
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombined experimental and theoretical study of long-range H–F interactions in α-fluoro amides†
Elena
Cosimi
 ,
Nils
Trapp
,
Marc-Olivier
Ebert
* and
Helma
Wennemers
*
,
Nils
Trapp
,
Marc-Olivier
Ebert
* and
Helma
Wennemers
*
Laboratory of Organic Chemistry, D-CHAB, ETH Zurich, Vladimir-Prelog-Weg 3, CH-8093 Zurich, Switzerland. E-mail: Marc-Olivier.Ebert@org.chem.ethz.ch; Helma.Wennemers@org.chem.ethz.ch
First published on 16th January 2019
Abstract
A combined experimental and computational approach provided insight into the nature and conformational dependence of long-range 4JHF couplings in α-fluoro amides. The dependence of 4JHF on substituents and the solvent was investigated. H–F coupling constants determined by NMR spectroscopy are in agreement with DFT calculations. NBO analysis revealed that a favourable nF→σNH* interaction correlates with the magnitude of 4JHF.
The introduction of fluorine into organic compounds is a valuable strategy to modify their physico-chemical properties and improve, e.g., the pharmacokinetic profile of drugs.1–5 One example are α-fluorinated amides that have been used to tune ligand–protein interactions6–9 and modulate the activity of small molecules with, for example, anticonvulsant,10 antibacterial,11 and cholesterol-lowering properties.12 α-Fluorination has also enabled control over the backbone conformation of β-amino acids and β-peptides.13–19 Previous studies showed that α-fluoropropionamides adopt mainly an antiperiplanar (ap) conformation, and to a lesser degree a gauche conformation (Fig. 1).20–22 The driving force for the preferred ap conformation is the opposing dipoles of the vicinal C–F and C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.‡22–24 Jaun and Seebach observed in α-fluoro β-peptides a conformation dependent NH–F (4JHF) coupling between fluorine and the adjacent amide proton.17–19 This long-range coupling is reminiscent of hydrogen bond-mediated fluorine couplings,§25,26 as e.g. in ortho fluorinated anilides or benzamides.27,28 Its presence suggests that the ap conformation is not only driven by electrostatic dipole–dipole interactions but might be supported by an additional weak interaction between fluorine and the amide group.¶29–31
O bonds.‡22–24 Jaun and Seebach observed in α-fluoro β-peptides a conformation dependent NH–F (4JHF) coupling between fluorine and the adjacent amide proton.17–19 This long-range coupling is reminiscent of hydrogen bond-mediated fluorine couplings,§25,26 as e.g. in ortho fluorinated anilides or benzamides.27,28 Its presence suggests that the ap conformation is not only driven by electrostatic dipole–dipole interactions but might be supported by an additional weak interaction between fluorine and the amide group.¶29–31
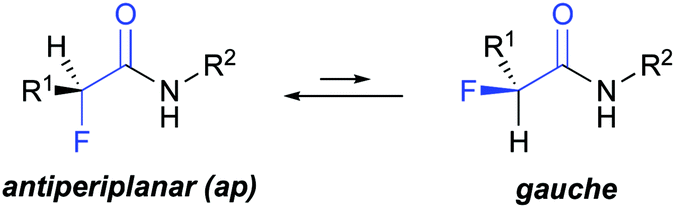 | ||
| Fig. 1 Equilibrium between antiperiplanar (ap) and gauche conformers of α-fluoro amides. | ||
Herein, we probed the nature of the long-range 4JHF coupling constant by studying a series of α-fluoro amides bearing different substituents with computational and experimental tools. We investigated the α-fluoro amides in different solvents by NMR spectroscopy and in the solid state by X-ray crystal structure analysis. We show that the value of 4JHF can be accurately predicted by DFT calculations and that a weak through-space stereoelectronic n→σ* interaction between the F and N–H moieties correlates with the magnitude of the 4JHF coupling constant.
