
Recent progress and strategies on the design of catalysts for electrochemical ammonia synthesis from nitrate reduction
Wei
Song
 a,
Luchao
Yue
*a,
Xiaoya
Fan
b,
Yongsong
Luo
b,
Binwu
Ying
b,
Shengjun
Sun
c,
Dongdong
Zheng
c,
Qian
Liu
d,
Mohamed S.
Hamdy
e and
Xuping
Sun
*bc
a,
Luchao
Yue
*a,
Xiaoya
Fan
b,
Yongsong
Luo
b,
Binwu
Ying
b,
Shengjun
Sun
c,
Dongdong
Zheng
c,
Qian
Liu
d,
Mohamed S.
Hamdy
e and
Xuping
Sun
*bc
aSchool of Chemistry and Chemical Engineering & Shanxi Provincial Key Laboratory for High Performance Battery Materials and Devices, North University of China, Taiyuan 030051, Shanxi, China. E-mail: ylctyut@163.com
bInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, China. E-mail: xpsun@uestc.edu.cn; xpsun@sdnu.edu.cn
cCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, Shandong, China
dInstitute for Advanced Study, Chengdu University, Chengdu 610106, Sichuan, China
eCatalysis Research Group (CRG), Department of Chemistry, College of Science, King Khalid University, P.O. Box 9004, 61413 Abha, Saudi Arabia
First published on 2nd May 2023
Abstract
Ammonia (NH3) is an essential raw material in the production of fertilizers and a promising carbon-free energy carrier, however, its synthesis still depends on the energy- and capital-intensive Haber–Bosch process. Recently, the electrochemical N2 reduction reaction has attracted significant interest as an emerging method for NH3 synthesis under ambient conditions. However, the limited solubility of N2 in aqueous electrolyte and the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds result in a low NH3 yield rate, inferior faradaic efficiency and unsatisfactory selectivity, impeding its further practical application. Considering the high water solubility of nitrate (NO3−), the electrochemical NO3− reduction reaction (NO3−RR) has become a fascinating route for achieving sustainable production of NH3, and enormous progress has been made in this field. As a consequence, this review discusses the reaction mechanism of the electrochemical reduction of NO3− and systematically summarizes the recent development of electrocatalysts for the NO3−RR, including noble-metal-based materials, single-atom metal catalysts, and transition-metal-based catalysts. Diverse design strategies of the catalysts to boost the NO3−RR performance, such as defect engineering, rational structure design, strain engineering and constructing heterostructures, are discussed. This is followed by an illustration of how a robust understanding of the optimization strategies affords fundamental insights into the NH3 yield rate, faradaic efficiency, and selectivity of the electrocatalysts. Finally, we conclude with future perspectives on the critical issues, challenges and research directions in the design of high-efficiency electrocatalysts for selective reduction of NO3− to NH3.
N bonds result in a low NH3 yield rate, inferior faradaic efficiency and unsatisfactory selectivity, impeding its further practical application. Considering the high water solubility of nitrate (NO3−), the electrochemical NO3− reduction reaction (NO3−RR) has become a fascinating route for achieving sustainable production of NH3, and enormous progress has been made in this field. As a consequence, this review discusses the reaction mechanism of the electrochemical reduction of NO3− and systematically summarizes the recent development of electrocatalysts for the NO3−RR, including noble-metal-based materials, single-atom metal catalysts, and transition-metal-based catalysts. Diverse design strategies of the catalysts to boost the NO3−RR performance, such as defect engineering, rational structure design, strain engineering and constructing heterostructures, are discussed. This is followed by an illustration of how a robust understanding of the optimization strategies affords fundamental insights into the NH3 yield rate, faradaic efficiency, and selectivity of the electrocatalysts. Finally, we conclude with future perspectives on the critical issues, challenges and research directions in the design of high-efficiency electrocatalysts for selective reduction of NO3− to NH3.
 Xuping Sun | Xuping Sun received his PhD degree from the Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences, in 2006. From 2006 to 2009, he carried out postdoctoral research at Konstanz University, the University of Toronto, and Purdue University. In 2010, he started his independent research career as a Full Professor at CIAC and then moved to Sichuan University in 2015. In 2018, he joined the University of Electronic Science and Technology of China where he founded the Research Center of Nanocatalysis & Sensing. He was recognized as a highly cited researcher (2018–2020) in both areas of chemistry and materials science by Clarivate Analytics. He has published over 600 papers with total citations of over 63 |
1. Introduction
Ammonia (NH3) as a high-value-added chemical exerts a significant influence in the synthesis of fertilizers for sustaining the rising global population, and is also being considered as a promising alternative fuel for hydrogen storage in the future.1–3 At present, the synthesis of NH3 in industry mainly hinges on the traditional Haber–Bosch process (HBP). Such a reaction process is accomplished under tough operating conditions, including high temperature (400–550 °C) and high pressure (15–30 MPa), which is extremely energy-consuming.4,5 Taking the enormous requirements into consideration (∼170 Mt per year, over 80% of total content for fertilizers), the HBP consumes 1–2% of the world's energy supply and is accompanied by extensive CO2 emissions.6 Furthermore, the extensive centralized infrastructures involved in the HBP have to spend substantial capital, leading to a large innovation barrier and uneven regional distribution.7 In this regard, exploring a clean and sustainable strategy for highly-efficient NH3 production is highly desired, and presents great challenges in both fundamental science and engineering.Recently, electrochemical NH3 synthesis has provided an alluring research direction in the search for a substitute for the traditional HBP due to its moderate production conditions and ability to integrate with renewable energy resources.8,9 Among them, the electrochemical N2 reduction reaction (NRR) has drawn tremendous interest and achieved substantial progress. In NRR systems, the electrochemical synthesis of NH3 directly originates from the reduction of N2 and the dissociation of H2O under ambient conditions, in which the driving force is regulated by the applied voltage.10–12 Consequently, the thermodynamic energy efficiency of the NRR is about 20% higher than that of the HBP.1 Meanwhile, this method can achieve the decentralized and on-site/-demand production of NH3, further supporting the fabrication of distributed fertilizers and reducing the cost of transportation. Nevertheless, the highly stable NN bond with a bond energy of 941 kJ mol−1, limited solubility of N2, and the competing hydrogen evolution reaction (HER) cause the extremely low NH3 yield rate, selectivity, and faradaic efficiency (FE), which are far from meeting the practical demands, and even result in unreliable quantifications experimentally owing to the trace amounts of contaminants.13,14
Recently, the electrochemical nitrate reduction reaction (NO3−RR) has been demonstrated as an alluring method for NH3 synthesis at room temperature and atmospheric pressure, and its good performance may originate from the following aspects: (i) NO3− is highly soluble in water, has a relatively low N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O dissociation energy of 204 kJ mol−1, and has a more positive potential than N2. These characteristics are beneficial to alleviate the competing HER and attain high NH3 selectivity with only a small overpotential required, indicating that the NO3−RR process is more energy-efficient than the HBP and NRR; (ii) NO3− is abundant in industrial wastewater and polluted groundwater, with a maximum concentration up to 2.0 mol L−1, and these can be considered as NO3− sources, providing a promising opportunity for large-scale fabrication of NH3; (iii) converting NO3− into NH3 alleviates the environmental degradation caused by excessive nitrate emission and maintains the balance of the perturbed nitrogen cycle. The reason is attributed to the fact that the accumulation of NO3− in drinking water will induce illness and jeopardize human health; (iv) this process utilized water as the proton source, eliminating fossil-fuel consumption and CO2 emission. Consequently, there has been a dramatic growth in research efforts to study the ambient electrochemical reduction of NO3− to NH3.15–17 However, the NO3−RR is an eight-electron reaction process and generates various by-products, such as NO2−, NO, N2O, N2, and NH2OH, resulting in low NH3 selectivity, FE and yield rate.18–20 Such electrocatalytic performance is mainly determined by the electrocatalysts; hence, the major challenge in large-scale production of NH3via the NO3−RR lies in finding a suitable catalyst. Recently, a series of electrocatalysts, including noble metals, signal-atom catalysts, and catalysts of transition metals and their compounds have been developed and/or designed for the NO3−RR under ambient conditions.21–24 Meanwhile, extensive research efforts have pointed out that their electrocatalytic activities can be significantly ameliorated by elaborate structural design, defect engineering (oxygen vacancies and heteroatom doping), strain engineering, and constructing heterostructures. Thus, a systematic discussion on the recent progress of electrocatalysts for the NO3−RR and an analysis of corresponding materials design principles could provide a specific direction for rationally developing efficient NO3−RR electrocatalysts.
O dissociation energy of 204 kJ mol−1, and has a more positive potential than N2. These characteristics are beneficial to alleviate the competing HER and attain high NH3 selectivity with only a small overpotential required, indicating that the NO3−RR process is more energy-efficient than the HBP and NRR; (ii) NO3− is abundant in industrial wastewater and polluted groundwater, with a maximum concentration up to 2.0 mol L−1, and these can be considered as NO3− sources, providing a promising opportunity for large-scale fabrication of NH3; (iii) converting NO3− into NH3 alleviates the environmental degradation caused by excessive nitrate emission and maintains the balance of the perturbed nitrogen cycle. The reason is attributed to the fact that the accumulation of NO3− in drinking water will induce illness and jeopardize human health; (iv) this process utilized water as the proton source, eliminating fossil-fuel consumption and CO2 emission. Consequently, there has been a dramatic growth in research efforts to study the ambient electrochemical reduction of NO3− to NH3.15–17 However, the NO3−RR is an eight-electron reaction process and generates various by-products, such as NO2−, NO, N2O, N2, and NH2OH, resulting in low NH3 selectivity, FE and yield rate.18–20 Such electrocatalytic performance is mainly determined by the electrocatalysts; hence, the major challenge in large-scale production of NH3via the NO3−RR lies in finding a suitable catalyst. Recently, a series of electrocatalysts, including noble metals, signal-atom catalysts, and catalysts of transition metals and their compounds have been developed and/or designed for the NO3−RR under ambient conditions.21–24 Meanwhile, extensive research efforts have pointed out that their electrocatalytic activities can be significantly ameliorated by elaborate structural design, defect engineering (oxygen vacancies and heteroatom doping), strain engineering, and constructing heterostructures. Thus, a systematic discussion on the recent progress of electrocatalysts for the NO3−RR and an analysis of corresponding materials design principles could provide a specific direction for rationally developing efficient NO3−RR electrocatalysts.
Currently, several high-quality reviews focusing on the electrochemical removal of NO3− have been presented.18,19,25,26 Nevertheless, reviews that exclusively focus on the rational design of electrocatalysts for the reduction of NO3− to NH3 are lacking. As a consequence, we review the recent advancements of electrocatalysts toward the NO3−RR for attaining large NH3 yields, high FE, as well as high selectivity under ambient conditions. Firstly, this review briefly discusses the fundamental reaction mechanisms of the electrocatalytic NO3−RR. Secondly, the most recent advancements of electrocatalysts for electrochemically converting NO3− to NH3 have been summarized, covering noble metals, single-atom catalysts, and catalysts of transition metals and their compounds (Fig. 1a). Meanwhile, several strategies to regulate the apparent activity or intrinsic activity of the electrocatalysts for the NO3−RR to form NH3 are highlighted. Finally, the perspective and challenges in this emerging area are also presented.
 | ||
| Fig. 1 (a) Element list of reported NO3−RR electrocatalysts to date. (b) The electron-mediated pathway of the electrochemical reduction of NO3−. | ||
2. Reaction pathways of electrocatalytic nitrate reduction
Owing to the multivalent nitrogen element, various nitrogen-containing species like NO2−, N2O, NH2ON, N2, N2H4, and NH3 will be generated during the electrochemical NO3− reduction procedure. Among them, N2 and NH3 feature the highest thermodynamic stability and are regarded as final products under standard conditions.27,28 The corresponding reactions can be expressed through the following equations:27| 2NO3− + 12H+ + 10e− → N2 + 6H2O, E° = 1.17 V vs. SHE | (1) |
| NO3− + 9H+ + 8e− → NH3 + 3H2O, E° = −0.12 V vs. SHE. | (2) |
Generally, the electroreduction of NO3− has two different pathways, including an indirect autocatalytic reduction pathway and a direct electrocatalytic reduction pathway.29–31 In terms of the former, NO3− does not participate in the electron-transfer process and the operating conditions are a large concentration of NO3− (>1 M) and high acidity (pH < 0). The latter can also be divided into two pathways: one is the regulation of active adsorbed hydrogen atoms (Hads); the second one is electron reduction at the cathode (Fig. 1b). In the adsorbed-hydrogen-mediated pathway, the first process is the generation of Hadsvia decomposition of the adsorbed H2O on the surface of the cathode. The second process is that Hads directly reduces NO3− and generates intermediates to give the final-product NH3 rather than N2, which is attributed to the fact that formation of an N–N bond is kinetically less favorable than that of an N–H bond. This process usually requires a small overpotential to promote the conversion from NO3− to NH3, which can efficiently suppress the competing HER, as well as attain high NH3 selectivity and FE.32,33 As for the electron-mediated pathway, electrons directly reduce NO3− adsorbed on the surface of the cathode to NO2− (eqn (3) and (4)).34 Notably, converting NO3− to NO2− generally requires a high activation energy, and this process is considered as the rate-determining step to regulate the reaction kinetics of the whole NO3−RR process. Then, the generated NO2−(ads) reduces to NO(ads), which is a decisive intermediate as a branch for the generation of N2 or NH3/NH4+. On the one hand, NO(ads) can be reduced to HNO(ads) and H2NO(ads), and quickly followed by hydrogenation to form hydroxylamine, which finally reduces to NH3.35 On the other hand, NO(ads) can be desorbed from the electrode surface to generate NO in solution. When NO(aq) presents in the solution, a weakly adsorbed NO dimer can be formed, which is the precursor of N2O(ads). The produced N2O(ads) further reduces to N2 according to eqn (9), and this process plays a dominant role in the pathway of N2 evolution.36,37
