
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into Ni active site coordination in nickel–manganese spinels for methanol electrooxidation catalysis†
Ruiying Guoa,
Chunru Liua,
Yun Yang *b,
Shuli Wanga and
Ligang Feng*a
*b,
Shuli Wanga and
Ligang Feng*a
aSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, P. R. China. E-mail: ligang.feng@yzu.edu.cn; fenglg11@gmail.com
bNanomaterials and Chemistry Key Laboratory, Wenzhou University, Wenzhou, P. R. China. E-mail: bachier@163.com
First published on 26th June 2025
Abstract
As non-precious catalysts, Ni-based catalysts play a significant role in methanol oxidation for energy conversion technologies. At the same time, the effect of the complicated chemical environment on catalytic efficiency remains unclear. Here, the coordination environment of Ni active sites in spinel nickel–manganese (NiMn2O4 and MnNi2O4) is investigated as a platform to elucidate the correlation with catalytic performance in methanol electro-oxidation. The occupation of Ni2+ ions in these structures modulates the intrinsic activity of Ni active sites in NiMn spinels, resulting in different catalytic mechanisms and intrinsic active site efficiency, although they have similar morphology and structure. The high-symmetry NiO6 octahedral structure in inverse spinel MnNi2O4 exhibits superior catalytic performance and stability compared to the NiO4 tetrahedral structure in normal NiMn2O4 spinel. Specifically, at 1.50 V vs. RHE, the MnNi2O4 inverse spinel delivers mass activity and specific activity for methanol oxidation that are 1.9 and 3.5 times those of the normal NiMn2O4 spinel, respectively. Furthermore, it also maintains a stable current density of 33.5 mA cm−2 at 1.56 V vs. RHE for 25 hours. Theoretical calculations reveal that Ni sites in MnNi2O4 exhibit a significantly lower activation energy barrier and enhanced CO anti-poisoning capability compared to those in NiMn2O4. The Ni site-dependent coordination environment in spinel structures provides useful insights into catalyst development and the methanol oxidation mechanism.
Introduction
Electrocatalytic water splitting holds a pivotal position in sustainable hydrogen generation.1–3 Nevertheless, the oxygen evolution reaction (OER), a multielectron transfer process, suffers from sluggish kinetics and high energy demands.4–6 Recent attention has shifted toward the anodic oxidation of small organic molecules to facilitate hydrogen production, with a particular focus on the methanol oxidation reaction (MOR).7–9 This is mainly due to its low overpotential (0.016 V) and high energy efficiency (∼60%), making it a promising alternative.10,11To fulfil efficient MOR catalysis, attention is directed to non-noble metal catalysts since platinum group metals (PGMs) are restricted by high cost and susceptibility to CO poisoning.12,13 Ni-based materials, in particular, show great promise as MOR catalysts, including Ni-hydroxides, perovskites, and layered double hydroxides.14,15 During the methanol reaction, the active high-valent NiOOH intermediate is inevitably formed from the pre-catalyst and serves as a chemical oxidant that converts methanol to carbon dioxide while being reduced to Ni(OH)2.16 Tuning the coordination environment of Ni sites has proven to be an effective strategy to improve catalytic efficiency, as it not only determines the electronic structures of Ni active sites but also governs the reaction mechanisms.17,18 For example, Mo-doped Ni(OH)2 with ultralow Ni–Ni coordination enhanced the interaction between OH− and CH3OH at the Ni active center, thereby promoting both the kinetics and durability of methanol electrocatalysis.19 A stable NiSnPH perovskite hydroxide, obtained via selective Sn leaching, optimized the local coordination environment of NiOOH active sites, resulting in enhanced selectivity for methanol oxidation to formate.20 Among Ni-based catalysts, particularly NiMn-based spinel oxides were effective for the MOR due to their stable crystal structures, controllable compositions, and polymetallic synergies.21,22 For instance, multi-walled carbon nanotube (MWCNT)- or reduced graphene oxide (rGO)-supported MnNi2O4 showed favourable MOR activity benefiting from the binary transition metal oxides' synergism.23,24 While Ni sites in the spinel structure serve as active sites to trigger methanol oxidation, their precise catalytic mechanism remains unclear. Actually, Ni sites would be an ideal platform to probe intrinsic activity and reveal the effect of the Ni coordination environment in the spinels on catalytic performance. For example, the Ni active sites in spinel nickel–manganese (NiO4 tetrahedral structure for NiMn2O4 and NiO6 octahedral structure for MnNi2O4) could be used to elucidate the connection between Ni site coordination and catalytic performance. However, to the best of our knowledge, no relevant reports have specifically addressed this concern.
In this study, we take NiMn-based spinel catalysts (NiMn2O4 and MnNi2O4) as a platform to explore the correlation between the Ni coordination environment in various spinel structures and catalytic performance for the MOR. MnNi2O4 spinel, with Ni sites in an octahedral configuration, exhibits significantly enhanced performance compared to the normal NiMn2O4 spinel. Specifically, at 1.50 V vs. RHE, the MnNi2O4 inverse spinel delivers mass activity and specific activity that are 1.9 and 3.5 times those of the normal NiMn2O4 spinel, respectively. Furthermore, it also maintains a stable current density of 33.5 mA cm−2 at 1.56 V vs. RHE for 25 hours. This enhancement stems from the presence of highly active Ni sites in the NiO6 octahedral coordination environment of MnNi2O4, contributing to excellent intrinsic catalytic efficiency. Theoretical analysis demonstrates a lower activation threshold and superior CO anti-poisoning ability of Ni sites in MnNi2O4 compared to NiMn2O4. The Ni site-dependent coordination environment in spinel structures provides new insights into catalyst development and the methanol oxidation mechanism.
