Synthesis of biodegradable polymers from renewable resources
Mathieu J.-L.
Tschan
,
Emilie
Brulé
,
Pierre
Haquette
and
Christophe M.
Thomas
*
Chimie ParisTech, UMR CNRS 7223, 11 rue Pierre et Marie Curie, 75231, Paris Cedex 05. France. E-mail: christophe-thomas@ens.chimie-paristech.fr; Web: http://www.enscp.fr/spip.php?article278
First published on 19th December 2011
Abstract
The vast majority of commodity materials are obtained from fossil fuels. However, many studies predict that all fossil resources will be depleted within a few centuries. Biomass represents an abundant carbon-neutral renewable resource for the production of materials. Using biomass for the production of new polymers can have both economic and environmental benefits. This review focuses on the use of biomass for the synthesis of biodegradable polymers.
 Mathieu J.-L. Tschan | Mathieu Tschan obtained his PhD degree in 2006 under the supervision of Prof. G. Süss-Fink (Switzerland). Then, he moved to Rennes (France), and worked one year as a post-doctoral fellow with Prof. J.-F. Carpentier in collaboration with Cray-Valley (Total Petrochemicals). In 2008, he joined the group of Prof. P. W. N. M. van Leeuwen at the ICIQ (Spain) for two years, working in collaboration with The Dow Chemical Company. In 2010, he obtained an ANR “retour post-doc” grant and joined the group of Prof. C. Thomas at the Ecole Nationale Supérieure de Chimie de Paris (France). |
 Emilie Brulé | Emilie Brulé received her Master degree at the University of Rennes in 2000 before moving to London to complete her PhD at King's College London with Y. de Miguel and K.K. (Mimi) Hii in 2003. In 2004, she joined the group of Prof. C. Moberg at KTH, Stockholm as a postdoctoral fellow. After spending one year at the University of Pierre and Marie Curie in the group of E. Rose, she was appointed in 2006 as Assistant Professor at the Ecole Nationale Supérieure de Chimie de Paris. |
 Pierre Haquette | Pierre Haquette received a PhD in 1996 in organometallic chemistry under the guidance of Prof. Pierre Dixneuf (Rennes, France). Then he completed a postdoctoral fellowship at the University of Utah (Salt Lake City, US) under the supervision of Prof. John Gladysz. In 1998, he was appointed Assistant Professor at the Ecole Nationale Supérieure de Chime de Paris (Paris, France) in the research group of Prof. Gérard Jaouen where he studied the synthesis and catalytic activity of artificial metalloenzymes. He joined the group of Prof. Christophe Thomas in 2009 where his research interests are directed towards the conception of new catalysts for the polymerization of cyclic polar monomers. |
 Christophe M. Thomas | Christophe Thomas obtained his PhD degree in 2002 working under the supervision of Professor Süss-Fink. He then joined Professor Coates' group at Cornell University as a postdoctoral fellow supported by the Swiss National Science Foundation (2002–2003). After spending one year in Professor Ward's laboratories, he was appointed as Assistant Professor at the University of Rennes in 2004. In 2008 he was promoted to Professor at the Ecole Nationale Supérieure de Chimie de Paris. His research interests comprise the study of fundamental processes in organometallic chemistry with an emphasis on catalytic transformations and the control of stereochemistry. |
Introduction
Commodity polymers such as polyolefins are ubiquitous in our societies. A combination of factors, including monomer cost and availability, synthetic ease, and excellent properties, have incited the widespread use of these materials. The vast majority of these commodity materials are obtained from fossil fuels and nowadays the utilization of non-renewable resources in the manufacture of plastics accounts for approximately 7% of worldwide oil and gas.1,2 However, these resources are limited and many studies predict that all fossil resources will be depleted within a few centuries.3–5 The growing environmental awareness over limited fossil fuel reserves (and consequently the increase of oil prices) has stimulated the search for novel polymeric materials and production processes drawn from sustainable, renewable feedstocks and which minimize the detrimental environmental effects associated with their usage.6 In addition, from the widespread use of synthetic polymeric materials has emerged another major concern; indeed, despite the increasing popularity of plastic recycling, disposal of these undegradable materials has led to serious environmental pollution. From this point of view, the continuous depletion of landfill space available for discarded plastic wastes leads to the need for biodegradable polymeric materials to be used as substitutes for non-degradable conventional plastics. Such biocompatible and biodegradable polymers are thus currently emerging as valuable alternatives to conventional synthetic (petroleum-based) polymers and might be produced from raw materials.Biodegradable polymers are defined as polymers that are degraded7 and catabolized, eventually to carbon dioxide and water, by naturally occurring microorganisms such as bacteria, fungi or algae. In addition, when they are degraded, these polymers should not generate any substances that are harmful to the natural environment.8 Generally, natural materials (i.e., polysaccharides, proteins and bacterial polyesters) or synthetic polymers which contain hydrolyzable bonds in the backbone such as polyamides, polyurethanes, polyureas, polyethers, polyanhydrides, polypeptides (and the corresponding copolymers) are interesting candidates for biodegradation.9 Among various families of biodegradable polymers, aliphatic polyesters have a leading position since hydrolytic and/or enzymatic chain cleavage yields hydroxy-carboxylic acids, which in most cases are ultimately metabolized.10 Several parameters have been reported to influence the degradation behavior of biodegradable polymers; the most important factors are the chemical composition, the molecular weight and molecular-weight distribution, the crystallinity, and the (micro)structure of the polymer. Recent studies have also evidenced the strong influence of ordered monomer sequences on degradation properties.11 Other important properties of the polymer matrix that depend on the polymer composition, such as the glass transition temperature (Tg), have additional indirect effects on degradation rates.
Although the biodegradability of a material is independent of the origin of the starting raw materials used, biomass represents an abundant carbon-neutral renewable resource for the production of biodegradable materials. Nature produces over 200 billion tons of biomass by photosynthesis each year, 75% of which can be assigned to the class of carbohydrates. However, only 3.5% of these compounds are used by mankind.12 It is crucial to exploit the vast biofeedstocks provided by Nature, through broad-scale basic research toward the development of efficient, environmentally benign, and economical process methodologies for the large-scale conversion of biomass (carbohydrates, proteins, fats, terpenoids) into industrially viable polymeric organic materials. There exists a real opportunity to discover new ways to produce novel materials within the context of sustainability issues that are beginning to permeate recent industrial thinking. Many of these renewable resource polymers can also be rendered biodegradable under the appropriate conditions.1,2 However, except for polylactide, their high cost hampers their widespread use as bulk polymeric materials, relative to conventional petroleum-based plastics. More economically viable processes and the synthesis of new types of biodegradable polymers would clearly increase the number of applications for these polymers, as well as lower their cost. Recognizing that the raw material cost accounts for up to 50% of the overall production cost of biodegradable polymers,13 several research groups have directed investigative efforts toward the synthesis of new renewable monomers and the conversion of these monomers into their corresponding polymers.
The present review is concerned with those catalytic reactions that can help to transform carbohydrates and vegetable oils into valuable or potentially valuable materials. To avoid duplicity with the existing literature, herein we will comment on recent selected papers that have described methodologies and strategies that allow the synthesis of (potentially) biodegradable polymers from renewable resources.14,15 While most potential catalytic chemical routes have been considered, we have focused our attention on chemical routes through homogeneous catalysis; enzymatic processes are treated only marginally.
Lactide
Of the variety of biodegradable polymers synthesized from renewable resources, poly(lactide) (PLA) is certainly the most promising polymer today.1,16,17 These polymers, derived from 100% renewable resources such as corn or sugar beets, have recently become commercial materials, with the goal of supplanting traditional polyolefin-based materials in several applications.3 Indeed, polylactides are recyclable and compostable18 and their physical and mechanical properties can be manipulated through the polymer architecture.19–22 Tremendous progress has been made during the past decade in controlling the polymerization of synthetic PLA. Although several methods exist for the synthesis of PLAs,16 the most promising is the ring-opening polymerization of lactide.23 Different strategies have been proposed for the ring-opening polymerization (ROP) of lactide involving anionic, nucleophilic, or cationic initiators. However, the controlled ring-opening polymerization of lactide by well defined metal-based catalysts is by far the most widely studied method. Therefore PLA can be obtained with homoleptic tin, aluminium and zinc-based systems but several drawbacks (e.g., complicated equilibria phenomena and multiple nuclearities) have limited the control of the polymerization and the structural influence on catalyst activity. Homoleptic yttrium alkoxides were also studied in lactide polymerization. McLain and Drysdale were the first to demonstrate the high potential of yttrium complexes for the synthesis of poly(lactide) with a homoleptic complex described as “Y(OCH2CH2NMe2)3”, which was shown to polymerize rac-lactide in a rapid and controlled manner.24 Also, Feijen et al. reported that Y5(μ-O)(OiPr)13 has a high activity in LA polymerization.25 Finally, Tolman and co-workers reported that Fe5(μ-O)(OEt)13 displayed very high rates and excellent molecular weight control in lactide polymerization.26These complexes have contributed significantly to a better understanding of the factors that govern the polymerization, and spectacular improvements have thereby been achieved in terms of catalytic activity as well as polymerization control. However, although tin, aluminium and zinc-based systems proved to be quite convenient in the preparation of aliphatic polyesters, there is also no possible control of stereochemistry. As stereochemistry plays an important role in PLAs, determining their mechanical properties, biodegradability and ultimately the end use of the material, a second generation of heteroleptic metal-based complexes with fine ligand adjustment attracted interest for a better control, activity, and selectivity of the polymerization reaction.27 Therefore, recent advances in catalyst design have led to a variety of PLA microstructures from the enantiomerically pure monomer, racemic mixture, or meso lactide. These lactide feedstocks can be used to construct various polymer architectures (i.e. atactic, isotactic, heterotactic and syndiotactic) (Scheme 1).
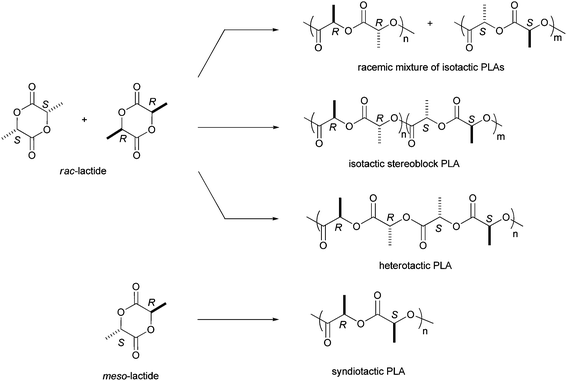 | ||
| Scheme 1 Lactide stereochemistry and typical PLA architectures. | ||
Two types of control exist for the stereoselection in the coordination–insertion ring-opening polymerization: the chain end control and the enantiomorphic site control. In a chain-end controlled mechanism, the chirality of the last unit in the growing polymer chain influences the chirality of the next monomer to be inserted. Enantiomorphic site control, however, is demonstrated when the chirality of the catalyst, and not the chain end, dictates the chirality of the next insertion. Despite recent developments of achiral and chiral complexes for the ROP of lactide,28 relatively few well-defined metal catalysts are able to achieve high stereochemical control in the ROP of meso- or rac-lactide. We will only discuss examples that allow the synthesis of highly stereocontrolled polymers.
