
Abiotic transformations of nitrogen mediated by iron sulfides and related species from early Earth to catalyst design
C. Felipe
Garibello
,
Daniel S.
Eldridge
 ,
Francois
Malherbe
and
Rosalie K.
Hocking
*
,
Francois
Malherbe
and
Rosalie K.
Hocking
*
Department of Chemistry and Biotechnology, School of Science, Computing and Engineering Technologies, Swinburne University of Technology, Victoria 3122, Australia. E-mail: rhocking@swin.edu.au
First published on 7th October 2023
Abstract
Nitrogen fixation and the cycles of nitrogen on Earth are key to life as we know it and are critical to both modern agriculture and sustaining natural ecosystems. Despite its importance, there is still much we do not know about the processes of transforming nitrogen in natural systems. Nitrogen transformation into ammonia is fundamentally a chemical reduction reaction. It can occur catalytically where there is a source of electrons and a catalyst, or directly where a substrate is concomitantly oxidized, providing the source of electrons directly. In this review we explore the chemistry of iron sulfides to understand the reactions they mediate when they interact with nitrogen species, both as direct reductants and as catalysts, as well as the relationship between catalysis and reduction. In parallel it also describes the chemistry of Earth from a protoplanet through to modern times of relevance to understanding nitrogen speciation and historical drivers of these transformations, with a focus on how the pressures and temperatures on Earth may have impacted nitrogen, iron, sulfur and related species. We explore how iron sulfides can both directly and catalytically mediate some chemical reduction reactions and explore how this may have been significant in life's origins.
Introduction
Nitrogen is an essential component of amino acids, hence proteins, and DNA; therefore understanding abiotic nitrogen fixation is a key process in life's origin theories.1 Despite its importance, there is still much that is unknown about the processes of transforming nitrogen. One thing is clear though: for life to evolve, it needed nitrogen in an accessible form, which is not the most abundant form on Earth i.e., N2. On Earth, in modern times, before the advent of the industrial scale Haber–Bosch process,2 nitrogen fixation primarily occurred through biological processes facilitated by nitrogenases.3,4 This leads to the question: did we have fixed nitrogen before nitrogenase evolved and if so, how did it arise? One place to look for systems capable of this fixation in early times was iron sulfides. In modern enzymes, most notably nitrogenase, iron sulfides form key components, and interestingly they are also amongst the strongest “natural” reductants on Earth today. The two have a striking structural similarity5 (Fig. 1).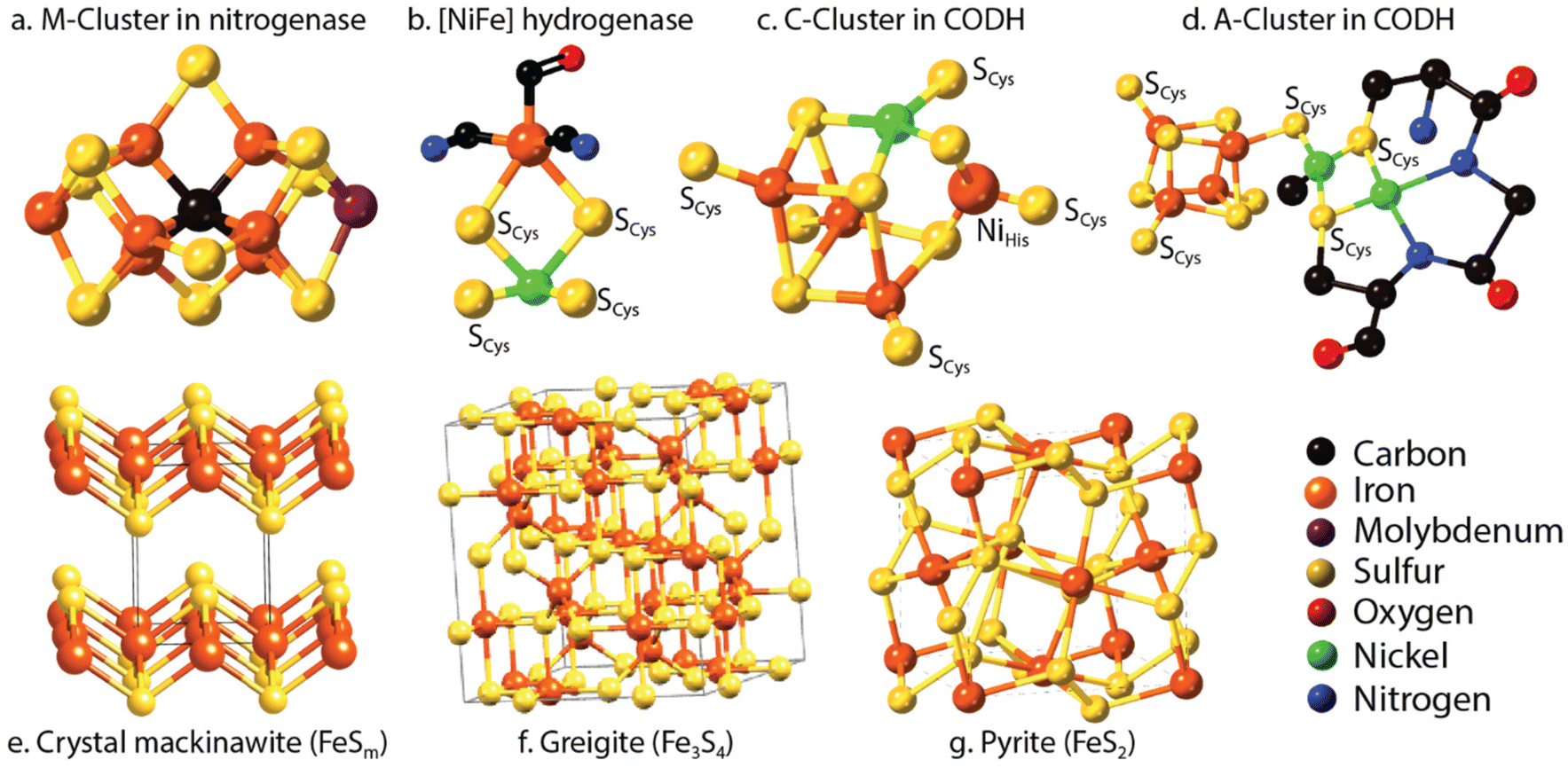 | ||
| Fig. 1 Upper row: (a) the M cluster in Fe–Mo nitrogenase, responsible for the fixation of nitrogen to ammonia, (b) Ni–Fe hydrogenase responsible for both proton reduction and oxidation, (c) C-cluster in carbon monoxide dehydrogenase (CODH), responsible for CO oxidation to CO2 in nature however capable of both reactions and (d) A-cluster in CODH. Lower row: crystal structures of well-known iron sulfide minerals: (e) mackinawite (FeSam), (f) greigite (Fe3S4), and (g) pyrite (FeS2). | ||
While nitrogen constitutes 78% of the Earth's atmosphere, changing it to either a more oxidized (NO3−, NO2−) or reduced state (NH3) is challenging (Fig. 2). As the triple bond of dinitrogen is very stable and strong, it requires significant activation energy to break.6,7 Nitrogen (N2) reduction to ammonia (NH3) is a reaction that requires both protons (H+) and electrons (e−). If conducted electrochemically, the redox equivalents of protons and electrons will come from the electrochemical system (eqn (1)).8,9 In the Haber–Bosch process (eqn (2)), the reducing agents are hydrogen atoms which are more oxidized in NH3 than in H2.2,10 Fundamentally, the source of electrons and protons in a system determines the mechanisms and selectivity of how reactions proceed.11,12 The stability of the nitrogen phases and iron sulfides as a function of Eh and pH is given by the Pourbaix diagrams in Fig. 3.
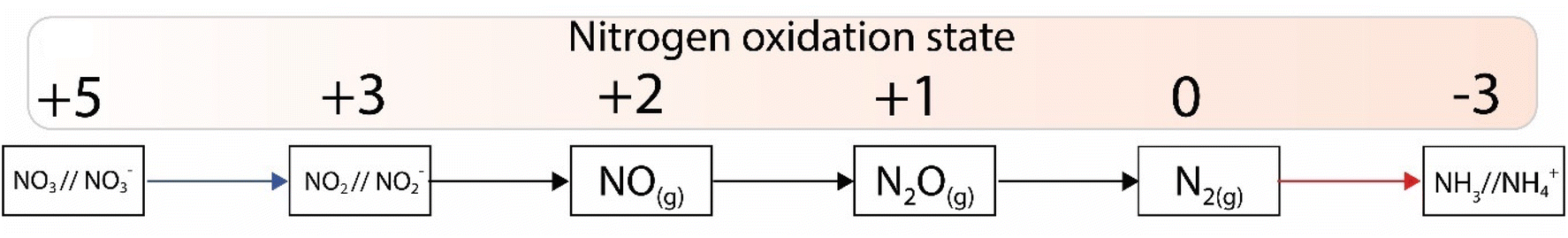 | ||
| Fig. 2 Nitrogen reduced products with their respective oxidation states. | ||
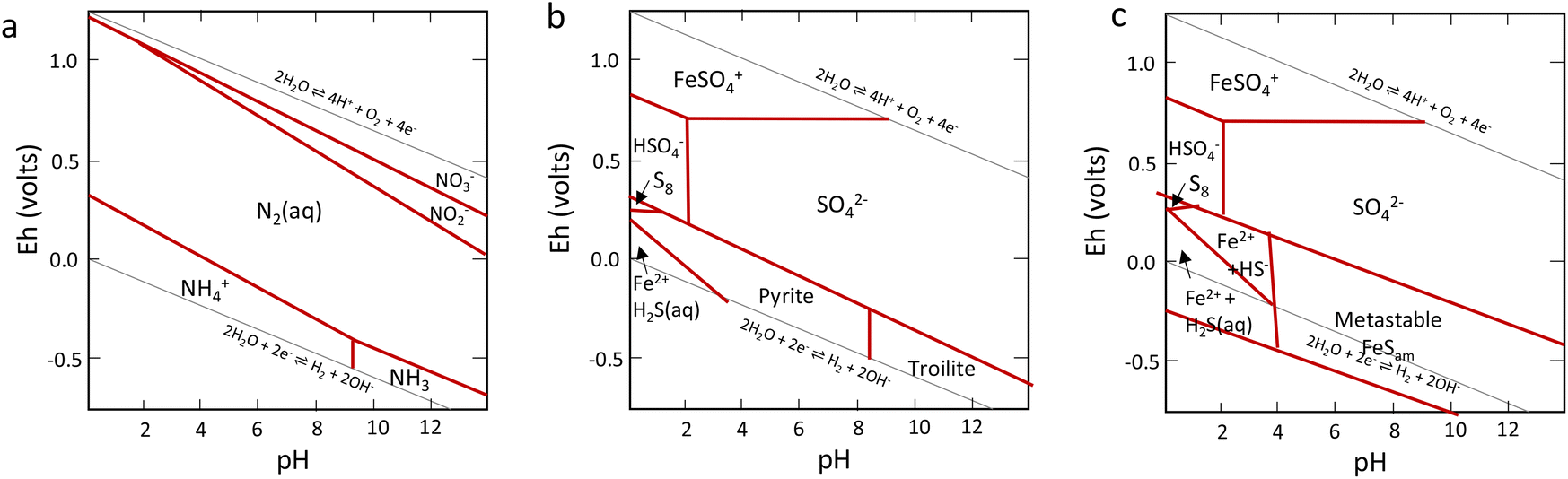 | ||
| Fig. 3 Key Pourbaix diagrams of relevance to this work. (a) Shows the nitrogen system, (b) the Fe/S system with thermodynamically stable products, and (c) the Fe/S system with metastable mackinawite shown. Reproduced from ref. 96 with permission from John Wiley and Sons, copyright 2022. | ||
Electrochemical nitrogen reduction for ammonia production
| N2 + 6H+ + 6e− ⇌ 2NH3 E0 = 0.057 V vs. SHE (T = 25 °C) | (1) |
Haber–Bosch process reactions for nitrogen reduction to produce ammonia
| N2 + 3H2 ⇌ 2NH3 (400–450 °C; 200 atm), ΔH = −92.4 kJ mol−1 | (2) |
In the present review, we highlight the chemical importance and complexity of iron sulfides mediating nitrogen fixation from two perspectives: direct reduction and catalytically mediated reduction where there is a secondary source of electrons. First, we explore critical differences in thermodynamics affecting nitrogen through the evolution of Earth's atmosphere, and the redox processes, along with the thermodynamic and chemical landscape at Earth's beginning. Furthermore, the fundamental physicochemical attributes intrinsic to iron sulfide materials are presented along with the known mechanisms involving the transformation of nitrogen species i.e., N2, NOx, and NH3 mediated by iron sulfide materials as both reductants and catalysts. Finally, we present an experimental approach for the study of direct and catalytic reduction reactions through the case study of the selectivity of the reduction products in the nitrite reduction reaction mediated by iron sulfide materials. Thus, we expect this review to consolidate the central role of iron sulfide materials in redox/catalytic reactions and thereby their contribution to the chemical transformations that could have had key roles in early life and the potential of iron sulfides in the development of effective catalyst design.