| NO3−(aq) → NO3−(ads) | (3) |
| NO3−(ads) + 2H+ + 2e− → NO2−(ads) + H2O | (4) |
| NO2−(ads) + 2H+ + e− → NO(ads) + H2O | (5) |
| NO(ads) + 6H+ + 5e− → NH4+ + H2O | (6) |
| NO(ads) → NO(aq) | (7) |
| NO(ads) + NO(aq) + 2H+ + 2e− → N2O(ads) + H2O | (8) |
| N2O(ads) + 2H+ + 2e− → N2 + H2O | (9) |
3. Efficient NO3−RR electrocatalysts
Exploring advanced electrocatalysts with a high selectivity, FE, and yield rate are keenly desired for the electrosynthesis of NH3 by converting NO3−. Recently, a series of electrocatalysts have been investigated for the NO3−RR process under ambient conditions, like noble-metal-based materials, single-atom metal catalysts, and transition-metal-based materials. In the following sections, the recent advances of those catalysts in the NO3−RR toward NH3 and the corresponding optimization strategies for electrocatalytic activity will be systematically discussed.3.1 Noble-metal catalysts
Noble metals have been widely utilized as electrocatalysts for diverse electrochemical conversion reactions including the HER, the oxygen evolution reaction, the oxygen reduction reaction, and the NRR, owing to their alluring electronic conductivity, moderate capturing ability for various reactants, and high density of under-coordinated surface atoms. Recently, both experiments and theoretical calculations have suggested that noble-metal materials (Pt, Pd, Ru, and Rh) are promising electrocatalysts for the NO3−RR under ambient conditions, as listed in Table 1.20,38–42,45–48,50,52 For example, Li et al.42 designed Ru/oxygen-doped Ru core/shell nanoclusters (Fig. 2a and b) as an NO3−RR electrocatalyst for the production of NH3, in which the introduction of oxygen can increase the size of the Ru unit cell to induce tensile strains (Fig. 2c). The strains suppressed the HER but benefit H* production by expanding the barrier of H–H coupling. As a result, this catalyst achieved a large NH3 formation rate of 5.56 mol g−1 h−1 with a nearly 100% selectivity at 120 mA cm−2. In addition, Chen et al.20 dispersed Ru nanoparticles into a Cu-nanowire matrix (Ru-CuNW) through a simple cation exchange method for electrochemical NH3 synthesis (Fig. 2d). This catalyst presented an industrial-level NO3− reduction current of ∼1 A cm−2 (Fig. 2e) accompanied by a maximum NH3 FE of 96% (Fig. 2f) when operating with a low NO3− concentration of 2000 ppm (typical industrial wastewater). Meanwhile, the voltage of Ru-CuNW does not significantly change during the long-term electrolysis of 100 h under the current density of 400 mA cm−2, with a high NH3 FE of 90% maintained, indicating that such a catalyst features excellent durability. More importantly, it showed an ultrahigh NO3− conversion ratio of 99% for NH3 production, making industrial wastewater reach a drinkable water level (concentration of NO3− < 50 ppm) (Fig. 2g), and the solid NH4Cl and liquid NH3 solution products were collected by coupling the NO3−-reduction effluent stream with the air-stripping process, as displayed in Fig. 2h. In another study, Jiang et al.41 investigated the effect of the noble-metal crystal structure on electrochemical NO3−RR activity. They chose Ru as the model material and fabricated amorphous Ru nanoclusters anchored on carbon nanotubes (aRu-CNTs) for electrochemical NH3 production from NO3−. The experimental results demonstrated that aRu-CNTs delivered an NH3 yield of 145 μg h−1 mg−1 with a FE of 80.62% at −0.2 V vs. RHE, and the achieved yield was 3.1 times larger than that of crystalline Ru. Therefore, the amorphization of noble metals can be recognized as a promising route for increasing the amount of active sites on the catalysts to a certain degree.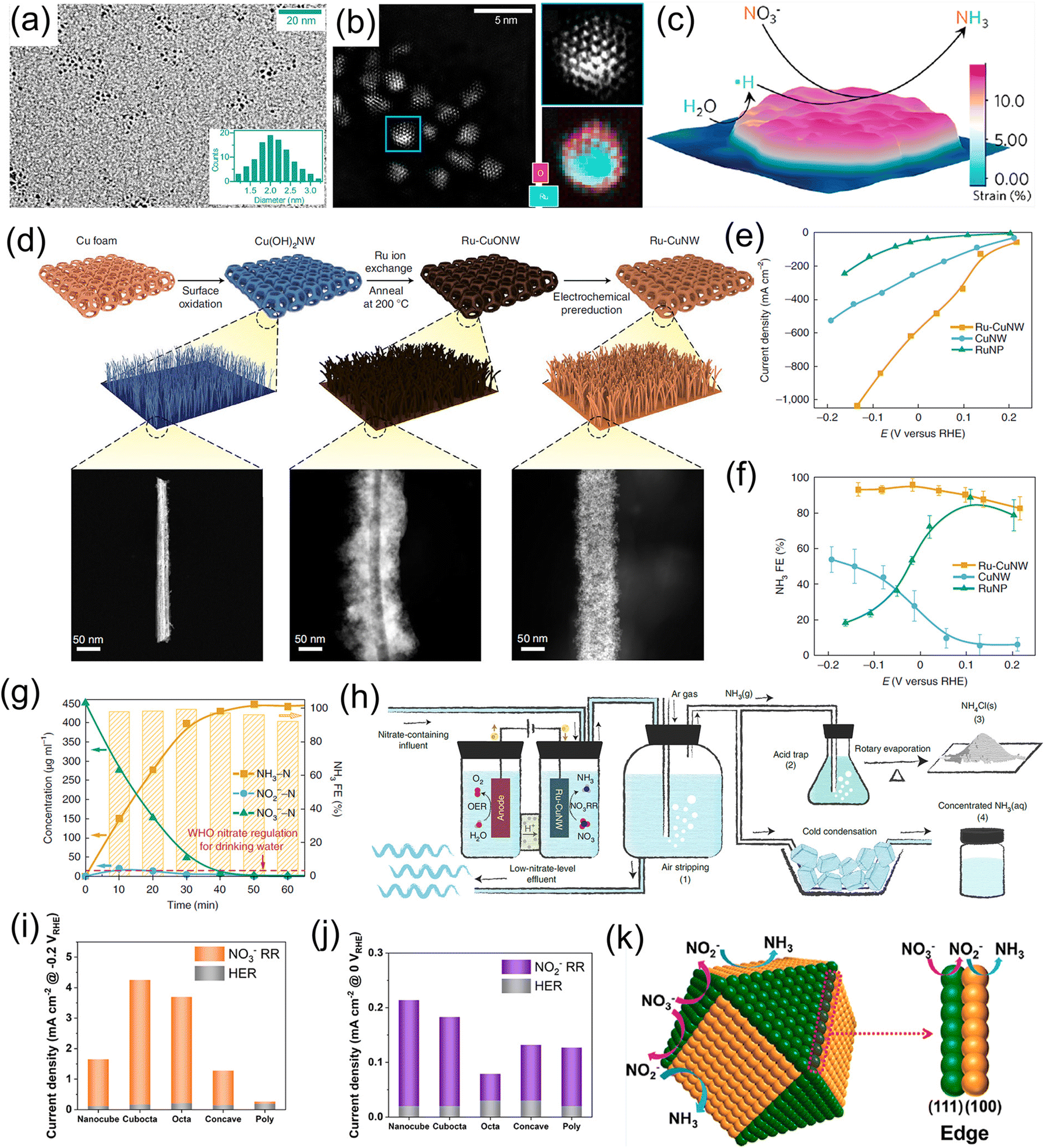 | ||
| Fig. 2 (a) TEM image and (b) aberration-corrected HAADF-STEM/EELS elemental map images of Ru/oxygen-doped Ru core/shell nanoclusters. (c) Schematic illustration of the reaction mechanism for the NO3−RR on the strained Ru nanoclusters. Reproduced from ref. 42 with permission from the American Chemical Society, copyright 2020. (d) Synthesis process of the Ru-CuNW catalyst. (e and f) I–V plots and corresponding NH3 FEs of Ru-CuNW and counterparts. (g) Complete NO3− removal using Ru-CuNW catalyst. (h) Schematic diagram of NH4Cl(s) and concentrated NH3(aq) products from nitrate-containing influent. Reproduced from ref. 20 with permission from the Nature Publishing Group, copyright 2022. (i and j) The electrocatalytic activity of Pd catalysts with various structures. (k) Schematic illustration of the reaction mechanism for the NO3−RR on the different crystal facets of Pd catalysts. Reproduced from ref. 43 with permission from the American Chemical Society, copyright 2021. | ||
| Catalyst | Electrolyte | NH3 yield rate | Faradaic efficiency | Potentials (V vs. RHE) | Ref. |
|---|---|---|---|---|---|
| a NH3 yield rate. b Faradaic efficiency. | |||||
| Ru-dispersed Cu nanowire | 1 M KOH + 2000 ppm KNO3 | 76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 μg h−1 cm−2 500 μg h−1 cm−2 |
90% | 0.04 | 20 |
| BC2N/Pd | 0.1 M KOH + 250 mM KNO3 | 1730 μg h−1 cm−2 | 97.42% | −0.7a/−0.3b | 40 |
| Amorphous Ru nanoclusters | 5 mM Cs2CO3 + 500 ppm NO3− | 145.1 μg h−1 mg−1 | 80.62% | −0.2 | 41 |
| Strained Ru nanoclusters | 1 M KOH + 1 M KNO3 | 5.56 mol gcat−1 h−1 | 96% | −0.3 | 42 |
| Pd/Cu2O octahedra | 0.5 M K2SO4 + 50 ppm NO3− | 925.11 μg h−1 mg−1 | 96.56% | −0.645 | 45 |
| Pd/Co3O4 | 0.5 M K2SO4 + 200 ppm NO3− | 0.204 mmol h−1 cm−2 | 88.6% | −0.645 | 46 |
| CuPd aerogels | 0.5 M K2SO4 + 50 mg L−1 NO3− | 784.37 μg h−1 mg−1 | 90.02% | −0.46 | 47 |
| CuPd nanocubes | 1 M KNO3 + 1 M KOH | 6.25 mol h−1 g−1 | 92.5% | −0.6a/−0.5b | 48 |
| PdBP nanothorn arrays | 0.5 M K2SO4 + 100 ppm NO3− | 0.109 mmol h−1 cm−2 | 64.73% | −0.66 | 50 |
| RuO2 nanosheets | 0.1 M K2SO4 + 200 ppm NO3− | 0.1158 mmol h−1 cm−2 | 97.46% | −0.35 | 52 |
Besides, revealing the electrocatalytic behaviors on noble metals with different facets for the NO3−RR is essential to explore rational design strategies for electrosynthesis of NH3. Lim et al.43 employed Pd as a model catalyst to understand the structure-sensitivity of NO3− reduction to NH3 on a Pd catalyst. Specifically, they fabricated Pd nanoparticles with diverse morphologies, such as nanocubes containing six (100) facets, cuboctahedrons containing six (100) and eight (111) facets, octahedrons containing eight (111) facets, and concave nanocubes containing (100) and (hk0) facets. Based on the experimental results, the Pd (111) facet is favorable to catalyze the reduction of NO3− to NO2−, while the (100) facet is inclined to catalyze the reduction of NO2− to NH3 (Fig. 2i–k). Hence, the activity for NO3− reduction decreases in the order of Pd (111) > Pd (100) > Pd (hk0) and the activity for NO2− reduction decreases in the order of Pd (100) > Pd (hk0) > Pd (111) under the alkaline electrolyte. As a consequence, NH3 production using noble-metal catalysts can be enhanced through controlling their structure and facets.
Numerous research studies have demonstrated that the electrochemical performance of noble metals for converting NO3− to NH3 can be significantly modified by constructing a heterostructure with two materials, with the modified performance originating from the unique physical properties induced by the charge distribution and energy-band bending at the heterointerface. For instance, Li et al.40 constructed a hybrid material consisting of Pd nanoparticles and a boron–carbon–nitrogen material (BC2N/Pd) for the NO3−RR, which showed a superior NH3 production rate of 1730 μg h−1 cm−2 at −0.7 V vs. RHE using 250 mM KNO3 solution as the nitrogen source. Theoretical calculations revealed that the free energy accumulation of the NO3−RR on BC2N/Pd was higher than that of individual Pd or BC2N, and the corresponding value from NO3− to *NH could conquer the reaction energy barriers from *NH to *NH2 and *NH2 to NH3. As exhibited in Fig. 3a–d, Ren et al.44 constructed Cu/Pd/CuOx heterostructures with abundant pores for electrochemical conversion of NO3− to NH3. In terms of the Cu/Pd/CuOx heterostructure, the electronic interactions between the Cu, Pd, and CuOx components lead to electron transfer from Pd to Cu, which can increase the number of reactive sites and thus modulate the adsorption ability for intermediates, meanwhile suppressing the competitive hydrogen evolution reaction process. Moreover, the abundant channels provided sufficient contact area between electrolyte and catalyst. Benefiting from interfacial engineering and a unique porous structure, the designed Cu/Pd/CuOx heterostructure afforded a superior NH3 production rate of 1510.3 μg h−1 mg−1, FE of 86.1%, and NH3 selectivity of 90.06% (Fig. 3e–f). Similarly, Xu et al.45 applied Cu2O corner-etched octahedra with cavities and oxygen defects as the substrate to support Pd nanoparticles (Pd-Cu2O), in which the loading content of Pd active materials was only 2.93%. For Pd-Cu2O catalyst system, Pd sites were regarded as the active center for capturing *H and generating Pd–H intermediate; while parts of Cu2O electrochemically reduced to Cu0 and in situ formed Cu/Cu+, which could provide active sites for NO3− electroreduction. Meanwhile, the oxygen defects in Cu2O were beneficial for the capture of NO3− and to weaken the N–O bond. As expected, Pd/Cu2O heterostructure catalyst exhibited an excellent electrocatalytic activity of NO3− to NH3, including NH3 formation rate of 925.11 μg h−1 mg−1, selectivity of 95.31%, and FE of 96.56%. Pd-PdO-modified Co3O4 nanowire arrays were fabricated and applied as a catalyst to electrochemically convert NO3− to NH3. In K2SO4 solution containing 200 mg L−1 of NO3− electrolyte, such catalyst showed a high NH3 FE of 88.6% and selectivity of 95.3%.46
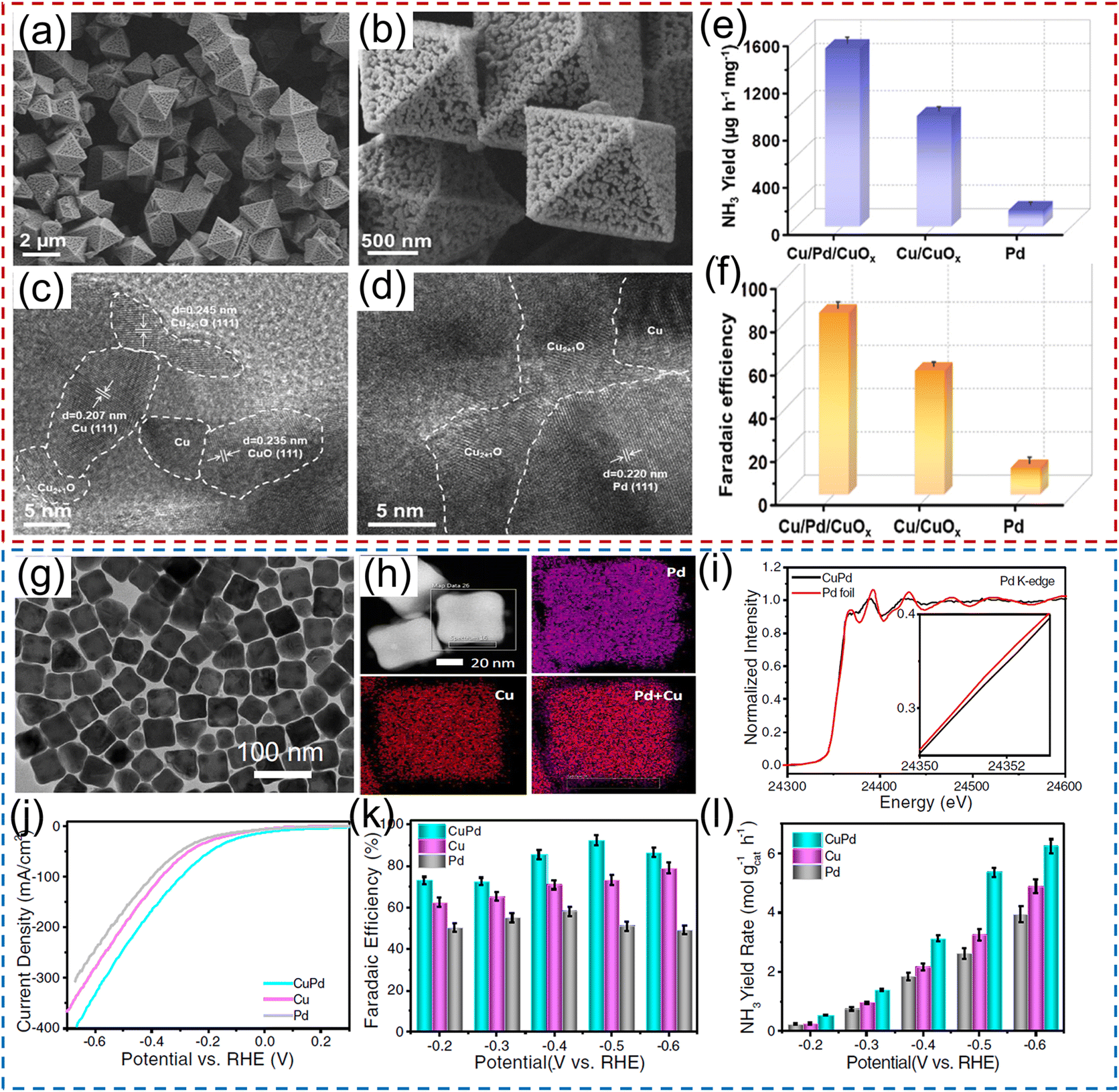 | ||
| Fig. 3 (a–d) TEM and HRTEM images of Pd/CuO material. (e) NH3 yield rate and (f) the corresponding FE of Pd/CuO catalyst. Reproduced from ref. 44 with permission from Elsevier, copyright 2022. (g and h) TEM and mapping images of the CuPd alloy. (i) Pd K-edge XANES spectra of CuPd and a Pd foil reference. (j) Linear scan voltammetry (LSV) curves, (k) NH3 FE and (l) NH3 yield rate under different potentials for CuPd, Cu, and Pd. Reproduced from ref. 48 with permission from the Nature Publishing Group, copyright 2022. | ||
Tailoring the catalytic sites of noble-metal materials by alloying with another metal is another fascinating strategy to further improve their catalytic activity for the NO3−RR to NH3. For example, Xu et al.47 rationally chose Pd as an active metal and Cu as a promoting metal to construct a CuPd bimetallic catalyst for electrochemical conversion of NO3− to NH3. In terms of the CuPd catalyst system, Pd sites serve as active centers to adsorb *H and promote the hydrogenation reaction for NH3 production. Consequently, the CuPd alloy catalyst delivered a large NH3 yield rate of 784.37 μg h−1 mg−1, and a high NH3 FE of 90.02% at −0.46 V vs. RHE, which was superior to those of pure Cu and Pd catalysts. Furthermore, Gao et al.48 employed density functional theory (DFT) calculations and machine learning to deduce that the upshifted d-band center of the Cu sites of the CuPd alloy favored the adsorption of *NO3, and *N was destabilized owing to the dominant role of Pauli repulsion from the subsequent Pd d orbitals, promoting the protonation of N-bonded species toward NH3. As demonstrated in Fig. 3g–l, they also experimentally synthesized CuPd nanocube alloy catalysts, and confirmed the existence of charge transfer between Pd and Cu via X-ray absorption near-edge spectroscopy (XANES). In 1.0 M NaOH solution containing 1.0 M KNO3, the PdCu nanocube catalyst showed an NH3 yield rate of 6.25 mol h−1 g−1 at −0.6 V vs. RHE and an NH3 FE of 92.5% at −0.5 V vs. RHE, respectively. Furthermore, the current density remained stable over 12 h of continuous operation, with a high NH3 FE of ∼85.1% maintained. Similarly, Zhang and his co-authors49 also employed metallic Ni as a promoter catalyst to regulate the electronic structure of Pd, and synthesized PdNi alloys for the NO3−RR. PdNi nanosheets displayed an NH3 formation rate of 16.7 mg h−1 mg−1 (at −1.2 V vs. RHE) and a FE of 87.9% (at −0.6 V vs. RHE). After that, a ternary PdBP nanothorn-array catalyst was also designed and utilized for converting NO3− to NH3. In terms of the ternary system, B and P doping could induce the lattice strain, thus regulating the electronic structure and increasing the number of active sites of Pd; in addition, the doping sites also served as the Lewis acid to improve the adsorption ability for NO3−. Thus, the electrochemical performance for reducing NO3− to NH3 was significantly enhanced after B and P doping.50
Several noble-metal oxides were also used as electrocatalysts for highly efficient NH3 formation by electrochemical conversion of NO3− at room temperature. Liu et al.51 fabricated oxide-derived silver and investigated its electrochemical activity for the NO3−RR. It is noted that this catalyst delivered excellent electrocatalytic activity of NO3− to NO2− and thus promoted the reduction reaction of NO2− to NH4+, which was well controlled by the applied potential and they obtained an NH4+ FE of 89%. Qin et al.52 designed and synthesized RuxOy clusters anchored on nickel metal–organic frameworks (MOF) for the NO3−RR. Such a catalyst could provide a nearly 100% NH4+ selectivity with an NH4+ yield rate of 274 μg h−1 mg−1. Wang et al.53 fabricated carbon-supported RuO2 nanosheets with abundant oxygen vacancies for electrochemical NO3− to NH3 conversion. The abundant oxygen vacancies within the RuO2 nanosheets could regulate the d-band center and improve the hydrogen affinity, thus reducing the reaction-energy barrier of the potential-determining step (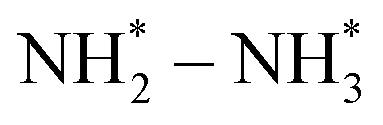 ). As a result, this catalyst displayed a superior electrocatalytic activity for the conversion of NO3−to NH3 (NH3 FE of 97.46% and selectivity of 96.42%) than that of the crystalline counterparts.
