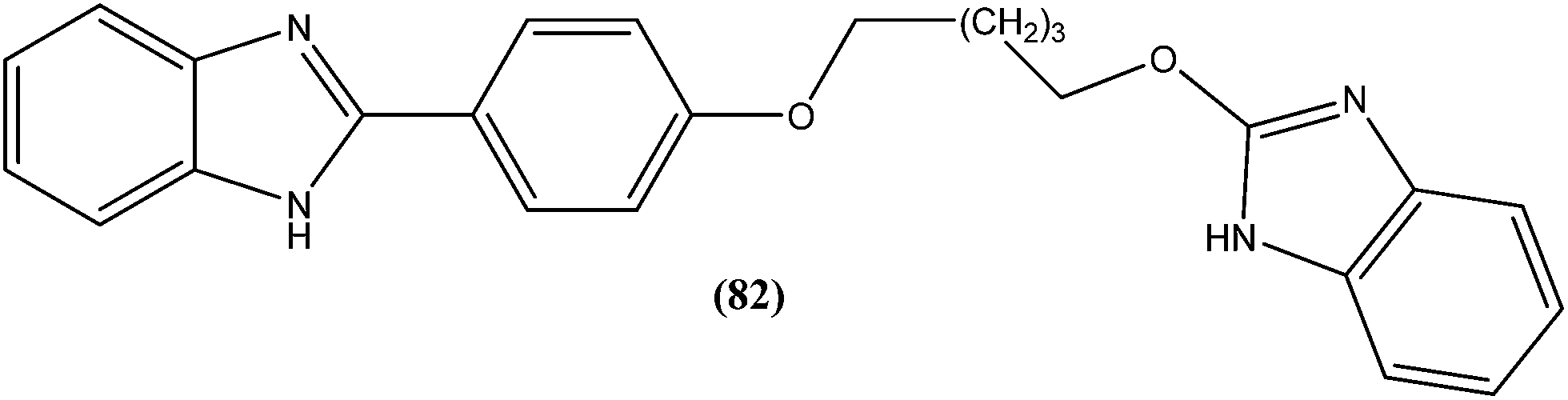
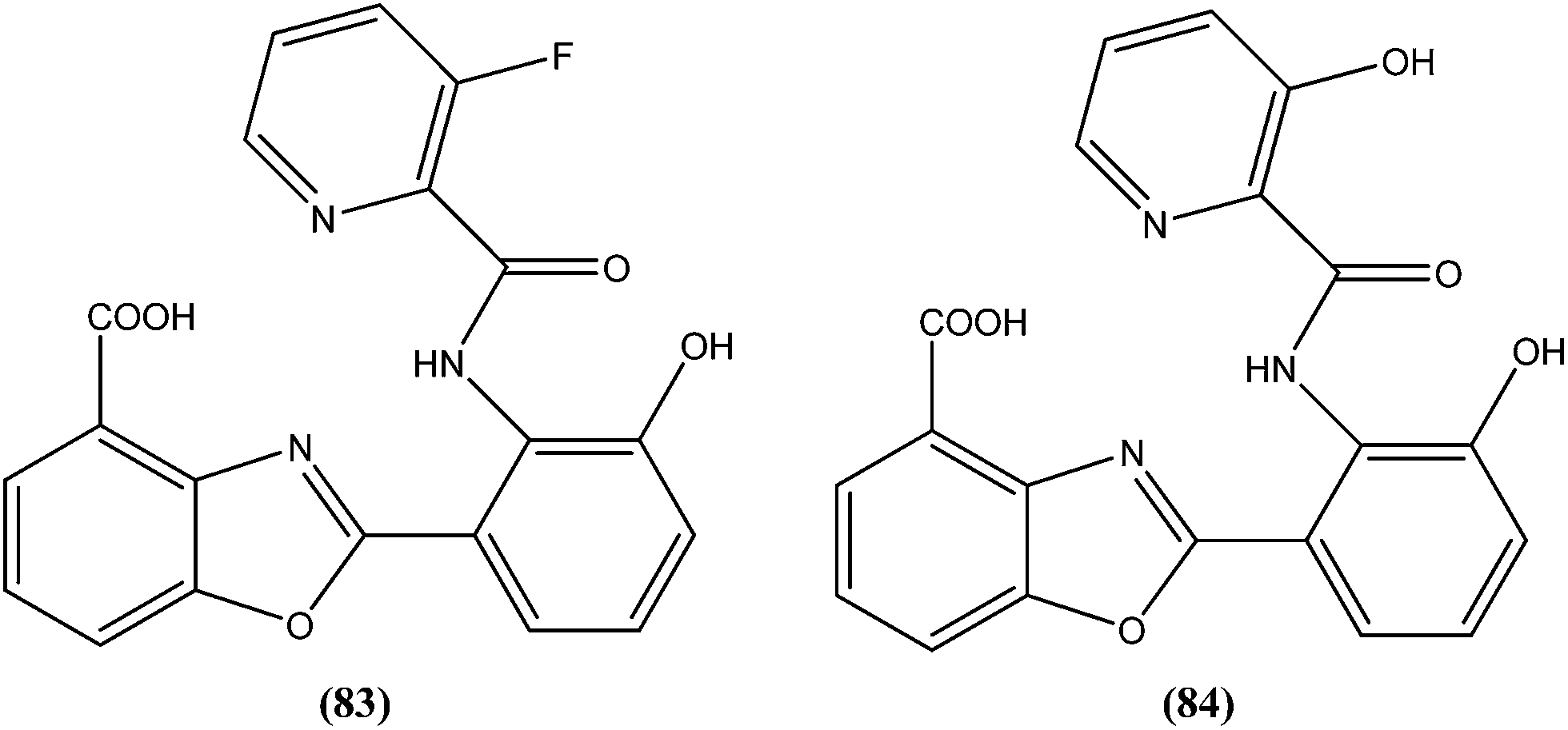
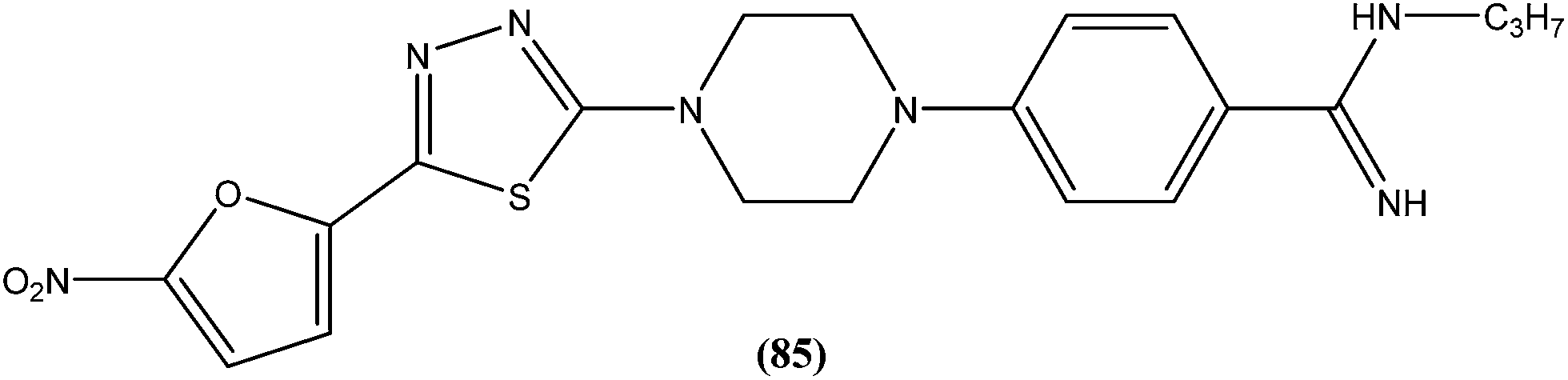
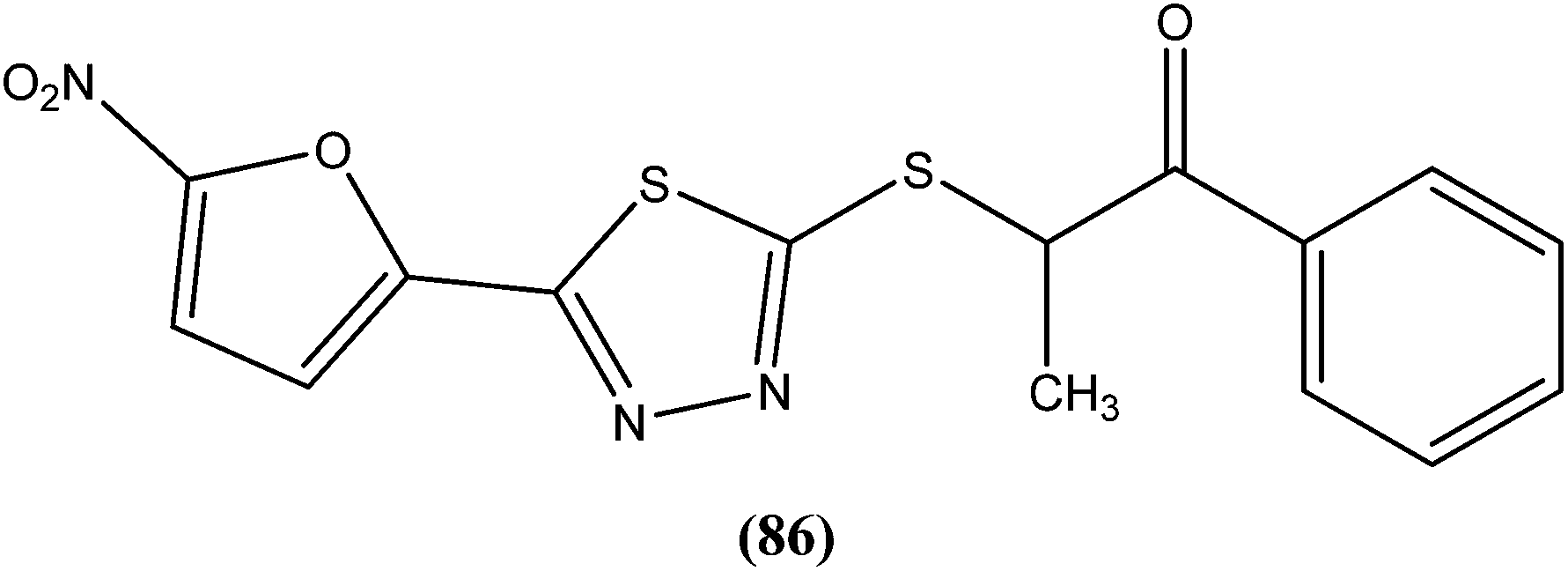
The same researchers also reported synthesis and antileishmanial activity of 5-(5-nitroaryl)-2-substituted-thio-1,3,4-thiadiazoles against the promastigote form of L. major using a tetrazolium bromide salt (MTT) colorimetric assay. Compound (86) appeared to be most potent with a lowest IC50 of 1.11 μM. A structure–activity relationship study indicated that the S-pendant group attached to the 2-position of the thiadiazole ring has high flexibility for structural alteration, therefore retaining good antileishmanial activity.177
5. Brief summary of promising scaffolds
From the above reported synthetic derivatives, we selected the five most promising scaffolds (32, 46, 47, 84, and 85) for antileishmanial activities (Fig. 4). Herein, we discuss these compounds in detail.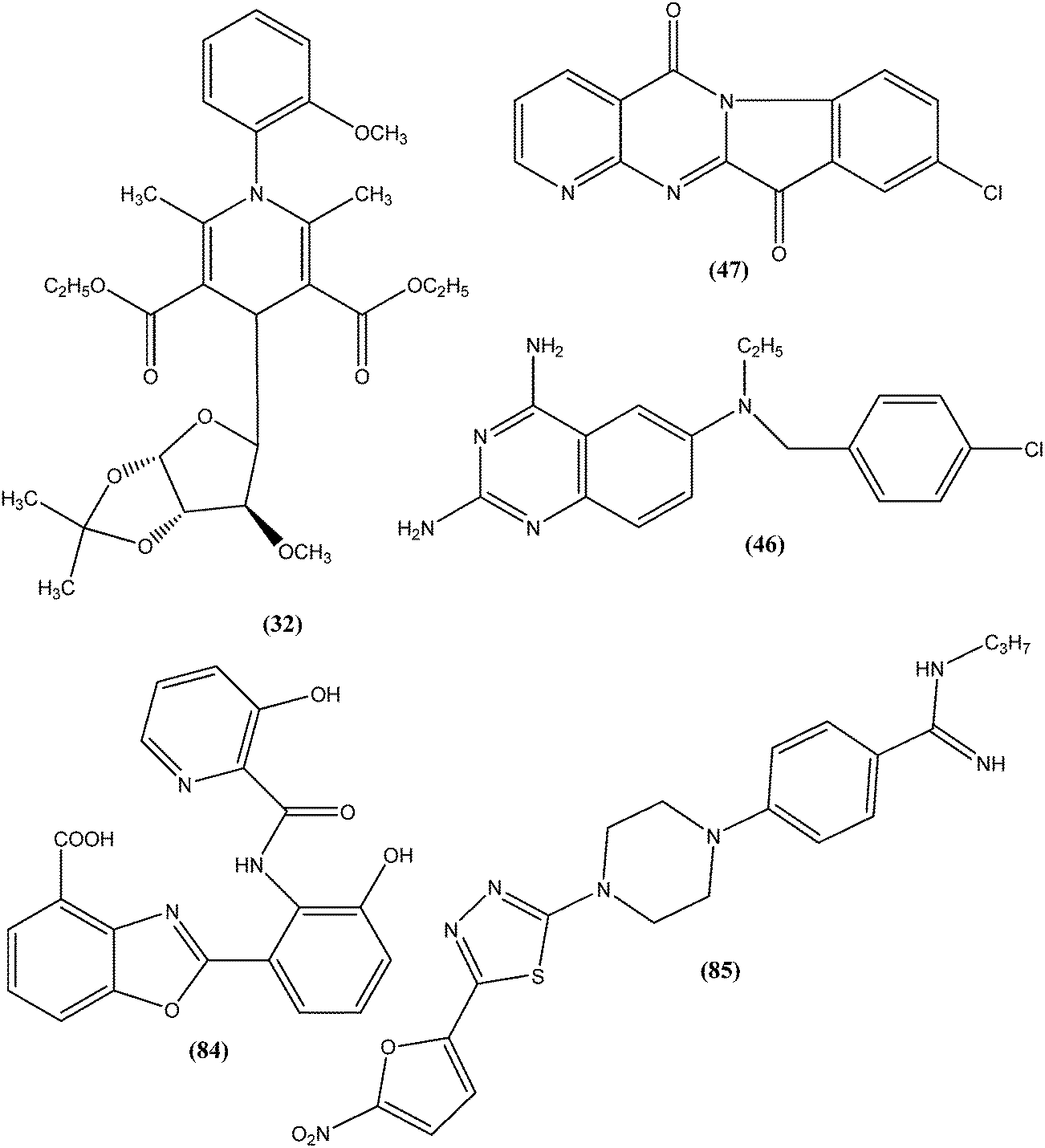 | ||
| Fig. 4 Structure of potential scaffolds for antileishmanial activities. | ||
Potent antileishmanial activities have been reported for a dihydropyridine class of compounds bearing phenyl and other sugar residues at the fourth position of the dihydropyridine ring. To optimize these dihydripyridine derivatives, Tripathi et al. extended the work and synthesized a series of 1-phenyl-4-glycosyl-dihyropyridines, evaluating the in vitro and in vivo activities of these against L. donovani. Compound (32) emerged as a potent antileishmanial agent. It had promising antileishmanial activities against L. donovani with IC50 values of 0.04 μM (anti-promastigote) and 1.16 μM (anti-amastigote) when compared with standard drugs like pentamidine and miltefosine. Compound (32) was also evaluated against J-774A.1 growing cells for cytotoxic activity, and showed good selective index (SI = 8.04). The compound was screened for in vivo activity against L. donovani in a hamster model, showing 49.73% inhibition. A molecular docking study revealed that such compounds inhibit PTR1 (Pteridine reductase 1) enzyme of leishmanial parasites. Thus, compound (32) has potential for further exploration in development of safe and effective antileishmanial drugs.125
Another compound (46) that showed potent antileishmanial activity is from the 2,4-diaminoquinazoline class. The ED50 value of compound (46) was found to be 0.00365 μM against L. major amastigotes. The activity was structurally specific because it depended on an unconstrained tertiary aromatic amine attached directly to the benzyl group of the quinazoline. Compound (46) has been suggested to be primarily leishmaniastatic in nature. It showed inhibition of 88% of L. mexicana promastigote DHFR at a concentration of 7.5 μg mL−1. The remarkable activity of compound (46) suggests that such analogs have potential for investigation as novel antileishmanial agents.141 The compound (47) tryptanthrin derivative has shown promising antileishmanial activity against L. donovani amastigotes (IC50 = 0.000013 μM). In vitro toxicity studies indicate that compound (47) is fairly well tolerated in both murine J774 macrophages and rat neuronal NG-108-15 cell lines. The carbonyl groups of the five- and six-membered rings in the indolo[2,1-b]quinazoline-6,12-dione skeleton and the electron transfer ability to the carbonyl atom appear to be crucial for activity. Compound (47) is found to be less toxic to mammalian cell lines than to Leishmania in vitro. Thus, this compound shows remarkable promise for further study as a potential antileishmanial candidate.142,143
Compound (84), a benzoxazole derivative, has shown potent activity against L. donovani amastigote (IC50 = 0.08 μM) with low cytotoxicity against L6 cells (CC50 = 14.2 μM). This compound is threefold more active than miltefosine (IC50 = 0.26 μM). Thus, discovery of compound (84) underscores the importance of the N-(2-benzoxazole-2-ylphenyl)benzamides as an important lead scaffold in design and synthesis of antileishmanial agents.175 Compound (85) has shown promising antileishmanial activity against L. major promastigotes (IC50 = 0.08 μM) with a very low level of toxicity against macrophages (CC50 = 785 μM and SI = 78.5). The potent activity of compound (85) indicates that propyl substitution on the amidine residue improve antileishmanial activity. Thus, compound (85) is a promising new hit for development of antileishmanial chemotherapy.176
6. Antileishmanial natural products
6.1. Plant origin
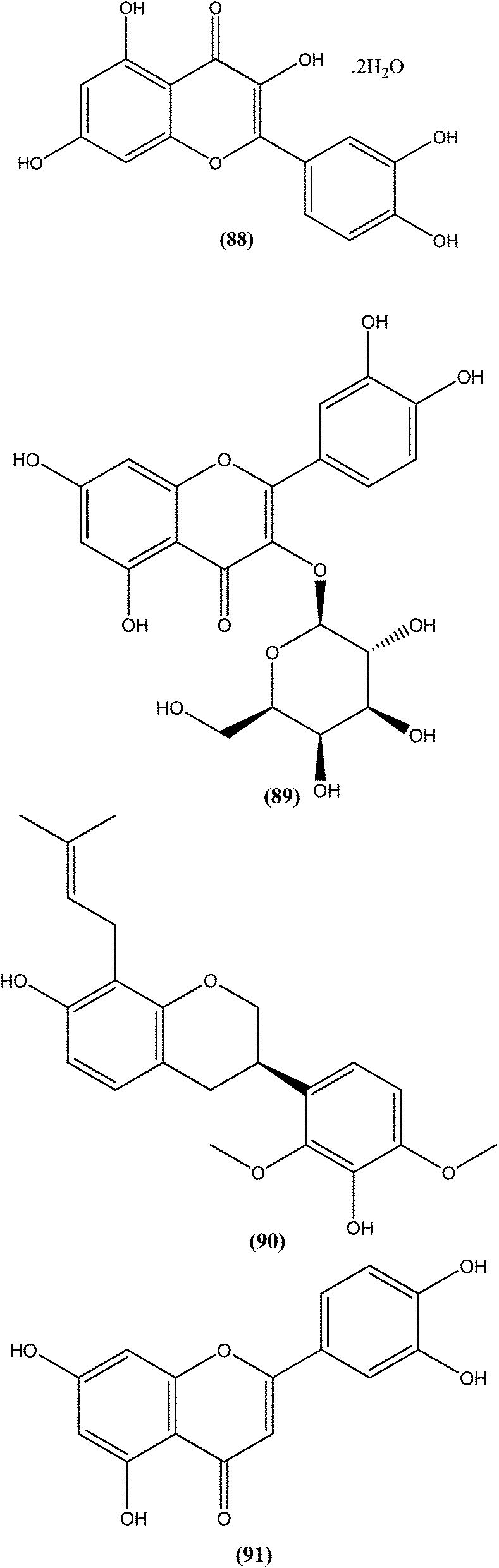
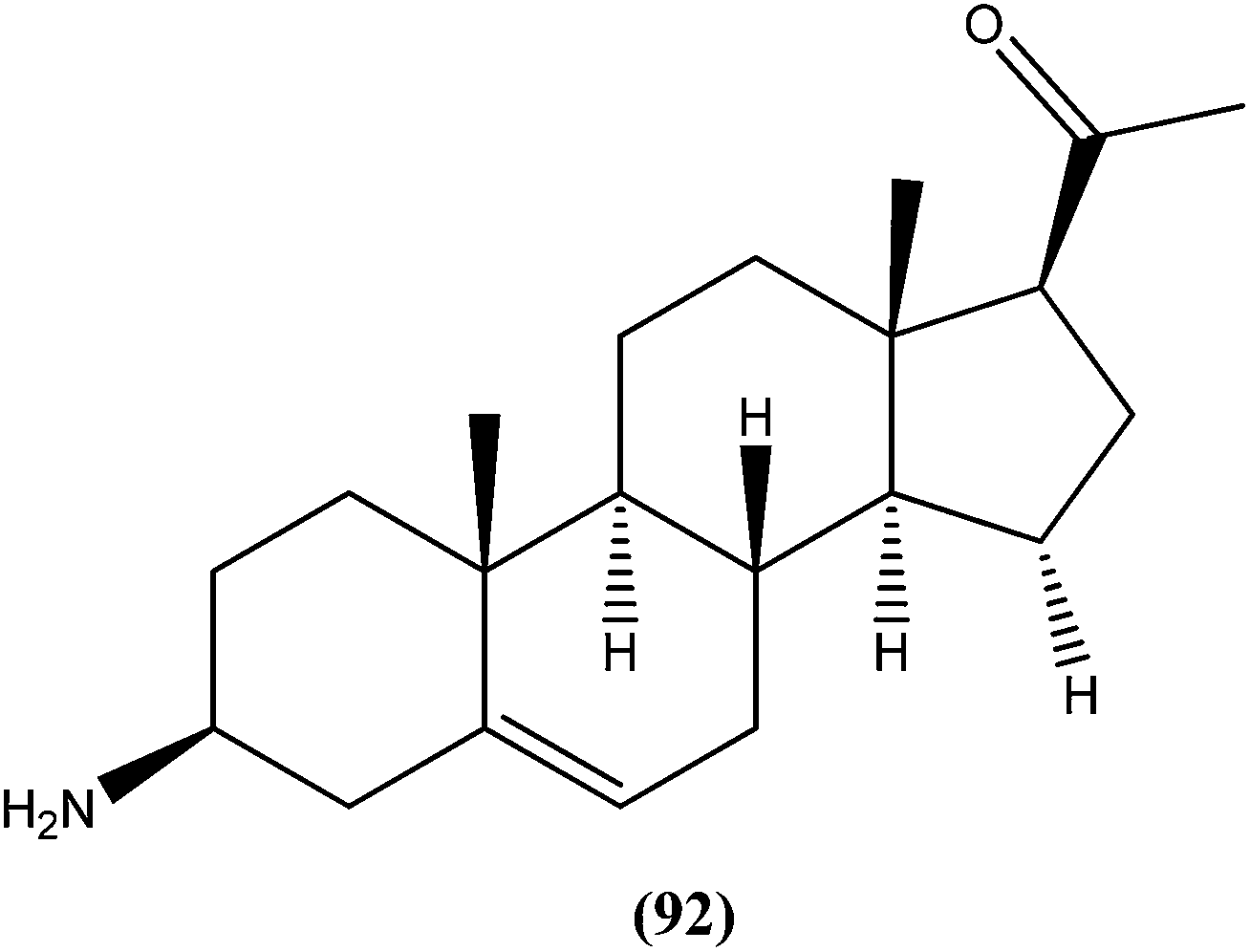
An indole alkaloid corynantheine (93), present in the bark of Corynanthe pachyceras (Rubiaceae), exhibited antileishmanial activity against L. major promastigotes with an IC50 value of about 3 μM.186 A macroline-derived indole alkaloid (94) obtained from stem bark of Alstonia angustifolia (Apocynaceae) was found to be potent against L. mexicana promastigotes, with an IC50 value of 57.8 μM.187
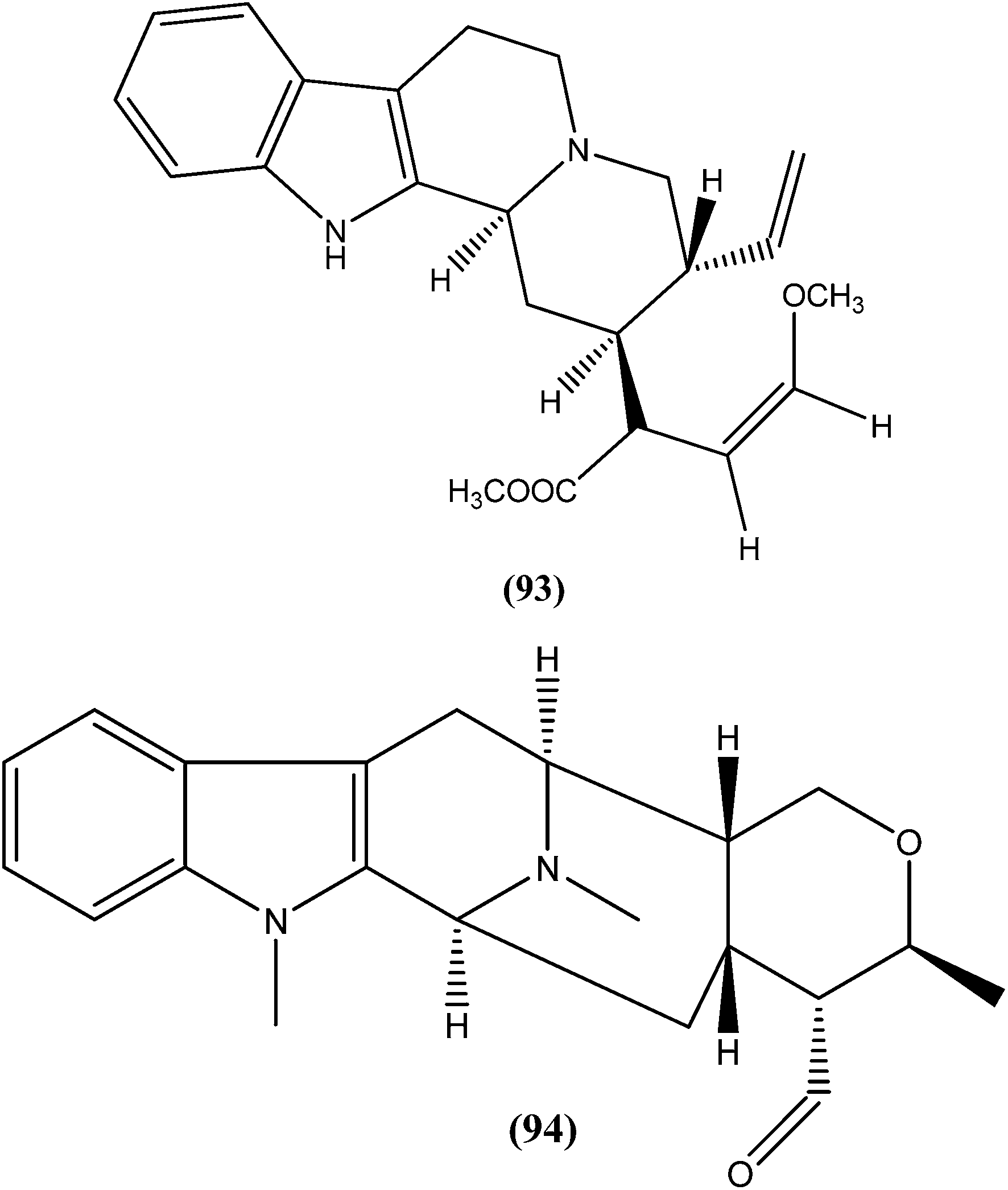
Another alkaloid named rhodesiacridone (95), which contains an acridone ring, was obtained from Thamnosma rhodesica (Rutaceae). After performing antileishmanial assay against L. major, it was found that at 10 μM concentration rhodesiacridone inhibited 69% of promastigote forms, whereas against amastigote form of the same species, over 90% and 50% inhibition were observed at concentrations of 10 μM and 1 μM, respectively. Compound (95) was found to be non-toxic to murine macrophages at the same concentrations.188
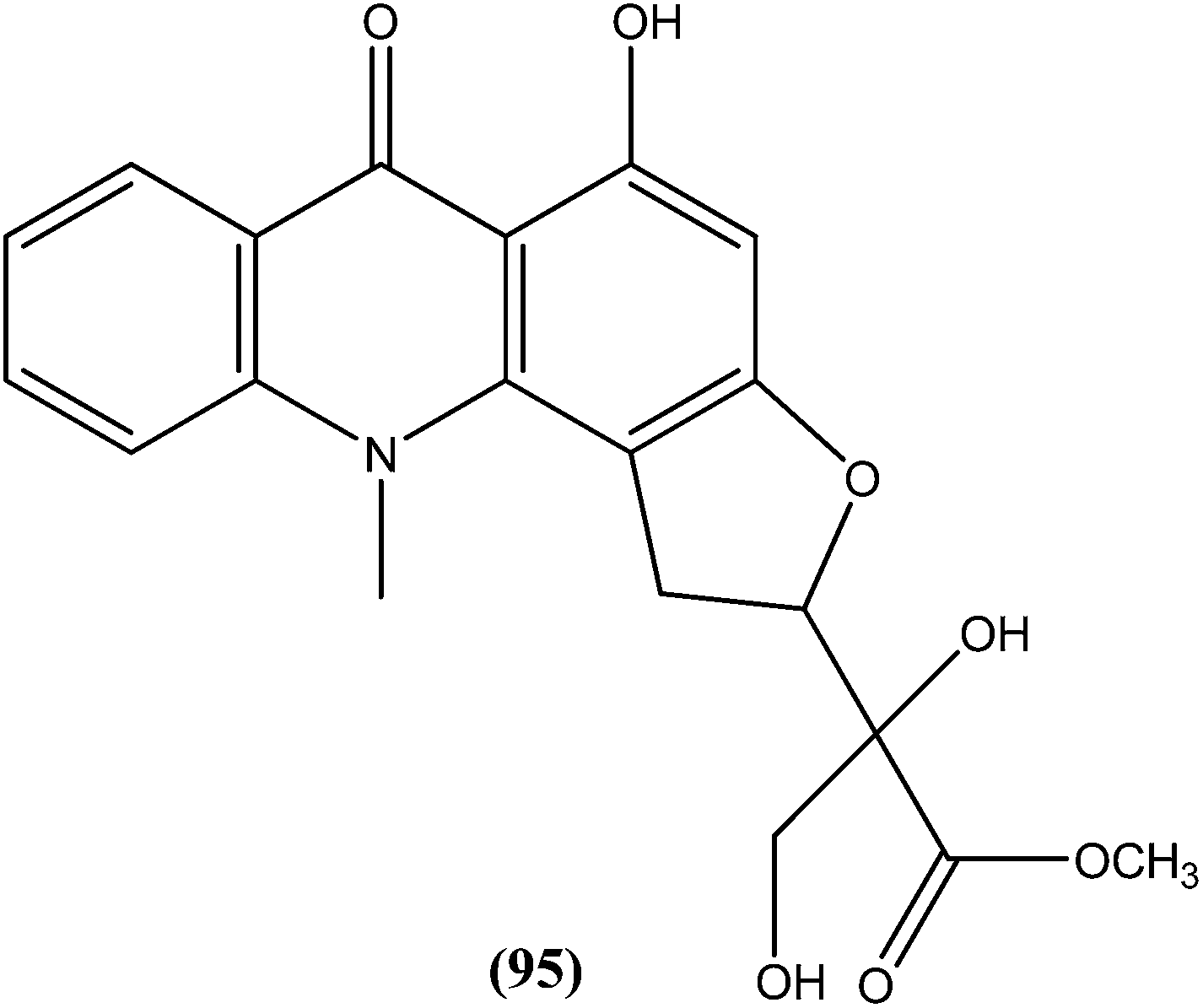
Among the compounds that were tested against L. donovani promastigotes, ancistrotanzanine B (96), an isoquinoline alkaloid isolated from the plant Ancistrocladus tanzaniensis (Ancistrocladaceae), was found to be most potent with an IC50 value of 3.81 μM.189
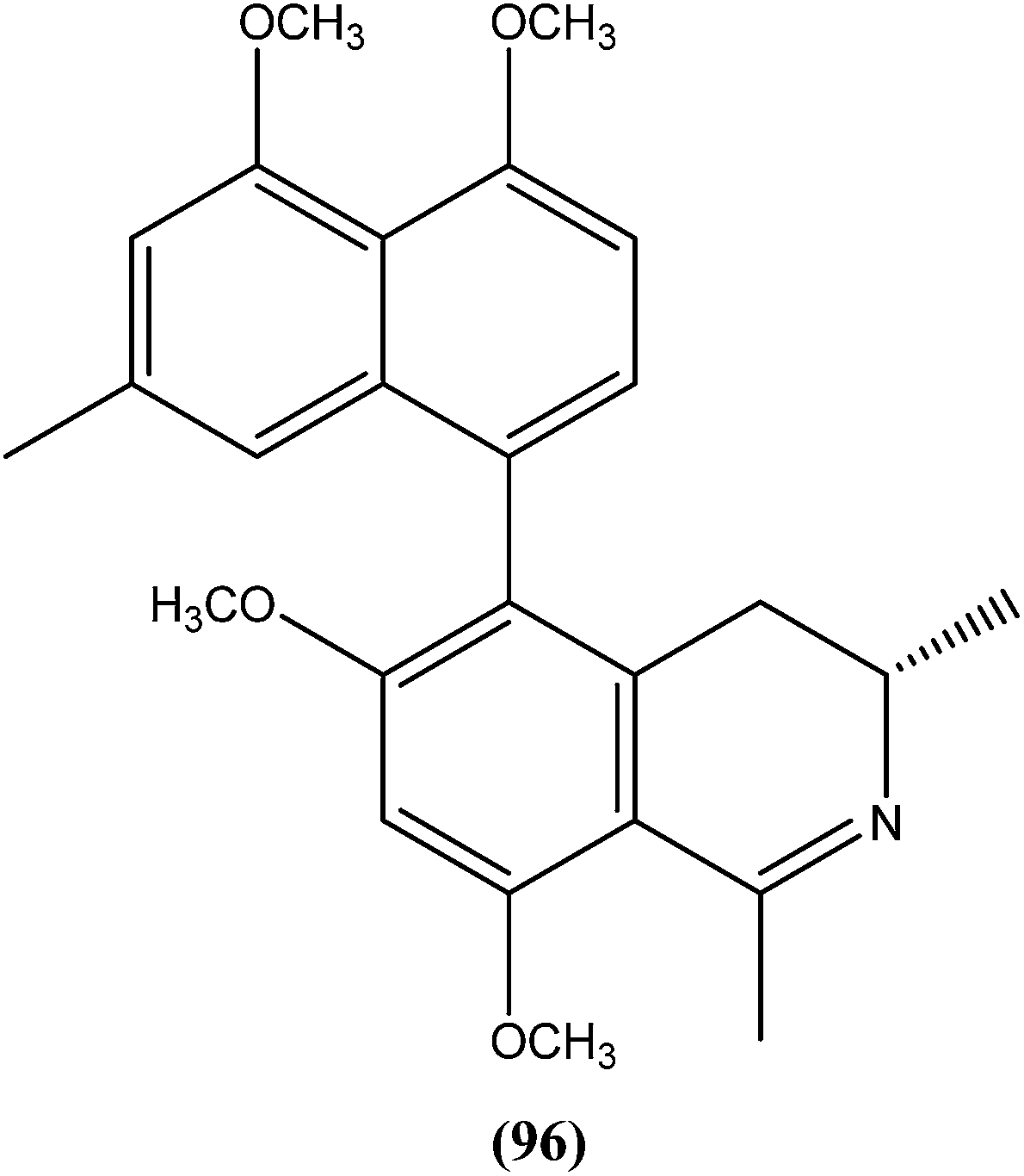
Dihydrochelerythrine (97), having a phenanthridine ring coupled with a benzene ring, isolated from the stem bark of Garcinia lucida (Clusiaceae), was found to be the most active from compounds tested against L. donovani axenic amastigotes, showing an IC50 value of 2.0 μM.190
