
Rhodium-catalyzed asymmetric arylation of N- and O-containing cyclic aldimines: facile and efficient access to highly optically active 3,4-dihydrobenzo[1,4]oxazin-2-ones and dihydroquinoxalinones†
Xu
Zhang
ab,
Bin
Xu
*a and
Ming-Hua
Xu
*b
aDepartment of Chemistry, Innovative Drug Research Center, Shanghai University, Shanghai 200444, China
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China. E-mail: xumh@simm.ac.cn; Tel: +86 21-50807388
First published on 31st May 2016
Abstract
A highly enantioselective rhodium-catalyzed arylation of benzoxazinones and quinoxalinones with arylboroxines was realized for the first time by employing a simple sulfur-olefin as the ligand. This protocol provides an efficient access to chiral 3,4-dihydrobenzo[1,4]oxazin-2-ones and dihydroquinoxalinones with exceptionally high enantiomeric purity (up to 99.9% ee).
3,4-Dihydrobenzo[1,4]oxazin-2-ones and dihydroquinoxalinones are two intriguing classes of nitrogen-containing heterocycles.1 These heterocycles are not only important intermediates for the synthesis of many pharmaceutical compounds, but also widely used in the agro-industry.2 As reported, optically active compounds containing these core structural units often show a wide range of biological activities. For example, compound I is a pyruvate kinase activator and can increase the lifetime of red blood cells (Fig. 1).3 Compound II possesses potent activity against retroviruses such as HIV.4 Dihydroquinoxalinone derivative III is a newly developed CETP inhibitor and is expected to be utilized in the treatment of atherosclerosis.5 Besides, other interesting bioactivities of such benzo-fused heterocycles are also demonstrated, including 5-HT2c agonist binding activity,6 antibacterial and anticancer effects,7 and euglycemic and hypolipidemic activities.8 Therefore, highly enantioselective synthesis of these chiral heterocycle structural motifs has been a compelling research topic.
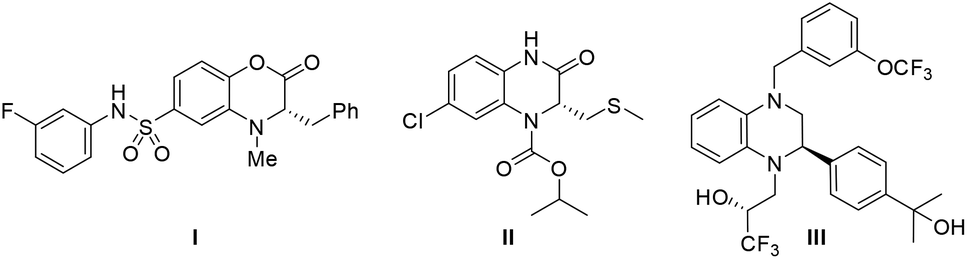 | ||
| Fig. 1 Bioactive 3,4-dihydrobenzo[1,4]oxazin-2-one and dihydroquinoxalinone derivatives. | ||
Over the past decade, several asymmetric approaches including chiral auxiliary9 and chiral pool10 strategies have been developed to construct enantiomerically enriched 3,4-dihydrobenzo[1,4]oxazin-2-one and dihydroquinoxalinone scaffolds. The most frequently used method to construct these chiral heterocycles is asymmetric reduction of aryl-substituted benzoxazinones and quinoxalinones mainly via transfer hydrogenation,11a–c hydrosilylation11d,e and biomimetic hydrogenation,11f–h catalyzed by Brønsted acids (chiral phosphoric acids) or Lewis base organocatalysts.11i Although asymmetric addition to imines is an efficient and direct method to furnish nitrogen-containing chiral organic molecules, there is only limited success involving the chiral Brønsted acid-catalyzed Friedel–Crafts reaction of indoles and pyrroles in which ester- or trifluoromethyl-substituted benzoxazinones were employed for the construction of highly enantiomerically enriched dihydrobenzoxazinones.12 In the case of a reaction with pyrrole,12b a sole example of addition to non-substituted benzoxazinone was described, however, giving only 45% yield and 9% ee without the activation and stereoeffect of the CF3-group. While the transition-metal-catalyzed asymmetric addition approach has been widely applied, surprisingly, successful examples of the synthesis of these heterocycles remain scarce,13,14 and enantioselective 1,2-addition of organoboron reagents to benzoxazinones and quinoxalinones has not yet been successfully developed. Therefore, there is still an unmet need for highly efficient asymmetric synthesis of these appealing heteroaromatic compounds. Herein, we report the first rhodium-catalyzed highly enantioselective arylation of cyclic benzoxazinones and quinoxalinones with arylboroxines using a simple sulfur-olefin as the ligand.