Aluminium-catalyzed polymerization of lactide
Some of the most significant advances in stereocontrolled polymerization of lactide have been demonstrated using aluminium alkoxides stabilized by tetradentate bis(iminophenoxide) (salen-type)29 or tetradentate bis(aminophenoxide) (salan-type)30 ligands. Spassky et al. were the first to demonstrate that the aluminium methoxide complex bearing a binaphthyl Schiff-base ligand 1 induced a highly stereocontrolled polymerization of rac-LA to produce at low conversions (<19%) isotactic PLAs with 88% enantiomeric enrichment in (R,R) units (Fig. 1).31 At high conversions a stereocomplex between (R,R)- and (S,S)-enriched stereocopolymers was formed (Mn up to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 g mol−1). The polymerization reaction showed living type features, and narrow molecular weight distributions (Mw/Mn = 1.05–1.30), indicating that transesterification reactions do not occur significantly with this sterically hindered initiator.
700 g mol−1). The polymerization reaction showed living type features, and narrow molecular weight distributions (Mw/Mn = 1.05–1.30), indicating that transesterification reactions do not occur significantly with this sterically hindered initiator.
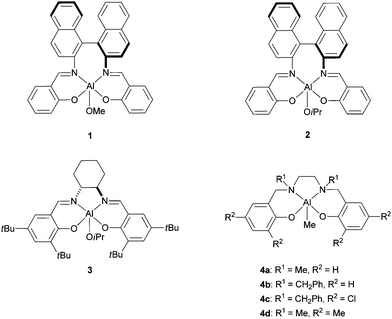 | ||
| Fig. 1 Stereoselective aluminium-based systems for the ROP of rac-lactide. | ||
The next reported initiators were essentially aluminium Schiff base systems and the corresponding polymerizations proceeded in a highly stereoselective manner and with a good control of the molecular weights (Fig. 1). It was shown that these initiators can be used to prepare PLA stereocomplexes and PLA stereoblocks, which showed enhanced thermal stability compared to the homochiral PLAs (Tm up to 210 °C). For instance, parallel stereocontrolled ROP of (R,R)- and (S,S)-LA from rac-LA was carried out with aluminium catalysts 232 and 333 to give isotactic stereoblock PLA. The formation of a stereoblock PLA was explained by a polymer exchange mechanism where growing chains switch between (R)- and (S)-species.32a,34
Other Al-based systems are able to generate isotactic PLAs from rac-lactide.35 Some of them are achiral and operate via a chain-end control mechanism. Gibson and co-workers reported the formation of isotactic stereoblock PLA (Pm up to 0.79) with tetradentate N,N'-disubstituted bis(aminophenoxide) aluminium complexes 4a and 4b.35c Remarkably, these aluminium initiators were demonstrated to generate a wide range of microstructures. Interestingly it was shown that isotactic PLAs were obtained in the presence of complexes bearing unsubstituted phenoxide groups whereas heterotactic PLAs were produced when the phenoxide units of the salan ligand contain substituents in the 3 and 5 positions (e.g. for complexes 4c and 4d). The authors proposed that the monomer selectivity is influenced by the alkylamino backbone substituents that can closely approach the site of polymer chain growth.
Also, syndiotactic, semicrystalline PLA can be formed by ROP of meso-lactide in the presence of the chiral aluminium isopropoxide complex 2, which preferentially ring-opened one acyl-oxygen site, leading to a highly alternating arrangement of stereocenters in the polymer (Mn up to 15400 g mol−1, Mw/Mn = 1.04–1.06).36 The resulting PLA (syndiotacticity up to 96%) exhibited a melting temperature at 152 °C.
Indium-catalyzed polymerization of lactide
Hillmyer et al. have recently reported the ROP of rac-LA using a catalyst prepared in situ from indium trichloride, benzyl alcohol and triethylamine to prepare highly heterotactic PLA (0.86 < Pr < 0.94 at 25 °C, Pr = 0.97 at 0 °C).37 The resulting robust system was found to be active under a variety of reaction conditions to give heterotactic PLA with controlled molecular weight and a narrow molecular weight distribution (Mn up to 159000 g.mol−1, Mw/Mn = 1.06–1.62). Unlike the other stereoselective catalysts which contain sophisticated ligands critical to obtain highly tactic PLA, this system allowed the induction of stereocontrol without an added directing multidentate ligand.38
Mehrkhodavandi and co-workers have recently published a chiral alkoxy-bridged dinuclear indium catalyst capable of active, living, and selective ROP of lactide (Fig. 2).39 The authors demonstrated a possibility of functional-group-tolerant polymerization and catalyst recovery. In addition, even if the racemic catalyst 5 revealed modest isoselectivity in LA polymerization, the enantiopure catalyst 5 showed significantly decreased enantioselectivity in lactide ROP, thus highlighting the importance of a site-control mechanism.
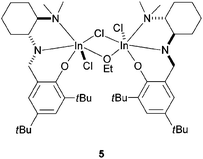 | ||
| Fig. 2 Stereoselective indium-based initiator for the synthesis of isotactic PLA. | ||
Zinc-catalyzed polymerization of lactide
In the past decade, a new class of β-diiminate zinc complexes for ROP of lactides was described by Coates et al.40 Such catalysts, like complexes 6 (Fig. 3), were found to act as single-site, living initiators for the polymerization of (S,S)-lactide, rac-lactide and meso lactide to PLA. These achiral complexes featured particularly high activities and selectivities, leading to the synthesis of highly heterotactic PLA (Pr up to 0.94 at 0 °C) from rac-LA by alternately incorporating the (R,R)- and (S,S)-LA. Among them, complex 6b exhibited the highest activity and stereoselectivity for the polymerization of rac-lactide to PLA. Changing the ligand substituents from isopropyl to n-propyl groups resulted in a decrease of heterotacticity (Pr = 0.76).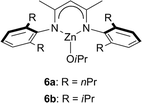 | ||
| Fig. 3 Zinc systems for the heterotactic ROP of rac-lactide. | ||
Group 3 metal-catalyzed polymerization of lactide
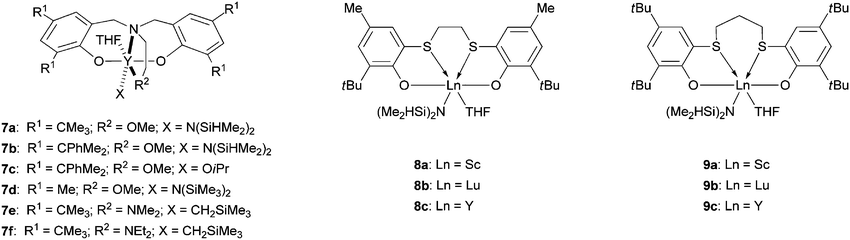
In 2002 the catalytic behavior of a new salen yttrium alkoxide complex was investigated by Ovitt and Coates in the ROP of lactide.41 Although this complex revealed relatively higher activity than the parent aluminium derivative, no stereoselectivity was observed for the polymerization of rac-LA. However, from this early study, other research groups investigated the synthesis and reactivity of well-defined yttrium complexes supported by bis(phenoxide) ligands in order to achieve effective ROP of rac-LA.42,43 Among these, the yttrium amido and alkoxo derivatives 7a–d (Fig. 4) have shown interesting performance for the heterotactic living polymerization of rac-LA. For instance, the PLA produced by 7b and c was found to be highly heterotactic, with a Pr of 0.90 (Mn up to 87000 g mol−1, Mw/Mn = 1.07–1.34). Subsequently analogous alkyl yttrium derivatives 7e and 7f were synthesized and displayed higher stereoselectivity for the polymerization of rac-lactide to give heterotactic poly(lactide) with a Pr ranging from 0.97 to 0.99.44 Finally, the yttrium systems 7a–c are also able to act as catalytic-like species in the presence of excess alcohol, producing larger quantities of heterotactic PLA.45
 | ||
| Fig. 4 Lanthanoid complexes for the heterotactic ROP of rac-lactide. | ||
Okuda and co-workers reported the synthesis of several lanthanoid complexes such as 8 and 9 supported by 1,ω-dithiaalkanediyl-bridged bis(phenoxide) ligands (Fig. 4).46 Among these dichalcogen-bridged bis(phenoxide) derivatives, scandium complexes 8a and 9a exhibited high heterotactic selectivity (Pr up to 0.95) during the controlled ROP of rac-LA (Mn up to 28500 g mol−1, Mw/Mn = 1.06–1.89). The authors attributed the high selectivity of these complexes to a dynamic monomer-recognition process involving interconversion of the ligand configuration.
Organocatalyzed polymerization of lactide
Although metal-based catalysts can produce highly selective PLA, the contamination of the polymer by traces of metals can be a major drawback for polymers with biomedical applications. Therefore organocatalysis may be an alternative for the stereoselective ROP of lactide.47 Most studies using organocatalysts for the ROP of lactide were carried out by Hedrick et al.48 In particular, stereoselective ROP of rac- and meso-lactide was accomplished at low temperature with organocatalysts such as N-heterocyclic carbenes49 and phosphazene base50 (Pm = 0.95 at −75 °C). The use of chiral organocatalysts presents the advantage of potentially inducing kinetic resolution of rac-lactide. Indeed, Miyake and Chen reported recently the stereoselective ROP of rac-lactide and its first successful kinetic resolution using a cinchona alkaloid, β-isocupreide (ICD).51 In the presence of benzyl alcohol, ICD converted 86% of rac-lactide to form an isotactic-enriched polymer (Pm = 0.74) with a good Mw/Mn of 1.12 at room temperature in dichloromethane (Mn up to 23300 g mol−1). Furthermore, it was shown that the ROP of (S,S)-lactide proceeded much faster than that of rac-lactide, indicating that the polymerization of (R,R)-LA was less favored. Indeed analysis of the unreacted monomer revealed a resolution of (R,R)-LA with 72% ee. The selectivity factor remains moderate but could be enhanced with further investigations.
Butyrolactone-based vinyl monomers
Polar vinyl molecules are interesting monomers for polymerization and methyl methacrylate (MMA) has been the most studied monomer to produce iso-, syndio- or atactic poly(methyl methacrylate) (PMMA) depending on the catalytic system used.52 Some renewable cyclic analogs of MMA (vinyl butyrolactones) are emerging as very interesting monomers. Indeed α-methylene-γ-butyrolactone (MBL) also called Tulipalin A is a natural product isolated from tulips.53 A recent study reported its synthesis in high yield via enzyme-mediated conversion of tuliposide A, found in large quantities in tulip tissues (0.2–2% w/w fresh weight).53b Its methyl derivative, γ-methyl-α-methylene-γ-butyrolactone (MMBL) is obtained in two steps using a method developed by DuPont54 starting from levulinic acid, derived from biomass and produced in 450 tons per year.55 Contrary to MMA, these vinyl butyrolactones are fixed in their s-cis form because of the ring strain, which increases their reactivity in free radical polymerization.56 Polymerization of MBL and MMBL to produce PMBL and PMMBL respectively occurs via their double bond (Scheme 2).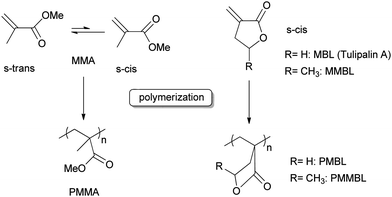 | ||
| Scheme 2 Polymerization of polar vinyl monomers. | ||
PMBL and PMMBL present some advantages over PMMA due to the presence of the butyrolactone ring: they tend to have a better durability, a higher refractory index (interesting for optical applications) and glass transition temperatures Tg (194 °C and 227 °C respectively) and therefore can be used for the synthesis of thermoplastic elastomers. The first reports of MBL polymerization by radical and anionic mechanisms date back to the early 80's.57 Group transfer polymerization58 and copolymerization59 have also been investigated. More recently, MBL was copolymerized with poly(ethyleneoxide) (PEO) by a radical mechanism60 and with a cyclic ketene acetal, 2-methylene-1,3-dioxepane (MDO) by mixing the two monomers between 70 °C and 120 °C to produce high molecular weight copolymers. Two parallel zwitterionic and radical mechanisms are involved but a moderate control was obtained with Mw/Mn values around between 2.4 and 2.8 in solvent-free conditions.61
MMBL, less studied, was polymerized by free-radical and anionic reactions62 and more recently PMMBL was obtained by radical,63 controlled mini-emulsion polymerization64 and copolymerization by emulsion with glycidyl methacrylate.65 Kinetic studies of the free radical copolymerization of MMBL with MMA and styrene were also reported by Hutchinson et al.66 However it is only recently that controlled polymerization of MBL and MMBL was achieved. The first example was conducted by atom transfer radical polymerization (ATRP) by Mosnáček and Matyjaszewski.67 It involved the homopolymerization of MBL as well as its copolymerization with PMMA and polybutylacrylate (PBA) to form well defined diblock or triblock copolymers. As MMA and BA are not obtained from renewable resources, their copolymerization with MBL will not be further discussed here.68,69 The catalytic system for the homopolymerization of MBL consisted of CuBr/CuBr2 with 2,2′-bipyridine (bpy) and bromopropionitrile as the initiator. The reactivity of MBL was found to be little higher than that of MMA because of the nearly planar five-membered ring. The choice of the solvent for polymerization of MBL is crucial as PMBL is poorly soluble in several organic solvents: in this study, DMF was chosen as it solubilises PMBL. After 100 min 90% MBL was converted into the polymer with an experimental molecular weight of 18200 g mol−1 according to GPC using MMA standards, and of 21100 g mol−1 calculated from the 1H NMR spectrum, the latter being closer to the theoretical value of 21090 g mol−1. A narrow molecular weight distribution of 1.09 was also obtained.