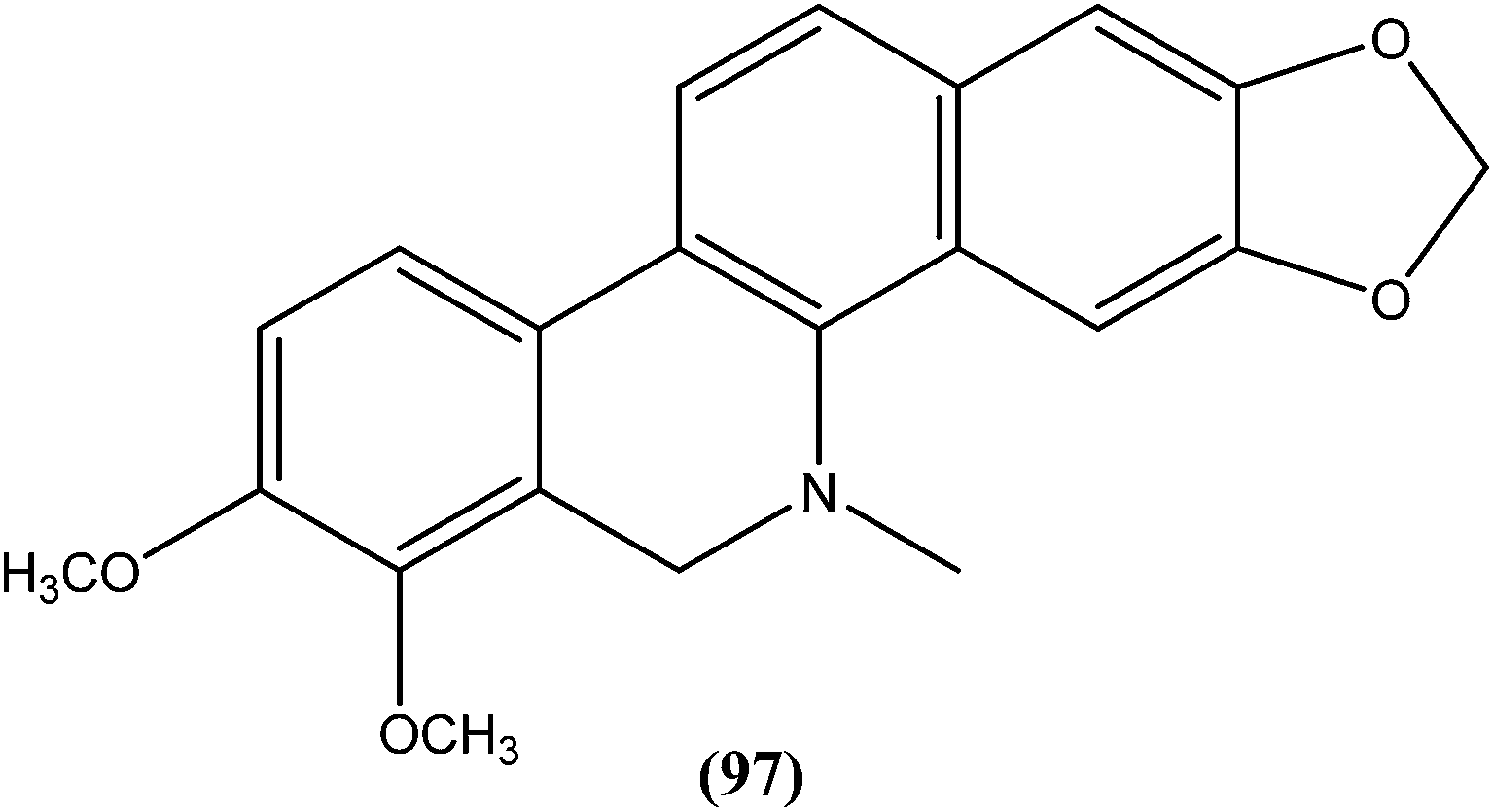
From the seeds of plant known as Peganum harmala, peganine hydrochloride dihydrate (98), a quinazoline alkaloid (identified as an orally active antileishmanial lead molecule), showed in vitro anti-promastigote and anti-amastigote activity against L. donovani, with IC50 values, respectively, of 16.99 μM and 18.30 μM. On testing its in vivo activity, peganine hydrochloride dihydrate (98) showed 79.6 ± 8.07% inhibition against the same species of established VL in hamster models at a dose of 100 mg per kg b.wt.191
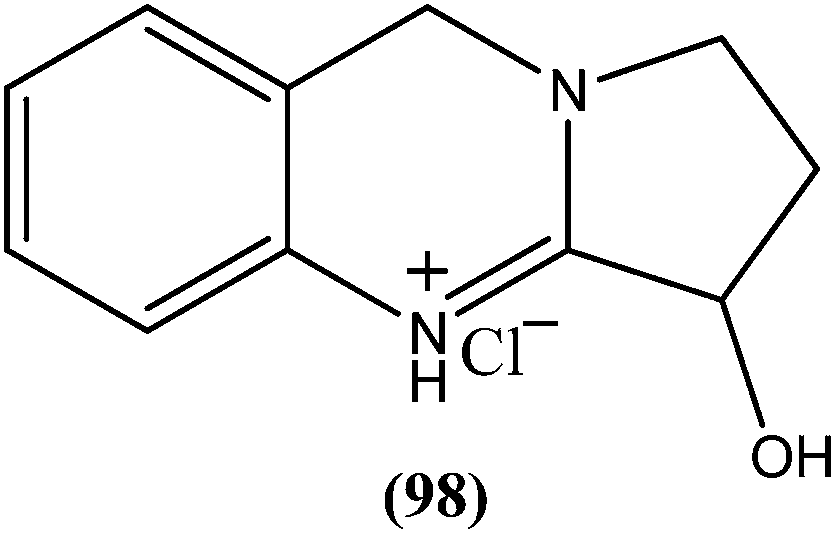
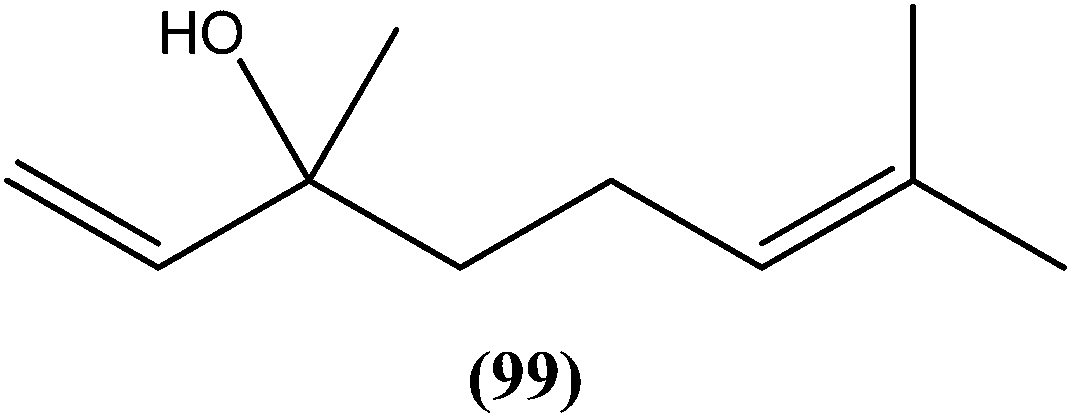
Agarofuran derivative (1S,4R,5R,6R,7R,8S,9R,10R)-8-acetoxy-1,9-dibenzoyloxy-4-hydroxy-6-nicotynoyloxy-dihydro-b-agarofuran (100a), a sesquiterpene obtained from root and barks of Maytenus apurimacensis, showed antileishmanial activity against L. tropica amastigotes with an IC90 value of 7 μM.193 One sesquiterpene furanoeremophil-1-on-13-oic acid (100b), obtained from the woody shrub Drypetes chevalieri Beille (Euphorbiaceae), was screened against the L. major promastigotes and showed significant antileishmanial activity (IC50 = 15.27 μM) compared with control drug pentamidine (IC50 = 11.18 μM).193 Another sesquiterpene known as nerolidol (a mixture of cis- and trans-nerolidol) (101), present in essential oil of several plants, showed antileishmanial activity when tested against promastigote forms of various Leishmania species such as L. amazonensis (IC50 = 85 μM), L. braziliensis (IC50 = 74 μM), and L. chagasi (IC50 = 75 μM). It also exhibited antileishmanial activity against L. amazonensis amastigote form, with IC50 value of 67 μM.194
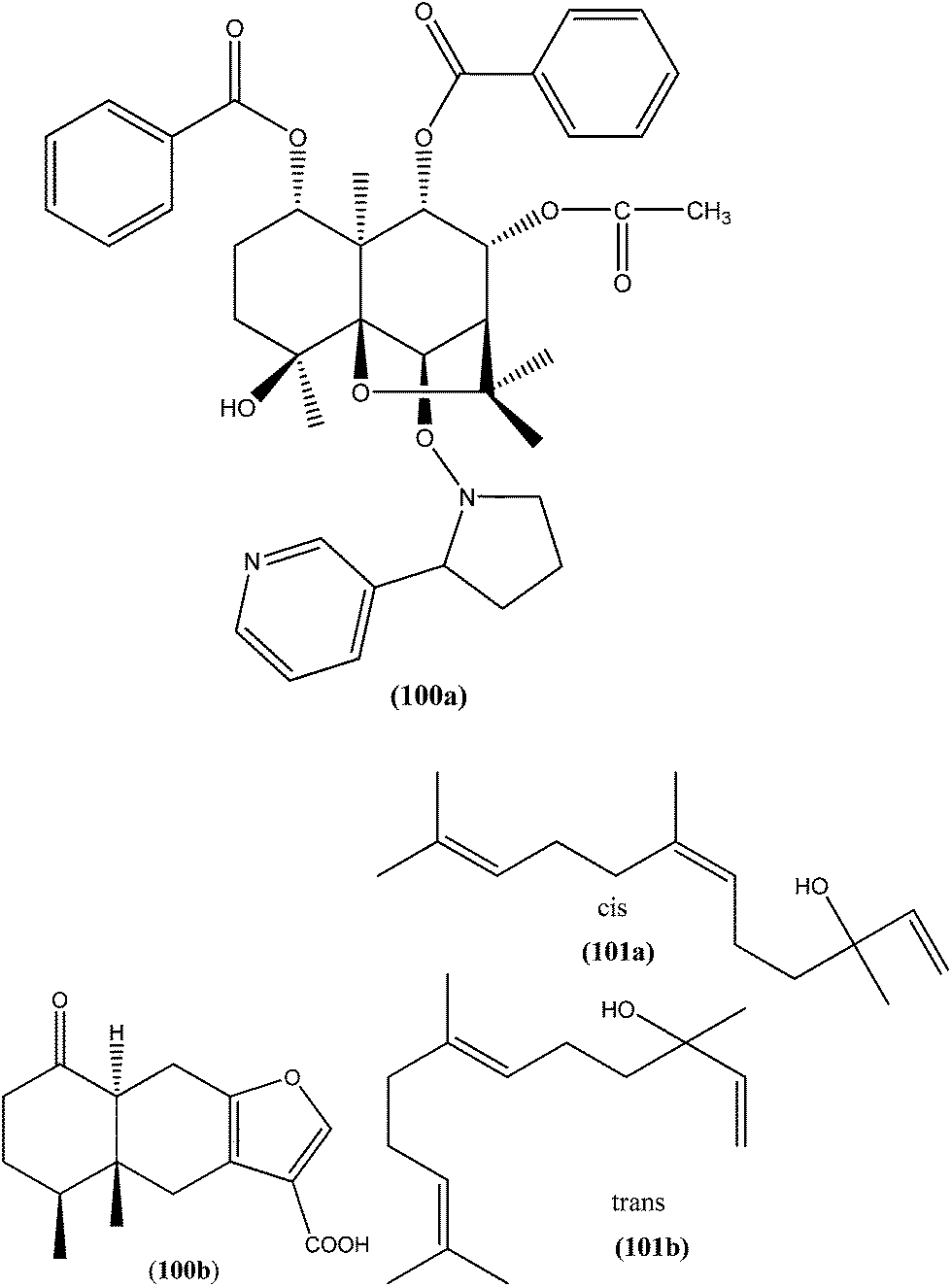
10-Deacetylbaccatin III (a precursor of the well-known drug taxol), a diterpenoid (102) isolated from Taxus baccata, showed an IC50 value of 0.07 μM against L. donovani intracellular amastigotes.195
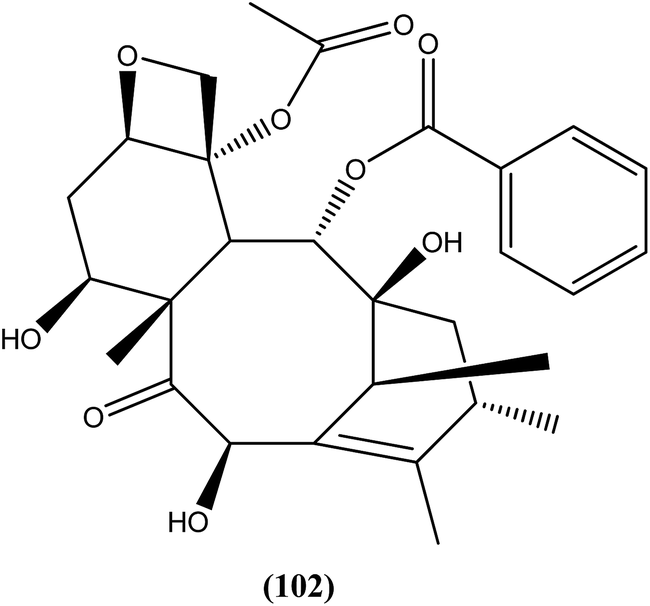
On testing their in vitro antileishmanial activity against amastigote form of L. major, the pentacyclic triterpenoids ursolic acid (103) and Me ursolate (104) obtained from the aerial parts of the plant Mitracarpus frigidus, were found to possess IC50 values of 0.28 μM and 0.45 μM, respectively.196 Compound (105), a nor-triterpene isolated from Lophanthera lactescens, showed antileishmanial activity against amastigote form of L. amazonensis with IC50 value of 0.50 μM.197
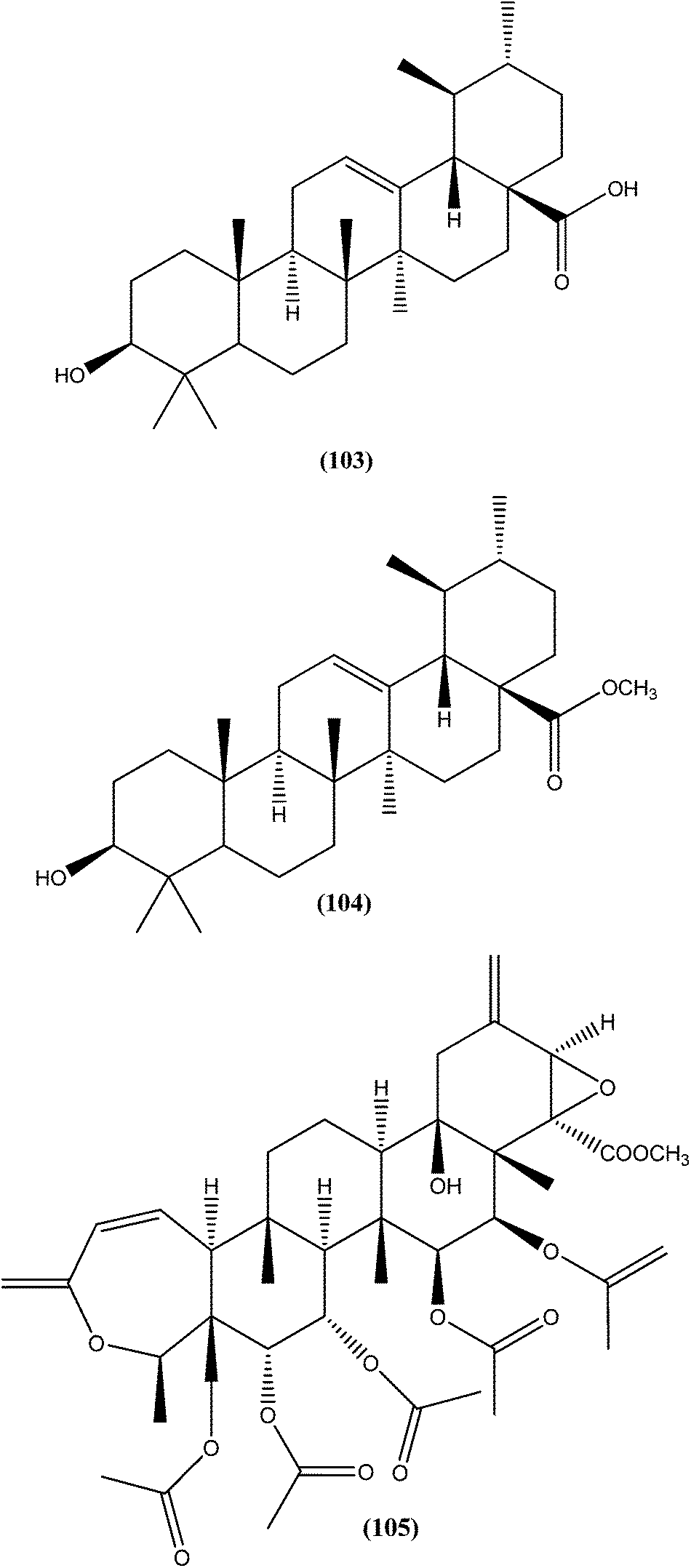
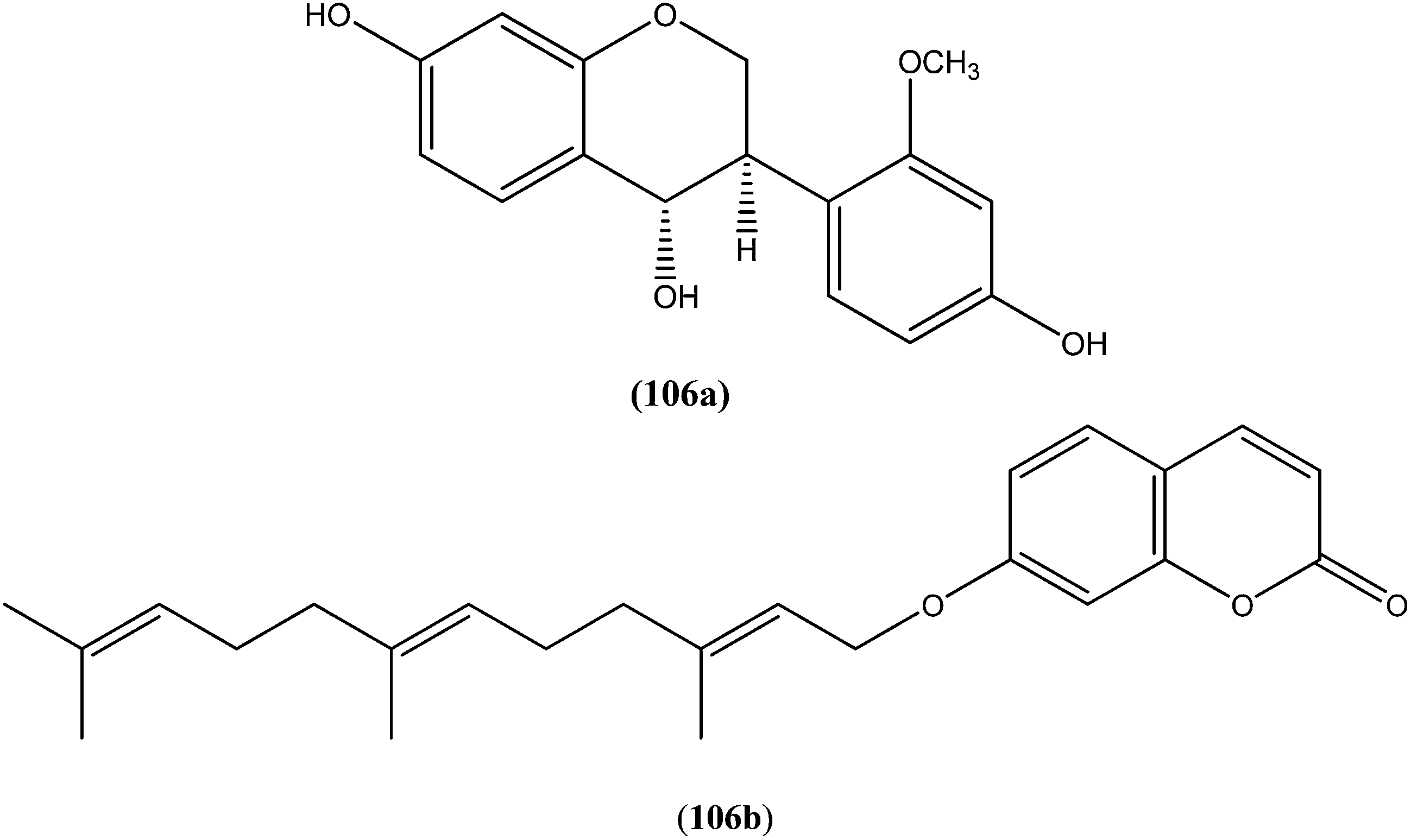
6.2. Marine origin
Almiramide C (107), a peptide present in the crude extract of marine cyanobacterium Lyngbya majuscule (isolated from mangrove roots), exhibited a strong antileishmanial property with an EC50 of 1.9 μM against L. donovani amastigotes.199 Dragonamide E (108), another marine peptide obtained from the same source, showed an EC50 of 5.1 μM when tested in vitro against L. donovani axenic amastigotes.200 Viridamide A (109), a marine peptide isolated from Oscillatoria nigro-viridis, displayed potent in vitro antileishmanial activity (EC50 = 1.5 μM) against amastigote form of L. mexicana.201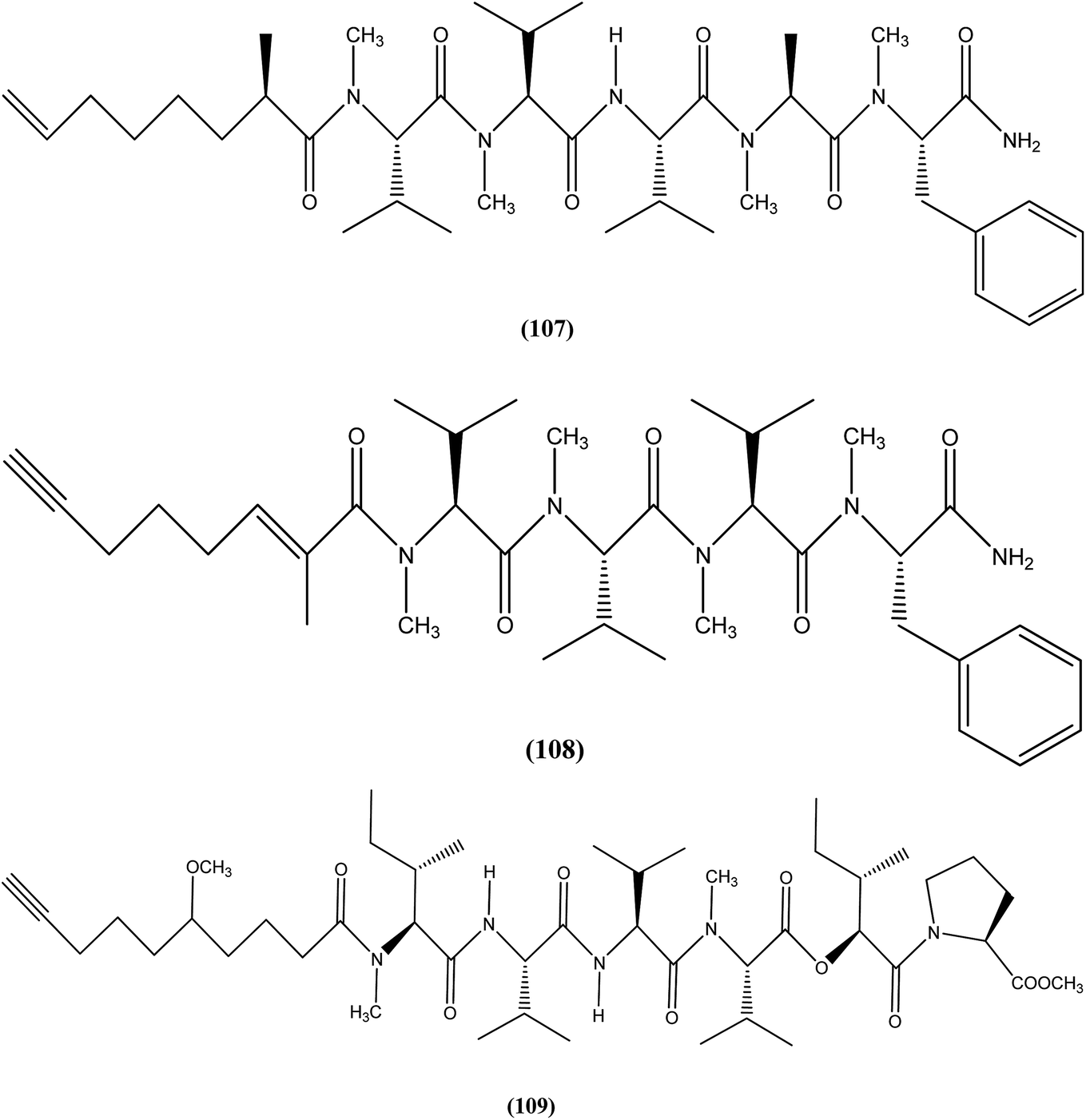
Isoaaptamine (110), a marine alkaloid obtained from marine sponge, that is Aaptos sponge, was found to have good antileishmanial activity with an EC50 of 0.31 μM when assayed in vitro against L. donovani promastigote form.202 Plakortide P (111), a polyketide isolated from marine sponge Plakortis angulospiculatus, exhibited in vitro antileishmanial activity against L. chagasi promastigotes with an EC50 value of 0.52 μM.203
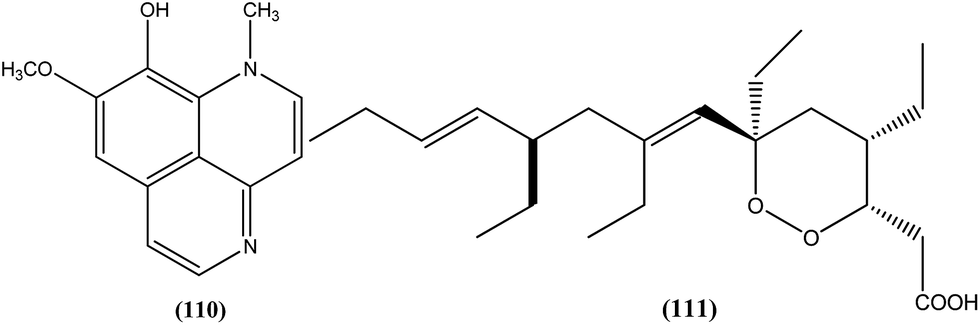
7. Recent patents on antileishmanial drug moieties
In a recent patent, Satoskar et al. discussed sterol compounds (isolated from P. andrieuxii and/or obtained hemi-synthetically using appropriate sterol precursor) as useful therapeutic agents for leishmaniasis. Among the compounds isolated from the roots of P. andrieuxii, the IC50 values of compounds (112) and (113) against amastigote form of L. mexicana were observed to be 0.03 and 1.4 μM, respectively, with compound (113) found to be most active against promastigote form of the same species with IC50 value of 9.2 μM.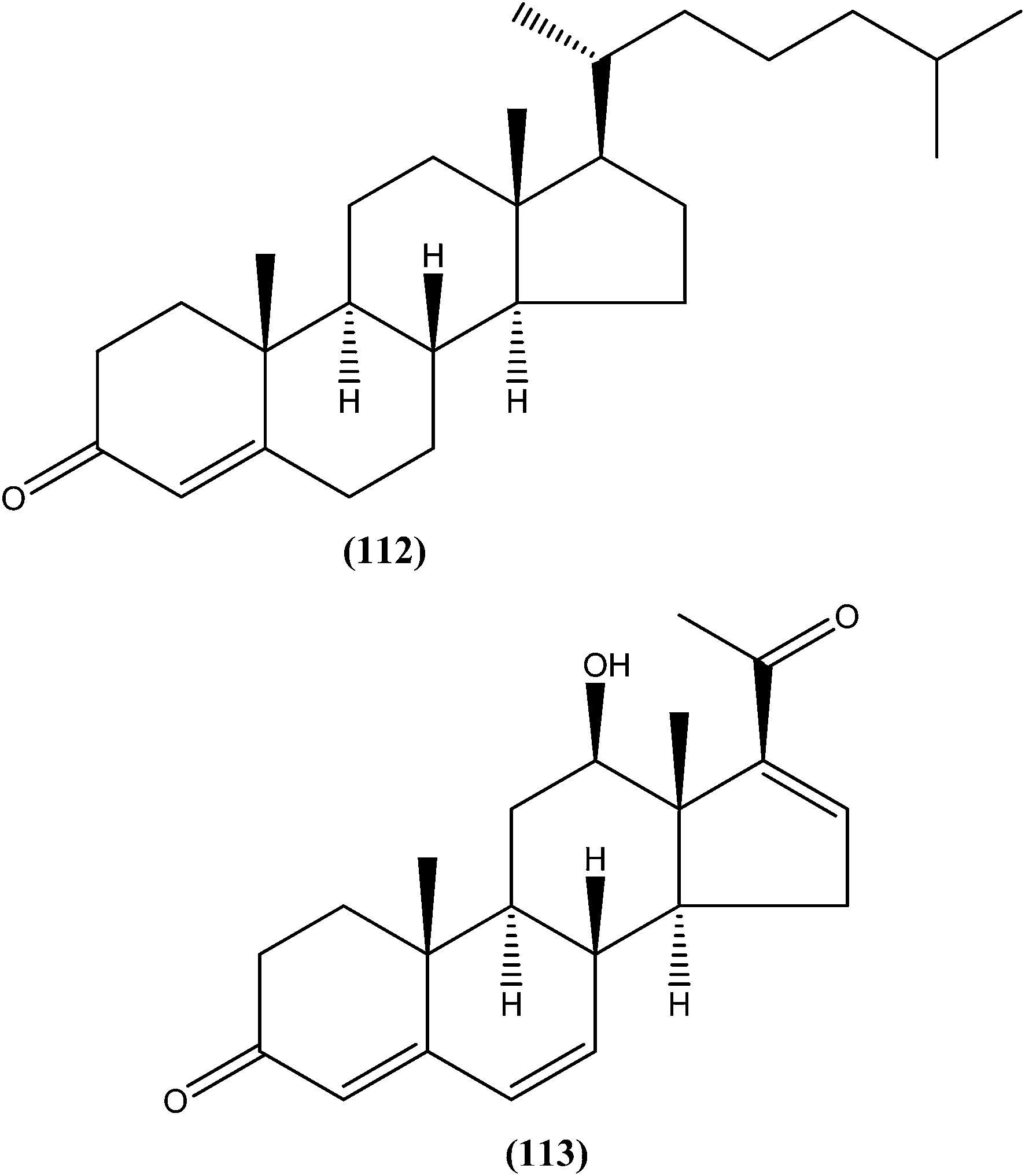
Out of the nine stem fractions that were isolated from P. andrieuxii, the fraction PASD3F2 showed potent activity against promastigote form (IC50 = 21.5 μg mL−1). None of the compounds were cytotoxic to the non-infected bone marrow-derived macrophages (IC50 = >100 μg mL−1), suggesting that such compounds are selective for protozoal cells.204 In the invention by Vasquez et al., use of quaternary ammonium salts is described for treatment of Leishmania infections. After screening the compounds against axenic amastigotes of L. panamensis, the authors commented that compound (114) was the most effective of the tested compounds (EC50 = 14.0 ± 0.9 μM).205
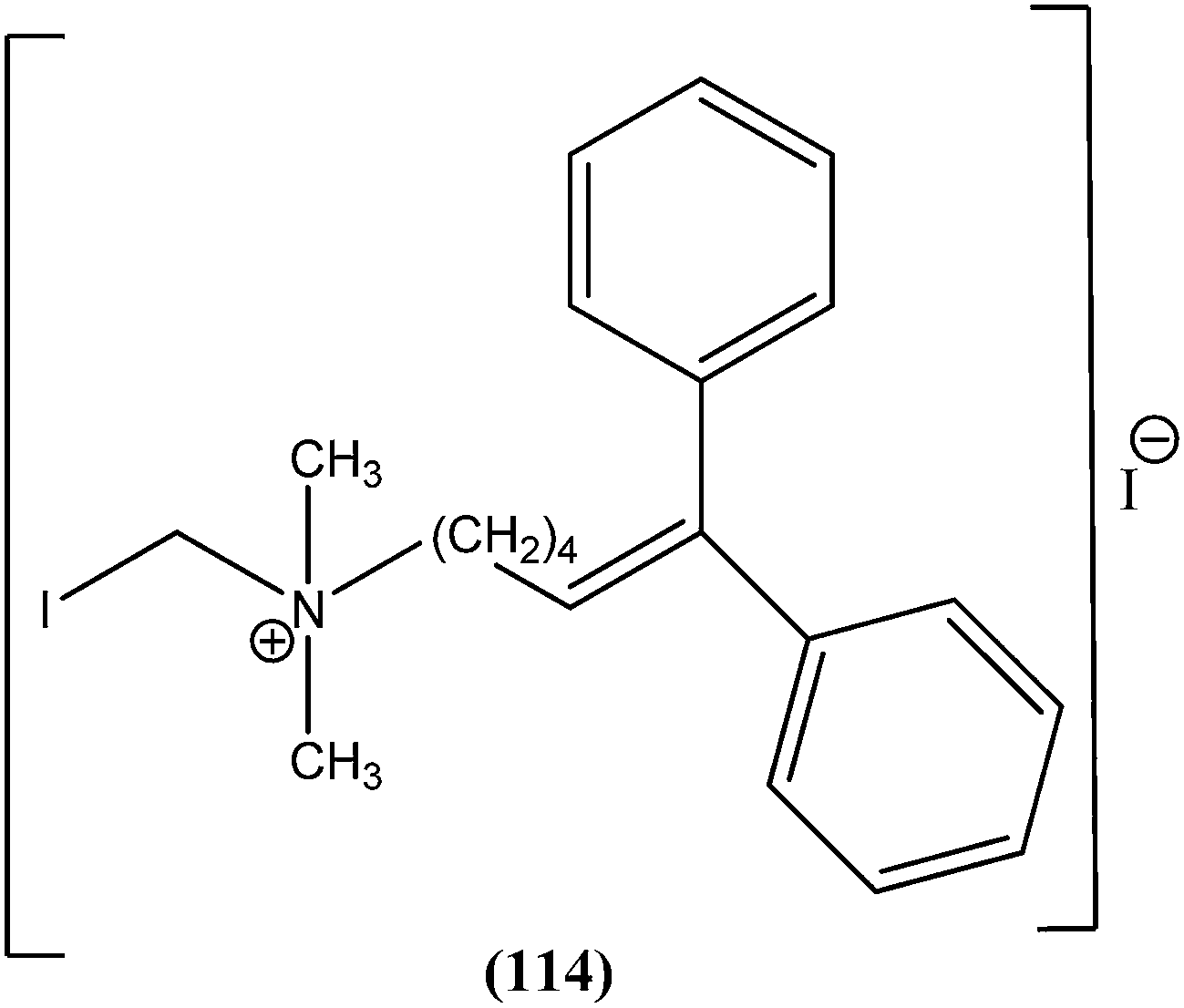
The patent by Boechat et al. refers to new 1,2,3-triazoles and imidazoles included in families of compounds represented by general formula (115), and also to a pharmaceutical composition comprising at least one of the azole compounds represented by the same general formula (115), to the use of such compositions, and to the method of treatment or inhibition of leishmaniasis.
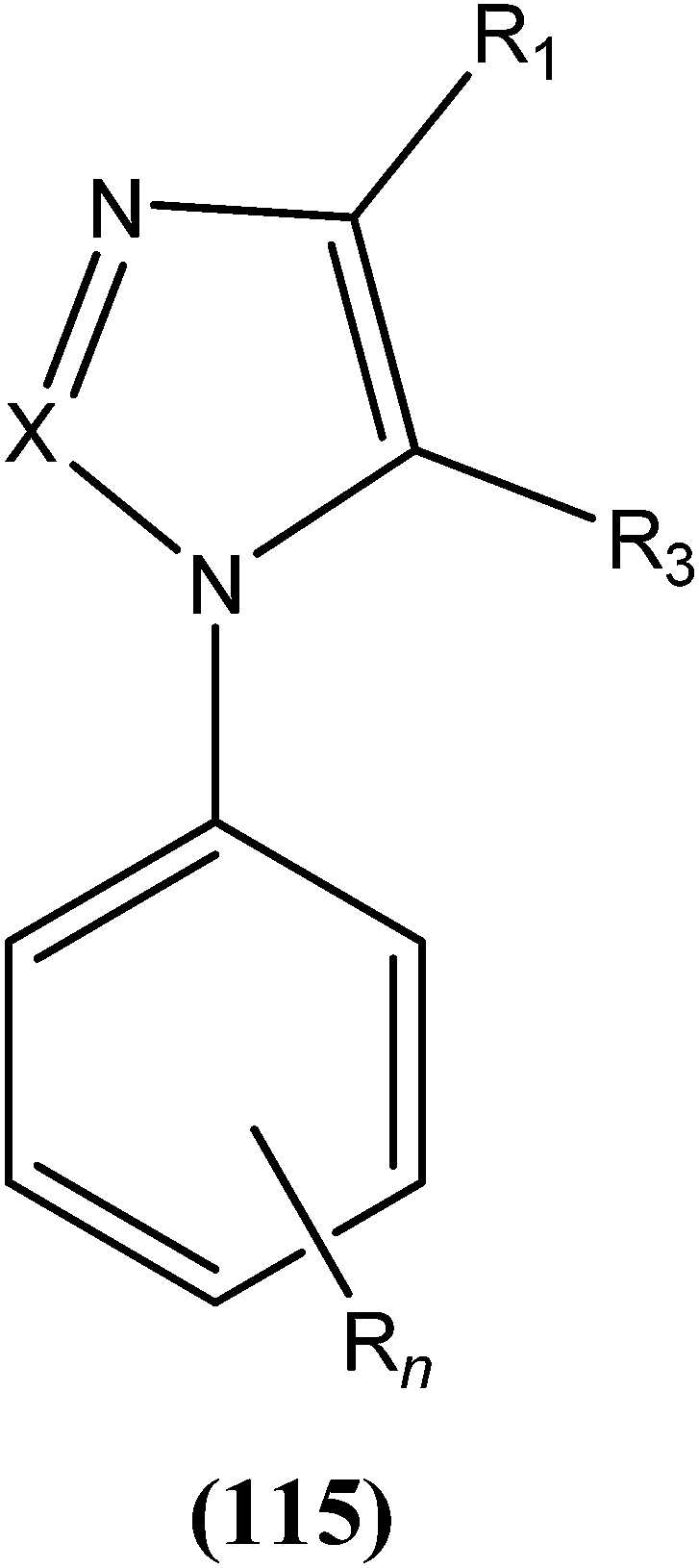
On in vitro analysis against promastigote form of L. amazonensis, the difluoromethyl derivative (116) showed potent activity (inhibition of parasite = 93%) at 10 μg mL−1 concentration when compared with the standard drug pentamidine (at 160 μg mL−1 concentration inhibits 53% of parasites).206
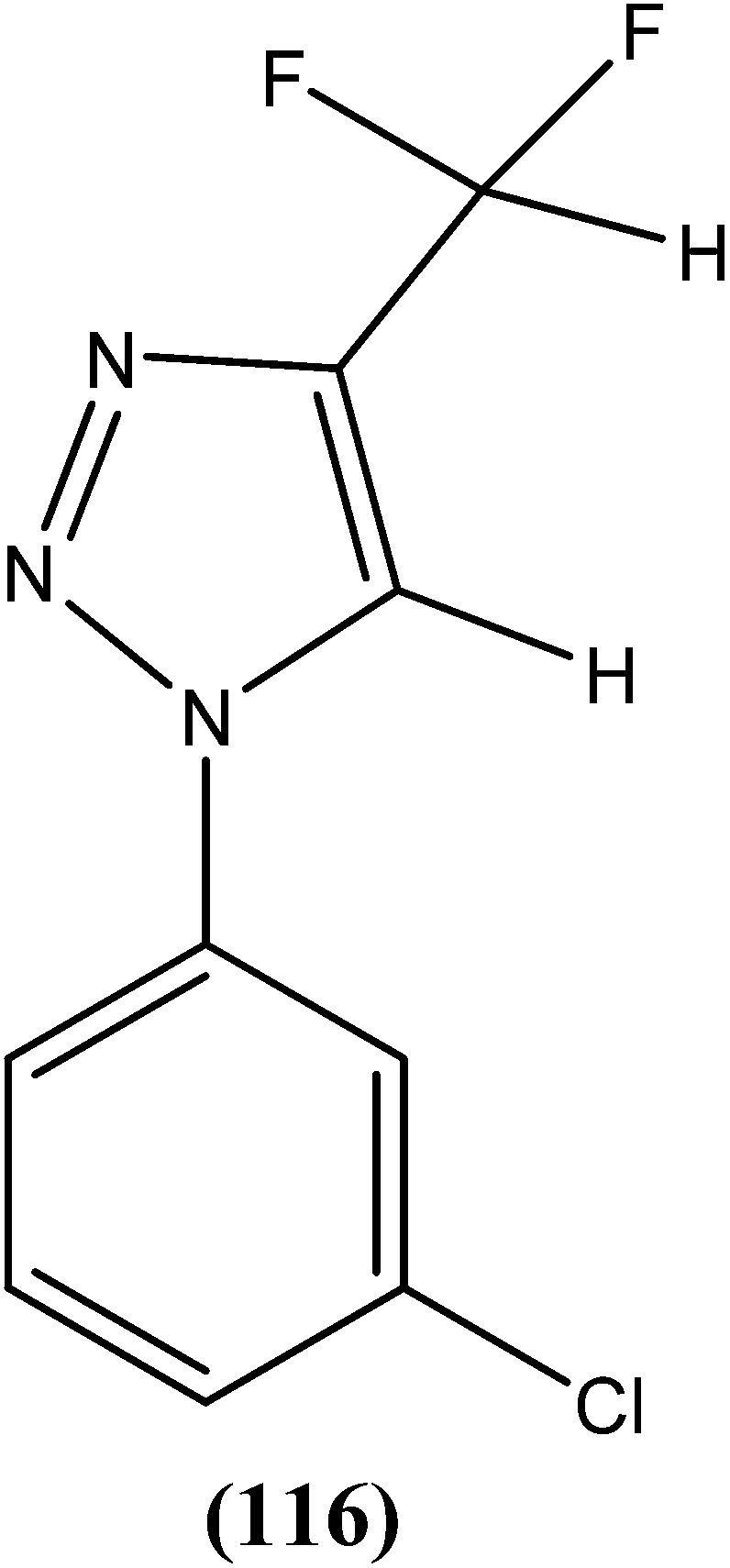
Kimura et al. found that an extract from Sargassum yamade, a brown alga (family: Sargassaceae; order: Fucales) had high antileishmanial activity. From the compounds that were subjected to in vitro analysis against promastigote form of L. major, compound (117) showed almost equal growth inhibition rate to that of amphotericin B, which was used as a positive control. The authors also evaluated in vivo analysis of the same compound on a leishmaniasis mouse model by administering 200 μg via a peritoneal route once a day for 3 weeks. It was found that compound (117) again exhibited activity equal to that of amphotericin B.207
