
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Convenient synthesis of pentafluoroethyl thioethers via catalytic Sandmeyer reaction with a stable fluoroalkylthiolation reagent†
C.
Matheis‡
,
B.
Bayarmagnai‡
,
K.
Jouvin
and
L. J.
Goossen
*
FB Chemie-Organische Chemie, TU Kaiserslautern, Erwin-Schrödinger-Str. Geb. 54, D-67663 Kaiserslautern, Germany. E-mail: goossen@chemie.uni-kl.de; Fax: +49 631 205 3921
First published on 6th June 2016
Abstract
Aromatic and heteroaromatic diazonium salts were smoothly converted into the corresponding pentafluoroethyl thioethers by reaction with Me4NSC2F5 in the presence of catalytic amounts of elemental copper. This Sandmeyer-type reaction proceeds at room temperature under mild conditions and is applicable to a wide range of functionalised molecules. It enables the late-stage introduction of pentafluoroethylthio groups, a promising but largely unexplored substituent, into bioactive molecules.
Fluorine-containing groups are of exceptional importance in modern bioactive molecules. Approximately 40% of currently marketed agrochemicals and 25% of pharmaceuticals contain fluorine atoms.1 The systematic introduction and screening of fluorinated residues has become a standard procedure in drug discovery. Thus, methods for the late-stage introduction of fluorinated substituents into functionalised molecules are highly sought-after. In the past decade, various powerful fluoroalkylation methods have been developed.2 The attention has recently shifted towards fluoroalkyl thioethers, since the SCF3 group induces even higher lipophilicity (Hansch constant 1.44 for SCF3vs. 0.88 for CF3) and membrane permeability.3
Contemporary trifluoromethylthiolation reactions of arenes are based on electrophilic,4 nucleophilic,5 radical,6 or oxidative processes,7 usually starting from arylboronic acids or aryl halides.
Our contribution to the field of fluoroalkyl(thiol)ations has been the development of several Sandmeyer-type processes.8 We have demonstrated that a Sandmeyer-thiocyanation followed by a Langlois-type nucleophilic CN/CF3- or CF2H-exchange allows the convenient synthesis of fluoroalkylthioethers.8f,9 For laboratory-scale applications, the use of preformed reagents such as (bpy)CuSCF3,10 AgSCF3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5a and Me4NSCF3 are more convenient. The bench-stable reagent Me4NSCF3 was first synthesised by Roeschenthaler and Yagupolskii11 and has successfully been employed in trifluoromethylthiolations of vinyl iodides,12 boronic acids,7d aryl halides,13 aryl triflates,14 and aryl C–H bonds15 catalysed by Cu, Ni, or Pd complexes.
5a and Me4NSCF3 are more convenient. The bench-stable reagent Me4NSCF3 was first synthesised by Roeschenthaler and Yagupolskii11 and has successfully been employed in trifluoromethylthiolations of vinyl iodides,12 boronic acids,7d aryl halides,13 aryl triflates,14 and aryl C–H bonds15 catalysed by Cu, Ni, or Pd complexes.
In medicinal chemistry, C2F5 derivatives have repeatedly been found to exhibit properties that are superior to those of their CF3 counterparts. Whereas several methods have been reported for the introduction of pentafluoroethyl groups, there are only few reports on the corresponding pentafluoroethylthio compounds.16 Pentafluoroethyl thioarenes cannot be prepared by classical halogen/fluorine exchange reactions, e.g. Swarts-type processes. Traditional syntheses of SC2F5 moieties are based on the reaction of C2F5 radicals or carbanions with disulfides or thiols.17 However, these methods suffer from harsh reaction conditions and limited availability of sulfur-containing substrates.
Modern methods suitable for the late-stage introduction of SC2F5 groups include the Friedel–Crafts-type reaction of electron-rich arenes with a pentafluoroethyl sulfenamide reagent described by Billard et al.18 and the electrophilic perfluoroalkylthiolation of indoles with perfluoroalkyl sulfinate salts in the presence of stoichiometric copper chloride reported by Zhang et al.19 However, these methods are limited to electron-rich arenes and indoles. A generally applicable, regiospecific method for the introduction of SC2F5 groups within a single step, based on widely available substrates and an inexpensive fluoroalkylation reagent, would be highly desirable.