In recent years, our laboratory has been exploring the design of new chiral olefin ligands and successfully introduced a series of sulfur- and phosphorus-based olefins as well as bisolefins for transition-metal-catalyzed asymmetric reactions.15 We found that readily available, sulfinamide-based branched olefin ligands showed great promise in the Rh-catalyzed asymmetric addition of arylboronic acids to cyclic N-sulfonyl imines, typically exhibiting excellent efficiency and the ideal enantioselectivity.16,17 On the basis of these results, we presumed that these simple sulfur-olefins might also act as effective ligands for asymmetric organoboron addition to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of benzoxazinones and quinoxalinones for the construction of chiral 3,4-dihydrobenzo[1,4]oxazin-2-one and dihydroquinoxalinone scaffolds (Scheme 1).
N bond of benzoxazinones and quinoxalinones for the construction of chiral 3,4-dihydrobenzo[1,4]oxazin-2-one and dihydroquinoxalinone scaffolds (Scheme 1).
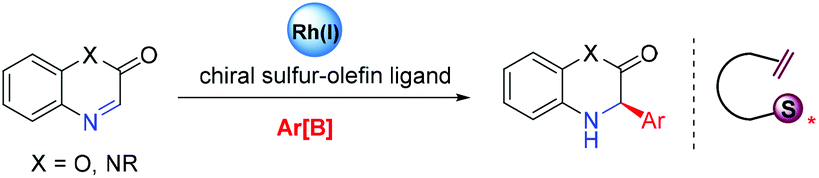 | ||
| Scheme 1 Asymmetric arylation strategy under rhodium catalysis. | ||
We initiated our studies by evaluating the reaction of cyclic aldimine benzoxazinone 1a with 4-methoxyphenylboronic acid (2a) in KF (1.5 M)/toluene at 60 °C using branched sulfur-olefin L1 as the ligand in the presence of the [Rh(COE)2Cl]2 catalyst (1.5 mol%) (Table 1, entry 1). To our delight, the reaction gave the desired addition product 3b with the expected excellent enantioselectivity (97% ee), albeit in a relatively low yield (39%). In contrast, sulfur-olefin L2 with a linear structure could only afford moderate enantioselectivity (64% ee) with a similar low yield (27%) (entry 2). Encouraged by this result, the effects of solvent and additive were carefully examined. The employment of KHF2 would largely promote the substrate conversion (entry 6), while utilization of bases such as K2CO3, K3PO4 and KOH led to an apparent decrease of yield due to the decomposition of the substrate (entries 3–5). Adding Et3N resulted in moderate yield but a dramatic decrease in enantioselectivity (entry 7). To avoid hydrolysis of the imine substrate, we examined the use of a solid salt as the additive (entries 8–10). Gratifyingly, we found that the reaction could afford the product in 70% yield with an outstanding enantioselectivity (99% ee) under the conditions of solid K3PO4 (1 equiv.)/dioxane (entry 9). Inspired by this result, the corresponding more reactive boroxine (1.2 equiv.) instead of boronic acid was employed with the addition of an alcohol (4 equiv.) as the proton source. Through further evaluation of the new reaction conditions, we were pleased to discover that the yield could be substantially improved to over 90% (entries 11–12). Notably, when the reaction was performed in the solid K3PO4/dioxane in the presence of methanol, a surprisingly high 98% yield and 99.9% ee were obtained (entry 11).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Additive | Yieldb (%) | eec (%) |
|
a Conditions: 1a (0.1 mmol), 2a (2 equiv.), [Rh(COE)2Cl]2 (1.5 mol%), ligand (3.3 mol%) and base (1.0 equiv.) in 1.0 mL of solvent at 60 °C for 2 h unless otherwise noted.
b Isolated yields.
c Determined by chiral HPLC analysis.
d 2b (1.2 equiv.) was used.
e Solid salt (1.0 equiv.) was used.
f 4.0 equiv. of alcohol were used.