Experimental
Materials fabrication
In the synthesis of MnNi2O4 spinel (Fig. S1†), 1.49 g (5.98 mmol) of Ni(CH3COO)2·4H2O, 1.47 g (5.95 mmol) of Mn(CH3COO)2·4H2O, and 80 mL of ultrapure water were added into a 100 mL beaker under magnetic stirring. The pH of the solution was adjusted to 10 by the dropwise addition of 5 mol per L NaOH solution, followed by stirring at room temperature for about 15 minutes. Then, the mixture was transferred to a 150 mL Teflon-lined autoclave and heated at 150 °C for 12 hours. After natural cooling to room temperature, the synthesized product was collected by centrifuging, washed several times with ultrapure water and anhydrous ethanol, and then dried under vacuum at 60 °C for 8 hours. Finally, the dried powder was annealed in air at 800 °C for 2 hours and cooled to room temperature to obtain MnNi2O4 spinel. The NiMn2O4 spinel was synthesized by a similar method with different molar ratios of nickel acetate tetrahydrate and manganese acetate tetrahydrate. The NiO and Mn2O3 samples were fabricated for performance comparisons (Table S1†).Materials characterization
The crystal structure was probed using powder X-ray diffraction (XRD) patterns which were obtained on a Bruker D8 Advance with a Cu Kα radiation source (λ = 1.5405 Å) under operating conditions of 40 kV and 40 mA, with a scan rate of 5° min−1. The morphology of the NiMn2O4 and MnNi2O4 spinels was analyzed by using a scanning electron microscope (SEM, Hitachi, S-4800 II, Japan) and transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) measurements (TECNAI G2 operating at 300 kV). The energy-dispersive X-ray detector spectrum (EDS) was obtained on a TECNAI G2 transmission electron microscope equipped with an EDXA detector. The chemical state of the surface was probed using X-ray photoelectron spectroscopy (XPS) with a Kα radiation source.All the electrochemical tests were done using a Bio-Logic VSP electrochemical workstation (Bio-Logic Co., France) in a conventional three-electrode system. The working electrode was obtained by dropping catalyst ink over a glassy carbon electrode (3.0 mm diameter, 0.07 cm2). The catalyst ink was prepared by ultrasonically mixing 4.5 mg of catalyst, 0.5 mg of carbon black, 950 μL of ethanol, and 50 μL of Nafion solution. 10 μL of the catalyst ink was coated over the electrode and dried naturally before electrochemical testing. A graphite rod and saturated calomel electrode (SCE, Hg/Hg2Cl2) were used as the counter and reference electrodes, respectively. All the potentials were converted to the reversible hydrogen electrode (RHE).
