
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew conjugated polymer nanoparticles with high photoluminescence quantum yields for far-red and near infrared fluorescence bioimaging
Panagiota
Koralli
ab,
Alkmini
D. Nega
a,
Lida Evmorfia
Vagiaki
b,
Aristea
Pavlou
bc,
Michael G.
Siskos
c,
Antonia
Dimitrakopoulou-Strauss
a,
Vasilis G.
Gregoriou
 d and
Christos L.
Chochos
*b
d and
Christos L.
Chochos
*b
aClinical Cooperation Unit Nuclear Medicine, German Cancer Research Center, 69120 Heidelberg, Germany
bInstitute of Chemical Biology, National Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, Athens, 11635, Greece. E-mail: chochos@eie.gr
cDepartment of Chemistry, Section of Organic Chemistry and Biochemistry, University of Ioannina, Ioannina, 45110, Greece
dNational Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, Athens, 11635, Greece
First published on 4th June 2020
Abstract
The development of new aqueous conjugated polymer nanoparticles with high photoluminescence quantum yields (PLQYs) at the far red and near infrared (NIR) spectral regions (>650 nm) as alternative polymer probes for fluorescence imaging is reported. This is achieved through an appropriate chemical design by the introduction of up to four fluorine atoms in the different positions of a donor–acceptor (D–A) polymer backbone consisting of thiophene as the electron donating unit and quinoxaline as the electron withdrawing building block. The resulting series of low synthetic complexity conjugated polymers enable us to perform a detailed structure–properties relationship study between the synthesized conjugated polymers and their corresponding aqueous nanoparticles as prepared by the nanoprecipitation and encapsulation methods. We manage to achieve PLQY for aqueous NIR conjugated polymer probes of 0.1 and a PLQY of 0.18 for the far-red emitting conjugated polymer probes. Furthermore, our work paves the way for the rational design of new conjugated polymers with enhanced PLQY, especially in the NIR spectral region, through better control of the intramolecular and intermolecular polymer chain interactions.
Introduction
Fluorescence imaging techniques with high sensitivity, low cost and temporal resolution have become indispensable for biomedical imaging applications.1,2 Fluorescent probes emitting in the far-red and near-infrared (NIR) spectral regions (650–1000 nm) are currently one of the main focuses for successful fluorescence-based bioimaging applications. Very recently, NIR fluorescence has been utilized for clinical image-guided cancer surgery, which holds great promise for improving cancer surgery outcomes.3 However, a limited choice of fluorophores has been a major obstacle to applying fluorescence imaging to a wide range of biological specimens.4,5 Within the NIR window, most biological species such as oxyhemoglobin, deoxyhemoglobin, water, melanon, and lipid absorb minimal light and exhibit low scattering.6,7 Moreover, tissues have almost no autofluorescence inside the NIR spectrum, and thus a high signal-to-noise contrast can be achieved using NIR-responsive fluorescent probes. Finally, whereas visible light can travel only several micrometers in tissues, NIR light (700–900 nm) can penetrate several millimeters (up to centimeters) through blood or tissues and cause minimal tissue damage to precisely guide surgeons in real time.For these reasons, different types of fluorophores have been developed toward this goal, including small molecule-based fluorophores,8–10 single-walled carbon nanotubes,11,12 semiconductor quantum dots (QD),13,14 and rare earth-doped nanoparticles.15–17 Each material, however, suffers its own limitations, such as the aggregation-caused quenching effect, low quantum yield (QY), poor photobleaching resistance, high cytotoxicity, or long-term retention in the body.18–20 For example, most FDA-approved clinical agents3 (e.g., indocyanine green, 5-aminolevulinic acid, and methylene blue) suffer from irreversible photobleaching and issues of low brightness and small Stokes shifts. As a result, the development of well-designed NIR fluorophores with high brightness, good photostability, and large Stokes shifts is a profound need in modern bioimaging and clinical diagnosis. Conjugated polymer nanoparticles (CPNs) have recently emerged as a new class of fluorescent nanoparticles because their fluorescence properties can be fine-tuned by an appropriate molecular design of conjugated polymers (CPs).21–24 Given their superior fluorescence brightness and photostability as well as low cytotoxicity, the applicability of CPNs as fluorescent tags and sensors has been expanding significantly during the last few years.25–29 Recent studies have demonstrated that, in some cases, the performance of CPNs as fluorescent tags surpasses that of conventional organic dyes and QDs.30,31
A major obstacle in the development of CPNs is that fluorescence quenching sites (i.e., energy trap sites) are generated through aggregate formation by strong interchain or intrachain interactions (e.g., π–π stacking) inside the CP nanoparticles, which results in severe fluorescence quenching (reduction of fluorescence quantum yields).32–35 Fluorescence quenching is more severe in the far-red and NIR emitting conjugated polymers since their extended π-conjugation system often needs fused and polycyclic molecules, leading to increased inter- and intra-chain interactions. To prevent the formation of fluorescence quenching sites and to obtain particles with high fluorescence quantum yields, different approaches have been utilized, such as aggregation-induced emission (AIE),36 the incorporation of a small amount of narrow bandgap monomers into a polyfluorene copolymer37 and the use of conjugated polymers that have bent and twisted conformations.38 While relatively large fluorescence quantum yields have been reported using the AIE approach, incorporating AIE moieties into conjugated polymers is difficult and restricts the flexibility of the molecular design. Similarly, the introduction of the exact composition of a narrow band gap monomer in a polymer backbone from batch to batch is not controllable.
In this contribution, we developed a new family of aqueous CPNs as potential far-red and NIR fluorescence polymer probes with high photoluminescence quantum yields by utilizing simultaneously unfused, less rigid polycyclic monomer building blocks and reducing the planarity of the polymer chains by anchoring fluorine atom(s) as substituents. The synthesized donor–acceptor (D–A) CPs comprise thiophene (T) as the electron donating unit and quinoxaline (Q) as the electron deficient segment in the repeat unit and vary with regards to the number of substituted fluorine atoms (Fig. 1). The formation of CPNs was carried out via both nanoprecipitation and encapsulation methods.39 As an encapsulation polymer, poly(ethylene glycol)methyl ether-poly(lactide-co-glycolide) (mPLGA-b-PEG) diblock copolymer was used. mPLGA-b-PEG is a biodegradable amphiphilic block copolymer approved by both the Food & Drug Administration (FDA) and European Medicine Agency (EMA).40 The size, the morphology and the optical properties of the nanoprecipitated and encapsulated CPNs are thoroughly characterized. This series of CPNs allowed us to perform an extensive structure–property relationship study, by investigating the effect caused by a relatively small change on the chemical structure within the optoelectronic properties of the CPs, as well as on the morphological and optical properties of the produced CPNs. In particular, to the best of our knowledge, a comparison between the two formulation methods for the preparation of CPNs (nanoprecipitation versus encapsulation) has not been performed up to now. The obtained results provide a clear understanding of how the number and the position of the fluorine atoms on the polymer backbone and the formulation method modify the maximum absorption wavelengths, the fluorescence emission characteristics and the photoluminescence quantum yields.
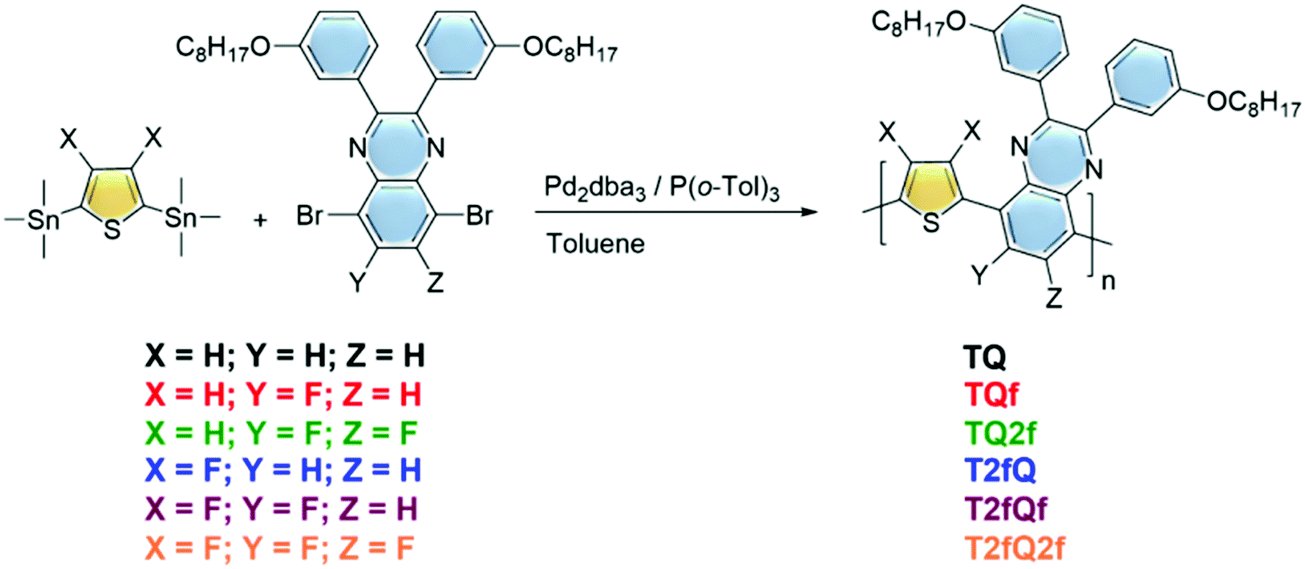 | ||
| Fig. 1 Synthesis of the TQ-based conjugated polymers. | ||
Experimental
Materials
All reactions are air and light sensitive and, therefore, were performed under argon and in the dark. All glassware was washed using detergent (Teepol), rinsed with excess water, acetone and methylene dichloride and dried in an oven at 120 °C. All solvents and reagents were purchased from Aldrich. Toluene was distilled using calcium hydride (CaH2) and benzophenone prior to polymerization. The syntheses of 5,8-dibromo-2,3-bis(3-(octyloxy)phenyl)quinoxaline,5,8-dibromo-6-fluoro-2,3-bis(3-(octyloxy)phenyl)quinoxaline, 5,8-dibromo-6,7-difluoro-2,3-bis(3-(octyloxy)phenyl)quinoxaline and bis(trimethylstannyl)thiophene were performed according to already published procedures.41–43 (3,4-Difluorothiophene-2,5-diyl)bis(trimethylstannane)44 was purchased from Derthon Optoelectronic Materials Science Technology Co., Ltd and poly(ethylene glycol)methyl ether-poly(lactide-co-glycolide) (mPLGA-b-PEG) [average molecular weight per number of PLGA is![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) nPLGA = 7000 g mol−1, and that of PEG is nPEG = 5000 g mol−1] was purchased from Sigma Aldrich.
nPLGA = 7000 g mol−1, and that of PEG is nPEG = 5000 g mol−1] was purchased from Sigma Aldrich.
