Advanced materials from corn: isosorbide-based epoxy resins†
J.
Hong
a,
D.
Radojčić
a,
M.
Ionescu
a,
Z. S.
Petrović
*a and
E.
Eastwood
b
aKansas Polymer Research Center, Pittsburg State University, Pittsburg, KS, USA. E-mail: zpetrovic@pittstate.edu
bHoneywell FM&T, LLC, Kansas City, MO, USA
First published on 15th May 2014
Abstract
Water soluble epoxy resins were prepared from diglycidyl ethers of isosorbide (DGEI) and isosorbide diamine (ISODA). We investigated the effect of synthesis methods on the structure of DGEI and ISODA and their influence on properties of cured resins, and compared with bisphenol A commercial epoxy resins (DGEBA). DGEI and DGEBA were cured with diethylene triamine (DETA) and ISODA to give optically clear yellow to brown epoxy resins of different degrees of crosslinking. All systems containing isosorbide either on the epoxy or amine side were faster than the standard petrochemical epoxy resin (DGEBA/DETA). The glass transition of isosorbide-based resins was about 60 °C lower than those of DGEBA/DETA. The effect of DGEI molecular weight on properties of cured resins was tested. Isosorbide-based epoxy resins displayed equal or better tensile and impact strength than commercial epoxy resins and are a viable replacement for DGEBA resins but extensive research is necessary to improve hydrolytic stability.
Introduction
Utilization of bio-based resources for creating new polymeric materials is important both from ecological and sustainability considerations. Carbohydrates derived from corn have potential for use as biopolymers. Corn is one of the most abundant commodity crops and a source of starch which can be converted to sugars and fermented to fuel or used as a platform for new chemicals such as isosorbide (1,4:3,6-dianhydro-D-glucitol).1–5 The value of isosorbide arises from its properties: the attractive price, rigidity of its molecule, high thermal stability, biodegradability, renewability, solubility in water and non-toxicity. Isosorbide or 1,4:3,6-dianhydrosorbitol is a solid compound with a melting point of 60–62 °C and reported to be thermally stable up to 270 °C.6 It is a V-shaped diol consisting of two cis-fused tetrahydrofuran rings with a 120° angle between rings which imparts a degree of rigidity to polymers, Fig. 1.7,8 There are three stereoisomers of 1,4:3,6-dianhydro-hexitols differing in the positions of hydroxyl groups: isosorbide has an exo (sticking out) and an endo (hydrogen bonded inside) group, isoidide two exo and isomanide two endo hydroxyls. Isoidide would be the most desirable monomer for most applications in polymers but unfortunately it is fairly expensive. Endo hydroxyls are fairly unreactive and require more aggressive conditions to react.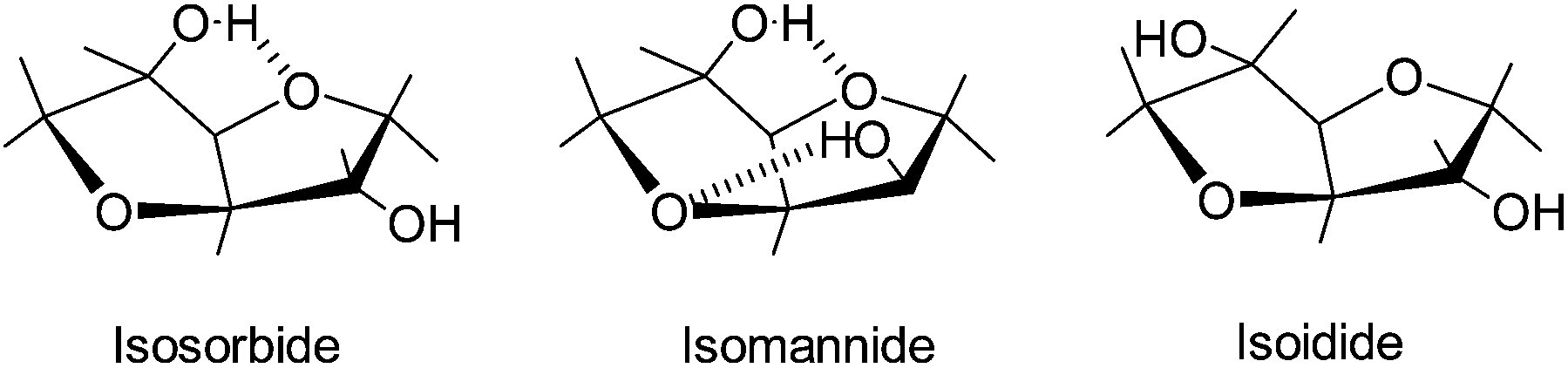 | ||
| Fig. 1 The three diastereoisomers of dianhydrohexitols. | ||
Isosorbide is obtained from starch in three steps. In the first step, starch is hydrolyzed to glucose. Glucose is then hydrogenated to sorbitol which is dehydrated to isosorbide as shown in Fig. 2.
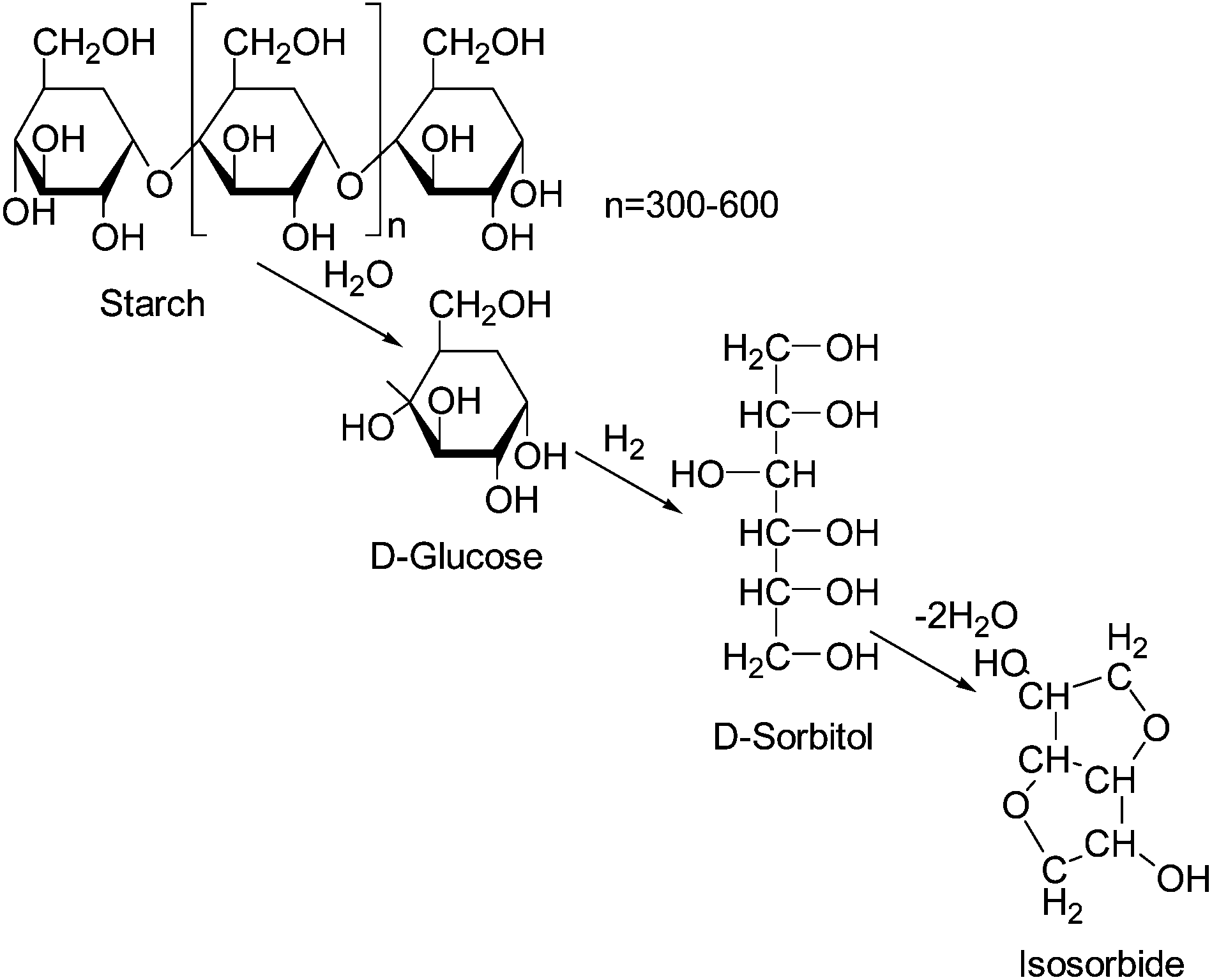 | ||
| Fig. 2 Scheme of the conversion of starch to isosorbide. | ||
Isosorbide was considered as a replacement for bisphenol A in polycarbonates and epoxy resins especially for applications in contact with food.3,5,9 Cured epoxy resin properties depend on the structure of diglycidyl ethers of isosorbide (DGEI) as well as on the curing agent structure. Typically oligomerization occurs during synthesis of DGEI, reducing the epoxy and increasing the hydroxyl content. The structure of DGEI is shown in Fig. 3.
