
Concise asymmetric total synthesis of catunaregin†
Hideki
Abe
,
Takuma
Hikichi
,
Kosuke
Emori
,
Toyoharu
Kobayashi
and
Hisanaka
Ito
*
School of Life Sciences, Tokyo University of Pharmacy and Life Sciences, 1432-1 Horinouchi, Hachioji, Tokyo 192-0392, Japan. E-mail: itohisa@toyaku.ac.jp
First published on 28th June 2016
Abstract
The asymmetric total synthesis of catunaregin isolated from the Chinese mangrove is described. The synthesis involves an asymmetric syn-selective aldol reaction and the successive ketalization of a furan diol derivative under acidic conditions. This methodology is very concise and highly stereoselective. The asymmetric total synthesis of the optically pure catunaregin was accomplished in 7 steps from a known methyl ester.
Lignans, a class of natural products, are metabolites constructed from two phenylpropanoic acids by oxidative dimerization.1,2 Catunaregin (1) and epicatunaregin (2) were isolated from the stem bark of Catunaregam spinosa Tirveng, a Chinese mangrove associate, by the Zhang group.3 These natural products 1 and 2 possess an unprecedented norneolignan skeleton, involving an oxygen-bridged furopyran skeleton (Fig. 1). They were initially found to exhibit inhibition against mammary cancer F10 cell lines. Later, it was reported that the novel norneolignan catunaregin (1) inhibited different VEGF-induced angiogenic phenotypes of HUVECs in vitro and vessel formation in transgenic zebrafish.4 The optical rotation value of the natural catunaregin (1) is very low ([α]20D = +1.4 (c 0.8, MeOH)), leading Zhang to surmise that the natural catunaregin is virtually racemic as a result of its biosynthetic pathway, which includes the production of benzofuran norneolignans with no enantioselective reduction. The structural features and biological properties of 1 attracted our attention, and we began a synthetic study of unique norneolignan 1 in the optically pure form. This report describes the asymmetric synthesis of catunaregin (1), which includes a syn-selective aldol reaction and construction of the oxygen-bridged furopyran skeleton by successive ketalization.
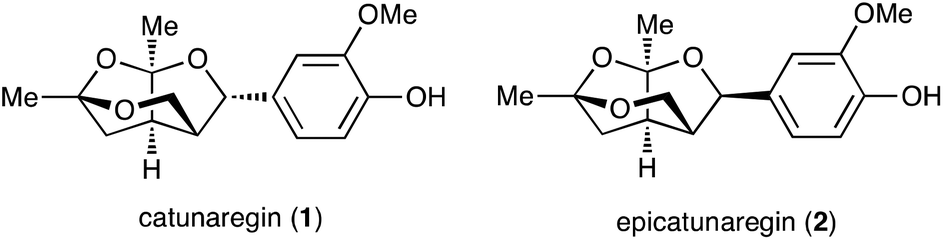 | ||
| Fig. 1 Structures of catunaregin (1) and epicatunaregin (2). | ||
The retrosynthetic analysis of catunaregin (1) is outlined in Scheme 1. The target norneolignan could be obtained by the construction of the tricyclic skeleton by successive ketalization of furanyl diol derivative 3, followed by cleavage of the protecting group of the phenolic hydroxyl group. The ketalization precursor 3 would be synthesized from furan derivative 5 and vanillin derivative 6 in two steps involving the asymmetric syn-selective aldol reaction of 5 with 6, and reduction of the syn-aldol product 4.
 | ||
| Scheme 1 Retrosynthetic analysis of catunaregin (1). | ||
Our investigation started with the synthesis of the donor 11 with a chiral auxiliary for the subsequent syn-selective Evans aldol reaction. Saponification of methyl ester 8, which was prepared from tert-butyl acetoacetate (7) in 3 steps according to the reported procedure,5 with sodium hydroxide gave the carboxylic acid 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 in 99% yield (Scheme 2). Installation of the (S)-4-benzyloxazolidinone group as the chiral auxiliary was achieved by a 2-step operation: transformation of the carboxylic acid to the mixed anhydride, followed by addition of the lithiated oxazolidinone to the resulting mixed anhydride to afford 11 in 93% yield. The syn-selective Evans aldol reaction7 of 11 and O-o-nitrobenzenesulfonyl (Ns)8,9 vanillin derivative 12 with di-n-butylboryl trifluoromethanesulfonate and diisopropyl(ethyl)amine gave the desired Evans syn product 13 exclusively in 98% yield. The absolute stereochemistry at C1 of 13 was determined by the improved Mosher's method10,11 as S.12
6 in 99% yield (Scheme 2). Installation of the (S)-4-benzyloxazolidinone group as the chiral auxiliary was achieved by a 2-step operation: transformation of the carboxylic acid to the mixed anhydride, followed by addition of the lithiated oxazolidinone to the resulting mixed anhydride to afford 11 in 93% yield. The syn-selective Evans aldol reaction7 of 11 and O-o-nitrobenzenesulfonyl (Ns)8,9 vanillin derivative 12 with di-n-butylboryl trifluoromethanesulfonate and diisopropyl(ethyl)amine gave the desired Evans syn product 13 exclusively in 98% yield. The absolute stereochemistry at C1 of 13 was determined by the improved Mosher's method10,11 as S.12
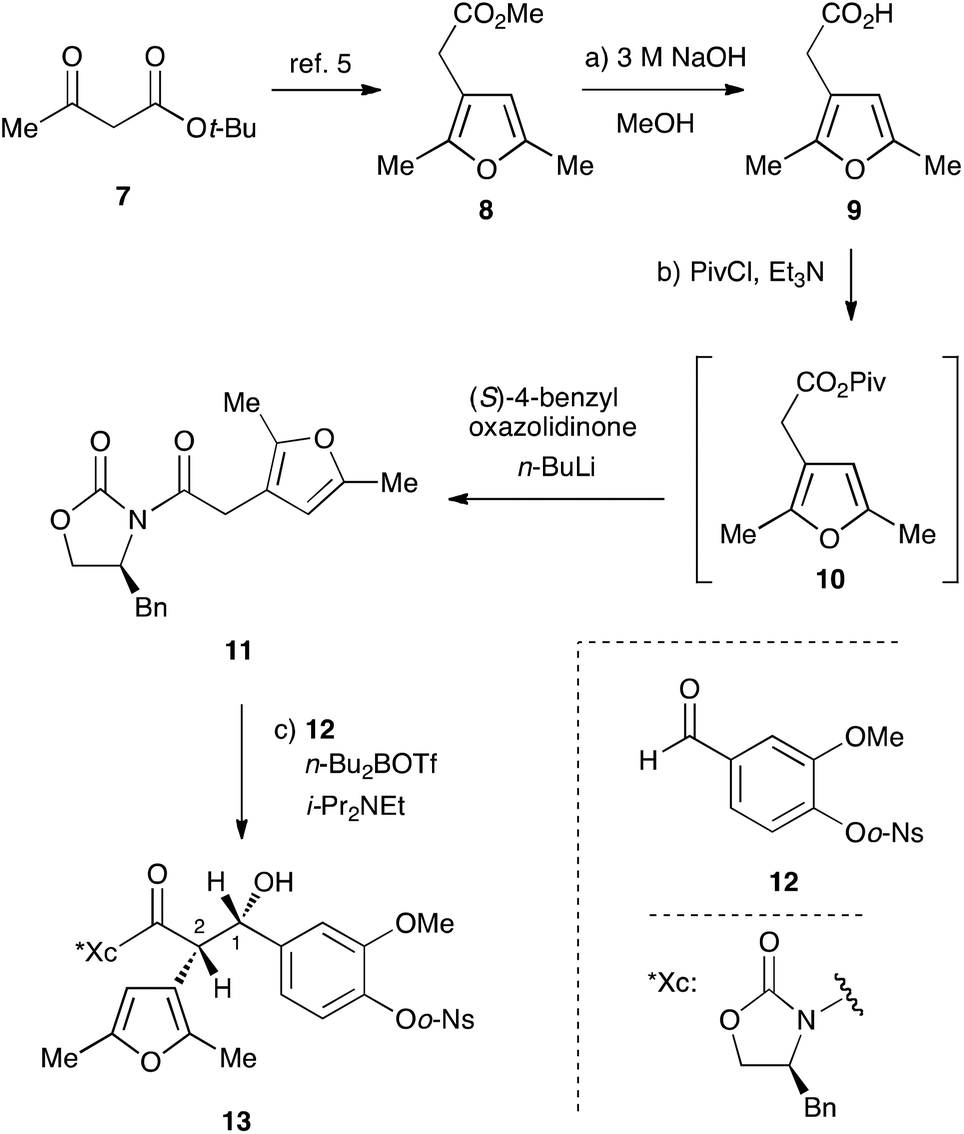 | ||
| Scheme 2 Synthesis of key intermediate 13via syn-selective Evans aldol reaction. Reagents and conditions: (a) 3 M NaOH aq., MeOH, rt, 2 h, 99%; (b) PivCl, Et3N, THF, −78 °C to 0 °C, 1 h, then (S)-4-Bn-oxazolidinone, n-BuLi, THF, −78 °C to 0 °C, 1 h, 93%; (c) n-Bu2BOTf, i-Pr2NEt, 12, CH2Cl2, −78 °C, 2.5 h, 98%, dr >95:5. Piv: pivaloyl; Bn: benzyl; n-Bu2BOTf: di-n-butylboryl trifluoromethanesulfonate. | ||
With the desired Evans syn product 13 in hand, we focused our efforts on the construction of the tricyclic fragment. As shown in Scheme 3, all attempts for the removal of the chiral auxiliary of 13 by reduction with hydride reagents, LiBH4, LiAlH4, and DIBALH, failed with the decomposition of 13. Since we suspected that the naked hydroxyl group at the benzylic position caused a retro aldol reaction as a side reaction, the hydroxyl group was protected with a triethylsilyl group. Protection of the hydroxyl group with the TES group allowed cleavage of the oxazolidinone part. Thus, after treatment of 13 with TESOTf and DBU at −50 °C, hydride reduction of the resulting TES ether with LiBH4 under mild conditions gave the primary alcohol 15, the precursor to successive ketalization, in 69% yield over 2 steps. The key successive ketalization of 15 was then carried out. After many reaction conditions (acids, solvents, and temperature) were attempted, treatment of 15 with concentrated sulfuric acid in THF at room temperature for 8 h gave the best result to afford the tricyclic compound 16 in 62% yield along with a small amount of diol 14 (4%). Construction of the tricyclic skeleton by this key reaction was achieved without generating the benzyl cation due to the electron withdrawing effect of the o-Ns group.
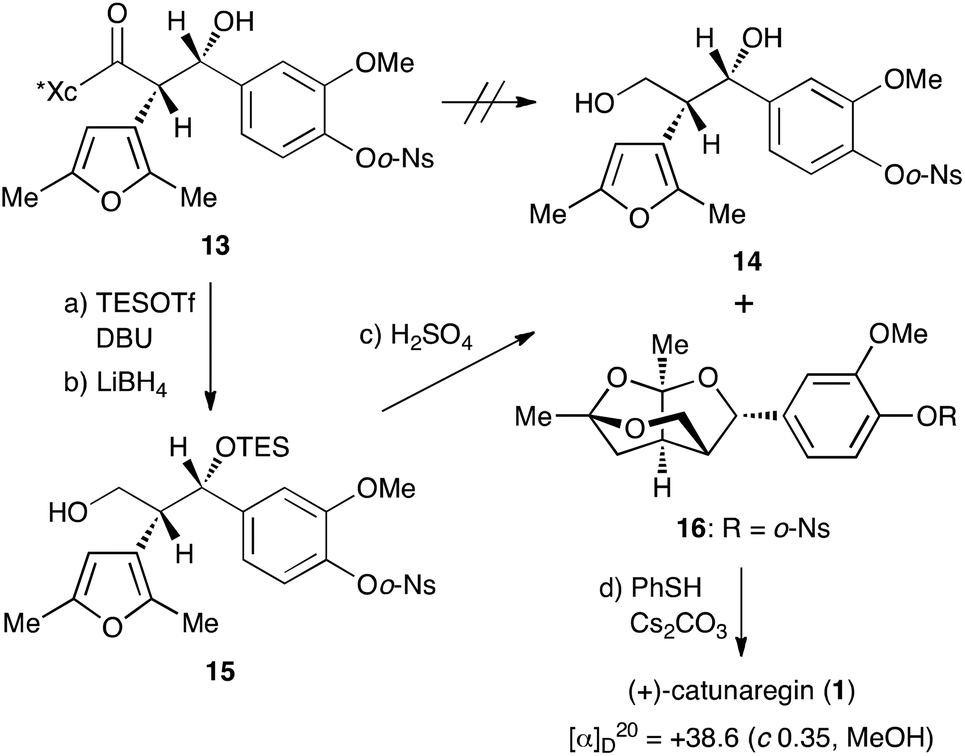 | ||
| Scheme 3 Total synthesis of catunaregin (1). Reagents and conditions: (a) TESOTf, DBU, CH2Cl2, −50 °C, 20 h, 90%; (b) LiBH4, THF, 50 °C, 26 h, 77%; (c) conc. H2SO4–THF (1:5), rt, 8 h, 62% for 16, and 4% for 14; (d) PhSH, Cs2CO3, MeCN, rt, 1.5 h, quant. TESOTf: triethylsilyl trifluoromethanesulfonate; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene. | ||
