
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A sample-to-answer electrochemical biosensor system for biomarker detection†
Kruthika
Kikkeri
 a,
Dan
Wu
b and
Joel
Voldman
*a
a,
Dan
Wu
b and
Joel
Voldman
*a
aDepartment of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: voldman@mit.edu; Fax: +1 617 258 5846; Tel: +1 617 253 2094
bDepartment of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA
First published on 10th December 2021
Abstract
Biomarker detection is critical for the diagnosis and treatment of numerous diseases. Typically, target biomarkers in blood samples are measured through tests conducted at centralized laboratories. Testing at central laboratories increases wait times for results, in turn increasing healthcare costs and negatively impacting patient outcomes. Alternatively, point-of-care platforms enable the rapid measurement of biomarkers, expand testing location capabilities and mitigate manual processing steps through integration and automation. However, many of these systems focus on sample detection rather than the equally important sample preparation. Here we present a fully integrated and automated sample-to-answer electrochemical biosensing platform which incorporates each aspect of the biomarker testing workflow from blood collection to sample preparation to assay operation and readout. The system combines a commercial microneedle blood sampling device with membrane-based plasma filtration upstream of a bead-based electrochemical immunoassay. We characterize the high separation efficiency (>99%) and low non-specific binding of the whole blood-to-plasma filtration membrane under a range of operating conditions. We demonstrate a full sample-to-answer workflow through the analysis of interlukin-6-spiked blood samples.
Introduction
The detection and measurement of protein biomarkers from blood is a powerful tool for the diagnosis and management of various diseases.1–4 Typically, blood samples are acquired at medical facilities and then shipped to a centralized facility for testing.2,5 Identification and quantitative analysis of target biomarkers is then typically performed with automated equipment by trained personnel.2,6,7 However, this process often results in multi-day turnaround times (TATs)—which can be detrimental to the management of diseases with fast dynamics, requires phlebotomy, and restricts the location of testing.1,2,8–12Thus, portable biomarker-detection systems for testing at the point-of-care (PoC) settings (i.e. hospitals, bedside) have grown in popularity as promising alternatives to central laboratories for a variety of analytes due to their low-cost, low-complexity, and analytical capabilities.6,8,13–16 Advances in these PoC systems have shown expedited assay times (minutes), expanded testing locations (portability), and reduced sample volume needs (μLs). Moreover, these systems maintain clinically relevant limit of detections (LoDs), limit of quantifications (LoQs), dynamic ranges, testing linearity, and analytical sensitivities and specificities. Recent PoC systems have also reduced manual steps through integration of assay modules and automation.1,7,8,19 PoC platforms have also reduced TATs and enabled testing for a variety of biomarkers by introducing bead-based assays and on-chip fluid handling.1,6,8,17 Given the challenges of quantitative analysis in complex microliter samples, much of the work on PoC systems has focused on analysis rather than preparation.15,18,19 However, ultimate adoption of these technologies will in many cases require development of systems that incorporate sample acquisition and preparation.
To address the challenge of making measurements from whole blood, a number of on-chip sample preparation techniques have been developed. In particular, microfluidic channel-based separation techniques have shown high blood-to-plasma separation capabilities through the use of hydrodynamics, hemodynamics, or external forces,5–12 albeit at the cost of additional system complexity. Alternatively, filtration membranes are advantageous to incorporate with PoCs as they do not require external forces or peripheral equipment, have simple designs, and are easily integrated with biosensor modules.14–16 Yet, these integrated PoC platforms still have difficulty going from sample-to-answer on-site as they often require centralized sample acquisition (i.e. venipuncture) or manual loading (pipetting) of sample onto the device.20
Here, we present a sample-to-answer PoC system which incorporates sample acquisition, and integrates and automates on-chip blood-to-plasma separation with an electronic bead-based biomarker detection assay.15,17,21 In contrast to our previous work,15,17 this system accounts for the full blood testing pipeline by interfacing with a painless blood collection device, TAP (Seventh Sense Biosystems), to enable true sample-to-answer detection of an immune biomarker at clinically relevant levels in a 30-minute PoC assay. We evaluated the performance of the on-chip sample preparation through characterization of the separation efficiency and non-specific binding levels. We then examined the full workflow of the platform using whole blood samples spiked with interleukin-6 (IL-6), a marker relevant to numerous diseases. Finally, we explored optimization of operation parameters to further reduce sample-to-answer time while maintaining sensitivity. The resulting system demonstrates a feasible approach for incorporating upstream sample acquisition and preparation into PoC analytical platforms.