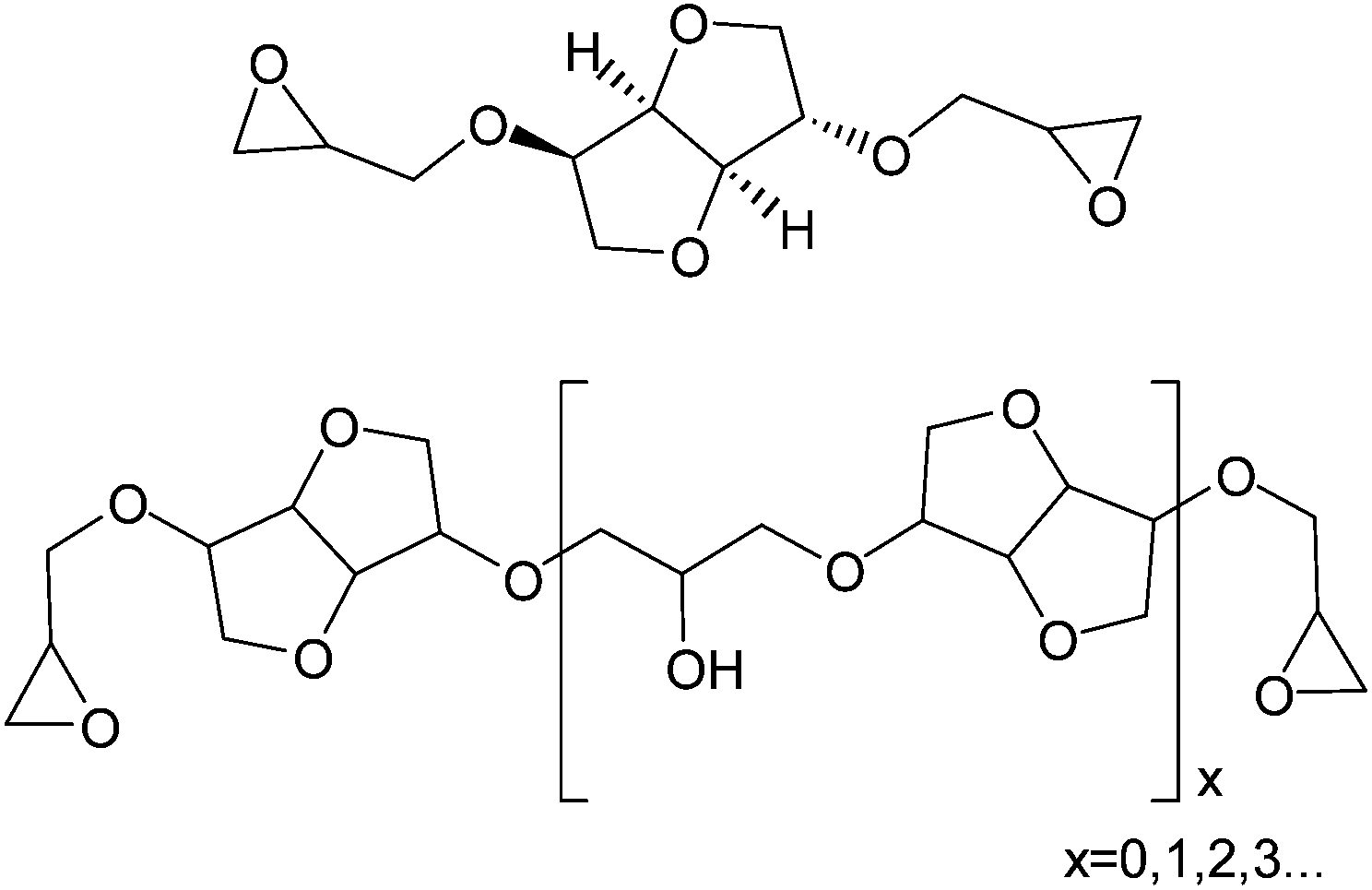 | ||
| Fig. 3 Monomeric and oligomeric forms of DGEI. | ||
For further discussion and comparison, the pure monomeric DGEI has a molecular weight of 258, an epoxy equivalent (EEQ) of 129 g eq.−1, an epoxy content of 33.33% (or an epoxy oxygen content of 12.4%), 0.775 equivalents per 100 g of resin and no hydroxyls. A previous patent10 on the synthesis of DGEI produced resin with EEQ/100 g of 0.467 or 60% of the theoretical value, primarily due to oligomerization. Several papers dealt with the synthesis and properties of epoxy resins from isosorbide to a limited extent.7,9,11,12
This paper provides a critical analysis of four methods of synthesis of diglycidyl ether of isosorbide with respect to the yield, molecular weight distribution and properties. Amine cured epoxy resins of different crosslinking densities were prepared from DGEI of different molecular weights and their properties correlated with the structure of networks. To understand the value of new resins, they were compared with standard petrochemical epoxy resin-DGEBA cured with the same amines. All resins were cured with isosorbide diamine and diethylene triamine. DGEI and ISODA are water soluble, which allowed preparation of films from water solutions. All resins were cured at 100 °C for 24 hours. While time and temperature were not optimized, the results clearly show the effect of structure on properties.
Experimental
Materials
High purity isosorbide (99.5%) was kindly donated by the Iowa Corn Association. Epichlorohydrin (ECH), allyl bromide, tetrabutyl ammonium bromide, and m-chloroperbenzoic acid were purchased from Fisher. DER-331 was acquired from Dow Chemical. It is a diglycidyl ether of bisphenol A (DGEBA) having EOC% = 8.48 (epoxy content 33.3%), epoxide equivalent weight 189 g eq.−1, viscosity 14 Pa s at 25 °C and OH number (TSI method) = 19.8 mg KOH per g. Diethylenetriamine (DETA) of 97% purity was obtained from AVOCADO Research Chemicals.Synthesis of DGEI
Four synthesis methods of DGEI were examined: epoxidation of isosorbide diallyl ether, acid catalyzed addition of ECH to isosorbide, NaOH catalyzed addition of ECH to isosorbide and Na-alcoholate of isosorbide reaction with ECH, as shown in Fig. 4. | ||
| Fig. 4 Different pathways to DGEI from isosorbide. | ||
Isosorbide diallyl ether (50 g) was epoxidized in 300 mL methylene chloride as a solvent with 105 g m-chloroperbenzoic acid (70–75%), at room temperature for around 5 hours. During the reaction, the resulting m-chloroperbenzoic acid precipitated as white crystals. The reaction mass was filtered. The organic layer was condensed to around 200 mL with a distilling solvent. Then the organic layer was cooled in a refrigerator and the precipitated m-chloroperbenzoic acid and residue m-chloroperbenzoic acid were removed by filtration. The cycle of distillation–cooling–filtration was repeated twice. In the second cycle, all solvents were removed under high vacuum and 36 g of the product was obtained (63.1% yield). The epoxy oxygen content (EOC) of the product was 10.9% (theoretical EOC is 12.4%), epoxy equiv. = 147 g eq.−1 or 0.70 eq./100 g. A higher purity product with 11.6% of EOC was obtained by vacuum distillation (150–160 °C, 1 mm Hg). Several investigators who used epoxidation of diallyl ethers of isosorbide reported a yield of 60% and an epoxy equivalent of 142 g eq.−1 after purification by preparative chromatography9 or simply a high purity product.11,12 Our method avoided using water since DGEI is soluble in water, resulting in higher yield and purity of DGEI than those reported.