We started by synthesising α-fluoro amides 1–5 as model compounds for our experimental studies (Scheme 1). These propionamide and malonamide derivatives were readily accessed from the appropriate α-fluoro thioester by reaction with methyl amine, benzyl amine, and aniline. Notably, the thioester allowed even for effective amidation with the poor nucleophile aniline.32
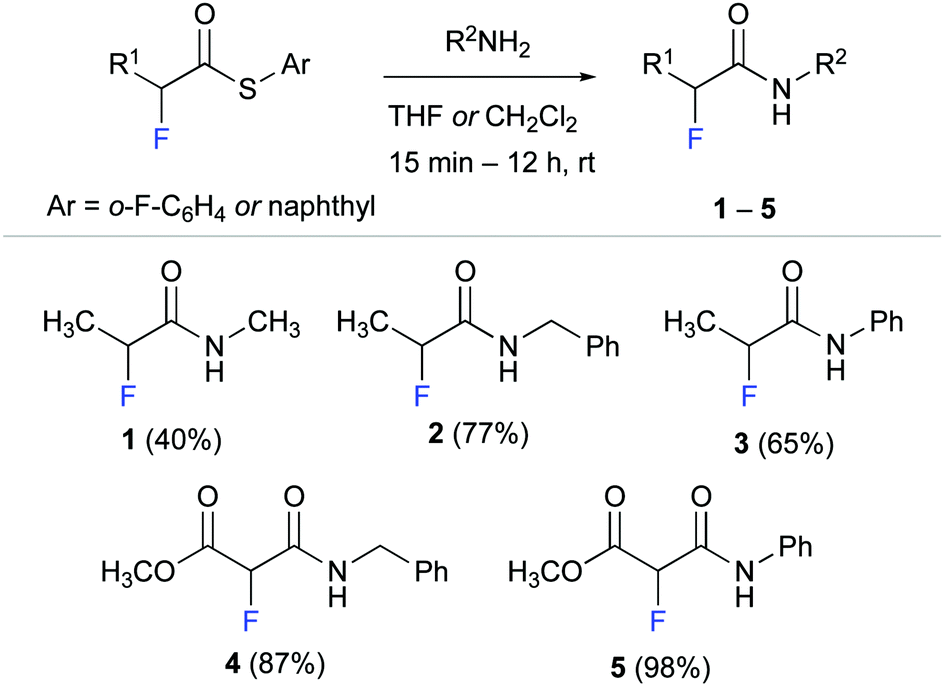 | ||
| Scheme 1 Synthesis of α fluoro amides 1–5. | ||
α-Fluoro amide 1 with methyl substituents at C(α) and at NH served as reference for our studies. Methyl ester derivative 4 was designed to probe the effect of an electron withdrawing group (EWG) at C(α). We envisioned that an EWG would lower the electron density at the fluorine atom and thereby weaken the putative NH–F interaction. Such a weakened interaction would be reflected in a smaller long-range 4JHF coupling. Conversely, the phenyl moieties of anilides 3 and 5 were expected to increase the acidity of the N–H group33 and thus strengthen the F⋯H–N interaction. Amide 2 with a benzyl group at the NH was used to explore the effect of an aromatic moiety that is not directly attached to the amide moiety.
Before starting the spectroscopic analysis, we performed DFT calculations with representative α-fluoro amides at the B3LYP/AUG-cc-pVTZ level of theory using the program Gaussian. For α-fluoro amide 1 restrained geometry optimizations (in steps of 15°) in vacuo were performed for a series of F–C–CO dihedral angles (Θ). The ap conformer was found to be energetically favoured by 5 kcal mol−1 over the gauche (Θ = −60°) conformer (Fig. 2, black curve). This is ∼1 kcal mol−1 less than reported previously by Banks et al. and Tormena et al. who used the same functional but smaller basis sets.21,22 In agreement with these reports, we also found that in vacuo the gauche conformer is not a local minimum on the potential energy surface. In contrast, the gauche conformer becomes a local minimum when the calculations were carried out with implicit solvation.21 Unrestrained geometry optimization converged to a local minimum of Θ = −55.3° with the polarizable continuum model (PCM) parametrized for CHCl3 and Θ = −46.3° with PCM parametrized for DMSO. Frequency calculations in implicit solvent for both ap and gauche conformations of 1 led to thermally corrected free energy differences (ΔGap-g) of 2.8 kcal mol−1 in CHCl3 and 1.8 kcal mol−1 in DMSO. This computational finding suggests that the ap conformer is favoured over the gauche conformer regardless of the solvent.