Cyclic voltammetry (CV) tests were conducted in 1 M KOH solution or 1 M KOH + 1 M CH3OH solution with a voltage range of 1.06–1.66 V vs. RHE at 50 mV s−1. The electrochemical impedance spectra (EIS) were recorded at frequencies ranging from 1000 kHz to 10 mHz with an amplitude of 5 mV in the above electrode system. The electrochemical surface area (ECSA) was calculated using the redox peak of Ni3+/Ni2+ in the backward scan of CV curves in the KOH solution. The charge during the cathodic reduction peak of Ni3+/Ni2+ was calculated using a factor of 0.257 mC cm−2 (0.257 mC cm−2 is the charge required to form a monolayer of Ni2+). The chronoamperometry (CA) experiment was performed at 1.56 V vs. RHE for 25 hours in 1 M KOH + 1 M CH3OH solution. The materials, some formulae for the performance calculation, and the theoretical analysis can be found in the ESI.†
Results and discussion
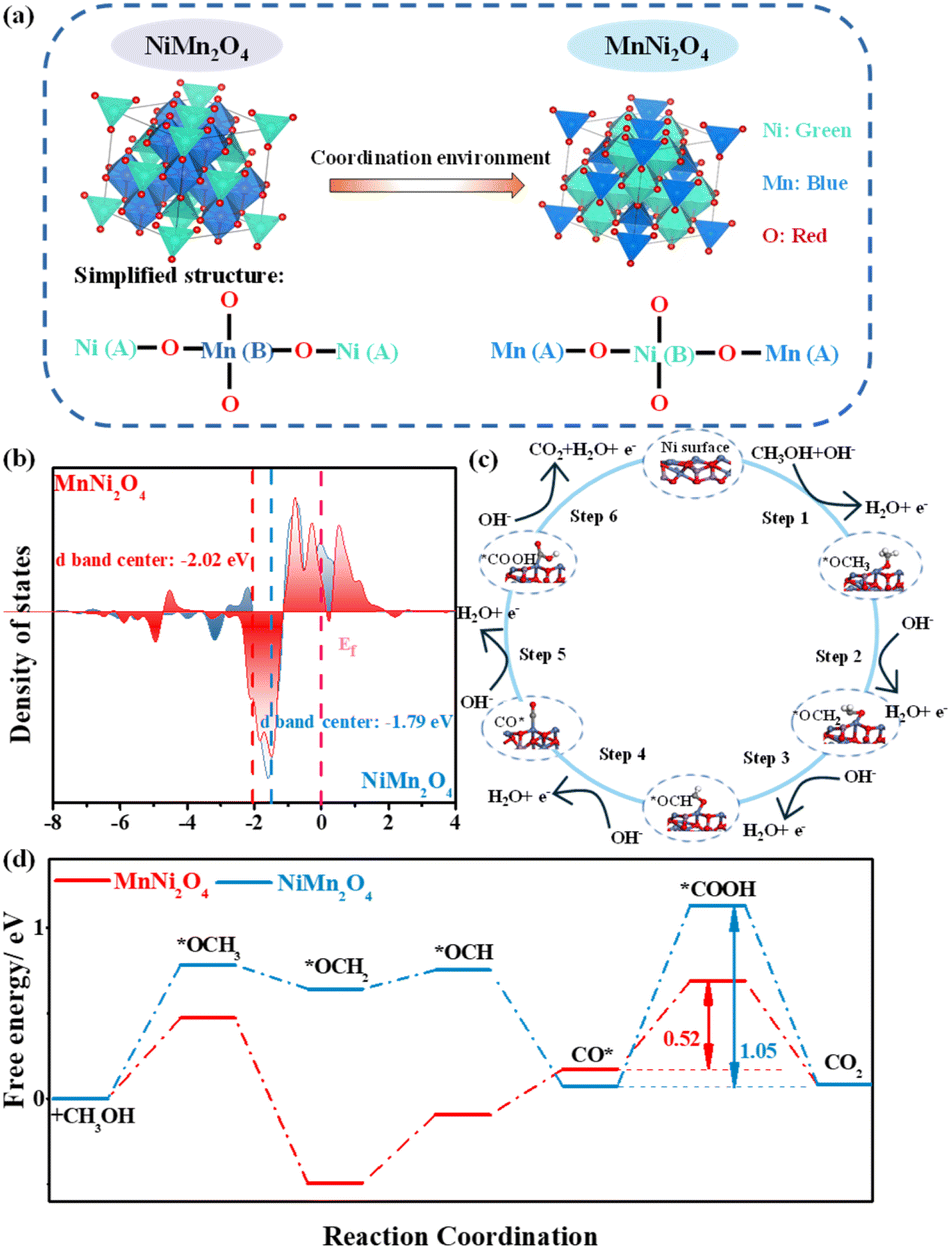
The three-dimensional (3D) spinel structures of NiMn2O4 and MnNi2O4 are illustrated in Fig. 1a. Both crystallize in the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and feature a characteristic structure where metal cations (Ni and Mn) are distributed across tetrahedral and octahedral coordination sites. The coordination structure in these compounds varies slightly with oxidation states due to their mixed valency. In NiMn2O4, Ni2+ ions typically occupy tetrahedral (A) sites, where each metal ion is coordinated to four oxygen atoms, while Mn3+ ions reside in octahedral (B) sites, coordinated to six oxygen atoms. These metal cations are bridged by oxygen ions (O2−). In the simplified structure, the A sublattice contains Ni2+ ions in tetrahedral sites (A), while the B sublattice contains Mn3+ ions in octahedral sites (B). MnNi2O4 adopts a similar spinel-type structure to NiMn2O4 but exhibits an inverse cation distribution because of its different stoichiometry. In this simplified structure, Ni2+ ions preferentially occupy octahedral (B) sites, while Mn4+ ions reside in tetrahedral (A) sites.
m space group and feature a characteristic structure where metal cations (Ni and Mn) are distributed across tetrahedral and octahedral coordination sites. The coordination structure in these compounds varies slightly with oxidation states due to their mixed valency. In NiMn2O4, Ni2+ ions typically occupy tetrahedral (A) sites, where each metal ion is coordinated to four oxygen atoms, while Mn3+ ions reside in octahedral (B) sites, coordinated to six oxygen atoms. These metal cations are bridged by oxygen ions (O2−). In the simplified structure, the A sublattice contains Ni2+ ions in tetrahedral sites (A), while the B sublattice contains Mn3+ ions in octahedral sites (B). MnNi2O4 adopts a similar spinel-type structure to NiMn2O4 but exhibits an inverse cation distribution because of its different stoichiometry. In this simplified structure, Ni2+ ions preferentially occupy octahedral (B) sites, while Mn4+ ions reside in tetrahedral (A) sites.