Epichlorohydrin (2.2 mol) was added continuously from the addition funnel over a 2-hour period to a molten isosorbide (1 mol) containing SnF2 (0.02 mol) as a Lewis acid catalyst under nitrogen at 130 °C. Unreacted epichlorohydrin was distilled off from isosorbide dichlorohydrin ether under high vacuum (∼2 mm Hg). The resulting product was diluted with toluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by weight) followed by continuous addition of a 50% NaOH solution (2 mol) to form epoxy groups. After three hours, the reaction mixture was washed three times with distilled water. NaCl, unreacted isosorbide and isosorbide monoglycidyl ether, which are soluble in water, formed the bottom layer that was removed from the product. Distillation of toluene under vacuum leads to a relatively low yield of DGEI (25–30%) due to losses during the water washing steps. The epoxy oxygen content was around 6.5–6.8%, OH value = 35–60 mg KOH per g and hydrolyzable chlorine content = 0.15%. The DGEI resin had a high content of oligomer (∼80%) which was formed in the first step of epichlorohydrin addition to isosorbide, probably by cationic oligomerization. The disadvantages of this method were a darker colored resin, higher hydrolyzable chlorine content and lower yield. Also, due to different reactivities of hydroxyl groups of isosorbide, epichlorohydrin was most likely preferentially added to the more reactive exo hydroxyl and to the hydroxyl groups in the formed chlorohydrin rather than to the less reactive endo hydroxyl. As a result, some hydroxyl groups may have been alkoxylated with two or more epichlorohydrin units.
:1 by weight) followed by continuous addition of a 50% NaOH solution (2 mol) to form epoxy groups. After three hours, the reaction mixture was washed three times with distilled water. NaCl, unreacted isosorbide and isosorbide monoglycidyl ether, which are soluble in water, formed the bottom layer that was removed from the product. Distillation of toluene under vacuum leads to a relatively low yield of DGEI (25–30%) due to losses during the water washing steps. The epoxy oxygen content was around 6.5–6.8%, OH value = 35–60 mg KOH per g and hydrolyzable chlorine content = 0.15%. The DGEI resin had a high content of oligomer (∼80%) which was formed in the first step of epichlorohydrin addition to isosorbide, probably by cationic oligomerization. The disadvantages of this method were a darker colored resin, higher hydrolyzable chlorine content and lower yield. Also, due to different reactivities of hydroxyl groups of isosorbide, epichlorohydrin was most likely preferentially added to the more reactive exo hydroxyl and to the hydroxyl groups in the formed chlorohydrin rather than to the less reactive endo hydroxyl. As a result, some hydroxyl groups may have been alkoxylated with two or more epichlorohydrin units.
The method is based on the stepwise addition of an aqueous 50% NaOH solution to a mixture of isosorbide and a high excess of epichlorohydrin (molar ratio [epichlorohydrin]/[isosorbide] was 10/1), at 110–115 °C over a 3-hour period. During the addition of the aqueous NaOH solution, the epichlorohydrin–water azeotrope (molar concentration: 63.1% water and 36.9% epichlorohydrin, boiling point of the azeotrope was 88 °C) was continuously distilled and collected in a Dean Stark trap. Water (the upper layer) was removed and epichlorohydrin (the bottom layer) was returned to the reactor. NaCl as the reaction product precipitated as white crystals. After adding NaOH solution, the reaction was maintained at around 110–115 °C for one hour. The precipitated solid NaCl was removed by filtration and the residual epichlorohydrin was distilled under vacuum. The resin was a yellow, transparent, viscous liquid having an EOC of 6.9% (EEQ – 230 g eq.−1). The product had around 70–73% oligomers with hydroxyl groups and 27–30% monomeric DGEI. The method was characterized by a high yield of 96–98%; it was reproducible and suitable for large scale production.
This method had several advantages such as high yield, lower oligomer content than method 3 and improved reactivity of endo OH groups in the alcoholate form. Unfortunately the hydrolyzable chlorine content was high (2.6%), indicating that ring closure of chlorohydrins to epoxies was not complete, possibly because of the heterogeneous nature of the system. We believe that this synthesis can be improved using longer reaction times, higher content of catalysts, finer particles of solid dialcoholate or selecting other phase transfer catalysts such as crown ethers.
Monomeric DGEI with an EOC of 11.6% (94% of theoretical) was obtained by vacuum distillation at 1–2 mmHg and 200–220 °C of the resin prepared by methods 1 and 3, which was used for further studies.
Synthesis of isosorbide-based diamine (ISODA)
Isosorbide di[3-aminopropyl ether] was synthesized in two steps as shown in Fig. 5, involving cyanoethylation of isosorbide with acrylonitrile in the presence of a basic catalyst and hydrogenation of di-cyanoethylated isosorbide in the presence of Raney Ni and an excess of ammonia.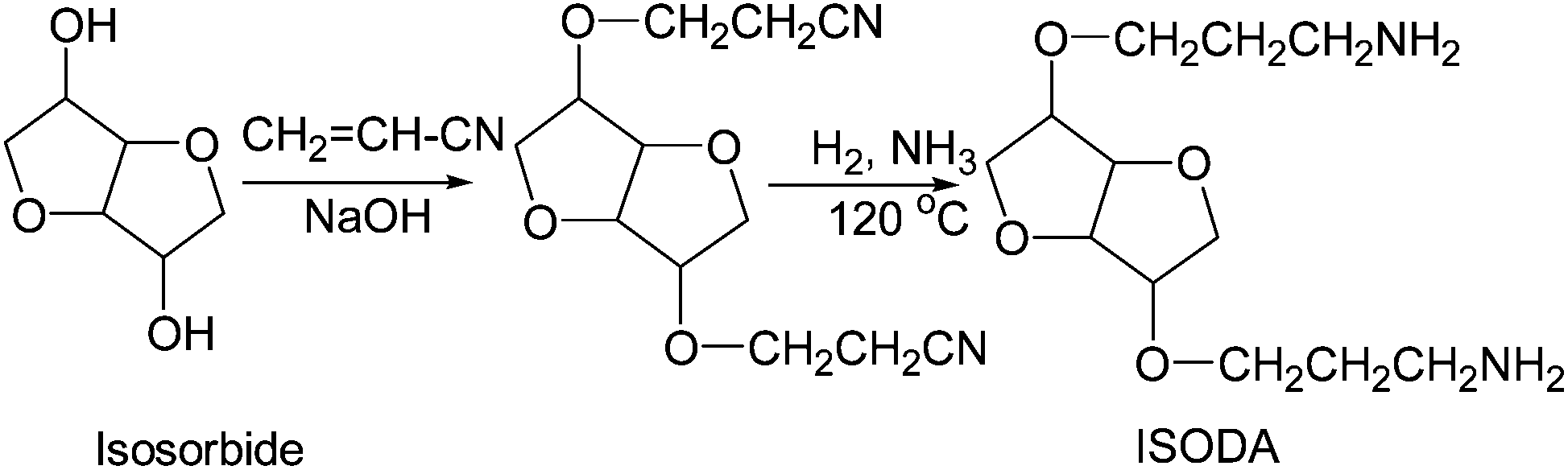 | ||
| Fig. 5 Preparation of isosorbide di[3-aminopropyl ether] (ISODA) by the cyanoethylation/hydrogenation method. | ||
Cyanoethylation of isosorbide
Cyanoethylation of isosorbide was carried out by a stepwise addition of acrylonitrile to molten isosorbide (m.p. = 60–62 °C) in the presence of 0.5% (relative to isosorbide) of an aqueous 50% NaOH solution at 65–70 °C over a period of 1.5–2 hours followed by additional stirring for around 3 hours. The traces of unreacted acrylonitrile were removed by vacuum distillation at 80–90 °C and 60–65 mm Hg. Generally, the product contained di- and mono-cyanoethylated isosorbide and unreacted isosorbide. Unreacted isosorbide and monocyanoethylated isosorbide are soluble in water (the upper layer) and were removed by triple water washing. The bottom organic layer contained relatively pure (92–95%) di-cyanoethylated isosorbide (yield 73–75%). Sometimes, the formation of a low concentration of polyacrylonitrile by anionic polymerization of acrylonitrile is possible. Fortunately, this small quantity of polyacrylonitrile can be easily removed by simple filtration.Hydrogenation of di-cyanoethylated isosorbide
Dicyanoethylated isosorbide (100 g) and 10 g of wet Ni catalyst were placed in a 450 mL Parr reactor. Ammonia (15 g) (50 g of 30% aqueous solution) was introduced at ambient temperature (NH3/CN = 0.55 molar ratio). The reaction mass was heated at 120 °C. The total pressure was brought to 2.76 MPa (400 psi) by introducing hydrogen. The pressure and temperature were maintained for around 5 hours. The crude product was separated from the catalyst by filtration. Water and ammonia were removed by vacuum distillation. The yield of diamine was 86.3% with an amine value of 420 mg KOH per g (the theoretical amine value is 431 mg KOH per g) and a viscosity of 6 Pa s.Characterization methods
The epoxy oxygen content (EOC) in epoxy resins was determined by direct titration of epoxy groups with HBr according to the standard method for oils and fats.14 The hydroxyl number was determined by the p-toluene sulfonyl isocyanate (TSI) method (ASTM-E1899). The amine value was determined using the DIN16945 method. Hydrolyzable chlorin was determined according to ASTM D1726-11. Viscosity was measured at 25 °C on the Rheometrics Scientific Inc. (Piscataway, NJ) SR-500 Dynamic Stress Rheometer, with parallel plates of 25 mm diameter and a gap of 0.2 mm.An AR 2000 dynamic stress rheometer (TA Instruments, New Castle, DE, USA), equipped with a 2° cone plate having 25 mm diameter and 55 μm truncation height, was used to measure viscosities at 25 °C.