Finally, cleavage of the o-Ns group of 16 with thiophenol and cesium carbonate8,9 gave the target molecule in quantitative yield. The spectral data of the synthetic sample 1 were identical to those of the reported natural catunaregin (1). The optical rotation of the synthetic sample was [α]25D +38.6 (c 1.00, MeOH). This result confirms that the isolated natural catunaregin is in the racemic form as speculated by Zhang.
Conclusions
The first asymmetric total synthesis of catunaregin (1) was accomplished. The key steps of this synthesis involved syn-selective Evans aldol reaction and successive ketalization to construct the oxygen-bridged tricyclic furopyran framework. This result established that the naturally occurring catunaregin was isolated in the racemic form. Studies of biological activity of the optically pure catunaregin are currently underway in our laboratory.Acknowledgements
This work was supported by JSPS KAKENHI (Grant No. 26460016) and the Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- J.-Y. Pan, S.-L. Chen, M.-H. Yang, J. Wu, J. Sinkkonen and K. Zou, Nat. Prod. Rep., 2009, 26, 1251 RSC.
- M. Saleem, H. J. Kim, M. S. Ali and Y. S. Lee, Nat. Prod. Rep., 2005, 22, 696 RSC.
- G.-C. Gao, X.-M. Luo, X.-Y. Wei, S.-H. Qi, H. Yin, Z.-H. Xiao and S. Zhang, Helv. Chim. Acta, 2010, 93, 339 CrossRef CAS.
- J.-X. Liu, M.-Q. Luo, M. Xia, Q. Wu, S.-M. Long, Y. Hu, G.-C. Gao, X.-L. Yao, M. He, H. Su, X.-M. Luo and S.-Z. Yao, Mar. Drugs, 2014, 12, 2790 CrossRef PubMed.
- F. Stauffer and R. Neier, Org. Lett., 2000, 2, 3535 CrossRef CAS PubMed.
- D. Mackay, E. G. Neeland and N. J. Taylor, J. Org. Chem., 1986, 51, 2351 CrossRef CAS.
- D. A. Evans, J. V. Nelson, E. Vogel and T. R. Taber, J. Am. Chem. Soc., 1981, 103, 3099 CrossRef CAS.
- T. Kan and T. Fukuyama, Chem. Commun., 2004, 353 RSC.
- T. Kan and T. Fukuyama, J. Synth. Org. Chem., Jpn., 2001, 59, 779 CrossRef CAS.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092 CrossRef CAS.
- J. M. Seco, E. Quiñoá and R. Riguera, Chem. Rev., 2004, 104, 17 CrossRef CAS.
- The detailed experimental results are described in the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00213g |
| This journal is © the Partner Organisations 2016 |