Synthesis of the TQ based polymers
The polymerization reactions are analytically described below:The (3,4-difluorothiophene-2,5-diyl)bis(trimethylstannane) (0.5 mmol) was combined with either the 5,8-dibromo-2,3-bis(3-(octyloxy)phenyl)quinoxaline (0.5 mmol) for the T2fQ or the 5,8-dibromo-6-fluoro-2,3-bis(3-(octyloxy)phenyl)quinoxaline (0.5 mmol) for the T2fQf or the 5,8-dibromo-6,7-difluoro-2,3-bis(3-(octyloxy)phenyl)quinoxaline (0.5 mmol) for the T2fQ2f and the bis(trimethylstannyl)thiophene (0.5 mmol) was combined with either the 5,8-dibromo-2,3-bis(3-(octyloxy)phenyl)quinoxaline (0.5 mmol) for the TQ or the 5,8-dibromo-6-fluoro-2,3-bis(3-(octyloxy)phenyl)quinoxaline (0.5 mmol) for the TQf or the 5,8-dibromo-6,7-difluoro-2,3-bis(3-(octyloxy)phenyl)quinoxaline (0.5 mmol) for the TQ2f. Then, dry toluene (0.025 M) was added to the reaction mixtures. Finally, tris(dibenzylideneacetone)dipalladium(0) [Pd2(dba3)] (0.02 equiv.) and tri(o-tolyl)phosphine [P(o-tol)3] (0.08 equiv.) were added and the reaction mixture was stirred at 120 °C under an argon atmosphere for 24 h. The polymers were purified by precipitation in methanol, filtered and washed using a Soxhlet apparatus with methanol, acetone, hexane and chloroform. The chloroform fractions were evaporated under reduced pressure and the polymers were precipitated in methanol, filtered through a 0.45 mm PTFE filter and finally dried under high vacuum, rendering a dark blue solid for the TQ, TQf and TQ2f and red, orange and yellow solids for the T2fQ, T2fQf and T2fQ2f, respectively. The yields of the resulting polymers are the following: TQ = 88%,TQf = 83%, TQ2f = 76%, T2fQ2 = 91%, T2fQf = 87% and T2fQ2f = 75%.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1, Đ = 3.2.
400 g mol−1, Đ = 2.3.
600 g mol−1, Đ = 2.3.
200 g mol−1, Đ = 1.5.
900 g mol−1, Đ = 3.2.
400 g mol−1, Đ = 2.3.
600 g mol−1, Đ = 2.3.
200 g mol−1, Đ = 1.5.
Instrumentation
n) and polydispersity indices (Đ) were determined with GPC at 30 °C on a Shimadzu liquid chromatography (LC-20AD) system consisting of a DGU-20A5R degassing unit, a SIL-20AC HT auto sampler, a CTO-20AC column oven, a SPD-20AV UV-Vis detector and a RID-20A refractive index detector connected in series. The system contains a PL-GEL 10 μm guard column, two PL-GEL 10 μm Mixed-B columns. The instrument was calibrated with narrow polystyrene standards with Mp ranging from 4730 g mol−1 to 3187000 g mol−1. Tetrahydrofuran was used as the solvent for the GPC measurements.
where RH is the hydrodynamic radii of nanodroplets, kB is the Boltzmann constant, T is the absolute temperature, η is the viscosity of the microemulsion (in specific temperature) and D is the diffusion constant.45
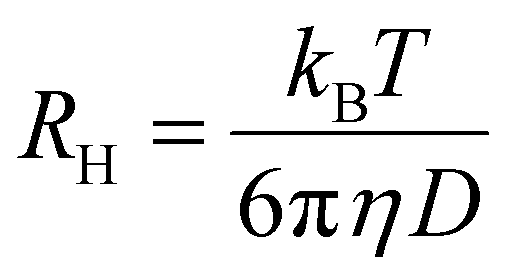
After their preparation the microemulsions were placed in a suitable glass cell under dust-free conditions. The experimental data were processed using version 6.32 of the Malvern Zetasizer Nano software (Malvern Panalytical Ltd, Enigma Business Park, UK). The temperature during the measurements was constant at 25 °C. Experiments were performed in triplicate for each sample, and the results were presented as average ± S.D.
| Φx = Φr(Fx/Fr)·(Ar/Ax)·(nx2/nr2). |
The indexes ‘r’ and ‘x’ are referred to the reference and the unknown sample, respectively.
Results and discussion
Synthesis and characterization of the TQ conjugated polymers
A family of conjugated polymers comprising of thiophene (T) as the electron donating unit and quinoxaline (Q) as the electron withdrawing unit by varying the number and the position of the fluorine atoms in the repeat unit (Fig. 1) were designed and synthesized. In particular TQ, TQf, and TQ2f contain unsubstituted thiophene and zero, one and two fluorine atom(s) on the quinoxaline, respectively whereas T2fQ, T2fQf, and T2fQ2f contain a substituted thiophene with two fluorine atoms and zero, one and two fluorine atom(s) on the quinoxaline, respectively. The scope is to investigate the correlation between structure and optoelectronic properties as regards the addition of fluorine atoms. The polymers in Fig. 1 were synthesized via metal catalyzed aromatic cross-coupling Stille polymerization54 reaction utilizing tris(dibenzylideneacetone)dipalladium(0) [Pd2(dba)3] in 2% per mole and tri(o-tolyl)phosphine [P(o-tol)3], 8% per mole, as the catalytic system in toluene solution. The final polymers were purified and fractionated using sequential Soxhlet extraction with methanol, acetone, hexane, and chloroform. The average molecular weights per number (n) and dispersity (Đ) of the chloroform-soluble fractions of the TQ-based polymers as measured by gel permeation chromatography (GPC) based on monodispersed polystyrene standards are shown in Fig. 2 and are summarized in Table 1.
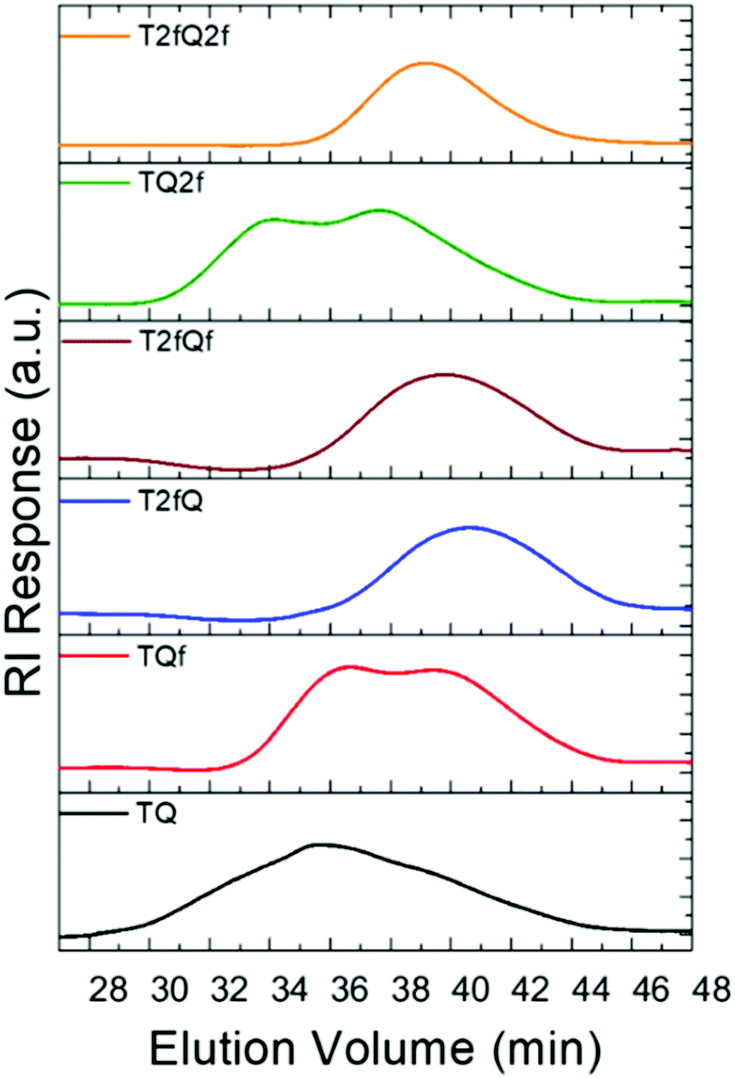 | ||
| Fig. 2 GPC chromatographs of the TQ-based conjugated polymers. | ||
|
n (g mol−1) |
w (g mol−1) |
Đ | λ solmax (nm) | PLsolmax (nm) | PLQY | |
|---|---|---|---|---|---|---|
| TQ | 19900 |
64200 |
3.2 | 354, 600 | 668 | 0.59 ± 0.04 |
| TQf | 11400 |
25800 |
2.3 | 338, 574 | 618 | 0.51 ± 0.02 |
| TQ2f | 22600 |
52500 |
2.3 | 319, 557 | 592 | 0.47 ± 0.03 |
| T2fQ | 6000 | 11300 |
1.9 | 538 | 600 | 0.37 ± 0.01 |
| T2fQf | 8200 | 15600 |
1.9 | 387, 498 | 571 | 0.30 ± 0.01 |
| T2fQ2f | 11200 |
16200 |
1.5 | 392, 456 | 544 | 0.22 ± 0.01 |
From the acquired GPC graphs, we can observe that TQ, TQf and TQ2f reveal multiple distinct peaks which correspond to high- and low-molecular-weight components in contrast to T2fQ, T2fQf, and T2fQ2f where only one peak is evident at higher elution times (Fig. 2). This points out that the TQ, TQf and TQ2f polymer chains are adopting mixed extended and aggregated forms in tetrahydrofuran (THF) solution at 25 °C while under the same conditions the T2fQ, T2fQf, and T2fQ2f polymer chains are adopting only the extended form. It seems that the presence of the two fluorine atoms on the thiophene of T2fQ, T2fQf, and T2fQ2f facilitates the solubility of the polymers in THF solution (25 °C) regardless of the number of fluorine atoms present on the quinoxaline moiety.
The absorption and the emission spectra of the TQs in THF solution are presented in Fig. 3, and the corresponding optoelectronic properties are summarized in Table 1. It is revealed that all synthesized polymers exhibit light absorption and fluorescence emission in the UV-Vis region of the spectra. Their optical properties are tunable and are significantly affected by the number and the position of the fluorine atoms anchored on the polymer backbone. The shape of the absorption spectra is qualitatively similar for all polymers, except TQ2f and T2fQ2f which display a different allocation. For each polymer, two major absorption bands are observed in solution, a phenomenon commonly observed for D–A copolymers. The absorption at low-wavelengths can be attributed to the π–π* transition whilst the high-wavelength peak is related to an intramolecular D–A charge transfer.55 All polymers exhibit absorption maxima at low-wavelengths in the range 319–394 nm, whereas the high-wavelength transition is detected in the range between 557 nm and 600 nm for TQ, TQf and TQ2f and between 456 nm and 538 nm for T2fQ, T2fQf, T2fQ2f (Table 1).