 | ||
| Fig. 1 (a) Structure models of NiMn2O4 and MnNi2O4 spinels: the transformation of Ni2+ from tetrahedral to octahedral coordination. (b) The partial density of states (PDOS) of NiMn2O4 and MnNi2O4 surfaces. (c) Diagram of the reaction process of the MOR on the MnNi2O4–Ni site. (d) The Gibbs free energy diagram for the MOR of NiMn2O4 and MnNi2O4. | ||
During fabrication, as Ni is added, Ni2+ ions progressively replace Mn3+ ions at octahedral positions, facilitating the formation of the inverse spinel structure. Upon transformation of NiMn2O4 into MnNi2O4, the MnNi2O4 inverse spinel phase with a high-coordination NiO6 octahedral structure exhibits enhanced crystal symmetry and stabilizes the spinel structure. Moreover, the increased number of Ni2+ ions in octahedral geometry generates more active Ni oxidation sites. Conversely, the low-coordination NiO4 tetrahedral structure in NiMn2O4 destabilizes the spinel structure. Thus, the occupation of Ni2+ ions in these structures modulates the intrinsic activity of Ni active sites in NiMn spinels.25
Given the superior MOR performance of MnNi2O4 to NiMn2O4 (vide infra), density functional theory (DFT) calculations were performed on optimized surface models of Ni and Mn sites in MnNi2O4 to evaluate the catalytic efficiency of distinct active sites (Fig. S2–S4†).26 Both Ni2+ ions preferentially occupied octahedral (B) sites, forming a Mn4+[Ni22+]O42− valence structure.27 The octahedral NiO6 structure showed significantly higher MOR performance than the MnO4 structure, consistent with previous studies (Fig. S5†).28 To elucidate the influence of the Ni site coordination environment on electrocatalytic performance, we compared the Ni d-band centers of MnNi2O4 and NiMn2O4. The Ni d-band center in MnNi2O4 (−2.02 eV) was more negative than that in NiMn2O4 (−1.79 eV), moving further from the Fermi level owing to the robust synergistic effect between Ni d orbitals and O p orbitals (Fig. 1b). This shift enhances charge transfer efficiency and reduces intermediate adsorption.29 The carbon monoxide intermediate (CO*) adsorption energy (ECO*) is also a key descriptor of MOR catalytic performance. MnNi2O4 exhibited a lower ECO* value (−1.13 eV) relative to NiMn2O4 (−1.24 eV), indicating enhanced CO poisoning resistance capacity (Fig. S6†). The widely accepted mechanism for the MOR catalyzed by Ni-based catalysts follows a stepwise process (Fig. 1c): Ni sites first serve as active centers for methanol adsorption, followed by a sequence of deprotonation steps leading to the formation of multiple bonded intermediates, all of which are formed at Ni sites and eventually convert to CO2.30 The overall MOR pathway can be described as follows: *OCH3 → *OCH2 → *OCH → CO* → *COOH → CO2. Gibbs free energy analysis revealed that the CO* → *COOH conversion was the rate-determining step (RDS) in the MOR pathway (Fig. 1d). The energy barrier for this step in MnNi2O4 (0.52 eV) was significantly lower than that in NiMn2O4 (1.05 eV), indicating more favorable MOR kinetics. Consequently, changes in the coordination environment of Ni active sites not only modulate MOR activation energies but also influence CO poisoning resistance.
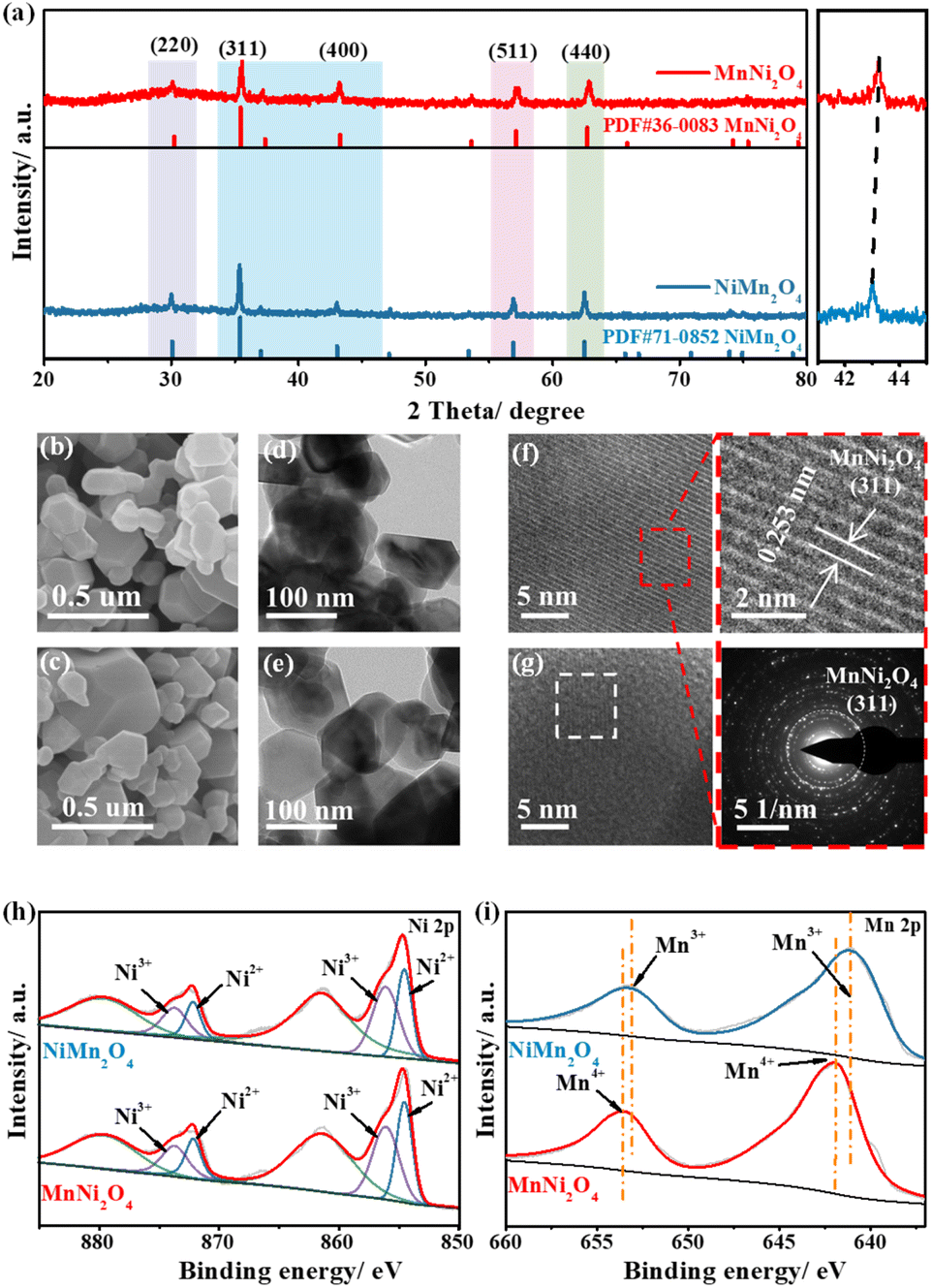
The crystal structures of NiMn2O4 and MnNi2O4 were confirmed by powder X-ray diffraction (XRD), with both belonging to the face-centered cubic (fcc) structure and Fdm (227) space group (Fig. 2a). The characteristic peaks of MnNi2O4 closely resembled those of NiMn2O4, but were slightly shifted to high angles at 43.2°, 57.1°, and 62.7°, indicating that Ni2+ ions successfully occupied octahedral positions. Scanning electron microscopy (SEM) images revealed that both NiMn2O4 and MnNi2O4 exhibited irregular aggregated particles (Fig. 2b and c). Transmission electron microscopy (TEM) images showed a similar morphology, with irregular hexagonal planes observed for both samples (Fig. 2d and e). High-resolution TEM (HRTEM) images showed distinct lattice fringes with a d-spacing of 0.253 nm, assigned to the (311) planes of MnNi2O4 and NiMn2O4 spinel structures (Fig. 2f and g). Energy dispersive X-ray spectroscopy (EDS) confirmed the Ni, Mn, C, and O within the samples (Fig. S7†). The elemental mapping images showed the uniform distribution of Ni, Mn, and O elements in the MnNi2O4 spinel (Fig. S8†).