). As a result, this catalyst displayed a superior electrocatalytic activity for the conversion of NO3−to NH3 (NH3 FE of 97.46% and selectivity of 96.42%) than that of the crystalline counterparts.
3.2 Single-atom catalysts
Single-atom catalysts, a group of emerging frontier materials comprising isolated metal atoms dispersed into support materials, have triggered explosive research interest within the catalysis field owing to their remarkably high catalytic activity and selectivity. When applied to the NO3−RR, single-atom catalysts can provide the following desirable advantages, including (i) the specific atomic structure can expose abundant adsorption sites and homogenous catalytic active centers; (ii) the strong interactions between single atoms and the surrounding atoms enable superior long-term electrolysis; and (iii) the absence of multiple neighboring active sites required for coupling N–N bonds in their structure can efficiently suppress the generation of by-product N2, thus enhancing NH3 selectivity. Currently, both experimental and theoretical calculations have suggested that single-atom catalysts are effective toward electroreduction of NO3− to NH3.For example, Niu and co-workers54 used first-principle calculations to systematically investigate the electrocatalytic activity of various transition-metal single-atoms (from Ti to Au) anchored on carbon nitride (TM/g-CN) for electrochemical NO3− to NH3 conversion, as shown in Fig. 4a. Firstly, the adsorption energies of NO3−, a H proton, and an N2 molecule on TM/g-CN were systematically calculated. As revealed from Fig. 4b, the adsorption ability for NO3− was stronger than for H proton or N2 on TM/g-CN catalysts, except for Pt and Au, indicating that the NO3−RR is more favorable than the HER and NRR. By combining with detailed pathways of NO3− reduction on TM/g-CN, they established a volcano plot of limiting potential selecting the adsorption energy of NO3− as a descriptor (Fig. 4c), where Ti and Zr appeared near the top of the volcano. Based on the above analysis, Ti/g-CN and Zr/g-CN possessed stronger adsorption abilities for NO3− compared with those of other TM/g-CN catalysts, making them fascinating electrocatalysts with high activity and selectivity for the NO3−RR. Similarly, Lv et al.55 explored the NO3−RR performance of a set of transition-metal single-atom (Ti, Os, Ru, Cr, Mn, and Pt) catalysts supported on g-C3N4 by performing DFT calculations. The calculation results suggested that Ru/g-C3N4 featured the highest activity and selectivity for the conversion of NO3− to NH3 with a limiting potential of −0.34 V, as presented in Fig. 4d. Thus, theoretical calculations have provided an advanced direction for the application of single-atom catalysts and paved the way for the electrochemical conversion of NO3− to NH3.
 | ||
| Fig. 4 (a) Atomic structure of TM/g-CN and corresponding element list (from Ti to Au). (b) Comparison of adsorption energies of NO3−, N2, and H proton on TM/g-CN. (c) NO3−RR volcano plot of TM/g-CN using the adsorption energy of NO3− as the descriptor. Reproduced from ref. 54 with permission from Wiley-VCH, copyright 2021. (d) Volcano correlation curve between the limiting potential and adsorption energy of NO3− of TM/g-C3N4. Reproduced from ref. 55 with permission from the American Chemical Society, copyright 2021. (e) NH3 FE of Fe-MoS2 under various potentials. (f) Reaction pathway for the NO3−RR on Fe-MoS2. (g) Schematic diagram of the interaction between NO and M-MoS2 nanosheets. Reproduced from ref. 57 with permission from Wiley-VCH, copyright 2022. | ||
Up to now, only a few single-atom catalysts have been experimentally fabricated and employed for the electrosynthesis of NH3 from NO3−, including Fe, Cu, Ni, Mo, and their alloys, as listed in Table 2. For example, Zhu et al.56 prepared a single-atom Cu-catalyst supported on nitrogenated carbon nanosheets (Cu–N–C) and investigated its catalytic performance for the NO3−RR for the first time. The strong binding between Cu and N (Cu–N2) was responsible for the good adsorption ability of the catalyst for NO3− adsorption, promoting the fast conversion from NO3− to NH3 as well as exhibiting excellent stability. Furthermore, Chen et al.24 demonstrated that Cu–N–C could effectively inhibit the generation of toxic NO2− and by-product N2, as well as facilitate the reduction of 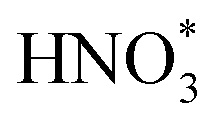 to
to  , and
, and  to
to  . It is well known that the nitrate reductase enzyme has a Mo(IV) atom coordinated with sulfur coordinating ligands, whereas nitrogenase is a multinuclear enzyme with MoFe7 clusters as the active sites. Inspired by this point, Voiry's group57 developed a novel heterogeneous catalyst composed of Fe single-atoms anchored on two-dimensional MoS2 (Fe-MoS2) for electrochemical NH3 synthesis by the reduction reaction of NO3−. Fe-MoS2 delivered a remarkably high FE of 98% for the NO3−RR to NH3 at an onset potential of −0.48 V using 0.1 M Na2SO4 containing 0.1 M NaNO3 electrolyte (Fig. 4e). Under the above testing conditions, this catalyst showed a 7-hour average NH3 formation rate of 431.8 μg h−1 cm−2. DFT calculations revealed that the Fe-MoS2 catalyst featured a superior ability for activating NO3− by virtue of the strong interaction between the d-band orbitals of the Fe atoms and the 2π* orbitals of the NO species, lowering the energy barrier for conversion of *NO to *N (the rate-determining step) (Fig. 4f and g). At the same time, Wu et al.58 chose an N-doped porous carbon matrix to anchor Fe single-atoms (Fe SAC) and applied them for electrochemical NO3− to NH3 conversion (Fig. 5a and b). A large NH3 yield rate (up to 0.46 mmol h−1 cm−2 at −0.85 V vs. RHE) and a high NH3 FE of 75% at −0.66 V vs. RHE were achieved in K2SO4 with 0.5 M KNO3, and remained stable during 20 consecutive electrolysis cycles (Fig. 5c–e). These results indicated that Fe SAC featured superior electrocatalytic activity and outstanding durability for the NO3−RR. The favorable catalytic activity of the as-designed Fe SAC originated from the unique structure, which can not only effectively suppress the N–N coupling and enhance the selectivity of NH3 production, but also enable the intrinsic high-efficiency active sites (Fe–N4) to possess lower thermodynamic barriers. However, the detailed reaction mechanism of Fe SAC was only revealed through theoretical modeling. During the electrocatalytic reaction process, the M (metal)–Nx catalyst may experience structural evolution induced by the applied potential and/or the interaction with reactants or electrolytes, which complicates the comprehension of the structure–performance relationship and seriously blocks the rational design of efficient catalysts. Consequently, revealing the dynamic transformation of the M–Nx structure under operating conditions is essential to recognize the real active sites. To achieve the above-mentioned target, Li et al.59 employed in situ characterisation techniques to establish the reaction pathway and evolution mechanism of the catalysts, selecting a catalyst of Fe single atoms anchored on carbon derived from polypyrrole as an example. They proposed the preoccupied NO3−RR mechanism presented in Fig. 5f that the exclusive existence of nitrate-preoccupied Fe(II)–Nx sites prior to the formation of Fe (0), which could effectively eliminate the competing adsorption of water under aqueous conditions. Subsequently, Yang et al.60 discovered the restructuring of Cu–N4 sites during the electrochemical production of NH3 by converting NO3− through in situ X-ray adsorption spectroscopy coupled with advanced electron microscopy. Specifically, as depicted in Fig. 5g–i, the Cu–N4 structure experienced the sequential evolution from Cu–N3 to near-free Cu0 single atoms and finally to aggregated Cu0 nanoparticles during the electroreduction of NO3− to NH3. Moreover, the formed Cu0 nanoparticles can be dismantled into single atoms and again recovered to give the Cu–N4 structure upon being exposed to an ambient atmosphere after the electrolysis.
. It is well known that the nitrate reductase enzyme has a Mo(IV) atom coordinated with sulfur coordinating ligands, whereas nitrogenase is a multinuclear enzyme with MoFe7 clusters as the active sites. Inspired by this point, Voiry's group57 developed a novel heterogeneous catalyst composed of Fe single-atoms anchored on two-dimensional MoS2 (Fe-MoS2) for electrochemical NH3 synthesis by the reduction reaction of NO3−. Fe-MoS2 delivered a remarkably high FE of 98% for the NO3−RR to NH3 at an onset potential of −0.48 V using 0.1 M Na2SO4 containing 0.1 M NaNO3 electrolyte (Fig. 4e). Under the above testing conditions, this catalyst showed a 7-hour average NH3 formation rate of 431.8 μg h−1 cm−2. DFT calculations revealed that the Fe-MoS2 catalyst featured a superior ability for activating NO3− by virtue of the strong interaction between the d-band orbitals of the Fe atoms and the 2π* orbitals of the NO species, lowering the energy barrier for conversion of *NO to *N (the rate-determining step) (Fig. 4f and g). At the same time, Wu et al.58 chose an N-doped porous carbon matrix to anchor Fe single-atoms (Fe SAC) and applied them for electrochemical NO3− to NH3 conversion (Fig. 5a and b). A large NH3 yield rate (up to 0.46 mmol h−1 cm−2 at −0.85 V vs. RHE) and a high NH3 FE of 75% at −0.66 V vs. RHE were achieved in K2SO4 with 0.5 M KNO3, and remained stable during 20 consecutive electrolysis cycles (Fig. 5c–e). These results indicated that Fe SAC featured superior electrocatalytic activity and outstanding durability for the NO3−RR. The favorable catalytic activity of the as-designed Fe SAC originated from the unique structure, which can not only effectively suppress the N–N coupling and enhance the selectivity of NH3 production, but also enable the intrinsic high-efficiency active sites (Fe–N4) to possess lower thermodynamic barriers. However, the detailed reaction mechanism of Fe SAC was only revealed through theoretical modeling. During the electrocatalytic reaction process, the M (metal)–Nx catalyst may experience structural evolution induced by the applied potential and/or the interaction with reactants or electrolytes, which complicates the comprehension of the structure–performance relationship and seriously blocks the rational design of efficient catalysts. Consequently, revealing the dynamic transformation of the M–Nx structure under operating conditions is essential to recognize the real active sites. To achieve the above-mentioned target, Li et al.59 employed in situ characterisation techniques to establish the reaction pathway and evolution mechanism of the catalysts, selecting a catalyst of Fe single atoms anchored on carbon derived from polypyrrole as an example. They proposed the preoccupied NO3−RR mechanism presented in Fig. 5f that the exclusive existence of nitrate-preoccupied Fe(II)–Nx sites prior to the formation of Fe (0), which could effectively eliminate the competing adsorption of water under aqueous conditions. Subsequently, Yang et al.60 discovered the restructuring of Cu–N4 sites during the electrochemical production of NH3 by converting NO3− through in situ X-ray adsorption spectroscopy coupled with advanced electron microscopy. Specifically, as depicted in Fig. 5g–i, the Cu–N4 structure experienced the sequential evolution from Cu–N3 to near-free Cu0 single atoms and finally to aggregated Cu0 nanoparticles during the electroreduction of NO3− to NH3. Moreover, the formed Cu0 nanoparticles can be dismantled into single atoms and again recovered to give the Cu–N4 structure upon being exposed to an ambient atmosphere after the electrolysis.
 | ||
| Fig. 5 (a) Schematic illustration of the preparation of Fe SAC. (b) Aberration-corrected medium-angle annular dark-field scanning TEM (HAADF STEM) and mapping images of Fe SAC. (c) NH3 FE. (d) NH3 yield rate and partial current density of Fe SAC under various potentials. (e) Cycling durability of Fe SAC at −0.66 V vs. RHE. Reproduced from ref. 58 with permission from the Nature Publishing Group, copyright 2021. (f) The proposed preoccupied NO3−RR mechanism for Fe SAC. Reproduced from ref. 59 with permission from The Royal Society of Chemistry, copyright 2021. (g) In situ XANES spectra of Cu–N4 at each given potential. (h) Linear combination fitting result of the Cu K-edge XANES spectra and (i) corresponding Cu K-edge FT-EXAFS spectra at different potentials. Reproduced from ref. 60 with permission from the American Chemical Society, copyright 2022. | ||
| Catalyst | Electrolyte | NH3 yield rate | Faradaic efficiency | Potentials (V vs. RHE) | Ref. |
|---|---|---|---|---|---|
| a NH3 yield rate. b Faradaic efficiency. | |||||
| Fe-MoS2 SAC | 0.1 M Na2SO4 + 0.1 M NaNO3 | 431.8 μg h−1 cm−2 | 98% | −0.48 | 57 |
| Fe SAC | 0.1 M K2SO4 + 0.5 M NO3− | 0.46 mmol h−1 cm−2 | 75% | −0.85a/−0.66b | 58 |
| Fe-PPy SAC | 0.1 M KOH + 0.1 M NO3− | 2.75 mg h−1 cm−2 | 100% | −0.7/−0.3 | 59 |
| Cu SAC | 0.1 M KOH + 0.1 M NO3− | 4.5 mg cm−2 h−1 | 84.7% | −1.0 | 60 |
| Cu-cis-N2O2 SAC | 0.5 M K2SO4 + 1000 ppm NO3− | 28.73 ± 1.25 mg h−1 cm−2 | 80% | −1.6 | 61 |
| FeN2O4 SAC | 0.1 M K2SO4 + 0.5 M NO3− | 46 mg h−1 mg−1 | 92% | −0.88a/−0.68b | 62 |
| Ni-Cu SAC | 0.5 M K2SO4 + 200 ppm NO3− | 326.7 μmol h−1 cm−2 | 100% | −0.55 | 63 |
| FeMo SAC | 0.05 M PBS + 0.16 M KNO3 | 18.0 μmol cm−2 h−1 | 94% | −0.45 | 64 |
Although single-atom catalysts deliver outstanding electrocatalytic activity for the conversion of NO3− to NH3, the isolated metal centers usually coordinate with four N atoms in C4V symmetry. Such a coordination structure features relatively weak adsorption ability for NO3−, leading to sluggish ionic migration and low NH3 production rate. Related literature has demonstrated that introducing weakly coordinated heteroatoms to substitute some of the coordinated N is an admirable strategy for breaking the coordination symmetry of the metal centers, consequently increasing the site polarity and improving NO3− accumulation. For instance, Cheng et al.61 broke the coordination symmetry of Cu SAC by replacing the local coordination atoms from 4N to 2N + 2O (Cu-cis-N2O2). First-principle calculations were preferentially employed to reveal the coordination symmetry-breaking in Cu SAC and investigate the reaction pathways of Cu-cis-N2O2 and Cu–N4 catalysts, as indicated in Fig. 6a–e. In terms of Cu-cis-N2O2, Cu is coordinated by two N and two O atoms and the catalyst possesses polar active sites, which are prone to enrich NO3− on the surface of the catalyst and promote the generation of the key reaction intermediate *ONH, further facilitating hydrogenation to NH3. Motivated by this, a Cu-cis-N2O2 catalyst was fabricated by pyrolysis of a Cu–Salen complex under an Ar atmosphere (Fig. 6g–h). When applied to the NO3−RR, the NH3 formation rate reached 27.84 mg h−1 cm−2 at an industrial-level current density of 366 mA cm−2. Moreover, the electrochemical activity of Cu-cis-N2O2 was well maintained after continual operation for 2000 h (Fig. 6i–k). Analogously, Zhang et al.62 fabricated an Fe single-atom catalyst with unique FeN2O2 coordination via direct pyrolysis of metal–organic frameworks possessing a preorganized FeN2O4 environment. When applying the designed Fe SAC for the NO3−RR, it showed a high NH3 production rate of 46 mg h−1 mg−1 with a FE of 92% in neutral electrolytes. Combined theoretical calculations revealed that the O atoms in FeN2O2 could regulate the d-band center of Fe and consequently enhance the adsorption energies of the NO3−RR intermediates. In comparison with FeN4, FeN2O2 features superior conductivity, NH3 selectivity and a lower reaction energy barrier from *NOH to *N, thus promoting the progress of the NO3−RR.
 | ||
| Fig. 6 (a) Illustration of the Cu-cis-N2O2 catalyst. (b–d) The molecular dynamic simulation of Cu-cis-N2O2 and counterparts. (e and f) Reaction pathways for the NO3−RR on the surface of Cu-cis-N2O2 and Cu-N4. (g) Synthesis process of Cu-cis-N2O2. (h) HAADF STEM and corresponding mapping images of Cu-cis-N2O2. (i) LSV curves, (j) NH3 formation rate and FE at each given potential. (k) Cycling stability test of Cu-cis-N2O2. Reproduced from ref. 61 with permission from Wiley-VCH, copyright 2022. | ||
To further enhance the catalytic activity of single-atom catalysts, tuning the electronic structures of active sites through introducing foreign atoms in the metal matrix can be regarded as an alluring approach to increase the NH3 production rate, selectivity and FE. For example, Cai et al.63 reported a single-atom Ni-alloyed Cu catalyst that achieved an NH3 yield rate of 326.7 μmol h−1 cm−2 at −0.55 V vs. RHE and a maximum FE of 100% in 0.5 M K2SO4 with 200 ppm NO3−, in which the yield rate was nearly 10.7 times superior to that of a bare Cu catalyst. Theoretical calculations suggested that the single Ni atom on the Cu catalyst regulated the third protonation reaction of the electrocatalytic NO3−RR and increased the adsorption energy of the crucial NOOH* intermediate, thus decreasing the limiting potential and inhibiting the formation of by-product. Murphy et al.64 reported a bimetallic FeMo-based single-atom catalyst for electroreduction of NO3− to NH3, in which Mo and Fe served as the dissociative and associative sites of the initial adsorption of NO3−, respectively. Benefiting from the synergistic effect of both Mo and Fe sites, this bimetallic catalyst achieved an NH3 production rate of 18.0 μmol cm−2 h−1 (153 μgNH3 mg−1 h−1) with a FE of 94%, as well as outstanding long-term durability with a well-maintained FE above 90% for over 60 h of electrolysis.
In the above-mentioned two parts, we have discussed noble-metal and single-atom metal catalysts for conversion of NO3− to NH3 under ambient conditions. Although an extensive number of electrocatalysts deliver desirable activity, high FE and superior selectivity, their large-scale practical application is still impeded by their expensive cost, rare resources (noble metals) and low yield (single-atom catalysts). In contrast, non-noble transition metals, such as Cu, Co, Ni, Fe, and their alloys, have drawn extensive attention as promising alternatives in the field of electrochemical conversion of NO3− to NH3 by virtue of their abundant resources and favorable catalytic activity. Transition-metal-based materials including metals, metal oxides, metal phosphides, and so on, have been widely investigated as highly efficient catalysts for the NO3−RR. In the following section, the recent advances in transition-metal-based electrocatalysts for the NO3−RR will be systematically discussed.