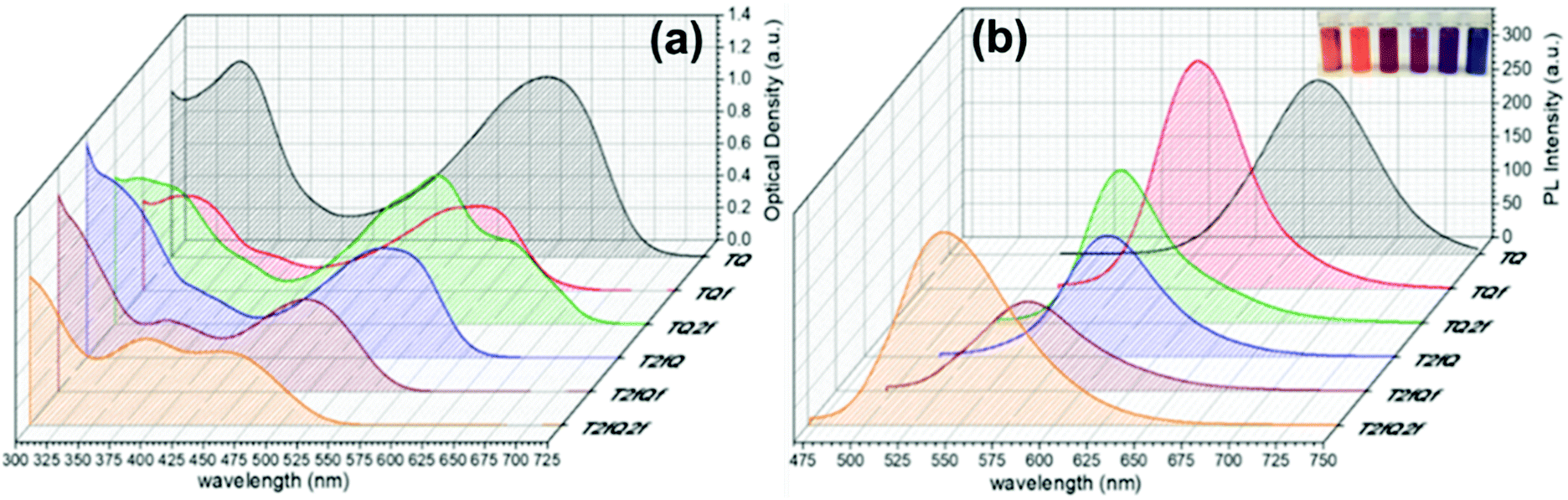 | ||
| Fig. 3 (a) Absorption and (b) photoluminescence spectra of TQs in THF solutions. In the inset of (b) the TQ polymers in THF solution are shown (from left to right; T2fQ2f, T2fQf, T2fQ, TQ2f, TQf, TQ). | ||
A general trend is detected for the high wavelength absorption maxima of the polymers. We noticed a blue shift of the absorption maxima for the high-energy transitions of all polymers upon the incorporation of fluorine atoms on the polymer chain, as compared to the corresponding absorption peak of the non-fluorinated TQ polymer (Fig. 3a). On the other hand, the low-wavelength absorption peak of the polymers presents a different trend. While TQ, TQf and TQ2f show a blue shift of the low wavelength absorption peak upon addition of fluorine atoms on the quinoxaline, T2fQf and T2fQ2f demonstrate a slight (∼5 nm) red shift (Table 1). This indicates that the gradual increase of the fluorine atoms weakens the intramolecular interaction of the D–A charge transfer and for the case of TQ, TQf and TQ2f the stepwise addition of the fluorine atoms in the quinoxaline is altering the locally excited (LE) state, whereas in T2fQ, T2fQf and T2fQ2f the increase of the fluorine atoms on the quinoxaline does not affect the LE state significantly. Interestingly, comparing the two related polymers TQ2f and T2fQ which differ as regards the positioning of the two fluorine atoms (thiophene and quinoxaline) it shows that the absorption maximum of TQ2f is red shifted versus T2fQ. It seems that when the thiophene is substituted with the two fluorine atoms the electron donating property is reduced due to the high electronegativity of the fluorine atoms.56 In addition, the electron withdrawing property of the fluoro-substituted quinoxaline is decreased upon removing the two fluorine atoms. It is therefore expected that the high wavelength absorption peak maxima, assigned to the ICT of the TQ2f, will be red-shifted versus T2fQ. Furthermore, it is obvious that the anchoring of the fluorine atoms on the polymer backbone blue shifts the emission maxima (Table 1). More specifically, TQ, TQf and TQ2f show emission maxima at 668 nm, 618 nm and 592 nm, respectively while T2fQ, T2fQf and T2fQ2f reveal emission maxima at 600 nm, 571 nm and 544 nm, respectively. Contrary to what has been observed for the variation of the absorption maxima between TQ2f and T2fQ, the emission maximum of the T2fQ in solution is red shifted versus TQ2f. Finally, we have calculated the PLQY of the CPs in the THF solution and the results are shown on Fig. 4 and depicted in Table 1. A general trend is observed indicating that as the number of fluorine atoms in the repeat unit increases, the PLQY drops. For instance, the PLQY of the TQ is 0.59 (no fluorine atoms in the repeat unit) and gradually drops to 0.22 for the case of T2fQ2f (four fluorine atoms in the repeat unit).
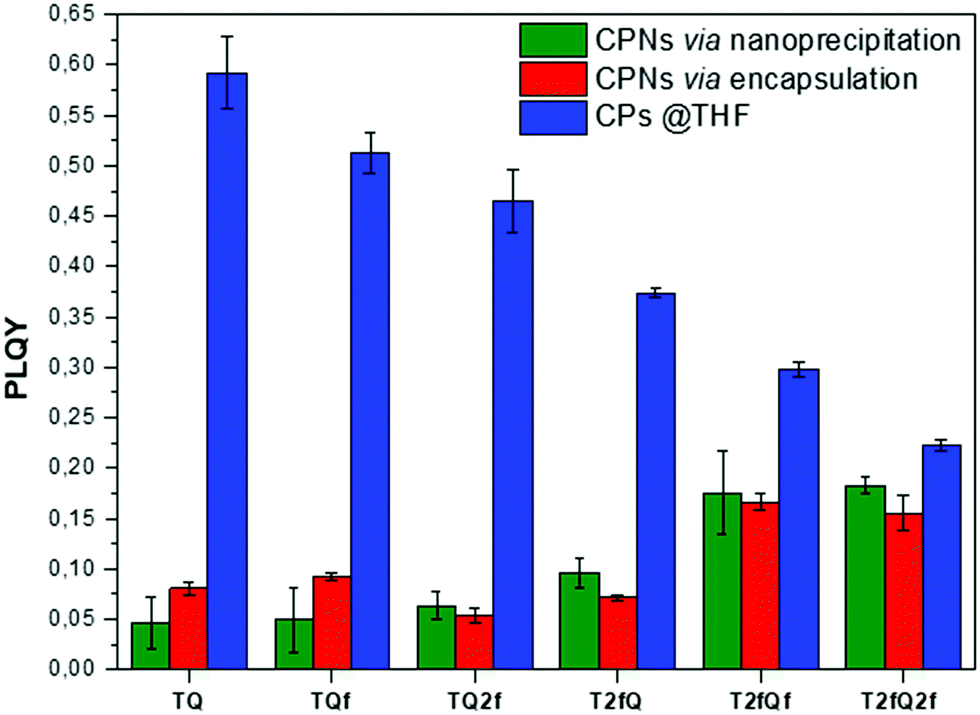 | ||
| Fig. 4 Comparison of the photoluminescence quantum yields for the TQ-based polymers in THF and their corresponding aqueous CPNs. | ||
Preparation of conjugated polymer nanoparticles and characterization of the properties
Generally, CPs are hydrophobic, and thus requires significant improvement of their water solubility to be applied for in vitro and in vivo applications. The two main approaches that are followed for the preparation of aqueous CPNs are through nanoprecipitation and encapsulation.39 In the first approach, also often termed the reprecipitation or injection method, the conjugated polymer is dissolved in a good (non-polar) solvent. This solution is then dispersed in a (polar) solvent, which is a poor solvent for the conjugated polymer, where either droplets of the polymer solution are formed or the polymer precipitates. In the second approach, amphiphilic PEGylated block copolymers are popularly utilized to encapsulate conjugated polymers in order to enhance the biocompatibility of nanomaterials and to further employ them in imaging and therapy applications.In this work, we choose to prepare the CPNs with both methods since to the best of our knowledge there are only limited studies that systematically compare the optical properties between the CPNs formed with the nanoprecipitation and encapsulation methods.57–59 For the formation of the CPNs via the nanoprecipitation method (Fig. 5), firstly, each conjugated polymer was dissolved in tetrahydrofuran (THF) which is a low boiling point aprotic polar solvent miscible with water at a concentration of 0.1 mg mL−1. At such low concentrations, the polymer chains usually adopt their extended form. Then, a different portion (from 1 mL to 1.5 mL on a case-by-case basis) of each different polymer THF solution (stock solution) was added dropwise to 7 mL of deionized water followed by sonication at room temperature. For the encapsulation method, 1 mg of each TQ conjugated polymer and 9 mg of poly(ethylene glycol)methyl ether-block-poly(lactide-co-glycolide) (mPEG-b-PLGA) were dissolved in 1 mL of THF. The choice of mPEG-b-PLGA was based on the fact that it is an FDA approved and metabolizable copolymer.40,60 The resulting solutions are added to 7 mL of deionized water following the same procedure as described for the nanoprecipitation method (Fig. 5). In both cases, the solutions were left overnight to allow complete evaporation of the THF and the volume of water lost was replaced. After this procedure, stable aqueous CPNs are formed through aggregate formation of the hydrophobic conjugated polymer chains in the case of nanoprecipitation, whilst by applying the encapsulation method, the hydrophobic PLGA segments are liable to entangle with TQ polymer chains and the hydrophilic PEG chains should extend into the aqueous phase. The obtained CPNs were then filtered through a 0.2 μm cellulose acetate filter. The resulting concentrations (before filtration) of the CPNs prepared via the nanoprecipitation method were 2.8 ppm for the TQ polymer and 4.3 ppm for all the others, and 143 ppm for the CPNs prepared via the encapsulation method.