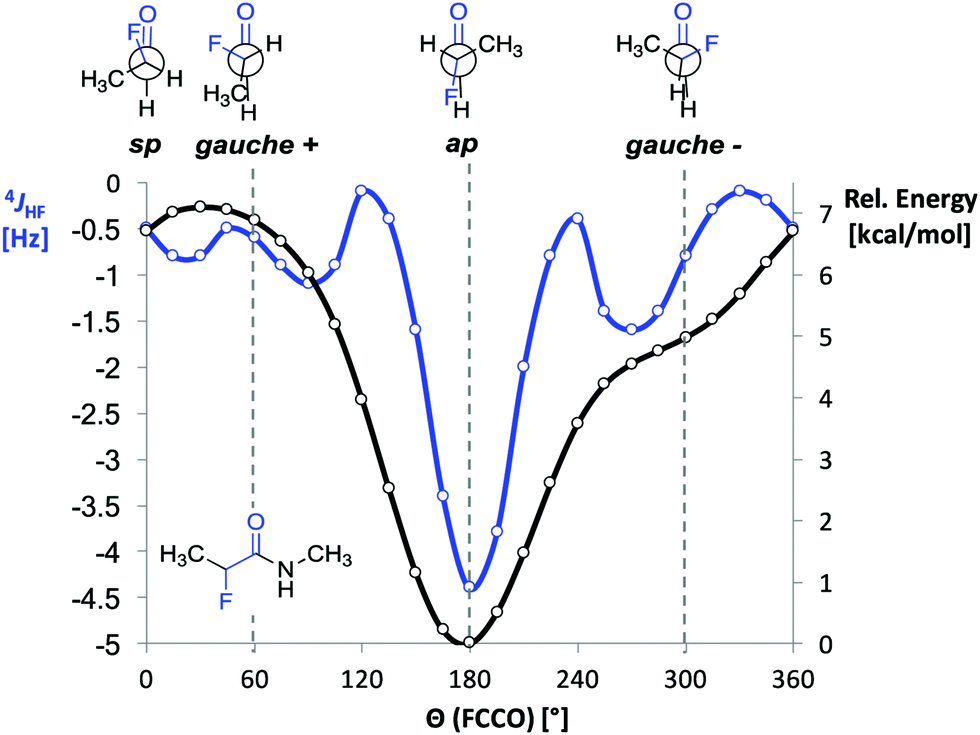 | ||
| Fig. 2 DFT calculated 4JHF coupling constant and relative B3LYP/AUG-cc-pVTZ energies as functions of the dihedral angle F–C–CO (Θ) in 1. | ||
Next, we computed the dependence of the 4JHF coupling constant on the dihedral angle Θ for α-fluoro amide 1 (Fig. 2, blue curve).34 The calculated 4JHF coupling curve shows a sharp minimum (strongest coupling) at the ap conformation where NH and F are closest in space (4JHF = −4.3 Hz, Θ = 180°). For the gauche conformation the coupling strength is significantly attenuated (4JHF = −0.8 Hz, Θ = 300°). The calculated dependence of 4JHF on Θ is in agreement with experimental observations by Jaun and Seebach for 2-fluoro-β3-alanine in peptides.17 For Θ close to 180° the absolute value of 4JHF was reported to be 3.7 Hz whereas for Θ close to 90° no resolved coupling could be detected (|4JHF| < 0.5).
We also performed unrestrained geometry optimization in vacuo of the ap conformations for 1, 3, and the N-methyl analogue of 4 using the same basis set and functional. The resulting minimum energy conformations were then used to predict the values of 4JHF. These calculated 4JHF coupling constants of 1, 3, and 4 differ significantly from each other. The absolute value for anilide 3 is greater (4JHF = −7.0 Hz) and that of 4 with an electron withdrawing ester group next to the C–F bond is smaller (4JHF = −3.3 Hz) compared to the one of reference compound 1 (4JHF = −4.3 Hz, Table 1, entry 1).