3.3 Transition-metal catalysts
| Catalyst | Electrolyte | NH3 yield rate | Faradaic efficiency | Potentials (V vs. RHE) | Ref. |
|---|---|---|---|---|---|
| a NH3 yield rate. b Faradaic efficiency. | |||||
| Cu nanosheets | 0.1 M KOH + 10 mM KNO3 | 390.1 μg mg−1 h−1 | 99.7% | −0.15 | 70 |
| Cu polycrystalline | 0.5 M Na2SO4 + 0.1 M KNO3 | 101.4 μmol h−1 cm−2 | 93.91% | −0.266 | 71 |
| dr-Cu nanoplates | 0.5 M K2SO4 + 50 ppm KNO3− | 781.25 μg h−1 mg−1 | 85.47% | −0.654 | 72 |
| Cu with grain boundaries | 0.1 M KOH + 10 mM NO3− | 487.8 mmol g−1 h−1 | 94.2% | −0.2 | 74 |
| Cu@C | 0.1 M KOH + 1 mM NO3− | 469.5 μg h−1 cm−2 | 72.0% | −0.9a/−0.3b | 77 |
| Cu/TiO2−x | 0.5 M Na2SO4 + 500 ppm NO3− | 0.1143 mmol h−1 mg−1 | 81.34% | −0.75 | 78 |
| Cu-CuO | 0.1 M KOH + 0.1 M NO3− | 3.17 mol h−1 g−1 | 98.7% | −0.8 | 79 |
| Co nanosheets | 1 M KOH + 0.1 M NO3− | 10.4 mmol h−1 cm−2 | 98% | −0.24 | 89 |
| Fe-cyano NSs | 1 M KOH + 0.1 M NO3− | 42.1 mg h−1 cm−2 | 90% | −0.5 | 90 |
| Ni-NCNTs | 0.5 M Na2SO4 + 0.3 M NO3− | 5.1 mg h−1 cm−2 | 99% | −0.5 | 92 |
| CuFe alloys | 0.1 M Na2SO4 + 100 ppm NO3− | — | 81.1% | −0.7 | 93 |
| CuNi@C alloy | 0.1 M PBS + 50 mg L−1 NO3− | — | 79.6% | −1.0 | 95 |
| Co0.5Cu0.5 | 1 M KOH + 50 mM KNO3 | — | 95% | −0.03 | 96 |
Meanwhile, an NH3 yield rate of 82.4 μmol h−1 cm−2 was still achieved after six continuous cycles, with a high FE above 92.85% retained, indicating an alluring stability for electroreduction of NO3− to NH3.71
On the other hand, interface engineering, such as defect engineering,72 heteroatom doping,73 coupling with carbon,65 grain-boundary engineering,74 and constructing heterostructures75,76 has been proposed to further enhance the electrochemical NO3−RR activity of metallic Cu catalysts. As presented in Fig. 7a and b, Xu et al.72 fabricated the atomic-defect-rich metallic Cu nanoplates (dr-Cu NPs) and investigated their catalytic activity for the NO3−RR. After the introduction of a large number of defects in the lattice, the electrochemically active surface area of the Cu nanoplates was remarkably enhanced (dr-Cu NPs: 1.28 mF cm−2vs. Cu NPs: 0.38 mF cm−2), thus increasing the surface-active sites and facilitating the adsorption of various intermediates during the electrochemical process. As expected, dr-Cu NPs displayed a large NO3− conversion rate of 93.26%, favorable NH3 selectivity of 81.99%, as well as a high NH3 FE of 85.47%, which were superior to those of Cu nanoplates without defects (Fig. 7c and d). Song et al.77 designed Cu nanoparticles encapsulated in a porous carbon matrix for NO3− to NH3 conversion (Fig. 7e and f). Under an ultralow concentration of 1 mM NO3−, Cu@C delivered a high NH3 FE of 72.0% and a yield rate of 469.5 μg h−1 cm−2 at −0.3 and −0.9 V vs. RHE, respectively, which were approximately 3.6 times larger than those of Cu nanoparticles (Fig. 7g and h). To reveal such impressive electrocatalytic activity, they adopted the finite-element method to simulate the enrichment effect of NO3− on the surface of Cu@C and Cu. The structure model of a Cu slab coated with or without porous carbon shown in Fig. 7i and j suggested that the porous carbon skeleton within Cu@C was beneficial to the concentration of NO3−, thereby expediting the mass transfer of NO3− for efficient electroreduction into NH3 at ultralow concentrations. In addition, Cu nanoparticles with abundant grain boundaries encapsulated by hollow carbon (Cu@C) were constructed and regarded as an electrocatalyst for thee conversion of NO3− to NH3 in alkaline media. In terms of this catalyst system, apart from the enrichment effect of carbon, the grain boundaries within the Cu nanoparticles could appropriately regulate the adsorption energy of NO3− for dwindling reaction barriers and enhance the reaction activity for the NO3−RR. As a consequence, the constructed Cu@C catalyst exhibited a maximum FE of 94.2% and a large NH3 yield rate of 487.8 mmol g−1 h−1 at a low potential of −0.2 V vs. RHE in alkaline media, achieving an exceptional performance for the NO3−RR.74 Zhang et al.78 constructed a heterostructure catalyst composed of metallic Cu and oxygen-vacancy-rich TiO2−x, in which the Cu nanoparticles were homogenously anchored on TiO2−x nanosheets (Fig. 7k). As a catalyst for the NO3−RR, the designed heterostructure electrode exhibited an NH3 formation rate of 0.1143 mmol h−1 mg−1 along with a high FE of 81.34%, which obviously outperformed the individual Cu and TiO2−x counterparts (Fig. 7l). Such excellent electrocatalytic activity could be ascribed to the introduction of oxygen defects and metallic Cu clusters, which not only modified the electronic conductivity of the heterostructure electrode, but also optimized the adsorption energy of NO3− and hydrogenation manner that suppressed the generation of by-products (Fig. 7m and n). Similarly, Zhao et al.79 designed and fabricated a Cu-CuO heterostructure as an electrocatalyst for the NO3−RR, where the heterointerface between Cu and CuO was favorable for promoting the hydrogenation of *NO to *NOH and inhibiting the HER during the reduction process of NO3−. Thus, this heterostructure catalyst showed a molar-level NH3 yield rate of 3.17 mol h−1 g−1 and an ultrahigh FE of 98.7%. In addition, metallic Cu exhibits substantially high energy barriers to the dissociation of water in both neutral and alkaline electrolytes during electroreduction of NO3−, which controls the proton transfer rate and further leads to sluggish reaction kinetics for NH3 synthesis.80 To address the above-mentioned issue, Yu et al.81 employed DFT calculations to seek applicable ligands and confirmed that the uncoordinated carboxylate ligands could considerably promote water dissociation on Cu, accelerating the proton transfer and reaction kinetics of NO3−. They experimentally encapsulated Cu nanoparticles into the uncoordinated carboxylate-ligand-rich MOF matrix through a particle decomposition route. As expected, the designed Cu-based catalyst achieved an alluring electrochemical performance for the reduction of NO3− to NH3 in alkaline media, including a high NH3 yield rate of 496.4 mmol h−1 g−1 at an ultralow potential of −0.2 V vs. RHE and an outstanding stability of 20 h.
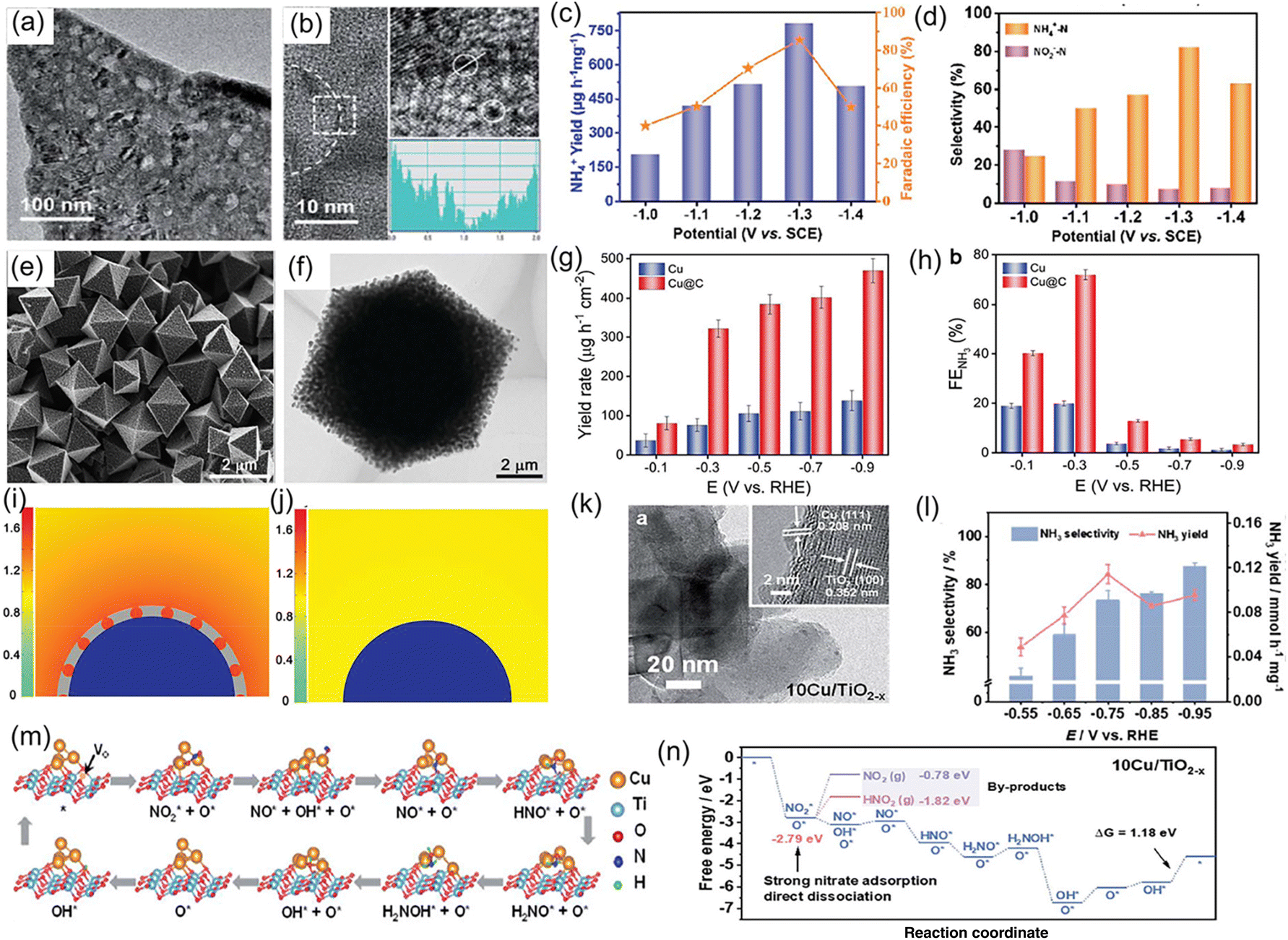 | ||
| Fig. 7 (a) TEM and (b) HRTEM images of a dr-Cu nanoplate. (c) NH4+ yield rate and FE, and (d) NH4+ selectivity of dr-Cu nanoplates at various potentials. Reproduced from ref. 72 with permission from The Royal Society of Chemistry, copyright 2021. (e) SEM and (f) TEM images of Cu@C. (g) NH3 yield rate and (h) FE of Cu@C under different potentials. Simulated concentrations and distribution of local NO3− on the surface of (i) Cu@C and (j) Cu at the diffusion time of 7 μs. The blue semicircle and the gray shell represent Cu and porous carbon, respectively. Reproduced from ref. 77 with permission from Wiley-VCH, copyright 2022. (k) TEM image of Cu/TiO2−x. (l) NH3 selectivity and yield rate of Cu/TiO2−x at each applied potential. (m) Reaction mechanism and (n) corresponding calculated free energy changes of the NO3−RR on the surface of Cu/TiO2−x. Reproduced from ref. 72 with permission from The Royal Society of Chemistry, copyright 2021. | ||
In addition to metallic Cu, other metal catalysts like Co, Fe, Ni, and Bi have been applied to highly effective reduction of NO3− to NH3.82–87 For instance, our group synthesized metallic Co–nitrogen-doped carbon nanotubes hybrid (Co–NCNTs) (Fig. 8a and b) and investigated their electrocatalytic activity for the NO3−RR. In 0.1 M NaOH with 0.1 M NO3−, the Co–NCNTs delivered a high activity for the NO3−RR with an NH3 production rate of 5996 μg h−1 cm−2 and FE of 92% at 0.6 V vs. RHE (Fig. 8c and d), and exhibited excellent durability with ∼8.7% attenuation of current density and well-maintained FE during the 12-h electrolysis. Furthermore, DFT calculations (Fig. 8e) revealed that the Co (111) facet is more favorable for the NO3−RR than the Co (200) and Co (220) facets, in which the rate-determining step is the hydrogenation of *NH to *NH2. The corresponding energy barrier was only 0.19 eV, indicating the impressive NO3−RR activity of metallic Co.88 Meanwhile, we also synthesized metallic Co nanoparticles embedded on carbon derived from corncob as an electrocatalyst for the NO3−RR to NH3, which achieved a large NH3 production rate of 0.6 mmol h−1 cm2 with a FE of 93.4%, as depicted in Fig. 8f and g.89 Fang and coworkers90 reported that metallic Fe anchored on cyano-coordination polymer porous nanosheets (Fe-cyano NSs) displayed an outstanding electrochemical NH3 synthesis through the reduction of NO3− in an alkaline electrolyte. Bi has also been employed as a highly efficient catalyst for electrochemical reduction of NO3− due to its unique atomic structure, in which the interlayer lattice compression shortens the Bi–Bi bond to broaden the 6p bandwidth for electronic delocalization, enhancing the adsorption energy for nitrogen intermediates.84 Iarchuk et al.91 synthesized Ni foam catalysts through a dynamic hydrogen-bubble-template-assisted electrodeposition process. An NH3 FE of more than 95% was obtained under the relatively low potential range from −0.1 to −0.3 V vs. RHE. Gao et al.92 constructed a Schottky heterostructure composed of metallic Ni and nitrogen-doped carbon nanotubes (Ni-NCNTs) for converting NO3− to NH3 at room temperature (Fig. 8h–j). In terms of heterostructure catalyst, the heterointerface between Ni nanoparticles and NCNTs could induce the formation of a built-in electric field (Fig. 8k), which facilitated the accumulation and fixation of NO3− on the surface of the catalyst and consequently promoting the reaction kinetics during the electrochemical process. As a result, the designed Ni-NCNTs enabled a high FE of 99% for the electrocatalytic reduction of NO3−, and a large NH3 formation rate of 5.1 mg h−1 cm−2 in the electrochemical conversion of NO3− (Fig. 8l and m).