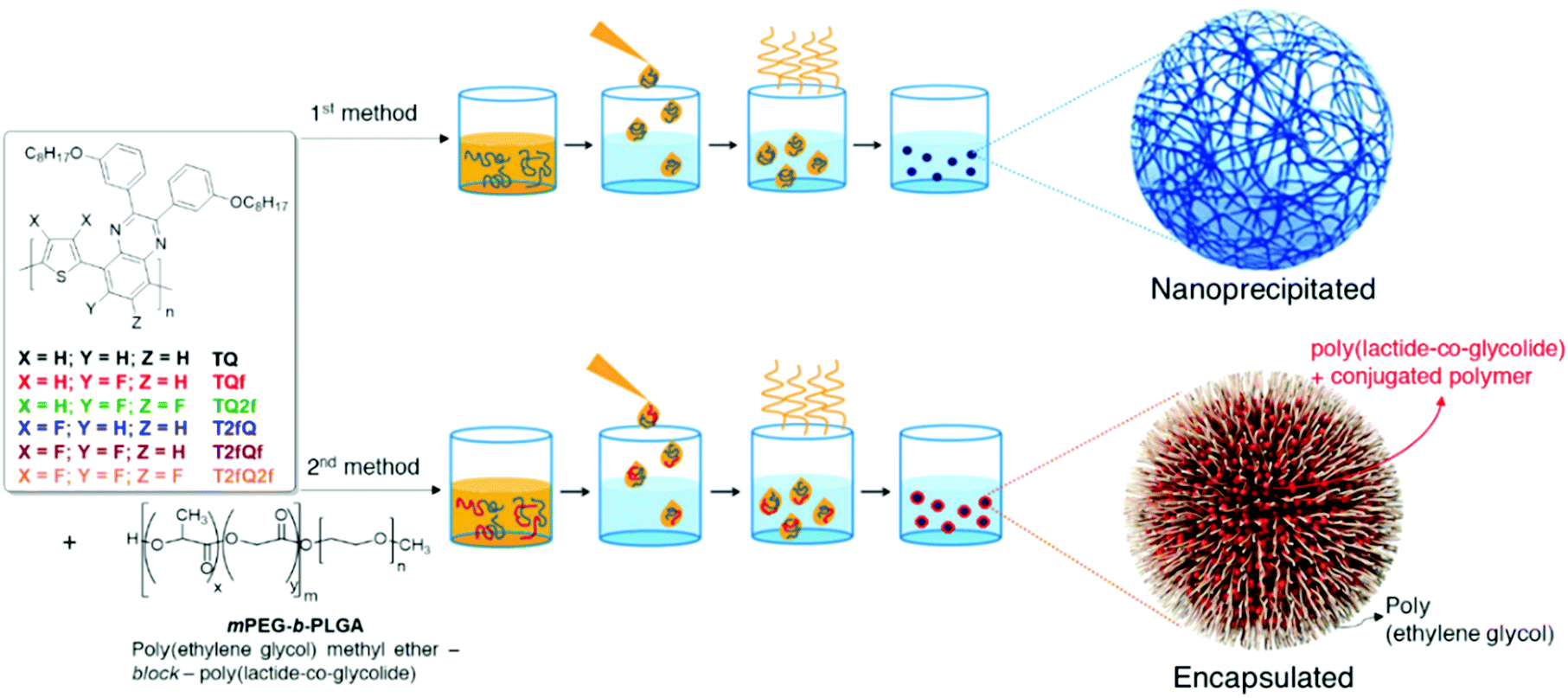 | ||
| Fig. 5 Schematic representation of the conjugated polymer nanoparticle preparation through nanoprecipitation (top) and encapsulation (bottom). | ||
It is important to use different characterization techniques to calculate the size and the distribution of the prepared nanoparticles in order to obtain a more complete view of the CPNs and their behavior. To evaluate the sizes of the formatted aqueous TQ-based polymer nanoparticles dynamic light scattering (DLS) measurements were performed at room temperature, 25 °C, a day after their preparation (Fig. 6 and 7) and the results are depicted in Table 2. The DLS measurements indicate that the aqueous TQ nanoparticles exhibit a unimodal size distribution regardless of their preparation method (Fig. 6 and 7). The majority of the nanoprecipitated CPNs had a hydrodynamic diameter between 30 and 50 nm (Table 2). Nanoparticles that were prepared via the encapsulation method were slightly larger, ranging in size from ca. 50 to 60 nm (Table 2). This observation was expected because of the use of the mPEG-b-PLGA block copolymer.
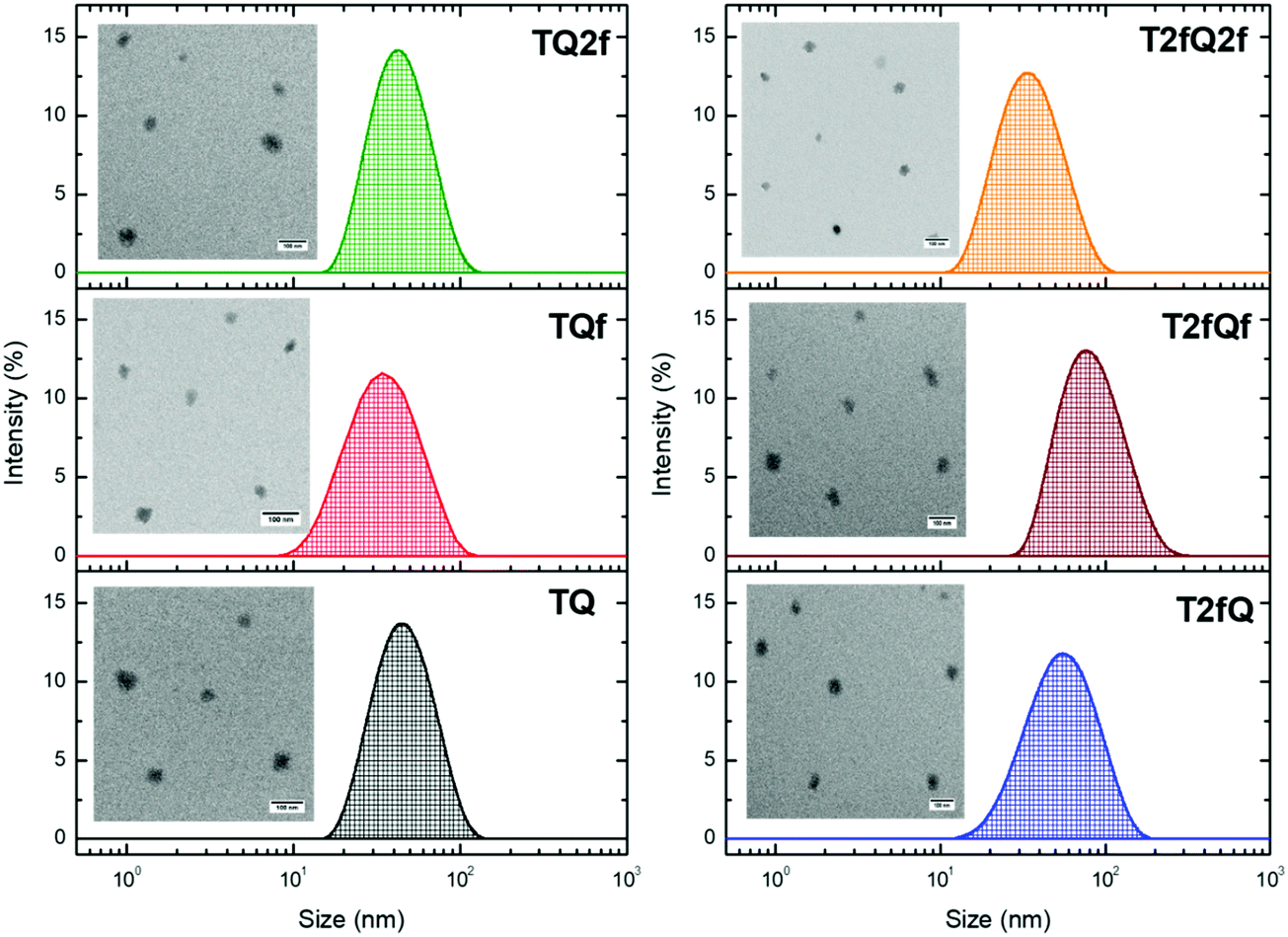 | ||
| Fig. 6 Size distribution of aqueous TQ nanoparticles prepared via the nanoprecipitation method determined by DLS measurements and their morphology using TEM. | ||
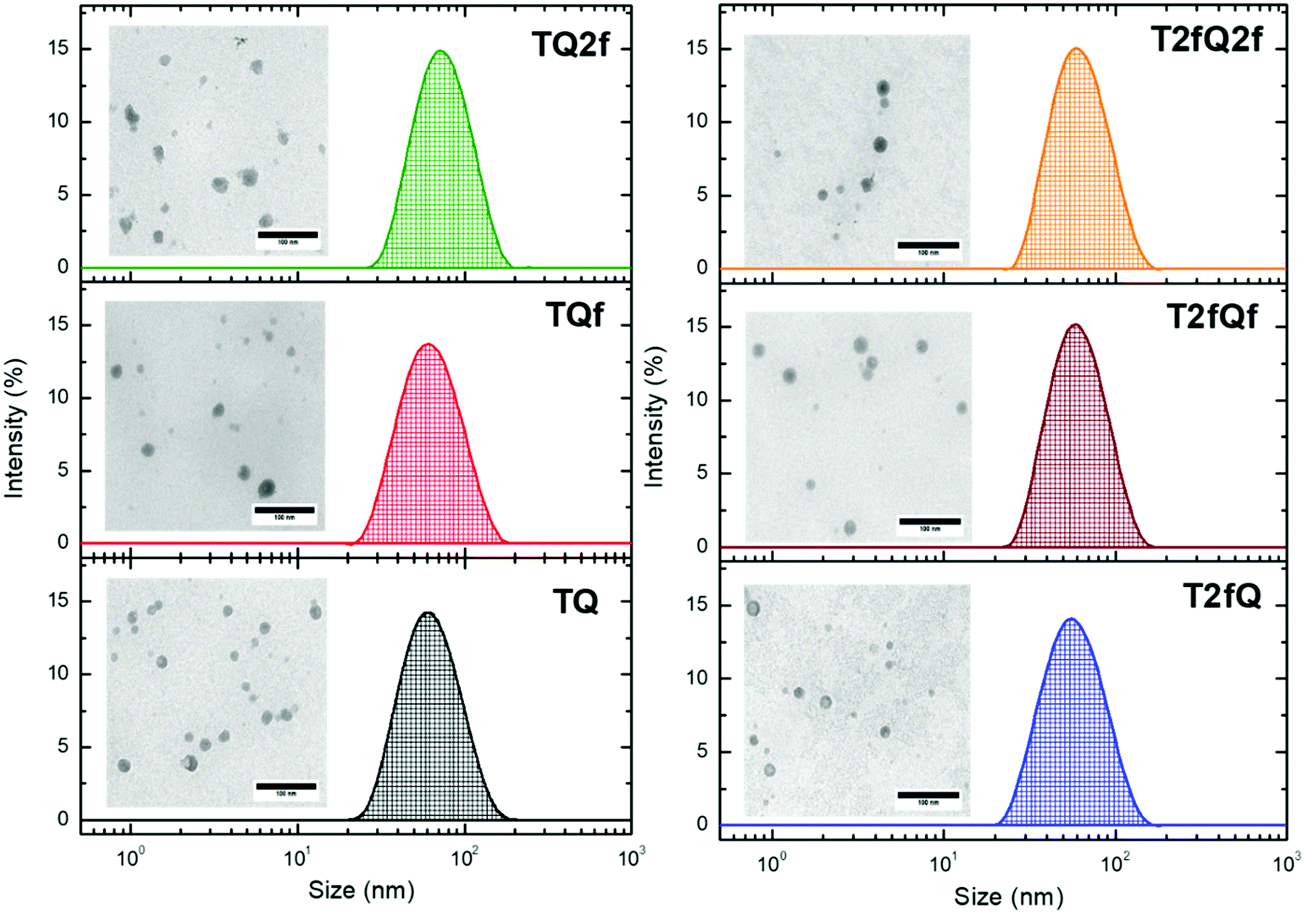 | ||
| Fig. 7 Size distribution of aqueous TQ nanoparticles prepared via the encapsulation method determined by DLS measurements and their morphology using TEM. | ||
| CPNs | Nanoprecipitation | ||||
|---|---|---|---|---|---|
| Z-Ave. (nm) | PDI | PK1 mean (nm) | Z potential (mV) | TEM | |
| TQ | 40.98 ± 0.12 | 0.13 ± 0.01 | 47.26 ± 0.56 | −20.3 | 30.85 ± 2.12 |
| TQf | 31.67 ± 0.09 | 0.21 ± 0.01 | 37.80 ± 1.07 | −19.6 | 24.04 ± 1.01 |
| TQ2f | 40.55 ± 0.12 | 0.18 ± 0.01 | 46.51 ± 0.48 | −17.7 | 33.74 ± 2.61 |
| T2fQ | 48.13 ± 0.31 | 0.17 ± 0.02 | 53.87 ± 3.58 | −21.0 | 33.50 ± 1.45 |
| T2fQf | 75.03 ± 1.22 | 0.16 ± 0.02 | 84.65 ± 4.94 | −18.2 | 39.77 ± 3.16 |
| T2fQ2f | 33.34 ± 0.15 | 0.21 ± 0.01 | 37.97 ± 0.60 | −15.5 | 27.45 ± 3.11 |
| Encapsulation | |||||
|---|---|---|---|---|---|
| Z-Ave. (nm) | PDI | PK1 mean (nm) | Z potential (mV) | TEM | |
| TQ | 57.71 ± 0.20 | 0.11 ± 0.01 | 65.64 ± 0.46 | −10.5 | 17.41 ± 1.82 |
| TQf | 57.86 ± 0.20 | 0.12 ± 0.01 | 66.78 ± 0.40 | −11.0 | 19.01 ± 2.06 |
| TQ2f | 69.40 ± 0.42 | 0.11 ± 0.01 | 75.54 ± 9.46 | −11.2 | 19.28 ± 1.06 |
| T2fQ | 52.46 ± 0.15 | 0.12 ± 0.01 | 60.13 ± 0.41 | −10.7 | 17.40 ± 0.74 |
| T2fQf | 52.26 ± 0.16 | 0.11 ± 0.01 | 63.93 ± 0.96 | −10.7 | 19.55 ± 0.91 |
| T2fQ2f | 58.32 ± 0.13 | 0.13 ± 0.01 | 69.13 ± 7.22 | −10.3 | 18.65 ± 1.90 |
Interestingly, we notice that the TQ2f nanoparticles prepared using the encapsulation method and the T2fQf nanoparticles prepared via the nanoprecipitation method exhibit hydrodynamic diameters around 70 nm and 75 nm, respectively (Table 2). This observation occurred for all the prepared batches that we examined probably due to their higher tendency to form aggregates based on the reported TEM images. According to the distribution of nanoparticle sizes, those prepared via the encapsulation method demonstrate relatively narrow size distributions with low polydispersity indexes (PDIs) of 0.10–0.13 compared to the PDI of those prepared via the nanoprecipitation method which presented higher PDI values (Table 2).
Simultaneously with the DLS measurements, the zeta potential of the CPNs was measured. The colloidal stability of our aqueous CPNs is confirmed by the substantial and negative zeta potential, as shown in Table 2. It is obvious that the nanoparticles prepared via the nanoprecipitation method display zeta potential values ranging from −15.5 to −21 mV, while the corresponding ones prepared via the encapsulation method exhibit a more neutral zeta potential of approximately −11 mV. The origin of a negative zeta potential has been addressed in previous studies61,62 and it is attributed to oxidative defects on the surface of the CPNs or to the presence of the π-polarizations of the polymer rings that enable stronger attractive van der Waals forces and hydrophobic interactions. Therefore, when the conjugated polymer is encapsulated inside the amphiphilic copolymer, those interactions are minimized resulting in lower zeta potential values.