We approached this challenge by investigating Sandmeyer-type pentafluoroethylthiolations (Scheme 1). Me4NSC2F5 appeared to be the reagent of choice, because according to a patent by Roeschenthaler, it is easily accessible from tetramethylammonium fluoride, elemental sulfur and TMSC2F5.11a,20
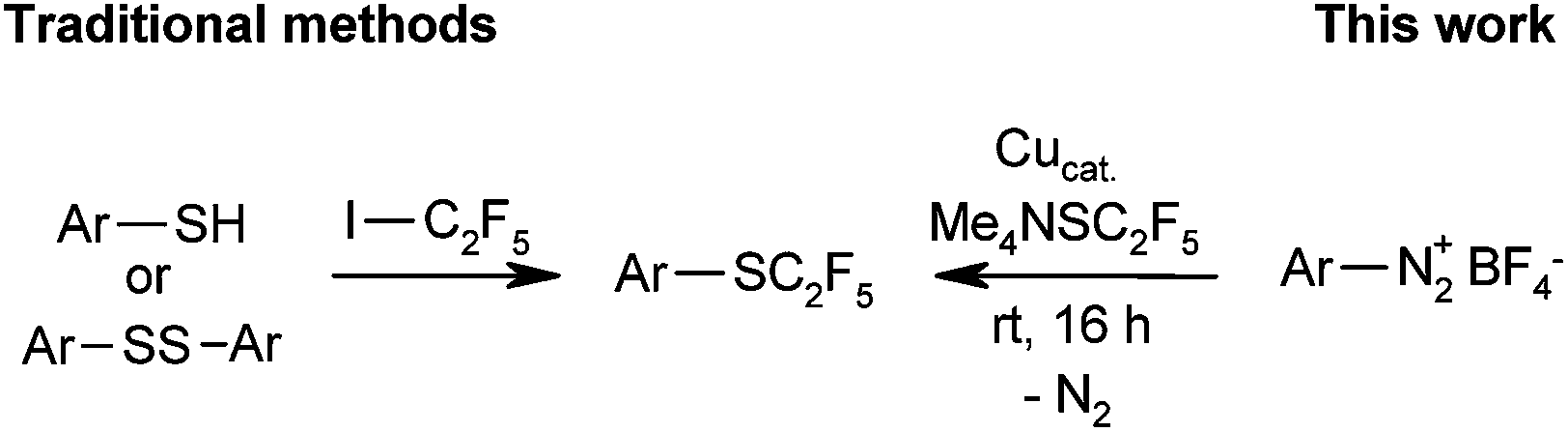 | ||
| Scheme 1 Syntheses of pentafluoroethyl thioethers. | ||
In order to probe the viability of our approach, we treated 4-methoxybenzenediazonium tetrafluoroborate with Me4NSC2F5 in the presence of 10 mol% CuSCN in acetonitrile at room temperature, conditions previously optimised for Sandmeyer trifluoromethylthiolations.8e The pentafluoroethyl thioether was indeed observed, albeit in unsatisfactory yield. The main products were 4-methoxyphenyl thiocyanate and the protodediazotisation product anisole (Table 1, entry 1). It soon became clear that C2F5S− is substantially less nucleophilic than SCF3−, so that pentafluoroethylthiolation takes place only in reaction media free of other nucleophiles. Thus, most counter-ions of copper(I) precursors led to unwanted side product formation. However, the desired product was formed in high yield in the presence of elemental copper (entries 2–4).
|
|
||
|---|---|---|
| Entry | Cu-source | Yield 2a [%] |
| a Reaction conditions: dropwise addition of 0.5 mmol of 1a in 1 mL acetonitrile to 1.5 equiv. Me4NSC2F5 and the copper source in 1 mL acetonitrile, 15 h at room temperature. Yields were determined by 19F NMR using trifluoroethanol as an internal standard. b 1 h reaction time. | ||
| 1 | 10 mol% CuSCN | 70 |
| 2 | 10 mol% CuOAc | 15 |
| 3 | 10 mol% CuI | 20 |
| 4 | 10 mol% Cu | 99 |
| 5 | 5 mol% Cu | 62 |
| 6 | 0.5 equiv. Cu | 89 |
| 7 | 1.0 equiv. Cu | 75 |
| 8b | 1.0 equiv. Cu | 12 |
| 9 | — | 0 |

The best results were obtained with 10 mol% of Cu (entries 5–7). This is remarkable, since there are only few examples of Sandmeyer reactions catalytic in copper. The markedly lower nucleophilicity of the pentafluoroethylthio group in comparison to the trifluoromethylthio group is reflected in the increased reaction times; the pentafluoroethylthiolation requires 15 hours to go to completion, whereas Sandmeyer trifluoromethylthiolations occur within less than one hour at room temperature (entry 8).8e Without copper, no product formation was observed (entry 9).