|
|||||
| 1 | L1 | Toluene | KF (1.5 M) | 39 | 97 |
| 2 | L2 | Toluene | KF (1.5 M) | 27 | 64 |
| 3 | L1 | Toluene | K2CO3 (1 M) | 23 | 97 |
| 4 | L1 | Toluene | K3PO4 (1 M) | 25 | 95 |
| 5 | L1 | Toluene | KOH (1 M) | Trace | — |
| 6 | L1 | Toluene | KHF2 (1 M) | 68 | 99 |
| 7 | L1 | Toluene | Et3N | 66 | 65 |
| 8 | L1 | Dioxane | KHF2e |
19 | 97 |
| 9 | L1 | Dioxane | K2CO3e |
66 | 98 |
| 10 | L1 | Dioxane | K3PO4e |
70 | 99 |
| 11d | L1 | Dioxane | K3PO4e/MeOHf |
98 | 99.9 |
| 12d | L1 | Dioxane | K3PO4e/t-amyl alcoholf |
94 | 99 |
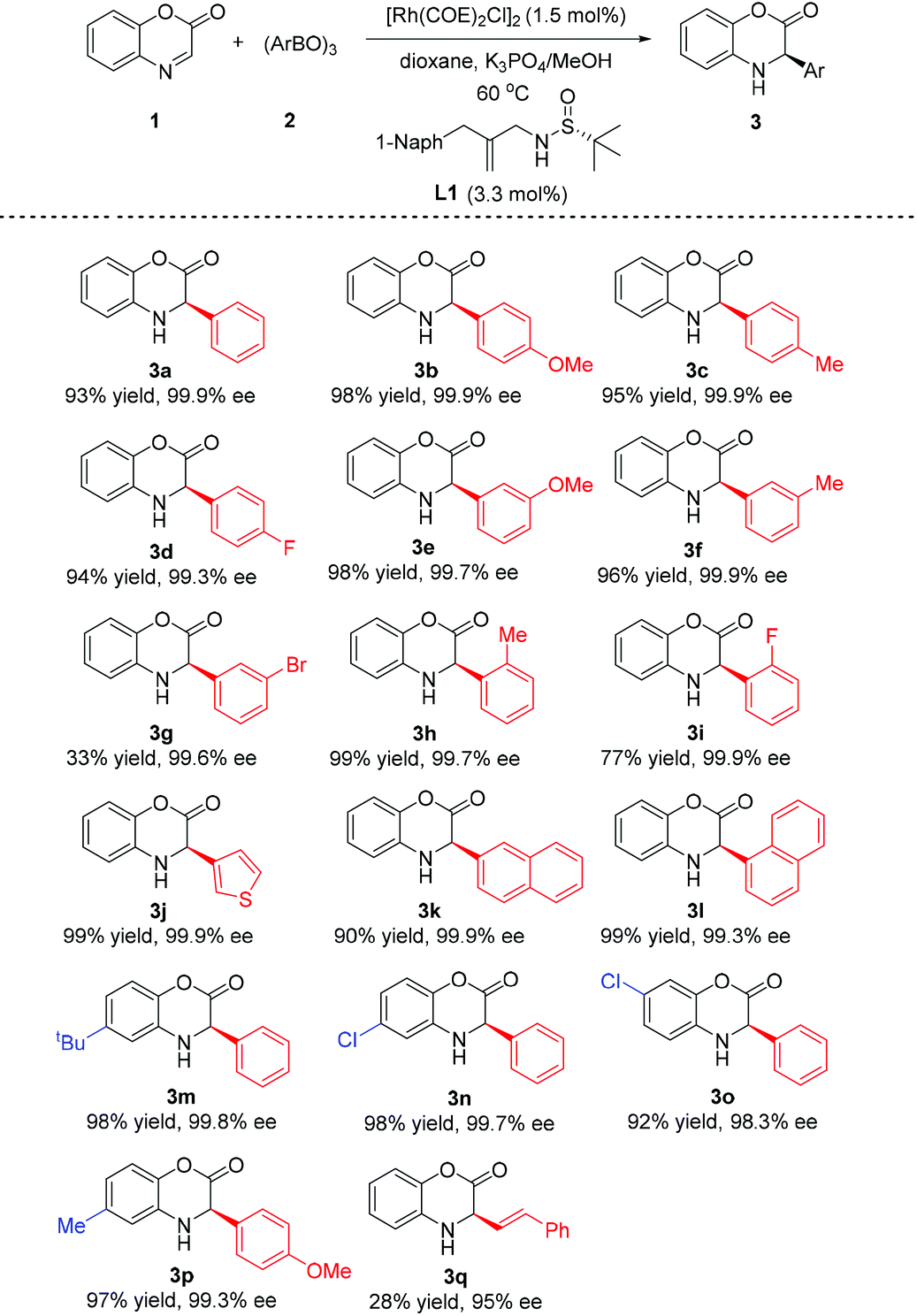
With the optimal conditions established, we proceeded to investigate the substrate scope of the reaction. As shown in Table 2, regardless of the substitution pattern, a wide variety of arylboroxines reacted smoothly with benzoxazinone 1 and gave all nearly enantiopure 3-aryl-3,4-dihydrobenzo[1,4]oxazin-2-ones (with 99.3–99.9% ee). The electronic effect of the para-substituent on the phenyl ring of boroxine barely showed the influence on the reactivity and enantioselectivity (3b–d). However, while electron donating group substituted phenylboroxines gave addition products in great yields (3e, 3f, 3h), the utilization of boroxines possessing phenyl rings with electron-withdrawing groups at the ortho- and meta-position showed an obvious decrease in yields (3g, and 3i). The reaction can be extended to heteroaryl boroxine and a 3-thieny-group could be introduced to form product 3j in 