Geochemistry
Nitrogen is not just essential for life as we know it; it was also key for the evolution of life on Earth.13 But how did the first abiotic nitrogen fixation and the consequent ammonia formation occur, and under what conditions? To understand this, we need to go back to Earth's beginnings and consider two different environments where ammonia could potentially be abiotically formed: the oceans and the atmosphere.14,15 Key geological changes in Earth are outlined in Fig. 4, alongside what is known about the atmosphere.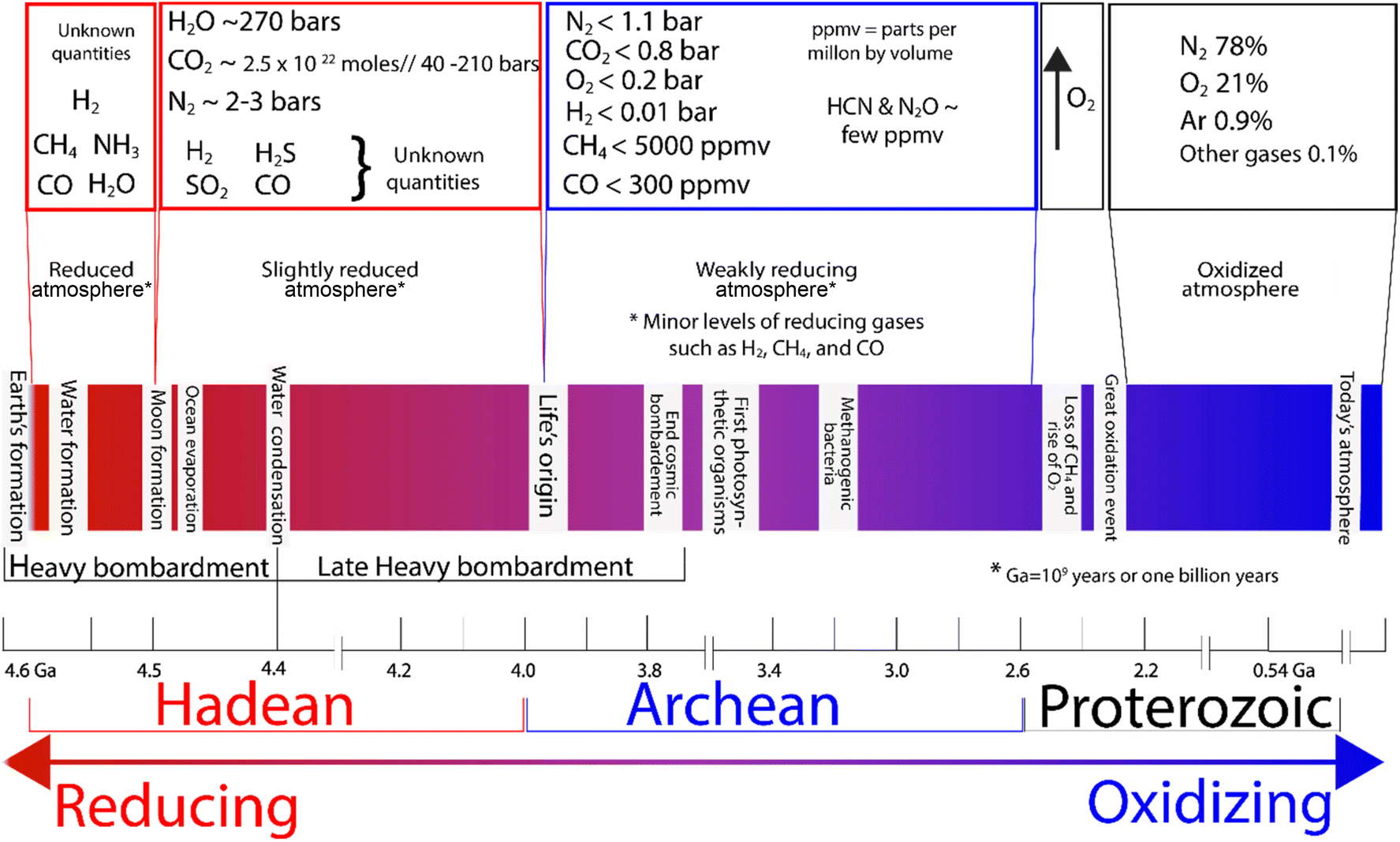 | ||
| Fig. 4 Atmospheric composition in the early Earth and changes over time.14,19–24 | ||
The Hadean atmosphere and oceans offered a wide variety of chemical elements in amounts that, along with a range of different conditions of pressure and temperature, could have provided far more thermodynamically favourable conditions for nitrogen fixation, compared to those of today.16–18 A study of the conditions where iron sulfides have been postulated to play a role in nitrogen fixation could offer us a different perspective in the design of a new generation of catalysts. Therefore, consideration of the chemistry of early Earth provides us insights into how primitive nitrogen fixation may have evolved. It is also important to note that there were extreme differences in terms of pressure that altered the thermodynamics and solubility of nitrogen, and with the amount of dissolved nitrogen being proportional to pressure, there was much more of it in the oceans in the Hadean era, and also at high ocean depths, where pressures are substantially higher and solubility is greater.14,19
Nitrogen and early Earth
The Hadean Eon, which is the earliest eon in Earth's geological history, is estimated to have lasted from approximately 4.6 billion years ago (4.6 Ga, the formation of the Earth) to about 4 billion years ago (4 Ga), and it is speculated that this was a dynamic time chemically. Physically, it featured constant bombardment by extra-terrestrial bodies and high volcanic activity.25 The chemistry of Earth in the Hadean era was likely subject to extreme conditions including plasmas, high temperatures, and pressures that may have promoted NH3 formation.17,26 However, it is speculated that ammonia in the atmosphere may not have persisted long due to photolysis by solar UV radiation,27–30 which breaks ammonia down to nitrogen and hydrogen.25,31The chemistry in the Hadean atmosphere was probably highly reductive compared to today.32 Early atmosphere is believed to have had significantly higher atmospheric pressure compared to today, with a dominant composition of CO2 (around 40–210 bar)33 and N2 (2–3 bar),34 H2O, CO, and small quantities of H2, CH4, P4O10, SO2, S8, H2S and NOx35,36 (Fig. 4). Although H2S would be emitted by oceanic volcanic activity, because of its high solubility in water only a small fraction would have reached the atmosphere. Given that there was a high concentration of Fe2+ in the ocean, any H2S formed would have likely formed insoluble/soluble phases of the iron sulfide family.28
The Archean atmosphere (∼3.7 Ga ago, after the evolution of life) featured the rise of atmospheric nitrogen, the appearance of oxygenic photosynthesis and the consequent increase in oxygen levels,37,38 producing a more oxidizing atmosphere (Fig. 4). Recent studies of basalts from the mid-ocean ridge suggested that as a consequence of the shift from a “chemically reduced” mantle to a “more oxidised mantle”, nitrogen becomes insoluble and was degassed in high proportions.39–41 The pressure of the atmosphere also plays a significant role in dissolved gases with the amount of gases dissolved being proportional to atmospheric pressure.42 Thus, under the extreme conditions of the primitive atmosphere, it is speculated that dissolved N2 would have been present in much higher amounts, which would have rendered any naturally occurring chemical reduction process more thermodynamically favourable.
Could an early Earth atmosphere fix nitrogen?
Different natural events such as lightning, photochemical reactions, meteorite strikes, hot lava flows, and extremes of pressure and temperature could support abiotic nitrogen fixation in the primitive atmosphere. From these events, lightning coupled with photochemical reactions is considered the most plausible.39–41,43High temperatures and plasmas produced by lightning were capable of dissociating CO2 into CO and O˙. The reactive O˙ radical is capable of breaking the triple bond of N2 forming NO and N, and then the N radical could react with CO2 to produce more CO and more NO (Fig. 5).43–46 Thus, the Hadean atmosphere had a sink reservoir of NO, which could be converted into other NOx species by photochemical reactions (Fig. 5).
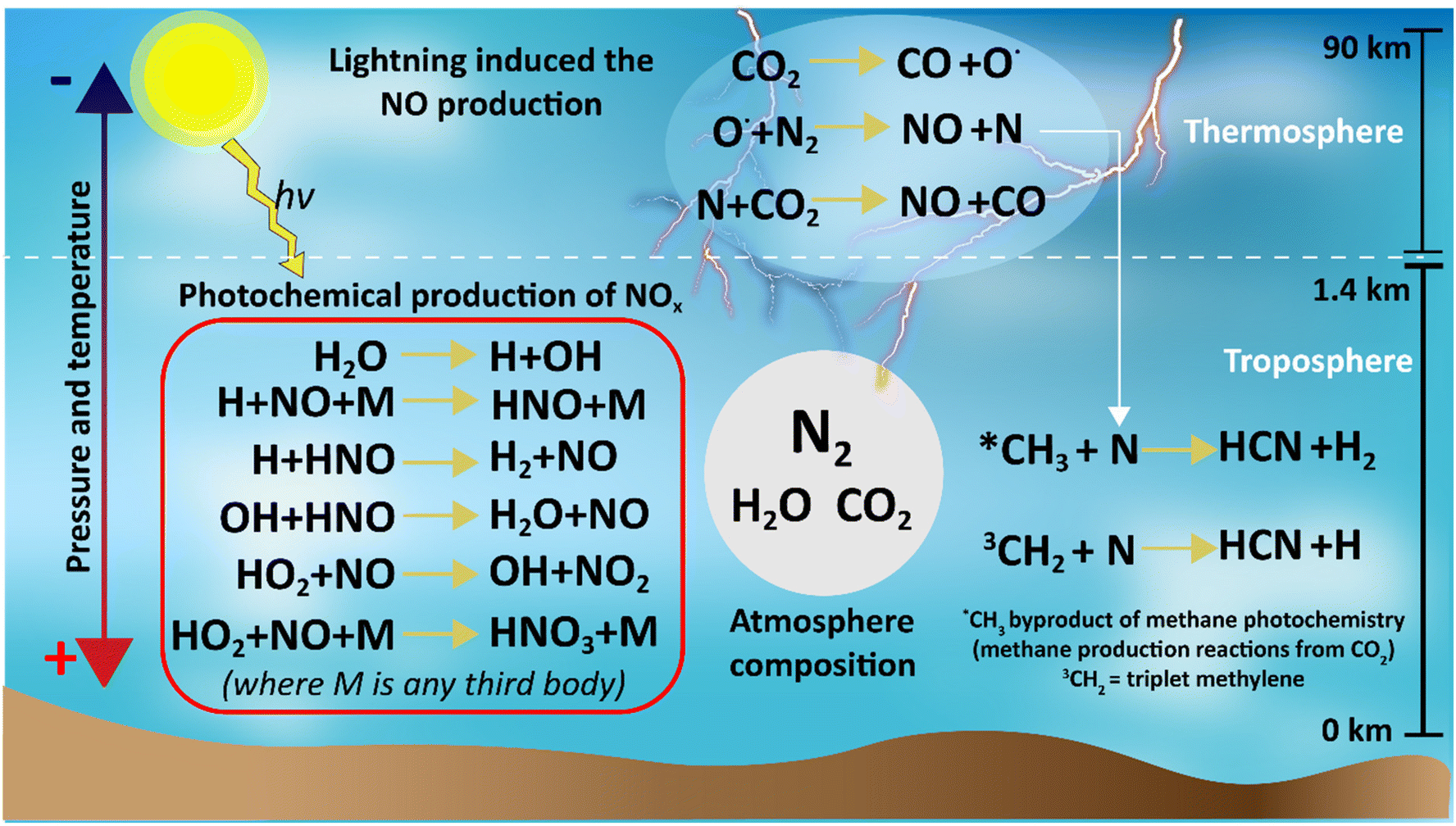 | ||
| Fig. 5 Summary of the reactions for nitrogen species production under a reduced atmosphere in the Hadean time. | ||
To be useful to life, the NOx species needed to reach the oceans. The profile of the NOx species (at different pressures of 0.1, 1 and 10-bar pCO2) produced by photocatalysis along with the rainout rates in the Hadean atmosphere was simulated by Wong et al.43 using the 1D Caltech/JPL chemical transport model.47 This study found that among all the NOx species, HNO has the highest initial formation rates followed by HNO3 and HNO2.
Additionally, the calculated amount of NOx rainfall into the Hadean waters was 2.4 × 105 (for the 0.1-bar pCO2), 6.5 × 108 (1-bar pCO2) and 1.9 × 108 molecules per cm2 per s (10-bar pCO2).43 The high pressures and reducing atmosphere of early Earth would have made ammonia formation more thermodynamically favourable, which may have led to additional forms of nitrogen being present. It is plausible that not enough nitrogen was produced by this method to impact the rock record.16,48,49
Recent investigations into photocatalysts utilizing nanomaterials have shown a promising avenue for the reduction of N2 to NH3 through a multistep reaction pathway. The photoinduced conduction band electrons and valence band holes reduce nitrogen and oxidize water, respectively.50 This process involves the formation of a sequence of lower-stability intermediate compounds.51 Additionally, photoreduction on the mineral surfaces under early Earth conditions has been previously reported,52,53 and the conditions during the Hadean era, characterized by specific combinations of pressure and temperature, may have enabled photocatalytic transformations mediated by iron sulfide minerals. Therefore, these recent findings open a new avenue for the photochemical nitrogen fixation mediated by iron sulfides in the early Earth's atmosphere, without the NO formation described above.
Could the chemistry of early oceans have promoted nitrogen fixation?
The aqueous composition of the Hadean waters is likely key to understanding the underlying chemistry and possible origins of biological nitrogen fixation. The high pressures of the atmosphere would have directly affected the dissolved gases and the reactions which took place. Hadean waters were acidic (around pH 5–6) due to the high concentration of atmospheric CO2,17,54,55 and the salinity was at least twice the current values.56 There is no consensus on the temperature, but it is believed to have been in the range of 50–100° C, the higher temperature than today being the result of high hydrothermal activity.57,58 Marine sedimentary rocks collected from Archean times (just after the evolution of photosynthesis) were reported to have high concentrations of iron and silica. These findings, along with the presence of Fe2+(aq) in large amounts in the deep Archean ocean, imply anoxic ocean conditions.20,59 Additionally, it is thought that there was a substantial amount of dissolved phosphate in the Hadean ocean in the form of orthophosphate mixed with iron (around 6–9 mmol kg−1) supplied by volcanic emissions into the ocean.60,61The prevailing chemistry of the Hadean waters before the advent of oxygen was very different from today. Oxygen substantially influences metal speciation in aqueous environments. For example, dissolved Fe2+ disappeared after the evolution of oxygenic photosynthesis precipitating as Fe(OH)3 species and forming the banded Fe formations that we know today.17,25,62 Prior to the emergence of photosynthesis, molybdenum would have existed in the ocean in the form of sulfide species. With the rise of atmospheric oxygen, molybdenum would have oxidized, changing the dominant species to oxyanions, such as molybdates (MoO42−) and related species. From lightning strikes noted above, it is thought that over a long time period, NOx species were dissolved in the oceans, along with the presence of reducing agents like H2S/HS− species and H2(aq) in different microenvironments (e.g., hydrothermal vents). These conditions, along with the effect of high pressures and temperature gradients, would have made ammonia formation in Hadean waters much more thermodynamically favourable than today.62–64 A comparison of the aqueous chemistry of the Hadean is summarised and compared to hydrothermal vents in Table 1. It is clear from the pressures and temperatures alone that ammonia would have been thermodynamically easier to form than in the oceans of today. This is also clear from the Pourbaix diagrams shown in Fig. 3.
| Hadean ocean | Hydrothermal vents (on Earth today) | Modern ocean | ||
|---|---|---|---|---|
| Atmosphere | Primitive crust and mantle | Type I: lost city field (LCF) | Type II: black smokersb | |
| Temperature and pH | Temperature and pH | Temperature and pH | Temperature and pH | Temperature and pH |
| ∼85 °C; slightly acidic.67 | ≲20 °C; pH ∼5–6 | ∼40–90 °C; pH ∼ 9–11.68 | ∼405 °C; pH ∼ 2–3.68 | ∼2 °C; pH 7.5.65 |
| Hydrogen | Hydrogen | Hydrogen | Hydrogen | |
| Small quantities of H2.35 | around 20 mmol kg−1.69 | 0.1–50 mmol kg−1.70 | 0.3 nmol kg−1.65 | |
| Composition | Composition | Composition (mmoles per litre) | Composition (mmoles per litre) | Composition (mmoles per litre) |
| H2O, N2, CO2 (around 1–10 bar17,55), CO and small quantities of H2, CH4, NO, P4O10, SO2 and S8.35 | Ag0, Au0, Cu0, Ni0, Fe0 and Al silicates with traces of metals.71 | Na+ 479–485 | Na+ 543–584 | Na+ 461 |
| K+ N/A | K+ 17–20 | K+ 9.9 | ||
| Ca2+ 21.0–23.3 | Ca2+ 26–31 | Ca2+ 10.31 | ||
| Cl− 546–549 | Cl− 633.5659 | Cl− 541.5 | ||
| CO2 not registered | CO2 3.9–215 | CO2 2.3 | ||
| CO2 ∼ 100–1000 times more than today | Composition (μmoles per litre) | Composition (μmoles per litre) | Composition (μmoles per litre) | |
| Li+ not registered | Li+ 367–411 | Li+ 27.5 | ||
| Mn2+ ∼ 4.1 ppm | Mn2+ 666–1000 | Mn2+ 10−3 | ||
| Zn2+ traces | Zn2+ > 36–46 | Zn2+ < 10−3 | ||
| Cu2+ ∼ 1.24 ppm | Cu2+ > 83–150 | Cu2+ < 10−3 | ||
| P (as PO42−) traces | P (as PO42−) 2.5 | P (as PO42−) 0.5 | ||
| CH4 0.13–0.28.65,66,72 | CH4 5000–45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.65,66 000.65,66 |
CH4 10−365,66 | ||
| Mineral composition | Mineral composition | Mineral composition | ||
| The ocean crust in the mafic, felsic and komatik lavas was composed by various minerals such as ferrous iron silicates, olivine, pyroxene, plagioclase and calcic feldspars, magnetite and pyroclastic, ultramafic, serpentinite and other metal precipitates.36,57 | Aragonite (CaCO3), brucite (Mg(OH)2), Olivine ((Mg, Fe)2 SiO4) and calcite (CaCO3) | Sulfide-complexing metals (Fe, Mn, Cu, Cd Pb and Zn),73,74 made principally of pyrite (FeS2) and chalcopyrite (CuFeS2). Carbonates, amorphous silica, ferrous and ferric oxyhydroxides, zeolites and various sulfide (Fe, Ni, Zn) amorphous silica.57 | ||
| Species of interest to Nitrogen fixation | |||
|---|---|---|---|
| Hadean ocean | Hydrothermal vents (on Earth today) | Modern oceand | |
| Primitive crust and mantlec | Lost city field (LCF)a,e | Black smokersb | |
| a Data from Kelley, S. D. et al., 2001.76 b Data from Beatty, J. T. et al., 2005.77 c Estimated mean concentrations of trace metals in the Archean ocean, based on trace element concentrations in Archean marine pyrite data from Large, R. R. et al.75 d Data values of the modern ocean in ppt data from Nozaki, 2010.78 e Data collected from black smoker fluids, associated with hydrothermal plumes at the Rodriguez Triple Junction, Central Indian Ridge.79 | |||
| Molybdenum (Mo) | Molybdenum (Mo) | Molybdenum (Mo) | Molybdenum (Mo) |
| ∼100 ppm as MoSx | ∼10000 ppm |
Traces | ∼10000 ppm, it forms a soluble ionic species, MoO42−, with a long residence time of 760000 years75 |
| Nickel (Ni) | Nickel (Ni) | Nickel (Ni) | Nickel (Ni) |
| ∼1200 ppm as Ni2+ | ∼20 ppm as Ni2+ | Traces metal | ∼600 ppm as Ni2+ with residence time, from 5000 to 50000 years75 |
| Iron (Fe) | Iron (Fe) | Iron (Fe) | Iron (Fe) |
| Substantial concentrations of Fe2+.71 | 50 ppm as a mixture of Fe2+/Fe3+ | 300 ppm (mostly Fe2+) | 30 ppm mostly as Fe(OH)3 |
| Rich in iron sulfide minerals | Rich in iron sulfide minerals | ||
| Sulfur speciation | Sulfur speciation | Sulfur speciation | Sulfur speciation |
| H2S ∼ 66% | H2S ∼ 4% | H2S ∼ 56% (3–110 mmol kg−1) | H2S not detected |
| HS− ∼ 33% | HS− ∼ 94% | HS− ∼ 43% | SO42− ∼ 898 × 106 corresponding to 98% of sulfur species |
| S2− ∼ 0.0%70 | S2− ∼ 1%70 | S2− ∼ 0.0%70 | |
Hydrothermal vents
Before the exploration of the deep ocean floors in 1977 by John Edmond and Jack Corliss on the crest of the Galapagos Rift (2500 m depth),80 the idea of a poor life environment in the ocean was generally accepted. This assumption was based on the fact that most life requires a direct source of energy, which for terrestrial life is provided by sunlight, and this light can only reach ∼300 m down into the oceans meaning there would not be life at the bottom of the ocean.This deep ocean exploration challenged the prevalent idea at that time, unveiling an ocean full of life. The important question that arose with this new finding is where and how those organisms fulfilled their energy requirements and whether understanding these processes could help us better comprehend primitive metabolism.