| 4 J HF [Hz] | ||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | 1 | 2 | 3 | 4 | 5 |
| a Determined at 50 mM. b The calculations were performed on the N-methyl analogue of 4. | ||||||
| 1 | Calc. (ap) | −4.3 | n.d. | −7.0 | −3.3b | n.d. |
| 2 | CDCl3 | −4.4 | −4.6 | −6.8 | −3.3 | −4.5 |
| 3 | CD2Cl2 | −4.3 | −4.3 | −6.5 | −2.7 | −4.2 |
| 4 | d6-Acetone | −2.9 | −2.8 | −3.9 | −1.5 | −1.8 |
| 5 | CD3OH | −2.4 | >−2 | n.d. | >−2 | n.d. |
| 6 | d6-DMSO | −1.7 | −1.7 | −1.8 | −0.7 | >−1 |
Next, we determined absolute values of the 4JHF coupling constants experimentally by recording 1H and 19F NMR spectra and started by using CDCl3 as a solvent. As suggested by the calculations, the sign of 4JHF was found to be negative by analysis of the cross-peak fine-structure in a constant-time 13C-HMBC spectrum.|| Reassuringly, the experimental 4JHF values in CDCl3 do not deviate by more than 0.2 Hz from the predicted values (Table 1, entries 1 and 2). Such a good agreement is remarkable, in particular in light of the well-known difficulties of DFT calculations to predict coupling constants involving fluorine.35,36
Since amide moieties can form intermolecular H-bonds that could disturb the conformational properties of α-fluoro amides, we recorded NMR spectra of 1–5 at different concentrations in CD2Cl2 (0.8–200 mM). These studies showed that the experimental values of 4JHF hardly change upon increasing the concentration.|| In contrast, the NH chemical shift has a significant concentration dependence, which suggests that intermolecular interactions between the amides occur (Kd ∼ 0.1–1 M).|| This finding shows that possible intermolecular interactions between the amides do not affect the 4JHF coupling constant to a significant extent.
Next, we explored the effect of substituents at the α-fluoro amide on the value of the 4JHF coupling. The NMR spectra of anilides 3 and 5 in CDCl3 revealed stronger 4JHF couplings (−6.8 Hz and −4.5 Hz) compared to those of the analogous benzyl amides 2 and 4 (−4.6 Hz and −3.3 Hz). This observation is in agreement with the expected higher N–H acidity of 3 and 5 in comparison to 2 and 4, respectively, and indicates a stronger F⋯H–N interaction. Conversely, the absolute 4JHF values are lower in case of 4 and 5 that bear ester moieties compared to 2 and 3 with methyl groups at Cα. This indicates that an electron withdrawing group (ester) at Cα lowers the electron density at fluorine and weakens the F⋯H–N interaction. Thus, in accordance with our expectations, changes in molecular structure designed to modulate the strength of the putative F⋯H–N hydrogen bond are mirrored in the measured values of the 4JHF coupling constant.
We then investigated the influence of the solvent on the value of the 4JHF coupling constant by recording 1H and 19F NMR spectra of 1–5 in CD2Cl2, d6-acetone, CD3OH, and d6-DMSO. In the more polar solvents the strength of the 4JHF coupling in 1–5 decreases (Table 1, entries 4–6). Considering the significant differences in the calculated 4JHF coupling constants of gauche and ap conformations (Fig. 2), these data show that the ap conformer becomes less populated in polar solvents, which is expected based on the higher dipole moment of the gauche compared to the ap conformation.