Transmission electron microscopy was further used to investigate the shape and dimensions of the CPNs (inset of Fig. 6 and 7). The measurements revealed distinguishable approximately spherical shape for all nanoparticles. The median diameters range from 24.04 ± 1.01 nm to 39.77 ± 3.16 nm for the nanoprecipitation method and from 17.40 ± 0.74 nm to 19.55 ± 0.91 nm for the encapsulation method, being in agreement with the DLS measurements according to the narrow size distributions exhibited by the encapsulated CPNs. The difference between the sizes of the nanoparticles observed by TEM versus the DLS measurements can be attributed to the fact that the hydrated size of CPNs in water is typically larger than size measurements obtained in the dry state. This discrepancy has been previously reported.63,64 In both cases, the average particle size was determined to be lower than 100 nm, which is an acceptable size for biological applications, since nanoparticles with sizes >100 nm may have issues such as poor mass transfer, tissue penetration and non-specific absorption.61,65 The resulting sizes and shapes of the prepared CPNs are sufficiently small to be internalized by the cells. Moreover, it has been demonstrated that CPNs larger than 200 nm are excreted via the liver and spleen while for sizes smaller than 10 nm the excretion is performed through the kidneys,66 highlighting the potential application of these CPNs as fluorescent contrast agents.
However, apart from the appropriate size and dimension of the nanoparticles, it is essential to show high photoluminescence quantum yields (PLQYs). It is well known that CPs exhibit inherent hydrophobicity and when they are formulated into nanoparticles in aqueous solution, the tight packing of the polymer chains in the particles results in severe fluorescence quenching, which significantly decreases the PLQY and the brightness of the probes. In this work, we attempted to prevent the strong π–π stacking of the CP chains by decreasing the intramolecular planarity of the polymer backbone through increase of the torsion angle that will occur via the gradual incorporation of fluorine atoms on the thiophene and quinoxaline units. Fig. 8 and 9 present the UV-Vis absorption and fluorescence spectra of the CPNs formulated by nanoprecipitation and encapsulation, and the corresponding UV-Vis absorption and fluorescence maxima are summarized in Table 3 along with the measured PLQY.
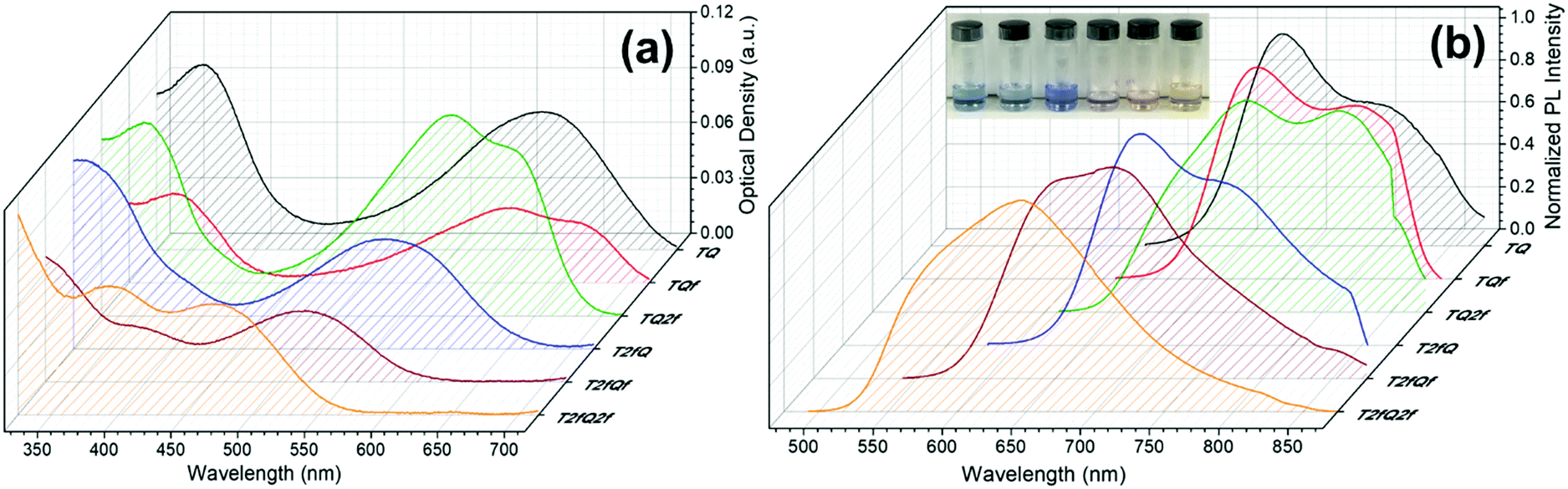 | ||
| Fig. 8 UV-Vis absorption spectra and normalized emission spectra of TQ-based CPNs prepared via the nanoprecipitation method. In the inset of Fig. 8b the nanoprecipitated CPNs in water are shown (from left to right; TQ, TQf, TQ2f, T2fQ, T2fQf, T2fQ2f). | ||
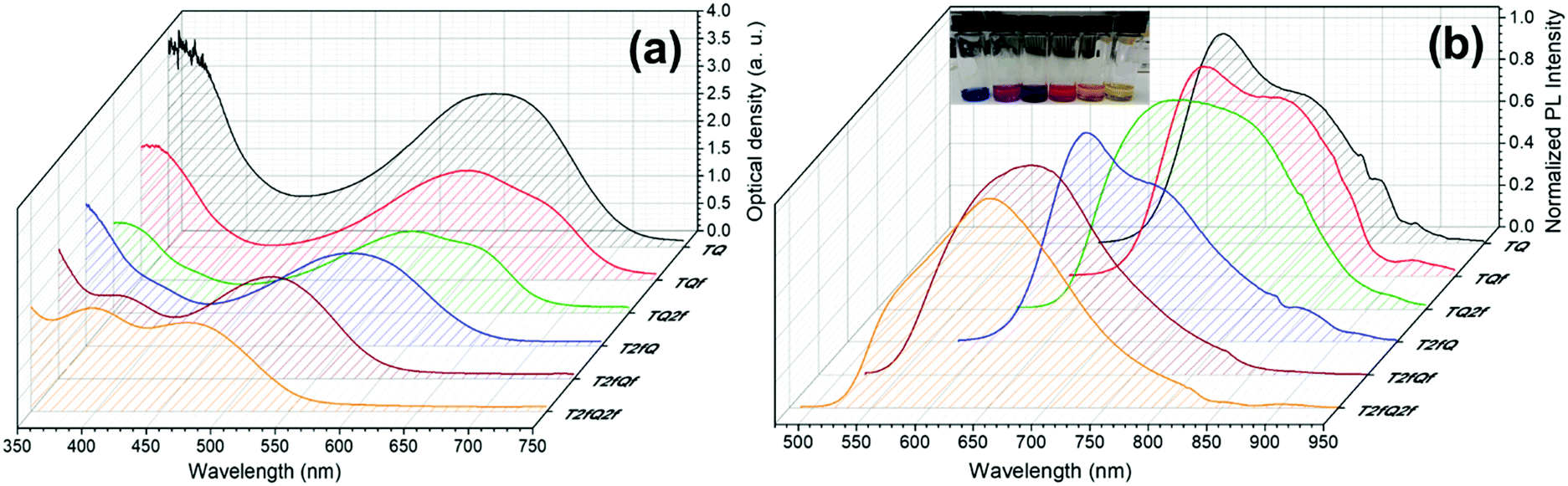 | ||
| Fig. 9 UV-Vis absorption spectra and normalized emission spectra of TQ-based CPNs prepared via the encapsulation method. In the inset of Fig. 9b the encapsulated CPNs in water are shown (from left to right; TQ, TQf, TQ2f, T2fQ, T2fQf, T2fQ2f). | ||
| CPNs via nanoprecipitation | CPNs via encapsulation | |||||
|---|---|---|---|---|---|---|
| λ absmax (nm) | Plmax (nm) | PLQY | λ absmax (nm) | Plmax (nm) | PLQY | |
| TQ | 359, 614 | 728 | 0.05 ± 0.03 | 604 | 726 | 0.08 ± 0.01 |
| TQf | 358, 612 | 732, 806 | 0.05 ± 0.03 | 362, 603 | 734 | 0.09 ± 0.01 |
| TQ2f | 357, 587 | 744, 812 | 0.06 ± 0.01 | 353, 582 | 744 | 0.05 ± 0.01 |
| T2fQ | 555 | 690 | 0.10 ± 0.02 | 555 | 683 | 0.07 ± 0.01 |
| T2fQf | 518 | 692 | 0.18 ± 0.04 | 397, 515 | 662 | 0.17 ± 0.01 |
| T2fQ2f | 394, 474 | 647 | 0.18 ± 0.01 | 397, 471 | 650 | 0.16 ± 0.02 |
It is revealed that all CPNs demonstrate a red shift in both absorption and emission maxima in comparison to the CPs in THF. This supports evidence of effective π–π stacking of the polymer chains passing from the extended form in THF to a more aggregate conformation, in the form of spherical species, in the aqueous nanoparticles.61,67 In general, we observe that on the one hand the CPNs substituted by the two fluorine atoms on the thiophene unit (T2fQ, T2fQf and T2fQ2f) exhibit absorption at the UV-Vis and fluorescence emission at the far-red part of the UV-Vis region. On the other hand, the CPNs not substituted with fluorine atoms on the thiophene unit (TQ, TQf, and TQ2f) present absorption maxima at the UV-Vis to far-red wavelengths and fluorescence peak maxima in the far-red and NIR spectral regions with emission tails extending up to 900 nm (Fig. 8 and 9). These observations are irrespective of the preparation method followed for the CPNs.68