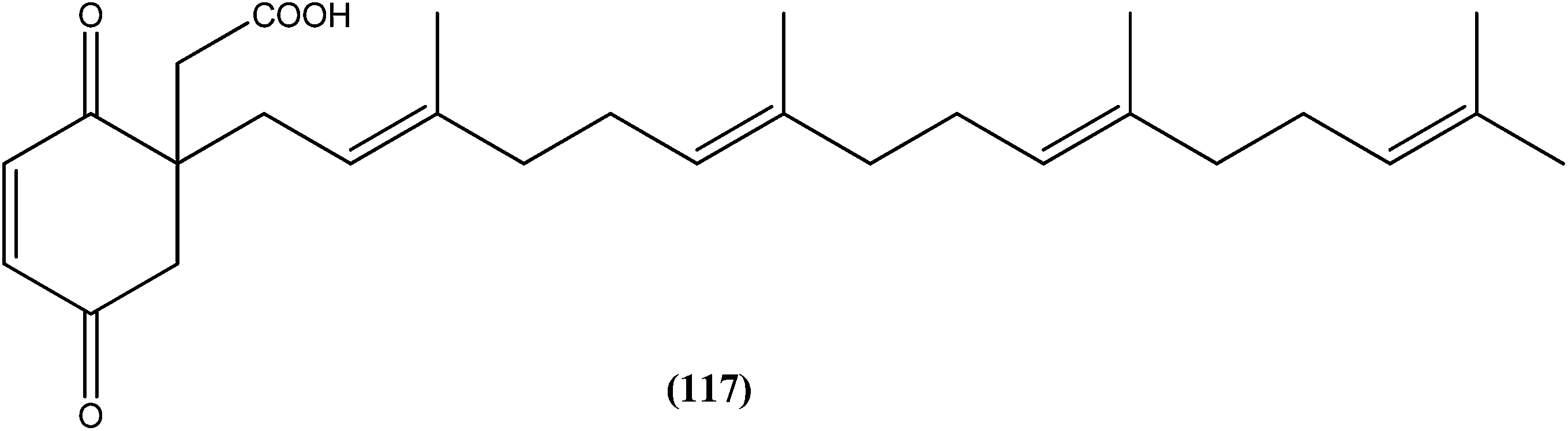
Sevilla et al. claimed that N6-(ferrocenmethyl)quinazoline-2,4,6-triamine compound (118) presents leishmanicidal activity at a concentration starting from 0.1 μg mL−1. On in vitro analysis against L. mexicana, they reported that compound (118) is lethal (in less than 5 h) at concentrations greater than 5 μg mL−1. They observed that the parasite structure was modified such that it lost its characteristic form, lost refringence, became spherical, and was incapable of multiplying. Although the mechanism could not be identified, the authors suggested that a necrosis process was likely to be involved. The same compound was found to be up to 10-fold faster at killing the total number of parasites when compared with other compounds such as metronidazole and hydroxyurea (having leishmanicidal activity). There was also no occurrence of cytotoxicity with murine cells in in vitro analysis, nor in mice (the in vivo analysis model) when administered orally, parenterally, or dermally.208
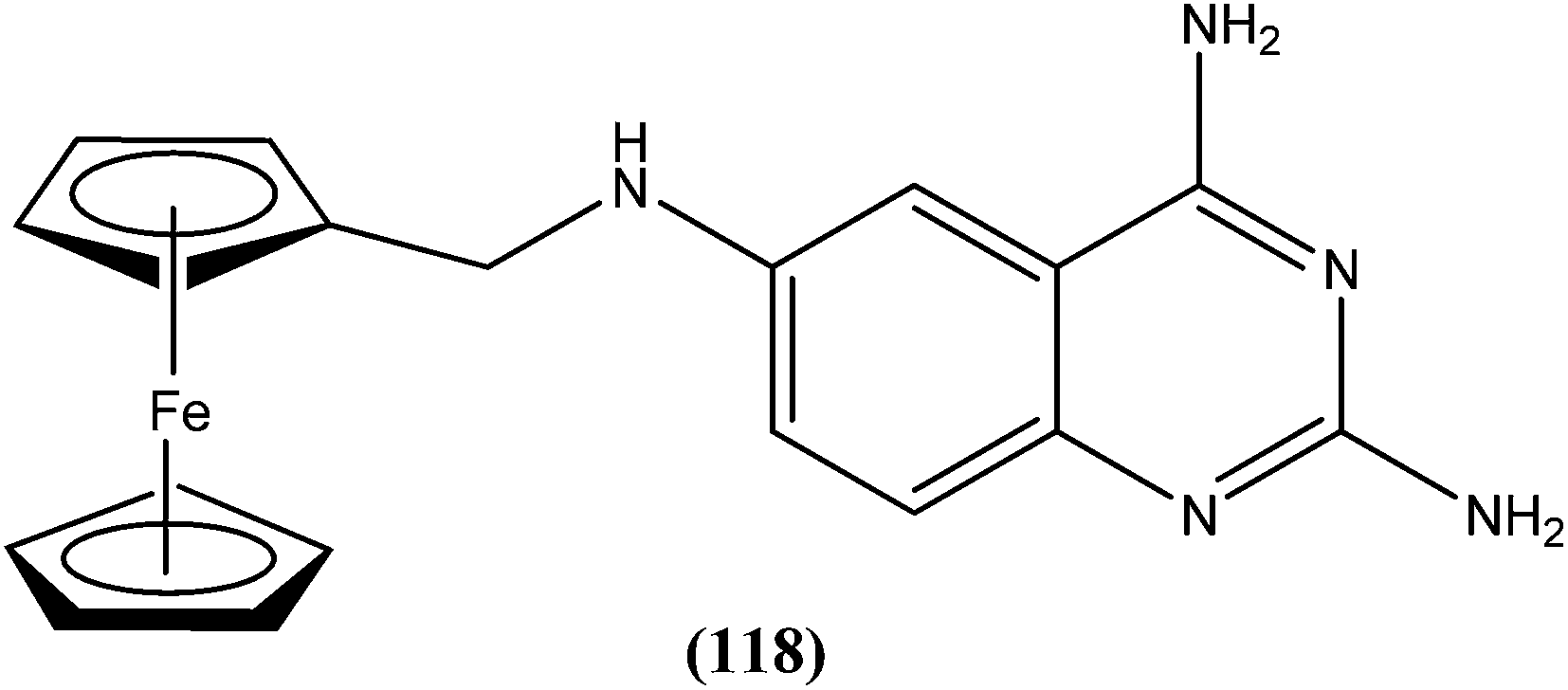
In their patent, Shairah et al. commented that certain metronidazole derivatives (2-methyl-5-nitro-imidazolyl compounds) are useful against L. donovani and L. tropica promastigotes. On their in vitro assay, compound (119) showed IC50 values of 109 μM and 54.54 μM against L. donovani and L. tropica, respectively.209
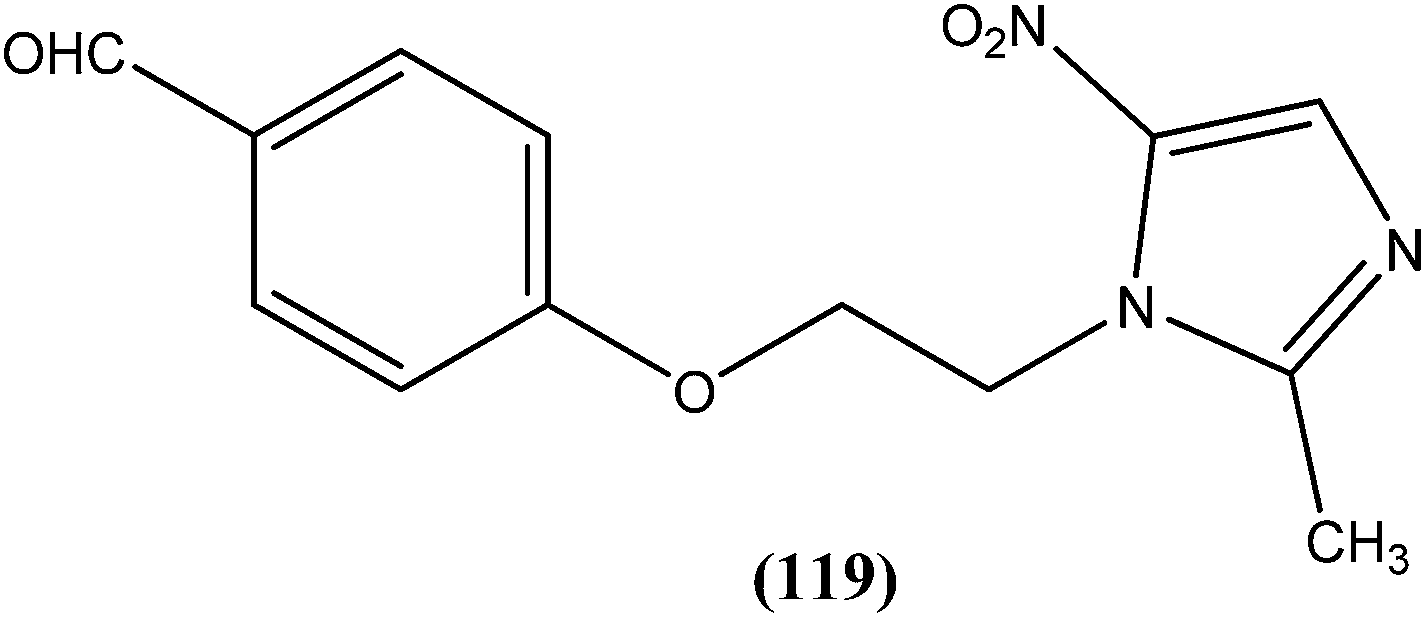
Curtis et al. reported that compounds, for example (120), that contain a substituted five- or six-membered ring core containing one or two oxygen, nitrogen, or sulfur atoms as constituent atoms of the ring, can be used to treat leishmaniasis or its symptoms by inhibiting sirtuin (e.g. SIRT1) present in the parasite. They further reported that the described SIRT1 inhibitor (e.g. 120) decreases the ability of parasite to develop resistance to conventional treatments, and/or decreases the viability and/or infectivity of the parasite. They tested various possible compounds for activity against SIRT1. Different compounds were found to have different activities (IC50), even lower than 1 μM.210
Cohen et al. reported their invention represented by the general formula (121), related to thiosemicarbazone and semicarbazone inhibitors of cysteine proteases, and methods of using such compounds to prevent and treat protozoan infections such as leishmaniasis.211
Searle et al. from GlaxoSmithKline, USA, reported sitamaquine tosylate (122) for treatment of leishmaniasis.212
Werbovetz et al., in their patent “Antileishmanial dinitroaniline sulfanoamides with activity against parasite tubulin,” highlighted usefulness of these compounds particularly in the treatment of leishmaniasis. Compound (123) showed good activity against L. donovani. Its IC50 values were found to be 8.02 ± 0.42 and 9.0 ± 0.7 μM against promastigote and amastigote forms, respectively.213
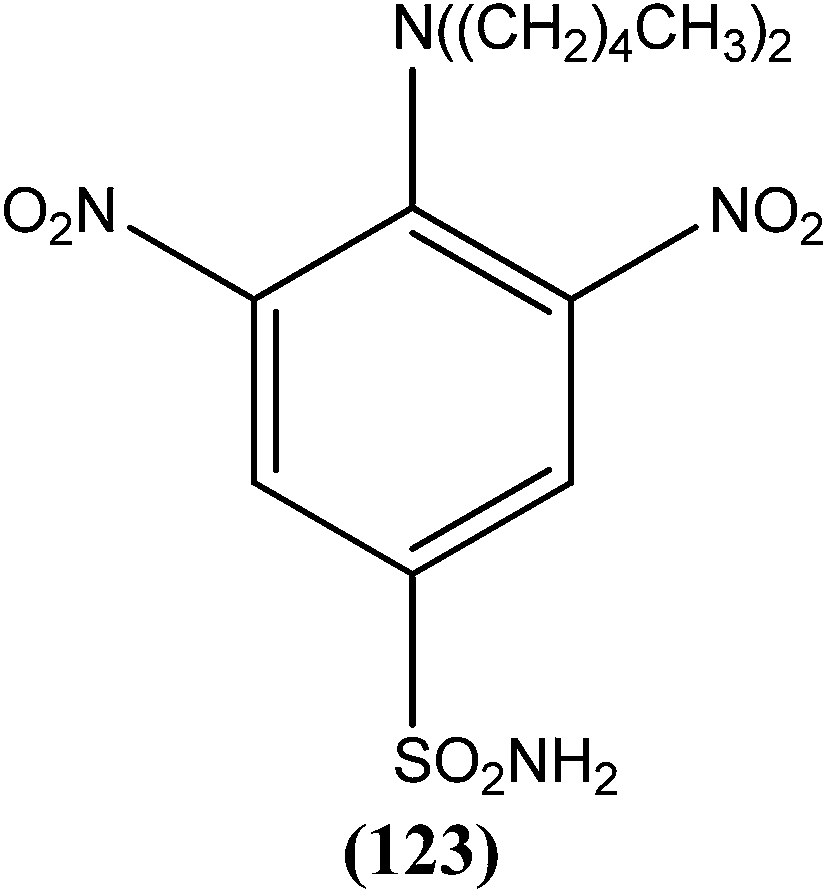
From the invention by Masataka et al. (Japan Science and Technology Corporation, Japan), compound (124) was found to be most potent, having an IC50 value of 0.0018 μM, more than that of standard drug amphotericin B (IC50 = 0.015 μM).214
Rios et al. in their patent had claimed that none of the current treatments for leishmaniasis use compounds with chemical structure comparable with the compounds of their present invention, which belong to the class of substances known as aporphine alkaloids. Compound (125) was found to be most potent of the tested compounds, with IC50 values against L. mexicana and L. panamensis of 3 ± 0.27 and 6 ± 0.07 μM, respectively. The compound was also found to have 37-fold higher toxicity towards L. mexicana than macrophages (IC50 = 112 ± 0.2 μM).215
8. Possible antileishmanial drug targets
8.1. Sterol pathway (enzymes of sterol biosynthesis)
Unlike cholesterol which is present in mammals, the Leishmania parasite has endogeneous ergosterol (counterpart of cholesterol) and stigmasterol in its cell membrane. This feature may be useful in drug targeting of antileishmanial agents. For example, azasterols inhibit 24-methyltransferase, an enzyme vital for ergosterol biosynthesis.216 Inhibitors of 14-α-methylsterol-14-demethyase, such as some azoles and triazoles, are effective against Leishmania.2178.2. Thiol pathway (enzymes of thiol metabolism)
Some reports indicated that a characteristic thiol metabolic defense mechanism developed by the parasite was involved in neutralization of host oxidative outcome (harmful to the parasite), explaining why the Leishmania parasite can withstand and proliferate in a toxic environment developed by macrophages of mammalian host. For example, Gradoni et al. reported that trypanothion [T(SH)2], a dithiol found in L. infantum, is capable of reducing nitric oxide (generated in mammals) and iron into a harmless stable dinitrosyl iron complex with 600 time more affinity than mammalian GSH (glutathione) reductase system. This is the mechanism by which the parasite protects itself from such lethal environments. In homology modelling of L. infantum, TR (trypanthione reductase, one of the antioxidant enzymes present in Leishmania) and mammalian glutathione reductase (GR) have shown remarkable differences in their three-dimensional and catalytic active sites.218–2208.3. Hypusine pathway
Hypusine (derived from the polyamine spermidine) is synthesized in two enzymatic steps as a result of post-translational modification in all eukaryotes. The first step is catalysed by the enzyme deoxyhypusine synthase (DHS). Recently, Chawala et al. showed that hypusine biosynthesis occurs in L. donovani and they identified two genes from this containing DHS domains. They further concluded that the gene DHS34 (DHS-like gene from chromosome 34) is essential for functional activity in vitro in L. donovani.221–2238.4. GPI pathway