Fourier transform Infrared (FT-IR) spectra were recorded with a Shimadzu IR Affinity-1 instrument (Kyoto, Japan) set to 16 scans and a resolution of 4 cm−1.
A Bruker DPX-300 spectrometer (Billerica, MA) was used to acquire the proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra. Deuterated chloroform (CDCl3) with tetramethyl silane (TMS) as internal reference was used as a solvent. The experiments were carried out at 300 MHz and 16 scans for proton NMR, and at 75 MHz and 1000 scans for carbon NMR, at room temperature.
Molecular weights (MW) and MW distribution were determined using a Waters Gel Permeation Chromatograph (Waters Corporation, Milford, MA, USA) consisting of a 510 pump, 410 differential refractometer and data collection system. Tetrahydrofuran (THF) was used as the eluent at 1.00 mL min−1 at 30 °C. Four Phenogel 5 μm columns (50, 100, 1000 and 10000 Å) plus a Phenogel guard column from Phenomenex (Torrance, CA, USA) covering a MW range of 102 to 106 were used.
Tensile properties were measured on Qtest-2 Tensile Tester from MTS, according to ASTM D882-97. Notched Izod impact strength was measured on a Resil Impactor, type 6957 from CEAST. Flexural properties were studied according to ASTM D790. The test was conducted on a Q-Test 2-tensile machine equipped with loading anvil (a crosshead speed of 0.85 mm min−1 and 2 point 38 mm support span).
DSC measurements were performed with a differential scanning calorimeter model Q100, from TA Instruments (New Castle, DE, USA) in a nitrogen atmosphere (50 mL min−1 purge flow) using standard aluminum pans, at a heating rate of 10°C min−1 from −80 °C to 200 °C. A thermogravimetric analyzer (TGA) model Q50 (TA Instruments, New Castle, DE, USA) was used for examining thermal stability. All experiments were carried out under nitrogen (60 mL min−1) with a heating rate of 10 °C min−1 from room temperature to 600 °C. Dynamic Mechanical Analysis (DMA) was performed using a DMA 2980 from TA instruments (New Castle, DE, USA). Tests were performed with a heating rate of 3 °C min−1 from −80 °C to 200 °C at 10 Hz. Thermomechanical (TMA) analysis was performed on a model Q400 (TA Instruments, New Castle, DE, USA) at a heating rate of 10 °C min−1 from −80 °C to 200 °C.
Results and discussion
Evaluation of synthesis methods
Isosorbide has two hydroxyl groups of very different reactivities. While exo hydroxyl groups react readily, forcing endo hydroxyl groups to react requires harsher reaction conditions (higher temperatures and longer time) and the underlying issue is whether the reaction is complete. The difference in reactivity was demonstrated by the reaction kinetics studies with isocyanates.15 Four methods used to synthesize DGEI gave different yields, epoxy contents and molecular weight distributions as shown in Table 1. The acid number of resins by methods 1 and 2 (prepared by epoxidation with m-chloroperbenzoic acid or Lewis acid, respectively) was below 1%. The color of products obtained from different methods is shown in Fig. 6. The density of DGEI by method 1 was 1.2 g cm−3 and 1.3 g cm−3 for products by other three methods. All resins were non-crystallizing liquids with a glass transition temperature (Tg) of −55 °C for pure DGEI and about −45 °C for oligomeric DGEI. | ||
| Fig. 6 Color of DGEI samples prepared by methods 1–4 (from left to right). | ||
The synthesis of DGEI by epoxidation of diallyl isosorbide gave a somewhat lower yield but the product was almost pure monomeric DGEI. Epoxidation in the presence of Lewis acid (method 2) was reported to give a low yield.11 Molecular weight distribution of such products was not reported in the literature but our tests showed that oligomerization occurred in the first step (chlorohydrin formation). An epoxy number of 6.5–6.8 is in a similar range to other methods but preferential addition of epichlorohydrin to the exo OH group may have occurred.
Direct epoxidation of isosorbide with ECH in the presence of a base is a standard procedure for preparation of commercial epoxy resins. It yields oligomers even at a very high excess of ECH but the process is economical and the yields are high. Literature data on the analysis of the resin by mass spectrometry showed the presence of small amounts of mono- and triglycidyl ethers of isosorbide in the mixture, confirming the complexity of epoxidation of compounds with OH groups of different reactivities.9
Method 4 gave an EOC content of 8.0–8.3% and a somewhat higher monomeric DGEI content (44%) than method 3 (30%). The reaction was carried out in the suspension of isosorbide alcoholate in the presence of a phase transfer catalyst but the product was darker, had good properties and yield around 90%. The reactivity of the endo hydroxyl in isosorbide was improved allowing more uniform addition of ECH to isosorbide.
Fig. 7 shows the overlay of GPC curves of DGEI samples prepared by all four methods. The oligomeric species of higher molecular weights were obtained even with very large excess of epichlorohydrin.
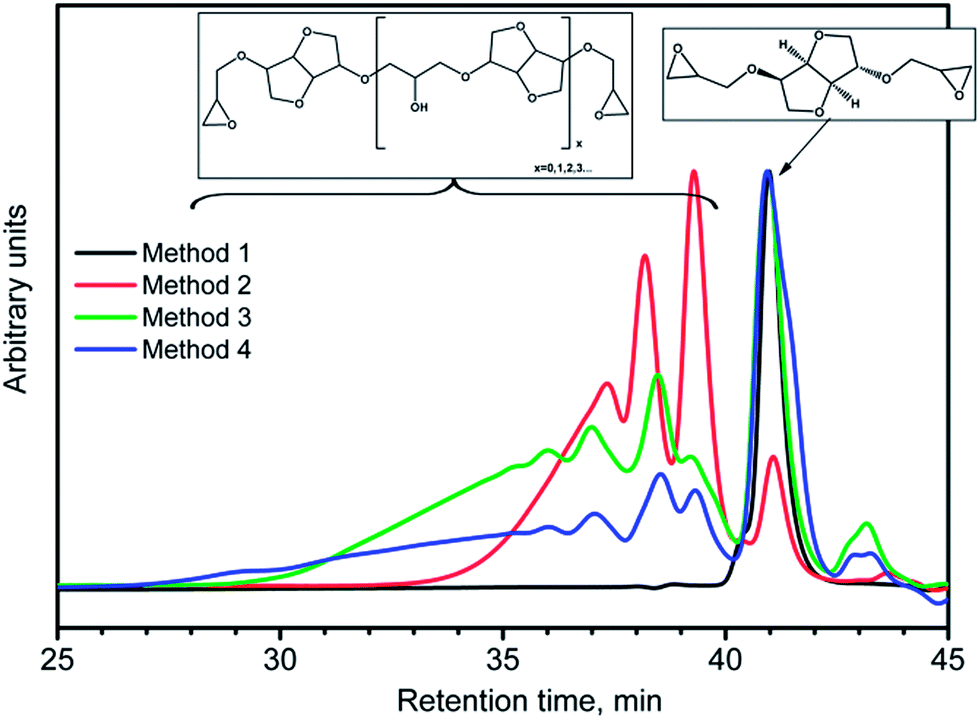 | ||
| Fig. 7 GPC curves of DGEI. Method 1 gives essentially pure DGEI. | ||
The overlay of infrared spectra of DGEI shown in Fig. 8 displays bands marked by arrows at 854 and 910 cm−1, characteristic of epoxy groups and C–O band at 1257 cm−1. A small peak at 3055 cm−1 was assigned to C–H vibrations in CH2 of the epoxy group. The presence of OH groups, confirmed by a strong OH peak at about 3450 cm−1, is an indication of higher oligomers. Spectra of DGEI by methods 2, 3 and 4 are identical but DGEI by method 1 does not have bands indicative of the hydroxyl group. The spectrum of DGEI cured with DETA shows some residual epoxy bands at 854 and 910 cm−1 and a new peak at 1645 cm−1, possibly due to residual N–H.