Results
Sample-to-answer workflow
To bridge the gap between biomarker detection and pre-processing, we designed a PoC system with a sample-to-answer workflow to incorporate each step in the blood testing pipeline. The sample-to-answer workflow, as shown in Fig. 1A, starts from with the collection of a blood sample (Fig. 1Ai). We used a TAP microneedle collection device (Seventh Sense Biosystems) to acquire samples due to its capacity to draw sufficient volumes of blood (up to 100 μL, though we use much smaller volumes here), ability to passively mix with anticoagulant, pain-free microneedle design, easy access for sample extraction and commercial availability.20 However, it should be noted that other sample collection devices exist with similar specifications that could alternatively be used for the blood acquisition step.22 For our preliminary investigations, we spiked blood sample into the TAP device, which was then placed on a tip connection to allow a peristaltic pump to extract the specimen from the TAP's internal storage reservoir and inject into the microfluidic device's separation channel.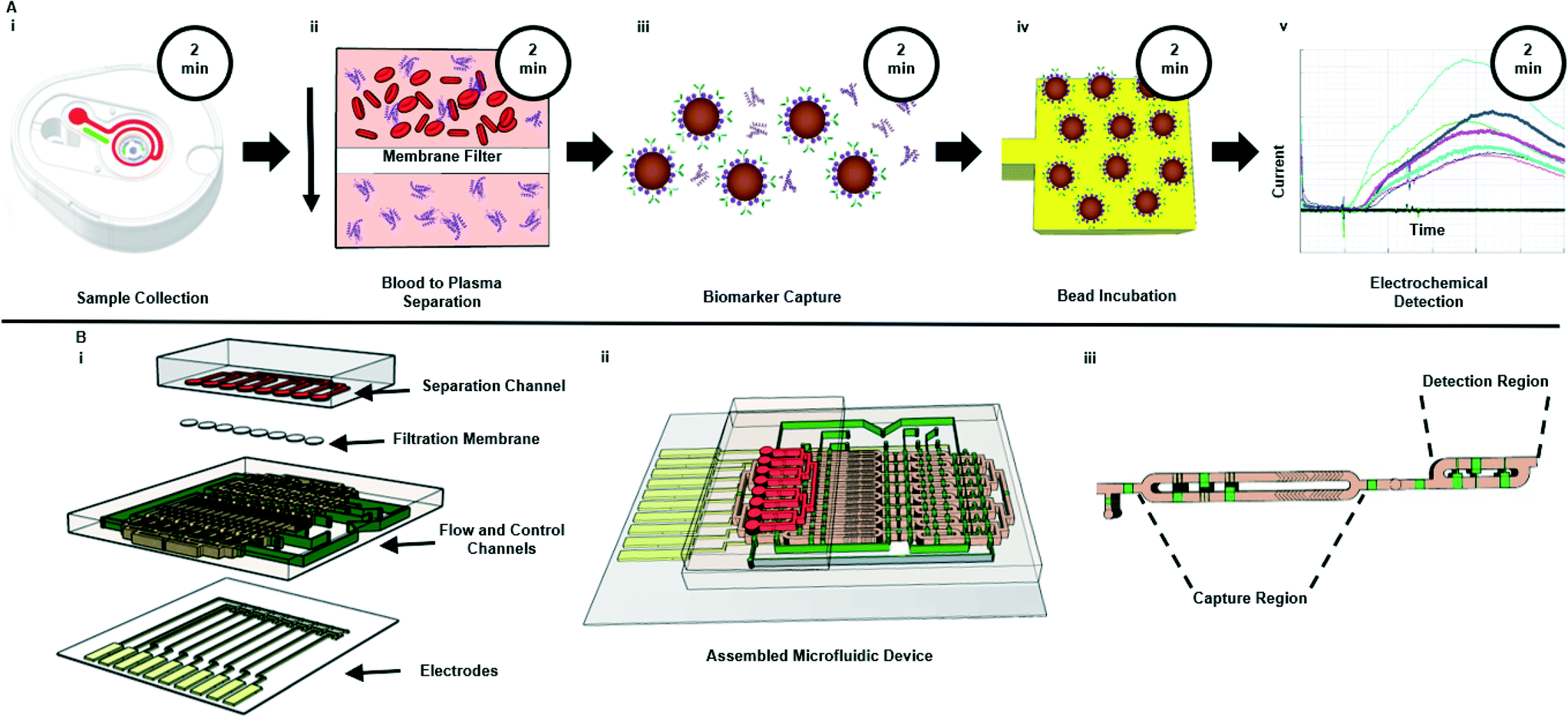 | ||
| Fig. 1 Schematic of (A) assay workflow and (B) microfluidic device. (A) Displays the POC platform workflow and associated durations from sample collection (step Ai) to electrochemical detection (step Av). The system uses a bead-based electrochemical sandwich assay20 to measure analyte concentration from samples following blood to plasma separation (step Aii). Total time from sample collection to electro-chemical detection is 30 minutes. (Bi) Depicts an exploded view of the multi-layer microfluidic system. The fully assembled system is shown in (Bii), while (Biii) shows a close-up of the flow channel highlighting the capture region (step Aiii) and detection region (step Aiv). | ||
Within the microfluidic device, the sample was filtered to remove erythrocytes (red blood cells), leukocytes (white blood cells) and platelets with the use of a Vivid membrane (Fig. 1Aii). This membrane was chosen for its low non-specific binding characteristics, hydrophilic nature, and asymmetric design, which enabled sufficient (μL) separation volumes.13,23,24 The filtered sample passively flowed into the capture region of the microfluidic device. Here, the sample was mixed with functionalized magnetic beads to capture target biomarkers (Fig. 1Aiii). The functionalized beads were then pulled to the detection region with the use of a magnet. Functionalized beads were then incubated on the electrodes for 10 minutes prior to a wash step used to flush out beads that did not bind to the functionalized electrodes (Fig. 1Aiv). Finally, a TMB substrate was flowed through the entire flow channel for amperometry measurement as shown in Fig. 1Av.
Characterization of plasma separation
One critical aspect of the PoC workflow is sample pre-processing, such as blood-to-plasma separation or dilution steps. Separation of whole blood to plasma is a vital step in a wide range of protein assays. Specifically, the need for this step stems from the concern over interference from red blood cells, white blood cells, and platelets.25 Additionally, the complex plasma matrix itself can alter inferred levels of biomarkers.26–28 Thus, it is common practice to separate plasma from whole blood, and to dilute the plasma to reduce matrix effects. To incorporate such sample preparation steps into a PoC system, we integrated a commercial Vivid membrane at the front end of the microfluidic device (Fig. 1Bi) to enable the removal of red blood cells, white blood cells, and platelets. We also investigated the characteristics of the membrane and dilution ratios (see Fig. S1† for linearity experiments) to ensure fit-for-purpose usage. It was found that the minimal dilution ratio needed to minimize matrix effects was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (sample:reagent diluent). While for this work we dilute blood samples separately and then directly load them into the TAP device, in practice the blood collection apparatuses can be pre-loaded with diluent.
:3 (sample:reagent diluent). While for this work we dilute blood samples separately and then directly load them into the TAP device, in practice the blood collection apparatuses can be pre-loaded with diluent.