In more detail, it is demonstrated that the low- and high-wavelength absorption peaks of all CPNs and the shape of the absorption profiles do not alter significantly independent of the preparation method, only the optical density is affected. The encapsulated CPNs present higher optical density than the nanoprecipitated ones due to the significantly higher concentration of the CPs loaded in the mPLGA-b-PEG (143 ppm) versus the lower concentration of the nanoprecipitated CPNs (2.8 or 4.3 ppm). Moreover, the same trend observed in solution for all CPs regarding the variation of the absorption maxima of both the low- and high-wavelength peaks as a function of the number of fluorine atoms is also present in the CPNs as well.69,70
Studying the photoluminescence properties of the CPNs as compared to the CPs in THF a controversial effect is revealed. The emission maxima of the CPs in THF are blue shifted upon the addition of fluorine atom(s) on the quinoxaline, regardless of the presence or not of fluorine atom(s) on the thiophene. However, the TQ, TQf, and TQ2f nanoparticles present red-shifted emission maxima whereas the T2fQ, T2fQf and T2fQ2f nanoparticles show blue shifted emission maxima with the gradual increase of the fluorine atom(s) on the quinoxaline independent of the preparation method (Tables 1 and 3). For example, the gradual introduction of fluorine atoms on the quinoxaline moiety of the TQ polymer red shifts the emission maxima of the TQ, TQf and TQ2f CPNs from 726 nm to 744 nm, while by anchoring two fluorine atoms on the thiophene and further adding fluorine atoms to the polymer chain blue shifts the emission peaks at lower wavelengths; from 690 nm to 647 nm in the case of nanoprecipitation and from 683 nm to 650 nm for the encapsulation method. This indicates that even though the self-assembly of the CPs in the CPNs leads to efficient π–π stacking, the number and the positioning of the fluorine atoms plays an essential role in the intra- and inter-chain interactions.
In parallel, we proceed to the measurement of the photoluminescence quantum yield values (PLQY) of the CPNs which are presented in Fig. 4 and depicted in Table 3. The PLQY of the TQ, TQf, TQ2f and T2fQ in THF is significantly higher and the PLQY of the T2fQf and T2fQ2f in THF is higher versus the corresponding CPNs in water (both in the encapsulation and the nanoprecipitation method) as shown in Fig. 4. This confirms our statement that the π–π* stacking of the polymer chains during the nanoparticle formation leads in an increased number of fluorescence quenching sites, especially in the case of TQ, TQf, TQ2f and T2fQ and to a lesser extent in T2fQf and T2fQ2f. In particular, the far-red emitting T2fQf and T2fQ2f nanoparticles exhibit the highest PLQYs independent of the preparation method with values of 0.16–0.18, slightly lower than the corresponding CPs in THF solution (0.30 and 0.22) and blue shifted emission maxima upon gradual increase of the fluorine atoms on the quinoxaline independent of the preparation method. These observations suggest that as the fluorine atoms on the quinoxaline increase the intramolecular disorder increases (in accordance with the theoretically predicted calculations on the dihedral angles in Fig. 10) and the intermolecular interactions between the polymer chains decreases. On the contrary, the NIR emitting CPNs exhibit PLQY up to 0.1. This may be attributed to the fact that the fluorescence quantum yield for NIR emitters are inherently lower than those of UV-visible and far-red emitters as a result of a smaller energy gap between the ground state and emitting state, often making non-radiative relaxation pathways competitive with radiative relaxation.61
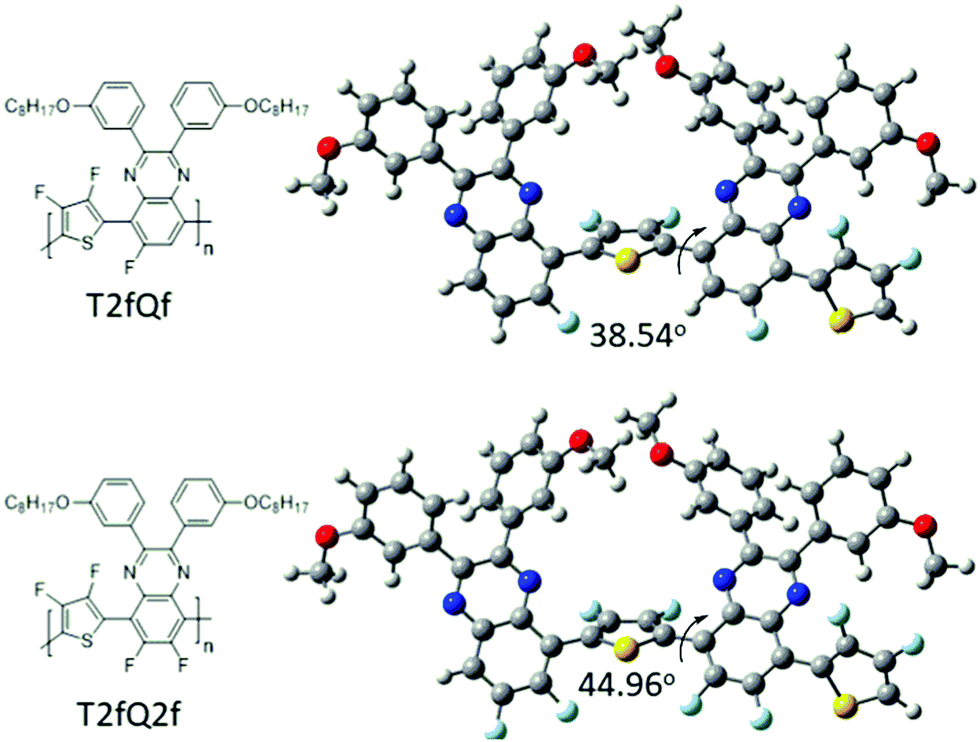 | ||
| Fig. 10 Theoretical calculated dihedral angles of the T2fQf and T2fQ2f polymers. | ||
In general, the PLQYs of the TQ, TQf and TQ2f nanoparticles are similar within the statistical error regardless of the preparation method. Moreover, the PLQYs of the T2fQ, T2fQf and T2fQ2f nanoparticles are higher than those of TQ, TQf and TQ2f nanoparticles in both preparation methods, except from the T2fQ based nanoparticles prepared via the encapsulation method. Between the T2fQ, T2fQf and T2fQ2f nanoparticles, the PLQYs of the T2fQf and T2fQ2f are similar regardless of the preparation method, whereas the PLQY of the nanoprecipitated T2fQ based nanoparticles is higher versus the encapsulated T2fQ based nanoparticles. These observations lead to the point that the TQ, TQf, TQ2f and T2fQ based nanoparticles exhibit comparable interchain interactions, while the T2fQf and T2fQ2f reveal slightly weaker interchain interactions independent of the preparation method. The fact that the absorption and emission spectra of the nanoparticles prepared from both methods are red-shifted versus the corresponding CPs in THF solution supports the above observation.