Recently, the catalytic polymerization of (M)MBL (referring to MBL and MMBL) with metal derivatives has also been explored by the group of Chen.70 Mainly rare earth metal catalysts have been used for the first coordination polymerization studies. The neutral samarium(II) Cp*2Sm(THF)2 (Cp* = η5-C5Me5) appeared to be a highly active catalyst for the polymerization of MBL and MMBL in DMF, in which the polymers are soluble.70a The total conversion of monomers induced atactic polymers in 10 minutes at room temperature with a good control of the molecular weight and a relatively narrow Mw/Mn (1.39 for PMBL and 1.19 for PMMBL with [(M)MBL]0/[Cp*2Sm(THF)2]0 = 100). The calculation of the initiator efficiency I* (Mn(calcd)/Mn(exptl) × 100) revealed a bimetallic mechanism, the Mn(calcd) being twice the Mn(exptl). Similar polymerization behavior was previously observed for the polymerization of MMA with lanthanocene Cp*2Ln(THF)2 (Ln = Yb, Sm) by Yasuda et al.71 and by Boffa and Novak72 who later proposed a redox-then-radical-coupling process. A similar mechanism was hypothesized by Chen and co-workers for the MBL polymerization (Scheme 3). The cationic samarium(III) and MBL radical anion species 10 is formed via an electron transfer from Cp*2Sm(II) to MBL before it is combined to afford the trivalent samarium active species 11. The radicals then combine to afford the bimetallic active initiator 12, followed by propagation with addition of monomers. Copolymerization of MBL with MMBL and MMA has also been carried out with Cp*2Sm(THF)2 to form statistical and block copolymers PMBL-ran-PMMBL and PMBL-b-PMMBL as well as PMMA-ran-PMBL and PMMA-b-PMBL in 10 minutes with Mw/Mn of 1.60, 1.36, 1.41 and 1.61 respectively. Differential scanning calorimetry analysis of the polymers showed that the glass transition temperature of PMMBL was much higher than the one of PMBL (227 °C vs. 194 °C) for polymers with similar Mn of 60000 g mol−1 and that PMMBL displayed increased thermal stability, while PMBL-b-PMMBL exhibits two glass transition temperatures at 192 and 218 °C.
 | ||
| Scheme 3 Proposed mechanism for the polymerization of MBL with Cp*2Sm(THF)2. | ||
Apart from the lanthanocene catalyst, less studied half-sandwich indenyl rare earth dialkyl complexes (scandium,73 lutetium,74 dysprosium74 and yttrium74) have also been investigated as (M)MBL polymerization catalysts (Fig. 5).70b As for the polymerization of MMA, dysprosium catalyst 13c displayed the highest activity for (M)MBL compared to 13a, 13b and 13d, following the trend of the ionic radii of the metal from the largest (Dy) to the smallest (Sc). Moreover, while conversions of MMA and MBL reached 84–88% after 24 h at room temperature in DMF, complex 13c appeared to be a remarkable catalyst for the polymerization of MMBL: the polymer was formed quantitatively within less than a minute with a TOF > 30000 h−1 in the presence of 400 equivalents of monomer (ten times better than with the sandwich Cp*2Sm(THF)2 complex) though a moderate Mw/Mn of 1.64 was obtained. MBL is often considered more active than MMBL but in this case the authors explained that the excellent activity of the methylated analogue is due to the higher electron density of the monomer which can bind more tightly to the rare earth metal catalyst. Moreover, catalyst 13c being less sterically hindered than the sandwich catalyst, the access of the larger monomer is facilitated.
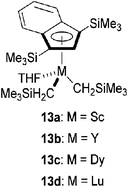 | ||
| Fig. 5 Half-sandwich indenyl rare earth metal dialkyl catalysts. | ||
Contrary to PMBL, PMMBL is soluble in CH2Cl2 and the polymerization of MMBL can therefore be carried out in this solvent. Results also followed the same trend as in DMF: 13c was the most active catalyst and gave a syndiotactic-enriched polymer (42.4% rr, 44.8% mr, 12.8% mm) with a Tg of 218 °C, similar to that obtained for atactic PMMBL with Cp*2Sm(THF)2 (221 °C). Kinetic studies in CH2Cl2 revealed a second order reaction law in respect to the catalyst concentration, suggesting a bimolecular propagation. A catalytic cycle involving a Michael addition was proposed, similar to the mechanism described for polymerization of methacrylates with non-bridged group 4 metallocene catalysts.75 MALDI-TOF mass spectrum analysis of an unpurified low molecular weight PMMBL ([MMBL]/[13c] = 20) revealed that one of the two mass distributions corresponded to PMMBL with one chain-end group being the alkyl CH2SiMe3 and the other a proton resulting from the acidic work-up procedure. Furthermore, the efficiency coefficient for the polymerization with these catalysts was found to be over 100%, suggesting the two alkyl species CH2SiMe3 participated in chain initiation but only one chain grew at a time since a first order dependence to MMBL was observed.
It can also be mentioned that homoleptic lanthanide silylamide Ln[N(SiMe3)2]3 (Ln = Sm, La, Nd, Er), cationic zirconocene and half titanocene complexes, which are efficient for the polymerization of MMA, were chosen as catalysts for the polymerization of (M)MBL. However the polymerization was much slower than with Cp*2Sm(THF)2 and not well controlled.70a
Polymerization of MBL and MMBL catalyzed by Cp*2Sm(THF)2 and dysprosium catalyst 13c respectively was studied in the presence of an external chain-transfer agent (CTA). A CTA is supposed to cleave the growing polymer from the active species and reinitiate the polymerization by anchoring to the polymer chain.52 Enolizable organo acid 3-methyl-2-butanone (MBO), methyl isobutyrate (MIB) and dimethyl malonate (DMM) were chosen as potential CTAs but only MBO can be considered as a suitable agent in the polymerization of MBL with Cp*2Sm(THF)2 giving an efficiency coefficient of 1060% with a turn over number (TON) of 10 with 20 eq. of the CTA.70a On the contrary, both MBO and MIB were ineffective for the polymerization of MMBL in the presence of Dy catalyst 13c.70b
Although most studies dealing with the polymerization of (M)MBL involved metal-based catalysts, Chen and co-workers have also reported the use of ambiphilic silicon propagators76 and frustrated Lewis pairs.77 The ambiphilic silicon catalyst system was recently successfully developed for (meth)acrylate polymerization78 and consequently has been applied to the methylene butyrolactones polymerization. Ambiphilic silicon propagating species consist of both the nucleophilic silyl ketene acetal (RSKA) initiating moiety and the electrophilic silylium catalyst. The first step consists in the oxidative activation of a silyl ketene acetal by a catalytic amount of [trityl tetrakis(pentafluorophenyl)borate] (TTPB) to form an electrophilic silylium cation 14 which undergoes a Michael addition of a second RSKA to lead to the active propagation species 15 (Scheme 4). In the proposed propagation mechanism, the monomer captured a silylium cation in a fast step followed by the rate determining step consisting in the intermolecular Michael addition of the polymeric SKA to the silylated monomer. With iBuSKA as the initiator, MBL polymerization in CH2Cl2 gave a bimodal polydispersity because of the insolubility of PMBL in that solvent. As already mentioned, PMMBL is soluble in CH2Cl2; thus its polymerization was controlled with quantitative yields obtained in 10 min at room temperature with excellent Mw/Mn between 1.01 and 1.06 depending on the ratio [MMBL]/[iBuSKA]. For [MMBL]/[iBuSKA] = 400, a syndio-enriched PMMBL was produced (45.8% rr, 39.9% mr, 14.3% mm). Moreover, when increasing to 600 equivalents of MMBL, characteristics of living polymerization were observed and for 800 equivalents, a high molecular weight of 548000 g mol−1 was produced. Investigations with MeSKA (best initiator for MMA polymerization) and activators other than TTPB (TRISPHAT and HB(C6F5)4) did not lead to better catalytic systems.
 | ||
| Scheme 4 Proposed mechanism for the polymerization of (M)MBL with ambiphilic silicon propagators (R′ = H or CH3). | ||
Copolymerization studies with the best catalytic system iBuSKA/TTPB produced the block copolymer PMMBL-b-PMBL displaying two glass transition temperatures of 197 and 212 °C (similar to the ones of isolated PMBL and PMMBL, respectively) and statistical PMMBL-co-PMBL with one Tg at 213 °C.
In the context of developing frustrated Lewis pair (FLP) catalytic systems,79 Chen and co-workers have reported the use of the sterically encumbered Lewis acid and Lewis base Al(C6F5)3 and 1,3-di-tert-butylimidazolin-2-ylidene (tBuNHC) respectively, as a catalytic system for the polymerization of (M)MBL in CH2Cl2 at room temperature.77 MBL and MMBL polymerized quantitatively but again, PMMBL was produced more rapidly than PMBL (in 1 min vs. 1 h) with narrower Mw/Mn (1.15 vs. 1.28) and a TOF almost 70 times higher (48000 vs. 704 h−1). However, atactic PMMBL is formed while in the same conditions, syndiotactic PMMA is observed.