Compounds 2, 3, and 4 crystallized and allowed for their conformational analysis by X-ray crystallography (Fig. 3).22 α-Fluoro amides 2 and 3 are in ap conformations in the crystalline state. Their interatomic NH–F distances of approximately 2.2 Å are significantly shorter than the sum of the van der Waals radii (2.5–2.7 Å). The F⋯H–N angles are 102° and 105° for 2 and 3, respectively. Both, the inter-atom distances and the bond angles, match the criteria for H–F hydrogen bonds as defined by Dunitz and Taylor (F–H distance < 2.3 Å, F⋯H–X angle > 90°).37 In contrast, the α-fluoro amido moiety of 4 adopts a near gauche conformation in the solid state (Θ = 42°). The crystal packing of 2, 3, and 4 reveals that the amide groups are involved in intermolecular chain-type N–H–O interactions. In 4, these intermolecular hydrogen bonds in the solid state override the favourable ap orientation of the F–C–CO moiety observed in solution.38
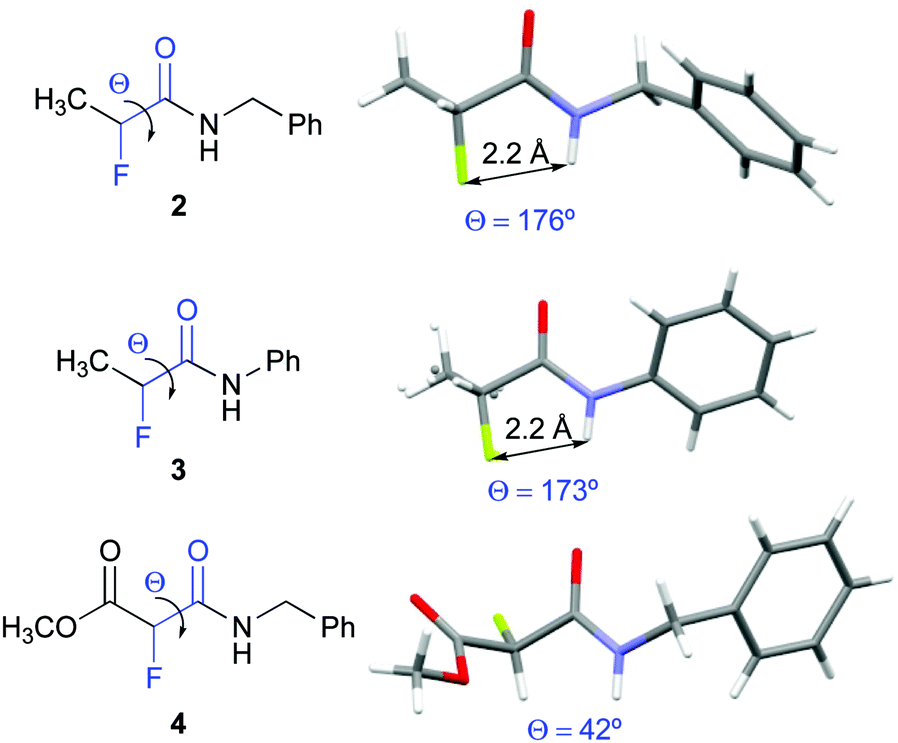 | ||
| Fig. 3 X-ray crystal structures of 2–4. | ||
Finally, we investigated the origin of the observed 4JHF long-range coupling constant by NBO calculations.39 Second order perturbation analysis revealed the presence of a favourable electron donation from the fluorine lone pair (nF) to the antibonding N–H orbital (σNH*) (Fig. 4). This finding suggests that this weak through-space interaction gives rise to the observed 4JHF coupling. The calculations predict a contribution of this nF→σNH* interaction of 0.8 kcal mol−1 for 1, 1.3 kcal mol−1 for 3, and 0.6 kcal mol−1 for 4 to the stability of the ap conformer of these α-fluoro amides.** Thus, the strength of this interaction increases progressively from the N-benzyl amides 4 and 1 to the N-phenyl amide 3, in agreement with the observed increase of the 4JHF coupling constants.
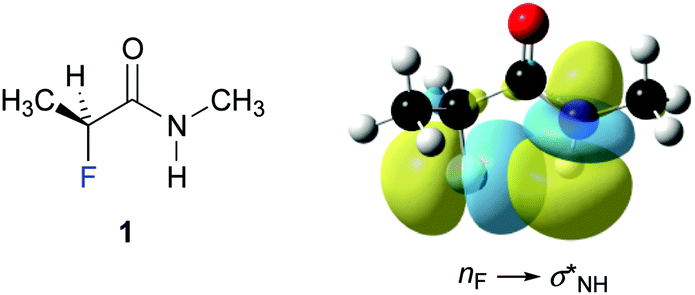 | ||
| Fig. 4 Graphical representation of the nF–σNH* interaction in 1. (iso value = 0.02 e Å−3).†† | ||
In conclusion, our results show that the 4JHF coupling in α-fluoro amides depends on the substituents and the solvent. Calculated 4JHF couplings for the ap conformations of a series of α-fluoro amides match the experimental values. The 4JHF coupling is strongest in the ap conformation when F and HN are closest in space. In this conformation the strengths of the observed 4JHF couplings and predicted nF→σNH* interactions correlate. This suggests that the 4JHF coupling is a genuine through-space coupling concomitant with a F⋯H–N hydrogen bond.