It is believed that in the Hadean times, hydrothermal vents could also provide suitable conditions for abiotic nitrogen fixation, which is considered one of the cornerstone reactions in most theories on the origins of life.43,81–83 Thus, knowing the composition of these hydrothermal systems could offer some critical insights into the chemistry of nitrogen fixation reactions.
A hydrothermal vent originates from faults and fissures caused by the subduction of tectonic plates in basaltic crusts, which allow the flux of cold seawater to the roof of magmatic centres. Then, as the heated fluids are chemically modified by the interaction with surrounding rocks, and expelled, this heated fluid leaks through fissures at ∼350 °C full of chemical reductive species such as sulfide and hydrogen.84 The interactions between the expelled material and their surroundings produce gradients of pH, temperatures, and a variety of chemical sources, which provide a stock of reactants for relevant redox reactions.85
Due to the multiplicity of the host rock and the diversity of tectonic conditions, two types of hydrothermal vents have been described, ‘black smokers’ and ‘LCF’ hydrothermal systems. While black smokers are fuelled by volcanic activity, the LCF hydrothermal system is the result of the interaction between sea water and minerals in the Earth's mantle along with serpentinization (i.e., hydrolysis and transformation of ferromagnesian minerals to produce H2-rich fluids). Although the exact composition of hydrothermal vents in the Hadean oceans is unknown, recent studies have measured the composition of hydrothermal vents on Earth today. These are described in Table 1 in comparison with the key chemistry of relevance for known nitrogen fixation.
A comparison between the composition of hydrothermal vents (black smokers and LCF) and the modern ocean is given in Table 1. Clear differences are evident in terms of the speciation and amounts of key elements including iron, molybdenum, and sulfur. Differences between the pH and temperatures of the hydrothermal vent systems and the surrounding waters offer states of redox disequilibria or redox gradients, and these gradients, along with the depth–pressure effect, could be relevant in terms of speciation and solubility and more importantly could shift the equilibrium of the chemical reactions under the conditions of hydrothermal vents that would ignite some of the first and key redox reactions for the origin of life.86,87
An interesting aspect of the chemistry of hydrothermal vents is the presence of chemolithotrophic life. It is thought that these types of organisms are amongst the most ancient on Earth. A chemolithotrophic microorganism draws its energy of metabolism from redox reactions with inorganic compounds/minerals. In these reactions, it is often compounds like iron sulfur species which act as reducing agents, providing the critical electrons that are necessary to support metabolism.88,89 Chemolithotrophs can use different compounds including Fe2+, CO, N2, H2, H2S, HS and other sulfur compounds as a source of electrons.90 Although separately both iron and sulfur compounds can supply the electrons in a wide range of sulfur-oxidizing bacteria (e.g. Thiobacillus thiooxidans)91 and iron-oxidizing bacteria (e.g. Thiobacillus ferriooxidans),92 recent studies show that the reaction of FeS with H2S to produce FeS2 may be the energy source to support chemolithotrophic metabolism in some bacteria.93
Chemolithotrophic bacteria have been harnessing the redox capability of the omnipresent iron sulfides to proliferate under extreme conditions (pH ≤ 3 and temperatures above 100 °C). Some of the key reactions of chemolithotrophic metabolism are present in modern enzymes, not as a primary energy source, but as an important regulator or modulator of metabolism. Consideration of chemolithotrophic processes in mediating reduction reactions is important as they are not catalytic, rather they represent direct redox processes often capable of changing key small molecules. It is possible that the direct cycles of oxidation and reduction mediated by the chemolithotrophs are evolutionary precursors to modern enzymes.
Redox gradients as a source of energy through redox reactions
The theories of the dawn of life on Earth are deeply connected to a constant supply of energy. The flow of energy is the engine of any chemical reactions and it is thought that these reactions first arose with primitive organisms.68,94 Thermodynamically, pH, temperature, and redox gradients can be used as ways to generate energy. Thus, the micro-environments such as hydrothermal vents and their interactions with the early ocean crust and ocean waters harbor those gradients. The use of reduction power from hydrogen sulfide (H2S) has been hypothesized to be one resource to drive the chemical reactions in the Hadean waters.81,95 Additionally, the redox chemistry of iron-based compounds with hydrogen, water, and oxygen under hydrothermal conditions could have been involved in some of the key reactions in life's origin theories.Recent studies show that iron sulfides can reduce nitrite under both catalytic and direct redox conditions. Of the materials tested, amorphous mackinawite showed the highest nitrite reduction under both conditions.96 Additionally, the incorporation of ∼10% of molybdenum into the amorphous mackinawite structure increased the selectivity of nitrite reduction to ammonia.97
Reductive fluid: H2S/HS−
Some of the most common reductants in natural systems are H2S, HS−, and H2. It is likely that they existed in considerable amounts in hydrothermal vents and in the Hadean ocean representing a likely electron source for the chemical reduction equivalents necessary for early metabolism. Hydrogen sulfide (H2S) is the most reduced form of sulfur. Sulfide in all its forms S2−, HS− and H2S is often an overlooked mediator of reduction but it is a strong reductant capable of shuffling 8 electron steps between S2− and SO42−. One of the greatest contrasts between the Hadean ocean and the ocean of today is the sulfur speciation. For comparison, in the modern ocean, there are large amounts of sulfate (oxidation state +6, Table 1). At the black smoker vents, the absence of oxidants, the high temperature and pressure along with low pH favour high concentrations of both H2S and other gases like N2.74,98There is much speculation on the origins of life and theories including the “iron–sulfur world”,99 an “iron–sulfur membrane”100 or a “zinc world”66,101 and more recently “geo-electrochemistry theory”,83,102,103 all of which propose that H2S is the main provider of electrons to drive the fixation of key molecules, including ammonia and carbon dioxide.104,105
Iron sulfide materials (FexSy)
As a result of anoxic and specific thermodynamic and redox conditions, an initially formed iron sulfide precipitate can evolve in a wide range of phases, which span from amorphous nanosized colloids to well-defined crystalline phases.106–108 From their precursors, i.e., Fe2+ and HS−, iron sulfides usually initially precipitate in a compound known as a mackinawite-like nanoparticulate (nFeSam, with n > 150), which is subsequently transformed into amorphous mackinawite by the ageing process. Fe2+ and HS− can also produce more ordered phases via oxidative transformations. Two of the most well-known are greigite (where iron oxidizes from Fe2+ to Fe3+) and pyrite (where S oxidizes from S2− to S−) (Fig. 6).109,110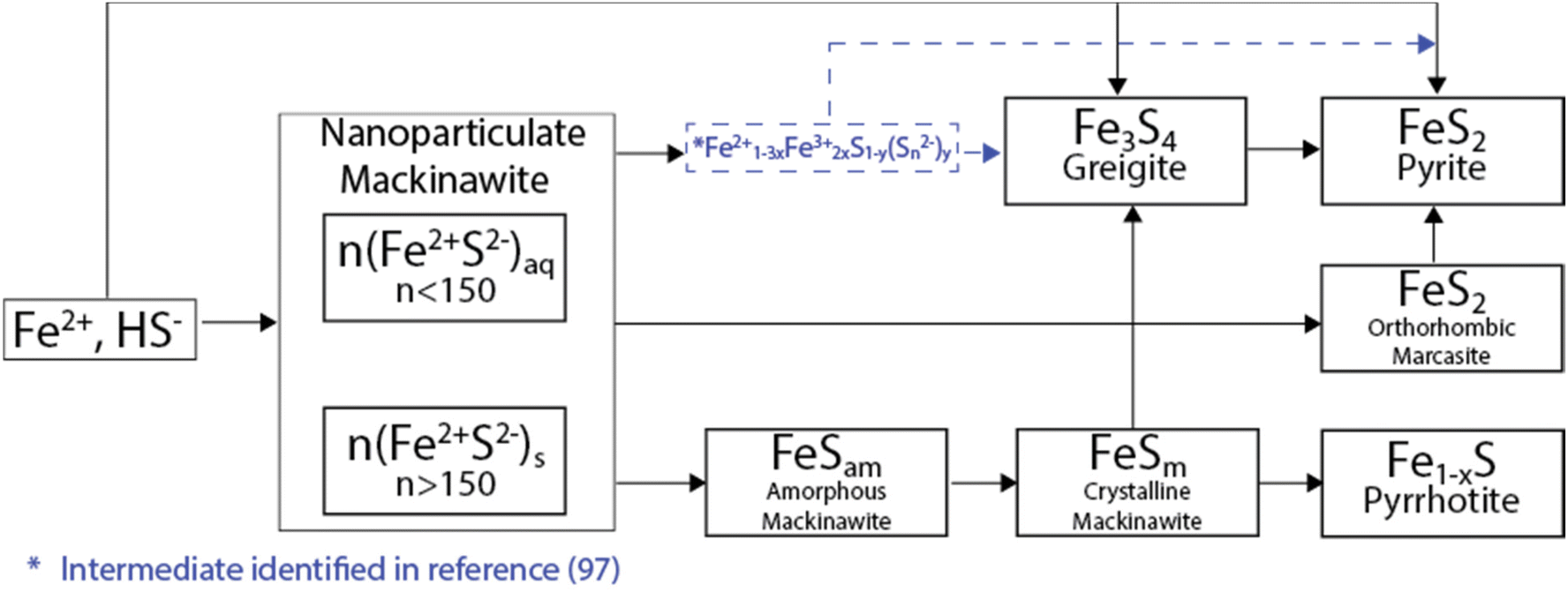 | ||
| Fig. 6 Oxidation pathways for iron sulfide materials. Figure adapted from ref. 116. | ||
Additionally, the initially precipitated amorphous mackinawite (FeSam) can evolve into other iron sulfide phases including pyrrhotite/troilite and marcasite.106–108 In terms of the natural distribution, pyrite is the most abundant form of iron sulfide, followed by pyrrhotite, marcasite (FeS2, polymorph of pyrite) and troilite.111
There is a large amount of research demonstrating that iron sulfides are among nature's strongest chemical reductants.109 The selectivity and scope of the reduction chemistry mediated by iron sulfide minerals are not well understood.112 Much of the existing research into the geochemical properties of iron sulfides focuses on their crystal structure, and thermodynamic high-pressure phase transitions rather than their reactivity.111,113 Furthermore, it is often the case that highly disordered nanoparticles (or nanomaterials) have a distinct reaction chemistry from their well-ordered cousins.114,115 This may mean that disordered forms of iron sulfide minerals are even stronger reducing agents than their more ordered cousins.112
Importantly, recent studies into materials for water oxidation have found that minerals are often most reactive in their amorphous, or disordered forms, and the same could be true for catalysts for the reduction of small molecules. The extent to which disorder affects the reduction chemistry of iron sulfides and whether it affects their ability to chemically reduce molecules like CO2 are not known.