To evaluate the separation performance (i.e. separation efficiency and separation volume capacity) of the membranes, diluted whole blood samples were infused across the membrane and into the separation channel at various volumes and flow rates. Specifically, sample volumes of 1–5 μL were flowed through 1.5 mm Vivid membrane discs to determine the maximum quantity that could be filtered without saturating the membrane, while ensuring that sufficient volume could be utilized for the bead-based assay. As seen in Fig. 2A, low flow rates or direct loading of blood onto the membrane showed high blood cell separation, indicating that minimal numbers of red blood cells passed into the flow channel. In contrast, higher flow rates increased the number of blood cells that passed through the filtration membrane (see Fig. S2† for details). Separation efficiencies were assessed by particle counting in the images, and calculated as below:
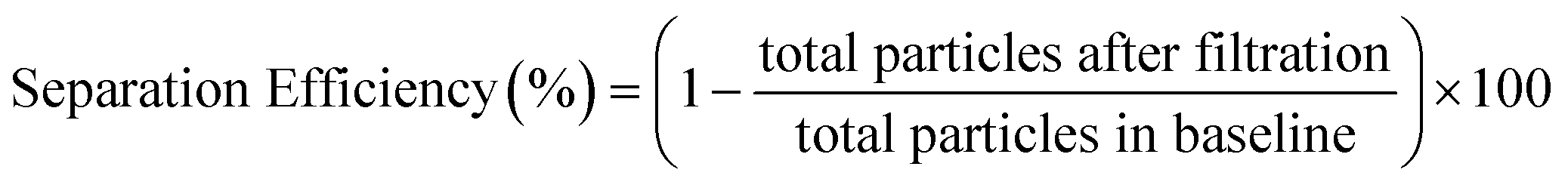 | (1) |
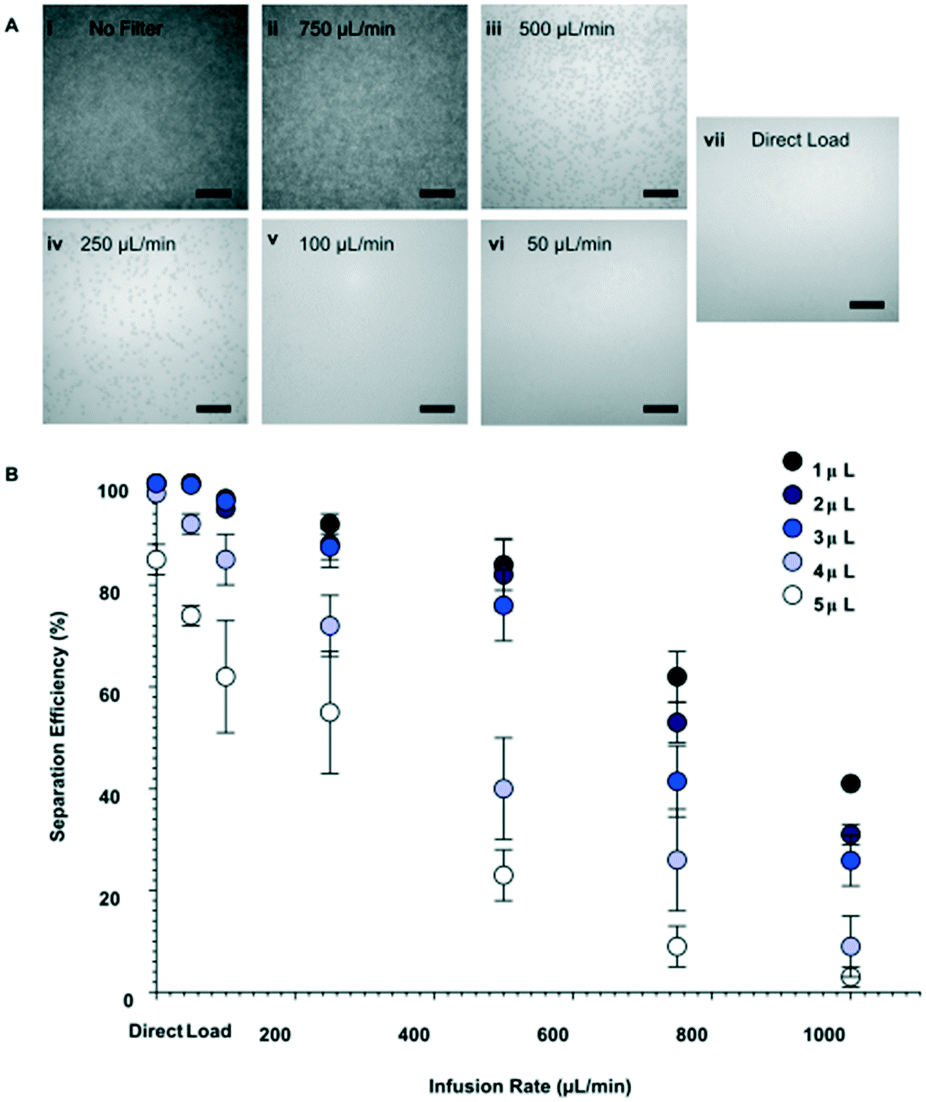 | ||
| Fig. 2 Characterization of blood-to-plasma separation membrane. (A) Bright-field images of diluted (1:3) whole blood to plasma separation, following passage of 4 μL sample across the membrane and into the flow channel at varying flow rates. (Ai) Depicts the baseline of diluted whole blood in the flow channel when there is no filtration membrane. (B) Effect of infusion flow rate and sample volume on plasma separation efficiency through 1.5 mm radius discs. Note that loading of whole blood with no filter resulted 0% separation efficiency. Separation efficiency was determined as a relative measure to (Ai). Scale bars in (A) represent 100 μm. | ||
In addition to its separation capabilities, we assessed the filtration membrane's binding affinity to protein, which could cause errors in the downstream analytical assay. We used IL-6, which is a marker for numerous diseases,29–32 as a model analyte. Enzyme-linked immunosorbent assays (ELISA) measurements were conducted to determine any reduction of IL-6 after filtration through the Vivid membrane. Fig. 3 illustrates the correlation of spiked IL-6 samples in human plasma in comparison to samples that were spiked in whole blood and then filtered. The results show that there are minimal discrepancies between the two sample suspensions with the largest deviation being 9.5% from linearity, which is within the allowable deviation from linearity (ADL) ±10% based on guidelines by the Clinical and Laboratory Standards Institute.33 It should be noted that the standard deviation for the commercial ELISA kit is specified as ≤20% by the manufacturer.34 Deviation of measured values from linearity can be attributed to non-specific binding and cross reactivity of matrix endogenous components. Overall, our results indicate that the membrane has low binding affinity or low non-specific binding for this particular biomarker.