99% yield with 99.3% ee. It is particularly notable that great yield and enantiomeric control were obtained with the use of more sterically hindered arylboroxines (3h, 3i, and 3l). To our knowledge, these results are otherwise difficult to be achieved by the known hydrosilylation or hydrogenation reactions. Benzoxazinone substrates bearing substitutents on the benzene ring can be well tolerated to furnish the corresponding products 3m–3p in equally excellent yields and enantioselectivities. Besides, it is also interesting to find that the reaction gave a promising enantioselectivity (95% ee) with challenging styrylboroxine, although the yield was low (25%, 3q).
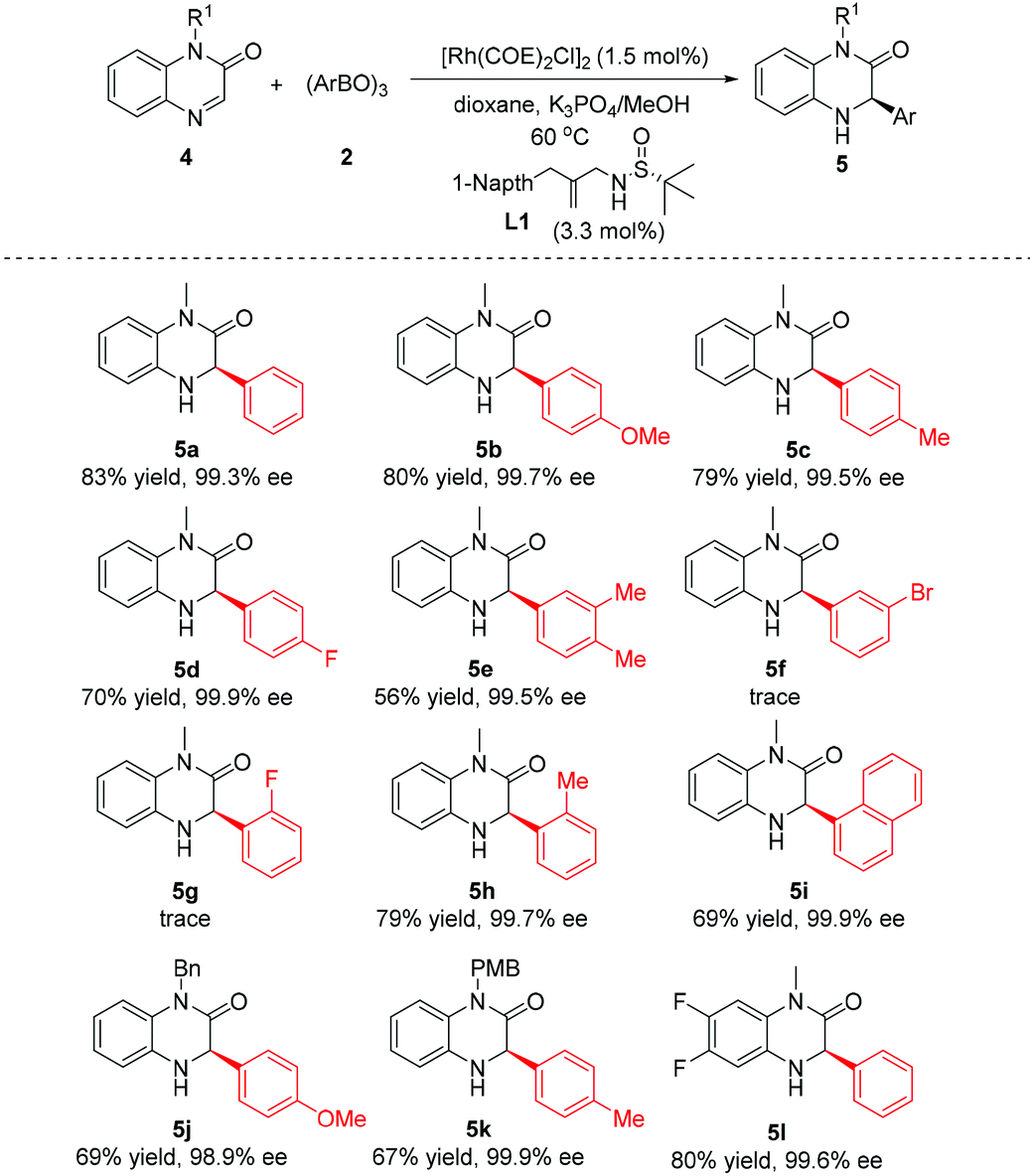
Encouraged by the above success, the current catalytic system was extended to the asymmetric addition of quinoxalinones 4 for construction of dihydroquinoxalinones (Table 3). Generally, the para-substituted phenylboroxines displayed a good reactivity as well as high enantioselectivity irrespective of the electronic nature of substituents (5b–d). Nevertheless, the use of arylboroxines bearing the electron-withdrawing group at the ortho- and meta-position of the phenyl ring failed to afford the product (5f, and 5g). Fortunately, satisfactory yields and enantioselectivities were obtained with sterically hindered 2-methylphenylboroxine and 1-naphthylboroxine (5h, 5i). Changing the N-substituent of substrate 4 from methyl to benzyl and p-methoxybenzyl did not affect the reaction enantioselectivity (5j, 5k). Finally, a difluoro-substituted quinoxalinone substrate was employed, and the same high level of enantiocontrol was observed (5l).