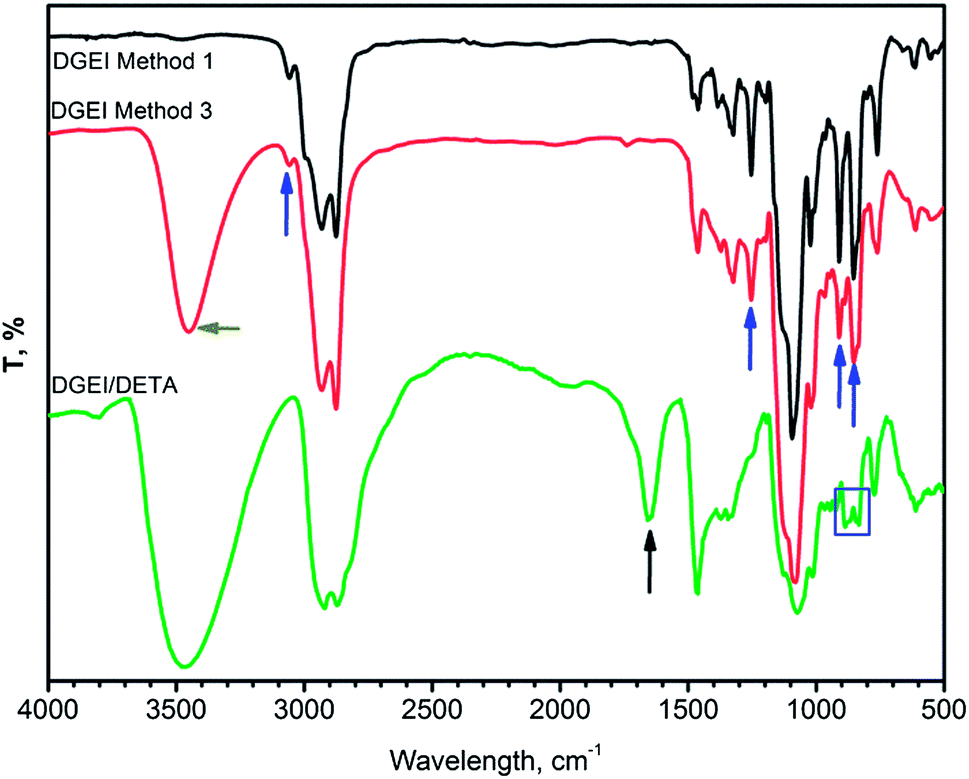 | ||
| Fig. 8 Overlay of infrared spectra of DGEI by methods 1 and 3, and cured DGEI/DETA resin. | ||
NMR characterization of DGEI
Both proton and carbon NMR spectra (Fig. S1 and 2, ESI†) of DGEI from method 1 (DGEI-1) are distinct since DGEI-1 is a pure monomer. The epoxy ring protons, CH2 and CH, gave peaks at δ = 2.50–2.70 ppm and 3.04–3.08 ppm, respectively. The protons common to the two isosorbide cycles (CH) gave peaks at 4.40–4.58 ppm. The peaks between 3.24 and 4.07 ppm are assigned to other protons. The 13C NMR spectrum shows very well-defined peaks of DEGI-1: the carbons of the epoxy ring, CH2 and CH, show signals at 44.1 and 50.5 ppm, respectively. The peaks at 69.8–73.3 ppm, 90.1–80.6 ppm, and 84.8–86.1 ppm correspond to other carbons. Due to oligomers, proton NMR spectra of DGEIs prepared by methods 2–4 have similar broad peaks to DGEI-1. However, in carbon NMR spectra, besides similar broad peaks with DGEI-1, new peaks appear at 69.5 ppm and 78.6 ppm. The peak at 69.8 ppm is tentatively assigned to the carbon of the hydroxy ether segment that links two isosorbide (–OCH2CH(OH)CH2O–). It shifts to 78.6 ppm with an epichlorohydrin recombination (–OCH2CH(OCH2CH(O)CH2)CH2O–). Some minor peaks found in the NMR spectra are from starting materials or catalysts, such as isosorbide diallyl ether, ECH and tetrabutyl ammonium bromide (TBAB). Hydrolysable chlorine has been detected in the DGEIs prepared by methods 2–4 (Table 1). Unfortunately, due to mass distribution, protons and carbons of isosorbide dichlorohydrin ether (O–CH2–CH(OH)–CH2Cl) could not be identified in the NMR spectra.Analysis of ISODA NMR spectra
A single broad peak of the 1H NMR spectrum (Fig. S3, ESI†) at 3.22 ppm can be attributed to amine protons (NH2). Trimethylene protons, CH2CH2CH2, CH2NH2, and OCH2CH2, gave peaks at 1.51–1.61 ppm, 2.54–2.59 ppm, and 2.63–2.68 ppm, respectively. Peaks at δ = 3.39–4.52 ppm belong to isosorbide cycle protons. The carbon next to amine gave a peak at 40.12 ppm in carbon NMR (Fig. S4, ESI†).Reactivity of DGEI and DGEBA resins with different amines
Reactivity of DGEI and DGEBA resins with DETA and ISODA was compared by rheological methods. DGEI had EOC around 8.0% and DGEBA had an EOC of 8.25%, but the former had an OH number of 121 mg KOH per g vs. 25 for the latter. Oligomers have hydroxyl groups which may participate in curing reactions and also catalyze them.16Fig. 9 shows a faster increase of viscosity and earlier gelation of isosorbide than bisphenol A-based resins when cured with DETA at 25 °C. The gel time, taken to be arbitrarily at 500 Pa s, was reached within 5000 s for DGEI/DETA while that for DGEBA/DETA was around 8000 s. Faster curing was aided by the higher content of hydroxyl groups in the former. The fastest curing system was based on isosorbide epoxy resin and isosorbide diamine while the second fastest was DGEBA also with isosorbide diamine.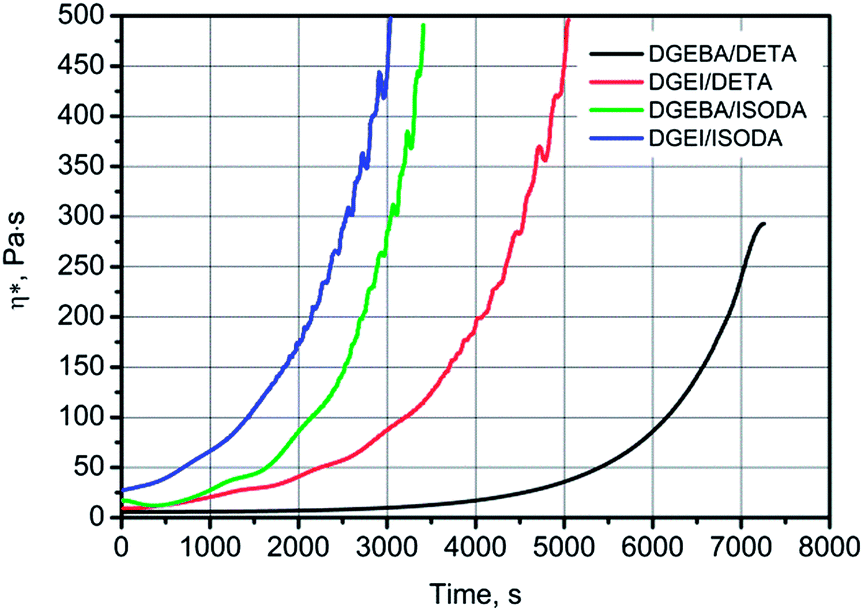 | ||
| Fig. 9 Increase of viscosity with time during the curing at 25 °C of indicated epoxy resin/amine systems. | ||
Effect of molecular weight distribution on properties of cured resins
In order to examine the effect of molecular weight of DGEI, i.e., oligomers, on the properties of crosslinked systems, two DGEI resins were used: one with EOC = 10.9% (essentially a pure monomer by method 1) and the second with an EOC = 6.9% (prepared by method 2 – designated as “DGEI polymeric”). They were cured with diethylene triamine (DETA) and ISODA to give rigid, 1 mm thick sheets for testing. For comparison, a commercial DGEBA resin was also cured with DETA and ISODA. Resins were prepared by mixing amine and epoxy at a 1:1 equivalent ratio at room temperature under high vacuum. The mixtures were poured in a cold mold and cured at 100 °C for 24 h. After testing, the resins were additionally cured at 150 °C for 2 h, and retested by DSC. The structure of the network prepared by reacting isosorbide epoxy resin cured with isosorbide diamine is given in Fig. 10.