Conclusions
To summarize, in this study we successfully synthesized a series of conjugated polymers combining thiophene as the electron donating and quinoxaline as the electron deficient component (TQs) by varying the number and the positioning of the fluorine atoms in the repeat unit. Water-soluble nanoparticles comprising the synthesized TQ derivatives were prepared using two different methods (nanoprecipitation and encapsulation within a mPLGA-b-PEG block copolymer), enabling their potential use as low-bandgap fluorescent probes for bioimaging. DLS measurements indicate that all CPNs in aqueous suspension have a unimodal size distribution less than 80 nm and TEM measurements revealed their spherical shape. The optical properties of the CPNs show light absorption in the UV-Vis region, while they demonstrated fluorescence emission in the far-red/NIR region, depending on the structure, the position and the number of fluorine atoms on the polymer backbone, as well as on the preparation method of the nanoparticles. The far-red emitting TQ-based polymer nanoparticles with three and four fluorine atoms on the repeat unit of the polymers demonstrate higher quantum yield values regardless of the preparation method as compared to the NIR emitting TQ-based polymer nanoparticles with none, one and two fluorine atoms on the repeat unit. Overall, the CPNs show great promise as new NIR fluorescent probes and in vitro studies are underway to evaluate their cytotoxicity effects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Helmholtz European partnering program for the cooperation between German Cancer Research Center (DKFZ) and National Hellenic Research Foundation (NHRF) to build the Athens Comprehensive Cancer Center (ACCC). The authors would like to thank Dr Karsten Richter (DKFZ) for acquiring the TEM images.Notes and references
- R. Weissleder and M. J. Pittet, Imaging in the Era of Molecular Oncology, Nature, 2008, 425, 580 CrossRef PubMed.
- K. Park, S. Lee, E. Kang, K. Kim, K. Choi and I. C. Kwon, New Generation of Multifunctional Nanoparticles for Cancer Imaging and Therapy, Adv. Funct. Mater., 2009, 19, 1553 CrossRef CAS.
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. H. van de Velde and J. V. Frangioni, Image-guided cancer surgery using near-infrared fluorescence, Nat. Rev. Clin. Oncol., 2013, 10, 507 CrossRef CAS PubMed.
- E. Hemmer, A. Benayas, F. Legare and F. Vetrone, Exploiting the Biological Windows: Current Perspectives on Fluorescent Bioprobes Emitting Above 1000 nm, Nanoscale Horiz., 2016, 1, 168 RSC.
- S. Q. He, J. Song, J. L. Qu and Z. Cheng, Crucial Breakthrough of Second Near-Infrared Biological Window Fluorophores: Design and Synthesis Toward Multimodal Imaging and Theranostics, Chem. Soc. Rev., 2018, 47, 4258 RSC.
- E. M. Sevick-Muraca, J. P. Houston and M. Gurfinkel, Fluorescence-Enhanced, Near Infrared Diagnostic Imaging with Contrast Agents, Curr. Opin. Chem. Biol., 2002, 6, 642 CrossRef CAS PubMed.
- V. Ntziachristos, C. Bremer and R. Weissleder, Fluorescence Imaging with Near-Infrared Light: New Technological Advances that Enable In Vivo Molecular Imaging, Eur. Radiol., 2003, 13, 195 CrossRef PubMed.
- A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. S. Hong, C. R. Qu, S. Diao, Z. X. Deng, X. M. Hu, B. Zhang, X. D. Zhang, O. K. Yaghi, Z. R. Alamparambil, X. C. Hong, Z. Cheng and H. J. Dai, A Small-Molecule Dye for NIR-II Imaging, Nat. Mater., 2016, 15, 235 CrossRef CAS PubMed.
- H. Wan, H. L. Ma, S. J. Zhu, F. F. Wang, Y. Tian, R. Ma, Q. L. Yang, Z. B. Hu, T. Zhu, W. Z. Wang, Z. R. Ma, M. X. Zhang, Y. T. Zhong, H. T. Sun, Y. Y. Liang and H. J. Dai, Developing a Bright NIR-II Fluorophore with Fast Renal Excretion and Its Application in Molecular Imaging of Immune Checkpoint PD-L1, Adv. Funct. Mater., 2018, 28, 1804956 CrossRef PubMed.
- P. F. Sun, Q. Wu, X. L. Sun, H. Miao, W. X. Deng, W. S. Zhang, Q. L. Fan and W. J. Huang, Aggregate Squaraine Nanoparticles with Bright NIR-II Fluorescence for Imaging Guided Photothermal Therapy, Chem. Commun., 2018, 54, 13395 RSC.
- G. S. Hong, S. Diao, J. L. Chang, A. L. Antaris, C. X. Chen, B. Zhang, S. Zhao, D. N. Atochin, P. L. Huang, K. I. Andreasson, C. J. Kuo and H. J. Dai, Through-Skull Fluorescence Imaging of the Brain in a New Near-Infrared Window, Nat. Photonics, 2014, 8, 723 CrossRef CAS PubMed.
- K. Welsher, Z. Liu, S. P. Sherlock, J. T. Robinson, Z. Chen, D. Daranciang and H. J. Dai, A Route to Brightly Fluorescent Carbon Nanotubes for Near-Infrared Imaging in Mice, Nat. Nanotechnol., 2009, 4, 773 CrossRef CAS PubMed.
- G. S. Hong, J. T. Robinson, Y. J. Zhang, S. Diao, A. L. Antaris, Q. B. Wang and H. J. Dai, In Vivo Fluorescence Imaging with Ag2S Quantum Dots in the Second Near-Infrared Region, Angew. Chem., Int. Ed., 2012, 51, 9818 CrossRef CAS PubMed.
- D. Franke, D. K. Harris, O. Chen, O. T. Bruns, J. A. Carr, M. W. B. Wilson and M. G. Bawendi, Continuous Injection Synthesis of Indium Arsenide Quantum Dots Emissive in the Short-Wavelength Infrared, Nat. Commun., 2016, 7, 12749 CrossRef CAS PubMed.
- D. J. Naczynski, M. C. Tan, M. Zevon, B. Wall, J. Kohl, A. Kulesa, S. Chen, C. M. Roth, R. E. Riman and P. V. Moghe, Rare-Earth-Doped Biological Composites as in Vivo Shortwave Infrared Reporters, Nat. Commun., 2013, 4, 2199 CrossRef CAS PubMed.
- W. Shao, G. Y. Chen, A. Kuzmin, H. L. Kutscher, A. Pliss, T. Y. Ohulchanskyy and P. N. Prasad, Tunable Narrow Band Emissions from Dye-Sensitized Core/Shell/Shell Nanocrystals in the Second Near-Infrared Biological Window, J. Am. Chem. Soc., 2016, 138, 16192 CrossRef CAS PubMed.
- L. Liu, S. F. Wang, B. Z. Zhao, P. Pei, Y. Fan, X. M. Li and F. Zhang, Er3+ Sensitized 1530 nm to 1180 nm Second Near-Infrared Window Upconversion Nanocrystals for InVivo Biosensing, Angew. Chem., Int. Ed., 2018, 57, 7518 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Aggregation-induced emission: together we shine, united we soar!, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Probing the cytotoxicity of semiconducting quantum dots, Nano Lett., 2004, 4, 11 CrossRef CAS PubMed.
- W. Wu, R. Li, X. Bian, Z. Zhu, D. Ding, X. Li, Z. Jia, X. Jiang and Y. Hu, Covalently combining carbon nanotubes with anticancer agent: preparation and antitumor activity, ACS Nano, 2009, 3, 2740 CrossRef CAS PubMed.
- K. Li and B. Liu, Polymer Encapsulated Conjugated Polymer Nanoparticles for Fluorescence Bioimaging, J. Mater. Chem., 2012, 22, 1257 RSC.
- C. Wu and D. T. Chiu, Highly Fluorescent Semiconducting Polymer Dots for Biology and Medicine, Angew. Chem., Int. Ed., 2013, 52, 3086 CrossRef CAS PubMed.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Water-Soluble Conjugated Polymers for Imaging, Diagnosis, and Therapy, Chem. Rev., 2012, 112, 4687 CrossRef CAS PubMed.
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, Multicolor Conjugated Polymer Dots for Biological Fluorescence Imaging, ACS Nano, 2008, 2, 2415 CrossRef CAS PubMed.
- C. Wu, S. J. Hansen, Q. Hou, J. Yu, M. Zeigler, Y. Jin, D. R. Burnham, J. McNeill, J. M. Olson and D. T. Chiu, Design of highly emissive polymer dot bioconjugates for in vivo tumor targeting, Angew. Chem., Int. Ed., 2011, 50, 3430 CrossRef CAS PubMed.
- E. Ahmed, S. W. Morton, P. T. Hammond and T. M. Swager, Fluorescent multiblock pi-conjugated polymer nanoparticles for in vivo tumor targeting, Adv. Mater., 2013, 25, 4504 CrossRef CAS PubMed.
- C. F. Wu, B. Bull, K. Christensen and J. McNeill, Ratiometric single nanoparticle oxygen sensors for biological imaging, Angew. Chem., Int. Ed., 2009, 48, 2741 CrossRef CAS PubMed.
- K. Y. Pu, A. J. Shuhendler and J. H. Rao, Semiconducting polymer nanoprobe for in vivo imaging of reactive oxygen and nitrogen species, Angew. Chem., Int. Ed., 2013, 52, 10325 CrossRef CAS PubMed.
- Q. Zhao, X. Zhou, T. Cao, K. Y. Zhang, L. Yang, S. Liu, H. Liang, H. Yang, F. Li and W. Huang, Fluorescent/phosphorescent dual-emissive conjugated polymer dots for hypoxia bioimaging, Chem. Sci., 2015, 6, 1825 RSC.
- H.-Y. Liu, P.-J. Wu, S.-Y. Kuo, C.-P. Chen, E.-H. Chang, C.-Y. Wu and Y.-H. Chan, Quinoxaline-based polymer dots with ultrabright red to near-infrared fluorescence for in vivo biological imaging, J. Am. Chem. Soc., 2015, 137, 10420 CrossRef CAS PubMed.
- I. C. Wu, I.-C. Wu, J. Yu, F. Ye, Y. Rong, M. E. Gallina, B. S. Fujimoto, Y. Zhang, Y.-H. Chan, W. Sun, X.-H. Zhou, C. Wu and D. T. Chiu, Squaraine-based polymer dots with narrow, bright near-infrared fluorescence for biological applications, J. Am. Chem. Soc., 2015, 137, 173 CrossRef CAS PubMed.
- J. K. Grey, D. Y. Kim, B. C. Norris, W. L. Miller and P. F. Barbara, Size-Dependent Spectroscopic Properties of Conjugated Polymer Nanoparticles, J. Phys. Chem. B, 2006, 110, 25568 CrossRef CAS PubMed.