A major component of the Leishmania surface coat is the glycosylphosphatidylinositol (GPI)-anchored polysaccharide called lipophosphoglycan (LPG), having some free GPIs which protect the parasite from the alternate complement pathway and external hydrolases. Sacks et al. reported that LPG is essential for infectivity of L. major promastigotes in both mammalian and insect hosts. They further concluded that LPG is required to maintain infection in the fly during excretion of the digested blood meal.224,2258.5. Glycolytic pathway
The unique compartmentalization of glycolytic enzymes (in glycosomes of Leishmania) and their large phylogenetic distance from the mammalian hosts provides them with unique features that can be targeted.226 Specific inhibitors have been designed for the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which is an intermediate enzyme for conversion of glucose to pyruvate in glycolysis occuring in Leishmania. For example, N6-(1-naphthalenemethyl)-2′-(3-methoxybenzamido) adenosine inhibited growth of L. mexicana with an IC50 of 0.28 μM.227 In another study, it was reported that two different types of iron superoxide dismutases FeSOD (absent in mammalian counterpart), Lcfesodb1 and Lcfesodb2, were characterized in L. chagasi (within glycosomes), and these were found to be responsible for survival and protection from lethal superoxide radicals.2288.6. Purine salvage pathway
Like other hemoflagellates, Leishmania parasites are incapable of synthesizing the purine nucleus. Therefore to utilize purine bases from their mammalian hosts, they depend solely on an exogenous supply of preformed purines by means of a purine “salvage” pathway. The enzyme phosphoribosyl transferase (PRT) is a mediator in salvage of purines. Adenine phosphoribosyl transferase (APRT), hypoxanthine guanine phosphoribosyl transferse (HGPRT), and xanthine phosphoribosyl transferse (XPRT) are the three PRTs identified as present in Leishmania species.223,229 Because of differences in substrate specificity of parasitic purine salvage enzymes from host enzymes, various inhibitors could be designed or developed to target them (e.g. allopurinol that targets HGPRT, gets phosphorylated therein and thus incorporated into parasital nucleic acid leading to its leishmanicidal action).2308.7. Nucleoside transporters
LdNT1 (present in promastigotes as well as amastigotes responsible for transportation of adenosine and pyrimidine nucleosides) and LdNT2 (present in amastigotes that transport purine nucleosides such as inosine, guanosine, etc.) are the two transporters documented from L. donovani. The parasitic transporters are different from mammalian transporters in terms of their higher specificity towards the substrate, making them vital targets as these transporters also uptake toxic nucleosides, which are inhibitory in action to the cell growth.231,2328.8. Cyclin dependent kinase
Cyclin dependent kinases (CDKs) are important in cell division, transcription, etc. In Leishmania, the cdc-2 related kinase (CRK) family have attracted attention as potential drug targets which are homologous to CDKs and are thought to be vital for cell cycle progression. For example, CRK3 was found to be active throughout the life cycle of L. mexicana, and it inhibitors of CRK3 inhibited the growth and replication of L. donovani amastigotes in peritoneal macrophages. Most potent inhibitors of CRK3 belongs to the indirubin class.233,2348.9. Mitogen activated protein kinase (MAPK)
In L. mexicana, MAPK was found to be important for transformation and cellular growth. MAPKs are not only important to amastigotes but also promastigotes.235 Therefore, they have potential as antileishmanial agents.8.10. Enzymes of polyamine biosynthesis
The putrescine, spermidine, and spermine-like polyamines and their metabolic pathways have important roles in growth and differentiation of parasites from promastigote to amastigote stages, and also downregulate lipid peroxidation generated by oxidant compounds and make the environment compatible for parasite survival. Arginase and ornithine decarboxylase are the two enzymes present in Leishmania involved in synthesis of putrescine and thereby spermine and spermidine, offering potential as targets.85,236–238 The intracellular polyamine transporters (LmPOT1) that transport both putrescine and spermidine could also be explored as drug targets.239,2408.11. Dihydrofolate reductase (DHFR)
Thymidylate synthase (TS) and dihydrofolate reductase (DHFR) are enzymes involved in the folate pathway during DNA biosynthesis. Classic inhibitors of DHFR were found to be ineffective against Leishmania.241 Another enzyme, pteridine reductase (PTR1) was viewed in some Leishmania mutants resistant to methotrexate, an inhibitor of DHFR-TS.242 Hardey et al. screened a number of compounds against PTR1 in L. major, after which four such compounds were identified that inhibited both the enzymes DHFR-TS and PTR1, and also the growth of the parasite. This indicated that an inhibitor is required that targets both the enzymes simultaneously or two compounds that can be used in combination to specifically inhibit both enzymes.2438.12. Peptidase
A total of 154 peptidases were found to be present in the L. major genome. Secretary endosomal system consists of subtilisin like serine peptidase, which participates in processing of secreted proteins and may be useful as a drug target. It was reported that TPCK (N-tosyl-l-lysyl-chloromethylketone) and benzamidine, the serine peptidase inhibitors, reduce the viability and induce morphological changes in L. amazonensis promastigotes, suggesting that serine peptidases could be useful potential drug targets. In vitro study revealed that proteasome was essential for growth of both parasitic forms, that is promastigotes and amastigotes. Hence, the proteasome of Leishmania is a potential therapeutic target.244–2468.13. Topoisomerase