 | ||
| Fig. 2 (a) XRD patterns of NiMn2O4 and MnNi2O4. (b) SEM image, (d) TEM image, (f) HRTEM image and SAED pattern of MnNi2O4. (c) SEM image, (e) TEM image, and (g) HRTEM image of NiMn2O4. Ni 2p (h) and Mn 2p XPS spectra (i) of NiMn2O4 and MnNi2O4. | ||
Though they have similar morphology and structure, variations in the coordination environment likely alter the chemical states of Ni sites. X-ray photoelectron spectroscopy (XPS) was thus conducted to explore the chemical states. The XPS survey spectra revealed the presence of C, O, Ni, and Mn, with the binding energy calibrated to the C–C bond reference peak at 284.8 eV (Fig. S9†). The high-resolution Ni 2p spectrum displayed two distinct peaks, corresponding to Ni 2p1/2 and Ni 2p3/2 (Fig. 2h). MnNi2O4 showed prominent double peaks of Ni2+ located at 854.5 and 872.1 eV, while the weak double peaks at 856.1 and 873.7 eV were attributed to Ni3+ (Table S2†). The Ni peak shifted from 854.8 eV in NiMn2O4 to 854.5 eV in MnNi2O4, indicating that more Ni2+ species occupied the octahedral sites.31 The Ni3+/Ni2+ intensity ratio increased from NiMn2O4 (0.89) to MnNi2O4 (1.38), which was attributed to easier surface oxidation and an increased oxidation state at the octahedral sites (Table S2†). For the Mn 2p spectrum, the deconvoluted spectrum of MnNi2O4 displayed two major peaks at 644.8 and 656.4 eV, corresponding to Mn4+, whereas for NiMn2O4, the peak positions were shifted to lower energies at 642.7 and 654.3 eV, assigned to Mn3+ (Fig. 2i and Table S3†).32 The high-resolution O 1s spectrum exhibited three characteristic peaks at 529.8, 531.0, and 532.4 eV, assigned to metal–oxygen bonds, adsorbed hydroxide groups, and adsorbed water molecules, respectively (Fig. S10†).
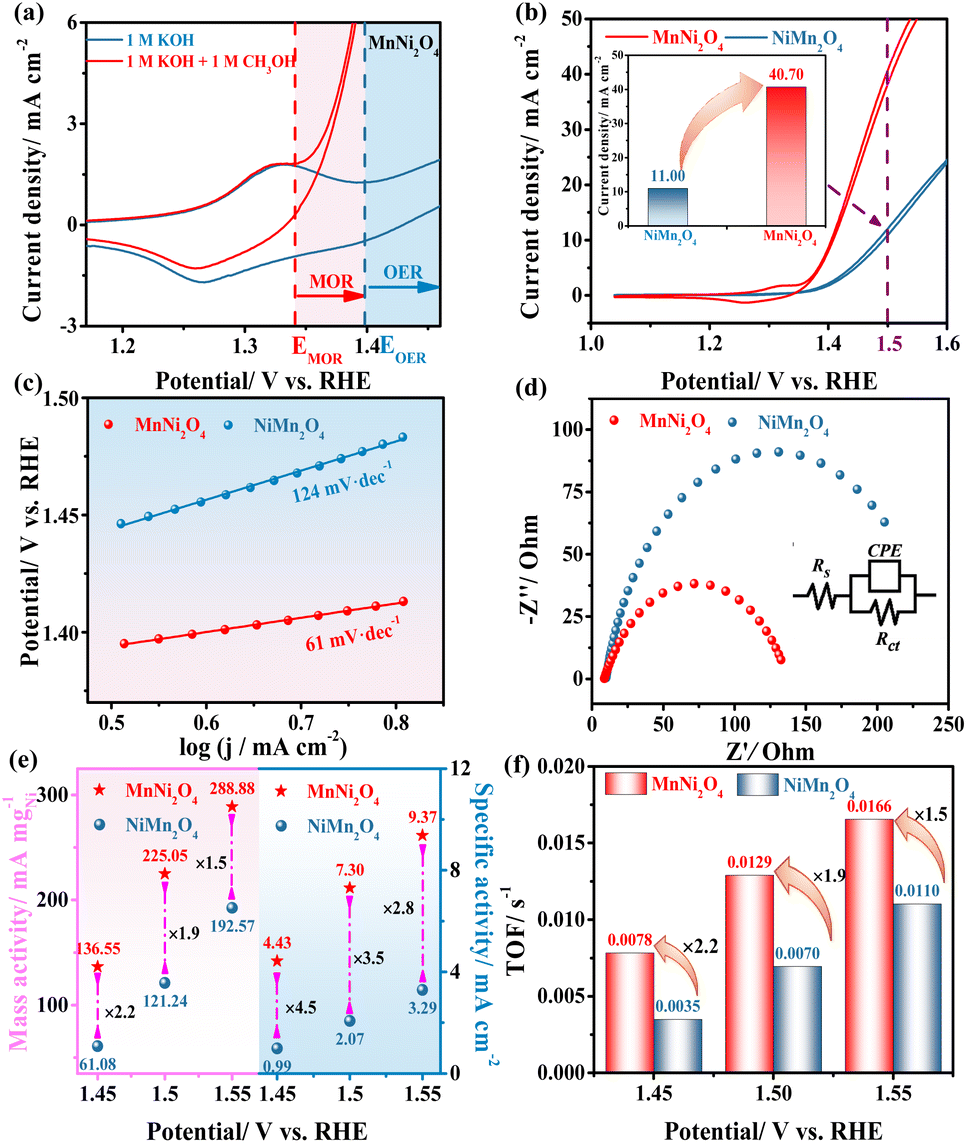
To investigate the catalytic performance in different coordination environments, electrochemical experiments were conducted on the synthesized catalysts for the MOR. CV measurements of MnNi2O4, NiMn2O4, NiO, and Mn2O3 electrodes in 1 M KOH with and without 1 M CH3OH indicated that Ni-based electrodes exhibited higher MOR performance, while the Mn2O3 electrode showed negligible activity, suggesting that Ni sites served as the active sites for the MOR (Fig. S11†). For MnNi2O4, the oxidation peak was assigned to the Ni2+/Ni3+ redox transition, followed by a rapid increase in the anode current density for both OER and MOR processes (Fig. 3a). The onset potential for the MOR was more negative relative to