- S. Habuchi, S. Onda and M. Vacha, Mapping the Emitting Sites within a Single Conjugated Polymer Molecule, Chem. Commun., 2009, 4868 RSC.
- S. Habuchi, S. Onda and M. Vacha, Molecular Weight Dependence of Emission Intensity and Emitting Sites Distribution within Single Conjugated Polymer Molecules, Phys. Chem. Chem. Phys., 2011, 13, 1743 RSC.
- J. B. Yu, C. F. Wu, Z. Y. Tian and J. McNeill, Tracking of Single Charge Carriers in a Conjugated Polymer Nanoparticle, Nano Lett., 2012, 12, 1300 CrossRef CAS PubMed.
- J. C. Lin, X. D. Zeng, Y. L. Xiao, L. Tang, J. X. Nong, Y. F. Liu, H. Zhou, B. B. Ding, F. C. Xu, H. X. Tong, Z. X. Deng and X. C. Hong, Novel Near-Infrared II Aggregation-Induced Emission Dots for in Vivo Bioimaging, Chem. Sci., 2019, 10, 1219 RSC.
- C.-S. Ke, C.-C. Fang, J.-Y. Yan, P.-J. Tseng, J. R. Pyle, C.-P. Chen, S.-Y. Lin, J. Chen, X. Zhang and Y.-H. Chan, Molecular Engineering and Design of Semiconducting Polymer Dots with Narrow-Band, Near-Infrared Emission for in Vivo Biological Imaging, ACS Nano, 2017, 11, 3166 CrossRef CAS PubMed.
- H. Piwoński, T. Michinobu and S. Habuchi, Controlling photophysical properties of ultra small conjugated polymer nanoparticles through polymer chain packing, Nat. Commun., 2017, 8, 15256 CrossRef PubMed.
- L. Feng, C. Zhu, H. Yuan, L. Liu, F. Lv and S. Wang, Conjugated polymer nanoparticles: preparation, properties, functionalization and biological applications, Chem. Soc. Rev., 2013, 42, 6620 RSC.
- Y. Wang, P. Li, T. T.-D. Tran, J. Zhang and L. Kong, Manufacturing Techniques and Surface Engineering of Polymer Based Nanoparticles for Targeted Drug Delivery to Cancer, Nanomaterials, 2016, 6, 26 CrossRef PubMed.
- R. Singh, G. Pagona, V. G. Gregoriou, N. Tagmatarchis, D. Toliopoulos, Y. Han, Z. Fei, A. Katsouras, A. Avgeropoulos, T. D. Anthopoulos, M. Heeney, P. E. Keivanidis and C. L. Chochos, Polym. Chem., 2015, 6, 3098 RSC.
- E. Tatsi, M. Spanos, A. Katsouras, B. M. Squeo, S. Fall, T. Heiser, P. Lévêque, V. G. Gregoriou, A. Avgeropoulos, N. Leclerc and C. L. Chochos, Macromol. Chem. Phys., 2019, 220, 1800418 CrossRef.
- D. Zimmermann, C. Sprau, J. Schröder, V. G. Gregoriou, A. Avgeropoulos, C. L. Chochos, A. Colsmann, S. Janietz and H. Krüger, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1457 CrossRef CAS.
- Y. Sakamoto, S. Komatsu and T. Suzuki, Tetradecafluorosexithiophene: The First Perfluorinated Oligothiophene, J. Am. Chem. Soc., 2001, 123, 4643 CrossRef CAS PubMed.
- P. A. Hassan, S. Rana and G. Verma, Making sense of Brownian motion: Colloid characterization by dynamic light scattering, Langmuir, 2015, 31, 12 CrossRef PubMed.
- J. N. Demas and G. A. Crosby, Measurement of photoluminescence quantum yields, J. Chem. Phys., 1971, 75, 991 CrossRef.
- G09|Gaussian.com, http://gaussian.com/glossary/g09/, accessed May 7, 2019.
- H. Bronstein, D. S. Leem, R. Hamilton, P. Woebkenberg, S. King, W. Zhang, R. S. Ashraf, M. Heeney, T. D. Anthopoulos, J. de Mello and I. McCulloch, Indacenodithiopheneco-benzothiadiazole Copolymers for High Performance Solar Cells or Transistors via Alkyl Chain Optimization, Macromolecules, 2011, 44, 6649 CrossRef CAS.
- L. Biniek, C. L. Chochos, N. Leclerc, O. Boyron, S. Fall, P. Lévèque and T. Heiser, 3,6-Dialkylthieno[3,2-b]thiophene moiety as a soluble and electron donating unit preserving the coplanarity of photovoltaic low band gap copolymers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1861 CrossRef CAS.
- L. Biniek, S. Fall, C. L. Chochos, N. Leclerc, P. Lévêque and T. Heiser, Optimization of the side-chain density to improve the charge transport and photovoltaic performances of a low band gap copolymer, Org. Electron., 2012, 13, 114 CrossRef CAS.
- J. Ku, Y. Lansac and Y. H. Jang, Time-dependent density functional theory study on benzothiadiazole-based low-band gap fused-ring copolymers for organic solar cell applications, J. Phys. Chem. C, 2011, 115, 21508 CrossRef CAS.
- C. Risko, M. D. McGehee and J.-L. Brédas, A quantum-chemical perspective into low optical-gap polymers for highly-efficient organic solar cells, Chem. Sci., 2011, 2, 1200 RSC.
- C. L. Chochos, P. Chávez, I. Bulut, P. Lévêque, M. Spanos, E. Tatsi, A. Katsouras, A. Avgeropoulos, V. G. Gregoriou and N. Leclerc, Experimental and theoretical investigations on the optical and electrochemical properties of π-conjugated donor-acceptor-donor (DAD) compounds toward a universal model, J. Chem. Phys., 2018, 149, 124902 CrossRef PubMed.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Examining the effect of the dipole moment on charge separation in donor–acceptor polymers for organic photovoltaic applications, Chem. Rev., 2011, 133, 1493 CrossRef PubMed.
- S. Roquet, A. Cravino, P. Leriche, O. Alévêque, P. Frère and J. Roncali, Triphenylamine–Thienylenevinylene Hybrid Systems with Internal Charge Transfer as Donor Materials for Heterojunction Solar Cells, J. Am. Chem. Soc., 2006, 128, 3459 CrossRef CAS PubMed.
- Z. Fei, M. Shahid, N. Yaacobi-Gross, S. Rossbauer, H. Zhong, S. E. Watkins, T. D. Anthopoulos and M. Heeney, Thiophene fluorination to enhance photovoltaic performance in low band gap donor-acceptor polymers, Chem. Commun., 2012, 48, 11130 RSC.
- P. K. Kandel, L. P. Fernando, P. C. Ackroyd and K. A. Christensen, Incorporating functionalized polyethylene glycol lipids into reprecipitated conjugated polymer nanoparticles for bioconjugation and targeted labeling of cells, Nanoscale, 2011, 3, 1037 RSC.
- E. Kemal, T. F. Abelha, L. Urbano, R. Peters, D. M. Owen, P. Howes, M. Green and L. A. Dailey, Bright, near infrared emitting PLGA–PEG dye-doped CN-PPV nanoparticles for imaging applications, RSC Adv., 2017, 7, 15255 RSC.
- T. F. Abelha, T. W. Phillips, J. H. Bannock, A. M. Nightingale, C. A. Dreiss, E. Kemal, L. Urbano, J. C. deMello, M. Green and L. A. Dailey, Bright conjugated polymer nanoparticles containing a biodegradable shell produced at high yields and with tuneable optical properties by a scalable microfluidic device, Nanoscale, 2017, 9, 2009 RSC.
- Y. Jiang, P. K. Upputuri, C. Xie, Z. Zeng, A. Sharma, X. Zhen, J. Li, J. Huang, M. Pramanik and K. Pu, Metabolizable Semiconducting Polymer Nanoparticles for Second Near-Infrared Photoacoustic Imaging, Adv. Mater., 2019, 1808166 CrossRef PubMed.
- C. V. Rohatgi, T. Harada, E. F. Need, M. Krasowska, D. A. Beattie, G. D. Dickenson, T. A. Smithand and T. W. Kee, Low-Bandgap Conjugated Polymer Dots for Near-Infrared Fluorescence Imaging, ACS Appl. Nano Mater., 2018, 1, 4801 CrossRef CAS.
- S. N. Clafton, D. A. Beattie, A. Mierczynska-Vasilev, R. G. Acres, A. C. Morgan and T. W. Kee, Chemical defects in the highly fluorescent conjugated polymer dots, Langmuir, 2010, 26, 17785 CrossRef CAS PubMed.
- H. Fissan, S. Ristig, H. Kaminski, C. Asbach and M. Epple, Comparison of different characterization methods for nanoparticle dispersions before and after aerosolization, Anal. Methods, 2014, 6, 7324 RSC.
- T. F. Abelha, P. R. Neumann, J. Holthof, C. A. Dreiss, C. Alexander, M. Green and L. A. Dailey, Low molecular weight PEG–PLGA polymers provide a superior matrix for conjugated polymer nanoparticles in terms of physicochemical properties, biocompatibility and optical/photoacoustic performance, J. Mater. Chem. B, 2019, 7, 5115 RSC.
- F. Ye, C. Wu, Y. Jin, M. Wang, Y. H. Chan, J. Yu, W. Sun, S. Hayden and D. T. Chiu, A Compact and Highly Fluorescent Orange-emitting Polymer Dot for Specific Subcellular Imaging, Chem. Commun., 2012, 48, 1778 RSC.
- E. Blanco, H. Shen and M. Ferrari, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol., 2015, 33, 941 CrossRef CAS PubMed.
- P. C. Tapping, S. N. Clafton, K. N. Schwarz, T. W. Kee and D. M. Huang, Molecular Level Details of Morphology-Dependent Exciton Migration in Poly(3-hexylthiophene) Nanostructures, J. Phys. Chem. C, 2015, 119, 7047 CrossRef CAS.
- R. Weissleder, A clearer vision for in vivo imaging, Nat. Biotechnol., 2001, 19, 316 CrossRef CAS PubMed.
- C. Wu, C. Szymanski and J. McNeill, Preparation and encapsulation of highly fluorescent conjugated polymer nanoparticles, Langmuir, 2006, 22, 2956 CrossRef CAS PubMed.
- G. Padmanaban and S. Ramakrishnan, Conjugation length control in soluble poly[2-methoxy-5-((2′-ethylhexyl)oxy)-1,4-phenylenevinylene] (MEHPPV): synthesis, optical properties, and energy transfer, J. Am. Chem. Soc., 2000, 122, 2244 CrossRef CAS.
| This journal is © the Partner Organisations 2020 |