DNA topoisomerases are important enzymes required in many essential processes like DNA replication, transcription, recombination, and repair. Both types of enzymes viz. type I topoisomerase and type II topoisomerase, have been characterized from L. donovani. Anti-leishmanial compounds such as sodium stibogluconate and urea stibamine are inhibitors of type I topoisomerase. Camptothecin, a plant alkaloid, was also found to be an inhibitor of L. donovani.247,248 Topoisomerase II was overexpressed and showed increased activity in arsenite-resistant L. donovani.249 Antibacterial and anticancerous drugs like novobiocin, etoposide, and fluoroquinolones can be used to target topoisomerase II to inactivate genetic integrity and cell survival.250 A derivative of betulinic acid, a pentacyclic triterpenoid i.e. dihydrobetulinic acid (DHBA), was found to be active against both topoisomerase I and topoisomerase II of L. donovani.2518.14. Metacaspase
Two metacaspases (MCAs), LdMCA1 and LdMCA2, are reported in L. donovani promastigotes and amastigotes.252 It has been reported that parasites which overexpress metacaspases are more sensitive to H2O2-induced programmed cell death.253 In L. major, it was found that LmjMCA, a metacaspase is essential for proper segregation of the nucleus and kinetoplast.2548.15. Glyoxalase system
The function of the glyoxalase system is to detoxify cells by eliminating toxic and mutagenic methylglyoxal, which is mainly formed in glycolysis as a byproduct. Glyoxalase I (characterized from L. donovani and L. major) and glyoxalase II (characterized from L. donovani) are the two enzymes involved in glyoxalase system.255–257 L. donovani glyoxalase I, which is highly substrate-specific, has been found to be an essential gene in the parasite.258,2599. Conclusions
Leishmaniasis is a life-threatening disease that mainly affects people in developing countries. There has been significant progress in the treatment of leishmaniasis during recent decades. Various drugs like miltefosine, paromomycin, pentamidine, and liposomal amphotericin B have substantially improved the options for treatment. However, growing incidences of resistance and toxicities with available drugs warrant precise use of antileishmanial drugs as well as necessitating the search for and development of newer effective drugs and vaccine candidates.Several new synthetic molecules with interesting antileishmanial activity have been proposed. Various cores and derivatives like indole, coumarin, quinoline, azoles, triazine, thienopyridine, pyrimidine, etc., have been reported to possess potent antileishmanial activity with good selectivity indices. Screening of natural compounds seems to be an attractive approach for development of effective new lead compounds or drugs. Importantly, natural products, viz., quercetin (flavonoid), luteolin (flavonoid), holamine (steroid), corynantheine (indole), rhodesiacridone (acridone), dihydrochelerythrine (phenanthridine), peganine (quinazoline), linalool, agarofuran and nerolidol (terpenoide), conferol (coumarin), and isoaaptamine (alkaloid) demonstrate interesting in vitro antileishmanial activity. Also, a number of products with antileishmanial activity have been patented following different strategies old and new. Several interesting drug targets also have been proposed including many proteins and enzymes namely, sterol pathway, thiol pathway, hypusine pathway, glycolytic pathway, purine salvage pathway, polyamine pathway, protein kinase, dihydrofolate reductase, topoisomerase, etc., that differ from their mammalian counterparts.
Acknowledgements
The authors are thankful to Padmashri Mrs Fatma Rafiq Zakaria, Chairman, Maulana Azad Educational Trust and Dr Zahid Zaheer, Principal, Y. B. Chavan College of Pharmacy, Dr Rafiq Zakaria Campus, Aurangabad 431 001 (M.S.), India for providing the necessary facilities.References
- N. Singh, B. B. Mishra, S. Bajpai, R. K. Singh and V. K. Tiwari, Bioorg. Med. Chem., 2014, 22, 18–45 CrossRef CAS PubMed
.
- S. Kamhawi, Trends Parasitol., 2006, 9, 439–445 CrossRef PubMed
- M. A. Vannier-Santos, A. Martiny and W. De Souza, Curr. Pharm. Des., 2002, 8, 297–318 CrossRef CAS
- L. Monzote, Open Antimicrob. Agents J., 2009, 1, 9–19 CAS
- World Health Organization, Sustaining the drive to overcome the global impact of neglected tropical diseases, second WHO report on neglected tropical diseases, “Diseases”, 2013, Leishmaniasis, 67–71.
- P. Bhargava and R. Singh, Interdiscip. Perspect. Infect. Dis., 2012, 2012, 1–14 Search PubMed
- R. F. Rodrigues, E. F. da Silva, A. Echevarria, R. F. Bonin, V. F. Amaral, L. L. Leon and M. M. Canto-Cavalheiro, Eur. J. Med. Chem., 2007, 42, 1039–1043 CrossRef CAS PubMed
-
(a) S. L. Croft and G. H. Coombs, Trends Parasitol., 2003, 19, 502–508 CrossRef CAS PubMed
- J. Alvar, I. D. Velez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin and M. den Boer, PLoS One, 2012, 7, e35671 CAS
- F. Kheirandish, A. C. Sharafi, B. Kazemi, M. Mohebali, A. Sarlak, M. J. Tarahi, K. Holakouee and H. Hajaran, Iran. J. Parasitol., 2013, 8, 382–388 Search PubMed
- M. G. Ritting and C. Bogdan, Parasitol. Today, 2000, 16, 292–297 CrossRef
- D. O. Santos, C. E. Coutinho, M. F. Madeira, C. G. Bottino, R. T. Vieira, S. B. Nascimento, A. Bernardino, S. C. Bourguignon, S. Corte-Real, R. T. Pinho, C. R. Rodrigues and H. C. Castro, Parasitol. Res., 2008, 103, 1–10 CrossRef PubMed
- B. B. Mishra, R. R. Kale, R. K. Singh and V. K. Tiwari, Fitoterapia, 2009, 80, 81–90 CrossRef CAS PubMed
- S. Singh and R. Sivakumar, J. Infect. Chemother., 2004, 10, 307–315 CrossRef PubMed
- N. Singh, M. Kumar and R. K. Singh, Asian Pac. J. Trop. Med., 2012, 5, 485–497 CrossRef CAS
- R. Yavar, K. Hadi, A. M. Reza, M. Mohebali, B. Hasan, O. M. Ali, R. Sina, B. H. Habib, H. Abodolrahim and G. Manuchehr, Asian Pac. J. Trop. Biomed., 2013, 3, 825–829 CrossRef
-
(a) H. Hussain, A. Al-Hurrasi, A. Al-Rawahi, I. R. Green and S. Gibbons, Chem. Rev., 2014, 114, 10369–10428 CrossRef CAS PubMed
- W. Peter, Indian J. Med. Res., 1981, 73, 1–18 Search PubMed
-
(a) J. Roychoudhury and N. Ali, Indian J. Biochem. Biophys., 2008, 45, 16–22 CAS
- W. L. Roberts, W. J. Mc murray and P. M. Rainey, Antimicrob. Agents Chemother., 1998, 42, 1076–1082 CAS
- J. D. Berman and D. J. Wyler, J. Infect. Dis., 1980, 142, 83–86 CrossRef CAS PubMed
- B. B. Mishra, R. K. Singh, A. Srivastava, V. J. Tripathi and V. K. Tiwari, Mini-Rev. Med. Chem., 2009, 9, 107–123 CrossRef CAS
-
(a) H. Denton, J. C. McGregor and G. H. Coombs, Biochem. J., 2004, 381, 405–412 CrossRef CAS PubMed