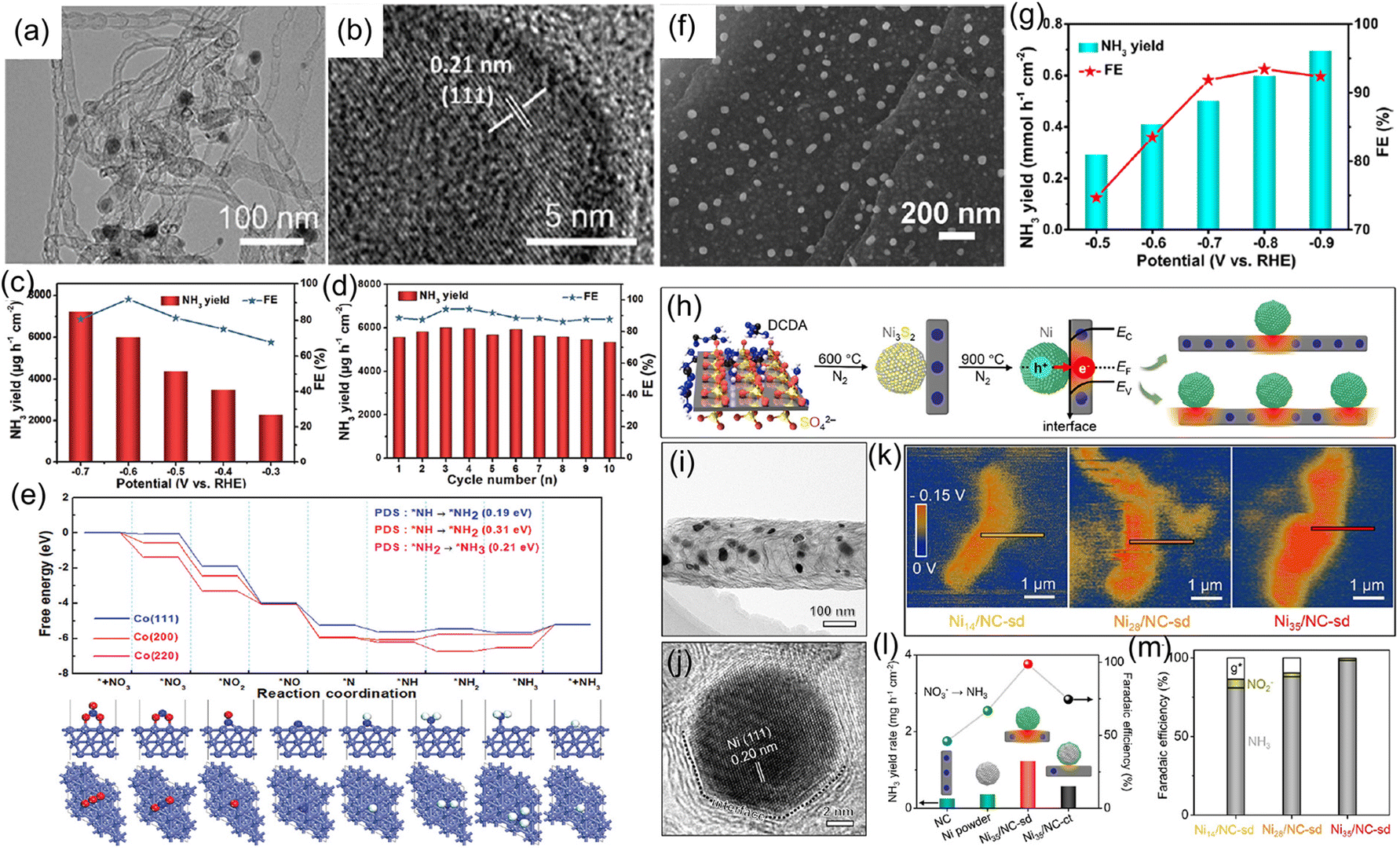 | ||
| Fig. 8 (a and b) TEM images of Co-NCNTs. (c) NH3 yield rate and FE of Co-NCNTs under various potentials. (d) Recycling test of Co-NCNTs at −0.6 V. (e) Free-energy profiles of the NO3−RR on different crystal facets of metallic Co. Reproduced from ref. 88 with permission from The Royal Society of Chemistry, copyright 2022. (f) SEM image, (g) NH3 yield rate and FE at different potentials of Co-carbon derived from corncob. Reproduced from ref. 89 with permission from the American Chemical Society, copyright 2022. (h) Fabrication process of Ni-NCNTs catalyst. (i and j) TEM and HRTEM images of Ni-NCNTs. (k) Surface electric field distribution of Ni-NCNTs sample. (l and m) NH3 yield rate and FE of Ni-NCNTs catalyst. Reproduced from ref. 92 with permission from Wiley-VCH, copyright 2021. | ||
:1 presented a high NH3 FE of 81.1% at −0.7 V vs. RHE within 6 h in 0.1 M Na2SO4 containing 100 ppm NO3−. Similarly, Sargent and coauthors94 indicated that Cu50Ni50 alloy catalysts only required an overpotential of 0.2 V to obtain the maximum NH3 FE under various concentrations of NO3−, and produced a 6-times increment in the NO3−RR activity compared to the case of pure Cu at 0 V vs. RHE. DFT calculations revealed that the introduction of Ni atoms led to the upshifting of the d-band center toward the Fermi level, which improved the adsorption energies of the intermediates and enhanced the selectivity for NH3. Recently, Liu et al.95 incorporated CuNi alloy nanoparticles into a nitrogen-doped carbon matrix with hierarchical pores by pyrolysis of bimetallic MOFs. A high NH3 selectivity of 94.4% and FE of 79.6% were achieved when utilizing the designed CuNi@C as a catalyst for the NO3−RR. Jeon et al.96 designed cobalt–copper (Co1−xCux) nanoparticles supported on a three-dimensional substrate for efficient and selective NH3 synthesis via an electrocatalytic NO3− reduction. Typically, the optimized Co0.5Cu0.5 catalyst performed at a high NH3 FE of over 95% at −0.03 V with an NH3 partial current density of ∼176 mA cm−2 at 50 mM nitrate, which is 7.3- and 1.7-fold higher than those of the pure Co and Cu counterparts, respectively. Importantly, replacing Co with Cu enabled tuning of the onset potential on the Co catalyst and maintained a high selectivity toward NH3.
3.4 Transition-metal compound catalysts
3.4.1.1 Copper-based oxides. Transition-metal oxides have been widely investigated as electrocatalysts for NH3 synthesis via converting NO3− under ambient conditions (Table 4). As summarized in the above section, metallic Cu has been intensively studied for the electroreduction of NO3− to NH3 owing to its favorable adsorption ability for NO3− and various intermediates (e.g., NO2− and NO). However, pure Cu catalysts still suffer from serious catalytic instability. For the purpose of overcoming this issue, substantial efforts have recently been made into the study of Cu-based oxide catalysts for highly-efficient electrochemical NO3− to NH3 conversion.
| Catalyst | Electrolyte | NH3 yield rate | Faradaic efficiency | Potentials (V vs. RHE) | Ref. |
|---|---|---|---|---|---|
| a NH3 yield rate. b Faradaic efficiency. | |||||
| Cu@Cu2+1O nanowires | 0.5 M K2SO4 + 50 mg L−1 NO3− | 576.53 μg h−1 mg−1 | 87.7% | −0.545 | 98 |
| CuO@MnO2 | 0.5 M K2SO4 + 100 mg L−1 NO3− | 0.240 mmol h−1 cm−2 | 94.92% | −0.645 | 104 |
| CuOx/TiO2 | 0.5 M Na2SO4 + 100 ppm NO3− | 1241.81 μg h−1 cm−2 | 92.34% | −0.75 | 105 |
| TiO2−x nanotubes | 0.5 M Na2SO4 + 50 ppm NO3− | 0.045 mmol h−1 mg−1 | 85% | −0.945 | 109 |
| Co-doped TiO2 nanosheet | 0.1 M NaOH + 0.1 M NO3− | 1127 μmol h−1 cm−2 | 98.2% | −0.9a/−0.5b | 111 |
| Co@TiO2 | 0.1 M PBS + 0.1 M NO3− | 800 μmol h−1 cm−2 | 96.7% | −1.0a/−0.7b | 112 |
| FeS2@TiO2 | 0.1 M NaOH + 0.1 M NaNO3 | 860.3 μmol h−1 cm−2 | 97.0% | −0.7a/−0.4b | 114 |
| Cu-doped Co3O4 nanowire | 0.1 M Na2SO4 + 500 ppm NO3− | 36.71 mmol h−1 g−1 | 86.5% | −0.6 | 117 |
| Co3O4 nanosheets with Co vacancies | 0.1 M NaOH + 0.1 M NaNO3 | 517.5 μmol h−1 cm−2 | 97.2% | −0.6a/−0.4b | 118 |
| NiCo2O4 nanowire | 0.1 M KOH + 0.1 M NaNO3 | 973.2 μmol h−1 cm−2 | 99.0% | −0.6a/−0.3b | 121 |
| ZnCo2O4 nanoarray | 0.1 M KOH + 0.1 M NaNO3 | 634.74 mmol h−1 cm−2 | 98.33% | −0.8a/−0.6b | 122 |
| BCDs/NiCo2O4 nanowire | 0.5 M K2SO4 + 200 ppm NO3− | 173.9 μmol h−1 cm−2 | 100% | −0.55 | 124 |
| CuO@Co3O4 | 1 M KOH + 1400 ppm NO3− | 1.915 mmol h−1 cm−2 | 99.17% | −0.23 | 125 |
| Co-doped Fe/Fe2O3 | 0.1 M Na2SO4 + 50 ppm NO3− | 1505.9 μg h−1 cm−2 | 85.2% | −0.95 | 135 |
| CoTiO3−x nanofibers | 0.1 M NaOH + 0.1 M NaNO3 | 30.4 mg h−1 mgcat−1 | 92.6% | −1.1a/−1.0b | 136 |
| CuWO4 nanospheres | 0.5 M Na2SO4 + 0.05 M NaNO3 | 5.84 mg h−1 mg−1 | 94.6% | −0.7 | 137 |
| Cu3P nanowires | 0.1 M PBS + 0.1 M NaNO3 | 1626.6 ± 36.1 μg h−1 cm−2 | 91.2 ± 2.5% | −0.5 | 141 |
| CoP nanosheets | 1.0 M NaOH + 1.0 M NaNO3 | 9.56 mol h−1 m−2 | 100% | −0.3 | 143 |
| Bi2S3/MoS2 | 0.1 M Na2SO4 + 0.1 M NaNO3 | 15.04 × 10−2 mmol h−1 cm−2 | 88.4% | −0.8 | 146 |
| Ni3N nanoparticles | 0.5 M Na2SO4 + 0.5 M NaNO3 | 9.185 mmol h−1 mg−1 | 89.5% | −0.795 | 148 |
| Fe3C nanoflakes | 1 M KOH + 75 mM KNO3 | 1.19 mmol h−1 mg−1 | 96.7% | −0.5 | 150 |
For example, Yuan et al.97 explored the influence of Cu oxidation state on the electrochemical reduction of NO3−, and found that the NH3 formation rate and FE of a Cu electrode could be significantly boosted after surface oxidation. After that, Ren et al.98 designed and fabricated core–shell structural Cu@Cu2+1O nanowires (Fig. 9a–c) for electrochemical conversion of NO3− to NH3. In terms of the Cu@Cu2+1O catalyst, the interior metallic Cu components could provide pathways for fast electron transfer due to the one-dimensional nanowire structure, while the exterior Cu2+1O layer affords a massive amount of catalytically active sites. Furthermore, DFT calculation results suggested that the introduction of a surface oxidation layer regulated the Cu d-band center and modulated the adsorption energies of various intermediates. Therefore, the constructed Cu@Cu2+1O catalyst exhibited a high NH3 yield rate of 576.53 μg h−1 mg−1 associated with a FE of 87.7% at −0.564 V vs. RHE, and NH3 selectivity of 76% (Fig. 9d). Qin et al.99 further illustrated the effects of the surface structure of Cu2O (exposing facets) on NO3− reduction to NH3. Both experimental and theoretical calculation results illustrated that the Cu2O (100) facet featured a relatively smaller energy barrier for NH3 formation than the Cu2O (111) facet, leading to a large NH3 formation rate (743 μg h−1 mg−1) and high FE (82.3%) at −0.6 V vs. RHE.
 | ||
| Fig. 9 (a) SEM, (b) TEM and (c) HRTEM images of Cu@Cu2+1O nanowires. (d) NH3 formation rate and FE of Cu@Cu2+1O nanowires under the applied potentials. Reproduced from ref. 98 with permission from Elsevier, copyright 2021. (e) Schematic illustration of NO3−-to-NH3 reduction over electrodes with nanotubular geometries. (f) NH3 FE and yield rate of CuOx/TiO2 at varying potentials. (g) Simulated NO2− concentration distribution on nanotubular and planar geometries. Reproduced from ref. 105 with permission from Elsevier, copyright 2022. (h) In situ electrochemical Raman spectra of CuO nanowires at given potentials. (i) Free-energy diagram for the NO3−RR over Cu nanowires. Reproduced from ref. 106 with permission from Wiley-VCH, copyright 2020. | ||
To further enhance the electrocatalytic activity of Cu2O toward the NO3−RR, various strategies have been employed, such as introducing oxygen defects100,101 and constructing heterostructures.79,102,103 For example, Xu et al.104 designed core–shell structural CuO@MnO2 hierarchical nanoarrays grown on Cu foam (CuO@MnO2/CF) for the NO3−RR. The heterointerface between the CuO nanowires and MnO2 nanosheets enabled abundant catalytically active sites and induced the formation of a built-in electric field, which were beneficial to the capture of NO3− and various intermediates during the electrochemical reactions, as well as accelerate ionic/electronic transfer at the interface. With these properties, CuO@MnO2/CF achieved an impressive electrochemical performance including a very-high NO3− conversion of 99.38%, NH3 FE of 94.92%, and selectivity of 96.67%. Meanwhile, this catalyst exhibits excellent stability, maintaining the NH3 yield rate and FE after 5 consecutive recycling tests. Qiu et al.105 incorporated CuOx nanoparticles into a TiO2-nanotube reactor for highly selective conversion of NO3− to NH3. In this CuOx/TiO2 catalyst system, TiO2 nanotubes could efficiently hinder the diffusion of NO2− intermediate and promote the conversion of NO3− to NH3 (Fig. 9e and g). The constructed CuOx/TiO2 heterostructure achieved a yield rate of 1241.81 μg h−1 cm−2, a high FE of 92.93% (Fig. 9f), and outstanding durability with a stable FE during the ten successive cycles of electrolysis.
Cu-based oxidation catalysts present outstanding electrochemical activity toward selective reduction of NO3− to NH3, but the origin of their activity and the structural evolution that occurs during the electrochemical reaction process were still experimentally unclear. In this regard, Zhang and his co-workers applied in situ characterization techniques to unveil the active phase of the CuO electrocatalyst. Experimental results suggested that CuO was transformed to Cu/Cu2O during the reduction process of NO3−, and served as an active phase for NO3− conversion (Fig. 9h). Then, online differential electrochemical mass spectrometry was adopted to analyze the reaction pathway. NO3− adsorbed on the surface of electrode was firstly reduced to *NO2 and *NO, in which *NO was hydrogenated to give *NHON and *NH2OH. Subsequently, *NH2OH was converted to *NH3 and further desorbed from the surface of the electrode generating NH3. DFT calculations also discovered that the origin of the activity enhancement was attributed to the reconstructed structure, in which electron transfer from Cu2O to Cu at the interface could promote the generation of the *NOH intermediate and limit the competing HER (Fig. 9i).106
3.4.1.2 Titanium-based oxides. Titanium oxide (TiO2) is a promising electrocatalyst candidate for the electrochemical reduction of NO3− owing to its advantages in terms of cost and robustness.107,108 For example, Jia et al.109 fabricated TiO2 nanotubes rich in oxygen vacancies as an electrocatalyst for the NO3−RR. An outstanding conversion rate of 95.2% for NH3 production from NO3− electroreduction associated with a FE of 85% was achieved. DFT calculations revealed that NO3− was adsorbed on the surface of the electrode and preferentially filled the oxygen defects existing in the TiO2 nanotubes, which weakened the N–O bonding, modulated the adsorption energies of the intermediates, and limited the generation of by-products. Analogously, oxygen-vacancy-TiO2 nanomaterials composed of rutile and anatase phases were fabricated as a catalyst for NH3 synthesis from the electroreduction of NO3−, which could deliver an NH3 FE of 78.0% and selectivity of 81.9%. Online differential electrochemical mass spectrometry and first-principle calculations revealed that the existence of oxygen vacancies (Ti3+) and the heterointerface between the rutile and anatase phases were favorable for modulating the adsorption energy of NO3− and facilitating the hydrogenation reaction to form *NOH, which led to a relatively high NH3 selectivity and FE.110 However, the limited selectivity for NH3 and sluggish reaction kinetics hinder their further application for NH3 electrosynthesis.
In this regard, our group proposed a series of modification strategies for improving the above-mentioned issues, such as heteroatom doping, and constructing Schottky junctions and p–n heterojunctions. For example, taking a Co-based catalyst with high catalytic activity into consideration, we introduced Co heteroatoms into a TiO2 nanoribbon array supported on Ti foil for electroreduction of NO3− (Fig. 10a and b). Co-doping can effectively improve the intrinsic electronic conductivity of TiO2 and increase the content of oxygen defects in TiO2, which further facilitates the adsorption of NO3− and transportation of charge at the interface, as well as decreasing the energy barrier of the potential-determining step (Fig. 10c). As a result, Co-doped TiO2 nanoribbon arrays delivered a large NH3 production rate of 1127 μmol h−1 cm−2 and a high FE of 98.2%, which was remarkably superior to that of its counterpart in alkaline media (88.5 μmol h−1 cm−2; 35.1%), as presented in Fig. 10d and e.111 Inspired by this, we further constructed a Schottky junction by integrating metallic Co nanoparticles into TiO2 nanobelt arrays (Co@TiO2) (Fig. 10f and g). A built-in electric field formed at the heterointerface between Co and TiO2, which was beneficial for the capture of NO3− on the surface of the electrocatalyst and thus facilitated mass transfer during the electroreduction process of NO3−. Consequently, in a neutral medium containing 0.1 M NO3−, the as-designed Co@TiO2 catalyst enabled a high NH3 FE of 96.7% at −0.7 V vs. RHE and a competitive NH3 formation rate of 800 μmol h−1 cm−2 at −1.0 V (Fig. 10h and i). Meanwhile, this catalyst also showed impressive durability during recycling tests and 50 h of bulk electrolysis (Fig. 10j).112 Following this, Fe3O4@TiO2,113 CoP@TiO2114 and FeS2@TiO2115 p–n heterojunctions were constructed by our team and utilized as electrocatalysts to convert NO3− to NH3, where the selectivity and efficiency of bare TiO2 for the NO3−RR were significantly enhanced.
 | ||
| Fig. 10 (a) SEM and (b) HRTEM images of Co-doped TiO2. (c) Calculated free-energy changes of the NO3−RR on the Co-doped TiO2. (d) NH3 yield rate and FE of Co-doped TiO2 under given potentials. (e) Comparison of NH3 yield rate and FE between TiO2 and Co-doped TiO2. Reproduced from ref. 111 with permission from The Royal Society of Chemistry, copyright 2022. (f) SEM and (g) HRTEM images of Co@TiO2 heterojunction catalyst. (h) LSV curves and (i) NH3 formation rate and FE at given potentials for Co@TiO2. (j) The long-term electrocatalytic performance of the Co@TiO2 catalyst. Reproduced from ref. 112 with permission from Wiley-VCH, copyright 2023. | ||
3.4.1.3 Spinel oxide. Spinel-type oxides feature unique advantages in terms of versatility, flexible ion arrangement, multivalence structure, and superior electronic conductivity, making them promising electrocatalysts for the NO3−RR.116 For example, Co3O4 has been extensively utilized as a catalyst for the electroreduction of NO3−, but its yield rate and selectivity of the target product NH3 are relatively low.117 To enhance the electrocatalytic activity of Co3O4, our group118 designed Co3O4 nanosheet arrays with Co vacancies on carbon cloth for converting NO3− to NH3 (Fig. 11a and b). As presented in Fig. 11c and d, Co3O4 with Co vacancies delivered a high NH3 yield rate of 517.5 μmol h−1 g−1 and a maximum FE of 97.2% at −0.6 and −0.4 V vs. RHE in alkaline electrolyte, respectively, which were higher than those of bare Co3O4 nanosheets (183.8 μmol h−1 g−1 with a FE of 85.9%). Furthermore, DFT calculations demonstrated that the introduction of Co vacancies regulated the electron structure of Co3O4, optimized the adsorption energy of NO3− and reduced the energy barrier of the potential-determining step (*NHO to *NHOH), leading to the high electrocatalytic activity (Fig. 11e and f). Similarly, we adopted Fe as a dopant to modulate the electron structure of Co3O4, further elevating its selectivity and NH3 yield rate during the electroreduction of NO3−.119 In line with the above-mentioned viewpoint, many bimetal spinel oxides, such as FeCo2O4,120 NiCo2O4,121 ZnCo2O4,122 AlCo2O4,77 and NiFe2O4,123 have been synthesized and investigated as electrocatalysts for a highly efficient NO3−RR by our team. As demonstrated in Fig. 11g and h, NiCo2O4 nanowire arrays grown on carbon cloth were synthesized for electrochemical NH3 production by conversion of NO3−. Owing to the synergistic effects of the two metal sites, NiCo2O4 nanowire arrays attained a large NH3 formation rate of 973.2 μmol h−1 cm−2 and large FE of 99.0% (Fig. 11i and j) in 0.1 M KOH with 0.1 M NaNO3. Impressively, the as-designed NiCo2O4 nanowires displayed exceptional durability with no significant fluctuations in both NH3 production rate and FE after 16 successive electrolysis experiments. After that, taking the Lewis-base property of NO3− into consideration, Lu et al.124 further introduced abundant Lewis acid sites on the surface of NiCo2O4 nanowire arrays for increasing the adsorption energy of NO3− by coupling with boron-doped carbon dots (BCDs/NiCo2O4). As revealed in Fig. 11k and l, the incorporation of BCDs enhanced the adsorption energy of NO3− on the surface of the BCDs/NiCo2O4 electrode. Meanwhile, in situ Raman spectra shown in Fig. 11m suggested that the intensity of the peak at 975 cm−1 associated with the N–O stretching vibration was boosted under the applied potentials, indicating that the Lewis acid sites induced by BCD doping were critically important for enhancing the adsorption ability of NO3−. As expected, the BCDs/NiCo2O4 catalyst provided a nearly ∼100% FE and a large NH3 production rate of 173.9 μmol h−1 cm−2 at −0.55 V vs. RHE (Fig. 11n).