In 2011 Chen et al. also investigated the anionic polymerization of (M)MBL using potassium salts (hydride, enolate and allyl) associated to tris(pentafluorophenyl)alane Al(C6F5)3.80 Potassium salts are less studied as anionic initiators than their lithium analogues but the authors justified their choice by describing the advantages of using a non-toxic (resorbable) and larger cation. Again MMBL was reported to be more active than MBL. A [(M)MBL]/[I] ratio of 400 was chosen for the study. The most efficient initiator KH/Al(C6F5)3 (in 1:2 ratio) produced PMMBL at room temperature in 50 min with high TOF (482 h−1), Mn (209000 g mol−1) and Mw/Mn of 1.56 whereas PMBL was obtained quantitatively after 24 h (TOF 4 times lower) with a broader Mw/Mn of 1.87. The enolate potassium salt Me2C = C(OiPr)OK with 2 equivalents of (C6F5)3Al led to longer reaction times (4–24 h), TOF around 5 times lower and similar Mw/Mn (1.79–1.84). Finally, the allyl salt K[(1,3-(SiMe3)2C3H3] only polymerized MMBL in 24 h with a narrower Mw/Mn of 1.42. Atactic P(M)MBL was produced with the three catalytic systems while syndiotactic PMMA was obtained in the same conditions.
All the catalytic systems described above enable the polymerization of α-methylene butyrolactone to proceed via its double bond without any ring opening polymerization. The first example of ROP with MBL was described only recently by copolymerization with ε-caprolactone (ε-CL) in the presence of bismuth trifluoromethanesulfonate.81 One of the interests of synthesizing poly(MBL-co-CL) resides in the exo-vinylidene moiety of MBL which can be used for instance for cross-link reactions with methacrylates to form bicomponent networks with a shape memory effect. The elasticity and the Tg of the polymers can be controlled from −26 °C to +29 °C (Scheme 5).
 | ||
| Scheme 5 Synthesis of bicomponent networks by radical cross-linking reaction using poly(MBL-co-CL). | ||
Polymers obtained from the ROP of MBL such as poly(MBL-co-CL) can be considered as biodegradable polymers due to the presence of the ester linkage in the backbone. In contrast, P(M)MBL obtained from the polymerization of the double bond without ring opening possesses the ester function only on the side chain (lactone). A preliminary degradation study of a copolymer poly(MBL–MDO) with a ratio MBL/MDO = 97:3 was carried out in a basic methanolic solution.61 A polymer containing hydroxy acid side chains resulting from the hydrolysis of the lactones was characterized by NMR spectroscopy. This hydrolyzed polymer is in part soluble in the alcoholic solution, suggesting a potentially slow degradation of the backbone.
In summary, ATRP can be used to successfully prepare copolymers containing the MBL moiety with thermal properties suitable for thermoplastic elastomers. Other catalytic systems showed that the polymerization of MMBL generally proceeds more rapidly and in a more controlled manner compared to that of MBL mainly because of the poorer solubility of PMBL in common polymerization solvents. It was also shown that both vinyl butyrolactones displayed different reactivity compared to MMA. Future investigations may focus on the selective polymerization of MBL and MMBL via their double bond to form iso- or syndio- rather than atactic polymers and on the ring-opening polymerization of the butyrolactones.
Fatty acids
Today plant oils are the most important renewable raw materials for the chemical industry. The annual global production of the major vegetable oils (from palm, soy, rapeseed, cotton, peanut, sunflower, palm kernel, olive, and coconut) amounted to 84.6 million tons in 1999/2000 and increased to 137.3 million tons in 2009/10.82 Vegetable oils are heavily used as raw materials for surfactants, cosmetic products, and lubricants. In addition, plant oils have been used for decades in paint formulations, as flooring materials and for coating and resin applications.83 Triglycerides contained in vegetable oils are triesters of glycerol and fatty acids. Only five major fatty acids are contained in vegetable oils as the triglyceride (Fig. 6): two saturated acids, stearic acid and palmitic acid and three unsaturated, oleic acid, linoleic acid and linolenic acid containing one, two and three carbon–carbon double bonds respectively. Ricinoleic acid, namely cis-12-hydroxyoctadeca-9-enoic acid produced from hydrolysis of castor oil is of interest, as it is a bifunctional fatty acid containing a hydroxyl group on the fatty chain.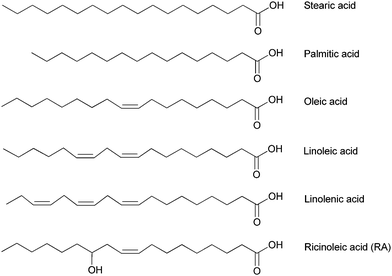 | ||
| Fig. 6 Main fatty acids issued from vegetable and castor oil. | ||
Fatty acids are good candidates for biocompatible and biodegradable polymers preparation, as they are natural body components containing hydrolyzable bonds.84 Several types of materials were prepared by direct copolymerization/insertion of bio-derived oil with other matrices to get bio-based nanocomposites85 or biodegradable materials (e.g., polyester and polyanhydrides).86,87 Due to the presence of a carbon–carbon double bond, triglycerides containing unsaturated fatty acids can be directly polymerized or copolymerized to get biopolymers, biocomposites and biocoatings.88 Also, biopolyamides derived from castor oil especially are industrially produced from chemical companies such as Arkema (Rilsan® PA11), Rhodia (Technyl® eXten) or BASF (Ultramid® Balance).89
However, well-defined polymers can only be prepared from pure monomers. Therefore further purification/separation and/or functionalization of the monomer issued from vegetable oils is always needed, increasing the final cost of the material. Nowadays, triglycerides and fatty acids can be obtained in high purities and are commercially available from chemical suppliers. A series of papers investigated the functionalization of fatty acids to get interesting monomers by epoxidation,90 hydroxylation,91 hydroformylation,92 ozonolysis93 and isomerization94 of the olefinic moiety. For instance, polymerization of epoxy fatty acids was reported by cationic ring opening polymerization of the epoxide function to form polyether–polyols.95 A focus on selected recent examples describing the synthesis of polymers from renewable fatty acids will be presented as follows. Firstly, polyester synthesis from fatty acids is discussed. Secondly, polyurethanes obtained by self-condensation of functionalized fatty acids are commented.
Polyesters by ring-opening polymerization of macrolactones
Increasing interest was given to polymers derived from long chain ω-hydroxy fatty acids, mainly produced by polycondensation methods.96 Enzyme polymerization, including ring-opening polymerization was already reviewed and won't be discussed in detail.97 The synthesis of macrolactones, not necessarily derived from bioresources, was reported and reviewed. For instance, efficient procedures were developed to prepare the C8 to C17 lactones by cyclization of the corresponding hydroxyacids.98 However, few examples of ring-opening polymerization of such monomers were reported. Chemical ring-opening polymerization and enzymatic ring-opening polymerization are methods of choice for controlling the polymer characteristics (molecular weight, polydispersity and microstructure). It is accepted that the driving force behind the chemical ROP is the release of the ring strain of the cyclic monomer to give the polymer chain. Thus an increase of the lactone ring size decreases the ring strain. Duda et al. compared thermodynamics and kinetics for chemical ROP and enzymatic ROP and demonstrated that contrary to chemical ROP, the rate of enzymatic ROP increases with the ring size of the lactone monomers from δ-valerolactone to 16-hexadecanolide: in enzymatic ROP, the rate determining step involves the formation of the lipase–lactone complex, promoted by the hydrophobicity of the lactone monomer, which is higher for larger lactone rings.99Thus, lipases were already reported as highly active initiators of macrolactone ROP affording high molecular weight polymers, the main drawback of the enzymes being their high price and temperature sensitivity.100 Pentadecalactone (PDL), a cyclic ω-hydroxy fatty acid used in the fragrance industry and belonging to the class of naturally occurring macrocyclic musks, is often used as a macrolactone model monomer. Polypentadecalactone (PPDL), obtained by ROP of pentadecalactone with Novozym 435, was reported having Mn up to 150000 g mol−1 (Mw/Mn = 2.1).100i In comparison, metal-catalyzed ROP of PDL generally gave polyesters with lower molecular weights ranging from 30000 to 40000 g mol−1 (Mw/Mn = 1.6) with rare-earth initiators, such as Y(OiPr)3 and Ln(BH4)3(THF)3.101 Anionic ROP of macrolactones102,103 such as PDL with tBuOK as the initiator was also successfully reported; a high molecular weight polymer was obtained (Mn = 92000 g mol−1, Mw/Mn = 2.1).103
To our knowledge, the first example of a polymer with high molecular weight (Mn = 155000 g mol−1, Mw/Mn = 2.0), obtained by chemical ROP of macrolactone, such as pentadecalactone was reported very recently by Heise and Duchateau et al. using an aluminium–salen complex as the initiator.104 However, these metal initiators were inactive at temperatures below 60 °C contrary to Novozym 435 and tBuOK, which polymerized PDL at room temperature.
An interesting work using ricinoleic acid (RA) was reported by Slivniak and Domb in 2005,105 taking advantage of the two functional groups of ricinoleic acid, a hydroxyl and a carboxylic function in order to form large macrolactones. The corresponding lactone was formed by intramolecular esterification of ricinoleic acid by the classical method (dicyclohexylcarbodiimide, dimethylaminopyridine and hydrochloric acid).98g Formation of the expected C12 lactone as well as macrocyclic lactones containing up to 6 ricinoleic units was observed (Scheme 6). The ratio of lactones is dependent on the initial concentration of the ricinoleic acid; low RA concentration (up to 10 mg mL−1) yielded mainly the C12, C24 and C36 lactones (the C12 lactone is obtained pure in 46% yield after purification) and higher RA concentration yielded larger rings.
 | ||
| Scheme 6 Formation of lactones and macrolactones mixture (up to n = 5). (i) Reaction conditions: dicyclohexylcarbodiimide, dimethylaminopyridine, hydrochloric acid in chloroform. | ||
Afterwards, ring-opening polymerization of the lactones C12, C24 or C12–C72 mixture was investigated with different initiators such as Y(OiPr)3 and Sn(octoate)2, which were inactive for the pure C12 lactone ROP but also for the lactone mixture (Scheme 7, (a)). However the tin initiator gave low molecular weight polymers by ROP of C24 lactone (Mn = 4400, Mw = 5700 g mol−1, representing up to 15–20 RA units). Unfortunately, only oligomers were also obtained using Y(OiPr)3.
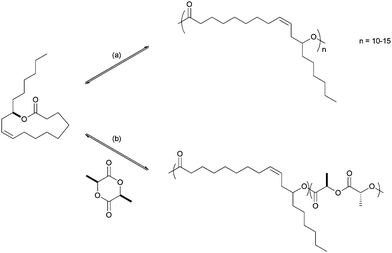 | ||
| Scheme 7 ROP of ricinoleic acid derived lactone. (a) Sn(octoate)2 or Me3SiONa, THF, 40 °C; (b) Sn(octoate)2, bulk, 135 °C. | ||
Copolymerization of ricinoleic lactone mixture and L-lactide (Scheme 7, (b)) was reported and low molecular weight polymers in the range of 5000–16000 g mol−1 were synthesized containing a low ratio of RA unit in the polymer chain as the ROP of ricinoleic lactone derivative is much slower than the one of lactide. The ratio of the RA unit in the polymer can be tuned by varying the initial RA lactone to lactide ratio; it was determined by 1H NMR spectroscopy that up to 17% of RA was inserted in the polymer chain using a 1:1 weight ratio of the two monomers. Domb also observed that the molecular weight of the polymer decreased with an increase in the content of the RA unit. They also studied the hydrolytic degradation properties of the P(RA–LA) copolymers obtained and pointed out that the material with a low RA content degraded slower than pure PLA of similar molecular weight.