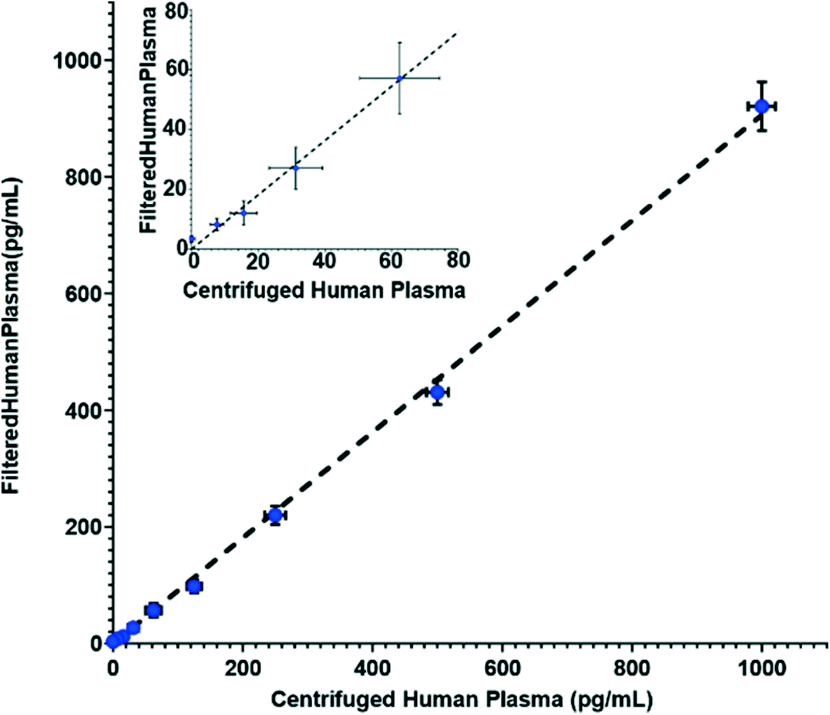 | ||
| Fig. 3 ELISA measurements of spiked IL-6 concentrations for samples suspended in human plasma vs. samples which were filtered using the Vivid membrane from IL-6 spiked whole blood to plasma at 50 μl min−1 infusion rate. Inset shows results at lower concentrations. Dotted line represents a linear curve fit of y = 0.91x with an R2 value of 0.95. | ||
Electrochemical sensing of IL-6 concentrations
Once the separation channel and membrane filter were incorporated with the biosensor, automated electrochemical measurements of IL-6 were acquired with the complete system. To provide an internal standard and mitigate effects due to device-to-device variation in amperometry measurements, we used a calibration curve for each run. Fig. 4 shows a sample amperometry measurement curve for a range of concentrations between 40 pg mL−1 and 3 ng mL−1 (Fig. S3† shows how current measurements are reported).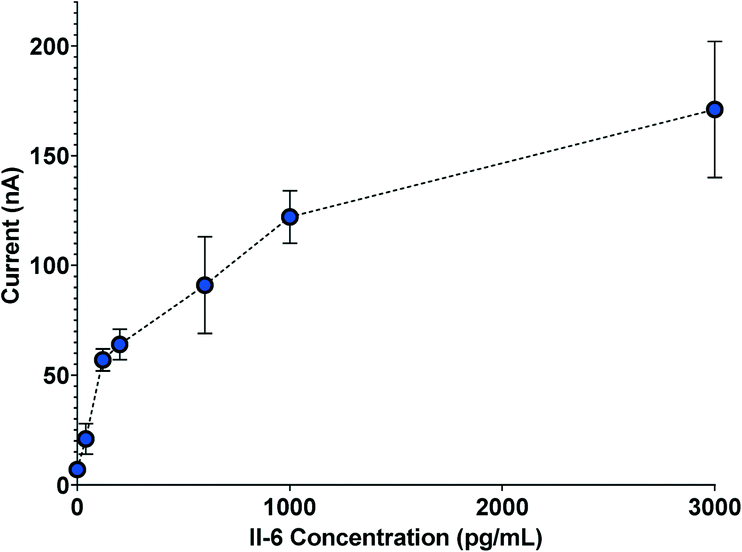 | ||
| Fig. 4 Sample amperometry measurement curve depicting electrochemical measurements from five separate biosensors. | ||
Following confirmation that target biomarkers could be measured using the sample-to-answer workflow shown in Fig. 1, we began to examine factors related to the PoC assay. One vital component we examined was to minimize the assay duration while maintaining performance. Thus, we investigated durations of each step within the workflow. We varied mixing and bead incubation times from 6 to 10 minutes and examined the inferred concentrations at three representative IL-6 concentrations (40, 120 and 200 pg mL−1).
From Fig. 5, it can be observed that measurements for 40, 120 and 200 pg mL−1 show some deviation from theoretical concentrations when incubation time is varied from 7 to 10 minutes. The largest variation in that time range was observed at 9 minutes where measurements for 40, 120 and 200 pg mL−1 showed maximum deviations of 12.5%, 11.7% and 2.5%, respectively. In fact, because calibration curves are used for each assay, minor deviations from standard operational parameters should be accounted for in the analysis. This indicates that variation of bead-sample incubation time has negligible impact on the performance of the electrochemical measurement across the 7 to 10 minute durations. However, a 6 minute bead-sample incubation time resulted in a deviation of 30.0%, 19.2%, and 12.5% from the theoretical values of 40, 120 and 200 pg mL−1, respectively. Thus, while bead-sample incubation time can be shortened somewhat, extreme reduction can reduce accuracy.