 | ||
| Fig. 11 (a) SEM and (b) TEM images of Co3O4 nanosheets with Co vacancies. (c) LSV curves and (d) NH3 yield rate, FE under different potentials for Co3O4 nanosheets with Co vacancies. (e) Charge density distribution of Co3O4 with/without Co vacancies. (f) Free-energy diagrams for the NO3−RR on Co3O4 with Co vacancies. Reproduced from ref. 118 with permission from the American Chemical Society, copyright 2022. (g and h) SEM and mapping images of NiCo2O4. (i) LSV curves and (j) yield rate and FE of NH3 under given potentials. Reproduced from ref. 121 with permission from the Wiley-VCH, copyright 2022. Charge-density difference for NO3− adsorption on NiCo2O4 (k) and BCDs/NiCo2O4 (l). (m) In situ Raman spectra of NO3−RR over BCDs/NiCo2O4 at different applied potentials. (n) Comparison of NH3 FEs and yield rate of BCDs/NiCo2O4 and NiCo2O4. Reproduced from ref. 124 with permission from the Elsevier, copyright 2022. | ||
Co3O4 as an electrocatalyst for the NO3−RR still suffers from the critical issue that it is difficult to electrochemically reduce NO3− to NO2− using this catalyst. As mentioned above, Cu-based materials possess excellent electrocatalytic activity for NO3− to NO2−, and thus constructing a Co3O4-based heterostructure with Cu-based materials could achieve promising electrochemical performance. Liu et al.125 fabricated Co3O4 grown on CuO nanowire arrays to construct a hierarchical heterostructure for an efficient NO3−RR. At −0.23 V vs. RHE, CuO@Co3O4 provided an NH3 yield rate of 1.915 mmol h−1 cm−2, which was higher than those of CuO and Co3O4. Fu et al.126 built dual active sites on a Co3O4/Cu electrode, in which Cu focused on the reduction of NO3− to NO2−, and then Co3O4 generated H* (active hydrogen) as a strong reducing agent to further convert NO2− to NH3. As a result, the Co3O4/Cu catalyst presented a large NH3 yield rate of 684 μg mg−1 h−1 with 94.6% FE. Recently, Fan et al.127 fabricated a Co3O4 nanosheet grown in situ on TiO2 nanosheet arrays for the NO3−RR, which gave a large NH3 yield rate of 875 μmol mg−1 h−1 and a high FE of 93.1% in alkaline electrolyte.
3.4.1.4 Other metal oxides. Other types of transition-metal oxides have also been investigated for the NO3−RR, such as Bi2O3, Mn3O4, BiFeO3, and La2CuO4.128–131 However, the inferior electronic conductivity of metal oxides hinders their electrocatalytic activity. Currently, regulating their electronic structure through oxygen-defect engineering is a promising strategy.132,133 For instance, Wang et al.134 fabricated ultrathin CoOx nanosheets with abundant surface oxygen as an NO3−RR catalyst, attaining a large NH3 yield of 82.4 ± 4.8 mg h−1 mg−1 with a FE of 93.4 ± 3.8% at −0.3 V vs. RHE. The surface oxygen on the Co sites was prone to stabilize the adsorbed hydrogen on CoOx, and thus efficiently suppressed the formation of H2 and achieved a high selectivity for NH3 synthesis. Zhang et al.135 reported a Co-doped Fe/Fe2O3 catalyst for electrochemical NH3 synthesis by reducing NO3− under ambient conditions. This catalyst afforded an NH3 production rate of 1505.9 μg h−1 cm−2 with a FE of 85.2% and a high NH3 selectivity of 99.0%. Recently, our group reported that CoTiO3−x nanofibers with oxygen vacancies showed an NH3 formation rate of 30.4 mg h−1 mg−1 and a large FE of 92.6% in 0.1 M NaOH solution containing 0.1 M NO3−.136 The CuWO4 hollow nanospheres with oxygen vacancies showed a high NH3 FE of 94.6% and yield rate of 5.84 mg h−1 mg−1 at −0.7 V vs. RHE.137
 | ||
| Fig. 12 (a) SEM and (b) TEM images of CoP. (c) NH3 yield rate of CoP at given potentials. (d) In situ XANES of the Co K-edge of CoP. (e) Gibbs free-energy diagram of the NO3−RR on CoP. (f) Mechanism of the NO3−RR on CoP. Reproduced from ref. 143 with permission from The Royal Society of Chemistry, copyright 2022. (g) SEM image of Cu3P. (h) LSV curve, (i) NH3 yield rate and FE of Cu3P in NaNO3. Reproduced from ref. 141 with permission from The Royal Society of Chemistry, copyright 2021. | ||
4. Conclusions
The electrochemical NO3−RR has opened up a green and sustainable route for NH3 synthesis under ambient conditions, which is associated with two advantages: (i) the electrochemical NH3 synthesis from NO3− utilizes water as a proton source and is powered by renewable energy, which means that this process avoids the utilization of fossil fuels and reduces the NH3 production cost; (ii) the benign reaction conditions of the conversion of NO3− would enable distributed NH3 production in smaller-scale devices, which facilitates the production of fertilizer on demand and realizes a neutral carbon footprint. The important electrochemical characteristics of NH3 yield rate, Faradaic efficiency and selectivity largely depend on the electrocatalysts. Therefore, this review briefly describes the electroreduction mechanism from NO3− to NH3 under mild environmental conditions and summarizes the recent development of various electrocatalysts including noble-metal-based materials, single-atom metal catalysts, and transition-metal-based materials. Meanwhile, various effective design strategies for enhancing the electrocatalytic activity are outlined. Furthermore, it provides profound insights into the knowledge behind various optimization strategies, which are imperative for the development of highly-efficient electrocatalysts for the electrochemical conversion of NO3− to NH3. Although considerable progress has been achieved so far, the following points should also be considered in this field:(i) As mentioned in the discussion above, ongoing research into electrochemical NH3 synthesis from NO3− mainly focuses on the design and investigation of metal-based materials; less attention has been given to the exploration of metal-free electrocatalysts. From the energy-saving and emission-reduction points of view, it is of great significance to explore metal-free electrocatalysts with high activity, large selectivity and excellent stability for enabling the electrocatalytic NO3−RR under ambient conditions. As a consequence, more attention should be given to elaborately developing carbon-based electrocatalysts for the NO3−RR, providing an alluring strategy for large-scale NH3 production.
(ii) The electrochemical NO3−RR, as an emerging strategy for NH3 production under ambient conditions, has attained a dramatic growth in interest and various catalysts have been investigated in this field. However, none of the suitable catalysts can be regarded as a benchmark catalyst for electrocatalytic NO3−RR research. Besides, the variety of experimental details, such as the pH value of the electrolyte and the concentration of the nitrogen resource, play a critical role in catalytic activity and selectivity, and their effects on the electrochemical performance remain to be thoroughly studied. Such issues lead to incomparable results and thus limit the mutual communication and promotion in the community. Therefore, finding a standard catalyst and unification of experimental parameters are urgently required in the field of the electrochemical NO3−RR.
(iii) Many catalysts have exhibited superior catalytic activity and high NH3 selectivity during the electrochemical NO3−RR process, but their catalytic mechanisms and reaction processes were only revealed by theoretical calculations and remain unclear experimentally. For this reason, in situ characterization to scrutinize the pristine catalyst surface evolution (surface structure, element valence state, and exposed active sites) and adsorbed intermediates should be elaborately developed to uncover the real catalytic sites and reaction pathways upon the electrochemical reaction process for the rational design of electrocatalysts for the NO3−RR.
(iv) From the point of view of practical application, besides the fact that the electrochemical NO3−RR to NH3 process is still developing and lacks a catalyst with excellent durability and performance for supporting long-term electrolysis at the moment, another challenge is that this process will generate a tremendous amount of H2 as a side-product during the electrolysis, which is directly vented off into the atmosphere, forming a safety issue. Therefore, tremendous efforts are required before the electrochemical NO3−RR to NH3 can be put into practical operation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding support through large group Research Project under Grant No. RGP2/199/44.References
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Catalysts for nitrogen reduction to ammonia, Nat. Catal., 2018, 1, 490–500 CrossRef.
- Y. C. Wan, J. C. Xu and R. T. Lv, Heterogeneous electrocatalysts design for nitrogen reduction reaction under ambient conditions, Mater. Today, 2019, 27, 69–90 CrossRef CAS.
- J. Liang, Q. Liu, A. A. Alshehri and X. Sun, Recent advances in nanostructured heterogeneous catalysts for N-cycle electrocatalysis, Nano Res. Energy, 2022, 1, e9120010 CrossRef.
- J. W. S. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, How a century of ammonia synthesis changed the world. W, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- R. Schlogl, Catalytic synthesis of ammonia—A “never-ending story”?, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- C. Philibert, Renewable energy for industry: from green energy to green materials and fuels, IEA Report, 2017.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Beyond fossil fuel–driven nitrogen transformations, Science, 2018, 360, eaar6611 CrossRef PubMed.
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Greening ammonia toward the solar ammonia refinery, Joule, 2018, 2, 1055–1074 CrossRef CAS.
- B. Ma, H. Zhao, T. Li, Q. Liu, Y. Luo, C. Li, S. Lu, A. M. Asiri, D. Ma and X. Sun, Iron-group electrocatalysts for ambient nitrogen reduction reaction in aqueous media, Nano Res., 2021, 14, 555–569 CrossRef CAS.
- X. Zhu, S. Mou, Q. Peng, Q. Liu, Y. Luo, G. Chen, S. Gao and X. Sun, Aqueous electrocatalytic N2 reduction for ambient NH3 synthesis: recent advances in catalyst development and performance improvement, J. Mater. Chem. A, 2020, 8, 1545–1556 RSC.
- C. Tang and S. Z. Qiao, How to explore ambient electrocatalytic nitrogen reduction reliably and insightfully, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- T. Xu, B. Ma, J. Liang, L. Yue, Q. Liu, T. Li, H. Zhao, Y. Luo, S. Lu and X. Sun, Recent progress in metal-free electrocatalysts toward ambient N2 reduction reaction, Acta Phys.–Chim. Sin., 2021, 37, 2009043 Search PubMed.
- Q. Liu, T. Xu, Y. Luo, Q. Kong, T. Li, S. Lu, A. A. Alshehri, K. A. Alzahrani and X. Sun, Recent advances in strategies for highly selective electrocatalytic N2 reduction toward ambient NH3 synthesis, Curr. Opin. Electrochem., 2021, 29, 100766 CrossRef CAS.
- D. Liu, M. Chen, X. Du, H. Ai, K. H. Lo, S. Wang, S. Chen, G. Xing, X. Wang and H. Pan, Development of electrocatalysts for efficient nitrogen reduction reaction under ambient condition, Adv. Funct. Mater., 2021, 31, 2008983 CrossRef CAS.
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Electrocatalytic nitrate reduction for sustainable ammonia production, Joule, 2021, 5, 290–294 CrossRef.
- Y. Xu, K. Shi, T. Ren, H. Yu, K. Deng, X. Wang, H. Wang and L. Wang, Electronic metal-support interaction triggering interfacial charge polarization over CuPd/N-doped-C nanohybrids drives selectively electrocatalytic conversion of nitrate to ammonia, Small, 2022, 18, 2203335 CrossRef CAS PubMed.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Nitrate electroreduction: mechanism insight, in situ characterization, performance evaluation, and challenges, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- D. Anastasiadou, Y. van Beek, E. J. M. Hensen and M. C. Figueiredo, Ammonia electrocatalytic synthesis from nitrate, Electrochem. Sci. Adv., 2022 DOI:10.1002/elsa.202100220.
- J. Theerthagiri, J. Park, H. T. Das, N. Rahamathulla, E. S. F. Cardoso, A. P. Murthy, G. Maia, D. V. N. Vo and M. Y. Choi, Electrocatalytic conversion of nitrate waste into ammonia: a review, Environ. Chem. Lett., 2022, 20, 2929–2949 CrossRef CAS.
- H. Xu, Y. Ma, J. Chen, W. X. Zhang and J. Yang, Electrocatalytic reduction of nitrate–A step towards a sustainable nitrogen cycle, Chem. Soc. Rev., 2022, 51, 2710 RSC.
- Z. Li, Z. Deng, L. Ouyang, X. Fan, L. Zhang, S. Sun, Q. Liu, A. A. Alshehri, Y. Luo, Q. Kong and X. Sun, CeO2 nanoparticles with oxygen vacancies decorated N-doped carbon nanorods: A highly efficient catalyst for nitrate electroreduction to ammonia, Nano Res., 2022, 15, 8914–8921 CrossRef CAS.
- Q. Liu, Q. Liu, L. Xie, L. Yue, T. Li, Y. Luo, N. Li, B. Tang, L. Yu and X. Sun, A 3D FeOOH nanotube array: an efficient catalyst for ammonia electrosynthesis by nitrite reduction, Chem. Commun., 2022, 58, 5160–5163 RSC.
- Y. Arikawa, Y. Otsubo, H. Fujino, S. Horiuchi, E. Sakuda and K. Umakoshi, Nitrite reduction cycle on a dinuclear ruthenium complex producing ammonia, J. Am. Chem. Soc., 2018, 140, 842–847 CrossRef CAS PubMed.
- H. Chen, C. Zhang, L. Sheng, M. Wang, W. Fu, S. Gao, Z. Zhang, S. Chen, R. Si, L. Wang and B. Yang, Copper single-atom catalyst as a high-performance electrocatalyst for nitrate-ammonium conversion, J. Hazard. Mater., 2022, 434, 128892 CrossRef CAS PubMed.
- Y. Zeng, C. Priest, G. Wang and G. Wu, Restoring the nitrogen cycle by electrochemical reduction of nitrate: progress and prospects, Small Methods, 2020, 4, 2000672 CrossRef CAS.
- X. Zhang, Y. Wang, C. Liu, Y. Yu, S. Lu and B. Zhang, Recent advances in non-noble metal electrocatalysts for nitrate reduction, Chem. Eng. J., 2021, 403, 126269 CrossRef CAS.
- S. Garcia-Segura, M. Lanzarini-Lopes, K. Hristovski and P. Westerhoff, Electrocatalytic reduction of nitrate: Fundamentals to full-scale water treatment applications, Appl. Catal., B, 2018, 236, 546–568 CrossRef CAS.
- M. D. V. Rosca, M. T. de Groot and M. T. M. Koper, Nitrogen cycle electrocatalysis, Chem. Rev., 2009, 109, 2209–2244 CrossRef PubMed.
- M. T. de Groot and M. T. M. Koper, The influence of nitrate concentration and acidity on the electrocatalytic reduction of nitrate on platinum, J. Electroanal. Chem., 2004, 562, 81–94 CrossRef CAS.
- R. Lange, E. Maisonhaute, R. Robin and V. Vivier, On the kinetics of the nitrate reduction in concentrated nitric acid, Electrochem. Commun., 2013, 29, 25–28 CrossRef CAS.
- D. Sicsic, F. Balbaud-Célérier and B. Tribollet, Mechanism of nitric acid reduction and kinetic modelling, Eur. J. Inorg. Chem., 2014, 2014, 6174–6184 CrossRef CAS.
- D. Xu, Y. Li, L. Yin, Y. Ji, J. Niu and Y. Yu, Electrochemical removal of nitrate in industrial wastewater, Front. Environ. Sci. Eng., 2018, 12, 9 CrossRef.
- J. Gao, B. Jiang, C. Ni, Y. Qi and X. Bi, Enhanced reduction of nitrate by noble metal-free electrocatalysis on P doped three-dimensional Co3O4 cathode: Mechanism exploration from both experimental and DFT studies, Chem. Eng. J., 2020, 382, 123034 CrossRef CAS.
- A. C. A. de Vooys, R. A. van Santen and J. A. R. van Veen, Electrocatalytic reduction of NO3− onpalladium/copper electrodes, J. Mol. Catal. A: Chem., 2000, 154, 203–215 CrossRef CAS.
- W. L. M. D. Bartberger, E. Ford, K. M. Miranda, C. Switzer, J. M. Fukuto, P. J. Farmer, D. A. Wink and K. N. Houk, The reduction potential of nitric oxide (NO) and its importance to NO biochemistry, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10958 CrossRef PubMed.
- A. C. A. de Vooys, M. T. M. Koper, R. A. van Santen and J. A. R. van Veen, Mechanistic study of the nitric oxide reduction on a polycrystalline platinum electrode, Electrochim. Acta, 2001, 46, 923–930 CrossRef CAS.
- T. Yoshioka, K. Iwase, S. Nakanishi, K. Hashimoto and K. Kamiya, Electrocatalytic reduction of nitrate to nitrous oxide by a copper-modified covalent triazine framework, J. Phys. Chem. C, 2016, 120, 15729–15734 CrossRef CAS.