Quinzler and Mecking reported a convenient route to high molecular weight poly(dodecyloate) (Mn up to 22000 g mol−1) by step growth polymerization of undec-10-en-1-ol and carbon monoxide.106 By using a cobalt catalyst system known for alkoxycarbonylation ([Co2(CO)8]/pyridine), the authors were able to prepare a semicrystalline polyester by copolymerization of CO and undec-10-en-1-ol which was first obtained in two high-yield steps from ricinoleic acid.107
Very recently, Mecking et al. published an interesting study on undecenoic acid, also produced from ricinoleic acid.108 1,20-Eicosanedioic acid was obtained by self-metathesis of undecenoic acid followed by hydrogenation. The corresponding diol, eicosane-1,20-diol, was formed by reduction of eicosanedioic acid. Subsequently, the diacid and the diol, obtained from the same renewable resource, were polycondensed to give a high molecular weight C20 polyester with a Tm of 108 °C. The reaction was carried out in bulk and catalyzed by titanium alkoxides (Scheme 8).
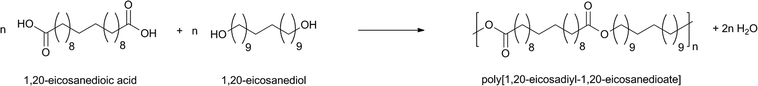 | ||
| Scheme 8 Synthesis of the 20,20-polyester by melt polycondensation. | ||
Gross et al. reported the melt-polycondensation of the ω-hydroxyl tetradecanoic acid (C14), a new monomer available by a fermentation process,109 catalyzed by titanium tetraisopropoxide. High molecular weight poly(ω-hydroxytetradecanoate) was prepared with good control (up to 140000 g mol−1 with a Mw/Mn of 1.8).110 The mechanical properties of the obtained polymer were studied and compared to linear high-density polyethylene (LHDPE) and authors concluded that such poly(ω-hydroxyalkanoate), such as C14, C16 and C18 polyesters, have the potential to function in similar ways to PE, possessing a potential biodegradability advantage. Meier et al. reported the synthesis of different polyethylene-like copolymers from renewable platform chemicals (castor oil derived chemicals: 11-bromo-1-undecene and 10-undecenol) using thiol–ene addition as well as Acyclic Diene METathesis (ADMET) polymerization.111
Polyurethane synthesis by self-condensation
Polyurethanes are an important class of polymers used in several different fields such as footwear, coatings and paintings, elastic fibers and medical devices.112 Recently several works dealing with polyurethanes based on vegetable oils were published and reviewed.93,95,113In 2010 Cramail and co-workers reported an interesting approach to synthesize polyurethane (PU) from methyl oleate (derived from sunflower oil) and ricinoleic acid using the AB-type self-polycondensation approach.114 In this work, three novel AB-type monomers, namely, a mixture of 10-hydroxy-9-methoxyoctadecanoyl azide/9-hydroxy-10-methoxyoctadecanoyl azide (HMODAz), 12-hydroxy-9-cis-octadecenoyl azide (HODEAz) and methyl-N-11-hydroxy-9-cis-heptadecen carbamate (MHHDC) were prepared from methyl oleate and ricinoleic acid using simple reaction steps (Scheme 9). Then, HMODAz and HODEAz monomers were polymerized by the acyl-azido and hydroxyl AB-type self-condensation approach. The acyl-azido and hydroxyl self-condensations were carried out at various temperatures (50, 60, 80 and 110 °C) in bulk with and without a catalyst. It was demonstrated that the polymerization of the acyl azide occurred through its thermal decomposition to isocyanate, known as the “Curtius rearrangement”, which further condensed with the hydroxyl group to form the polyurethane. A FT-IR study of the polymerization, using HMODAz at 80 °C without a catalyst, indicated in situ formation of an intermediate isocyanate group in the first minutes of the reaction. Also, MHHDC monomer was polymerized through AB-type self-condensation. In this case, a transurethane reaction was used to obtain a similar PU (which was obtained by AB-type acyl-azido and hydroxyl self-condensation of HODEAz) in the presence of titanium tetrabutoxide as a catalyst at 130 °C.
 | ||
| Scheme 9 Polyurethane prepared from functionalized oleic acid and ricinoleic acid (a) 80 °C, 24 h (for clarity only the 10-hydroxy-9-methoxy isomer of HMODaz is presented); (b) CH3OH, reflux, 4 h; (c) Ti(OBu)4, 130 °C, 6 h. | ||
The self-condensation of these bifunctional monomers led to polymers with low molecular weight. This was explained by the formation of macrocycles, detected by MALDI-TOF analyses of the polyurethanes. Due to the presence of soft and hard segments, poly(HMODAz) and poly(HODEAz) showed two glass transition temperatures respectively at −22 °C, 27 °C and −53 °C, 26 °C.
Also, Cramail et al. reported polyaddition of diamines with vegetable-based biscarbonates to prepare new polyurethanes (Scheme 10).115 The starting material 16a was obtained from 16b by a metathesis reaction with Hoveyda's catalyst. Then the epoxy ester dimers 18a-b were obtained from 16a-b in two steps (Scheme 10). The biscarbonates 19a-b were prepared from 18a-b and CO2 and further self-polycondensed with ethylene diamine and isophorone diamine to form polyurethanes. The optimal conditions for the carbonation reaction appeared to be in the presence of tetrabutylammonium bromide (TBABr) as the catalyst. The average molecular mass of the polyurethane reached 13500 g mol−1 with a good control over the chain length (Mw/Mn < 1.5) and their glass transition temperature Tg ranging from −25 to −13 °C.
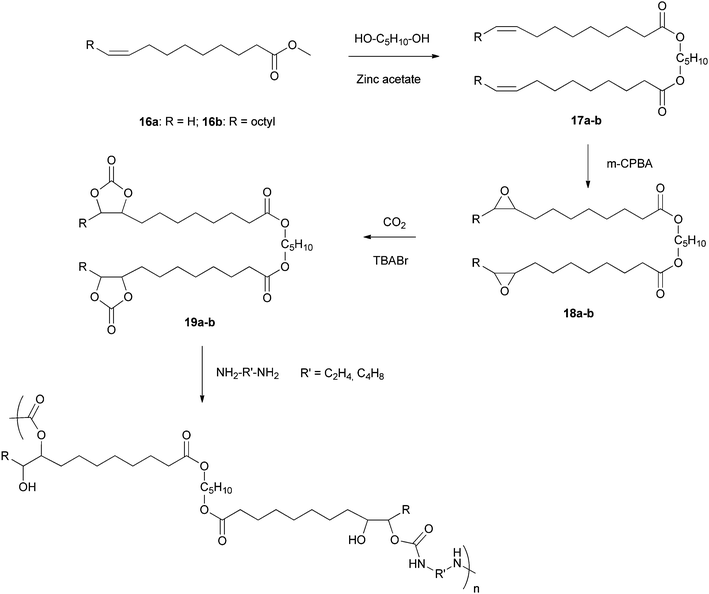 | ||
| Scheme 10 Polyurethane from methyloleates. | ||
Functionalized lactones
Poly(lactides) may lack chemical functional groups which can modify the hydrophilic properties and degradation rate;116 however, functionalized aliphatic polyesters issued from bioresources are not broadly available. Even if ring-opening polymerization of lactide, previously described, and lactones can be well controlled,23,117 only few examples of ring-opening of functionalized and/or bioderived lactones were reported.118 Moreover, syntheses of functionalized lactones are often multi-step reactions using complex procedures and/or purification methods. Carbohydrate 1,5-lactones seem to be interesting monomers, but very few studies were reported on their polymerization. In 1927, Drew and Haworth reported the formation of “polymeride” when tri-O-methyl-D-arabino-1,5-lactone was reacted with acids.119 More recently, in 2002, a methodology for the polymerization of D-gluconolactone was patented but the characterization of the resulting material was limited.120 An attempt to polymerize such monomers undertaken by Haider and Williams revealed a dimerization or trimerization of the previously mentioned monomers.118l Recently, Williams et al. reported the ROP of a carbohydrate lactone, obtained in 90% yield in two steps from the commercially available D-gluconolactone.121D-Gluconolactone is derived from gluconic acid (the latter being obtained from glucose) and is used as a food additive.12e,g The new lactone 20 was obtained as a racemic mixture of the two syn enantiomers S,S and R,R (Scheme 11).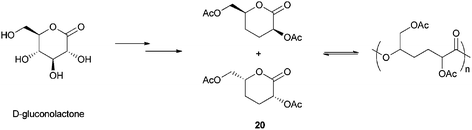 | ||
| Scheme 11 ROP of a carbohydrate derived lactone. | ||
The ring-opening polymerization was studied with “classical initiators” derived from Sn(OR)2 and LZn(OEt). The tin catalyst showed a slow conversion of lactone (from 140 to 680 h to convert 30–120 eq. of lactone) while the reaction was much faster with the Zn catalyst (1 h to convert 100 eq. of lactone). A large disparity between the expected mass and the calculated one was obtained for the corresponding polyesters due to the formation of cyclic polyesters (observed by MALDI-TOF analysis) and transesterification reaction.
Recently, dihydrocarvone obtained from carvone, a natural terpenoid found in both Mentha spicata (spearmint) (up to 60–70% of oil content) and Carum carvi (caraway) (only small percent) oils, was used as the starting material for the synthesis of lactone monomers.122 Compared to production of fatty acids, carvone stays quite marginal as spearmint and caraway oil world markets are estimated to be 1500 t per year and 10 t per year respectively.123 Thus, Hillmyer et al. prepared two new lactone monomers from dihydrocarvone (Scheme 12). Carvomenthide (22) was obtained in good yield (∼70%) in two steps consisting in hydrogenation of dihydrocarvone to carvomenthone followed by oxidation. In the case of dihydrocarvide (21), obtained by a direct selective Baeyer–Villiger oxidation of dihydrocarvone with Oxone®, purification of the product was more challenging as 21 was obtained in 42% yield still containing epoxide impurities. Both lactones were ring-opening polymerized with ZnEt2/nBuOH as the initiator system.
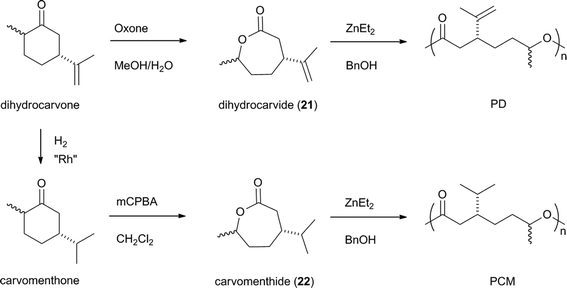 | ||
| Scheme 12 Formation of polyesters with lactones derived from dihydrocarvone. | ||
Polymers polycarvomenthide (PCM) with a Mn up to 50000 g mol−1 were obtained with good polymerization control (1.08 < Mw/Mn < 1.26). In the case of polydihydrocarvide (PD), the olefinic bond remained intact after the polymerization but a disparity was observed between the expected molar mass and the determined one. This was explained by the fact that 21 self-polymerizes due to the presence of polymerization initiating impurities, probably epoxides. No transesterification reactions were observed, as Mw/Mn remained low; thus high molecular weight can be reached contrary to that obtained with ε-caprolactone.124 Both polymers PCM and PD exhibited low glass transition temperatures (less than −20 °C). It was also shown that copolymerization of 21 and PD as well as cross-linking polymerization of the PD polymer after epoxidation of the olefinic moiety was possible. This points out the versatility of these monomers for the preparation of renewable materials by post-polymerization functionalization and/or copolymerization strategies. Block copolymers were also reported by copolymerization of carvomenthide and polylactide to give elastomers.125
Conclusion and perspectives
We have reviewed renewable structures that can be turned into viable biodegradable macromolecules. Using biomass for the production of new polymers can have both economic and environmental benefits. The discovery of efficient and selective processes for the synthesis of renewable polymers is a crucial requirement for the sustained growth of the chemical industry. In this area, promising results have been obtained using catalytic processes. However, despite recent significant advances, some major points remain to be addressed and improved.For instance, preparation of high molecular weight polyesters from renewable resources remains challenging due to the presence of functional groups on the monomers, relative stability of the monomer (large ring lactone for instance) and side-reactions (cyclization, transesterification). Therefore focus on the development of ligand and initiators, tolerant to functional groups, efficient and stable is needed to promote a wide use and applications of bioderived monomers and polymers.