Having thus found an effective protocol for the Sandmeyer pentafluoroethylthiolation, we next investigated its scope. Various arenediazonium tetrafluoroborates were smoothly converted into the corresponding pentafluoroethyl thioethers in high yields (Table 2).
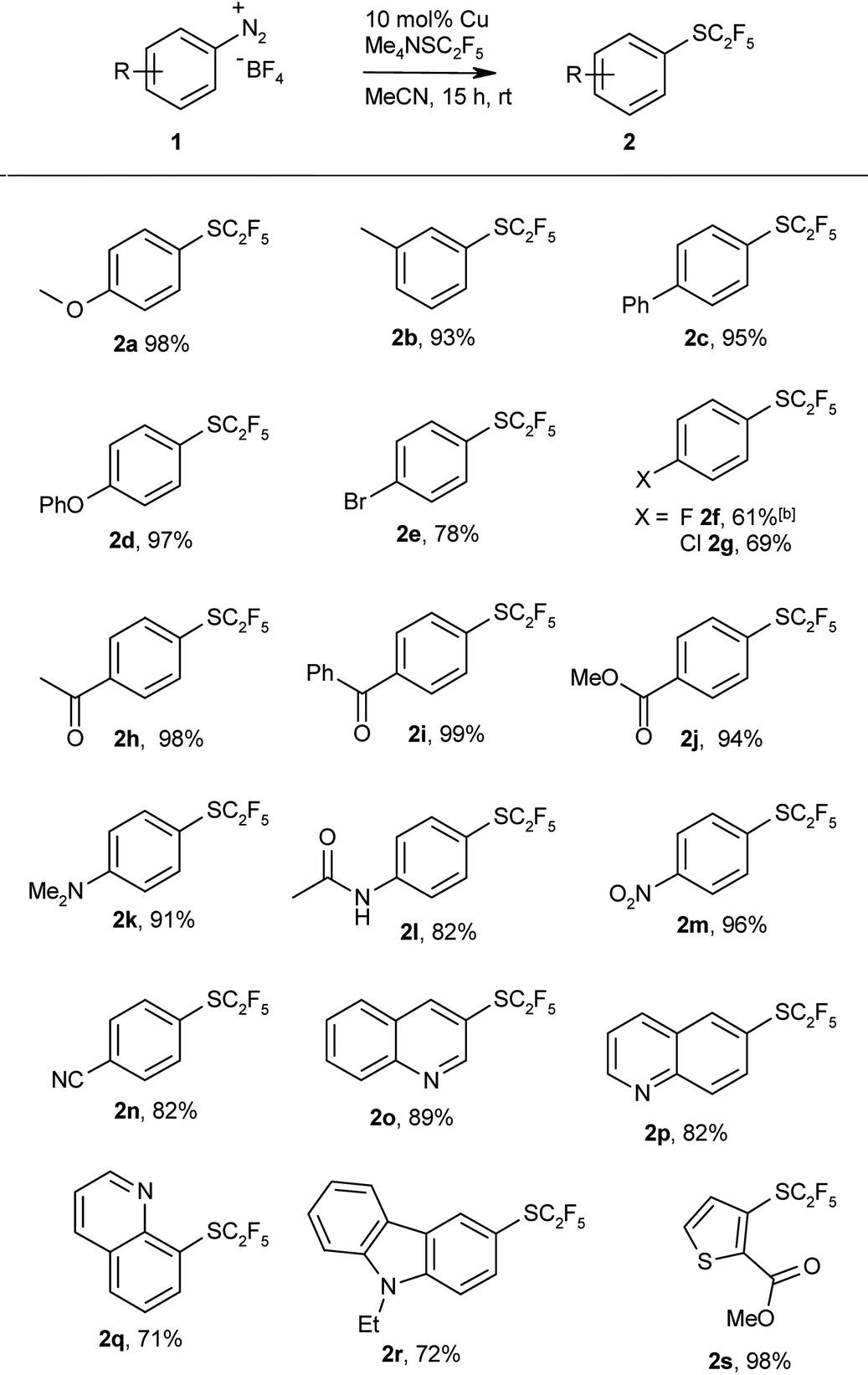
Both electron-rich and electron-deficient substrates give similarly high yields, and various functionalities are tolerated including ester, ether, amino, keto, carboxylate, cyano, and even bromo groups. Various heterocycles were also pentafluoroethylthiolated in good yields. These examples clearly demonstrate the utility of the protocol for late-stage pentafluoroethylthiolations of functionalised intermediates. The products are obtained in reasonable purity after simple aqueous workup, and can be further purified by column chromatography.