We are grateful to the Swiss National Science Foundation (grant 200020_169423) and the Laboratory of Organic Chemistry of ETH Zurich for financial support. We thank Rainer Frankenstein and Michael Solar for recording NMR spectra and X-ray crystal structures. This publication is dedicated to Prof. Bernhard Jaun on the occasion of his 70th birthday.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- A. A. Berger, J. S. Voller, N. Budisa and B. Koksch, Acc. Chem. Res., 2017, 50, 2093–2103 CrossRef CAS PubMed.
- C. Thiehoff, Y. P. Rey and R. Gilmour, Isr. J. Chem., 2017, 57, 92–100 CrossRef CAS.
- L. Unione, M. Alcala, B. Echeverria, S. Serna, A. Arda, A. Franconetti, F. J. Canada, T. Diercks, N. Reichardt and J. Jimenez-Barbero, Chem. – Eur. J., 2017, 23, 3957–3965 CrossRef CAS PubMed.
- M. Ettaoussi, A. Sabaouni, M. Rami, J. A. Boutin, P. Delagrange, P. Renard, M. Spedding, D. H. Caignard, P. Berthelot and S. Yous, Eur. J. Med. Chem., 2012, 49, 310–323 CrossRef CAS PubMed.
- D. P. Zlotos, N. M. Riad, M. B. Osman, B. R. Dodda and P. A. Witt-Enderby, MedChemComm, 2015, 6, 1340–1344 RSC.
- M. Winkler, T. Moraux, H. A. Khairy, R. H. Scott, A. M. Slawin and D. O'Hagan, ChemBioChem, 2009, 10, 823–828 CrossRef CAS PubMed.
- N. Pessah, M. Bialer, B. Wlodarczyk, R. H. Finnell and B. Yagen, J. Med. Chem., 2009, 52, 2233–2242 CrossRef CAS PubMed.
- G. O. Danelon and O. A. Mascaretti, J. Fluorine Chem., 1992, 56, 109–140 CrossRef CAS.
- J. W. Clader, D. A. Burnett, M. A. Caplen, M. S. Domalski, S. Dugar, W. Vaccaro, R. Sher, M. E. Browne, H. Zhao, R. E. Burrier, B. Salisbury and H. R. Davis, Jr., J. Med. Chem., 1996, 39, 3684–3693 CrossRef CAS PubMed.
- M. Salwiczek, E. K. Nyakatura, U. I. Gerling, S. Ye and B. Koksch, Chem. Soc. Rev., 2012, 41, 2135–2171 RSC.
- T. L. March, M. R. Johnston, P. J. Duggan and J. Gardiner, Chem. Biodiversity, 2012, 9, 2410–2441 CrossRef CAS PubMed.
- V. Peddie, R. J. Butcher, W. T. Robinson, M. C. Wilce, D. A. Traore and A. D. Abell, Chem. – Eur. J., 2012, 18, 6655–6662 CrossRef CAS PubMed.
- E. Cosimi, O. D. Engl, J. Saadi, M. O. Ebert and H. Wennemers, Angew. Chem., Int. Ed., 2016, 55, 13127–13131 CrossRef CAS PubMed.
- B. Jaun, D. Seebach and R. I. Mathad, Helv. Chim. Acta, 2011, 94, 355–361 CrossRef CAS.
- R. I. Mathad, F. Gessier, D. Seebach and B. Jaun, Helv. Chim. Acta, 2005, 88, 266–280 CrossRef CAS.
- R. I. Mathad, B. Jaun, O. Flögel, J. Gardiner, M. Löweneck, J. D. C. Codée, P. H. Seeberger, D. Seebach, M. K. Edmonds, F. H. M. Graichen and A. D. Abell, Helv. Chim. Acta, 2007, 90, 2251–2273 CrossRef CAS.