Whereas carbon dioxide is not plant-derived, the utilization of CO2 for the production of polymers could be critically important given its widespread abundance. In particular, there has been considerable interest in the development of catalysts for the alternating copolymerization of carbon dioxide with epoxides to produce polycarbonates. As a result, a significant amount of recent research has focused on the discovery and development of new catalysts for this process.126 While CO2 is an ideal synthetic feedstock since it is abundant, inexpensive and nontoxic, most epoxide/CO2 copolymerization systems focus on petroleum derivatives such as propylene oxide or cyclohexene oxide. However, a major breakthrough has recently been achieved by Coates et al. who reported the alternating copolymerization of CO2 with limonene oxide, which is derived from limonene, the major component of oils from citrus fruit peels (e.g. 95% of the oil from orange peels).127 Another way to produce polycarbonates issued from renewable resources is ring-opening polymerization of cyclic carbonates. Indeed, such carbonate monomers can be derived from biomass. For instance, trimethylene carbonate can be prepared from glycerol. Also, Carpentier et al. used levulinic and itaconic acids as a naturally occurring source to produce two 7-membered cyclic carbonates as monomers. Subsequent ring-opening polymerization using various (organo)metallic and organic catalysts afforded the corresponding polycarbonates with quite good control and activities.128
Finally a recent study reported a new strategy to obtain biodegradable polyesters.129 This was achieved by tandem catalysis, which confers great interest to this approach. Commercially available complexes were used as efficient catalysts for cyclization of dicarboxylic acids followed by alternating copolymerization of the resulting anhydrides with epoxides. Given an operationally simple method, this tandem catalysis is an attractive strategy for the production of new renewable materials.
References
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 Search PubMed.
- J. Rass-Hansen, H. Falsig, B. Jorgensen and C. H. Christensen, J. Chem. Technol. Biotechnol., 2007, 82, 329–333 CrossRef CAS.
- At the present utilization rate, the first fossil resource anticipated to be depleted is oil, followed by natural gas and finally coal, which is estimated to last about 200 years from now: G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond oil and Gas: The Methanol Economy, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2006, ISBN: 3-527-31275-7.
- P. B. Weisz, Phys. Today, 2004, 57, 47–52 Search PubMed.
- J. Zaldivar, J. Nielsen and L. Olsson, Appl. Microbiol. Biotechnol., 2001, 56, 17–34 CrossRef CAS.
- A number of standards authorities have sought to produce definitions for (bio)degradable plastics. Therefore degradation is defined as a deleterious change in the chemical structure, physical properties, or appearance of a plastic and a degradable plastic: a plastic designed to undergo a significant change in its chemical structure under specific environmental conditions, resulting in a loss of some properties that may vary as measured by standard test methods appropriate to the plastic and the application in a period of time that determines its classification. See: B. Shi, V. Topolkaraev and J. Wang, in Renewable and Sustainable Polymers, ed. G. F. Payne and P. B. Smith, ACS Symposium Series, American Chemical Society, Washington DC, 2011, pp. 117–132 and references therein.
- To be considered as biodegradable, a material shall pass several tests including, for instance, chemical composition (quantity of heavy metal present), complete degradation in laboratory and real-life conditions and ecotoxicity of the obtained compost. Moreover the plastic has to be degraded to fragments smaller than 2 mm size at more than 90% under real life conditions for instance. See: S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078–1085 Search PubMed and references therein.
- One exception of a biodegradable polymer with pure C–C bonds is poly(vinyl alcohol). In this case, degradation proceeds via primary oxidation of the hydroxyl groups, followed by polymer chain cleavage similar to fatty acid degradation.
- C. M. Thomas and J.-F. Lutz, Angew. Chem., Int. Ed., 2011, 50, 9244–9246 Search PubMed and references therein.
- J. Li, R. M. Stayshich and T. Y. Meyer, J. Am. Chem. Soc., 2011, 133, 6910–6913 CrossRef CAS.
- (a) M. Poliakoff and P. Licence, Science, 2006, 211, 810–812 Search PubMed; (b) A. J. Ragauskas, Science, 2006, 311, 484–489 CrossRef; (c) F. S. Güner, Y. Yagci and A. T. Erciyes, Prog. Polym. Sci., 2006, 31, 633–670 CrossRef; (d) J. F. Jenck, Green Chem., 2004, 6, 544–556 RSC; (e) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS; (f) U. Biermann, W. Friedt, S. Lang, W. Luhs, G. Machmuller, J. O. Metzger, M. R. Klaas, H. J. Schafer and M. P. Schneider, Angew. Chem., Int. Ed., 2000, 39, 2206–2224 CrossRef CAS; (g) F. W. Lichtenthaler, Acc. Chem. Res., 2002, 35, 728–737 CrossRef CAS.
- L. A. Lucia, D. S. Argyropoulos, L. Adamopoulos and A. R. Gaspar, Can. J. Chem., 2006, 84, 960–970 Search PubMed.
- Recently, excellent articles on the polymerization of renewable resources have been published. See for instance: (a) R. T. Mathers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1–15 Search PubMed; (b) Renewable and Sustainable Polymers, ed. G. F. Payne, and P. B. Smith, ACS Symposium Series, American Chemical Society, Washington DC, 2011 Search PubMed.
- Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008 Search PubMed.
- (a) R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS; (b) H. Danner and R. Braun, Chem. Soc. Rev., 1999, 28, 395–405 RSC.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef.
- R. E. Drumright, P. R. Gruber and E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- M. Hiljanen-Vainio, P. Varpomaa, J. Seppala and P. Tormala, Macromol. Chem. Phys., 1996, 197, 1503–1523 Search PubMed.
- G. Perego, G. D. Cella and C. Bastioli, Polymer, 1996, 59, 37–43 Search PubMed.
- H. Tsuji and Y. Ikada, Macromol. Chem. Phys., 1996, 197, 3483–3499 Search PubMed.
- J.-R. Sarasua, R. E. Prud'homme, M. Wisniewski, A. Le Borgne and N. Spassky, Macromolecules, 1998, 31, 3895–3905 CrossRef CAS.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- (a) S. McLain and N. Drysdale, US Pat., 5 028 667, 1991; (b) S. McLain, T. Ford and N. Drysdale, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1992, 33, 463–464 Search PubMed.
- W. M. Stevels, M. J. K. Ankone, P. J. Dijkstra and J. Feijen, Macromolecules, 1996, 29, 6132–6138 CrossRef CAS.
- B. J. O'Keefe, S. M. Monnier, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2001, 123, 339–340 CrossRef CAS.
- See for instance: (a) M. S. Reeve, S. P. McCarthy, M. J. Downey and R. A. Gross, Macromolecules, 1994, 27, 825–831 CrossRef CAS; (b) J. R. Sarasua, R. E. Prud'homme, M. Wisniewski, A. Le Borgne and N. Spassky, Macromolecules, 1998, 31, 3895–3905 CrossRef CAS; (c) Z. Zheng, G. Zhao, R. Fablet, M. Bouyahyi, C. M. Thomas, T. Roisnel, O. Casagrande, Jr and J.-F. Carpentier, New J. Chem., 2008, 32, 2279–2291 RSC; (d) B. Lian, C. M. Thomas, O. L. Casagrande, Jr, T. Roisnel and J.-F. Carpentier, Polyhedron, 2007, 26, 3817–3824 CrossRef CAS; (e) B. Lian, C. M. Thomas, O. L. Casagrande, Jr, C. W. Lehmann, T. Roisnel and J.-F. Carpentier, Inorg. Chem., 2007, 46, 328–340 CrossRef CAS.
- J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS.
- H2salen = N, N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexenediimine.
- The term salan was introduced by Atwood to refer to saturated salen ligands: D. A. Atwood, Coord. Chem. Rev., 1997, 165, 267–296 Search PubMed.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- (a) T. M. Ovitt and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4686–4692 CrossRef CAS; (b) C. P. Radano, G. L. Baker and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 1552–1553 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS.
- (a) N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS; (b) N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem.–Eur. J., 2007, 13, 4433–4451 CrossRef CAS; (c) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS; (d) A. Alaaeddine, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Organometallics, 2009, 28, 1469–1475 CrossRef CAS; (e) M. Bouyahyi, E. Grunova, N. Marquet, E. Kirillov, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Organometallics, 2008, 27, 5815–5825 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 4072–4073 CrossRef CAS.
- A. Pietrangelo, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2009, 2736–2737 RSC.
- A. Pietrangelo, S. C. Knight, A. K. Gupta, L. J. Yao, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2010, 132, 11649–11657 CrossRef CAS.
- A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290–2293 CrossRef CAS.
- (a) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS and references therein; (b) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS.
- A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem.–Eur. J., 2006, 12, 169–179 CrossRef CAS and references therein.
- A. Amgoune, C. M. Thomas and J.-F. Carpentier, Pure Appl. Chem., 2007, 79, 2013–2030 CrossRef CAS.
- X. Liu, X. Shang, T. Tang, N. Hu, F. Pei, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 2747–2757 CrossRef CAS.
- A. Amgoune, C. M. Thomas and J.-F. Carpentier, Macromol. Rapid Commun., 2007, 28, 693–697 CrossRef CAS.
- H. Ma, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2006, 45, 7818–7821 CrossRef and references therein.
- D. J. Coady, A. C. Engler, H. W. Horn, K. M. Bajjuri, K. Fukushima, G. O. Jones, A. Nelson, J. E. Rice and J. L. Hedrick, ACS Macro Lett., 2012, 1, 19–22 Search PubMed.
- L. Zhang, R. C. Pratt, F. Nederberg, H. W. Horn, J. E. Rice, R. M. Waymouth, C. G. Wade and J. L. Hedrick, Macromolecules, 2010, 43, 1660–1664 CrossRef CAS and references therein.
- (a) E. F. Connor, G. W. Nyce, M. Myers, A. Möck and J. L. Hedrick, J. Am. Chem. Soc., 2001, 124, 914–915 Search PubMed; (b) A. P. Dove, H. Li, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2006, 2881–2883 RSC.
- (a) L. Zhang, F. Nederberg, J. M. Messman, R. C. Pratt and J. L. Hedrick, J. Am. Chem. Soc., 2007, 129, 12610–12611 CrossRef CAS; (b) L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Macromolecules, 2007, 40, 4154–4158 CrossRef CAS.
- G. M. Miyake and E. Y.-X. Chen, Macromolecules, 2011, 44, 4116–4124 CrossRef CAS.
- E. Y.-X. Chen, Chem. Rev., 2009, 109, 5157–5214 CrossRef CAS.
- (a) M. W. P. C. V. Rossum, M. Alberda and L. H. W. Van der Plas, Phytochemistry, 1998, 49, 723–729 Search PubMed; (b) Y. Kato, H. Yoshida, K. Shoji, Y. Sato, N. Nakajima and S. Ogita, Tetrahedron Lett., 2009, 50, 4751–4753 Search PubMed.
- L. E. Manzer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS.