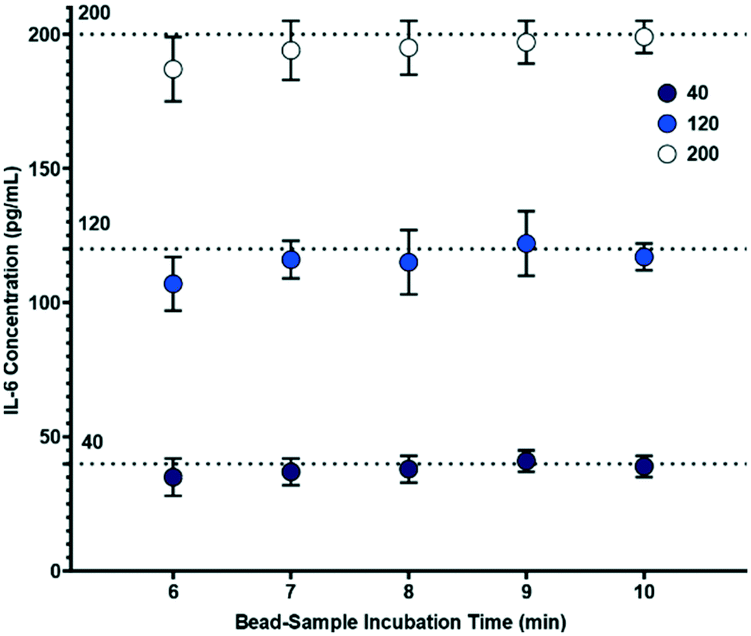 | ||
| Fig. 5 Effect of varying bead-sample incubation time on biosensor concentration measurements (n = 5), where the bead-electrode interaction time is 10 minutes. | ||
To evaluate the influence of blood cell separation efficiency on assay reproducibility, we ran the assay system with diluted whole blood samples infused such that 99.98%, 90, 80%, 70%, 60% and 50% of red blood cells were removed (counts verified through microscopy). Electrochemical readouts of the automated assay are portrayed in Fig. 6. By comparing the measured IL-6 concentrations to the theoretical spiked concentration, we found that the slope of the linear fits decrease as the separation efficiency decreases (Fig. 6A–F), and that at the lowest separation efficiency (50%, Fig. 6F) there is a substantial offset in the fit line. Furthermore, Fig. 6G shows that 99.98%, 90 and 80% separations had linearity variations that were within an ADL of 10%. However, separation efficiencies of 70% and lower showed higher linearity variations than the ADL. We observe that the discrepancies in the unity linear slope occur as the separation efficiency—and thus concentration of interfering red blood cells—changes, with decreasing linear slope as the concentration of red blood cells increases. This suggests that the cells, or, more likely protein components from the cells are causing the discrepancies from unity linear slope. This is consistent with prior literature that has identified components from blood cells as important inhibitors of molecular assays.25,35,36 It was also observed that red blood cells would become caught in the microfluidic structure and were potential clogging hazards. Thus, these results show that separation efficiencies of at least 80% are sufficient to ensure compliance with ADL standard.
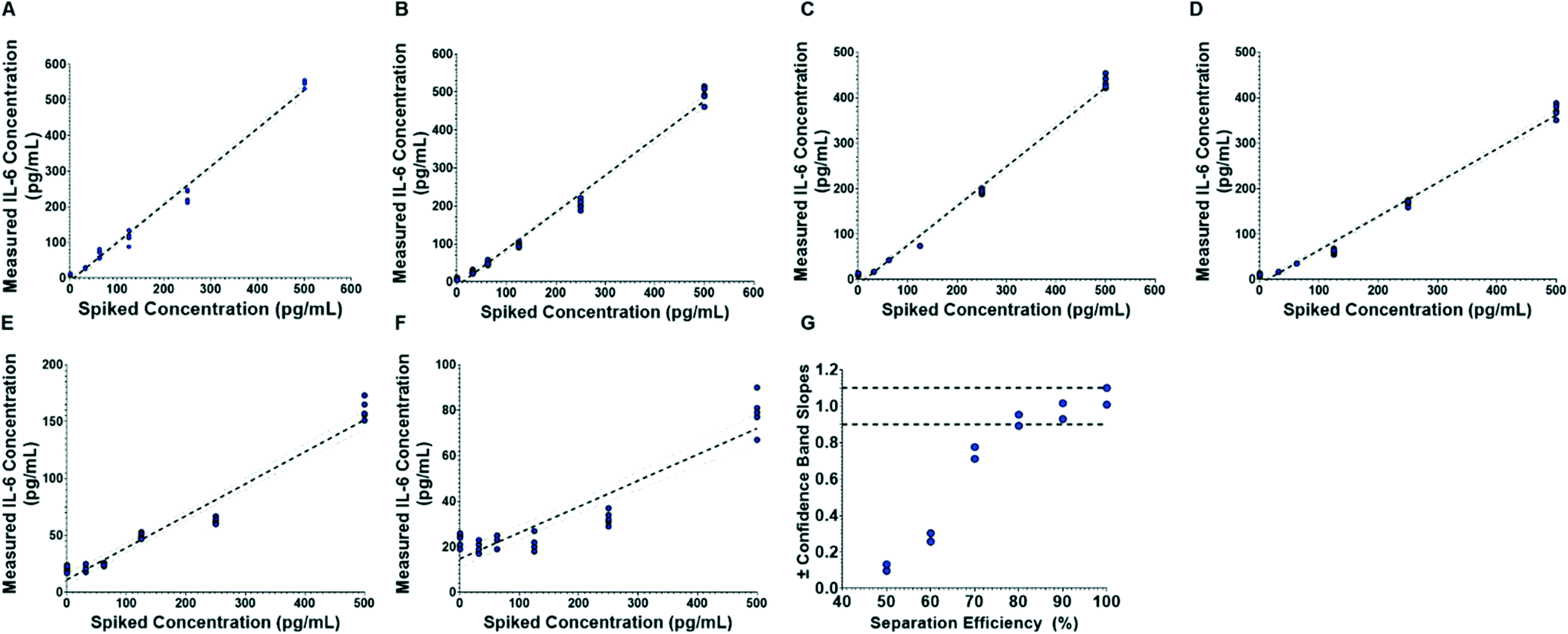 | ||
| Fig. 6 Comparison of biosensor IL-6 measurements (n = 5) to theoretical spiked concentrations plasma separation efficiencies of (A) 99.98%, (B) 90, (C) 80%, (D) 70%, (E) 60% and (F) 50%. The dotted lines show the linear regression and 95% confidence bands. (G) Shows the variation from linearity. | ||
Discussion
There has been a growing trend toward PoC testing for healthcare applications. Development of the upstream pipeline (sample acquisition and pre-processing steps) is important for the broad adoption of these systems.1,6 Here, we report an automated PoC platform that integrates sample acquisition and on-chip blood-to-plasma filtration with a biomarker detection assay. We demonstrate seamless incorporation of front-end, on-site sample acquisition and simple connection to our platform to enable complete sample-to-answer PoC testing. Furthermore, our results indicate that this workflow and PoC can be utilized for the sensitive and rapid measurement of proteins (in this case, IL-6), while reducing sources of error for downstream analysis.Through the design of this workflow and system, we found that a critical factor to consider is sample acquisition. While we utilized the TAP device for sample collection, there are a number of other commercial acquisition systems, such as those offered by Tasso Inc. (HemoLink) and Renephra Ltd. These devices can be advantageous to utilize due to their painless extraction of blood with microneedle arrays and easy access to samples. Furthermore, these devices extract sufficient quantities of blood for integration with assays which require small or large volumes (1–100 μL). This ensures samples are large enough to account for dead volume in devices, run multiplexed systems, and allow for simpler interface with PoC platforms. Alternatively, Dixon et al.13 and Swank et al.37 reported finger-prick sample acquisition that can be utilized for PoC testing. Usage of finger-prick collection could enable direct loading of sample without the need of extraction with a peristaltic pump. However, finger-prick acquisition can lead to other sources of error due to insufficient volume quantities or droplet-to-droplet variation. Thus, it is important to consider on-site sample acquisition methods and evaluate how to interface these techniques to ensure fully decentralized PoCs.