the OER, and the methanol oxidation closely coincided with the redox peak, while water oxidation occurred after the Ni3+ oxidation, suggesting the high energy efficiency of the MOR.33 By comparing the MOR performance of MnNi2O4 and NiMn2O4 electrodes, MnNi2O4 displayed a much higher forward current density of 40.7 mA cm−2 at 1.50 V vs. RHE compared to NiMn2O4 (11.0 mA cm−2) (Fig. 3b). Among state-of-the-art non-precious catalysts for methanol oxidation, the MnNi2O4 spinels ranked the top in their family (Table S4†).
 | ||
| Fig. 3 (a) Cyclic voltammetry curves of MnNi2O4 measured in 1 M KOH with and without 1 M CH3OH at 50 mV s−1. (b) CV curves of NiMn2O4 and MnNi2O4 measured in 1 M KOH with 1 M CH3OH at 50 mV s−1 (inset: current density at 1.50 V vs. RHE). (c) Tafel plots of NiMn2O4 and MnNi2O4. (d) Nyquist plots of NiMn2O4 and MnNi2O4 at 1.56 V vs. RHE (inset: an equivalent circuit model used for fitting the EIS data). (e) Mass activity and specific activity. (f) TOF of NiMn2O4 and MnNi2O4 at 1.45, 1.50, and 1.55 V vs. RHE. | ||
The MOR kinetics were evaluated by using the Tafel slope and electrochemical impedance spectroscopy (EIS). MnNi2O4 exhibited a lower Tafel slope of 61 mV dec−1 compared to NiMn2O4 (124 mV dec−1) (Fig. 3c), indicating different catalytic mechanisms, namely, Eley–Rideal (E–R) and Langmuir–Hinshelwood (L–H) mechanisms, respectively.34 Additionally, the E–R mechanism for the MnNi2O4 electrode supported the notion that the absorbed CO* interacted directly with OH− from the alkaline solution and was subsequently oxidized to CO2, which was consistent with the reaction mechanism discussed earlier. The charge transfer resistance (Rct) for the MnNi2O4 electrode was 128 Ω, much lower than that of NiMn2O4 (253 Ω) (Fig. 3d and Table S5†), indicating faster reaction kinetics due to the highly active octahedrally occupied Ni2+ in MnNi2O4. The electrochemical stability of the as-prepared catalysts was assessed through a long-term chronoamperometric (CA) test conducted at 1.56 V vs. RHE for 25 hours (Fig. S12†). Initially, the current density of both MnNi2O4 and NiMn2O4 declined rapidly due to the diffusion-controlled MOR, but MnNi2O4 maintained a higher current density of 33.5 mA cm−2 over time than NiMn2O4, indicating good stability for the MOR. Moreover, the structure and morphology of MnNi2O4 catalysts after the stability test exhibited no evident changes, confirming their good anti-corrosion ability for long-term operation (Fig. S13†).
