
High-pressure phase behavior of 1-ethyl-3-methylimidazolium tetrafluoroborate and carbon dioxide system
Abstract
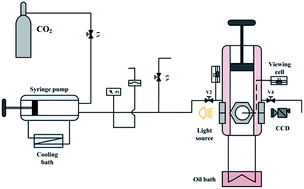
The phase behavior of the 1-ethyl-3-methylimidazolium tetrafluoroborate ([emim][BF4]) and carbon dioxide (CO2) system from atmospheric to supercritical state was investigated at 0.1–15.0 MPa and 308.15–343.15 K. The solubility data of CO2 in [emim][BF4] and the volumetric expansion ratios of CO2-saturated [emim][BF4] were provided. Experimental results indicated that although CO2 dissolved significantly in [emim][BF4] (from 0 to 62 mol%), the ionic liquid phase only had an expansion ratio less than 17 vol%. The density difference between CO2-saturated [emim][BF4] and pure [emim][BF4] was obvious at high pressure and low temperature, with a maximum of 16.11% at 308.15 K and 15.0 MPa. Based on the experimental results, a fitting equation applicable to precise data interpolation was proposed for the CO2-saturated [emim][BF4] density in this study.
 Please wait while we load your content...
Please wait while we load your content...