Another key step to evaluate was the integration of pre-processing techniques, in this case blood-to-plasma separation with biomarker-detection assays. We implemented a Vivid filter within a microfluidic device to integrate all processing aspects of the PoC. Our aim here was to quickly filter (<5 min) and integrate separation within our detection assay. Thus, the filtration membrane area was fixed based on the channel dimension and target sample volumes (∼1 μL). However, channel dimensions, and membrane area can be varied for processing of larger volumes. Additionally, our membrane was coated with BSA to fit the electronic electrochemical assay, but could also be coated with other materials for other assay designs such 3D μPADs for colorimetric assays.38 Furthermore, other pre-processing techniques could be utilized for sample preparation. For example, Poudineh et al. reported a real-time ELISA which employed deterministic lateral displacement (DLD) sorting to isolate target glucose and insulin analytes.39 This DLD technique and other microfluidic structure-based approaches have shown promise in filtering blood cells and detecting target biomarkers. However, they can be challenging to implement as they can need to be optimized for purpose and add complexity to device.
More fundamentally, when integrating these features, it is critical to determine what, if any, pre-processing steps are necessary. In fact, there have been several developed immunoassays which capture target biomarkers and test directly from whole blood.40,41 However, these methods must always be validated in the context of whole blood, since the blood cells can interfere with the analytical performance. To evaluate the need for sample preparation steps in our PoC, we investigated the minimum amount of blood-to-plasma separation efficiency needed to maintain analytical performance (ADL). Although we determined a minimum separation efficiency for this assay, it should be noted that this and the overall assay could be further optimized. In fact, operational parameters for the other aspects of our workflow such as wash time, flow rates, and detection times could be modulated to lower TAT and/or improve signal output. Moreover, because the assay can be modified to target other protein biomarkers with minimal alterations, we envision that this PoC system could be applied broadly to a variety of clinical applications for disease diagnostics and monitoring.
Conclusions
In this paper we reported a sample-to-answer PoC platform which incorporated each aspect of the blood testing pipeline. We validated the high separation efficiency and low non-specific binding of IL-6 for the separation membrane. In addition, we also showed that IL-6 levels could be measured from sample collection to results in 30 minutes, indicating that rapid biomarker detection can be accomplished with the integration of sample preparation steps. Finally, we explored the avenues for reducing assay time through optimization of the sample-bead incubation period. Thus, the described PoC platform demonstrates the capability to detect biomarkers from sample-to-answer through an integrated and automated system.Materials and methods
Blood and calibration samples
Human whole blood samples were acquired from Innovative Research Inc. in 10 mL K2EDTA vacutainer tubes. 100 μL of diluted (1:3) whole blood in 0.2% bovine serum albumin (BSA) was manually injected into TAP blood collection devices (Seventh Sense Biosystems; Medford, MA) for plasma separation testing of filtration membranes. To assess the performance of the integrated microfluidic biosensor, whole blood samples were spiked with human IL-6 standard (WHO International Standard) at varying concentrations and pipetted into individual TAP devices. Calibration samples were 1:3 dilutions of human plasma (Innovative Research Inc.; Novi, MI) in 0.2% BSA solution which were then spiked with various human IL-6 standard concentrations.
Integration with Seventh Sense Biosystems TAP blood collection device
The TAP blood collection device uses microneedles to pierce skin and vacuum draw blood. This painless and automated device is attached on a patient's arm and activated by the press of a button. Capillary blood is then drawn from a patient into the TAP device where the sample is immediately and passively mixed with an anticoagulant through a micromixer, and is then flown into a storage reservoir.20 To access the blood sample, the user needs to pierce the foil covered reservoir access port and collect the specimen. To simulate this process and avoid repeated exposure of samples to anticoagulants, spiked whole blood was directly injected into the TAP reservoir through the access port. The access port was then recovered with foil and directly placed on connector which linked to a peristaltic pump. This peristaltic pump was directly connected to the integrated microfluidic biosensor.Device fabrication
Negative molds for the separation channel, flow channel, and control channel (microvalves) were formed through tradition photolithography techniques and stereo-lithography (Protolabs; Maple Plain, MN). 10 nm of Ti and 200 nm of gold was deposited onto a Pyrex wafer to form the three electrodes (working [600 × 600 μm], counter [600 × 600 μm], reference [400 × 600 μm]) used for multiplexed amperometry. The flow channel and electrodes were designed to allow for eight parallel measurements.Microfluidic layers were fabricated using a 10:1 RTV silicone ratio. The middle layer was cured at 80 °C for 25 minutes before bonding, while the bottom layer was cured for 3.5 minutes. Once cool, the middle and bottom layer were aligned and thermally bonded on an 80 °C hot plate overnight. Prior to bonding, electrodes were cleaned with acetone, methanol, IPA and submerged under Nano-Strip (KMG Chemicals Inc.; Fort Worth, TX) for one hour. Following cleaning, electrodes were washed with deionized (DI) water and dried. Ag/AgCl ink (ALS Co., Ltd; Tokyo, Japan) was applied to reference electrodes and dried at 120 °C for two minutes. Electrodes and the two-layer device was then aligned and plasma bonded. It was then heated at 80 °C for 10 minutes to remove any trapped air bubbles between the fluidic layers and Pyrex substrate. Prior to assembly, commercial Vivid GR grade membrane (Pall Corporation; Ann Arbor, MI) sheets were dipped in 1% BSA and dried to improve sample flow through the surface. The Vivid GR grade membrane has a capacity to recover 40–50 μL cm−2. To ensure sufficient volume for microchannels with ∼1 μL capacity, the membrane was then cut into 1.25 mm radius discs. These discs were then placed directly over desired inlets on the two-layer device. The top RTV layer was then aligned and plasma bonded over the two-layer apparatus. It should also be noted that the top microchannel was designed to accommodate for 330 ± 20 μm thickness and void volume caused by blood cell aggregation on the filter. Fully assembled devices were primed under vacuum for at least 30 minutes prior to experimentation.