It is safe to assume that in analogy to classical Sandmeyer halogenations and trifluoromethylthiolations of diazonium salts, the reaction proceeds via a single-electron transfer mechanism as depicted in Scheme 2. The use of metallic copper as source of Cu(I) species in these processes is rare but not unprecedented.8e,21 The addition of radical quenchers such as 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) or p-benzoquinone suppressed the reaction, which confirms that the reaction involves radical intermediates. In order to exclude an alternative cationic pathway for extremely electron-poor substrates, analogous control experiments were conducted with 4-nitrobenzenediazonium tetrafluoroborate. In the absence of copper or in the presence of radical trapping reagents no product formation was detected, which supports a Sandmeyer type mechanism even for substrates in which other pathways are conceivable.
 | ||
| Scheme 2 Sandmeyer pentafluoroethylthiolation of aromatic amines. | ||
Conclusions
The Sandmeyer-type process reported herein allows the straightforward synthesis of pentafluoroethylthiolated compounds from the corresponding aromatic amines. The key advantages of this method are its mild reaction conditions (neutral, room temperature), the use of an inexpensive copper catalyst in only 10 mol% loading, and the exceptional functional group tolerance. As a result, this method is well-suited for the late-stage introduction of pentafluoroethylthio groups into drug-like molecules.Acknowledgements
We thank the Heinrich-Böll-Stiftung e.V. (scholarship to B. B.) for financial support.References
- (a) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (b) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- (a) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS PubMed; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (c) X.-F. Wu, H. Neumann and M. Beller, Chem. – Asian J., 2012, 7, 1744–1754 CrossRef CAS PubMed; (d) T. Liu and Q. Shen, Eur. J. Org. Chem., 2012, 6679–6687 CrossRef CAS; (e) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed; (f) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683–730 CrossRef CAS PubMed; (g) C. Alonso, E. Martínez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef CAS PubMed.
- (a) C. Hansch, A. Leo, S. H. Unger, K. H. Kim, D. Nikaitani and E. J. Lien, J. Med. Chem., 1973, 16, 1207–1216 CrossRef CAS PubMed; (b) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415–2428 CrossRef CAS.
- (a) A. Tlili and T. Billard, Angew. Chem., Int. Ed., 2013, 52, 6818–6819 CrossRef CAS PubMed; (b) X. Shao, X. Wang, T. Yang, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 3457–3460 CrossRef CAS PubMed; (c) Y.-D. Yang, A. Azuma, E. Tokunaga, M. Yamasaki, M. Shiro and N. Shibata, J. Am. Chem. Soc., 2013, 135, 8782–8785 CrossRef CAS PubMed; (d) R. Pluta, P. Nikolaienko and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 1650–1653 CrossRef CAS PubMed; (e) C. Xu, B. Ma and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 9316–9320 CrossRef CAS PubMed.
- (a) G. Teverovskiy, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 7312–7314 CrossRef CAS PubMed; (b) C.-P. Zhang and D. A. Vicic, J. Am. Chem. Soc., 2012, 134, 183–185 CrossRef CAS PubMed; (c) Z. Weng, W. He, C. Chen, R. Lee, D. Tan, Z. Lai, D. Kong, Y. Yuan and K.-W. Huang, Angew. Chem., Int. Ed., 2013, 52, 1548–1552 CrossRef CAS PubMed.
- L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237–18240 CrossRef CAS PubMed.
- (a) C. Chen, Y. Xie, L. Chu, R.-W. Wang, X. Zhang and F.-L. Qing, Angew. Chem., Int. Ed., 2012, 51, 2492–2495 CrossRef CAS PubMed; (b) C. Chen, L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2012, 134, 12454–12457 CrossRef CAS PubMed; (c) C.-P. Zhang and D. A. Vicic, Chem. – Asian J., 2012, 7, 1756–1758 CrossRef CAS PubMed; (d) S.-Q. Zhu, X.-H. Xu and F.-L. Qing, Eur. J. Org. Chem., 2014, 4453–4456 CrossRef CAS.