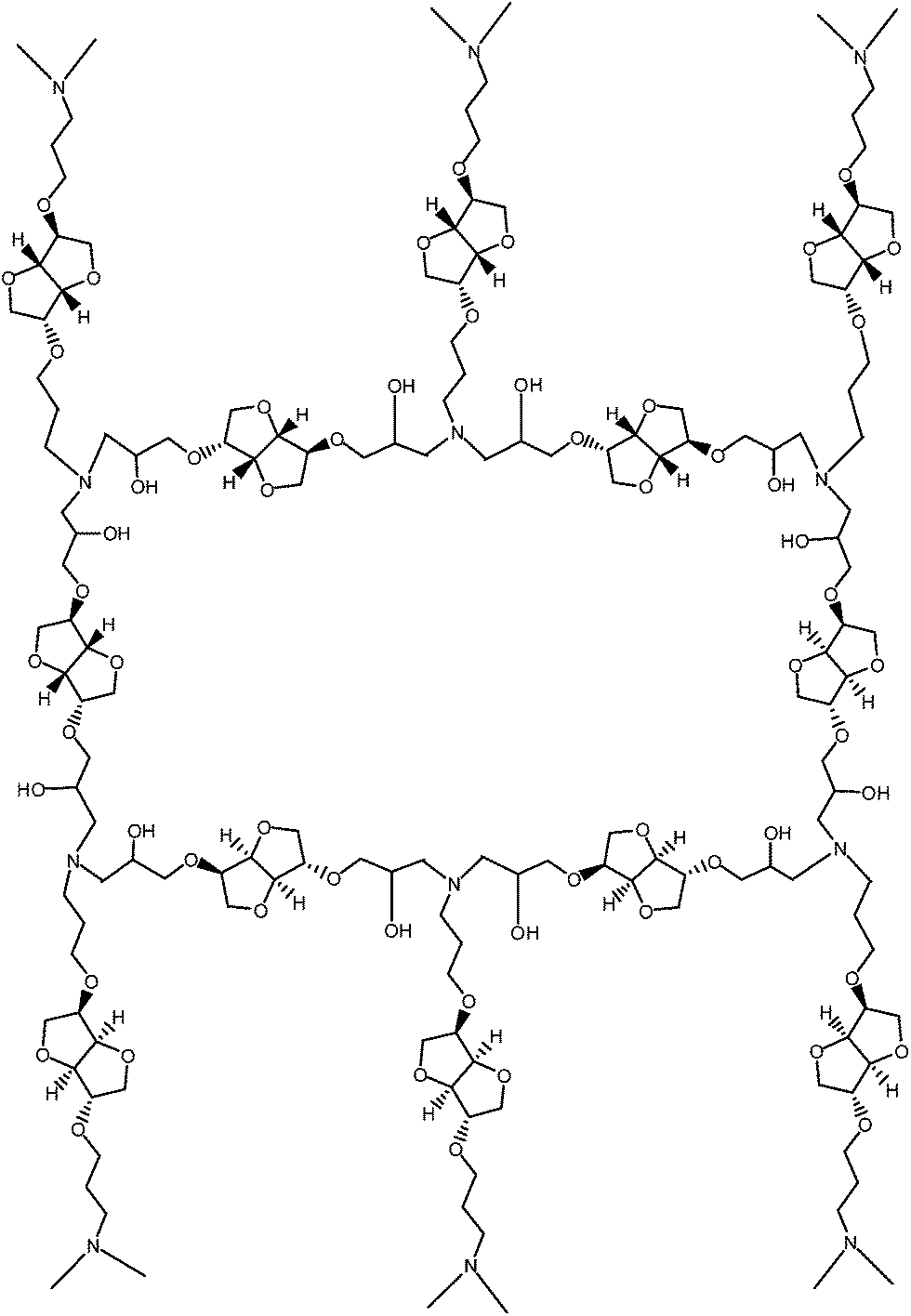 | ||
| Fig. 10 Section of the epoxy network from DGEI and ISODA. | ||
The molecular weight of network chains, Mc, in this structure is 257 and replacing ISODA with pentafunctional DETA gives Mc in the same range, characteristic of highly crosslinked systems. Curing with amines is complex, involving reactions of epoxy groups with primary amine, secondary amine and also hydroxyls. Taking into consideration these three reactions with relative rates 100:40:10 and assuming some cyclization gave us Monte Carlo simulation Mc = 670. However, since curing is never complete and the functionality of components is lower than theoretical, these values will be higher, which together with the presence of flexible isopropyl groups in ISODA/DGEI systems accounts for relatively low glass transition of cured resins.
DSC curves for DGEI and DGEBA cured with DETA and ISODA (Fig. S5, ESI†) show the highest Tg for DGEBA/DETA at 129 °C which after additional curing at 150 °C for 2 hours increased to 134 °C. DGEBA/ISODA and DGEI/DETA displayed Tg at 74 and 75 °C, which after post-curing increased to 79 and 76 °C, respectively. Thus, Tg of isosorbide containing systems have at least 50 °C lower glass transitions than DGEBA/DETA, which limits the applications' range of isosorbide epoxies. Both monomeric and polymeric DGEI cured with ISODA gave relatively low Tg at 43 °C after post-curing at 150 °C, reflecting the effect of flexible isopropyl structures in both components. The effect of DGEI molecular weight on glass transition was observed clearly when comparing DGEI-monomeric/DETA having Tg = 75 (76 °C post-cured at 150 °C) with DGEI polymeric/DETA (Tg = 48 °C or 63 °C after post-curing). A higher EOC content in the former produces higher crosslinking density, although the difference is not large. Tg values obtained by the thermomechanical method in the second cycle (after heating to 200 °C) correlate quite well with DSC data.
Higher glass transition temperatures of DGEI could be obtained by curing with aromatic and cycloaliphatic amines and anhydrides. It was reported that phthalic anhydride-cured DGEI displayed a Tg at 108 °C, tetrahydrophthalic anhydride at 95 °C,7 isophorone diamine at ca. 105 °C,9 methyl-5-norbornene-2,3-dicarboxylic anhydride at 113 °C and 4,4′-(hexafluoroisopropylidene)diphthalic anhydride at 200 °C.12
TMA is also used to test Tg and linear coefficients of thermal expansion (LCTE) (Fig. S6, ESI†). Tg values and LCTE measured about 50 degrees below and above glass transition are listed in Table 2. These values are from the second run performed after heating the samples to 200 °C in the first cycle. Somewhat higher Tg values from TMA than in DSC are due to thermal lag with larger samples. All samples displayed volume relaxation at the Tg manifested as shrinking in the first cycle, which was erased in the second cycle. LCTE for all samples was low both in the glassy and the rubbery state due to high crosslinking density, which was apparently higher than in the reference resins. In spite of the higher crosslinking density of DGEI resins, compared with DGEBA cured with DETA, higher Tg values for the latter reflect the more rigid nature of aromatic bisphenol rings.