- M. Sauer, D. Porro, D. Mattanovich and P. Branduardi, Trends Biotechnol., 2008, 26, 100–108 CrossRef CAS.
- J. W. Stansbury and J. M. Antonucci, Dent. Mater., 1992, 8, 270–273 CrossRef CAS.
- (a) M. K. Akkapeddi, Macromolecules, 1979, 12, 546–551 CrossRef CAS; (b) M. K. Akkapeddi, Polymer, 1979, 20, 1215–1216 CrossRef CAS; (c) M. Ueda, M. Takahashi, Y. Imai and C. U. Pittman, Jr, J. Polym. Sci., Part A: Polym. Chem., 1982, 20, 2819–2828 Search PubMed.
- D. Y. Sogah, W. R. Hertler, O. W. Webster and G. M. Cohen, Macromolecules, 1987, 20, 1473–1488 CrossRef CAS.
- (a) C. Lee and H. K. Hall, Jr, Macromolecules, 1989, 22, 21–25 CrossRef CAS; (b) M. Van den Brink, W. Smulders, A. M. Van Herk and A. L. German, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3804–3816 CrossRef; (c) D. L. Trumbo, Polym. Bull., 1991, 26, 271–275 CrossRef CAS; (d) H. Koinuma, K. Sato and H. Hirai, Makromol. Chem., Rapid Commun., 1982, 3, 311–315 CrossRef CAS.
- T. Uno, S. Kawaguchi, M. Kubo and T. Itoh, J. Power Sources, 2008, 178, 716–722 Search PubMed.
- S. Agarwal and R. Kumar, Macromol. Chem. Phys., 2011, 212, 603–612 Search PubMed.
- J. Suenaga, D. M. Sutherlin and J. K. Stille, Macromolecules, 1984, 17, 2913–2916 CrossRef CAS.
- C. U. Pittman, Jr and H. Lee, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1759–1777 Search PubMed.
- G. Qi, M. Nolan, F. J. Schork and C. W. Jones, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5929–5944 CrossRef CAS.
- C. J. Brandenbourg, WO 2004069926, 2004.
- (a) R. C. Cockburn, T. F. L. McKenna and R. A. Hutchinson, Macromol. Chem. Phys., 2010, 211, 501–509 CrossRef CAS; (b) R. C. Cockburn, R. Siegmann, K. A. Payne, S. Beuermann, T. F. L. McKenna and R. A. Hutchinson, Biomacromolecules, 2011, 12, 2319–2326 Search PubMed.
- J. Mosnáček and K. Matyjaszewski, Macromolecules, 2008, 41, 5509–5511 CrossRef CAS.
- J. Mosnáček, J. A. Yoon, A. Juhari, K. Koynov and K. Matyjaszewski, Polymer, 2009, 50, 2087–2094 CrossRef CAS.
- A. Juhari, J. Mosnáček, J. A. Yoon, A. Nese, K. Koynov, T. Kowalewskin and K. Matyjaszewski, Polymer, 2010, 51, 4806–4813 Search PubMed.
- (a) G. M. Miyake, S. E. Newton, W. R. Mariott and E. Y.-X. Chen, Dalton Trans., 2010, 39, 6710–6718 RSC; (b) Y. Hu, X. Xu, Y. Zhang, Y. Chen and E. Y.-X. Chen, Macromolecules, 2010, 43, 9328–9336 Search PubMed.
- H. Yasuda, H. Yamamoto, M. Yamashita, K. Yokoto, A. Nakamura, S. Miyake, Y. Kai and N. Kanehisa, Macromolecules, 1993, 26, 7134–7143 CrossRef CAS.
- L. S. Boffa and B. M. Novak, Macromolecules, 1994, 27, 6993–6995 CrossRef CAS.
- X. Xu, Y. Chen and J. Sun, Chem.–Eur. J., 2009, 15, 846–850 CrossRef CAS.
- X. Xu, Y. Chen, J. Feng, G. Zou and J. Sun, Organometallics, 2010, 29, 459–553 Search PubMed.
- G. Stojcevic, H. Kim, N. J. Taylor, T. B. Marder and S. Collins, Angew. Chem., Int. Ed., 2004, 43, 5523–5526 CrossRef CAS.
- G. M. Miyake, Y. Zhang and E. Y.-X. Chen, Macromolecules, 2010, 43, 4902–4908 CrossRef CAS.
- Y. Zhang, G. M. Miyake and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2010, 49, 10158–10162 CrossRef CAS.
- (a) Y. Zhang and E. Y.-X. Chen, Macromolecules, 2008, 41, 36–42 CrossRef CAS; (b) Y. Zhang and E. Y.-X. Chen, Macromolecules, 2008, 41, 6353–6360 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CAS.
- Y. Hu, L. O. Gustafson, H. Zhu and E. Y.-X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2008–2017 Search PubMed.
- J. Zhou, A. M. Schmidt and H. Ritter, Macromolecules, 2010, 43, 939–942 Search PubMed.
- Meier et al. reviewed the production and chemical transformations of oils and fats: U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 Search PubMed and references therein.
- (a) M. Eissen, J. O. Metzger, E. Schmidt and U. Schneidewind, Angew. Chem., Int. Ed., 2002, 41, 414–436 CrossRef CAS; (b) J. O. Metzger and M. Eissen, C. R. Chim., 2004, 7, 569–581 CrossRef CAS; (c) F. Sylvestre, J.-M. Aubry, T. Benvegnu, J. Brendle, M. Durand, A. Lavergne, C. Len, V. Molinier, Z. Mouloungui and D. Plusquellec, Actual. Chim., 2010, 338–339, 28–40 Search PubMed.
- Selected examples: (a) R. Langer, Acc. Chem. Res., 2000, 33, 94–101 CrossRef CAS; (b) A. J. Domb, O. Elmalak, V. R. Shastry, Z. Ta-Shma, D. M. Masters, L. Ringel, D. Teomim and R. Langer, in Handbook of Biodegradable Polymers, ed. A. J. Domb, J. Kost and D. M. Weisman, Harwood Academic, Chichester, England, 1997, p. 135 Search PubMed; (c) D. Stephens, L. Li, D. Robinson, S. Chen, H. C. Chang, R. M. Liu, Y. Q. Tian, E. J. Ginsberg, X. Y. Gao and T. J. Stultz, J. Controlled Release, 2000, 63, 305–317 Search PubMed; (d) A. J. Domb, S. Amselen and M. Maniar, Polym. Biomater., 1994, 399–433 Search PubMed; (e) A. J. Domb and R. Nudelman, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 717–725 Search PubMed.
- (a) H. Miyagawa, M. Misra, L. T. Drzal and A. K. Mohanty, Polymer, 2005, 46, 445–453 CrossRef CAS; (b) Y. Lu and R. C. Larock, Biomacromolecules, 2006, 7, 2692–2700 CrossRef CAS.
- (a) M. Y. Krasko, A. Shikynov, A. Ezra and A. J. Domb, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1059–1069 Search PubMed; (b) D. Teomim, A. Nyska and A. J. Domb, J. Biomed. Mater. Res., 1999, 45, 258–267 CrossRef CAS; (c) D. Teomin and A. J. Domb, Biomacromolecules, 2001, 2, 37–44 Search PubMed.
- Polyesters preparation from ricinoleic acid or castor oil was reported, leading to biodegradable polymers directly from the crude oils. See for instance: (a) S. Matsumura and K. Takahashi, Makromol. Chem. Rapid Commun., 1986, 7, 369–373 Search PubMed; (b) H. Ebata, K. Toshima and S. Matsumura, Macromol. Biosci., 2007, 7, 798–803 CrossRef CAS; (c) H. J. Wang, M. Z. Rong, M. Q. Zhang, H. W. Chen and T. Czigány, Biomacromolecules, 2008, 9, 615–623 Search PubMed.
- Selected recent examples, as numerous patents and articles refer to the topics: (a) U. Biermann, J. O. Juergen and M. A. R. Meier, Macromol. Chem. Phys., 2010, 211, 854–862 CAS; (b) N. Williams, C. Lampard, J. T. Carter, S. Quinn, PCT Int. Appl., WO 2009080598 A1 20090702, 2009; (c) D. Zulim Botage, S. Bastida, S. Marmesat, L. Perez-Olleros, B. Ruiz-Roso and F. J. Sanchez-Muniz, J. Am. Oil Chem. Soc., 2009, 86, 419–425 Search PubMed; (d) H. Esen, S. Kusefoglu and R. Wool, J. Appl. Polym. Sci., 2007, 103, 626–633 CrossRef CAS; (e) R. P. Wool, S. H. Kusefoglu, S. N. Khot, R. Zhao, G. Palmese, A. Boyd, C. Fisher, S. Bandypadhyay, A. Paesano and P. Dhurjat, Polym. Prepr., 1998, 39, 90 Search PubMed; (f) M. Galia, L. Montero de Espinosa, J. C. Ronda, G. Lligadas and V. Cadiz, Eur. J. Lipid Sci. Technol., 2010, 112, 87–96 CrossRef CAS; (g) J. T. Carter, S. Quinn, PCT Int. Appl., WO 2009080599 A1 20090702, 2009; (h) S. Yoshiko, F. Masahiro, Jpn. Kokai Tokkyo Koho, JP 2005200563, 2005; (i) R. L. Quirino and R. C. Larock, in Renewable and Sustainable Polymers, ed. G. F. Payne and P. B. Smith, ACS Symposium Series, American Chemical Society, Washington DC, 2011, pp. 37–59 Search PubMed.
- (a) V. Sharma and P. P. Kundu, Prog. Polym. Sci., 2006, 31, 983–1008 CrossRef CAS; (b) R. P. Wool, in Bio-Based Polymers and Composites, ed. R. P. Wool and X. S. Sun: Academic Press, Amsterdam, 2005; p. 114 Search PubMed; (c) A. S. Trevino and D. L. Trumbo, Prog. Org. Coat., 2002, 44, 49–54 Search PubMed.
- (a) Z. S. Petrovic, A. Zlatanic, C. C. Lava and S. Sinadinovic-Fiser, Eur. J. Lipid Sci. Technol., 2002, 104, 293–299 CrossRef CAS; (b) Z. S. Petrovic, W. Zhang and I. Javni, Biomacromolecules, 2005, 6, 713–719 Search PubMed.
- (a) Z. S. Petrovic, A. Guo and W. Thang, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4062–4069 CrossRef CAS; (b) Y. H. Hu, Y. Gao, D. N. Wang, C. P. Hu, S. Zu, L. Vanoverloop and R. Randall, J. Appl. Polym. Sci., 2002, 84, 591–597 CrossRef CAS; (c) L. Monteavaro, E. da Silva, A. Costa, D. Samios, A. Gerbase and C. Petzhold, J. Am. Oil Chem. Soc., 2005, 82, 365–371 CrossRef CAS; (d) Z. Lozada, G. J. Suppes, Y. C. Tu and F. H. Hsieh, J. Appl. Polym. Sci., 2009, 113, 2552–2560 CrossRef CAS.
- (a) A. Guo, D. Demydov, W. Zhang and Z. S. Petrovic, J. Polym. Environ., 2002, 10, 49–52 CrossRef CAS; (b) Z. S. Petrovic, A. Go, I. Javni, I. Cvetkovi and D. P. Hong, Polym. Int., 2008, 57, 275–281 CrossRef CAS.
- Z. S. Petrovic, W. Zhang and I. Javni, Biomacromolecules, 2005, 6, 713–719 Search PubMed.
- D. D. Andejelkovic, B. Min, D. Ahn and R. C. Larock, J. Agric. Food Chem., 2006, 54, 9535–9543 CrossRef CAS.