- L. H. Freitas-Junior, E. Chatelain, H. A. Kim and J. L. Siqueira-Neto, Int. J. Parasitol.: Drugs Drug Resist., 2012, 2, 11–19 CrossRef CAS PubMed
- B. L. Herwaldt and J. D. Berman, Am. J. Trop. Med. Hyg., 1992, 46, 296–306 CAS
- T. Polonio and T. Efferth, Int. J. Mol. Med., 2008, 22, 277–286 CAS
- S. Espuelas, D. Plano, P. Nguewa, M. Font, J. A. Palop, J. M. Irache and C. Sanmartin, Curr. Med. Chem., 2012, 19, 4259–4288 CrossRef CAS
-
(a) F. Frezard, C. Demicheli and R. R. Ribeiro, Molecules, 2009, 14, 2317–2336 CrossRef CAS PubMed
- M. A. Franco, A. C. Barbosa, S. Rath and J. G. Dorea, Am. J. Trop. Med. Hyg., 1995, 52, 435–437 CAS
- N. C. Hepburn, J. Postgrad. Med., 2003, 49, 50–54 CrossRef CAS PubMed
- R. A. Gasser Jr, A. J. Magill, C. N. Oster, E. D. Franke, M. Grogl and J. D. Berman, Clin. Infect. Dis., 1994, 18, 83–90 CrossRef CAS
- C. F. Brummitt, J. A. Porter and B. L. Herwaldt, Clin. Infect. Dis., 1996, 22, 878–879 CrossRef CAS PubMed
- I. Y. Zaghloul and M. Al-Jasser, Ann. Trop. Med. Parasitol., 2004, 98, 793–800 CrossRef CAS PubMed
- C. Bern, J. Adler-Moore, J. Berenguer, M. Boelaert, M. den Boer, R. N. Davidson, C. Figueras, L. Gradoni, D. A. Kafetzis, K. Ritmeijer, E. Rosenthal, C. Royce, R. Russo, S. Sundar and J. Alvar, Clin. Infect. Dis., 2006, 43, 917–924 CrossRef CAS PubMed
- A. K. Saha, T. Mukherjee and A. Bhaduri, Mol. Biochem. Parasitol., 1986, 19, 195–200 CrossRef CAS
- C. P. Thakur, R. K. Singh, S. M. Hassan, R. Kumar, S. Narain and A. Kumar, Trans. R. Soc. Trop. Med. Hyg., 1999, 93, 319–323 CrossRef CAS
- H. Ramos, E. Valdivieso, M. Gamargo, F. Dagger and B. E. Cohen, J. Membr. Biol., 1996, 152, 65–75 CrossRef CAS
- R. H. Johnson and H. E. Einstein, Ann. N. Y. Acad. Sci., 2007, 1111, 434–441 CrossRef CAS PubMed
- J. D. Berman, R. Badaro, C. P. Thakur, K. M. Wasunna, K. Behbehani, R. Davidson, F. Kuzoe, L. Pang, K. Weerasuriya and A. D. M. Bryceson, Bull. W. H. O., 1998, 76, 25–32 CAS
- J. P. Gangneux, A. Sulahian, Y. J. Garin and F. Derouin, Trans. R. Soc. Trop. Med. Hyg., 1996, 90, 574–577 CrossRef CAS
- V. Yardley and S. L. Croft, Antimicrob. Agents Chemother., 1997, 41, 752–756 CAS
- J. Adler-Moore and R. T. Proffitt, J. Antimicrob. Chemother., 2002, 49, 21–30 CrossRef CAS PubMed
- R. Balana-Fouce, R. M. Requera, J. C. Cubría and D. Ordóñez, Gen. Pharmacol., 1998, 30, 435–443 CrossRef CAS
- R. R. C. New, M. L. Chance and S. Heath, J. Antimicrob. Chemother., 1981, 8, 371–381 CrossRef CAS PubMed
- L. Gradoni, R. N. Davidson, S. Orsini, P. Betto and M. Giambenedetti, J. Drug Targeting, 1993, 1, 311–316 CrossRef CAS PubMed
- F. Meheus, M. Balasegaram, P. Olliaro, S. Sundar, S. Rijal, Md. A. Faiz and M. Boelaert, PLoS Neglected Trop. Dis., 2010, 4, e818 Search PubMed
- A. Meyerhoff, Clin. Infect. Dis., 1999, 28, 42–48 CAS
- S. Sundar, J. Chakravarty, D. Agarwal, M. Rai and H. W. Murray, N. Engl. J. Med., 2010, 362, 504–512 CrossRef CAS PubMed
- P. V. Bodhe, R. N. Kotwani, B. G. Kirodian, A. V. Pathare, A. K. Pandey, C. P. Thakur and N. A. Kshirsagar, Trans. R. Soc. Trop. Med. Hyg., 1999, 93, 314–318 CrossRef CAS
- S. Sundar, H. Mehta, A. V. Suresh, S. P. Singh, M. Rai and H. W. Murray, Clin. Infect. Dis., 2004, 38, 377–383 CrossRef CAS PubMed
- R. Laniado-Laborin and M. N. Cabralis Vergas, Rev. Iberoam. Microl., 2009, 26, 223–227 CrossRef PubMed
- S. Sundar, P. K. Sinha, M. Rai, D. K. Verma, K. Nawin, S. Alam, J. Chakravarty, M. Vaillant, N. Verma, K. Pandey, P. Kumari, C. S. Lal, R. Arora, B. Sharma, S. Ellis, N. Strub-Wourgaft, M. Balasegaram, P. Olliaro, P. Das and F. Modabber, J.-Lancet, 2011, 377, 477–486 CrossRef CAS
- J. D. Berman, Clin. Infect. Dis., 1997, 24, 684–703 CrossRef CAS PubMed
- C. P. Thakur, G. P. Sinha, V. Sharma, A. K. Pandey, M. Kumar and B. B. Verma, Indian J. Med. Res., 1993, 97, 170–175 CAS
- A. R. de Arias, E. Pandolfi, M. C. Vega and M. Rolón, Curr. Bioact. Compd., 2012, 8, 1–25 CrossRef
- C. Unger, M. Peukert, H. Sindermann, P. Hilgard, G. Nagel and H. Eibl, Cancer Treat. Rev., 1990, 17, 243–246 CrossRef CAS
- L. C. Rubiano, M. C. Miranda, S. Muvdi-Arenas, L. M. Montero, I. Rodríguez-Barraquer, D. Garcerant, M. Prager, L. Osorio, M. X. Rojas, M. Pérez, R. S. Nicholls and N. Gore-Saravia, J. Infect. Dis., 2012, 205, 684–692 CrossRef CAS PubMed
- S. L. Croft, K. Seifert and M. Duchene, Mol. Biochem. Parasitol., 2003, 126, 165–172 CrossRef CAS
- S. Sundar, T. K. Jha, H. Sindermann, K. Junge, P. Bachmann and J. Berman, Pediatr. Infect. Dis. J., 2003, 22, 434–438 Search PubMed
- H. Lux, N. Heise, T. Klenner and F. R. Opperdoes, Mol. Biochem. Parasitol., 2000, 111, 1–14 CrossRef CAS
- C. Paris, J. Bertoglio and J. Bréard, Apoptosis, 2007, 12, 1257–1267 CrossRef CAS PubMed
- P. Wadhone, M. Maiti, R. Agarwal, V. Kamat, S. Martin and B. Saha, J. Immunol., 2009, 182, 7146–7154 CrossRef CAS PubMed
- S. Sundar and H. W. Murray, Bull. W. H. O., 2005, 83, 394–395 Search PubMed
- F. J. Perez-Victoria, F. Gamarro, M. Ouellette and S. J. Castanys, Biol. Chem., 2003, 278, 49965–49971 CrossRef CAS PubMed
- Y. Fichoux, D. Rousseau, B. Ferrua, S. Ruette, A. Lelievre, D. Grousson and J. Kubar, Antimicrob. Agents Chemother., 1998, 42, 654–658 Search PubMed
- K. Seifert and S. L. Croft, Antimicrob. Agents Chemother., 2006, 50, 73–79 CrossRef CAS PubMed
- S. Sundar and J. Chakravarty, Expert Opin. Invest. Drugs, 2008, 17, 787–794 CrossRef CAS PubMed
- S. Sundar, N. Agrawal, R. Arora, D. Agarwal, M. Rai and J. Chakravarty, Clin. Infect. Dis., 2009, 49, 914–918 CrossRef CAS PubMed
- J. G. Hamilton, Parasite, 2008, 15, 252–256 CrossRef CAS PubMed
- G. Grimaldi Jr, R. B. Tesh and D. McMahon-Pratt, Am. J. Trop. Med. Hyg., 1989, 41, 687–725 CAS
- C. P. Thakur, T. P. Kanyok, A. K. Pandey, G. P. Sinha, C. Messick and P. Olliaro, Trans. R. Soc. Trop. Med. Hyg., 2000, 94, 432–433 CrossRef CAS
- A. B. Salah, P. A. Buffet, H. Louzir, G. Morizot, A. Zaatour, N. B. Alaya, B. Hajhmida, S. Elahmadi, S. Chlif, E. Lehnert, S. Doughty, K. Dellagi and M. Grogl, WR279396 an efficient non-toxic topical treatment of old world cutaneous leishmaniasis, Office of the Surgeon General, Department of the Army, U.S. Government, USA, Protocol no. A-9768.2, Version 9, 2005, pp. 1–39.