| Resin | DSC | TMA | DMA (tension mode) | Tensile strengthb, MPa | Elongationb, % | Young modulusb, MPa | Flexural modulusb, MPa | Impact strengthb (ASTM), J m−1 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T g , °C | T g, °C | LCTE -glassy, μm m−1 °C−1 | LCTE-rubbery, μm m−1 °C−1 | T(E′′max), °C |
T(tanδmax), °C |
||||||
| a Values in brackets are obtained after post-curing at 150 °C for 2 h. b Values in brackets – coefficient of variation. Those without coefficient of variation are based on one specimen. | |||||||||||
| DGEBA/DETA | 129(134) | 130 | 60 | 200 | 137 | 146 | 26(8.2%) | 3(5.7%) | 1389(4.7%) | 3061(1.6%) | 60(1.2%) |
| DGEBA/ISODA | 74(79) | 73 | 64 | 232 | 80 | 92 | 67(4%) | 5(13%) | 1825(4.5%) | 3364 | 94(65%) |
| DGEI(mono)/DETA | 75(76) | 77 | 50 | 201 | 79 | 88 | 62(9%) | 6(30%) | 1798(1.2%) | 4027 | 72(16.8%) |
| DGEI(mono)/ISODA | 32(43) | 35 | 56 | 207 | 34 | 47 | 41(21%) | 5(20%) | 1532(2.6%) | 1168 | 65(23%) |
| DGEI(polymeric)/ISODA | 36(43) | 52 | 64 | 200 | 36 | 53 | 52(8.1%) | 3(22%) | 2461(9.5%) | 3520 | 57(14.7%) |
| DGEI(polymeric)/DETA | 48(63) | 50 | 55 | 206 | 48 | 61 | 52(18%) | 5(6.7%) | 1774(8%) | 2747 | 113(33%) |
Dynamic mechanical analysis of amine cured epoxy resins was carried out in the temperature range from −80 to 200 °C. Fig. 11 displays the change of storage modulus (E′), loss modulus (E′′) and tanδ with temperature for the DETA cured DGEBA reference sample. E′′ and tanδ have two distinct peaks associated with Tg, α transition at 146 °C and β-transition at −21 °C. A small decrease in E′ at the β-transition during heating is associated with unfreezing of the motions of the glyceryl group,17 while a major drop of about two orders of magnitude is a consequence of unfreezing of segmental motions in the crosslinked epoxy resin. The nature of the beta relaxation in DGEBA epoxy resins was studied by several investigators.17,18 Tanδ maximum reached 0.7 in the DGEBA/DETA cured system.
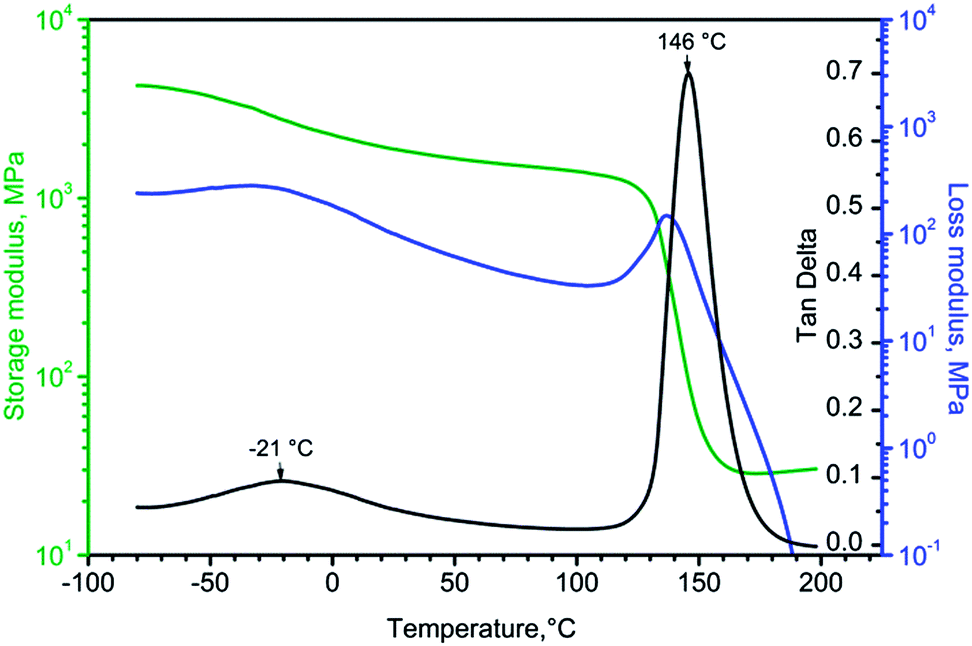 | ||
| Fig. 11 DMA curves for the DGEBA/DETA cured system. | ||
The overlay of storage modulus curves for four cured epoxy systems is shown in Fig. 12. Glassy moduli seem to be higher for DGEI systems than for DGEBA because of higher crosslinking density, whereas rubbery moduli of DETA cured systems were similar (within experimental error) since in highly crosslinked systems the method cannot discern the subtle differences. Only the DGEI/ISODA rubbery modulus was significantly lower.
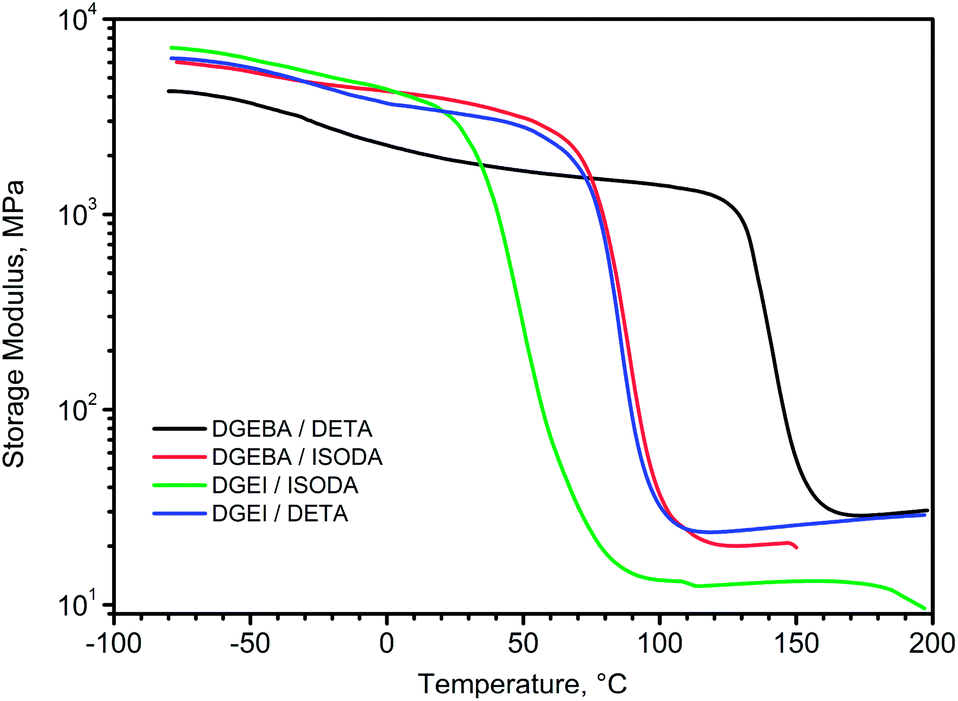 | ||
| Fig. 12 Storage modulus–temperature curves for DGEBA and DGEI cured with DETA and ISODA. | ||
DMA curves for isosorbide epoxy resins show the same features with a beta peak (tan delta) at −25 °C for DGEI-mono/DETA and −33 °C for DGEI-mono/ISODA but alpha peaks associated with glass transition shifted to lower temperatures as displayed in Fig. 13 and Table 2. Maximum values on tanδ curves were around 1.0, i.e., higher than that of DGEBA/DETA, indicating that DGEI resins were better energy absorbing materials than the reference resins. The temperature of the tan delta α-peak for DGEI/DETA was 88 °C compared to 146 for DGEBA/DETA, while DGEI-mono/ISODA gave an α-peak at 47 °C and the beta peak was not discernible. Evidently, ISODA gives less rigid epoxy resins than DETA cured analogs.
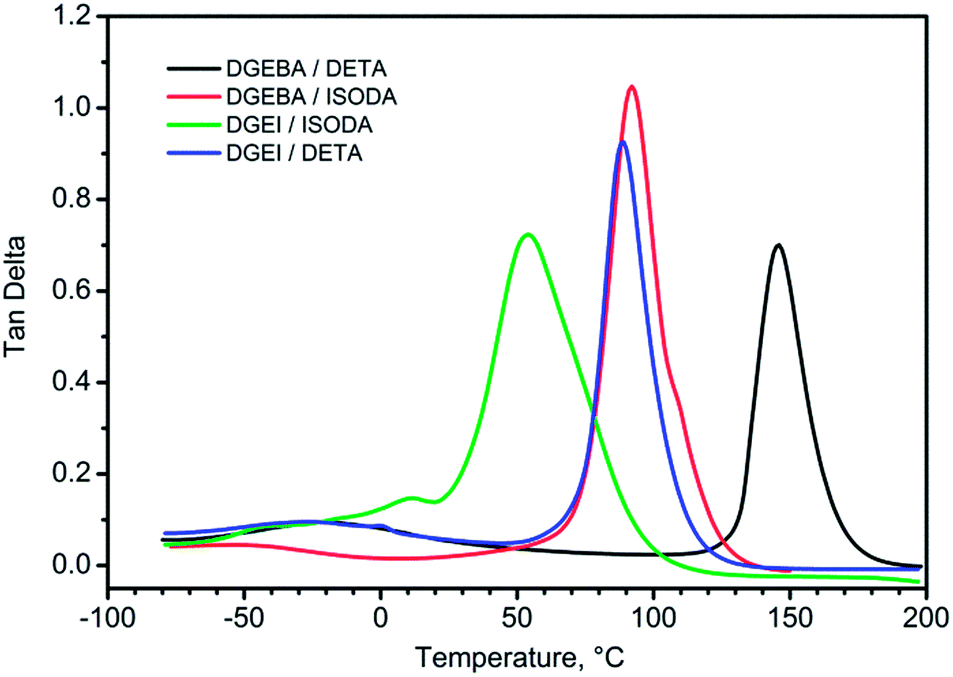 | ||
| Fig. 13 Tan delta curves for DGEBA and DGEI cured with DETA and ISODA. | ||
The glass transition temperature by DSC, TMA and DMA and mechanical properties of amine cured epoxy resins are summarized in Table 2.