Several attempts to describe the role and mechanisms of iron sulfide materials in the synthesis of prebiotic organic molecules have been explored in recent years.18,117–121 There remains uncertainty about the dual behaviour of these materials which have the ability to work as both catalysts and direct reducing agents. Studies in the reduction of nitrogen compounds in low pH solutions show that ammonia formation is linked with the oxidation of FeS to FeOOH compounds,118 while the use of iron sulfides as catalysts in ammonia production under hydrothermal conditions (temperature ∼300–800 °C and pressure ∼0.1–0.4 GPa) was achieved with yields of 21% to 89% (NH4+ was quantified in this study using the indophenol blue method).16
Mackinawite
Mackinawite is the first formed phase upon mixing Fe2+ and HS−. Mackinawite has received special attention in the last decade due to its geological relevance as a probable precursors of basic molecules in the abiotic theory for the origin of life, and also because of their sorption capacity.122–124 In addition, this material has a remarkable chemical reactivity, which even provides the capacity to transform halocarbons via chemical reduction.124–126 Generally, freshly precipitated mackinawite is an amorphous hydrated nanomaterial,127,128 which can act as an intermediate precursor of more stable crystalline phases such as pyrite (FeS2), marcasite (FeS2), and greigite (Fe3S4).129Synthetic mackinawite is usually prepared at room temperature and the most commonly used synthetic method involves mixing a sulfide solution with metallic iron or a ferrous iron solution. Mackinawite was the last of the major iron sulfides to be characterised by crystallography, as it was hard to grow crystals of it. The first experiments relied on electron diffraction130 rather than X-ray diffraction owing to its lack of crystallinity, as opposed to other crystalline iron sulfide phases, which were characterised crystallographically as early as the 1930s. Mackinawite in its crystal form has been described as a tetragonal crystal system with space group P4/nmm and with unit cell: a = 3.67 Å, c = 5.03 Å in its unit cell (Fig. 7).123,131
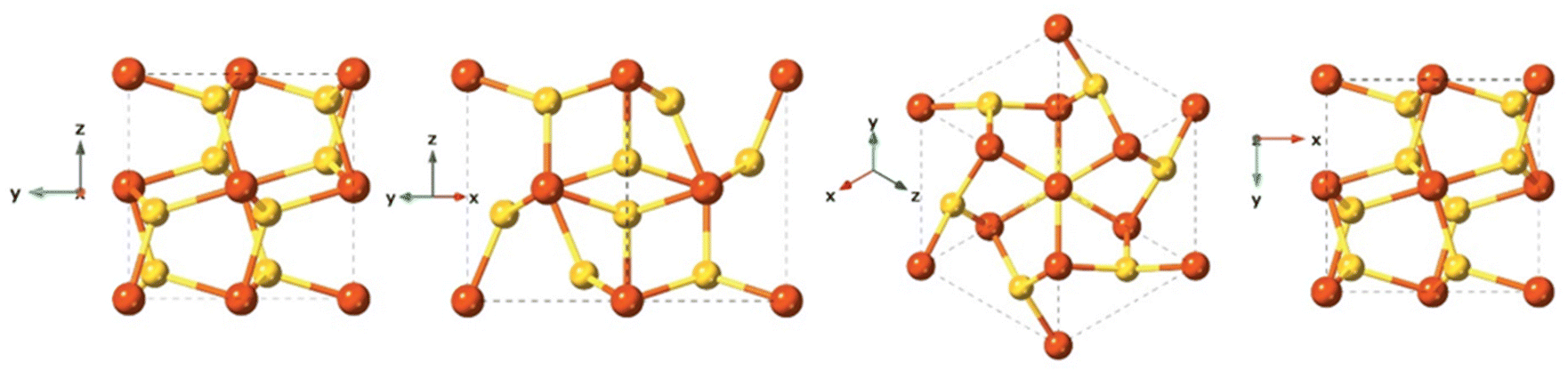 | ||
| Fig. 7 Different crystal face views of the mackinawite structure. | ||
Greigite (Fe3S4)
Greigite is known for its ferromagnetic properties and as the sulfur analogue of magnetite.132 This material is found in sedimentary rocks and its formation is commonly the result of the actions of sulfate-reducing bacteria. Additionally, greigite is an intermediate on the pathway from mackinawite to pyrite.109 The greigite structure has an inverse spinel structure with a cubic close-packed sulfur array with space group Fd3m and unit cell a = 9.876 Å. This arrangement (Fig. 8) shows significant similarity with the cubic, closely packed S-array of mackinawite. Greigite formation from mackinawite is an iron oxidation process, producing ferric (Fe3+) iron.109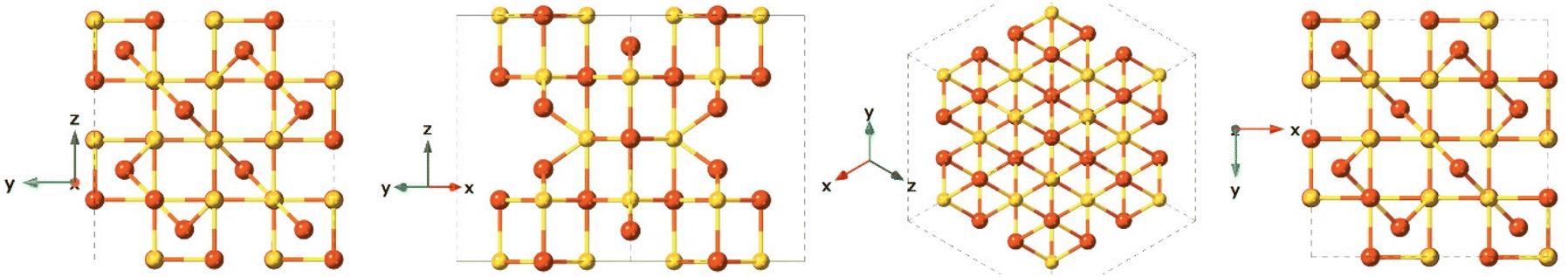 | ||
| Fig. 8 Different crystal face views of the greigite structure. | ||
Pyrite (FeS2)
Pyrite is the most thermodynamically stable and abundant compound on Earth among the iron-sulfides.109 Under anaerobic conditions, oxidation of iron sulfide(FeSx) to pyrite(FeS2) shows significant reducing power, which several studies suggest was involved in key reactions in the early carbon fixation process, for example, the reduction of carbonyl groups to methylene.133,134 As a consequence, it is postulated that pyrite could participate in reactions included in the chemoautotrophic origin of life theory.99,134,135 Pyrite formation is associated with metamorphic, igneous, and sedimentary rocks. The pyrite structure is typically cubic with space group Pa3 and cell parameters a = 5.142 Å (Fig. 9). Pyrite formation from FeSam is a complex, ligand oxidation-based process.109 Although pyrite is generally considered a non-reactive phase, its aqueous oxidation is an electrochemical process rather than a purely chemical reaction and this produces a range of compounds including sulfur, polysulfides, iron oxides and oxyhydroxides, sulfate and ferrous iron, which could contribute to the reactivity of pyrite.136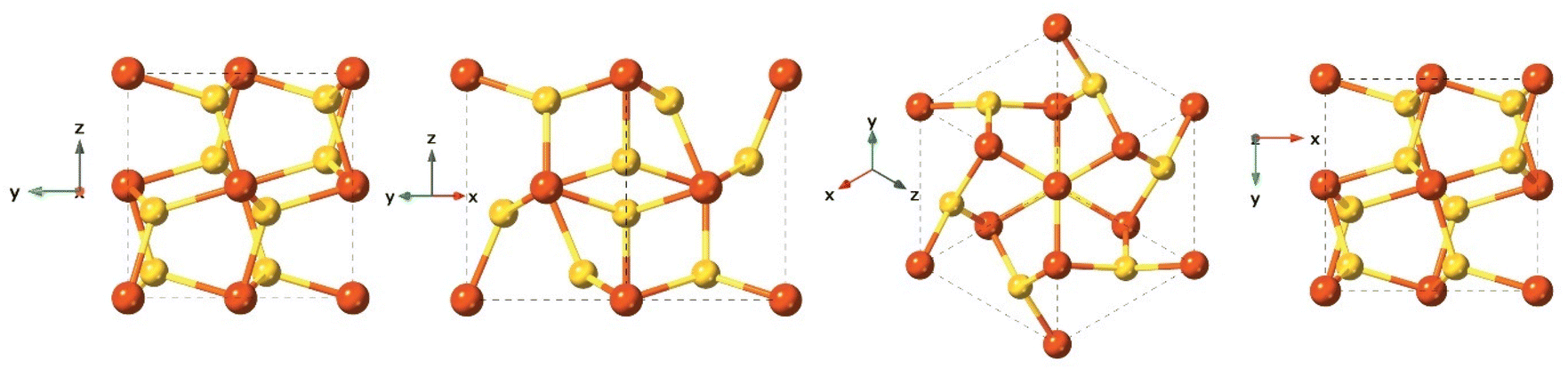 | ||
| Fig. 9 Different crystal face views of the pyrite structure. | ||
Marcasite (FeS2)
Marcasite and pyrite are polymorphs that have the same chemical structure and common chemical environment but differ in their crystal structure. While pyrite is typically a cubic structure, marcasite is orthorhombic,137 with lattice parameters: a = 4.443 Å, b = 5.424 Å, c = 3.387 Å, space group Pnnm. Although marcasite presents a higher conductivity and stronger Fe–S bonds along with weak S–S interactions.138Pyrrhotite (Fe1−xS)
After pyrite, pyrrhotite is the second most common iron sulfide in nature, it is a non-stochiometric compound with the calculated formula Fe1−xS, where x varies from 0 (FeS, known as troilite), to 0.125 (Fe7S8), and is often associated with pyrite and is found in diverse sulfidic ores.139 Under acidic conditions pyrrhotite is highly soluble and can produce Fe2+ and a mixture of polysulfides.139Troilite (FeS)
Although troilite is more thermodynamically stable than mackinawite, its occurrence on Earth is not common. It is more abundant on the Moon, Mars, and meteorites.140 Troilite is normally associated with serpentinized rocks and its formation is correlated with strongly reducing conditions.140 Troilite is the iron-rich end member of the pyrrhotite group. Their crystal form has been described as a hexagonal crystal system with space group P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 2c and with lattice parameters a = b = 5.962 Å, c = 11.750 Å (Fig. 10). It is not formed as a direct precipitate, rather it is the product of hydrothermal synthesis under anoxic conditions and at high pressures.109
2c and with lattice parameters a = b = 5.962 Å, c = 11.750 Å (Fig. 10). It is not formed as a direct precipitate, rather it is the product of hydrothermal synthesis under anoxic conditions and at high pressures.109
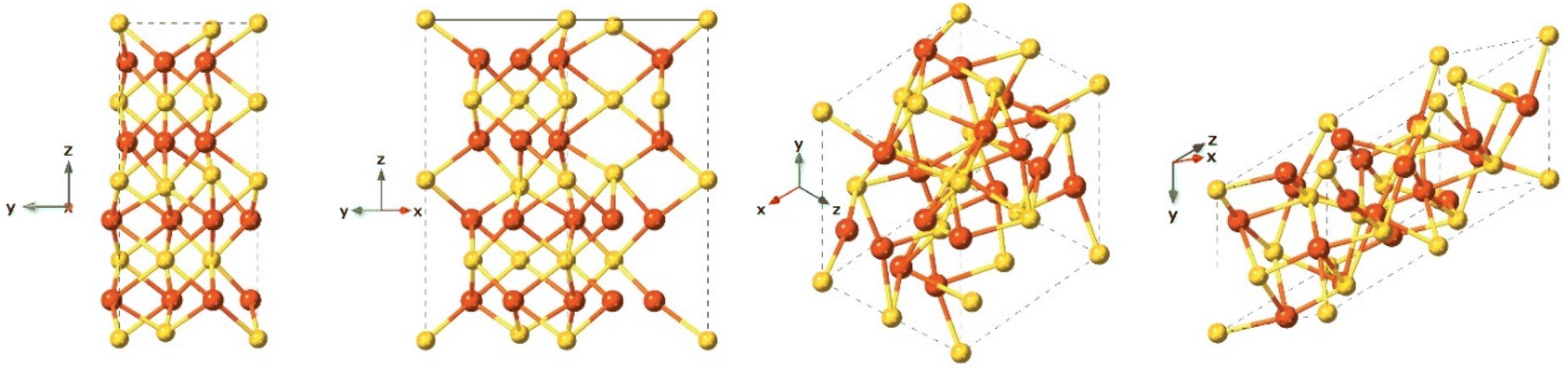 | ||
| Fig. 10 Different crystal face views of the troilite structure. | ||
Nitrogen compounds reduction reactions mediated by iron sulfides
Several attempts to describe the molecular-based mechanisms of the reduction of nitrogen compounds to produce ammonia mediated by iron-sulfide materials have been reported in recent years.18,117,118,120,121,141 Interestingly, no clear distinction is made in terms of the reduction mechanism. Thus, certain studies characterize iron sulfides as mediators that facilitate redox reactions, while others describe the role of iron sulfides as catalysts in the reduction of nitrogen compounds.Iron sulfides as reductants of nitrogen compounds
In 1993, Summer and Chang119 suggested that under early Earth conditions, the presence of iron(II) could be responsible for the reduction of nitrogen compounds such as nitrate (NO3−) and nitrite (NO2−) to ammonia (NH3) according to eqn (3). This reaction is favored by alkaline conditions (pH between 7.3 and 9.0) and temperatures above 25 °C.119| 2Fe2+ + 5H2O + NO2− ⇌ 2Fe(OH)3 + (OH)− + NH3 (T > 25 °C) | (3) |
These researchers also suggested that the reduction of nitrate to ammonia is slower by a factor of eight than with nitrite and the yield in ammonia is directly proportional to temperature. Also, it is postulated that the presence of anions, cations, and salts can be considered as interferences due to the possibility that these compounds create complexes with iron(II) and nitrite, thus blocking the active sites.119
In a later work, Summers118 experimented with the reduction of nitrite but this time performed the reaction under acidic conditions using different iron-sulfide materials (FexSy). He found that at low pH values the reduction reaction involved the iron sulfide surface and not the Fe2+ in solution. Furthermore, it was shown using energy dispersive X-ray spectroscopy (EDX) that after being exposed to nitrite, the bulk surface of iron-sulfide was more sulphur-rich compared to when nitrite was not present. Thus, this study suggested that a thin layer of pyrite (FeS2) was formed on the iron-sulfide materials, according to eqn (4). The remnant iron(III) can react with water to produce Fe(OH)3, which explains the brown supernatant that appears a couple of hours after the reduction reaction.118
| 2FeS ⇌ FeS2 + Fe(OH)3 + 3e− (T = 25 °C) | (4) |
Dörr et al.18 explored nitrogen reduction by freshly precipitated iron sulfide, this study highlighted that the main reaction driver is the oxidation of iron sulfide (FeS) to iron disulfide (FeS2) (eqn (5)). The proton supply came from hydrogen sulfide (H2S) acting as a reductant. They demonstrated that the reduction of dissolved dinitrogen to produce ammonia was thermodynamically affordable using the redox reaction shown in eqn (6).18
| FeS + H2S ⇌ FeS2 + H2; ΔG° = −38.6 kJ mol−1; E° = −600 mV; pH = 6.5 | (5) |
| N2 + 3FeS + 3H2S ⇌ 3FeS2 + 2NH3 (T < 90 °C) | (6) |
The chemical reduction of nitrite (NO2)− by Fe2+ was studied in an environmental context by Kampschreur.142 According to their results, the final product of the reduction of nitrite at pH = 6.5 was nitrous oxide (N2O) with nitric oxide (NO) as an intermediate.142 These reactions are shown in eqn (7) and (8).
| NO2− + Fe2+ + 2H+ ⇌ Fe3+ + NO + H2O (T = 35 °C) | (7) |
| 2NO + Fe2+ + 2H+ ⇌ Fe3+ + N2O + H2O (T = 35 °C) | (8) |
Singireddy et al.117 explained how pyrite (FeS2) in the Hadean can provide sites for the reduction of nitrate (NO3−) and nitrite (NO2−) to produce ammonia (NH3) in the temperature range of 20–120 °C for nitrite and at 120 °C for nitrate. The consumption of nitrite (NO2−) was explained by different reaction pathways. The first is the production of ammonia by Fe(II) according to eqn (3). Although this reaction is exclusive to alkaline pH, this study proved that the surface of pyrite releases Fe2+ under anoxic conditions and at acidic pH values. This free Fe2+ is responsible for a small amount of ammonia production.117
As part of this study Singireddy et al.117 proposed two alternative pathways for the nitrite reduction presented in eqn (9) and (10). All these reaction mechanisms are not exclusive; on the contrary, they can occur simultaneously.117
| 10NO2− + 2FeS2 + 8H+ ⇌ 5N2 + 2Fe3+ + 4SO42− + 4H2O (T = 20–120 °C) | (9) |
| 15NO2− + FeS2 + 14H+ ⇌ 15NO + Fe3+ + 2SO42− + 7H2O (T = 20–120 °C) | (10) |
Pyrite (FeS2) can facilitate the reduction of nitric oxide (NO) produced in eqn (7) and (10) to ammonia according to eqn (11). In this latest reaction, the authors proposed that pyrite acts as an electron donor, thus the N bound to the pyrite surface is more reactive due to electron donation from the iron atom. This flow of electrons promotes protonation of the N-site to form HNO. Consecutive protonations can result in the ONH2+ species, which activates the final cleavage of the O–N bond to finally produce ammonia (NH3).117
| 3NO + FeS2 + 5H2O + 2H+ ⇌ Fe3+ + 3NH4+ + 2SO42− (T = 70–120 °C) | (11) |
Gordon et al.143 investigated the kinetics of the reduction of NO3− and NO2− to NH3 using fresh mackinawite, and identified the main intermediates in the surface bonding process on the freshly precipitated nanocrystalline mackinawite at different temperatures and under alkaline conditions. This study suggested that the formation of NO is a key intermediate step in ammonia production and it is possible at 120 °C. The redox couple proposed by the authors are reported in eqn (12) and (13).
Interestingly, the activation energy calculated for the ammonia production at 120 °C on the surface of fresh mackinawite using NO2− is ∼62.76 kJ mol−1, almost half the energy required for NO2− reduction by Fe2+(aq).18,119 Thus, the FeSm surface represents a feasible way to produce ammonia with a lower energy barrier.143
| FeSm + H2O + 9NO2− ⇌ Fe3+ + SO42− + 9NO + 10OH− (T = 120 °C) | (12) |
| 16H2O + 9NO + 5FeSm ⇌ 9NH3 + 5Fe3+ + 5SO42− + 5OH− (T = 120 °C) | (13) |
Iron sulfides as a catalysts for redox transformation of nitrogen compounds
Brandes et al.16 demonstrated experimentally the feasibility of mineral catalysed reduction of nitrogen compounds to ammonia under conditions of the hydrothermal vents and the ocean crust, with temperatures between 300–800 °C and 987 atm (0.1 GPa) and 3948 atm (0.4 GPa).16 Additionally, they tested how changes in pressure affect the yield in ammonia for two systems, namely magnetite-formic acid-nitrogen (Fe3O4/HCO2H/N2) and metallic iron–water–nitrogen (Fe–H2O–N2). The increase in pressure from 98.7 atm (0.01 GPa) to 987 atm (0.1 GPa) was accompanied by ∼10% increase in ammonia for both systems and a further increase of the pressure did not show any significant change in yield.16Li et al.144 proposed that sulfur complexes with molybdenum (Mo) could act as a catalyst in the reduction of nitrogen compounds (NO2− and NO3−) to form ammonia. Under the conditions found in the hydrothermal environments, the electrochemical experiments performed by Li et al.144 revealed that at neutral pH and in the absence of NO2− (which was used as the substrate), MoS2 produced a cathodic current above −1.8 V vs. the reversible hydrogen electrode (RHE) often associated with the hydrogen evolution reaction (HER). After the addition of nitrite, the onset potential changed from −0.18 V to 0.07 V. The identity of the products generated was determined using online differential electrochemical mass spectroscopy (DEMS) which concluded that the reduction of nitrate to form ammonia produced intermediates such as NO and NO2.