- G. A. Cerrón-Calle, A. S. Fajardo, C. M. Sánchez-Sánchez and S. Garcia-Segura, Highly reactive Cu-Pt bimetallic 3D-electrocatalyst for selective nitrate reduction to ammonia, Appl. Catal., B, 2022, 302, 120844 CrossRef.
- C. A. Clark, C. P. Reddy, H. Xu, K. N. Heck, G. Luo, T. P. Senftle and M. S. Wong, Mechanistic insights into pH-controlled nitrite reduction to ammonia and hydrazine over rhodium, ACS Catal., 2020, 10, 494–509 CrossRef CAS.
- X. Li, X. Zhao, Y. Zhou, J. Hu, H. Zhang, X. Hu and G. Hu, Pd nanocrystals embedded in BC2N for efficient electrochemical conversion of nitrate to ammonia, Appl. Surf. Sci., 2022, 584, 152556 CrossRef CAS.
- M. Jiang, A. Tao, Y. Hu, L. Wang, K. Zhang, X. Song, W. Yan, Z. Tie and Z. Jin, Crystalline modulation engineering of Ru nanoclusters for boosting ammonia electrosynthesis from dinitrogen or nitrate, ACS Appl. Mater. Interfaces, 2022, 14, 17470–17478 CrossRef CAS PubMed.
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S. X. Dou, L. Zhang and J. C. Yu, Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed.
- J. Lim, C. Y. Liu, J. Park, Y. H. Liu, T. P. Senftle, S. W. Lee and M. C. Hatzell, Structure sensitivity of Pd facets for enhanced electrochemical nitrate reduction to ammonia, ACS Catal., 2021, 11, 7568–7577 CrossRef CAS.
- T. Ren, Z. Yu, H. Yu, K. Deng, Z. Wang, X. Li, H. Wang, L. Wang and Y. Xu, Interfacial polarization in metal-organic framework reconstructed Cu/Pd/CuOx multi-phase heterostructures for electrocatalytic nitrate reduction to ammonia, Appl. Catal., B, 2022, 318, 121805 CrossRef CAS.
- Y. Xu, K. Ren, T. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Ultralow-content Pd in-situ incorporation mediated hierarchical defects in corner-etched Cu2O octahedra for enhanced electrocatalytic nitrate reduction to ammonia, Appl. Catal., B, 2022, 306, 121094 CrossRef CAS.
- M. Liu, Q. Mao, K. Shi, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Electroreduction of nitrate to ammonia on palladium–cobalt–oxygen nanowire arrays, ACS Appl. Mater. Interfaces, 2022, 14, 13169–13176 CrossRef CAS PubMed.
- Y. Xu, K. Ren, T. Ren, M. Wang, M. Liu, Z. Wang, X. Li, L. Wang and H. Wang, Cooperativity of Cu and Pd active sites in CuPd aerogels enhances nitrate electroreduction to ammonia, Chem. Commun., 2021, 57, 7525–7528 RSC.
- Q. Gao, H. S. Pillai, Y. Huang, S. Liu, Q. Mu, X. Han, Z. Yan, H. Zhou, Q. He, H. Xin and H. Zhu, Breaking adsorption-energy scaling limitations of electrocatalytic nitrate reduction on intermetallic CuPd nanocubes by machine-learned insights, Nat. Commun., 2022, 13, 2338 CrossRef CAS PubMed.
- G. Zhang, X. Li, P. Shen, Y. Luo, X. Li and K. Chu, PdNi nanosheets boost nitrate electroreduction to ammonia, J. Environ. Chem. Eng., 2022, 10, 108362 CrossRef CAS.
- Y. Xu, Y. Sheng, M. Wang, T. Liu, H. Yu, K. Deng, Z. Wang, L. Wang and H. Wang, Lattice-strain and Lewis acid sites synergistically promoted nitrate electroreduction to ammonia over PdBP nanothorn arrays, J. Mater. Chem. A, 2022, 10, 16290–16296 RSC.
- H. Liu, J. Park, Y. Chen, Y. Qiu, Y. Cheng, K. Srivastava, S. Gu, B. H. Shanks, L. T. Roling and W. Li, Electrocatalytic nitrate reduction on oxide-derived silver with tunable selectivity to nitrite and ammonia, ACS Catal., 2021, 11, 8431–8442 CrossRef CAS.
- J. Qin, K. Wu, L. Chen, X. Wang, Q. Zhao, B. Liu and Z. Ye, Achieving high selectivity for nitrate electrochemical reduction to ammonia over MOF-supported RuxOy clusters, J. Mater. Chem. A, 2022, 10, 3963–3969 RSC.
- Y. Wang, H. Li, W. Zhou, X. Zhang, B. Zhang and Y. Yu, Structurally disordered RuO2 nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia, Angew. Chem., Int. Ed., 2022, 61, e202202604 CAS.
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Shao and Y. Guo, Theoretical insights into the mechanism of selective nitrate-to-ammonia electroreduction on single-atom catalysts, Adv. Funct. Mater., 2021, 31, 2008533 CrossRef CAS.
- L. Lv, Y. Shen, J. Liu, X. Meng, X. Gao, M. Zhou, Y. Zhang, D. Gong, Y. Zheng and Z. Zhou, Computational screening of high activity and selectivity TM/gC3N4 single-atom catalysts for electrocatalytic reduction of nitrates to ammonia, J. Phys. Chem. Lett., 2021, 12, 11143–11150 CrossRef CAS PubMed.
- T. Zhu, Q. Chen, P. Liao, W. Duan, S. Liang, Z. Yan and C. Feng, Single-atom Cu catalysts for enhanced electrocatalytic nitrate reduction with significant alleviation of nitrite production, Small, 2020, 16, 2004526 CrossRef CAS PubMed.
- J. Li, Y. Zhang, C. Liu, L. Zheng, E. Petit, K. Qi, Y. Zhang, H. Wu, W. Wang, A. Tiberj, X. Wang, M. Chhowalla, L. Lajaunie, R. Yu and D. Voiry, 3.4% Solar-to-ammonia efficiency from nitrate using Fe single atomic catalyst supported on MoS2 nanosheets, Adv. Funct. Mater., 2022, 32, 2108316 CrossRef CAS.
- Z. Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F. Y. Chen, J. Y. T. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- P. Li, Z. Jin, Z. Fang and G. Yu, A single-site iron catalyst with preoccupied active centers that achieves selective ammonia electrosynthesis from nitrate, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- J. Yang, H. Qi, A. Li, X. Liu, X. Yang, S. Zhang, Q. Zhao, Q. Jiang, Y. Su, L. Zhang, J. F. Li, Z. Q. Tian, W. Liu, A. Wang and T. Zhang, Potential-driven restructuring of Cu single atoms to nanoparticles for boosting the electrochemical reduction of nitrate to ammonia, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS PubMed.
- X. F. Cheng, J. H. He, H. Q. Ji, H. Y. Zhang, Q. Cao, W. J. Sun, C. L. Yan and J. M. Lu, Coordination symmetry breaking of single-atom catalysts for robust and efficient nitrate electroreduction to ammonia, Adv. Mater., 2022, 34, 2205767 CrossRef CAS PubMed.
- W. D. Zhang, H. Dong, L. Zhou, H. Xu, H. R. Wang, X. Yan, Y. Jiang, J. Zhang and Z. G. Gu, Fe single-atom catalysts with pre-organized coordination structure for efficient electrochemical nitrate reduction to ammonia, Appl. Catal., B, 2022, 317, 121750 CrossRef CAS.
- J. Cai, Y. Wei, A. Cao, J. Huang, Z. Jiang, S. Lu and S. Q. Zang, Electrocatalytic nitrate-to-ammonia conversion with ∼100% Faradaic efficiency via single-atom alloying, Appl. Catal., B, 2022, 316, 121683 CrossRef CAS.
- E. Murphy, Y. Liu, I. Matanovic, S. Guo, P. Tieu, Y. Huang, A. Ly, S. Das, I. Zenyuk, X. Pan, E. Spoerke and P. Atanassov, Highly durable and selective Fe- and Mo-based atomically dispersed electrocatalysts for nitrate reduction to ammonia via distinct and synergized NO2− pathways, ACS Catal., 2022, 12, 6651–6662 CrossRef CAS.
- D. Yin, Y. Liu, P. Song, P. Chen, X. Liu, L. Cai and L. Zhang, In situ growth of copper/reduced graphene oxide on graphite surfaces for the electrocatalytic reduction of nitrate, Electrochim. Acta, 2019, 324, 134846 CrossRef CAS.
- G. F. Chen, Y. Yuan, H. Jiang, S. Y. Ren, L. X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- E. Pérez-Gallent, M. C. Figueiredo, I. Katsounaros and M. T. M. Koper, Electrocatalytic reduction of nitrate on copper single crystals in acidic and alkaline solutions, Electrochim. Acta, 2017, 227, 77–84 CrossRef.
- X. Wang, M. Zhu, G. Zeng, X. Liu, C. Fang and C. Li, A three-dimensional Cu nanobelt cathode for highly efficient electrocatalytic nitrate reduction, Nanoscale, 2020, 12, 9385–9391 RSC.
- K. Wu, C. Sun, Z. Wang, Q. Song, X. Bai, X. Yu, Q. Li, Z. Wang, H. Zhang, J. Zhang, X. Tong, Y. Liang, A. Khosla and Z. Zhao, Surface reconstruction on uniform Cu nanodisks boosted electrochemical nitrate reduction to ammonia, ACS Mater. Lett., 2022, 4, 650–656 CrossRef CAS.
- X. Fu, X. Zhao, X. Hu, K. He, Y. Yu, T. Li, Q. Tu, X. Qian, Q. Yue, M. R. Wasielewski and Y. Kang, Alternative route for electrochemical ammonia synthesis by reduction of nitrate on copper nanosheets, Appl. Mater. Today, 2020, 19, 100620 CrossRef.
- Y. Zhao, Y. Liu, Z. Zhang, Z. Mo, C. Wang and S. Gao, Flower-like open-structured polycrystalline copper with synergistic multi-crystal plane for efficient electrocatalytic reduction of nitrate to ammonia, Nano Energy, 2022, 97, 107124 CrossRef CAS.
- Y. Xu, M. Wang, K. Ren, T. Ren, M. Liu, Z. Wang, X. Li, L. Wang and H. Wang, Atomic defects in pothole-rich two-dimensional copper nanoplates triggering enhanced electrocatalytic selective nitrate-to-ammonia transformation, J. Mater. Chem. A, 2021, 9, 16411–16417 RSC.
- L. Yang, J. Li, F. Du, J. Gao, H. Liu, S. Huang, H. Zhang, C. Li and C. Guo, Interface engineering cerium-doped copper nanocrystal for efficient electrochemical nitrate-to-ammonia production, Electrochim. Acta, 2022, 411, 140095 CrossRef CAS.
- Q. Hu, Y. Qin, X. Wang, H. Zheng, K. Gao, H. Yang, P. Zhang, M. Shao and C. He, Grain boundaries engineering of hollow copper nanoparticles enables highly efficient ammonia electrosynthesis from nitrate, CCS Chem., 2022, 4, 2053–2064 CrossRef CAS.
- X. Zhao, G. Hu, F. Tan, S. Zhang, X. Wang, X. Hu, A. V. Kuklin, G. V. Baryshnikov, H. Ågren, X. Zhou and H. Zhang, Copper confined in vesicle-like BCN cavities promotes electrochemical reduction of nitrate to ammonia in water, J. Mater. Chem. A, 2021, 9, 23675–23686 RSC.
- X. Zhu, H. Huang, H. Zhang, Y. Zhang, P. Shi, K. Qu, S. B. Cheng, A. L. Wang and Q. Lu, Filling mesopores of conductive metal–organic frameworks with Cu clusters for selective nitrate reduction to ammonia, ACS Appl. Mater. Interfaces, 2022, 14, 32176–32182 CrossRef CAS PubMed.
- Z. Song, Y. Liu, Y. Zhong, Q. Guo, J. Zeng and Z. Geng, Efficient Electroreduction of Nitrate into Ammonia at Ultralow Concentrations Via an Enrichment Effect, Adv. Mater., 2022, 34, 2204306 CrossRef CAS PubMed.
- X. Zhang, C. Wang, Y. Guo, B. Zhang, Y. Wang and Y. Yu, Cu clusters/TiO2-x with abundant oxygen vacancies for enhanced electrocatalytic nitrate reduction to ammonia, J. Mater. Chem. A, 2022, 10, 6448–6453 RSC.
- J. Zhao, Z. Shen, J. Yu, Y. Guo, M. Mushtaq, Y. Ding, Z. Song, W. Zhang, X. Huang, Y. Li, D. Liu and X. Cai, Constructing Cu-CuO heterostructured skin on Cu cubes to promote electrocatalytic ammonia production from nitrate wastewater, J. Hazard Mater., 2022, 439, 129653 CrossRef CAS PubMed.
- M. Luo, Z. Wang, Y. C. Li, J. Li, F. Li, Y. Lum, D. H. Nam, B. Chen, J. Wicks, A. Xu, T. Zhuang, W. R. Leow, X. Wang, C. T. Dinh, Y. Wang, Y. Wang, D. Sinton and E. H. Sargent, Hydroxide promotes carbon dioxide electroreduction to ethanol on copper via tuning of adsorbed hydrogen, Nat. Commun., 2019, 10, 5814 CrossRef CAS PubMed.
- J. Yu, Y. Qin, X. Wang, H. Zheng, K. Gao, H. Yang, L. Xie, Q. Hu and C. He, Boosting electrochemical nitrate-ammonia conversion via organic ligands-tuned proton transfer, Nano Energy, 2022, 103, 107705 CrossRef CAS.
- X. Deng, Y. Yang, L. Wang, X. Z. Fu and J. L. Luo, Metallic Co nanoarray catalyzes selective NH3 production from electrochemical nitrate reduction at current densities exceeding 2 A cm−2, Adv. Sci., 2021, 8, 2004523 CrossRef CAS PubMed.
- J. Wang, J. Liang, P. Liu, Z. Yan, L. Cui, L. Yue, L. Zhang, Y. Ren, T. Li, Y. Luo, Q. Liu, X. E. Zhao, N. Li, B. Tang, Y. Liu, S. Gao, A. M. Asiri, H. Hao, R. Gao and X. Sun, Biomass Juncus derived carbon decorated with cobalt nanoparticles enables high-efficiency ammonia electrosynthesis by nitrite reduction, J. Mater. Chem. A, 2022, 10, 2842–2848 RSC.
- N. Zhang, J. Shang, X. Deng, L. Cai, R. Long, Y. Xiong and Y. Chai, Governing interlayer strain in bismuth nanocrystals for efficient ammonia electrosynthesis from nitrate reduction, ACS Nano, 2022, 16, 4795–4804 CrossRef CAS PubMed.
- Q. Chen, J. Liang, Q. Liu, K. Dong, L. Yue, P. Wei, Y. Luo, Q. Liu, N. Li, B. Tang, A. A. Alshehri, M. S. Hamdy, Z. Jiang and X. Sun, Co nanoparticle-decorated pomelo-peel-derived carbon enabled high-efficiency electrocatalytic nitrate reduction to ammonia, Chem. Commun., 2022, 58, 4259–4262 RSC.
- K. Wang, R. Mao, R. Liu, J. Zhang and X. Zhao, Sulfur-dopant-promoted electrocatalytic reduction of nitrate by a self-supported iron cathode: selectivity, stability, and underlying mechanism, Appl. Catal., B, 2022, 319, 121862 CrossRef CAS.
- C. Wang, W. Zhou, Z. Sun, Y. Wang, B. Zhang and Y. Yu, Integrated selective nitrite reduction to ammonia with tetrahydroisoquinoline semi-dehydrogenation over a vacancy-rich Ni bifunctional electrode, J. Mater. Chem. A, 2021, 9, 239 RSC.
- J. Chen, Q. Zhou, L. Yue, D. Zhao, L. Zhang, Y. Luo, Q. Liu, N. Li, A. A. Alshehri, M. S. Hamdy, F. Gong and X. Sun, Co–NCNT nanohybrid as a highly active catalyst for the electroreduction of nitrate to ammonia, Chem. Commun., 2022, 58, 3787–3790 RSC.
- T. Xie, X. Li, J. Li, J. Chen, S. Sun, Y. Luo, Q. Liu, D. Zhao, C. Xu, L. Xie and X. Sun, Co nanoparticles decorated corncob-derived biomass carbon as an efficientelectrocatalyst for nitrate reduction to ammonia, Inorg. Chem., 2022, 61, 14195–14200 CrossRef CAS PubMed.
- Z. Fang, Z. Jin, S. Tang, P. Li, P. Wu and G. Yu, Porous two-dimensional iron-cyano nanosheets for high-rate electrochemical nitrate reduction, ACS Nano, 2022, 16, 1072–1081 CrossRef CAS PubMed.
- A. Iarchuk, A. Dutta and P. Broekmann, Novel Ni foam catalysts for sustainable nitrate to ammonia electroreduction, J. Hazard. Mater., 2022, 439, 129504 CrossRef CAS PubMed.
- P. Gao, Z. H. Xue, S. N. Zhang, D. Xu, G. Y. Zhai, Q. Y. Li, J. S. Chen and X. H. Li, Schottky barrier-induced surface electric field boosts universal reduction of NOx− in water to ammonia, Angew. Chem., Int. Ed., 2021, 60, 20711–20716 CrossRef CAS PubMed.
- Z. Tang, Z. Bai, X. Li, L. Ding, B. Zhang and X. Chang, Chloride-derived bimetallic Cu-Fe nanoparticles for high-selective nitrate-to-ammonia electrochemical catalysis, Processes, 2022, 10, 751 CrossRef CAS.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D. H. Nam, C. S. Tan, Y. Ding, J. Wu, Y. Lum, C. T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, Enhanced nitrate-to-ammonia activity on copper-nickel alloys via tuning of intermediate adsorption, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- Y. Liu, B. Deng, K. Li, H. Wang, Y. Sun and F. Dong, Metal-organic framework derived carbon-supported bimetallic copper-nickel alloy electrocatalysts for highly selective nitrate reduction to ammonia, J. Colloid Interface Sci., 2022, 614, 405–414 CrossRef CAS PubMed.