To demonstrate the practicality of this conversion, we conducted the reaction on a larger scale (3.42 mmol, 500 mg) under standard conditions, which furnished product 3b in 92% yield and 99% ee. The stereochemistry of the newly formed chiral center of product 3g was determined to be R by single-crystal X-ray diffraction analysis.18 Assuming the same stereoinduction pathway, the configuration of the other 3,4-dihydrobenzo[1,4]oxazin-2-one products as well as dihydroquinoxalinones was assigned by analogy. Based on this reaction stereochemical outcome, an empirical transition state model is proposed. As depicted in Fig. 2, the arylrhodium species prefer a specific geometry, in which the aryl group is in the trans-position to the olefin ligand. To avoid unfavorable steric interaction with the bulky R group on the double bond, the cyclic imine substrate could only coordinate to rhodium from the re-face of CN (B) to furnish the (R)-product.
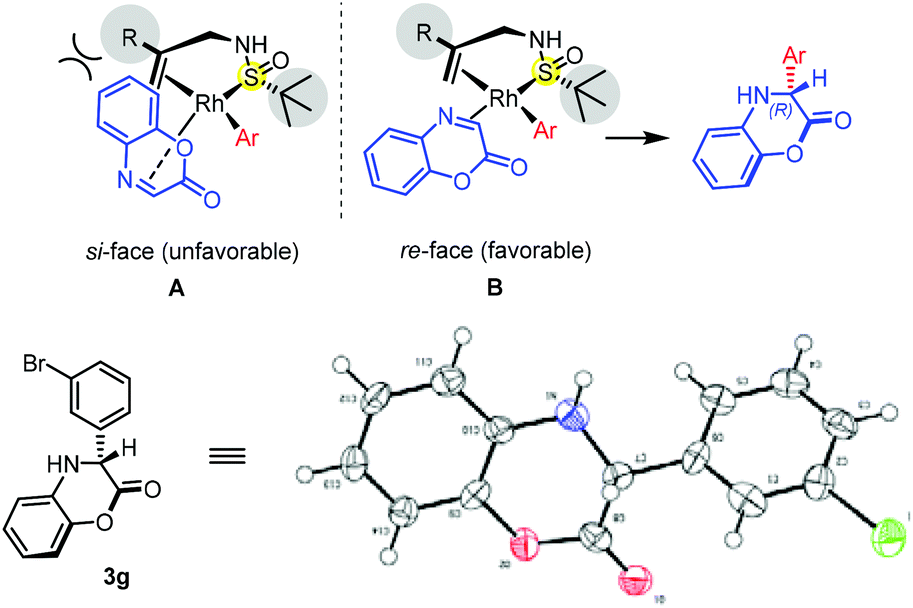 | ||
| Fig. 2 Proposed transition-state model and the X-ray crystallographic structure of 3g. | ||