Evidently, all mechanical properties are better with DETA-cured monomeric than with polymeric DGEI due to the higher crosslinking density. Such difference does not exist in ISODA cured mono- and polymeric DGEI due to higher crosslinking density of the latter, as a consequence of uneven curing conditions.
Swelling in water for DGEBA/DETA was 1%, for DGEBA/ISODA was 5% while polymeric DGEI/ISODA, DGEI-mono/DETA and DGEI-mono/ISODA swelled 30–50%. All DGEI/ISODA samples disintegrated into pieces after swelling, displaying limitations for their use in environments with high water content. Selected curing conditions (100 °C and 24 h) were not sufficient to obtain complete crosslinking and post-curing at elevated temperatures (150 °C or higher) would improve all properties.
TGA traces in nitrogen of epoxy resins cured with different amines are displayed in Fig. 14. Judging by the temperature of the knee on TGA curves, DGEBA resin is more thermally stable than isosorbide resins. The early weight loss in some systems with ISODA is a consequence of the presence of low molecular volatile compounds in the amine.
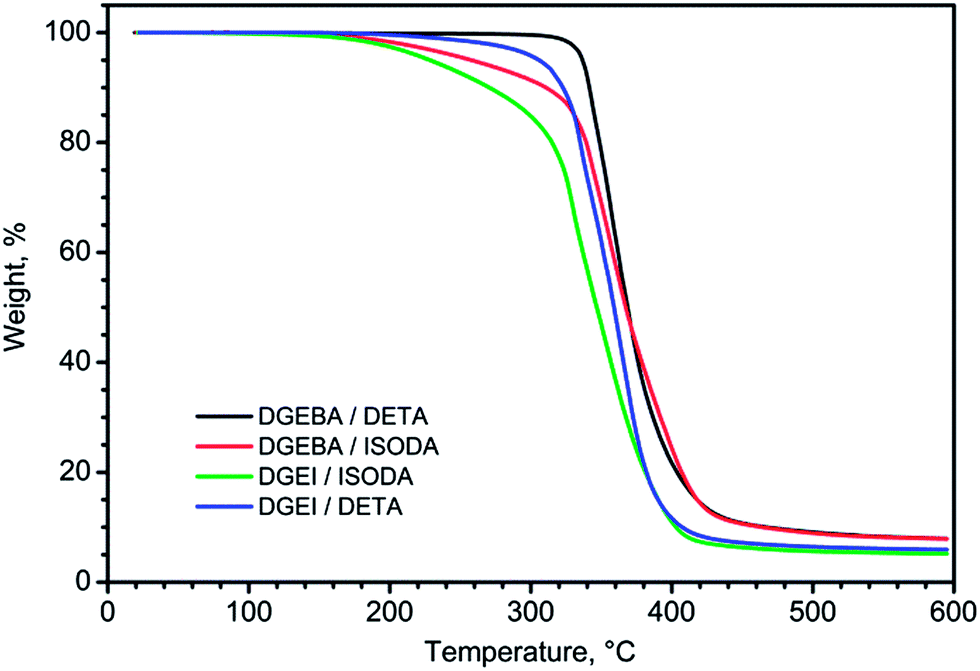 | ||
| Fig. 14 TGA curves for DGEBA and DGEI cured with DETA and ISODA. | ||
Conclusions
Four synthesis methods of diglycidyl ether of isosorbide were evaluated and compared. Only the method involving epoxidation of diallyl sorbide renders almost pure monomers, while other methods generate oligomers. Lewis acid catalyzed epoxidation gave a relatively good epoxy content but somewhat lower yield.Direct epoxidation with ECH in the presence of sodium hydroxide results in high yield but wider molecular weight distribution even at high excess of ECH. The use of sodium alcoholate gave a somewhat higher monomeric DGEI content, good yield and approximately the same epoxy content as direct epoxidation but with higher hydrolyzable chlorine.
The presence of oligomers increases the reactivity of epoxy resins with amines. The higher epoxy content (lower molecular weight) in DGEI results in higher crosslinking density of networks and higher glass transitions, tensile strength and modulus but lower impact strength.
Generally, DETA-cured isosorbide epoxy resins have about 60 °C lower glass transition than analogous DGEBA resins, limiting the application temperature to about 70 °C.
Isosorbide diamine was prepared in good yield and used to make all isosorbide water soluble resins. However, the glass transition of these resins was only about 40 °C, but isosorbide diamine-cured DGEBA displayed excellent mechanical properties.
Tensile and impact strength of isosorbide resins were generally better than those of DGEBA resins, and the moduli were in the same region. Water swelling of cured isosorbide epoxy resins was significant, which requires additional improvements if these resins are to be used in wet conditions.
Acknowledgements
We are indebted to Honeywell for supporting this project financially and Iowa Corn Association for supplying us with isosorbide.Notes and references
- S. V. Malhotra, V. Kumar, A. East and M. Jaffe, Bridge, 2007, 37, 17–24 Search PubMed.
- M. Paster, J. L. Pellegrino and T. M. Carole, Industrial Bioproducts: Today and Tomorrow, U.S. Department of Energy, 2003 Search PubMed.
- X. Feng, A. J. East, W. Hammond and M. Jaffe, in Contemporary Science of Polymeric Materials, ed. L. Korugic-Karasz, American Chemical Society, Washington, DC, 2010 Search PubMed.
- P. B. Smith, in Renewable and Sustainable Polymers, ed. G. Payne and P. B. Smith, American Chemical Society, Washington, DC, 2011, vol. 1063, ch. 6, pp. 95–109 Search PubMed.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- C. Rupp-Dahlem, presented in part at the Bio-Perspectives 2005, BREW Symposium, Wiesbaden, Germany, 11th May 2005 Search PubMed.
- J. Łukaszczyk, B. Janicki and M. Kaczmarek, Eur. Polym. J., 2011, 47, 1601–1606 CrossRef PubMed.
- R. M. Gohil, Polym. Eng. Sci., 2009, 49, 544–553 CAS.
- M. Chrysanthos, J. Galy and J.-P. Pascault, Polymer, 2011, 52, 3611–3620 CrossRef CAS PubMed.
- J. Morrison, Martin–Marietta Corp., US Pat., 3,041,300, 1962.
- A. East, M. Jaffe, Y. Zhang and L. H. Catalani, New Jersey Institute of Technology, US Pat., 7,619,056 B2, 2009.
- X. Feng, A. J. East, W. B. Hammond, Y. Zhang and M. Jaffe, Polym. Adv. Technol., 2011, 22139–22150 Search PubMed.
- J. D. Zech, Atlas Chemical Industries, US Pat., 3,272,845, 1966.
- IUPAC, Applied Chemistry Division, Commission on Oils Fats and Derivatives: Standard Methods for the Analysis of Oils, Fats and Derivatives, ed. C. Paquot and A. Hautfenne, Blackwell Scientific Publications, London, 1987 Search PubMed.
- M. Ionescu, Z. S. Petrović, M. D. Sandhu, I. Javni, N. Bilić and E. Eastwood, presented in part at the Polyurethane 2011 Technical Conference, September 26–28, Nashville, TN, 2011 Search PubMed.
- D. Ratna, in Handbook of Thermoset Resins, iSmithers – A Smithers Group Company, Shawbury, Shrewsbury, Shropshire, UK, 2009, pp. 155–174 Search PubMed.
- J. G. Williams, J. Appl. Polym. Sci., 2003, 23, 3433–3444 CrossRef.
- J. M. Charlesworth, Polym. Eng. Sci., 2004, 28, 221–229 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, DSC and TMA analysis curves. See DOI: 10.1039/c4py00514g |
| This journal is © The Royal Society of Chemistry 2014 |