- M. Maarouf, Y. Kouchkovsky, S. Brown, P. X. Petit and M. Robert-Gero, Exp. Cell Res., 1997, 232, 339–348 CrossRef CAS PubMed
- L. X. Liu and P. F. Weller, N. Engl. J. Med., 1996, 334, 1178–1184 CrossRef CAS PubMed
- M. Maarouf, M. T. Adeline, M. Solignac, D. Vautrin and M. Robert-Gero, Parasite, 1998, 5, 167–173 CrossRef CAS PubMed
- S. Sundar, T. K. Jha, C. P. Thakur, P. K. Sinha and S. K. Bhattacharya, N. Engl. J. Med., 2007, 356, 2571–2581 CrossRef CAS PubMed
- S. Sundar, J. Chakravarty, V. K. Rai, N. Agrawal, S. P. Singh, V. Chauhan and H. Murray, Clin. Infect. Dis., 2007, 45, 556–561 CrossRef CAS PubMed
- M. Maarouf, F. Lawrence, S. L. Croft and M. Robert-Gero, Parasitol. Res., 1995, 81, 421–425 CrossRef CAS
- A. Jhingran, B. Chawla, S. Saxena, M. P. Barrett and R. Madhubala, Mol. Biochem. Parasitol., 2009, 164, 111–117 CrossRef CAS PubMed
- D. Fourmy, S. Yoshizawa and J. D. Puglisi, J. Mol. Biol., 1998, 277, 333–345 CrossRef CAS PubMed
- W. Khan and N. Kumar, J. Drug Targeting, 2011, 19, 239–250 CrossRef CAS PubMed
- T. A. Patel and D. N. Lockwood, Trop. Med. Int. Health, 2009, 14, 1064–1070 CrossRef CAS PubMed
- D. S. Fries and A. H. Fairlamb, in Burger's Medicinal Chemistry and Drug Discovery, ed. D. J. Abraham, John Wiley & Sons, 6th edn, 2003, vol. 5, pp. 1033–1087 Search PubMed
- M. Kandpal, R. Balana-Fouce, A. Pal, P. J. Guru and B. L. Tekwani, Mol. Biochem. Parasitol., 1995, 71, 193–201 CrossRef CAS
- M. Kandpal, B. L. Tekwani, P. M. S. Chauhan and A. P. Bhaduri, Life Sci., 1996, 59, 75–80 CrossRef
- J. Mishra, A. Saxena and S. Singh, Curr. Med. Chem., 2007, 14, 1153–1169 CrossRef CAS
- S. Sundar and M. Chatterjee, Indian J. Med. Res., 2006, 123, 345–352 CAS
- M. Basselin, H. Denise, G. H. Coombs and M. Barrett, Antimicrob. Agents Chemother., 2002, 46, 3731–3738 CrossRef CAS
- S. L. Croft, S. Sundar and A. H. Fairlamb, Clin. Microbiol. Rev., 2006, 19, 111–126 CrossRef CAS PubMed
- C. M. Mesa-Valle, J. Castilla-Calvente, M. Sanchez-Moreno, V. Moraleda-Lindez, J. Barbe and A. Osuna, Antimicrob. Agents Chemother., 1996, 40, 684–690 CAS
- P. Alberti, J. Ren, M. P. Teulade-Fichou, L. Guittat, J. F. Riou, J. Chaires, C. Helene, J. P. Vigneron, J. M. Lehn and J. L. Mergny, J. Biomol. Struct. Dyn., 2001, 19, 505–513 CAS
- C. D. Giorgio, K. Shimi, G. Boyer, F. Delmas and J. P. Galy, Eur. J. Med. Chem., 2007, 42, 1277–1284 CrossRef PubMed
- F. Delmas, A. Avellaneda, C. D. Giorgio, M. Robin, E. D. Clercq, P. Timon-David and J. P. Galy, Eur. J. Med. Chem., 2004, 39, 685–690 CrossRef CAS PubMed
- J. Mauel, W. Denny, S. Gamage, A. Ransijn, S. Wojcik, D. Figgitt and R. Ralph, Antimicrob. Agents Chemother., 1993, 37, 991–996 CrossRef CAS
- C. Spino, M. Dodier and S. Sotheeswaran, Bioorg. Med. Chem. Lett., 1998, 8, 3475–3478 CrossRef CAS
- V. Arango, S. Robledo, B. Séon Méniel, B. Figadére, W. Cardona, J. Sáez and F. Otálvaro, J. Nat. Prod., 2010, 73, 1012–1014 CrossRef CAS PubMed
- M. E. Ferreira, A. R. Arias, G. Yaluff, N. V. Bilbao, H. Nakayama, S. Torres, A. Schinini, I. Guy, H. Heinzen and A. Fournet, Phytomedicine, 2010, 17, 375–378 CrossRef CAS PubMed
- M. Iranshahi, P. Arfa, M. Ramezani, M. R. Jaafari, H. Sadeghian, C. Bassarello, S. Piacente and C. Pizza, Phytochemistry, 2007, 68, 554–561 CrossRef CAS PubMed
- R. K. Verma, V. J. Prajapati, G. K. Verma, D. Chakraborty, S. Sundar, M. Rai, V. K. Dubey and M. S. Singh, ACS Med. Chem. Lett., 2012, 3, 243–247 CrossRef CAS PubMed
- J. T. Pierson, A. Dumetre, S. Hutter, F. Delmas, M. Laget, J. P. Finet, N. Azas and S. Combes, Eur. J. Med. Chem., 2010, 45, 864–869 CrossRef CAS PubMed
- M. Knockaert, K. Wieking, S. Schmitt, M. Leost, K. M. Grant, J. C. Mottram, C. Kunick and L. Meijer, J. Biol. Chem., 2002, 277, 25493–25501 CrossRef CAS PubMed
- R. L. Clark, K. C. Carter, A. B. Mullen, G. D. Coxon, G. Owusu-Dapaah, E. McFarlane, M. D. D. Thi, M. H. Grant, N. A. Justice Tettey and S. P. Mackay, Bioorg. Med. Chem. Lett., 2007, 17, 624–627 CrossRef CAS PubMed
- H. P. Avila, E. F. A. Smânia, F. D. Monache and A. Smânia, Bioorg. Med. Chem., 2008, 16, 9790–9794 CrossRef CAS PubMed
- M. Cabrera, M. Simoens, G. Falchi, M. L. Lavaggi, O. E. Piro, E. E. Castellano, A. Vidal, A. Azqueta, A. Monge, A. L. de Ceráin, G. Sagrera, G. Seoane, H. Cerecetto and M. González, Bioorg. Med. Chem., 2007, 15, 3356–3367 CrossRef CAS PubMed
- L. D. Chiaradia, R. dos Santos, C. E. Vitor, A. A. Vieira, P. C. Leal, R. J. Nunes, J. B. Calixto and R. A. Yunes, Bioorg. Med. Chem., 2008, 16, 658–667 CrossRef CAS PubMed
- M. Chen, S. B. Christensen, J. Blom, E. Lemmich, L. Nadelmann, K. Fich, T. G. Theander and A. Kharazmi, Antimicrob. Agents Chemother., 1993, 37, 2550–2556 CrossRef CAS
- M. Chen, S. B. Christensen, T. G. Theander and A. Kharazmi, Antimicrob. Agents Chemother., 1994, 38, 1339–1344 CrossRef CAS
- L. Zhai, J. Blom, M. Chen, S. B. Christensen and A. Kharazmi, Antimicrob. Agents Chemother., 1995, 39, 2742–2748 CrossRef CAS
- L. Zhai, M. Chen, S. B. Christensen, T. G. Theander and A. Kharazmi, Antimicrob. Agents Chemother., 2001, 45, 2023–2029 CrossRef PubMed
- Z. Nazarian, S. Emami, S. Heydari, S. K. Ardestani, M. Nakhjiri, F. Poorrajab, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2010, 45, 1424–1429 CrossRef CAS PubMed
- D. G. Rando, M. A. Avery, B. L. Tekwani, S. I. Khan and E. I. Ferreira, Bioorg. Med. Chem., 2008, 16, 6724–6731 CrossRef CAS PubMed
- S. F. Nielsen, S. B. Christensen, G. Cruciani, A. Kharazmi and T. Liljefors, J. Med. Chem., 1998, 41, 4819–4832 CrossRef CAS PubMed
- T. P. Barbosa, S. C. O. Sousa, F. M. Amorim, Y. K. S. Rodrigues, P. A. C. de Assis, J. P. A. Caldas, M. R. Oliveira and M. L. A. A. Vasconcellos, Bioorg. Med. Chem., 2011, 19, 4250–4256 CrossRef CAS PubMed
- C. R. Andrighetti-Fröhner, K. N. de Oliveira, D. Gaspar-Silva, L. K. Pacheco, A. C. Joussef, M. Steindel, C. M. O. Simões, A. M. T. de Souza, U. O. Magalhaes, I. F. Afonso, C. R. Rodrigues, R. J. Nunes and H. C. Castro, Eur. J. Med. Chem., 2009, 44, 755 CrossRef PubMed
- M. L. Bello, L. D. Chiaradia, L. R. S. Dias, L. K. Pacheco, T. R. Stumpf, A. Mascarello, M. Steindel, R. A. Yunes, H. C. Castro, R. J. Nunes and C. R. Rodrigues, Bioorg. Med. Chem., 2011, 19, 5046–5052 CrossRef CAS PubMed
- A. Foroumadi, S. Emami, M. Sorkhi, M. Nakhjiri, Z. Nazarian, S. Heydari, S. K. Ardestani, F. Poorrajab and A. Shafiee, Chem. Biol. Drug Des., 2010, 75, 590–596 CAS
- R. Shivahare, V. Korthikunta, H. Chandasana, M. K. Suthar, P. Agnihotri, P. Vishwakarma, T. K. Chaitanya, P. Kancharla, T. Khaliq, S. Gupta, R. S. Bhatta, J. V. Pratap, J. K. Saxena, S. Gupta and N. Tadigoppula, J. Med. Chem., 2014, 57, 3342–3357 CrossRef CAS PubMed
- S. Gupta, R. Shivahare, V. Korthikunta, R. Singh, S. Gupta and N. Tadigoppula, Eur. J. Med. Chem., 2014, 81, 359–366 CrossRef CAS PubMed
- S. B. Bharate, J. B. Bharate, S. I. Khan, B. L. Tekwani, M. R. Jacobb, R. Mudududdla, R. R. Yadav, B. Singh, P. R. Sharma, S. Maity, B. Singh, I. A. Khan and R. A. Vishwakarma, Eur. J. Med. Chem., 2013, 63, 435–443 CrossRef CAS PubMed
- L. T. D. Tonin, M. R. Panice, C. V. Nakamura, K. J. P. Rocha, A. O. dos Santos, T. Ueda-Nakamura, W. F. da Costaa and M. H. Sarragiotto, Biomed. Pharmacother., 2010, 64, 386–389 CrossRef CAS PubMed
- G. S. Singh, Y. M. S. A. Al-kahraman, D. Mpadi and M. Yasinzai, Bioorg. Med. Chem. Lett., 2012, 22, 5704–5706 CrossRef CAS PubMed
- S. S. Chauhan, L. Gupta, M. Mittal, P. Vishwakarma, S. Gupta and P. M. S. Chauhan, Bioorg. Med. Chem. Lett., 2010, 20, 6191–6194 CrossRef CAS PubMed
- C. S. Reid, A. A. Farahat, X. Zhu, T. Pandharkar, D. W. Boykin and K. A. Werbovetz, Bioorg. Med. Chem. Lett., 2012, 22, 6806–6810 CrossRef CAS PubMed
- A. Bouhlel, C. Curti, A. Dumètre, M. Laget, M. D. Crozet, N. Azas and P. Vanelle, Bioorg. Med. Chem., 2010, 18, 7310–7320 CrossRef CAS PubMed
- V. P. Pandey, S. S. Bisht, M. Mishra, A. Kumar, M. I. Siddiqi, A. Verma, M. Mittal, S. A. Sane, S. Gupta and R. P. Tripathi, Eur. J. Med. Chem., 2010, 45, 2381–2388 CrossRef CAS PubMed
- J. N. Sangshetti, R. I. Shaikh, F. A. K. Khan, R. H. Patil, S. D. Marathe, W. N. Gade and D. B. Shinde, Bioorg. Med. Chem. Lett., 2014, 24, 1605–1610 CrossRef CAS PubMed
- H. Liu and L. Nolan, Antileishmanial mode of action of derivatives of the quinoline alkaloid (QUININE) from the bark of Cinchona Ledgeriana, Acta Hortic., International Symposium on Medicinal and Aromatic Plants, 1996, vol. 1 Search PubMed
- R. Dietze, S. F. Carvalho, L. C. Valli, J. Berman, T. Brewer, W. Milhous, J. Sanchez, B. Schuster and M. Grogl, Am. J. Trop. Med. Hyg., 2001, 65, 685–689 CAS
- K. Lackovic, J. P. Parisot, N. Sleebs, J. B. Baell, L. Debien, K. G. Watson, J. M. Curtis, E. Handman, I. P. Street and L. Kedzierski, Antimicrob. Agents Chemother., 2010, 54, 1712–1719 CrossRef CAS PubMed
- R. Sharma, A. K. Pandey, R. Shivahare, K. Srivastava, S. Gupta and P. M. S. Chauhan, Bioorg. Med. Chem. Lett., 2014, 24, 298–301 CrossRef CAS PubMed
- S. Y. Ablordeppey, P. Fan, A. M. Clark and A. Nimrod, Bioorg. Med. Chem., 1999, 7, 343–349 CrossRef CAS
- S. Hazra, S. Ghosh, S. Debnath, S. Seville, V. K. Prajapati, C. W. Wright, S. Sundar and B. Hazra, Parasitol. Res., 2012, 111, 195–203 CrossRef PubMed
- S. Guglielmo, M. Bertinaria, B. Rolando, M. Crosetti, R. Fruttero, V. Yardley, S. L. Croft and A. Gasco, Eur. J. Med. Chem., 2009, 44, 5071–5079 CrossRef CAS PubMed
- L. Paloque, P. Verhaeghe, M. Casanova, C. Castera-Ducros, A. Dumètre, L. Mbatchi, S. Hutter, M. Kraiem-M'Rabet, M. Laget, V. Remusat, S. Rault, P. Rathelot, N. Azas and P. Vanelle, Eur. J. Med. Chem., 2012, 54, 75–86 CrossRef CAS PubMed
- A. M. L. Carmo, F. M. C. Silva, P. A. Machado, A. P. S. Fontes, F. R. Pavan, C. Q. F. Leite, S. R. de A. Leite, E. S. Coimbra and A. D. Da Silva, Biomed. Pharmacother., 2011, 65, 204–209 CrossRef CAS PubMed
- P. M. Loiseau, S. Gupta, A. Verma, S. Srivastava, S. K. Puri, F. Sliman, M. Normand-Bayle and D. Desmaele, Antimicrob. Agents Chemother., 2011, 55, 1777–1780 CrossRef CAS PubMed
- V. S. Gopinath, J. Pinjari, R. T. Dere, A. Verma, P. Vishwakarma, R. Shivahare, M. Moger, P. S. Kumar Goud, V. Ramanathan, P. Bose, M. V. Rao, S. Gupta, S. K. Puri, D. Launay and D. Martin, Eur. J. Med. Chem., 2013, 69, 527–536 CrossRef CAS PubMed
- V. S. Gopinath, M. Rao, R. Shivahare, P. Vishwakarma, S. Ghose, A. Pradhan, R. Hindupur, K. D. Sarma, S. Gupta, S. K. Puri, D. Launay and D. Martin, Bioorg. Med. Chem. Lett., 2014, 24, 2046–2052 CrossRef CAS PubMed
- S. Kumar, N. Shakya, S. Gupta, J. Sarkar and D. P. Sahu, Bioorg. Med. Chem. Lett., 2009, 19, 2542–2545 CrossRef CAS PubMed
- K. C. Agarwal, V. Sharma, N. Shakya and S. Gupta, Bioorg. Med. Chem. Lett., 2009, 19, 5474–5477 CrossRef CAS PubMed
- J. D. Berman, M. King and N. Edwards, Antimicrob. Agents Chemother., 1989, 33, 1860–1863 CrossRef CAS
- K. A. Werbovetz, J. J. Brendle and D. Sackett, Mol. Biochem. Parasitol., 1999, 98, 53–65 CrossRef CAS
- A. K. Bhattacharjee, D. J. Skanchy, B. Jennings, T. H. Hudson, J. J. Brendle and K. A. Werbovetz, Bioorg. Med. Chem., 2002, 10, 1979–1989 CrossRef CAS
- K. S. Van Horn, X. Zhu, T. Pandharkar, S. Yang, B. Vesely, M. Vanaerschot, J. C. Dujardin, S. Rijal, D. E. Kyle, M. Z. Wang, K. A. Werbovetz and R. Manetsch, J. Med. Chem., 2014, 57, 5141–5156 CrossRef CAS PubMed
- M. Sharma, K. Chauhan, R. Shivahare, P. Vishwakarma, M. K. Suthar, A. Sharma, S. Gupta, J. K. Saxena, J. Lal, P. Chandra, B. Kumar and P. M. S. Chauhan, J. Med. Chem., 2013, 56, 4374–4392 CrossRef CAS PubMed
- C. D. Buarque, G. C. G. Militão, D. J. B. Lima, L. V. Costa-Lotufo, C. Pessoa, M. O. de Moraesd, E. F. Cunha-Junior, E. C. Torres-Santos, C. D. Netto and P. R. R. Costa, Bioorg. Med. Chem., 2011, 19, 6885–6891 CrossRef CAS PubMed
- W. Peters, E. R. Trotter and B. L. Robinson, Ann. Trop. Med. Parasitol., 1980, 74, 289–298 CAS
- V. J. Ram, A. Goel, P. K. Shukla and A. Kapil, Bioorg. Med. Chem. Lett., 1997, 7, 3101–3106 CrossRef CAS
- A. Irshad, H. M. F. Madkour, Y. Farina, Y. M. S. A. Al-Kahraman and N. Baloch, Int. J. Sci. Eng. Res., 2014, 5, 198–204 Search PubMed
- K. Chauhan, M. Sharma, R. Shivahare, U. Debnath, S. Gupta, Y. S. Prabhakar and P. M. S. Chauhan, ACS Med. Chem. Lett., 2013, 4, 1108–1113 CrossRef CAS PubMed
- N. Sunduru, A. Agarwal, S. B. Katiyar, Nishi, N. Goyal, S. Gupta and P. M. S. Chauhan, Bioorg. Med. Chem., 2006, 14, 7706–7715 CrossRef CAS PubMed
- F. G. Braga, E. S. Coimbra, M. O. Matos, A. M. L. Carmo, M. D. Cancio and A. D. da Silva, Eur. J. Med. Chem., 2007, 42, 530–537 CrossRef CAS PubMed
- J. D. Berman, L. S. Lee, R. K. Robins and G. R. Revankar, Antimicrob. Agents Chemother., 1983, 24, 233–236 CrossRef CAS
- D. J. Nelson, S. W. LaFon, T. E. James, T. Spector, R. L. Berens and J. J. Marr, Biochem. Biophys. Res. Commun., 1982, 106, 349–354 CrossRef
- P. Rainy and D. V. Santi, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 288–292 CrossRef
- E. Casanova, D. Moreno, A. Gigante, E. Rico, C. M. Genes, C. Oliva, M.-J. Camarasa, F. Gago, A. Jimenez-Ruiz and M. Perez-Perez, Chem. Med. Chem., 2013, 8, 1161–1174 CrossRef CAS PubMed
- P. Zhang, D. E. Nicholson, J. M. Bujnicki, X. Su, J. J. Brendle, M. Ferdig, D. E. Kyle, W. K. Milhous and P. K. Chiang, J. Biomed. Sci., 2002, 9, 34–40 CrossRef CAS
- S. Nwaka and A. Hudson, Nat. Rev. Drug Discovery, 2006, 5, 941–955 CrossRef CAS PubMed
- X. Chen, C. R. Chong, L. Shi, T. Yashimoto, D. J. Sullivan and O. Liu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14548–14553 CrossRef CAS PubMed
- C. C. Musonda, G. A. Whitlock, M. J. Witty, R. Brun and M. Kaiser, Bioorg. Med. Chem. Lett., 2009, 19, 401–405 CrossRef CAS PubMed
- A. Agarwal, Ramesh, Ashutosh, N. Goyal, P. M. S. Chauhan and S. Gupta, Bioorg. Med. Chem., 2005, 13, 6678–6684 CrossRef CAS PubMed
- S. N. Suryawanshi, S. Kumar, R. Shivahare, S. Pandey, A. Tiwari and S. Gupta, Bioorg. Med. Chem. Lett., 2013, 23, 5235–5238 CrossRef CAS PubMed
- M. Taha, M. S. Baharudin, N. H. Ismail, K. M. Khan, F. M. Jaafar, Samreen, S. Siddiqui and M. I. Choudhary, Bioorg. Med. Chem. Lett., 2013, 23, 3463–3466 CrossRef CAS PubMed
- D. G. Rando, M. A. Avery, B. L. Tekwani, S. I. Khan and E. I. Ferreira, Bioorg. Med. Chem., 2008, 16, 6724–6731 CrossRef CAS PubMed
- Y. M. S. A. Al-Kahraman, H. M. F. Madkour, D. Ali and M. Yasinzai, Molecules, 2010, 15, 660–671 CrossRef CAS PubMed
- R. C. N. R. Corrales, N. B. de Souza, L. S. Pinheiro, C. Abramo, E. S. Coimbra and A. D. Da Silva, Biomed. Pharmacother., 2011, 65, 198–203 CrossRef CAS PubMed
- S. Marhadour, P. Marchand, F. Pagniez, M.-A. Bazin, C. Picot, O. Lozach, S. Ruchaud, M. Antoine, L. Meijer, N. Rachidi and P. L. Pape, Eur. J. Med. Chem., 2012, 58, 543–556 CrossRef CAS PubMed
- V. K. Marrapu, M. Mittal, R. Shivahare, S. Gupta and K. Bhandari, Eur. J. Med. Chem., 2011, 46, 1694–1700 CrossRef CAS PubMed
- A. Verma, S. Srivastava, S. A. Sane, V. K. Marrapu, N. Srinivas, M. Yadava, K. Bhandari and S. Gupta, Acta Trop., 2011, 117, 157–160 CrossRef CAS PubMed
- N. Srinivas, S. Palne, Nishi, S. Gupta and K. Bhandari, Bioorg. Med. Chem. Lett., 2009, 19, 324–327 CrossRef CAS PubMed
- S. N. Suryawanshi, A. Tiwari, S. Kumar, R. Shivahare, M. Mittal, P. Kant and S. Gupta, Bioorg. Med. Chem. Lett., 2013, 23, 2925–2928 CrossRef CAS PubMed
- M. S. dos Santos, M. L. V. Oliveira, A. M. R. Bernardino, R. M. de Léo, V. F. Amaral, F. T. de Carvalho, L. L. Leon and M.