The synthesis of ammonia from nitrate (NO3−) is kinetically more unfavourable than from nitrite (NO2−); in part due to the stability of nitrate.145 Li et al.144 also demonstrated that at neutral pH nitrate was reduced to ammonia and that the selectivity of this reaction over the HER was 100%. The authors applied potentials more negative than 0 V and found that the only intermediate detected was NO2−, suggesting that the reductive pathway is nitrate, nitrite, and ammonia.144
Li et al.144 proposed a model incorporating the previous findings in terms of spontaneous electron generation in the inner hydrothermal vents by H2S or H2 and tested the conductivity of the iron sulfide-bearing walls in the hydrothermal vents. Also, the temperature–pH gradients were observed between the hot alkaline hydrothermal fluids and cold acidic seawater and the stable catalytic activity of MoS2, where protonated Mo species act as a catalyst in the production of the intermediates and ammonia in a Proton Coupled Electron Transfer (PCET) mechanism (Fig. 11).144
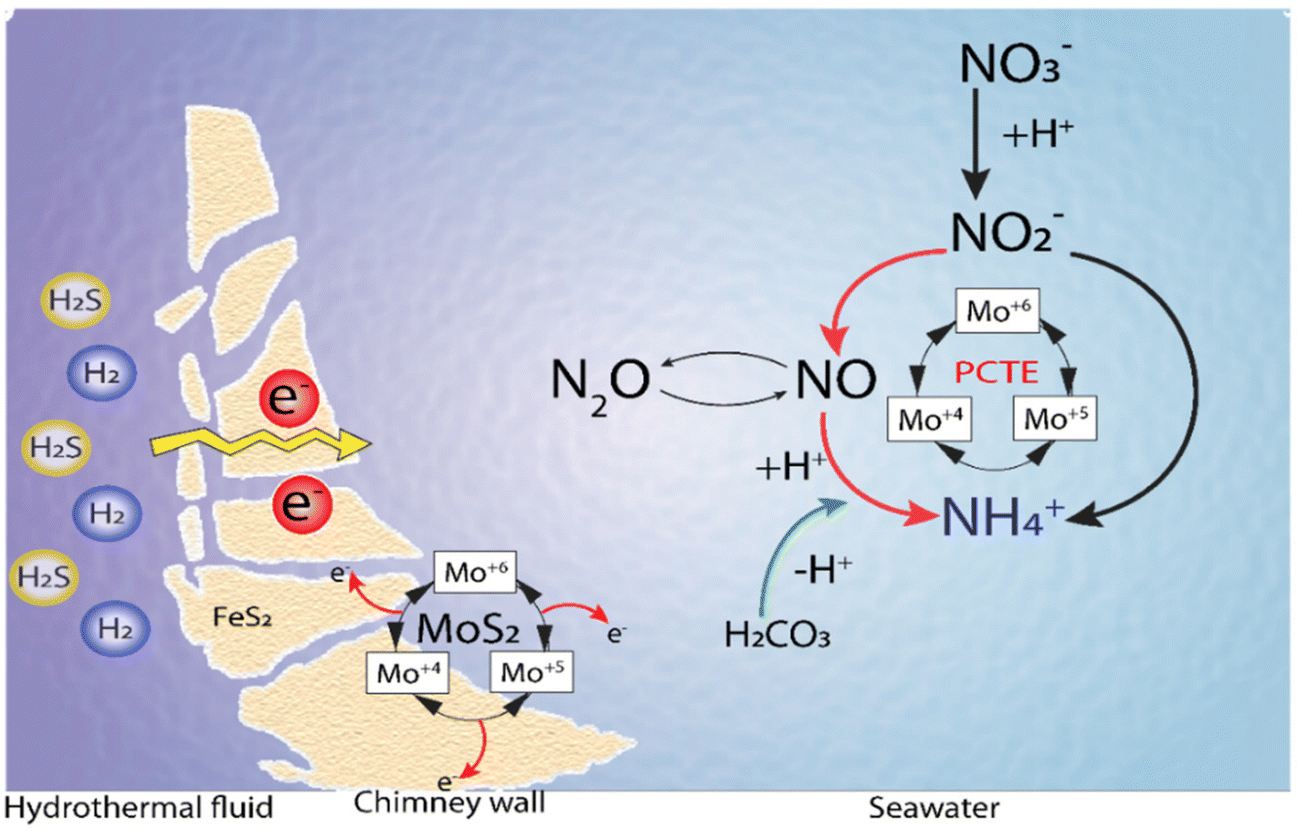 | ||
| Fig. 11 Ammonia synthesis by MoS2 under Hadean conditions. Adapted from Li et al.144 | ||
Thus, the ammonia production from either nitrate or nitrite in hydrothermal vent environments relies on the electron flow generated by the oxidation of H2/H2S, and the transfer of these electrons to nitrate/nitrite in a catalytic reaction mediated by Mo–S catalytic centers. The source of protons could be either the same reductant species (i.e., H2/H2S) or the acidic environment of the water surrounding the hydrothermal systems144 (Fig. 11).
Here it is important to highlight that most enzymes operate using PCET in their redox reactions.146,147 Also, the turnover frequencies (TOFs) found by nitrate/nitrite reduction using MoS2144 are similar to the TOF in the nitrate reductases (1.8–33 s−1),148,149 and the presence of the Mo–S bonds in denitrification enzymes suggests the link between Mo–sulfide materials and the evolution of the current enzymes.144
Nickel is also a metal which commonly co-exists with iron sulfide minerals. For instance, in hydrothermal systems it is common to find deposits of awaruite (Ni3Fe), and greigite (Fe3S4), and these minerals have been reported to effectively catalyse the reduction of CO2.150 Brandes et al.151 demonstrated that nickel sulfides are able to convert nitrates and nitrites to ammonia under hydrothermal vent conditions. Nickel is often associated with the active sites of key enzymes (i.e. hydrogenases, carbon monoxide dehydrogenase), and its presence in eight different enzymes suggests that this metal was a key element in the evolutive pathways of enzyme systems.152
Nickel compounds are also widely applied in catalysis. Nickel-based catalysts have proved to have significant efficiencies for key reactions. For example, the most efficient layer double hydroxides/metal oxo-hydroxides (LDH/MOOH) type water oxidation catalysts are made of NiFeCr/NiFeV.115,153,154 Notably, bulk iron nickel sulfides (Fe4.5Ni4.5S8) have been reported as highly reactive materials for the hydrogen evolution reaction (HER), reaching a current density (J) of 10 mA cm−2 and sustainable long term catalytic activities activity over a time (i.e., 170 h at J > 600 mA cm−2).155
Direct reduction vs. catalytic reduction
One interesting aspect of the iron sulfide materials is that they can act as both direct mediators of reduction as well as catalysts for reduction. We have recently studied the relationship between direct redox and catalytic chemistry as mediated by iron sulfides. Fig. 12 shows the selectivity of the reaction products obtained by iron sulfides as catalysts and contrasts it to that obtained from direct redox processes, at 1 day of reaction and pH = 4.5. Different iron sulfide members were tested, including the first precipitated product of Fe2+ and HS− commonly named amorphous/nanocrystalline mackinawite (FeSx) and some of more ordered phases such as greigite (Fe3S4), pyrite (FeS2) and troilite (FeS). In addition, we include a molybdenum sulfide (MoSx) as a reference for the evaluation of the effect of the incorporation of molybdenum into the FeSx structure. Three different colored bars divide the NO2− reduction products. Ammonium-produced (orange), other nitrogen-reduced products (i.e., ONRP = ΣN2O + NO + N2, grey) and the residual nitrite are given in blue.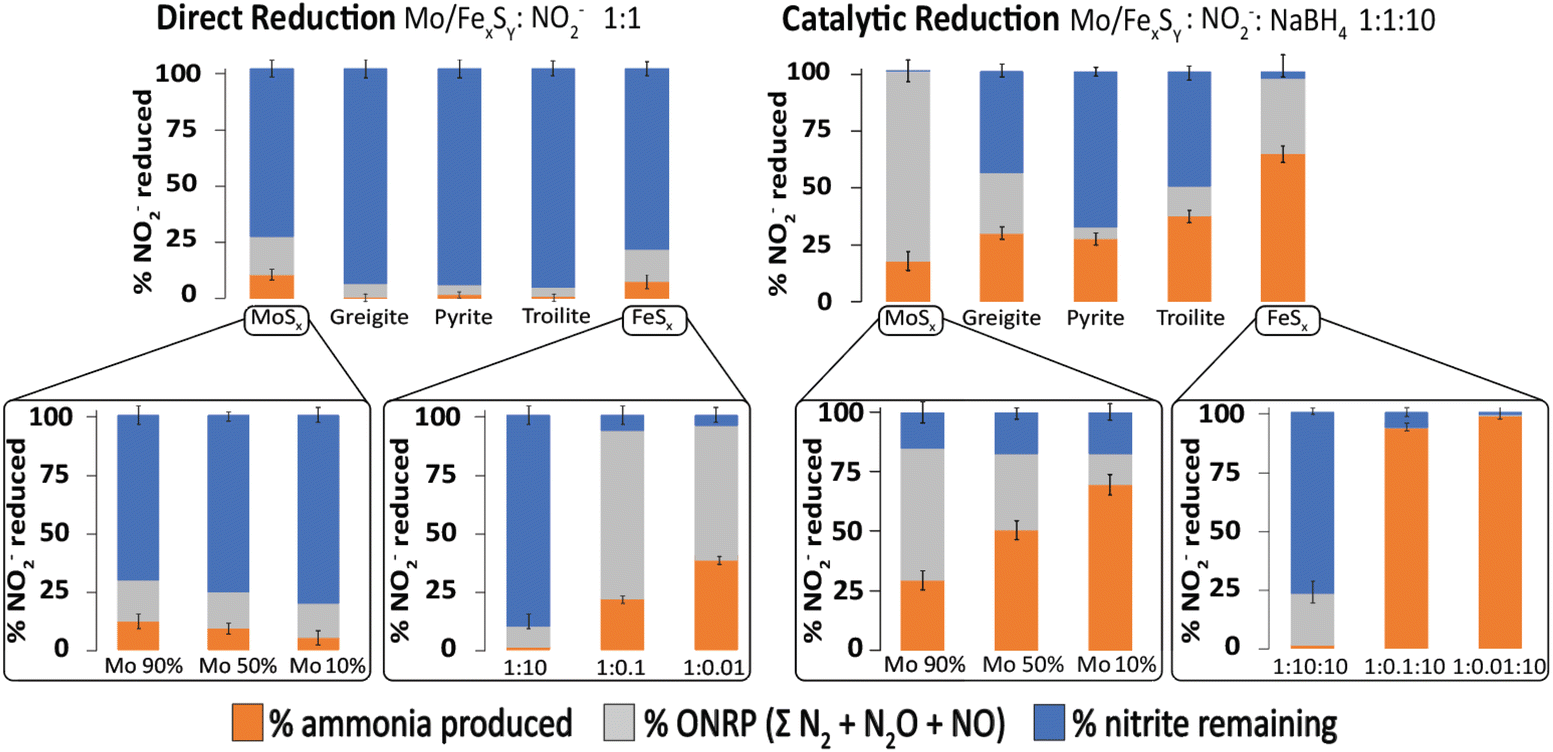 | ||
| Fig. 12 Comparison of catalytic to direct reduction of NO2− mediated by Mo/FexSy as observed at 1 day of reaction and pH = 4.5. Error bars represent the 95% confidence interval derived from three replicates. Adapted with permission from ref. 97, copyright 2023, American Chemical Society and from ref. 96 with permission from John Wiley and Sons, copyright 2022. | ||
In the context of the direct reduction reaction, it becomes evident that FeSx and MoSx samples exhibit high reactivity, compared to the more ordered phases. This could be explained as a consequence of the thermodynamic attributes inherent to disordered materials. It's important to note that this effect is not exclusive to iron sulfide samples; analogous phenomena have been reported for other materials.112,156 Moreover, the results shown in the insert graphs demonstrate that modulating the FeSx:NO2− ratio and the percentage of Mo integration into the FeSx structure shifts the distribution of reduction products.
In the catalytic reduction reaction, the same effects were observed, if the NO2− concentration is lower relative to FeSx, more ammonia is produced. An interesting observation is the role of Fe/Mo content in product selectivity, samples with high content of iron show selectivity for ONRP over ammonia, while the presence of iron favoured ammonia production over the ONRP. The Mo/FeSx samples with 10%Mo and 90% iron show the highest ammonia production and this is an interesting result because a similar ratio is found in nitrogenase.96,97
Thus, key thermodynamic factors play a role in the selectivity of products in nitrite reduction mediated by FexSy materials, including the concentration of the substrate relative to the catalyst, the amount of a secondary metal incorporated into the FexSy structure as well as the electron source and its chemistry.96,97
Perspectives in catalyst design
The electrochemical methods offer an attractive substitute for ammonia production over the Haber–Bosch process due to the fact that these can be performed at room temperature on a small scale and the energy required to power the process could be obtained by renewable electricity sources, particularly wind and solar.157–159 Promising improvements have been found in recent years in the field of electrochemical nitrogen reduction to produce ammonia under room temperature conditions, yet the challenge to find a material with the optimal balance of activity, selectivity, stability, and efficiency remains.160–162 No direct electrochemical process has been able to produce the significant amount of ammonia the Haber–Bosch process does. Several catalysts based on noble metals,163 metal nitrides,164 metal oxides,165 nitrogen and boron-doped carbon,166 and metal sulfides167 have been proposed for the electrochemical nitrogen reduction reaction. However, there have also been many problems with some of these catalysts with authors reporting false positives on selectivity for ammonia production.159,168Several aspects must be taken into account in the design of optimal catalysts for nitrogen fixation, the ideal catalyst should have high activity, selectivity for the desired products, stability and high efficiency (Fig. 13). The inherent activity of a given material is related to the electronic structure, and it could be tuned with heteroatom doping, or with a smart design of the defect/strain into the material, along with the pH. These could be the key to improvement of the activity of materials towards nitrogen fixation.162,169,170
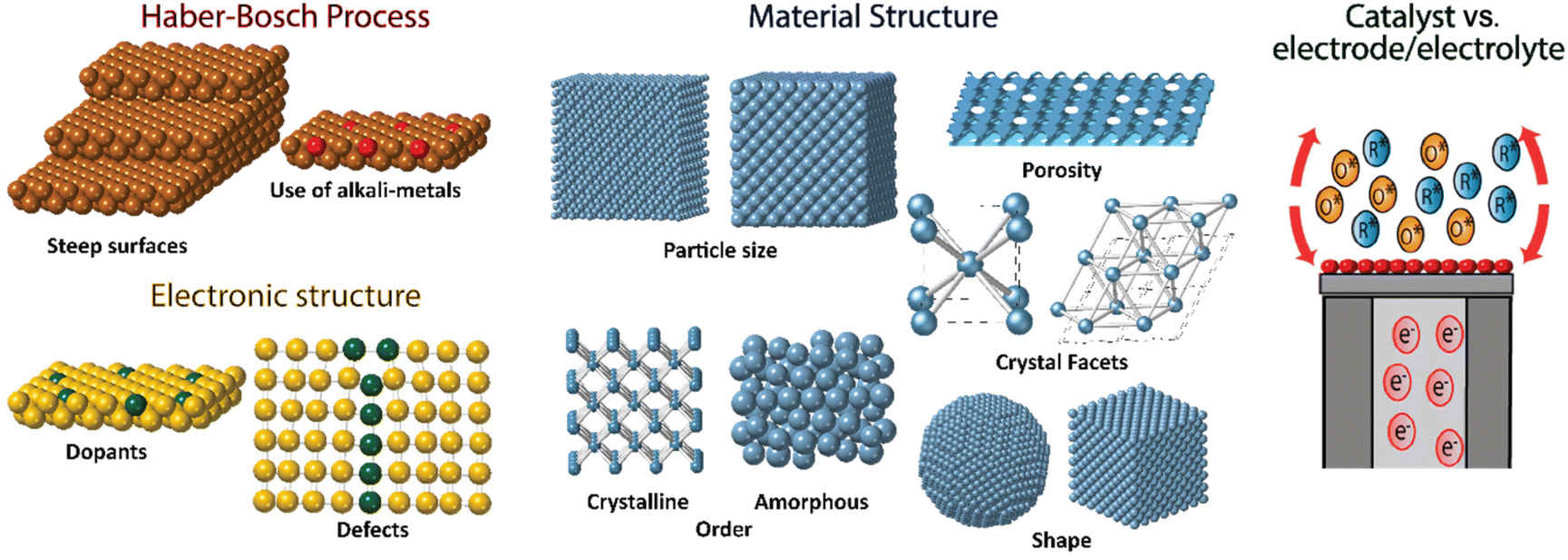 | ||
| Fig. 13 Proposed approaches to enhance heterogeneous electrochemical NH3 production from N2. | ||
Recent outcomes in the study of biological systems which mediated nitrogen fixation (enzymes), along with the pathways for abiotic natural ammonia formation in the early atmosphere and water bodies offer us a new perspective in the design of new materials able to produce ammonia under ambient conditions. Materials such as iron sulfides, which are present in both active sites of key enzymes for nitrogen fixation and in hydrothermal vents (most likely environments where key chemical reactions may happen for life's origin), could be good candidates for a new generation of catalysts.