Electrode surface modification
Control valves were filled with DI water, while the flow channel was initially primed with phosphate buffered saline (PBS) to prevent introduction of bubbles. 1 mL of 2 mM CT(PEG)12 (Thermo Fisher Scientific; Waltham, MA) was injected overnight into the flow channel at 0.1 mL h−1. Following a PBS wash step, working electrodes were then activated with a 25 mM NHS/25 mM sulfo-EDC in MES buffer solution for 15 minutes. After another PBS wash step, the working electrodes were incubated with a 0.2 mg mL−1 IL-6 capture antibody solution for three hours. The entire flow channel was then blocked with 3% BSA for one hour.Bead conjugation
10 μL of T1 steptavidin Dynabeads (Thermo Fisher Scientific; Waltham, MA) were suspended into a 200 μL solution of 0.2% BSA. Using a magnetic holder, the Dynabeads were washed three times and re-suspended into 200 μL of 0.2% BSA. The solution was mixed with a 200 μL solution of 6 μg of biotinylated IL-6 antibody (detection) and 4 μg of biotinylated horseradish peroxidase (HRP), to form a final volume of 400 μL. Beads were incubated for 30 minutes on a rocker, before being washed three times with 0.2% BSA, and re-suspended into a 200 μL solution of 1% BSA.Assay workflow
The ADUCM350 evaluation kit (Eval-ADuCM350EB1Z-Analog Devices, Inc.; Norwood, MA) and MATLAB was utilized for operation of the automated assay. Further details on multiplexed amperometry and integration of the automated system are outlined in.15,17,21 For assay operation, conjugated beads were injected into the analyte capture region. IL-6 calibration standards were then injected into individual channels in the flow layer. Spiked whole blood samples were extracted from the TAP device through a peristaltic pump and flowed into the separation channel. Separation of plasma was allowed to proceed for three minutes to ensure adequate volume of blood had flowed through the Vivid filtration membrane into the flow channel. Samples and beads were mixed with microvalves in the analyte capture region and incubated for ten minutes. It should be noted that the analyte capture region incorporated a herringbone design within the rotatory to ensure even suspension during mixing. Incubated beads were then pulled to the detection region by a magnet. Beads were then re-suspended and incubated over the working electrode for ten minutes. Following incubation, channels were flushed with PBS with 0.05% Tween-20 (PBST) at 20 μL min−1 for two minutes. Finally, a substrate bolus of 200 μM 3,3′,5,5′-Tetramethylbenzidine (TMB) and 2 mM hydrogen peroxide in PBS was injected at a rate of 20 μL min−1 for three minutes. This induced electrochemical reactions which generated current. The generated current was monitored and analyzed using MATLAB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Novartis Institutes of Biomedical Research and the MIT-HKUST Alliance. K. Kikkeri was supported by the NSF Graduate Research Fellowship (Grant No. 478969) and Takeda Fellowship.References
- B. Heidt, W. F. Siqueira, K. Eersels, H. Diliën, B. van Grinsven, R. T. Fujiwara and T. J. Cleij, Biosensors, 2020, 10, 133 CrossRef PubMed.
- A. I. Barbosa and N. M. Reis, Analyst, 2017, 142, 858–882 RSC.
- J. A. Stenken and A. J. Poschenrieder, Anal. Chim. Acta, 2015, 853, 95–115 CrossRef CAS.
- S. Nahavandi, S. Baratchi, R. Soffe, S.-Y. Tang, S. Nahavandi, A. Mitchell and K. Khoshmanesh, Lab Chip, 2014, 14, 1496–1514 RSC.
- M. Bull, D. Lee, J. Stucky, Y.-L. Chiu, A. Rubin, H. Horton and M. J. McElrath, J. Immunol. Methods, 2007, 322, 57–69 CrossRef CAS PubMed.
- J. Park, D. H. Han and J.-K. Park, Lab Chip, 2020, 20, 1191–1203 RSC.
- D. Xu, X. Huang, J. Guo and X. Ma, Biosens. Bioelectron., 2018, 110, 78–88 CrossRef CAS.
- D. Liu, J. Wang, L. Wu, Y. Huang, Y. Zhang, M. Zhu, Y. Wang, Z. Zhu and C. Yang, TrAC, Trends Anal. Chem., 2020, 122, 115701 CrossRef CAS.
- D. Gasperino, T. Baughman, H. V. Hsieh, D. Bell and B. H. Weigl, Annu. Rev. Anal. Chem., 2018, 11, 219–244 CrossRef PubMed.
- M. Ahmadi, H. Elmongy, T. Madrakian and M. Abdel-Rehim, Anal. Chim. Acta, 2017, 958, 1–21 CrossRef CAS PubMed.
- M. Sonker, V. Sahore and A. T. Woolley, Anal. Chim. Acta, 2017, 986, 1–11 CrossRef CAS.
- J. C. Cunningham, P. R. DeGregory and R. M. Crooks, Annu. Rev. Anal. Chem., 2016, 9, 183–202 CrossRef.
- C. Dixon, J. Lamanna and A. R. Wheeler, Lab Chip, 2020, 20, 1845–1855 RSC.
- J. Berger, E. Valera, A. Jankelow, C. Garcia, M. Akhand, J. Heredia, T. Ghonge, C. Liu, V. Font-Bartumeus, G. Oshana, J. Tiao and R. Bashir, Biomed. Microdevices, 2020, 22, 36 CrossRef CAS.