Finally, we set out to investigate the transformation of the highly enantiomerically enriched 3,4-dihydrobenzo[1,4]oxazin-2-ones and dihydroquinoxalinones into other useful molecules. As revealed in Scheme 2a, treatment of 3,4-dihydrobenzo[1,4]oxazin-2-one 3b with LiAlH4 at 0 °C, followed by the Mitsunobu reaction can give rise to benzomorpholine 6 in 83% yield without the loss of optical purity (99% ee). Accordingly, dihydroquinoxalinones can be readily converted into useful tetrahydroquinoxalines under the conditions of BH3·THF (2.5 equiv.) in refluxing THF with quantitative yield, as exemplified in Scheme 2b. It is worth noting that benzomorpholines and tetrahydroquinoxalines are also valuable heterocycles that often possess interesting biological activities.19 In another case, dihydrobenzoxazinone 3c was subjected to ring opening and transformed to aryl glycine amide 8 in the presence of pyridin-2-ol and benzyl amine in THF.
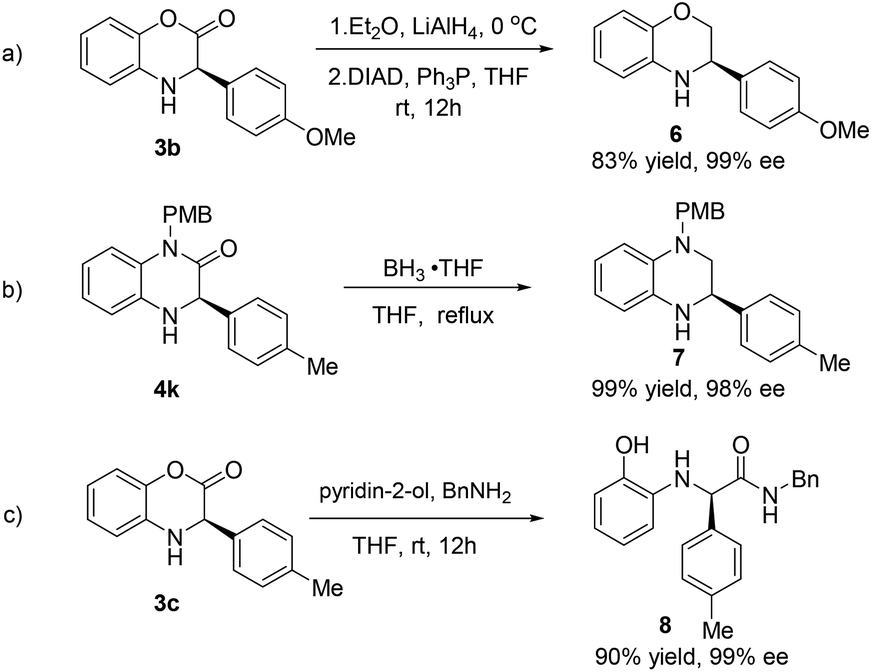 | ||
| Scheme 2 Transformation of addition products. | ||
In summary, we have developed an efficient and highly enantioselective Rh-catalyzed arylation of benzoxazinones and quinoxalinones with arylboroxines. This protocol provides a facile method to achieve chiral 3,4-dihydrobenzo[1,4]oxazin-2-ones and dihydroquinoxalinones with excellent stereocontrol (up to 99.9% ee) in the presence of a simple sulfur-olefin ligand. Moreover, the work reveals new opportunities to access other important heterocycles such as chiral benzomorpholines and tetrahydroquinoxalines through operationally simple procedures. It should find future applications in medicinal chemistry and organic synthesis.