Since MnNi2O4 contained two Ni sites, a fair comparison of intrinsic catalytic activity was necessary. To verify the correlation between the oxidation equivalence of Ni active sites and intrinsic catalytic activity for the MOR, commonly used methods for intrinsic activity comparison, including mass activity (MA), specific activity (SA), and turnover frequency (TOF), were employed. By normalizing the current to the Ni mass amount on the electrode, MnNi2O4 exhibited enhanced electrocatalytic mass activity compared to NiMn2O4 (Fig. S14a†).35,36 The electrochemical active surface area (ECSA) was calculated from the characteristic Ni redox peaks (Table S6†). MnNi2O4 showed a higher ECSA value of 0.39 cm2 than NiMn2O4 (0.37 cm2), indicating slightly greater exposure to active Ni species. More significantly, when normalizing the current to the ECSA, MnNi2O4 still showed higher specific activity than NiMn2O4 (Fig. S14b†). The typical performance comparison for both mass activity and specific activity at 1.45, 1.50, and 1.55 V vs. RHE is shown in Fig. 3e and Table S6.† Specifically, at 1.50 V vs. RHE, the mass and specific activities of MnNi2O4 were 1.9 and 3.5 times those of the NiMn2O4 electrode, respectively. Assuming that all the Ni sites were in the active phase, the TOF was calculated (Fig. S15†). At 1.45, 1.50, and 1.55 V vs. RHE, the MnNi2O4 electrode exhibited values of 0.0078, 0.0129, and 0.0165 s−1, respectively, about 2.2, 1.9, and 1.5 times those of NiMn2O4 (Fig. 3f and Table S7†). These results conclusively showed that Ni sites in MnNi2O4 have significantly higher intrinsic activity than those of NiMn2O4.
As mentioned in the above theoretical calculation, the Mn sites are not active for the MOR, but the different Mn oxidation states would influence the performance, as they contribute to the different coordination states of Ni active sites. Additionally, the XPS analysis revealed that the high-valent Mn4+ in MnNi2O4 induced a stronger electronic interaction with Ni sites, promoting the formation of high-valence Ni species and further improving the MOR catalytic performance. Furthermore, it should be pointed out that due to the surface oxidation driven by high potentials, the valence state of Ni sites is increased as can be seen in the Ni redox peaks. In the current study, we revealed the effect of the coordination environment of Ni active sites in nickel–manganese spinels on methanol electrooxidation. It would be more interesting to correlate the surface chemical state changes with the catalytic ability in future studies.
Conclusions
In summary, the Ni active sites in nickel–manganese spinels were probed for methanol oxidation catalysis. Though they have similar morphology and structure, changes in the coordination environment of Ni sites influence their catalytic performance in the MOR. The inverse spinel MnNi2O4, characterized by Ni active sites in octahedral coordination, offers a high density of exposed Ni sites with superior intrinsic activity, thereby achieving exceptional catalytic performance and durability. Notably, MnNi2O4 exhibits mass and specific activities more than twice those of NiMn2O4. Theoretical calculations reveal a significantly lower activation energy barrier and enhanced CO anti-poisoning capability of Ni sites in MnNi2O4 compared to NiMn2O4. The dependence of catalytic efficiency on the coordination environment of Ni sites provides critical insights into the catalytic mechanism of the methanol oxidation reaction.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ruiying Guo: conceptualization, methodology, investigation, validation, writing the original draft. Chunru Liu: formal analysis, investigation, validation. Yun Yang: formal analysis, investigation. Shuli Wang: methodology, investigation, and formal analysis. Ligang Feng: writing-review & editing, sources, and funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22272148, and 22202172).Notes and references
- Y. Guo, X. Zou, X. Wei, W. Bao, J. Zhang, J. Han and F. Jia, Chin. J. Struct. Chem., 2024, 43, 100206 CAS.
- C. Liu and L. Feng, Chin. J. Struct. Chem., 2023, 42, 100136 CAS.
- A. S. Jamadar, R. Sutar, S. Patil, R. Khandekar and J. B. Yadav, Mater. Rep.: Energy, 2024, 4, 100283 CAS.
- S. Pan, Z. Yang and F. Luo, Chin. J. Struct. Chem., 2024, 43, 100373 CAS.
- S. Pan, Z. Ma, W. Yang, B. Dongyang, H. Yang, S. Lai, F. Dong, X. Yang and Z. Lin, Mater. Rep.: Energy, 2023, 3, 100212 CAS.
- Y. Lim, S. Surendran, W. So, S. Shanmugapriya, C. Jo, G. Janani, H. Choi, H. S. Han, H. Choi, Y.-H. Yun, T.-H. Kim, M.-J. Kim, K. Jin, J. K. Kim and U. Sim, Mater. Chem. Front., 2023, 7, 5843–5857 RSC.
- D. Yan, C. Mebrahtu, S. Wang and R. Palkovits, Angew. Chem., Int. Ed., 2023, 62, e202214333 CrossRef CAS PubMed.
- S. Ali, P. M. Ismail, F. Wahid, A. Kumar, M. Haneef, F. Raziq, S. Ali, M. Javed, R. U. Khan, X. Wu, H. Xiao, G. Yasin, L. Qiao and H. Xu, Fuel Process. Technol., 2022, 236, 107427 CrossRef CAS.
- G. Bharath, G. Karthikeyan, A. Kumar, J. Prakash, D. Venkatasubbu, A. Kumar Nadda, V. Kumar Gupta, M. Abu Haija and F. Banat, Appl. Energy, 2022, 318, 119244 CrossRef CAS.
- J. Li, F. Yang and L. Feng, Coord. Chem. Rev., 2025, 534, 216603 CrossRef CAS.