Recent advancements in the investigation of biological systems, particularly those facilitating nitrogen fixation via enzymatic processes, along with the study of materials involved in abiotic routes to natural ammonia formation in early Earth, offer us a new perspective in the design of new materials able to produce ammonia under ambient conditions. Notably, iron sulfides, are present within the active sites of nitrogen-fixing enzymes and in the mineral structure of hydrothermal vents—environments likely key to primordial chemical reactions for life's origin—and emerge as promising materials for the next frontier of catalyst development.
Materials with the capacity to incorporate additional metals emerge as highly desirable candidates for the development of a new generation of catalysts. Multi-metal systems have received increased attention in catalysis in the last decade as they have been observed to exhibit higher activity/selectivity than the performance displayed by individual metals.171,172 Although many of the chemical properties of the single metals are preserved in a bimetallic system, the formation of heteroatom bonds changes the geometry and electronic structure of the metal surface, thus the chemical bonding between the metal catalysts and the key reaction intermediates is altered by the presence of the neighbours.173,174 This strategic incorporation of extra metals offers a versatile approach to fine-tuning reactions, thus enhancing the efficiency and selectivity of the catalytic process.
The complex interplay between a material's capacity to facilitate direct reduction and its role as a catalyst for reduction reactions could also play an important role in catalyst design. While direct reduction is commonly associated with thermodynamic effects and catalysis is governed by kinetic effects, both reactions are driven by electron transfer rates, which in turn rely on the underlying thermodynamic stability. Consequently, comprehending the thermodynamic stability of diverse materials and their modulation in response to factors such as pH and substrate concentration has a key importance in catalyst design.
Conclusions
In conclusion, we highlight that iron sulfide materials (FeSx) have a structural resemblance with metallo-enzymes (nitrogenases and hydrogenases) in terms of compositional structure and, to some extent, functionality. Both enzymes and iron sulfide materials are able to catalyse as well as directly mediate key reactions including the hydrogen evolution reaction and nitrate/nitrite reduction. The relationship between direct and catalytic reduction may be key for understanding some types of evolutionary processes. While we think about catalysis and direct reduction as distinct, if a material can be regenerated, as may be the case for some biological, nanoparticulate or metastable systems, a direct redox process can also be turned into a catalytic one. In the evolution of life, direct redox chemistry, in the form of chemolithotrophs whose metabolism comes from direct redox chemistry, likely preceded the catalytic processes of metalloenzymes (Fig. 14).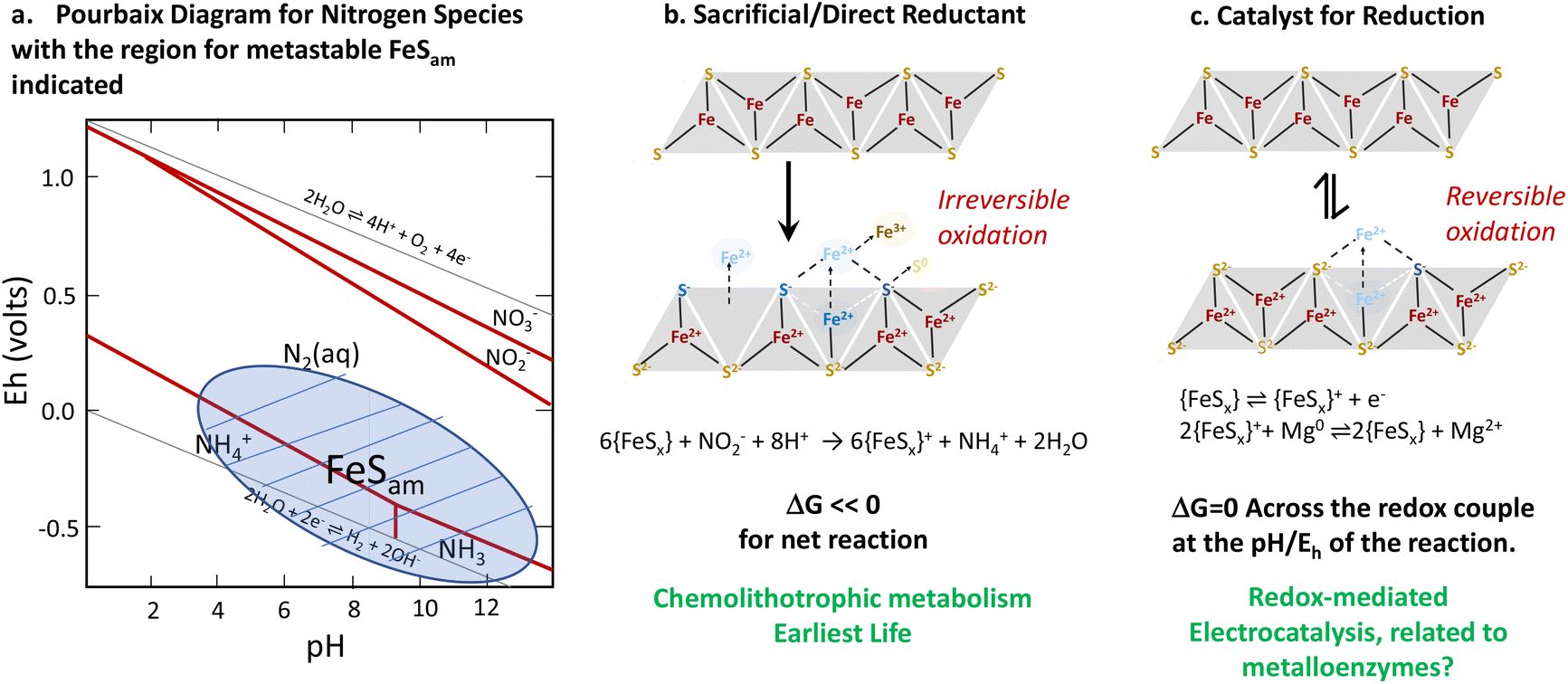 | ||
| Fig. 14 Summary of the thermodynamics of redox and catalytic processes involving iron sulfides (a). A relationship between chemolithotrophic life (b) and more complex life (c). Reproduced from ref. 96 with permission from John Wiley and Sons, copyright 2022. | ||
Despite the very interesting correlations with nitrogenase, the role of iron sulfide in the nitrogen reduction reaction is yet to be determined. It is possible that the highly disordered phases of iron sulfides are metastable with respect to some of the more established structures which may couple secondary redox processes in natural systems to perform nitrogen reduction. From our perspective, a clear and unambiguous system demonstrating this has not yet been developed.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge support for this project from the Australian Research Council via DP200101878. Dr. Rosalie Hocking is grateful to Swinburne University for a Vice Chancellors's Women in STEM Fellowship. Mr C. Felipe Garibello recognises a Swinburne SUPRA scholarship.References
-
V. Smil, Nitrogen in Agriculture, in Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, The MIT Press, 2000 Search PubMed
.
- T. Kandemir, M. E. Schuster, A. Senyshyn, M. Behrens and R. Schlögl, The Haber-Bosch Process Revisited: On the Real Structure and Stability of “Ammonia Iron” under Working Conditions, Angew. Chem., Int. Ed., 2013, 52(48), 12723–12726 CrossRef CAS PubMed
- B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, Nitrogenase: A Draft Mechanism, Acc. Chem. Res., 2013, 46(2), 587–595 CrossRef CAS PubMed
- B. Hoffman, D. Lukoyanov, Y. Zhi-Yong, D. Dean and L. Seefeldt, Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage, Chem. Rev., 2014, 114(8), 4041–4062 CrossRef CAS PubMed
- F. Möller, S. Piontek, R. G. Miller and U.-P. Apfel, From Enzymes to Functional Materials—Towards Activation of Small Molecules, Eur. J. Chem., 2018, 24(7), 1471–1493 CrossRef PubMed
- R. A. Alberty, Thermodynamics of the nitrogenase reactions, J. Biol. Chem., 1994, 269(10), 7099–7102 CrossRef CAS PubMed
- M. P. Shaver and M. D. Fryzuk, Activation of Molecular Nitrogen: Coordination, Cleavage and Functionalization of N2 Mediated By Metal Complexes, Adv. Synth. Catal., 2003, 345(9–10), 1061–1076 CrossRef CAS
- A. Tsuneto, A. Kudo and T. Sakata, Efficient Electrochemical Reduction of N2 to NH3 Catalyzed by Lithium, Chem. Lett., 1993, 22(5), 851–854 CrossRef
- V. Kordali, G. Kyriacou and C. Lambrou, Electrochemical Synthesis of Ammonia at Atmospheric Pressure and Low Temperature in a Solid Polymer Electrolyte Cell, Chem. Commun., 2000, 1673 RSC
- M. Coyne, Review of Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production by Vaclav Smil, Agric. Hist., 2005, 79(3), 383–384 Search PubMed
- R. A. Marcus and N. Sutin, Electron transfers in chemistry and biology, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811(3), 265–322 CrossRef CAS
-
J. J. Warren and J. M. Mayer, Proton-Coupled Electron Transfer, in Encyclopedia of Biophysics, ed. G. C. K. Roberts, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 2112–2114 Search PubMed
- J. F. Kasting and J. C. G. Walker, Limits on oxygen concentration in the prebiological atmosphere and the rate of abiotic fixation of nitrogen, J. Geophys. Res.: Oceans, 1981, 86(C2), 1147–1158 CrossRef CAS
- A. Henderson-Sellers and A. W. Schwartz, Chemical evolution and ammonia in the early Earth's atmosphere, Nature, 1980, 287(5782), 526–528 CrossRef CAS
- D. P. Summers and S. Chang, Prebiotic ammonia from reduction of nitrite by iron(II) on the early Earth, Nature, 1993, 365(6447), 630–633 CrossRef CAS PubMed
- J. Brandes, N. Boctor, G. Cody, B. A. Cooper, R. M. Hazen and H. Yoder, Abiotic nitrogen reduction on the early Earth, Nature, 1998, 395(6700), 365–367 CrossRef CAS PubMed
- J. Morse and F. Mackenzie, Hadean Ocean Carbonate Geochemistry, Aquat. Geochem., 1998, 4(3–4), 301–319 CrossRef CAS
- M. Dörr, J. Käßbohrer, R. Grunert, G. Kreisel, W. A. Brand, R. A. Werner, H. Geilmann, C. Apfel, C. Robl and W. Weigand, A Possible Prebiotic Formation of Ammonia from Dinitrogen on Iron Sulfide Surfaces, Angew. Chem., Int. Ed., 2003, 42(13), 1540–1543 CrossRef PubMed
-
H. D. Holland, The Chemical Evolution of the Atmosphere and Oceans, Princeton University Press, 1984, pp. 582 Search PubMed
- H. D. Holland, B. Lazar and M. McCaffrey, Evolution of the atmosphere and oceans, Nature, 1986, 320(6057), 27–33 CrossRef CAS PubMed
- J. F. Kasting, Theoretical constraints on oxygen and carbon dioxide concentrations in the Precambrian atmosphere, Precambrian Res., 1987, 34(3), 205–229 CrossRef CAS PubMed
- J. Kasting, K. Zahnle, J. Pinto and A. Young, Sulfur, ultraviolet radiation, and the early evolution of life, Origins Life Evol. Biospheres, 1989, 19(2), 95–108 CrossRef CAS PubMed
- J. F. Kasting, Bolide impacts and the oxidation state of carbon in the Earth's early atmosphere, Origins Life Evol. Biospheres, 1992, 20, 199–231 CrossRef CAS PubMed
- J. F. Kasting, D. H. Eggler and S. P. Raeburn, Mantle Redox Evolution and the Oxidation State of the Archean Atmosphere, J. Geol., 1993, 101(2), 245–257 CrossRef CAS PubMed
- G. H. Shaw, Earth's atmosphere – Hadean to early Proterozoic, Chem. Erde, 2008, 68(3), 235–264 CrossRef CAS
- B. S. Kamber, The evolving nature of terrestrial crust from the Hadean, through the Archaean, into the Proterozoic, Precambrian Res., 2015, 258, 48–82 CrossRef CAS
- J. F. Kasting, Stability of ammonia in the primitive terrestrial atmosphere, J. Geophys. Res., 1982, 87(C4), 3091–3098 CrossRef CAS
-
J. Kasting, Atmospheric composition of Hadean–early Archean Earth: The importance of CO, 2014, vol. 504, pp. 19–28 Search PubMed
- E. Nisbet and N. Sleep, The habitat and nature of early life, Nature, 2001, 409, 1083–1091 CrossRef CAS PubMed
-
D. L. Pinti, The Origin and Evolution of the Oceans, in Lectures in Astrobiology: Volume I, ed. M. Gargaud, B. Barbier, H. Martin and J. Reisse, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 83–112 Search PubMed
- K. Armstrong, D. J. Frost, C. A. McCammon, D. C. Rubie and T. Boffa Ballaran, Deep magma ocean formation set the oxidation state of Earth's mantle, Science, 2019, 365(6456), 903–906 CrossRef CAS PubMed
-
J. C. G. Walker, Evolution of the atmosphere/James C. G. Walker, Macmillan, New York, 1977 Search PubMed
- J. Farquhar, J. Savarino, S. Airieau and M. H. Thiemens, Observation of wavelength-sensitive mass-independent sulfur isotope effects during SO2 photolysis: Implications for the early atmosphere, J. Geophys. Res., 2001, 106(E12), 32829–32839 CrossRef CAS
- C. Goldblatt, M. W. Claire, T. M. Lenton, A. J. Matthews, K. J. Zahnle and A. J. Watson, Nitrogen-enhanced greenhouse warming on early Earth, Nat. Geosci., 2009, 2(12), 891–896 CrossRef CAS
-
J. T.-F. Wong and A. Lazcano, Prebiotic Evolution and Astrobiology, CRC Press LLC, Texas, UNITED STATES, 2009 Search PubMed
- M. J. Russell, A. J. Hall and W. Martin, Serpentinization as a source of energy at the origin of life, Geobiology, 2010, 8(5), 355–371 CrossRef CAS PubMed
- J. N. Bahcall, M. H. Pinsonneault and S. Basu, Solar Models: Current Epoch and Time Dependences, Neutrinos, and Helioseismological Properties, Astrophys. J., 2001, 555(2), 990–1012 CrossRef CAS
- C. Sagan and G. Mullen, Earth and Mars: Evolution of Atmospheres and Surface Temperatures, Science, 1972, 177(4043), 52–56 CrossRef CAS PubMed
- B. Marty, The origins and concentrations of water, carbon, nitrogen and noble gases on Earth, Earth Planet. Sci. Lett., 2012, 313–314, 56–66 CrossRef CAS
- S. Mikhail and D. A. Sverjensky, Nitrogen speciation in upper mantle fluids and the origin of Earth's nitrogen-rich atmosphere, Nat. Geosci., 2014, 7(11), 816–819 CrossRef CAS
- D. C. Catling and K. J. Zahnle, The Archean atmosphere, Sci. Adv., 2020, 6(9), eaax1420–eaax1420 CrossRef CAS PubMed