Acknowledgements
We thank the National Natural Science Foundation of China (21325209, 21472205, 81521005) and the Shanghai Municipal Committee of Science and Technology (Program of Shanghai Academic Research Leader, 14XD1404400) for financial support.Notes and references
- (a) D. D. Andrea and E. J. John, (Warner Lambert Co., USA), U.S. Patent6410536(B1), 2002 Search PubMed; (b) D.-S. Su, M. K. Markowitz, R. M. DiPardo, K. L. Murphy, C. M. Harrell, S. S. O'Malley, R. W. Ransom, R. S. L. Chang, F. J. Hess and D. J. Pettibone, J. Am. Chem. Soc., 2003, 125, 7516 CrossRef CAS PubMed; (c) P. E. Mahaney, M. B. Webb, F. Ye, J. P. Sabatucci, R. J. Steffan, C. C. Chadwick, D. C. Harnish and E. J. Trybulski, Bioorg. Med. Chem., 2006, 14, 3455 CrossRef CAS PubMed; (d) H. Daniel and P. William, (P & H Therapeutics Inc., USA), U.S. Patent2009311318, 2009 Search PubMed.
- (a) S. X. Cai, S. M. Kher, Z.-L. Zhou, V. Ilyin and S. A. Espitia, J. Med. Chem., 1997, 40, 730 CrossRef CAS PubMed; (b) M. Patel, R. J. McHugh, B. C. Cordova, R. M. Klabe, S. Erickson-Viitanen and G. L. Trainor, Bioorg. Med. Chem. Lett., 2000, 10, 1729 CrossRef CAS PubMed; (c) L. A. Cruz, E. Estébanez-Perpiña, S. Pfaff, S. Borngraeber, N. Bao, J. Blethrow and R. J. Fletterick, J. Med. Chem., 2008, 51, 5856 CrossRef CAS PubMed; (d) D. V. Smil, S. Manku, Y. A. Changtigny, S. Leit, A.-M. Lmieuz and A. NIcolescuz, Bioorg. Med. Chem., 2009, 19, 688 CrossRef CAS PubMed.
- S. S.-S. Michael, (Agios Pharm. Inc.), Patent WO2012151440, 2012 Search PubMed.
- M. Rosner, U.-M. Billhardt-Troughton, R. Kirsh, J.-P. Kleim, C. Meichsner, G. Riess and I. Winkler, U.S. Patent, 5723461, 1998 Search PubMed.
- R. Anand, V. Colandrea, J. M. Reiter, P. Vachal, A. Zwicker, J. E. Wilson, F. Zhang and K. Zhao, Patent WO2013028382, 2013 Search PubMed.
- G. Yoon, H. J. Jeong, J. J. Kim and S. H. Cheon, Arch. Pharm. Res., 2008, 31, 989 CrossRef CAS PubMed.
- (a) I. V. Mashaevskaya, R. R. Makhmudov, G. A. Aleksandrova, A. V. Duvalov and A. N. Maslivets, Pharm. Chem. J., 2001, 35, 69 CrossRef; (b) X. K. Li, N. Liu, H. N. Zhang, S. Knudson, R. A. Slayden and P. J. Tonge, Bioorg. Med. Chem. Lett., 2010, 20, 6306 CrossRef CAS PubMed; (c) M. Y. Abu Shuheil, M. R. Hassuneg, Y. M. Al-Hiari, A. M. Qaisi and M. M. El-Abadelah, Heterocycles, 2007, 71, 2155 CrossRef CAS; (d) S. Tanimori, T. Nishimira and M. Kirihata, Bioorg. Med. Chem. Lett., 2009, 19, 4119 CrossRef CAS PubMed.
- D. Gupta, N. N. Ghosh and R. Chandra, Bioorg. Med. Chem. Lett., 2005, 15, 1019 CrossRef CAS PubMed.
- Y. M. Lee and Y. S. Park, Heterocycles, 2009, 78, 2233 CrossRef CAS.
- S. Luo and J. K. De Brabander, Tetrahedron Lett., 2015, 56, 3179 CrossRef CAS.