- T. H. Jeon, Z. Y. Wu, F. Y. Chen, W. Choi, P. J. J. Alvarez and H. Wang, Cobalt-copper nanoparticles on three-dimensional substrate for efficient ammonia synthesis via electrocatalytic nitrate reduction, J. Phys. Chem. C, 2022, 126, 6982–6989 CrossRef CAS.
- J. Yuan, Z. Xing, Y. Tang and C. Liu, Tuning the oxidation state of Cu Electrodes for selective electrosynthesis of ammonia from nitrate, ACS Appl. Mater. Interfaces, 2021, 13, 52469–52478 CrossRef CAS PubMed.
- T. Ren, K. Ren, M. Wang, M. Liu, Z. Wang, H. Wang, X. Li, L. Wang and Y. Xu, Concave-convex surface oxide layers over copper nanowires boost electrochemical nitrate-to-ammonia conversion, Chem. Eng. J., 2021, 426, 130759 CrossRef CAS.
- Q. Hu, Y. Qin, X. Wang, Z. Wang, X. Huang, H. Zheng, K. Gao, H. Yang, P. Zhang, M. Shao and C. He, Reaction intermediate-mediated electrocatalyst synthesis favors specified facet and defect exposure for efficient nitrate–ammonia conversion, Energy Environ. Sci., 2021, 14, 4989–4997 RSC.
- Z. Gong, W. Zhong, Z. He, Q. Liu, H. Chen, D. Zhou, N. Zhang, X. Kang and Y. Chen, Regulating surface oxygen species on copper(I) oxides via plasma treatment for effective reduction of nitrate to ammonia, Appl. Catal., B, 2022, 305, 121021 CrossRef CAS.
- J. Geng, S. Ji, H. Xu, C. Zhao, S. Zhang and H. Zhang, Electrochemical reduction of nitrate to ammonia in a fluidized electrocatalysis system with oxygen vacancy-rich CuOx nanoparticles, Inorg. Chem. Front., 2021, 8, 5209–5213 RSC.
- H. Wang, Y. Guo, C. Li, H. Yu, K. Deng, Z. Wang, X. Li, Y. Xu and L. Wang, Cu/CuOx in-plane heterostructured nanosheetarrays with rich oxygen vacancies enhance nitrate electroreduction to ammonia, ACS Appl. Mater. Interfaces, 2022, 14, 34761–34769 CrossRef CAS PubMed.
- S. Liu, L. Cui, S. Yin, H. Ren, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Heterointerface-triggered electronic structure reformation: Pd/CuO nano-olives motivate nitrite electroreduction to ammonia, Appl. Catal., B, 2022, 319, 121876 CrossRef CAS.
- Y. Xu, Y. Sheng, M. Wang, T. Ren, K. Shi, Z. Wang, X. Li, L. Wang and H. Wang, Interface coupling induced built-in electric fields boost electrochemical nitrate reduction to ammonia over CuO@MnO2 core-shell hierarchical nanoarrays, J. Mater. Chem. A, 2022, 10, 16883–16890 RSC.
- W. Qiu, X. Chen, Y. Liu, D. Xiao, P. Wang, R. Li, K. Liu, Z. Jin and P. Li, Confining intermediates within a catalytic nanoreactor facilitates nitrate-to-ammonia electrosynthesis, Appl. Catal., B, 2022, 315, 121548 CrossRef CAS.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS PubMed.
- Y. Guo, R. Zhang, S. Zhang, Y. Zhao, Q. Yang, Z. Huang, B. Dong and C. Zhi, Pd doping-weakened intermediate adsorption to promote electrocatalytic nitrate reduction on TiO2 nanoarrays for ammonia production and energy supply with zinc-nitrate batteries, Energy Environ. Sci., 2021, 14, 3938–3944 RSC.
- J. Gao, B. Jiang, C. Ni, Y. Qi, Y. Zhang, N. Oturan and M. A. Oturan, Non-precious Co3O4-TiO2/Ti cathode based electrocatalytic nitrate reduction: preparation, performance and mechanism, Appl. Catal., B, 2019, 254, 391–402 CrossRef CAS.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, Boosting selective nitrate electroreduction to ammonium by constructing oxygen vacancies in TiO2, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- Z. Wei, X. Niu, H. Yin, S. Yu and J. Li, Synergistic effect of oxygen defects and hetero-phase junctions of TiO2 for selective nitrate electroreduction to ammonia, Appl. Catal., A, 2022, 636, 118596 CrossRef CAS.
- D. Zhao, C. Ma, J. Li, R. Li, X. Fan, L. Zhang, K. Dong, Y. Luo, D. Zheng, S. Sun, Q. Liu, Q. Li, Q. Lu and X. Sun, Direct eight-electron NO3−-to-NH3 conversion: using a Co-doped TiO2 nanoribbon array as a high-efficiency electrocatalyst, Inorg. Chem. Front., 2022, 9, 6412–6417 RSC.
- X. Fan, D. Zhao, Z. Deng, L. Zhang, J. Li, Z. Li, S. Sun, Y. Luo, D. Zheng, Y. Wang, B. Ying, J. Zhang, A. Alshehri, Y. Lin, C. Tang, X. Sun and Y. Zheng, Constructing Co@TiO2 nanoarray heterostructure with schottky contact for selective electrocatalytic nitrate reduction to ammonia, Small, 2023, 19, 2208036 CrossRef CAS PubMed.
- X. He, J. Li, R. Li, D. Zhao, L. Zhang, X. Ji, X. Fan, J. Chen, Y. Wang, Y. Luo, D. Zheng, L. Xie, S. Sun, Z. Cai, Q. Liu, K. Ma and X. Sun, Ambient ammonia synthesis via nitrate electroreduction in neutral media on Fe3O4 nanoparticles-decorated TiO2 nanoribbon array, Inorg. Chem., 2023, 62, 25–29 CrossRef CAS PubMed.
- H. Wang, D. Zhao, C. Liu, X. Fan, Z. Li, Y. Luo, D. Zheng, S. Sun, J. Chen, J. Zhang, Y. Liu, S. Gao, F. Gong and X. Sun, FeS2@TiO2 nanobelt array enabled high-efficiency electrocatalytic nitrate reduction to ammonia, J. Mater. Chem. A, 2022, 10, 24462–24467 RSC.
- Z. Deng, C. Ma, X. Fan, Z. Li, Y. Luo, S. Sun, D. Zheng, Q. Liu, J. Du, Q. Lu, B. Zheng and X. Sun, Construction of CoP/TiO2 nanoarray for enhanced electrochemical nitrate reduction to ammonia, Mater. Today Phys., 2022, 28, 100854 CrossRef CAS.
- X. Fan, L. Xie, J. Liang, Y. Ren, L. Zhang, L. Yue, T. Li, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, Q. Liu, Q. Kong and X. Sun, In situ grown Fe3O4 particle on stainless steel: A highly efficient electrocatalyst for nitrate reduction to ammonia, Nano Res., 2022, 15, 3050–3055 CrossRef CAS.
- Z. Niu, S. Fan, X. Li, Z. Liu, J. Wang, J. Duan, M. O. Tade and S. Liu, Facile tailoring of the electronic structure and the d-band center of copper-doped cobaltate for efficient nitrate electrochemical hydrogenation, ACS Appl. Mater. Interfaces, 2022, 14, 35477–35484 CrossRef CAS PubMed.
- Z. Deng, C. Ma, Z. Li, Y. Luo, L. Zhang, S. Sun, Q. Liu, J. Du, Q. Lu, B. Zheng and X. Sun, High-efficiency electrochemical nitrate reduction to ammonia on a Co3O4 nanoarray catalyst with cobalt vacancies, ACS Appl. Mater. Interfaces, 2022, 14, 46595–46602 CrossRef CAS PubMed.
- P. Wei, J. Liang, Q. Liu, L. Xie, X. Tong, Y. Ren, T. Li, Y. Luo, N. Li, B. Tang, A. M. Asiri, M. S. Hamdy, Q. Kong, Z. Wang and X. Sun, Iron-doped cobalt oxide nanoarray for efficient electrocatalytic nitrate-to-ammonia conversion, J. Colloid Interface Sci., 2022, 615, 636–642 CrossRef CAS PubMed.
- J. Li, D. Zhao, L. Zhang, L. Yue, Y. Luo, Q. Liu, N. Li, A. A. Alshehri, M. S. Hamdy, Q. Li and X. Sun, A FeCo2O4 nanowire array enabled electrochemical nitrate conversion to ammonia, Chem. Commun., 2022, 58, 4480–4483 RSC.
- Q. Liu, L. Xie, J. Liang, Y. Ren, Y. Wang, L. Zhang, L. Yue, T. Li, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, I. Shakir, P. O. Agboola, Q. Kong, Q. Wang, D. Ma and X. Sun, Ambient ammonia synthesis via electrochemicalreduction of nitrate enabled by NiCo2O4 nanowire array, Small, 2022, 18, 2106961 CrossRef CAS PubMed.
- Z. Li, J. Liang, Q. Liu, L. Xie, L. Zhang, Y. Ren, L. Yue, N. Li, B. Tang, A. A. Alshehri, M. S. Hamdy, Y. Luo, Q. Kong and X. Sun, High-efficiency ammonia electrosynthesis via selective reduction of nitrate on ZnCo2O4 nanosheet array, Mater. Today Phys., 2022, 23, 100619 CrossRef CAS.
- L. Xie, L. Hu, Q. Liu, S. Sun, L. Zhang, D. Zhao, Q. Liu, J. Chen, J. Li, L. Ouyang, A. Alshehri, Q. Kong and X. Sun, High-performance electrochemical nitrate reduction to ammonia under ambient conditions using NiFe2O4 nanosheet arrays, Inorg. Chem. Front., 2022, 9, 3392–3397 RSC.
- X. Lu, J. Yu, J. Cai, Q. Zhang, S. Yang, L. Gu, G. I. N. Waterhouse, S. Q. Zang, B. Yang and S. Lu, Exclusive nitrate to ammonia conversion via boron-doped carbon dots induced surface Lewis acid sites, Cell Rep. Phys. Sci., 2022, 3, 100961 CrossRef CAS.
- H. Liu, J. Li, F. Du, L. Yang, S. Huang, J. Gao, C. Li and C. Guo, A core-shell copper oxides-cobalt oxides heterostructure nanowire arrays for nitrate reduction to ammonia with high yield rate, Green Energy Environ., 2022 DOI:10.1016/j.gee.2022.03.003.
- W. Fu, Z. Hu, Y. Du, P. Su, Y. Su, Q. Zhang and M. Zhou, Building dual active sites Co3O4/Cu electrode to break scaling relations for enhancement of electrochemical reduction of nitrate to high-value ammonia, J. Hazard. Mater., 2022, 434, 128887 CrossRef CAS PubMed.
- X. Fan, C. Ma, D. Zhao, Z. Deng, L. Zhang, Y. Wang, Y. Luo, D. Zheng, T. Li, J. Zhang, S. Sun, Q. Lu and X. Sun, Unveiling selective nitrate reduction to ammonia with Co3O4 nanosheets/TiO2 nanobelt heterostructure catalyst, J. Colloid Interface Sci., 2023, 630, 714–720 CrossRef CAS PubMed.
- M. Chen, J. Bi, X. Huang, T. Wang, Z. Wang and H. Hao, Bi2O3 nanosheets arrays in-situ decorated on carbon cloth for efficient electrochemical reduction of nitrate, Chemosphere, 2021, 278, 130386 CrossRef CAS PubMed.
- H. Wang, Q. Mao, T. Ren, T. Zhou, K. Deng, Z. Wang, X. Li, Y. Xu and L. Wang, Synergism of interfaces and defects: Cu/oxygen vacancy-rich Cu-Mn3O4 heterostructured ultrathin nanosheet arrays for selective nitrate electroreduction to ammonia, ACS Appl. Mater. Interfaces, 2021, 13, 44733–44741 CrossRef CAS PubMed.
- J. Wang, D. Wu, M. Li, X. Wei, X. Yang, M. Shao and M. Gu, Bismuth ferrite as an electrocatalyst for the electrochemical nitrate reduction, Nano Lett., 2022, 22, 5600–5606 CrossRef CAS PubMed.
- Z. Gong, W. Zhong, Z. He, C. Jia, D. Zhou, N. Zhang, X. Kang and Y. Chen, Improving electrochemical nitrate reduction activity of layered perovskite oxide La2CuO4 via B-site doping, Catal. Today, 2022, 402, 259–265 CrossRef CAS.
- F. Du, J. Li, C. Wang, J. Yao, Z. Tan, Z. Yao, C. Li and C. Guo, Active sites-rich layered double hydroxide for nitrate-to-ammonia production with high selectivity and stability, Chem. Eng. J., 2022, 434, 134641 CrossRef CAS.
- X. Wan, W. Guo, X. Dong, H. Wu, X. Sun, M. Chu, S. Han, J. Zhai, W. Xia, S. Jia, M. He and B. Han, Boosting nitrate electroreduction to ammonia on NbOx via constructing oxygen vacancies, Green Chem., 2022, 24, 1090–1095 RSC.
- J. Wang, C. Cai, Y. Wang, X. Yang, D. Wu, Y. Zhu, M. Li, M. Gu and M. Shao, Electrocatalytic reduction of nitrate to ammonia on low-cost ultrathin CoOx nanosheets, ACS Catal., 2021, 11, 15135–15140 CrossRef CAS.
- S. Zhang, M. Li, J. Li, Q. Song and X. Liu, High-ammonia selective metal-organic framework–derived Co-doped Fe/Fe2O3 catalysts for electrochemical nitrate reduction, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2115504119 CrossRef CAS PubMed.
- X. Fan, J. Liang, L. Zhang, D. Zhao, L. Yue, Y. Luo, Q. Liu, L. Xie, N. Li, B. Tang, Q. Kong and X. Sun, Enhanced electrocatalytic nitrate reduction to ammonia using plasma-induced oxygen vacancies in CoTiO3-x nanofiber, Carbon Neutralization, 2022, 1, 6–13 CrossRef.
- D. Chen, S. Zhang, X. Bu, R. Zhang, Q. Quan, Z. Lai, W. Wang, Y. Meng, D. Yin, S. Yip, C. Liu, C. Zhi and J. C. Ho, Synergistic modulation of local environment for electrochemical nitrate reduction via asymmetric vacancies and adjacent ion clusters, Nano Energy, 2022, 98, 107338 CrossRef CAS.
- Q. L. Hong, J. Zhou, Q. G. Zhai, Y. C. Jiang, M. C. Hu, X. Xiao, S. N. Li and Y. Chen, Cobalt phosphide nanorings towards efficient electrocatalytic nitrate reduction to ammonia, Chem. Commun., 2021, 57, 11621–11624 RSC.
- Y. Jia, Y. G. Ji, Q. Xue, F. M. Li, G. T. Zhao, P. J. Jin, S. N. Li and Y. Chen, Efficient nitrate-to-ammonia electroreduction at cobalt phosphide nanoshuttles, ACS Appl. Mater. Interfaces, 2021, 13, 45521–45527 CrossRef CAS PubMed.
- Q. Yao, J. Chen, S. Xiao, Y. Zhang and X. Zhou, Selective electrocatalytic reduction of nitrate to ammonia with nickel phosphide, ACS Appl. Mater. Interfaces, 2021, 13, 30458–30467 CrossRef CAS PubMed.
- J. Liang, B. Deng, Q. Liu, G. Wen, Q. Liu, T. Li, Y. Luo, A. A. Alshehri, K. A. Alzahrani, D. Ma and X. Sun, High-efficiency electrochemical nitrite reduction to ammonium using a Cu3P nanowire array under ambient conditions, Green Chem., 2021, 23, 5487–5493 RSC.
- G. Wen, J. Liang, Q. Liu, T. Li, X. An, F. Zhang, A. A. Alshehri, K. A. Alzahrani, Y. Luo, Q. Kong and X. Sun, Ambient ammonia production via electrocatalytic nitrite reduction catalyzed by a CoP nanoarray, Nano Res., 2022, 15, 972–977 CrossRef CAS.
- S. Ye, Z. Chen, G. Zhang, W. Chen, C. Peng, X. Yang, L. Zheng, Y. Li, X. Ren, H. Cao, D. Xue, J. Qiu, Q. Zhang and J. Liu, Elucidating the activity, mechanism and application of selective electrosynthesis of ammonia from nitrate on cobalt phosphide, Energy Environ. Sci., 2022, 15, 760–770 RSC.
- Q. Yao, J. Chen, S. Xiao, Y. Zhang and X. Zhou, Selective electrocatalytic reduction of nitrate to ammonia with nickel phosphide, ACS Appl. Mater. Interfaces, 2021, 13, 30458–30467 CrossRef CAS PubMed.
- F. Ni, Y. Ma, J. Chen, W. Luo and J. Yang, Boron-iron nanochains for selective electrocatalytic reduction of nitrate, Chin. Chem. Lett., 2021, 32, 2073–2078 CrossRef CAS.
- X. Liu, X. Xu, F. Li, J. Xu, H. Ma, X. Sun, D. Wu, C. Zhang, X. Ren and Q. Wei, Heterostructured Bi2S3/MoS2 nanoarrays for efficient electrocatalytic nitrate reduction to ammonia under ambient conditions, ACS Appl. Mater. Interfaces, 2022, 14, 38835–38843 CrossRef CAS PubMed.
- Z. Li, G. Wen, J. Liang, T. Li, Y. Luo, Q. Kong, X. Shi, A. M. Asiri, Q. Liu and X. Sun, High-efficiency nitrate electroreduction to ammonia on electrodeposited cobalt–phosphorus alloy film, Chem. Commun., 2021, 57, 9720 RSC.
- X. Zhang, G. Ma, L. Shui, G. Zhou and X. Wang, Ni3N nanoparticles on porous nitrogen-doped carbon nanorods for nitrate electroreduction, Chem. Eng. J., 2022, 430, 132666 CrossRef CAS.
- Y. Shi, S. Xu and F. Li, Electrocatalytic nitrate reduction to ammonia via amorphous cobalt boride, Chem. Commun., 2022, 58, 8714–8717 RSC.
- Y. Wang, L. Zhang, Y. Niu, D. Fang, J. Wang, Q. Su and C. Wang, Boosting NH3 production from nitrate electroreduction via electronic structure engineering of Fe3C nanoflakes, Green Chem., 2021, 23, 7594–7608 RSC.
| This journal is © the Partner Organisations 2023 |