- D. Wu and J. Voldman, An integrated and automated electronic system for point-of-care protein testing, 2019 41st Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), IEEE, 2019, pp. 1571–1574 Search PubMed.
- E. A. Phillips, T. J. Moehling, K. F. K. Ejendal, O. S. Hoilett, K. M. Byers, L. A. Basing, L. A. Jankowski, J. B. Bennett, L.-K. Lin, L. A. Stanciu and J. C. Linnes, Lab Chip, 2019, 19, 3375–3386 RSC.
- D. Wu, D. Rios-Aguirre, M. Chounlakone, S. Camacho-Leon and J. Voldman, Biosens. Bioelectron., 2018, 117, 522–529 CrossRef CAS.
- J. Berger, E. Valera, A. Jankelow, C. Garcia, M. Akhand, J. Heredia, T. Ghonge, C. Liu, V. Font-Bartumeus and G. Oshana, Biomed. Microdevices, 2020, 22, 1–11 CrossRef.
- L. Ma, Y. Abugalyon and X. Li, Anal. Bioanal. Chem., 2021, 1–9 Search PubMed.
- T. M. Blicharz, P. Gong, B. M. Bunner, L. L. Chu, K. M. Leonard, J. A. Wakefield, R. E. Williams, M. Dadgar, C. A. Tagliabue and R. El Khaja, Nat. Biomed. Eng., 2018, 2, 151–157 CrossRef CAS.
- D. Wu and J. Voldman, Biosens. Bioelectron., 2020, 154, 112070 CrossRef CAS PubMed.
- G.-S. Liu, Y. Kong, Y. Wang, Y. Luo, X. Fan, X. Xie, B.-R. Yang and M. X. Wu, Biomaterials, 2020, 232, 119740 CrossRef CAS.
- C. Liu, S.-C. Liao, J. Song, M. G. Mauk, X. Li, G. Wu, D. Ge, R. M. Greenberg, S. Yang and H. H. Bau, Lab Chip, 2016, 16, 553–560 RSC.
- W. Liu, J. Das, A. H. Mepham, C. R. Nemr, E. H. Sargent and S. O. Kelley, Lab Chip, 2018, 18, 1928–1935 RSC.
- S. Vemulapati and D. Erickson, Lab Chip, 2018, 18, 3285–3292 RSC.
- J. W. Lee, V. Devanarayan, Y. C. Barrett, R. Weiner, J. Allinson, S. Fountain, S. Keller, I. Weinryb, M. Green and L. Duan, Pharm. Res., 2006, 23, 312–328 CrossRef CAS.
- M. Bull, D. Lee, J. Stucky, Y.-L. Chiu, A. Rubin, H. Horton and M. J. McElrath, J. Immunol. Methods, 2007, 322, 57–69 CrossRef CAS PubMed.
- E. Saar, D. Gerostamoulos, O. H. Drummer and J. Beyer, Anal. Bioanal. Chem., 2009, 393, 727–734 CrossRef CAS PubMed.
- D. M. Del Valle, S. Kim-Schulze, H.-H. Huang, N. D. Beckmann, S. Nirenberg, B. Wang, Y. Lavin, T. H. Swartz, D. Madduri, A. Stock, T. U. Marron, H. Xie, M. Patel, K. Tuballes, O. Van Oekelen, A. Rahman, P. Kovatch, J. A. Aberg, E. Schadt, S. Jagannath, M. Mazumdar, A. W. Charney, A. Firpo-Betancourt, D. R. Mendu, J. Jhang, D. Reich, K. Sigel, C. Cordon-Cardo, M. Feldmann, S. Parekh, M. Merad and S. Gnjatic, Nat. Med., 2020, 26, 1636–1643 CrossRef CAS PubMed.
- C. Lucas, P. Wong, J. Klein, T. B. Castro, J. Silva, M. Sundaram, M. K. Ellingson, T. Mao, J. E. Oh and B. Israelow, Nature, 2020, 584, 463–469 CrossRef CAS.
- A. Ng, W. W. Tam, M. W. Zhang, C. S. Ho, S. F. Husain, R. S. McIntyre and R. C. Ho, Sci. Rep., 2018, 8, 1–12 Search PubMed.
- C. Russell, A. C. Ward, V. Vezza, P. Hoskisson, D. Alcorn, D. P. Steenson and D. K. Corrigan, Biosens. Bioelectron., 2019, 126, 806–814 CrossRef CAS.
- P. R. J. McEnroe, Evaluation of Lineartiy of Quantitative Measurement Procedures, 2nd edn, 2020 Search PubMed.
- R. Systems, How to Analyze ELISA Data, https://www.rndsystems.com/resources/how-to-analyze-elisa-data, 2021 Search PubMed.
- A. Akane, K. Matsubara, H. Nakamura, S. Takahashi and K. Kimura, J. Forensic Sci., 1994, 39, 362–372 CAS.
- W. A. Al-Soud and P. Rådström, J. Clin. Microbiol., 2001, 39, 485–493 CrossRef CAS.
- Z. Swank, G. Michielin, H. M. Yip, P. Cohen, D. O. Andrey, N. Vuilleumier, L. Kaiser, I. Eckerle, B. Meyer and S. J. Maerkl, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(18), e2025289118 CrossRef CAS.
- C. Park, H.-R. Kim, S.-K. Kim, I.-K. Jeong, J.-C. Pyun and S. Park, ACS Appl. Mater. Interfaces, 2019, 11, 36428–36434 CrossRef CAS PubMed.
- M. Poudineh, C. L. Maikawa, E. Y. Ma, J. Pan, D. Mamerow, Y. Hang, S. W. Baker, A. Beirami, A. Yoshikawa, M. Eisenstein, S. Kim, J. Vučković, E. A. Appel and H. T. Soh, Nat. Biomed. Eng., 2021, 5, 53–63 CrossRef CAS PubMed.
- P. Von Lode, Clin. Biochem., 2005, 38, 591–606 CrossRef CAS PubMed.
- P. B. Luppa, C. Müller, A. Schlichtiger and H. Schlebusch, TrAC, Trends Anal. Chem., 2011, 30, 887–898 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00910a |
| This journal is © The Royal Society of Chemistry 2022 |