- For selected examples, see: (a) R. I. Storer, D. E. Carrera, Y. K. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84 CrossRef CAS PubMed; (b) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 6751 CrossRef CAS PubMed; (c) F. Shi, W. Tan, H.-H. Zhang, M. Li, Q. Ye, G.-H. Ma, S.-J. Tu and G.-G. Li, Adv. Synth. Catal., 2013, 355, 3715 CrossRef CAS; (d) A. V. Maikov, K. Vrankova, S. Stoncius and P. Kocovsky, J. Org. Chem., 2009, 74, 5839 CrossRef PubMed; (e) Z.-Y. Xue, Y. Jiang, X.-Z. Peng, W.-C. Yuan and X.-M. Zhang, Adv. Synth. Catal., 2010, 352, 2132 CrossRef CAS; (f) Q.-A. Chen, M.-W. Chen, C.-B. Yu, L. Shi, D.-S. Wang, Y. Yang and Y.-G. Zhou, J. Am. Chem. Soc., 2011, 133, 16432 CrossRef CAS PubMed; (g) Q.-A. Chen, K. Gao, Y. Duan, Z.-S. Ye, Y. Yang and Y.-G. Zhou, J. Am. Chem. Soc., 2012, 134, 2442 CrossRef CAS PubMed; (h) L.-Q. Lu, Y.-H. Li, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 2763 CrossRef CAS PubMed. Only limited success was achieved in the standard asymmetric hydrogenation, for a recent report using a chiral iridium complex as the catalyst, see: (i) J. L. Núňez-Rico and A. Vidal-Ferran, Org. Lett., 2013, 15, 2066 CrossRef PubMed.
- (a) T. Kano, R. Takechi, R. Kobayashi and K. Maruoka, Org. Biomol. Chem., 2014, 12, 724 RSC; (b) H. Lou, Y. Wang, E. Jin and X. Lin, J. Org. Chem., 2016, 81, 2019 CrossRef CAS PubMed.
- H. Miyabe, Y. Yamaoka and Y. Takemoto, J. Org. Chem., 2006, 71, 2099 CrossRef CAS PubMed.
- For a Pd-catalyzed non-asymmetric addition of arylboronic acids to quinoxalinones, see: A. Carrër, J.-D. Brion, M. Alami and S. Messaoudi, Adv. Synth. Catal., 2014, 356, 3821 CrossRef . For an oxidative arylation example, see: A. Carrër, J.-D. Brion, S. Messaoudi and M. Alami, Org. Lett., 2013, 15, 5606 CrossRef PubMed.
- (a) C.-G. Feng, M.-H. Xu and G.-Q. Lin, Synlett, 2011, 1345 CAS; (b) Y. Li and M.-H. Xu, Chem. Commun., 2014, 50, 3771 RSC; (c) Y.-N. Yu and M.-H. Xu, Org. Chem. Front., 2014, 1, 738 RSC; (d) H.-Q. Dong, M.-H. Xu, C.-G. Feng, X.-W. Sun and G.-Q. Lin, Org. Chem. Front., 2015, 2, 73 RSC; (e) D. Chen, X. Zhang, W.-Y. Qi, B. Xu and M.-H. Xu, J. Am. Chem. Soc., 2015, 137, 5268 CrossRef CAS PubMed.
- (a) H. Wang, T. Jiang and M.-H. Xu, J. Am. Chem. Soc., 2013, 135, 971 CrossRef CAS PubMed; (b) H. Wang, Y. Li and M.-H. Xu, Org. Lett., 2014, 16, 3962 CrossRef CAS PubMed; (c) T. Jiang, Z. Wang and M.-H. Xu, Org. Lett., 2015, 17, 528 CrossRef CAS PubMed.
- For recent examples of Rh-catalyzed arylation of cyclic imines using other chiral ligands, see: (a) T. Nishimura, A. Noishiki, Y. Ebe and T. Hayashi, Angew. Chem., Int. Ed., 2013, 52, 1777 CrossRef CAS PubMed; (b) Y. Luo, H. B. Hepburn, N. Chotsaeng and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 8309 CrossRef CAS PubMed; (c) Y. Li, Y.-N. Yu and M.-H. Xu, ACS Catal., 2016, 6, 661 CrossRef CAS and references cited therein.
- CCDC 1478345 (3g) contains the supplementary crystallographic data.
- (a) R. Pauwels, K. Andries, Z. Debyser, P. V. Daele, D. Schols, P. Stoffels, K. D. Vreese, R. Woestenborghs, A. M. Vandamme and E. D. Clercq, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1711 CrossRef CAS PubMed; (b) E. Perola, J. Med. Chem., 2010, 53, 2986 CrossRef CAS PubMed; (c) H. J. Sanganee, A. Baxter, S. Barber, A. J. H. Brown, D. Grice, F. Hunt, S. King, A. Laughton, G. Pairaudeau, B. Thong, R. Weaver and J. Unitt, Bioorg. Med. Chem. Lett., 2009, 19, 1143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1478345. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00191b |
| This journal is © the Partner Organisations 2016 |