- R. M. Rosenberg and W. L. Peticolas, Henry's Law: A Retrospective, J. Chem. Educ., 2004, 81(11), 1647 CrossRef CAS
- M. Wong, B. Charnay, P. Gao, Y. Yung and M. Russell, Nitrogen Oxides in Early Earth's Atmosphere as Electron Acceptors for Life's Emergence, Astrobiology, 2017, 17(10), 975–983 CrossRef CAS PubMed
- D. Nna Mvondo, R. Navarro-González, C. P. McKay, P. Coll and F. Raulin, Production of nitrogen oxides by lightning and coronae discharges in simulated early earth, venus and mars environments, Adv. Space Res., 2001, 27(2), 217–223 CrossRef CAS PubMed
- A.-L. Ducluzeau, R. van Lis, S. Duval, B. Schoepp-Cothenet, M. J. Russell and W. Nitschke, Was nitric oxide the first deep electron sink?, Trends Biochem. Sci., 2009, 34(1), 9–15 CrossRef CAS
- R. L. Mancinelli and C. P. McKay, The evolution of nitrogen cycling, Origins Life Evol. Biospheres, 1988, 18, 311–325 CrossRef CAS PubMed
- M. Allen, Y. L. Yung and J. W. Waters, Vertical transport and photochemistry in the terrestrial mesosphere and lower thermosphere (50–120 km), J. Geophys. Res., 1981, 86(A5), 3617–3627 CrossRef CAS
- R. Navarro-González, M. J. Molina and L. T. Molina, Nitrogen fixation by volcanic lightning in the early Earth, Geophys. Res. Lett., 1998, 25(16), 3123–3126 CrossRef
- D. N. Mvondo, R. Navarro-González and C. P. McKay, A possible nitrogen crisis for Archaean life due to reduced nitrogen fixation by lightning, Nature, 2001, 412(6842), 61–64 CrossRef PubMed
- H. Lu, J. Tournet, K. Dastafkan, Y. Liu, Y. H. Ng, S. K. Karuturi, C. Zhao and Z. Yin, Noble-Metal-Free Multicomponent Nanointegration for Sustainable Energy Conversion, Chem. Rev., 2021, 121(17), 10271–10366 CrossRef CAS PubMed
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith and T. W. Hamann, Recent Advances and Challenges of Electrocatalytic N2 Reduction to Ammonia, Chem. Rev., 2020, 120(12), 5437–5516 CrossRef CAS PubMed
- G. N. Schrauzer, N. Strampach, L. N. Hui, M. R. Palmer and J. Salehi, Nitrogen photoreduction on desert sands under sterile conditions, Proc. Natl. Acad. Sci. U. S. A., 1983, 80(12), 3873–3876 CrossRef CAS PubMed
- K. Ranjit and B. Viswanathan, Photocatalytic reduction of nitrite and nitrate ions over doped TiO2 catalysts, J. Photochem. Photobiol., A, 1997, 107(1–3), 215–220 CrossRef CAS
- M. Kusakabe, G. Z. Tanyileke, S. A. McCord and S. G. Schladow, Recent pH and CO2 profiles at Lakes Nyos and Monoun, Cameroon: implications for the degassing strategy and its numerical simulation, J. Volcanol. Geotherm., 2000, 97(1–4), 241–260 CrossRef CAS
- G. Macleod, C. McKeown, A. Hall and M. Russell, Hydrothermal and oceanic pH conditions of possible relevance to the origin of life, Origins Life Evol. Biospheres, 1994, 24(1), 19–41 CrossRef CAS PubMed
- J. Korenaga, N. J. Planavsky and D. A. D. Evans, Global water cycle and the coevolution of the Earth’s interior and surface environment, Philos. Trans. R. Soc., A, 2017, 375(2094), 20150393 CrossRef PubMed
- F. Westall, K. Hickman-Lewis, N. Hinman, P. Gautret, K. A. Campbell, J.-G. Bréhéret, F. Foucher, A. Hubert, S. Sorieul, A. V. Dass, T. P. Kee, T. Georgelin and A. Brack, A Hydrothermal-Sedimentary Context for the Origin of Life, Astrobiology, 2018, 18(3), 259–293 CrossRef CAS PubMed
- A. Hofmann and C. Harris, Silica alteration zones in the Barberton greenstone belt: A window into subseafloor processes 3.5–3.3 Ga ago, Chem. Geol., 2008, 257(3), 221–239 CrossRef
- H. D. Holland, Volcanic gases, black smokers, and the great oxidation event, Geochim. Cosmochim. Acta, 2002, 66(21), 3811–3826 CrossRef CAS
- Y. Yamagata, H. Watanabe, M. Saitoh and T. Namba, Volcanic production of polyphosphates and its relevance to prebiotic evolution, Nature, 1991, 352(6335), 516 CrossRef CAS PubMed
- W. Hagan, A. Parker, A. Steuerwald and M. Hathaway, Phosphate Solubility and
the Cyanate-Mediated Synthesis of Pyrophosphate, Origins Life Evol. Biospheres, 2007, 37(2), 113–122 CrossRef CAS PubMed
- D. Trail, E. Bruce Watson and N. D. Tailby, The oxidation state of Hadean magmas and implications for early Earth's atmosphere, Nature, 2011, 480(7375), 79–82 CrossRef CAS PubMed
- T. Shibuya, M. J. Russell and K. Takai, Free energy distribution and hydrothermal mineral precipitation in Hadean submarine alkaline vent systems: Importance of iron redox reactions under anoxic conditions, Geochim. Cosmochim. Acta, 2016, 175, 1–19 CrossRef CAS
- D. P. Summers, Sources and Sinks for Ammonia and Nitrite on the Early Earth and the Reaction of Nitrite with Ammonia, Origins Life Evol. Biospheres, 1999, 29(1), 33–46 CrossRef CAS PubMed
- D. S. Kelley, J. A. Baross and J. R. Delaney, VOLCANOES, FLUIDS, AND LIFE AT MID-OCEAN RIDGE SPREADING CENTERS, Annu. Rev. Earth Planet. Sci., 2002, 30(1), 385–491 CrossRef CAS
- A. Y. Mulkidjanian, On the origin of life in the Zinc world: 1. Photosynthesizing, porous edifices built of hydrothermally precipitated zinc sulfide as cradles of life on Earth.(Hypothesis)(Report), Biol. Direct, 2009, 4, 26 CrossRef
- J. F. Kasting and T. P. Ackerman, Climatic Consequences of Very High Carbon Dioxide Levels in the Earth's Early Atmosphere, Science, 1986, 234(4782), 1383–1385 CrossRef CAS
- W. Martin, J. Baross, D. Kelley and M. J. Russell, Hydrothermal vents and the origin of life, Nat. Rev. Microbiol., 2008, 6(11), 805–814 CrossRef CAS
- T. Shibuya, M. Yoshizaki, M. Sato, K. Shimizu, K. Nakamura, S. Omori, K. Suzuki, K. Takai, H. Tsunakawa and S. Maruyama, Hydrogen-rich hydrothermal environments in the Hadean ocean inferred from serpentinization of komatiites at 300 °C and 500 bar, Prog. Earth Planet. Sci., 2015, 2(1), 46 CrossRef
- K. R. Olson and K. D. Straub, The Role of Hydrogen Sulfide in Evolution and the Evolution of Hydrogen Sulfide in Metabolism and Signaling, Physiology, 2016, 31(1), 60 CrossRef CAS
-
P. Martina, C. X. Joana, L. S. Filipa, Z. Verena, N. Anna, Q. L. Susan, H. C. Greenwell, K. Karl, T. Harun, M. M. Tom, G. H. Nils and F. M. William, Serpentinization: Connecting Geochemistry, Ancient Metabolism and Industrial Hydrogenation. 2018, vol. 8, p. 41 Search PubMed
- K. A. Ludwig, D. S. Kelley, D. A. Butterfield, B. K. Nelson and G. Früh-Green, Formation and evolution of carbonate chimneys at the Lost City Hydrothermal Field, Geochim. Cosmochim. Acta, 2006, 70(14), 3625–3645 CrossRef CAS
- Y. Fouquet, U. V. Stackelberg, J. L. Charlou, J. P. Donval, J. Erzinger, J. P. Foucher, P. Herzig, R. Mühe, S. Soakai, M. Wiedicke and H. Whitechurch, Hydrothermal activity and metallogenesis in the Lau back-arc basin, Nature, 1991, 349(6312), 778 CrossRef CAS
- H. Hsu-Kim, K. Mullaugh, J. Tsang, M. Yucel and G. Luther, Formation of Zn- and Fe-sulfides near hydrothermal vents at the Eastern Lau Spreading Center: implications for sulfide bioavailability to chemoautotrophs, Geochem. Trans., 2008, 9(1), 1–14 CrossRef PubMed
- R. R. Large, I. Mukherjee, D. D. Gregory, J. A. Steadman, V. V. Maslennikov and S. Meffre, Ocean and Atmosphere Geochemical Proxies Derived from Trace Elements in Marine Pyrite: Implications for Ore Genesis in Sedimentary Basins, Econ. Geol., 2017, 112(2), 423–450 CrossRef
- S. D. Kelley, A. K. Jeffrey, K. B. Donna, L. F.-G. Gretchen, A. B. David, D. L. Marvin, J. O. Eric, O. S. Matthew, K. R. Kevin, T. L. Geoff, R. Pete and P. The At3-60 Shipboard, An off-axis hydrothermal vent field near the Mid-Atlantic Ridge at 30° N, Nature, 2001, 412(6843), 145 CrossRef PubMed
-
S. E. Humphris and M. K. Tivey, A synthesis of geological and geochemical investigations of the TAG hydrothermal field: Insights into fluid-flow and mixing processes in a hydrothermal system, in Ophiolites and oceanic crust: new insights from field studies and the Ocean Drilling Program, Geological Society of America, 2000, vol. 349 Search PubMed
-
Y. Nozaki, in Elemental Distribution: Overview, Elsevier Ltd, 2nd edn, 2009, pp. 255–260 Search PubMed
- T. Gamo, H. Chiba, T. Yamanaka, T. Okudaira, J. Hashimoto, S. Tsuchida, J.-I. Ishibashi, S. Kataoka, U. Tsunogai, K. Okamura, Y. Sano and R. Shinjo, Chemical characteristics of newly discovered black smoker fluids and associated hydrothermal plumes at the Rodriguez Triple Junction, Central Indian Ridge, Earth Planet. Sci. Lett., 2001, 193(3–4), 371–379 CrossRef CAS
-
D. Desbruyères and M. Segonzac, Handbook of Deep-sea Hydrothermal Vent Fauna, Quæ, Versailles, FRANCE, 1997 Search PubMed
- M. A. Schoonen and Y. Xu, Nitrogen reduction under hydrothermal vent conditions: implications for the prebiotic synthesis of C-H-O-N compounds, Astrobiology, 2001, 1(2), 133 CrossRef CAS PubMed
- E. L Shock and M. D. Schulte, Organic synthesis during fluid mixing in hydrothermal systems, J. Geophys. Res., 1998, 103(E12), 28513–28527 CrossRef CAS
- M. Yamamoto, R. Nakamura, T. Kasaya, H. Kumagai, K. Suzuki and K. Takai, Spontaneous and Widespread Electricity Generation in Natural Deep–Sea Hydrothermal Fields, Angew. Chem., Int. Ed., 2017, 56(21), 5725–5728 CrossRef CAS PubMed
- K. G. Bemis, R. P. Von Herzen and M. J. Mottl, Geothermal heat flux from hydrothermal plumes on the Juan de Fuca Ridge, J. Geophys. Res., 1993, 98(B4), 6351–6365 CrossRef
- R. Lowell, P. A. Rona and R. Vonherzen, SEA-FLOOR HYDROTHERMAL SYSTEMS, J. Geophys. Res.: Solid Earth, 1995, 100(B1), 327–352 CrossRef CAS
- W. Martin and M. J. Russell, On the origins of cells: a hypothesis for the evolutionary transitions from abiotic geochemistry to chemoautotrophic prokaryotes, and from prokaryotes to nucleated cells, Philos. Trans. R. Soc., B, 2003, 358(1429), 59–85 CrossRef CAS
-
R. de Graaf, V. Sojo and R. Hudson, Catalysis at the Origin of Life, 2022 Search PubMed
- T. Nunoura, Y. Chikaraishi, R. Izaki, T. Suwa, T. Sato, T. Harada, K. Mori, Y. Kato, M. Miyazaki, S. Shimamura, K. Yanagawa, A. Shuto, N. Ohkouchi, N. Fujita, Y. Takaki, H. Atomi and K. Takai, A primordial and reversible TCA cycle in a facultatively chemolithoautotrophic thermophile, Science, 2018, 359(6375), 559 CrossRef CAS PubMed
- S. Marakushev and O. G. Belonogova, The Divergence and Natural Selection of Autocatalytic Primordial Metabolic Systems, J. Int. Astrobiol. Soc., 2013, 43(3), 263–281 CAS
-
B. H. Kim, Bacterial physiology and metabolism, Cambridge, New York, 2008 Search PubMed
- I. Suzuki, Oxidation of elemental sulfur by an enzyme system of Thiobacillus thiooxidans, Biochim. Biophys. Acta, Gen. Subj., 1965, 104(2), 359–371 CrossRef CAS PubMed
- W. Sand, Ferric iron reduction by Thiobacillus ferrooxidans at extremely low pH-values, Biogeochemistry, 1989, 7(3), 195–201 CrossRef CAS
- J. Thiel, J. M. Byrne, A. Kappler, B. Schink and M. Pester, Pyrite formation from FeS and H2S is mediated through microbial redox activity, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(14), 6897 CrossRef CAS PubMed
- L. M. White, R. Bhartia, G. D. Stucky, I. Kanik and M. J. Russell, Mackinawite and greigite in ancient alkaline hydrothermal chimneys: Identifying potential key catalysts for emergent life, Earth Planet. Sci. Lett., 2015, 430, 105–114 CrossRef CAS
- E. L. Shock and M. D. Schulte, Organic synthesis during fluid mixing in hydrothermal systems, J.