- C. R. S. Briggs, D. O'Hagan, J. A. K. Howard and D. S. Yufit, J. Fluorine Chem., 2003, 119, 9–13 CrossRef CAS.
- C. F. Tormena, N. S. Amadeu, R. Ritter and R. J. Abraham, Perkin Trans. 2, 2002, 773–778 RSC.
- J. W. Banks, A. S. Batsanov, J. A. K. Howard, D. O'Hagan, H. S. Rzepa and S. Martin-Santamaria, J. Chem. Soc., Perkin Trans. 2, 1999, 2409–2411 RSC.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- D. C. Lankin, N. S. Chandrakumar, S. N. Rao, D. P. Spangler and J. P. Snyder, J. Am. Chem. Soc., 1993, 115, 3356–3357 CrossRef CAS.
- (a) P. A. Champagne, J. Desroches and J. F. Paquin, Synthesis, 2015, 306–322 CAS; (b) J. C. Hierso, Chem. Rev., 2014, 114, 48384867 CrossRef PubMed; (c) H. J. Schneider, Chem. Sci., 2012, 3, 1381–1394 RSC.
- (a) G. T. Giuffredi, V. Gouverneur and B. Bernet, Angew. Chem., Int. Ed., 2013, 52, 10524–10528 CrossRef CAS PubMed; (b) B. Linclau, F. Peron, E. Bogdan, N. Wells, Z. Wang, G. Compain, C. Q. Fontenelle, N. Galland, J. Y. Le Questel and J. Graton, Chem. – Eur. J., 2015, 21, 17808–17816 CrossRef CAS PubMed.
- I. Alkorta, J. Elguero, H. H. Limbach, I. G. Shenderovich and T. Winkler, Magn. Reson. Chem., 2009, 47, 585–592 CrossRef CAS PubMed.
- C. Li, S. F. Ren, J. L. Hou, H. P. Yi, S. Z. Zhu, X. K. Jiang and Z. T. Li, Angew. Chem., Int. Ed., 2005, 44, 5725–5729 CrossRef CAS PubMed.
- S. J. Grabowski, Chem. Rev., 2011, 111, 2597–2625 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CAS.
- R. W. Newberry and R. T. Raines, Nat. Chem. Biol., 2016, 12, 1084–1088 CrossRef CAS PubMed.
- T. Kanzian, T. A. Nigst, A. Maier, S. Pichl and H. Mayr, Eur. J. Org. Chem., 2009, 6379–6385 CrossRef CAS.
- F. G. Bordwell and G. Z. Ji, J. Am. Chem. Soc., 1991, 113, 8398 CrossRef CAS.
- W. Deng, J. R. Cheeseman and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 1028–1037 CrossRef CAS PubMed.
- T. Helgaker, M. Jaszuński and M. Pecul, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 53, 249–268 CrossRef CAS.
- J. Vaara, Phys. Chem. Chem. Phys., 2007, 9, 5399–5418 RSC.
- J. D. Dunitz and R. Taylor, Chem. – Eur. J., 1997, 3, 89–98 CrossRef CAS.
- C. R. Jones, P. K. Baruah, A. L. Thompson, S. Scheiner and M. D. Smith, J. Am. Chem. Soc., 2012, 134, 12064–12071 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.9, Theoretical Chemistry Institute, University of Wisconsin, Madison WI, 2012 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on the NMR spectroscopic analyses, DFT calculations, X-ray crystallography, and analytical data of 1–5. CCDC 1884973–1884976. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc09987a |
| ‡ For a similar charge-dipole effect, see ref. 24. |
| § For related examples on F⋯H–O bonds, see ref. 26. |
| ¶ Such a through space interaction is reminiscent of intraresidual hydrogen bonds within β-sheets formed by peptides, ref. 31. |
| || For details, see the ESI.† |
| ** In the gauche conformation of 1 this stabilizing contribution is absent. |
| †† Note, the relative signs of the n and σ* orbitals have no physical relevance. |
| This journal is © The Royal Society of Chemistry 2019 |