M. Canto-Cavalheiro, Bioorg. Med. Chem. Lett., 2011, 21, 7451–7454 CrossRef CAS PubMed
- M. Verma, A. Bhandari and R. M. Nema, J. Chem. Pharm. Res., 2010, 2, 244–250 CAS
- A. Mayence, A. Pietka, M. S. Collins, M. T. Cushion, B. L. Tekwani, T. L. Huang and J. J. V. Eynde, Bioorg. Med. Chem. Lett., 2008, 18, 2658–2661 CrossRef CAS PubMed
- S. K. Tipparaju, S. Joyasawal, M. Pieroni, M. Kaiser, R. Brun and A. P. Kozikowski, J. Med. Chem., 2008, 51, 7344–7347 CrossRef CAS PubMed
- A. Tahghighi, F. R. Marznaki, F. Kobarfard, S. Dastmalchi, J. S. Mojarrad, S. Razmi, S. K. Ardestani, S. Emami, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2011, 46, 2602–2608 CrossRef CAS PubMed
- E. Alipour, S. Emami, A. Yahya-Meymandi, M. Nakhjiri, F. Johari, S. K. Ardestani, F. Poorrajab, M. Hosseini, A. Shafiee and A. Foroumadi, J. Enzyme Inhib. Med. Chem., 2011, 26, 123–128 CrossRef CAS PubMed
- O. L. Adebayo, D. Suleman and A. A. Samson, J. Asian Sci. Res., 2013, 3, 157–173 Search PubMed
- T. Polonio and T. Efferth, Int. J. Mol. Med., 2008, 22, 277–286 CAS
-
(a) A. K. Sen, K. K. Sarkar, P. C. Majumder and N. Banerji, Phytochemistry, 1981, 20, 183–185 CrossRef CAS
- E. R. da Silva, C. C. Maquiaveli and P. P. Magalhaes, Exp. Parasitol., 2012, 130, 183–188 CrossRef CAS PubMed
- J. Sidana, D. Neeradi, A. Choudhary, S. Singh, W. J. Foley and I. P. Singh, J. Chem. Sci., 2013, 125, 765–775 CrossRef CAS
- M. Sairafianpour, O. Kayser, J. Christensen, M. Asfa, M. Witt, D. Staerk and J. W. Jaroszewski, J. Nat. Prod., 2002, 65, 1754–1758 CrossRef CAS PubMed
- B. Mittra, A. Saha, A. R. Chowdhury, C. Pal, S. Mandal, S. Mukhopadhyay, S. Bandyopadhyay and H. K. Majumder, J. Mol. Med., 2000, 6, 527–541 CrossRef CAS
- T. Kam, K. Sim, T. Koyano, M. Toyoshima, M. Hayashi and K. Komiyama, J. Nat. Prod., 1998, 61, 1332–1336 CrossRef CAS PubMed
- D. Staerk, E. Lemmich, J. Christensen, A. Kharazmi, C. E. Olsen and J. W. Jaroszewski, Planta Med., 2000, 66, 531–536 CrossRef CAS PubMed
- L. Pan, C. Terrazas, U. M. Acuna, T. N. Ninh, H. Chai, E. J. C. Blanco, D. D. Soejarto, A. R. Satoskar and A. D. Kinghorn, Phytochem. Lett., 2014, 10, 54–59 Search PubMed
- K. M. Ahua, J.-R. Ioset, A. Ransijn, J. Mauël, S. Mavi and K. Hostettmann, Phytochemistry, 2004, 65, 963–968 CrossRef CAS PubMed
- G. Bringmann, M. Dreyer, J. H. Faber, P. W. Dalsgaard, D. Staerk, J. W. Jaroszewski, H. Ndangalasi, F. Mbago, R. Brun and S. B. Christensen, J. Nat. Prod., 2004, 67, 743–748 CrossRef CAS PubMed
- J. Fotie, D. S. Bohle, M. Olivier, M. Adelaida Gomez and S. Nzimiro, J. Nat. Prod., 2007, 70, 1650–1653 CrossRef CAS PubMed
- T. Khaliq, P. Misra, S. Gupta, K. P. Reddy, R. Kant, P. R. Maulik, A. Dube and T. Narender, Bioorg. Med. Chem. Lett., 2009, 19, 2585–2586 CrossRef CAS PubMed
- M. S. S. Rosa, R. R. Mendonça-Filho, H. R. Bizzo, R. M. Soares, I. de Almeida Rodrigues, T. Souto-Padrón, C. S. Alviano and A. H. Lopes, Antimicrob. Agents Chemother., 2003, 47, 1895–1901 CrossRef CAS PubMed
-
(a) P. Delgadoendez, N. Herrera, H. Chavez, A. E. B. A. G. Ravelo, F. Cortes, F. Castanys and F. Gamarro, Bioorg. Med. Chem., 2008, 16, 1425–1430 CrossRef PubMed
- D. C. Arruda, F. L. D'Alexandri, A. M. Katzin and S. R. B. Uliana, Antimicrob. Agents Chemother., 2005, 49, 1679–1687 CrossRef CAS PubMed
- K. Georgopoulou, D. Smirlis, S. Bisti, E. Xingi, L. Skaltsounis and K. Soteriadou, Planta Med., 2007, 73, 1081–1088 CrossRef CAS PubMed
- R. L. Fabri, R. A. Garcia, J. R. Florencio, L. Oliveira de Carvalho, N. Pinto, E. S. Coimbra, E. M. de Souza-Fagundes, A. Ribeiro and E. Scio, Med. Chem. Res., 2014, 23, 5294–5304 CrossRef CAS
- M. G. M. Danelli, D. C. Soares, H. S. Abreu, L. M. T. Peçanha and E. M. Saraiva, Phytochemistry, 2009, 70, 608–614 CrossRef CAS PubMed
- B. Shumaila, A. Mahboob, A. Achyut, S. R. Lal, Y. Sammer, A. Bashir, P. Shama, A. Akhtar and I. Choudhary, Phytochem. Lett., 2014, 9, 46–50 CrossRef PubMed
- L. M. Sanchez, D. Lopez, B. A. Vesely, G. T. Togna, W. H. Gerwick, D. E. Kyle, R. G. Linington and A.-C. Almiramides, J. Med. Chem., 2010, 53, 4187–4197 CrossRef CAS PubMed
- M. J. Balunas, R. G. Linington, K. Tidgewell, A. M. Fenner, L. D. Urena, G. D. Togna, D. E. Kyle and W. H. Gerwick, J. Nat. Prod., 2010, 73, 60–66 CrossRef CAS PubMed
- T. L. Simmons, N. Engene, L. D. Urena, L. I. Romero, E. Ortega-Barria, L. Gerwick and W. H. Gerwick, J. Nat. Prod., 2008, 71, 1544–1550 CrossRef PubMed
- G. Gul, N. L. Hammond, M. Yousaf, J. J. Bowling, R. F. Schinazi, S. S. Wirtz, G. C. Andrews, C. Cuevas and M. T. Hamann, Bioorg. Med. Chem., 2006, 14, 8495–8505 CrossRef PubMed
- M. H. Kossuga, A. M. Nascimento, J. Q. Reimao, A. G. Tempone, N. N. Taniwaki, K. Veloso, A. G. Ferreira, B. C. Cavalcanti, C. Pessoa, M. O. Moraes, M. A. S. Mayer, E. Hajdu and R. G. S. Berlinck, J. Nat. Prod., 2008, 71, 334–339 CrossRef CAS PubMed
- A. R. Satoskar, J. R. Fuchs, A. D. Kinghorn, L. Pan, C. M. Lezama-Davila and E. Bachelder, US Pat., 0287030 A1, 2014
- L. A. R. Vasquez, R. O. Cardona, S. M. Doque, S. M. R. Restrepo, I. D. V. Bernal, D. L. C. Medina and M. A. Jones, US Pat., 0194640 A1, 2014
- N. Boechat, M. S. Costa, M. C. S. Lourenco, I. Neves, M. S. Genestra and V. F. Ferreira, US Pat., 0274298 A1, 2013
- J. Kimura, S. Horie, H. Marushima, Y. Matsumoto, C. Sanjoba and Y. Osada, US Pat., 0245288 A1, 2013
- N. C. Galindo Sevilla and F. Hernandez Luis, US Pat., 0109663 A1, 2013
- A. E. A. Mohammad, S. H. A. Mohammad, M. I. M. Ibrahim, A. M. T. Ayoub and M. M. Suleiman, EP Pat., 2 085 394 B1, 2012
- R. Curtis and P. Distefano, WO Pat., 2010054382 A1, 2010
- F. E. Cohen, X. Du, C. Guo and J. H. McKerrow, US Pat., 7495023 B2, 2009
- Searle, Andrew and David, WO Pat., 043726, 2008
- K. Werbovetz, D. L. Sackett and M. M. Salem, US Pat., 7211696 B2, 2007
- M. Ihara, K. S. M. Takasu, H. S. M. Terauchi, S. Sekita and M. Takahashi, EP Pat., 1 623 981 A1, 2006
- L. C. Rios, L. I. Romero, E. Ortega-Barria and T. Capson, WO Pat., 084801 A2, 2004
- S. O. Lorente, C. J. Jimenez, L. Gros, V. Yardley, K. de Luca-Fradley, S. L. Croft, J. Urbina, L. M. Ruiz-Perez, D. G. Pacanowska and I. H. Gilbert, Bioorg. Med. Chem., 2005, 13, 5435–5453 CrossRef CAS PubMed
- H. Sanati, P. Belanger, R. Fratti and M. Ghannoum, Antimicrob. Agents Chemother., 1997, 41, 2492–2496 CAS
- P. Ascenzi, A. Bocedi, P. Visca, G. Antonini and L. Gradoni, Biochem. Biophys. Res. Commun., 2003, 309, 659–665 CrossRef CAS PubMed
- M. L. Cunningham, R. G. Titus, S. J. Turco and S. M. Beverley, Science, 2001, 292, 285–287 CrossRef CAS PubMed
- R. L. Krauth-Siegel and O. Inhoff, Parasitol. Res., 2003, 90, S77–S85 CrossRef PubMed
- T. Shiba, H. Mizote, T. Kaneko, T. Nakajima and Y. Kakimoto, Biochim. Biophys. Acta, 1971, 244, 523–531 CrossRef CAS
- M. H. Park, H. L. Cooper and J. E. Folk, J. Biol. Chem., 1982, 257, 7217–7222 CAS
- B. Chawla, A. Jhingran, S. Singh, N. Tyagi, M. H. Park, N. Srinivasan, S. C. Roberts and R. Madhubala, J. Biol. Chem., 2010, 285, 453–463 CrossRef CAS PubMed
- G. F. Spath, L. Epstein, B. Leader, S. M. Singer, H. A. Avila, S. J. Turco and S. M. Beverley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9258–9263 CrossRef CAS PubMed
- D. L. Sacks, G. Modi, E. Rowton, G. Spath, L. Epstein, S. J. Turco and S. M. Beverley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 406–411 CrossRef CAS
- F. M. Vellieux, J. Hajdu, C. L. Verlinde, H. Groendijk, R. J. Read, T. J. Greenhough, J. W. Campbell, K. H. Kalk, J. A. Littlechild, H. C. Watson and W. G. J. Hol, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 2355–2359 CrossRef CAS
- A. M. Aronov, S. Suresh, F. S. Buckner, W. C. Van Voorhis, C. L. Verlinde, F. R. Opperdoes, W. G. Hol and M. H. Gelb, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4273–4278 CrossRef CAS
- K. A. Plewes, S. D. Barr and L. Gedamu, Infect. Immun., 2003, 71, 5910–5920 CrossRef CAS
-
(a) R. H. Glew, A. K. Saha, S. Das and A. T. Remaley, Microbiol. Rev., 1988, 52, 412–432 CAS
- S. Martinez and J. J. Marr, N. Engl. J. Med., 1992, 326, 741–744 CrossRef CAS PubMed
- G. Vasudevan, N. S. Carter, M. E. Drew, S. M. Beverley, M. A. Sanchez, A. Seyfang, B. Ullman and S. M. Landfear, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9873–9878 CrossRef CAS
- G. Vasudevan, B. Ullman and S. M. Landfear, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6092–6097 CrossRef CAS PubMed
- K. M. Grant, P. Hassan, J. S. Anderson and J. C. Mottram, J. Biol. Chem., 1992, 273, 10153–10159 CrossRef PubMed
- K. M. Grant, M. H. Dunion, V. Yardley, A. L. Skaltsounis, D. Marko, G. Eisenbrand, S. L. Croft, L. Meijer and J. C. Mottram, Antimicrob. Agents Chemother., 2004, 48, 3033–3042 CrossRef CAS PubMed
- M. A. Morales, O. Renaud, W. Faigle, S. L. Shorte and G. F. Spath, Int. J. Parasitol., 2007, 37, 1187–1199 CrossRef CAS PubMed
- M. Kandpal and B. L. Tekwani, Life Sci., 1997, 60, 1793–1801 CrossRef CAS
- J. Tavares, A. Ouaissi, P. K. Lin, A. Tomas and A. Cordeiro-da-Silva, Int. J. Parasitol., 2005, 35, 637–646 CrossRef CAS PubMed
- M. A. Vannier-Santos, D. Menezes, M. F. Oliveira and F. G. de Mello, Microbiology, 2008, 154, 3104–3111 CrossRef CAS PubMed
- O. Heby, L. Persson and M. Rentala, Amino Acids, 2007, 33, 359–366 CrossRef CAS PubMed
- R. M. Reguera, B. L. Tekwani and R. Balana-Fouce, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2005, 140, 151–164 CrossRef PubMed
- R. A. Neal and S. L. Croft, J. Antimicrob. Chemother., 1984, 14, 463–475 CrossRef CAS PubMed
- B. Nare, J. Luba, L. W. Hardy and S. Beverley, Parasitology, 1997, 114, S101–S110 Search PubMed
- L. W. Hardy, W. Matthews, B. Nare and S. M. Beverley, Exp. Parasitol., 1997, 87, 157–169 CrossRef CAS
- C. Witheres-Martinez, L. Jean and M. J. Blackman, Mol. Microbiol., 2004, 53, 55–63 CrossRef PubMed
- R. E. Silva-Lopez, J. A. Morgado-Diaz, M. A. Chavez and S. Giovanni-de-Simone, Parasitol. Res., 2007, 101, 1627–1635 CrossRef CAS PubMed
- H. Mahmoudzadeh-Niknam and J. H. McKerrow, Exp. Parasitol., 2004, 106, 158–163 CrossRef CAS PubMed
- A. Das, A. Dasgupta, T. Sengupta and H. K. Majumder, Trends Parasitol., 2004a, 20, 381–387 Search PubMed
- A. L. Bodley, M. C. Wani, M. E. Wall and T. A. Shapiro, Biochem. Pharmacol., 1995, 50, 937–942 CrossRef CAS
- G. Singh, K. G. Jayanarayan and C. S. Dey, Mol. Biochem. Parasitol., 2005, 141, 57–69 CrossRef CAS PubMed
- A. C. Rosypal, S. Tripp, S. Lewis, J. Francis, M. K. Stoskopf, R. S. Larsen and D. S. Lindsay, J. Parasitol., 2010, 96, 1230–1231 CrossRef PubMed
- A. R. Chowdhury, S. Mandal, A. Goswami, M. Ghosh, L. Mandal, D. Chakraborty, A. Ganguly, G. Tripathi, S. Mukhopadhyay, S. Bandyopadhyay and H. K. Majumder, J. Mol. Med., 2003, 9, 26–36 CAS
- N. Lee, S. Gannavaram, A. Selvapandian and A. Debarabant, Eukaryotic Cell, 2007, 6, 1745–1757 CrossRef CAS PubMed
- I. Gonzalez, C. Desponds, C. Schaff, J. Mottram and N. Fasel, Int. J. Parasitol., 2007, 37, 161–172 CrossRef CAS PubMed
- H. Denise, J. Poot, M. Jimenez, A. Ambit, D. C. Herrmann, A. N. Vermeulen, G. H. Coombs and J. C. Mottram, BMC Mol. Biol., 2006, 7, 42 CrossRef PubMed
- R. A. Cooper, Annu. Rev. Microbiol., 1984, 38, 49–68 CrossRef CAS PubMed
- T. J. Vickers, N. Greig and A. H. Fairlamb, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13186–13191 CrossRef CAS PubMed
- P. K. Padmanabhan, A. Mukherjee and R. Madhubala, Biochem. J., 2006, 393, 227–234 CrossRef CAS PubMed
- P. K. Padmanabhan, A. Mukherjee, S. Singh, S. Chattopadhyaya, V. S. Gowri, P. J. Myler, N. Srinivasan and R. Madhubala, Biochem. Biophys. Res. Commun., 2005, 337, 1237–1248 CrossRef CAS PubMed
- S. C. Chauhan and R. Madhubala, PLoS One, 2009, 4, e6805 Search PubMed
| This journal is © The Royal Society of Chemistry 2015 |