Geophys. Res.: Planets, 1998, 103(E12), 28513–28527 CrossRef CAS
- C. F. Garibello, A. N. Simonov, D. S. Eldridge, F. Mahlerbe and R. K. Hocking, Redox properties of iron sulfides: direct versus catalytic reduction and implications for catalyst design, ChemCatChem, 2022, 14(12), e202200270 CrossRef CAS
- C. F. Garibello, A. N. Simonov, S. L. Y. Chang, B. Johannessen, F. Malherbe, D. S. Eldridge and R. K. Hocking, Tuning Catalyst Selectivity for Ammonia vs Hydrogen: An Investigation into the Coprecipitation of Mo and Fe Sulfides, Inorg. Chem., 2023, 62(24), 9379–9390 CrossRef CAS PubMed
- W. Gilhooly, D. Fike, G. Druschel, F.-C. Kafantaris, R. Price and J. Amend, Sulfur and oxygen isotope insights into sulfur cycling in shallow-sea hydrothermal vents, Milos, Greece, Geochem. Trans., 2014, 15(1), 1–19 CrossRef PubMed
- G. Wächtershäuser, Before enzymes and templates: theory of surface metabolism, Microbiol. Rev., 1988, 52(4), 452–484 CrossRef PubMed
- M. J. Russell and A. J. Hall, The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front, J. Geol. Soc. London, 1997, 154(3), 377–402 CrossRef CAS PubMed
- A. Mulkidjanian and M. Galperin, On the origin of life in the Zinc world. 2. Validation of the hypothesis on the photosynthesizing zinc sulfide edifices as cradles of life on Earth, Biol. Direct, 2009, 4(27) DOI:10.1186/1745-6150-4-27
- R. Nakamura, T. Takashima, S. Kato, K. Takai, M. Yamamoto and K. Hashimoto, Electrical Current Generation across a Black Smoker Chimney, Angew. Chem., Int. Ed., 2010, 49(42), 7692–7694 CrossRef CAS PubMed
- N. Kitadai, R. Nakamura, M. Yamamoto, K. Takai, Y. Li, A. Yamaguchi, A. Gilbert, Y. Ueno, N. Yoshida and Y. Oono, Geoelectrochemical CO production: Implications for the autotrophic origin of life, Sci. Adv., 2018, 4(4), eaao7265 CrossRef PubMed
- K. Chu, H. Nan, Q. Li, Y. Guo, Y. Tian and W. Liu, Amorphous MoS3 enriched with sulfur vacancies for efficient electrocatalytic nitrogen reduction, J. Energy Chem., 2021, 53, 132–138 CrossRef CAS
- K. Chu, J. Wang, Y.-p. Liu, Q.-q. Li and Y.-l. Guo, Mo-doped SnS2 with enriched S-vacancies for highly efficient electrocatalytic N2 reduction: the critical role of the Mo–Sn–Sn trimer, J. Mater. Chem. A, 2020, 8(15), 7117–7124 RSC
- C.-S. Horng and A. P. Roberts, Authigenic or detrital origin of pyrrhotite in sediments?: Resolving a paleomagnetic conundrum, Earth Planet. Sci. Lett., 2006, 241(3), 750–762 CrossRef CAS
- J. C. Larrasoaña, A. P. Roberts, R. J. Musgrave, E. Gràcia, E. Piñero, M. Vega and F. Martínez-Ruiz, Diagenetic formation of greigite and pyrrhotite in gas hydrate marine sedimentary systems, Earth Planet. Sci. Lett., 2007, 261(3), 350–366 CrossRef
- S. Hunger and L. Benning, Greigite: a true intermediate on the polysulfide pathway to pyrite, Geochem. Trans., 2007, 8(1), 1–20 CrossRef PubMed
- D. Rickard and G. W. Luther, Chemistry of iron sulfides, Chem. Rev., 2007, 107(2), 514 CrossRef CAS PubMed
- S. A. Sanden, R. K. Szilagyi, Y. Li, N. Kitadai, S. M. Webb, T. Yano, R. Nakamura, M. Hara and S. E. McGlynn, Electrochemically induced metal- vs. ligand-based redox changes in mackinawite: identification of a Fe3+- and polysulfide-containing intermediate, Dalton Trans., 2021, 50(34), 11763–11774 RSC
- Y. Li, N. Kitadai and R. Nakamura, Chemical Diversity of Metal Sulfide Minerals and Its Implications for the Origin of Life, Life, 2018, 8(4), 46 CrossRef CAS
- M. Sabri, H. J. King, R. J. Gummow, X. Lu, C. Zhao, M. Oelgemöller, S. L. Y. Chang and R. K. Hocking, Oxidant or Catalyst for Oxidation? A Study of How Structure and Disorder Change the Selectivity for Direct versus Catalytic Oxidation Mediated by Manganese(III,IV) Oxides, Chem. Mater., 2018, 30(22), 8244–8256 CrossRef CAS
- R. R. Chianelli, Periodic Trends Transition Metal Sulfide Catalysis:Intuition and Theory, Oil Gas Sci. Technol., 2006, 61(4), 503–513 CrossRef CAS
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Electrosynthesis, functional, and structural characterization of a water-oxidizing manganese oxide, Energy Environ. Sci., 2012, 5(5), 7081–7089 RSC
- M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Layered manganese oxides for water-oxidation: alkaline earth cations influence catalytic activity in a photosystem II-like fashion, Chem. Sci., 2012, 3(7), 2330–2339 RSC
- R. Hudson, R. de Graaf, M. S. Rodin, A. Ohno, N. Lane, S. E. McGlynn, Y. M. A. Yamada, R. Nakamura, L. M. Barge, D. Braun and V. Sojo, CO2 reduction driven by a pH gradient, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(37), 22873–22879 CrossRef CAS PubMed
- S. Singireddy, A. Gordon, A. Smirnov, M. Vance, M. Schoonen, R. Szilagyi and D. Strongin, Reduction of Nitrite and Nitrate to Ammonium on Pyrite, Int. J. Astrobiol., 2012, 42(4), 275–294 CAS
- D. Summers, Ammonia Formation By The Reduction Of Nitrite/Nitrate By Fes: Ammonia Formation Under Acidic Conditions, Origins Life Evol. Biospheres, 2005, 35(4), 299–312 CrossRef CAS PubMed
- D. P. Summers and S. Chang, Prebiotic ammonia from reduction of nitrite by iron(II) on the early Earth, Nature, 1993, 365(6447), 630 CrossRef CAS PubMed
- C. Di Giovanni, W.-A. Wang, S. Nowak, J.-M. Grenèche, H. Lecoq, L. Mouton, M. Giraud and C. Tard, Bioinspired Iron Sulfide Nanoparticles for Cheap and Long-Lived Electrocatalytic Molecular Hydrogen Evolution in Neutral Water, ACS Catal., 2014, 4(2), 681–687 CrossRef CAS
- C. D. Giovanni, Á. Reyes-Carmona, A. Coursier, S. Nowak, J. M. Grenèche, H. Lecoq, L. Mouton, J. Rozière, D. Jones, J. Peron, M. Giraud and C. Tard, Low-Cost Nanostructured Iron Sulfide Electrocatalysts for PEM Water Electrolysis, ACS Catal., 2016, 6(4), 2626–2631 CrossRef CAS
- J. Liu, K. T. Valsaraj, I. Devai and R. D. Delaune, Immobilization of aqueous Hg(II) by mackinawite (FeS), J. Hazard. Mater., 2008, 157(2), 432–440 CrossRef CAS PubMed
- D. Renock, T. Gallegos, S. Utsunomiya, K. Hayes, R. C. Ewing and U. Becker, Chemical and structural characterization of As immobilization by nanoparticles of mackinawite (FeSm), Chem. Geol., 2009, 268(1–2), 116–125 CrossRef CAS
- A. Matamoros-Veloza, O. Cespedes, B. R. G. Johnson, T. M. Stawski, U. Terranova, N. H. de Leeuw and L. G. Benning, A highly reactive precursor in the iron sulfide system, Nat. Commun., 2018, 9(1), 3125 CrossRef PubMed
- H. Y. Jeong and K. F. Hayes, Reductive Dechlorination of Tetrachloroethylene and Trichloroethylene by Mackinawite (FeS) in the Presence of Metals: Reaction Rates (Book review), Environ. Sci. Technol., 2007, 41(18), 6390–6396 CrossRef CAS PubMed
- E. C. Butler and K. F. Hayes, Kinetics of the Transformation of Trichloroethylene and Tetrachloroethylene by Iron Sulfide (Book review), Environ. Sci. Technol., 1999, 33(12), 2021–2027 CrossRef CAS
- D. Rickard, A. Griffith, A. Oldroyd, I. B. Butler, E. Lopez-Capel, D. A. C. Manning and D. C. Apperley, The composition of nanoparticulate mackinawite, tetragonal iron(II) monosulfide, Chem. Geol., 2006, 235(3), 286–298 CrossRef CAS
- J. W. Morse, F. J. Millero, J. C. Cornwell and D. Rickard, The chemistry of the hydrogen sulfide and iron sulfide systems in natural waters, Earth-Sci. Rev., 1987, 24(1), 1–42 CrossRef CAS
- M. Wolthers, L. Charlet, P. R. van Der Linde, D. Rickard and C. H. van Der Weijden, Surface chemistry of disordered mackinawite (FeS), Geochim. Cosmochim. Acta, 2005, 69(14), 3469–3481 CrossRef CAS
- H. Ohfuji and D. Rickard, High resolution transmission electron microscopic study of synthetic nanocrystalline mackinawite, Earth Planet. Sci. Lett., 2006, 241(1–2), 227–233 CrossRef CAS
- H. Y. Jeong, J. H. Lee and K. F. Hayes, Characterization of synthetic nanocrystalline mackinawite: Crystal structure, particle size, and specific surface area, Geochim. Cosmochim. Acta, 2008, 72(2), 493–505 CrossRef CAS PubMed
- B. J. Skinner, R. C. Erd and F. S. Grimaldi, Greigite, the thio-spinel of iron; a new mineral, Am. Mineral., 1964, 49, 543–555 CAS
- C. Huber and G. Wächtershäuser, Activated Acetic Acid by Carbon Fixation on (Fe,Ni)S Under Primordial Conditions, Science, 1997, 276(5310), 245–247 CrossRef CAS PubMed
- G. Wächtershäuser, Evolution of the first metabolic cycles, Proc. Natl. Acad. Sci. U. S. A., 1990, 87(1), 200–204 CrossRef PubMed
- G. Wächtershäuser, Groundworks for an evolutionary biochemistry: The iron-sulphur world, Prog. Biophys. Mol., 1992, 58, 85–201 CrossRef PubMed
- A. P. Chandra and A. R. Gerson, The mechanisms of pyrite oxidation and leaching: A fundamental perspective, Surf. Sci. Rep., 2010, 65(9), 293–315 CrossRef CAS
- I. Uhlig, R. Szargan, H. W. Nesbitt and K. Laajalehto, Surface states and reactivity of pyrite and marcasite, Appl. Surf. Sci., 2001, 179(1), 222–229 CrossRef CAS
- H.-H. Fan, H.-H. Li, K.-C. Huang, C.-Y. Fan, X.-Y. Zhang, X.-L. Wu and J.-P. Zhang, Metastable Marcasite-FeS2 as a New Anode Material for Lithium Ion Batteries: CNFs-Improved Lithiation/Delithiation Reversibility and Li-Storage Properties, ACS Appl. Mater. Interfaces, 2017, 9(12), 10708–10716 CrossRef CAS PubMed
- N. Belzile, Y.-W. Chen, M.-F. Cai and Y. Li, A review on pyrrhotite oxidation, J. Geochem. Explor., 2004, 84(2), 65–76 CrossRef CAS
- H. T. Evans, Lunar Troilite: Crystallography, Science, 1970, 167(3918), 621 CrossRef CAS PubMed
- P. S. David and C. Sherwood, Prebiotic ammonia from reduction of nitrite by iron(II) on the early Earth, Nature, 1993, 365(6447), 630 CrossRef PubMed
- M. J. Kampschreur, R. Kleerebezem, W. W. J. M. de Vet and M. C. M. van Loosdrecht, Reduced iron induced nitric oxide and nitrous oxide emission, Water Res., 2011, 45(18), 5945–5952 CrossRef CAS PubMed
- A. Gordon, A. Smirnov, S. Shumlas, S. Singireddy, M. DeCesare, M. Schoonen and D. Strongin, Reduction of Nitrite and Nitrate on Nano-dimensioned FeS, Int. J. Astrobiol., 2013, 43(4), 305–322 Search PubMed
- Y. Li, A. Yamaguchi, M. Yamamoto, K. Takai and R. Nakamura, Molybdenum Sulfide: A Bioinspired Electrocatalyst for Dissimilatory Ammonia Synthesis with Geoelectrical Current, J. Phys. Chem. C, 2017, 121(4), 2154–2164 CrossRef CAS
- M. R. Waterland, D. Stockwell and A. M. Kelley, Symmetry breaking effects in NO 3 − : Raman spectra of nitrate salts and ab initio resonance Raman spectra of nitrate–water complexes, Chem. Phys., 2001, 114(14), 6249–6258 CAS
- L. B. Maia and J. J. G. Moura, How Biology Handles Nitrite, Chem. Rev., 2014, 114(10), 5273–5357 CrossRef CAS PubMed
- A. Magalon, M. Asso, B. Guigliarelli, R. A. Rothery, P. Bertrand, G. Giordano and F. Blasco, Molybdenum Cofactor Properties and [Fe-S] Cluster Coordination in Escherichia coli Nitrate Reductase A: Investigation by Site-Directed Mutagenesis of the Conserved His-50 Residue in the NarG Subunit, Biochemistry, 1998, 37(20), 7363–7370 CrossRef CAS PubMed
- V. V. Pollock, R. C. Conover, M. K. Johnson and M. J. Barber, Bacterial expression of the molybdenum domain of assimilatory nitrate reductase: production of both the functional molybdenum-containing domain and the nonfunctional tungsten analog, Arch. Biochem. Biophys., 2002, 403(2), 237–248 CrossRef CAS PubMed
- I. Lambeck, J. C. Chi, S. Krizowski, S. Mueller, N. Mehlmer, M. Teige, K. Fischer and G. Schwarz, Kinetic analysis of 14-3-3-inhibited arabidopsis thaliana nitrate reductase, Biochemistry, 2010, 49(37), 8177–8186 CrossRef CAS PubMed
- M. Preiner, K. Igarashi, K. B. Muchowska, M. Yu, S. J. Varma, K. Kleinermanns, M. K. Nobu, Y. Kamagata, H. Tüysüz, J. Moran and W. F. Martin, A hydrogen-dependent geochemical analogue of primordial carbon and energy metabolism, Nat. Ecol. Evol., 2020, 4(4), 534–542 CrossRef PubMed
- J. A. Brandes, R. M. Hazen and H. S. Yoder Jr., Inorganic nitrogen reduction and stability under simulated hydrothermal conditions, Astrobiology, 2008, 8, 1113–1126 CrossRef CAS PubMed
- S. W. Ragsdale, Nickel-based Enzyme Systems, J. Biol. Chem., 2009, 284(28), 18571–18575 CrossRef CAS PubMed
- H. J. King, S. A. Bonke, S. L. Y. Chang, L. Spiccia, B. Johannessen and R. K. Hocking, Engineering Disorder into Heterogenite-Like Cobalt Oxides by Phosphate Doping: Implications for the Design of Water-Oxidation Catalysts, ChemCatChem, 2017, 9(3), 511–521 CrossRef CAS
- C. K. Mavrokefalos and G. R. Patzke, Water Oxidation Catalysts: The Quest for New Oxide-Based Materials, Inorganics, 2019, 7(3), 29 CrossRef CAS
- C. L. Bentley, C. Andronescu, M. Smialkowski, M. Kang, T. Tarnev, B. Marler, P. R. Unwin, U. P. Apfel and W. Schuhmann, Local Surface Structure and Composition Control the Hydrogen Evolution Reaction on Iron Nickel Sulfides, Angew. Chem., Int. Ed., 2018, 57(15), 4093–4097 CrossRef CAS PubMed
- R. K. Hocking, H. J. King, A. Hesson, S. A. Bonke, B. Johannessen, M. Fekete, L. Spiccia and S. L. Y. Chang, Engineering Disorder at a Nanoscale: A Combined TEM and XAS Investigation of Amorphous versus Nanocrystalline Sodium Birnessite, Aust. J. Chem., 2015, 68(11), 1715–1722 CrossRef CAS
- N. Morlanés, S. P. Katikaneni, S. N. Paglieri, A. Harale, B. Solami, S. M. Sarathy and J. Gascon, A technological roadmap to the ammonia energy economy: Current state and missing technologies, Chem. Eng. J., 2021, 408, 127310 CrossRef
- A. Valera-Medina, H. Xiao, M. Owen-Jones, W. I. F. David and P. J. Bowen, Ammonia for power, Prog. Energy Combust. Sci., 2018, 69, 63–102 CrossRef
- B. H. R. Suryanto, H. L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Challenges and Prospects in the Catalysis of Electroreduction of Nitrogen to Ammonia, Nat. Catal., 2019, 2, 290 CrossRef CAS
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Progress in the Electrochemical Synthesis of Ammonia, Catal. Today, 2017, 286, 2–13 CrossRef CAS
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, A Universal Principle for a Rational Design of Single-Atom Electrocatalysts, Nat. Catal., 2018, 1, 339 CrossRef CAS
- X. Cui, C. Tang and Q. Zhang, A Review of Electrocatalytic Reduction of Dinitrogen to Ammonia under Ambient Conditions, Adv. Energy Mater., 2018, 8, 1800369 CrossRef
- D. Bao, Q. Zhang, F.-L. Meng, H.-X. Zhong, M.-M. Shi, Y. Zhang, J.-M. Yan, Q. Jiang and X.-B. Zhang, Electrochemical Reduction of N2 under Ambient Conditions for Artificial N2 Fixation and Renewable Energy Storage Using N2/NH3 Cycle, Adv. Mater., 2017, 29(3), 1604799 CrossRef PubMed
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, Mechanistic Insights into Electrochemical Nitrogen Reduction Reaction on Vanadium Nitride Nanoparticles, J. Am. Chem. Soc., 2018, 140(41), 13387–13391 CrossRef CAS PubMed
- J. Kong, A. Lim, C. Yoon, J. H. Jang, H. C. Ham, J. Han, S. Nam, D. Kim, Y.-E. Sung, J. Choi and H. S. Park, Electrochemical Synthesis of NH3 at Low Temperature and Atmospheric Pressure Using a γ-Fe2O3 Catalyst, ACS Sustainable Chem. Eng., 2017, 5(11), 10986–10995 CrossRef CAS
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Defect Engineering Metal–Free Polymeric Carbon Nitride Electrocatalyst for Effective Nitrogen Fixation under Ambient Conditions, Angew. Chem., Int. Ed., 2018, 57(32), 10246–10250 CrossRef CAS PubMed
- N. Furuya and H. Yoshiba, Electroreduction of nitrogen to ammonia on gas-diffusion electrodes loaded with inorganic catalyst, J. Electroanal. Chem. Interfacial Electrochem., 1990, 291(1), 269–272 CrossRef CAS
- J. Choi, B. H. R. Suryanto, D. Wang, H.-L. Du, R. Y. Hodgetts, F. M. Ferrero Vallana, D. R. MacFarlane and A. N. Simonov, Identification and elimination of false positives in electrochemical nitrogen reduction studies, Nat. Commun., 2020, 11(1), 5546 CrossRef CAS PubMed
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Review of electrochemical ammonia production technologies and materials, Int. J. Hydrogen Energy, 2013, 38(34), 14576–14594 CrossRef CAS
- C. Tang, H.-F. Wang and Q. Zhang, Multiscale Principles To Boost Reactivity in Gas-Involving Energy Electrocatalysis, Acc. Chem. Res., 2018, 51(4), 881–889 CrossRef CAS PubMed
- C. Especel, G. Lafaye and F. Epron, Bimetallic Catalysts for Sustainable Chemistry: Surface Redox Reactions For Tuning The Catalytic Surface Composition, ChemCatChem, 2023, 15(3), e202201478 CrossRef CAS
- W. Yu, M. D. Porosoff and J. G. Chen, Review of Pt-Based Bimetallic Catalysis: From Model Surfaces to Supported Catalysts, Chem. Rev., 2012, 112(11), 5780–5817 CrossRef CAS PubMed
- T. Bligaard and J. K. Nørskov, Ligand effects in heterogeneous catalysis and electrochemistry, Electrochim. Acta, 2007, 52(18), 5512–5516 CrossRef CAS
- J. Greeley and M. Mavrikakis, Alloy catalysts designed from first principles, Nat. Mater., 2004, 3(11), 810–815 CrossRef CAS PubMed
| This journal is © the Partner Organisations 2023 |