- Y. Kuang, W. Qiao, S. Wang, F. Yang and L. Feng, ACS Mater. Lett., 2024, 6, 1722–1731 CrossRef CAS.
- J. Li, M. Li, F. Yang and L. Feng, Chin. J. Chem., 2025, 43, 146–154 CrossRef CAS.
- M. Li, F. Yang, J. Chang, A. Schechter and L. Feng, Acta Phys.-Chim. Sin., 2023, 39, 2301005 Search PubMed.
- H. Cheng, B. Dong, Q. Liu and F. Wang, J. Am. Chem. Soc., 2023, 145, 26858–26862 CrossRef CAS PubMed.
- Y. Yi, J. Li and C. Cui, Chin. Chem. Lett., 2022, 33, 1006–1010 CrossRef CAS.
- C. Liu, F. Yang, A. Schechter and L. Feng, Adv. Sens. Energy Mater., 2023, 2, 100055 CrossRef.
- S. Li, R. Ma, J. Hu, Z. Li, L. Liu, X. Wang, Y. Lu, G. E. Sterbinsky, S. Liu, L. Zheng, J. Liu, D. Liu and J. Wang, Nat. Commun., 2022, 13, 2916 CrossRef CAS PubMed.
- G. Janani, S. Surendran, D. J. Moon, P. S. Ramesh, J. Y. Kim, Y. Lim, K. Veeramani, S. Mahadik, S. C. Jesudass, J. Choi, I. G. Kim, P. Jung, H. Choi, G. Kwon, K. Jin, J. k. Kim, Y. I. Park, J. Heo, K. Hong, Y. S. Kang and U. Sim, Adv. Sustainable Syst., 2024, 8, 2400059 CrossRef CAS.
- G. Fu, X. Kang, Y. Zhang, X. Yang, L. Wang, X.-Z. Fu, J. Zhang, J.-L. Luo and J. Liu, Nano-Micro Lett., 2022, 14, 200 CrossRef CAS PubMed.
- J. Shao, Y. Fang, X. Wu, M. I. Abdullah and Y. Tao, Nano Res., 2024, 17, 2388–2399 CrossRef CAS.
- H. Tian, X. Wang, W. Luo, R. Ma, X. Yu, S. Li, F. Kong, X. Cui and J. Shi, Chem. Sci., 2024, 15, 11013–11020 RSC.
- S. Cyril Jesudass, S. Surendran, G. Janani, T.-H. Kim, Y. I. Park and U. Sim, Appl. Surf. Sci., 2025, 688, 162352 CrossRef CAS.
- S. Hosseini, M. B. Askari and H. Beitollahi, Int. J. Hydrogen Energy, 2023, 48, 21240–21248 CrossRef CAS.
- M. B. Askari, P. Salarizadeh, A. Di Bartolomeo and F. Şen, Nanotechnology, 2021, 32, 325707 CrossRef CAS PubMed.
- T. Zhang, Y. Liu, L. Tong, J. Yu, S. Lin, Y. Li and H. J. Fan, ACS Nano, 2023, 17, 6770–6780 CrossRef CAS PubMed.
- S. Song, X. Huang, Y. Yang and L. Feng, Chem. Commun., 2024, 60, 10906–10909 RSC.
- A. B. Devale and D. K. Kulkarni, J. Phys. C: Solid State Phys., 1982, 15, 899 CrossRef CAS.
- M. Ahmad, Q. Riaz, M. Tabassum, S. S. Shafqat, A. t. Ayesha, M. Zubair, Y. Xiong, A. Syed, H. A. Al-Shwaiman, M. A. Nadeem, X. Jia, G. Xu and M. N. Zafar, RSC Adv., 2024, 14, 28285–28297 RSC.
- J. Li, C. Wu, Z. Wang, H. Meng, Q. Zhang, Y. Tang, A. Zou, Y. Zhang, H. Zhong, S. Xi, J. Xue, X. Wang and J. Wu, Angew. Chem., 2024, 136, e202404730 CrossRef.
- Q. Fang, S. Ye, L. Zheng, H. Wang, L. Hu, W. Gu, L. Wang, L. Shi and C. Zhu, ACS Catal., 2024, 14, 9235–9243 CrossRef CAS.
- H.-R. Zhao, C.-Z. Yuan, C. Zhou, W. Zhao, L. Zhang, C.-H. Li, L. Xin, F. Wu, S. Ye, X. Zhang and Y. Chen, J. Energy Chem., 2024, 94, 458–465 CrossRef CAS.
- P. W. Menezes, A. Indra, O. Levy, K. Kailasam, V. Gutkin, J. Pfrommer and M. Driess, Chem. Commun., 2015, 51, 5005–5008 RSC.
- T. Wang, L. Miao, S. Zheng, H. Qin, X. Cao, L. Yang and L. Jiao, ACS Catal., 2023, 13, 4091–4100 CrossRef CAS.
- M. Weber-Stockbauer, O. Y. Gutiérrez, R. Bermejo-Deval and J. A. Lercher, Catal. Sci. Technol., 2019, 9, 509–516 RSC.
- Y. Liu, L. Bai, Q. Jia, P. Li, Y. Yan, N. Yuan and X. Hao, Chin. Chem. Lett., 2023, 34, 107855 CrossRef CAS.
- J. Li, C. Yin, S. Wang, B. Zhang and L. Feng, Chem. Sci., 2024, 15, 13659–13667 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02883c |
| This journal is © The Royal Society of Chemistry 2025 |