- Industrial polyether–polyols are derived essentially from ethylene and propylene oxide and used to soften or tighten polyurethanes: (a) M. Ionescu, Z. S. Petrovic and X. Wan, J. Polym. Environ., 2007, 15, 237–243 CrossRef CAS; (b) Y. Lu and R. C. Larock, Biomacromolecules, 2008, 9, 3332–3340 CrossRef CAS; (c) Y. Lu and R. C. Larock, Biomacromolecules, 2007, 8, 3108–3114 CrossRef CAS; (d) X Pan and D. C. Webster, Macromol. Rapid Commun. , 2011, 32, 1324–1330 Search PubMed.
- J. P. Jain, M. Sokolski, N. Kumar and A. J. Domb, Polym. Rev., 2008, 48, 156–191 Search PubMed.
- (a) R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097–2124 CrossRef CAS; (b) S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS.
- (a) M. Bartra and J. Vilarrasa, J. Org. Chem., 1991, 56, 5132–5138 CrossRef CAS; (b) G. E. Keck, E. P. Boden and M. R. Wiley, J. Org. Chem., 1989, 54, 896–906 CrossRef CAS; (c) G. E. Keck, C. Sanchez and C. A. Wager, Tetrahedron Lett., 2000, 41, 8673–8676 CrossRef CAS; (d) G. Rousseau, Tetrahedron, 1995, 51, 2777–2849 CrossRef CAS; (e) C. J. Roxburgh, Tetrahedron, 1995, 51, 9767–9822 CrossRef CAS; (f) C. L. Ruddick, P. Hodge, A. Cook and A. J. McRiner, J. Chem. Soc., Perkin Trans. 1, 2002, 5, 629–637 Search PubMed; (g) E. P. Boden and G. E. Keck, J. Org. Chem., 1985, 50, 2394–2395 CrossRef CAS.
- A. Duda, A. Kowalski, S. Penczek, H. Uyama and S. Kobayashi, Macromolecules, 2002, 35, 4266–4270 CrossRef CAS.
- (a) I. van der Meulen, M. de Geus, H. Antheunis, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2008, 9, 3404–3410 CrossRef CAS; (b) H. Uyama, K. Takeya, N. Hoshi and S. Kobayashi, Macromolecules, 1995, 28, 7046–7050 CrossRef CAS; (c) K. S. Bisht, L. A. Anderson, R. A. Gross, D. L. Kaplan and G. Swift, Macromolecules, 1997, 30, 2705–2711 CrossRef CAS; (d) A. Taden, M. Antonietti and K. Landfester, Macromol. Rapid Commun., 2003, 24, 512–516 CrossRef CAS; (e) L. van der Mee, F. Helmich, R. De Bruijn, J. A. J. M. Vekemans, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2006, 39, 5021–5027 CrossRef CAS; (f) S. Namekawa, S. Suda, H. Uyama and S. Kobayashi, Int. J. Biol. Macromol., 1999, 25, 145 CrossRef CAS; (g) M. A. Veld, L. Fransson, A. R. A. Palmans, E. W. Meijer and K. Hult, ChemBioChem, 2009, 10, 1330–1334 Search PubMed; (h) M. L. Focarete, M. Scandola, A. Kumar and R. A. Gross, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 1721–1729 CrossRef CAS; (i) M. de Geus, I. van der Meulen, B. Goderis, K. Van Hecke, M. Dorschu, H. van der Werff, C. E. Koning and A. Heise, Polym. Chem., 2010, 1, 525–533 RSC; (j) M. Takwa, N. Simpson, E. Malmström, K. Hult and M. Martinelle, Macromol. Rapid Commun., 2006, 27, 1932–1936 CrossRef CAS; (k) B. Chen, J. Hu, E. M. Miller, W. Xie, M. Cai and R. A. Gross, Biomacromolecules, 2008, 9, 463–471 CrossRef CAS.
- Y. Nakayama, N. Watanabe, K. Kusaba, K. Sasaki, Z. Cai, T. Shiono and C. Tsutsumi, J. Appl. Polym. Sci., 2011, 121, 2098–2103 Search PubMed; Z. Zhong, P. J. Dijkstra and J. Feijen, Macromol. Chem. Phys., 2000, 201, 1329–1333 CrossRef CAS.
- R. Nomura, A. Ueno and T. Endo, Macromolecules, 1994, 27, 620–621 CrossRef CAS.
- Z. Jedlinski, M. Juzwa, G. Adamus and M. Kowalczuk, Macromol. Chem. Phys., 1996, 197, 2923–2929 Search PubMed.
- I. van. der Meulen, E. Gubbels, S. Huijser, R. Sablong, C. E. Koning, A. Heise and R. Duchateau, Macromolecules, 2011, 44, 4301–4305 Search PubMed.
- R. Slivniak and A. J. Domb, Biomacromolecules, 2005, 6, 1679–1688 CrossRef CAS.
- D. Quinzler and S. Mecking, Chem. Commun., 2009, 5400–5402 RSC.
- F. C. Naughton, J. Am. Oil Chem. Soc., 1974, 51, 65–71 CrossRef CAS.
- J. Trzaskowski, D. Quinzler, C. Bährle and S. Mecking, Macromol. Rapid Commun., 2011, 32, 1352–1356 Search PubMed.
- W. H. Lu, J. A. Ness, W. C. Xie, X. Y. Zhang, J. Minshull and R. A. Gross, J. Am. Chem. Soc., 2010, 132, 15451–15455 CrossRef CAS.
- C. Liu, F. Liu, J. Cai, W. Xie, T. E. Long, R. S. Turner, A. Lyons and R. A. Gross, Biomacromolecules, 2011, 12, 3291–3298 Search PubMed.
- O. T. ürünç, L. Montero de Espinosa and M. A. R. Meier, Macromol. Rapid Commun., 2011, 32, 1357–1361 Search PubMed.
- G. Woods, in The ICI Polyurethanes Book, Wiley, New-York, 2nd edn, 1990 Search PubMed.
- (a) M. N. Satheesh Kumar, K. S. Manjula and Siddaramaiah, J. Appl. Polym. Sci., 2007, 105, 3153–3161 Search PubMed; (b) Y. Lu, L. Tighzert, F. Berzin and S. Rondot, Carbohydr. Polym., 2005, 61, 174–182 CrossRef CAS; (c) D. P. Pfister, Y. Xia and R. C. Larock, ChemSusChem, 2011, 4, 703–717 Search PubMed; (d) Z. S. Petrovic, Polym. Rev., 2008, 48, 109–155 Search PubMed; (e) Y. Xia and R. C. Larock, Macromol. Rapid Commun., 2011, 32, 1331–1337 Search PubMed; (f) C. N. D. Neumann, W. D. Bulach, M. Rehahn and R. Klein, Macromol. Rapid Commun., 2011, 32, 1373–1378 Search PubMed.
- D. V. Palaskar, A. Boyer, E. Cloutet, C. Alfos and H. Cramail, Biomacromolecules, 2010, 11, 1202–1211 CrossRef CAS.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205–2213 RSC.
- (a) W. Amass, A. Amass and B. Tighe, Polym. Int., 1998, 47, 89–144 CrossRef CAS; (b) A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- Selected reviews: (a) J.-F. Carpentier, Macromol. Rapid Commun., 2010, 31, 1696–1705 CrossRef CAS; (b) A. P. Dove, Handbook of Ring-Opening Polymerization, 2009, p. 357 Search PubMed; (c) J. H. Khan, F. Schue and G. A. George, Polym. Int., 2009, 58, 296–301 CrossRef CAS; (d) A.-C. Albertsson and R. K. Srivastava, Adv. Drug Delivery Rev., 2008, 60, 1077–1093 CrossRef CAS; (e) N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS; (f) S. Agarwal, C. Mast, K. Dehnicke and A. Greiner, Macromol. Rapid Commun., 2000, 21, 195–212 CrossRef CAS; (g) N. Spassky, P. Dumas, A. Le Borgne, A. Momtaz and M. Sepulchre, Bull. Soc. Chim. Fr., 1994, 131, 504–514 Search PubMed.
- (a) C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC; (b) G. L. Baker, E. B. Vogel and M. R. Smith, Polym. Rev., 2008, 48, 64–84 Search PubMed; (c) X. D. Lou, C. Detrembleur and R. Jerome, Macromol. Rapid Commun., 2003, 24, 161–172 CrossRef CAS; (d) M. Vert, Polym. Degrad. Stab., 1998, 59, 169–175 CrossRef CAS; (e) K. M. Benabdillah, J. Coudane, M. Boustta, R. Engel and M. Vert, Macromolecules, 1999, 32, 8774–8780 CrossRef CAS; (f) K. Marcincinova-Benabdiillah, M. Boustta, J. Coudane and M. Vert, Biomacromolecules, 2001, 2, 1279–1284 CrossRef CAS; (g) I. M. Pinilla, M. B. Martinez and J. A. Galbis, Carbohydr. Res., 2003, 338, 549–555 CrossRef CAS; (h) O. Coulembier, L. Mespouille, J. L. Hedrick, R. M. Waymouth and P. Dubois, Macromolecules, 2006, 39, 4001–4008 CrossRef CAS; (i) P. G. Parzuchowski, M. Grabowska, M. Trynowski and G. Rokicki, Macromolecules, 2006, 39, 7181–7186 CrossRef CAS; (j) C. E. Fernandez, M. Mancera, E. Holler, J. A. Galbis and S. Munoz-Guerra, Polymer, 2006, 47, 6501–6508 CrossRef CAS; (k) T. Q. Liu, T. L. Simmons, D. A. Bohnsack, M. E. Mackay, M. R. Smith and G. L. Baker, Macromolecules, 2007, 40, 6040–6047 CrossRef CAS; (l) A. F. Haider and C. K. Williams, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2891–2896 CrossRef CAS; (m) H. Urakami and Z. Guan, Biomacromolecules, 2008, 9, 592–597 CrossRef CAS; (n) J. R. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC.
- H. D. K. Drew and W. N. Haworth, J. Chem. Soc., 1927, 775–777 RSC.
- M. Masato, JP 2002167430 A2 2002611, Japan, 2002.
- M. Tang, A. J. P. White, M. M. Stevens and C. K. Williams, Chem. Commun., 2009, 941–943 RSC.
- J. R. Lowe, M. T. Martello, W. B. Tolman and M. A. Hillmyer, Polym. Chem., 2011, 2, 702–708 RSC.
- C. C. C. R. de Carvalho and M. M. R. da Fonseca, Food Chem., 2006, 95, 413–422 CrossRef CAS.
- K. M. Stridsberg, M. Ryner and A.-C. Albertsson, Adv. Polym. Sci., 2002, 157, 41–65 CAS.
- (a) C. L. Wanamaker, L. E. O'Leary, N. A. Lynd, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2007, 8, 3634–3640 CrossRef CAS; (b) C. L. Wanamaker, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 443–448 CrossRef CAS.
- The early discoveries in this alternating copolymerization process have been recently covered in several reviews and will not be duplicated herein. See for instance: (a) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618 CrossRef CAS; (b) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS; (c) E. Brulé, J. Guo, G. W. Coates and C. M. Thomas, Macromol. Rapid Commun., 2011, 32, 169–185 Search PubMed.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS.
- P. Brignou, M. Priebe Gil, O. Casagrande, J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2010, 43, 8007–8017 CrossRef CAS.
- C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011 Search PubMed DOI: 10.1038/ncomms1596.
| This journal is © The Royal Society of Chemistry 2012 |