- (a) B. Bayarmagnai, C. Matheis, E. Risto and L. J. Goossen, Adv. Synth. Catal., 2014, 356, 2343–2348 CrossRef CAS; (b) G. Danoun, B. Bayarmagnai, M. Grünberg, C. Matheis, E. Risto and L. Gooßen, Synthesis, 2014, 2283–2286 CAS; (c) C. Matheis, K. Jouvin and L. J. Goossen, Org. Lett., 2014, 16, 5984–5987 CrossRef CAS PubMed; (d) B. Bayarmagnai, C. Matheis, K. Jouvin and L. J. Goossen, Angew. Chem., Int. Ed., 2015, 54, 5753–5756 CrossRef CAS PubMed; (e) C. Matheis, V. Wagner and L. J. Goossen, Chem. – Eur. J., 2016, 22, 79–82 CrossRef CAS PubMed; (f) G. Danoun, B. Bayarmagnai, M. F. Gruenberg and L. J. Goossen, Chem. Sci., 2014, 5, 1312–1316 RSC.
- (a) B. Exner, B. Bayarmagnai, F. Jia and L. J. Goossen, Chem. – Eur. J., 2015, 21, 17220–17223 CrossRef CAS PubMed; (b) K. Jouvin, C. Matheis and L. J. Goossen, Chem. – Eur. J., 2015, 21, 14324–14327 CrossRef CAS PubMed; (c) C. Matheis, M. Wang, T. Krause and L. Goossen, Synlett, 2015, 26, 1628–1632 CrossRef CAS.
- (a) Z. Weng, W. He, C. Chen, R. Lee, D. Tan, Z. Lai, D. Kong, Y. Yuan and K.-W. Huang, Angew. Chem., Int. Ed., 2013, 52, 1548–1552 CrossRef CAS PubMed; (b) C. Chen, Y. Xie, L. Chu, R.-W. Wang, X. Zhang and F.-L. Qing, Angew. Chem., Int. Ed., 2012, 51, 2492–2495 CrossRef CAS PubMed; (c) Y. Zhang, K. Gan and Z. Weng, Org. Process Res. Dev., 2016, 20, 799–802 CrossRef CAS.
- (a) P. Kirsch, G. V. Roeschenthaler, B. Bissky and A. Kolomeitsev, DE-A110254597, 2003, Merck GmbH Search PubMed; (b) W. Tyrra, D. Naumann, B. Hoge and Y. L. Yagupolskii, J. Fluorine Chem., 2003, 119, 101–107 CrossRef CAS.
- M. Rueping, N. Tolstoluzhsky and P. Nikolaienko, Chem. – Eur. J., 2013, 19, 14043–14046 CrossRef CAS PubMed.
- (a) G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164–4172 CrossRef CAS PubMed; (b) G. Yin, I. Kalvet and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 6809–6813 CrossRef CAS PubMed; (c) Y. Yang, L. Xu, S. Yu, X. Liu, Y. Zhang and D. A. Vicic, Chem. – Eur. J., 2016, 22, 858–863 CrossRef CAS PubMed.
- A. B. Dürr, G. Yin, I. Kalvet, F. Napoly and F. Schoenebeck, Chem. Sci., 2016, 7, 1076–1081 RSC.
- C. Xu and Q. Shen, Org. Lett., 2014, 16, 2046–2049 CrossRef CAS PubMed.
- (a) M. Andrzejewska, Eur. J. Med. Chem., 2002, 37, 973–978 CrossRef CAS PubMed; (b) A. Johansson, A. Poliakov, E. Åkerblom, K. Wiklund, G. Lindeberg, S. Winiwarter, U. H. Danielson, B. Samuelsson and A. Hallberg, Bioorg. Med. Chem., 2003, 11, 2551–2568 CrossRef CAS PubMed; (c) A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2013, 135, 12584–12587 CrossRef CAS PubMed.
- N. Roques, J. Fluorine Chem., 2001, 107, 311–314 CrossRef CAS.
- S. Alazet and T. Billard, Synlett, 2014, 76–78 Search PubMed.
- L. Jiang, J. Qian, W. Yi, G. Lu, C. Cai and W. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14965–14969 CrossRef CAS PubMed.
- (a) P. Kirsch, Modern fluoroorganic chemistry: synthesis, reactivity, applications, Wiley-VCH, Weinheim, 2004, p. 145 Search PubMed; (b) Me4NSC2F5 was commercially available by CF Plus Chemicals s. r. o.
- (a) N. Kornblum, G. D. Cooper and J. E. Taylor, J. Am. Chem. Soc., 1950, 72, 3013–3021 CrossRef CAS; (b) C. Galli, Chem. Rev., 1988, 88, 765–792 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00194g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |