
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic one-pot synthesis of 4-(hetero)aryl substituted 5-(2-oxoethyl) oxazol-2(3H)-ones by coupling–isomerization–elimination (CIE) sequence†‡
Christina
Görgen née Boersch
a,
Kiril
Lutsenko
a,
Eugen
Merkul
a,
Walter
Frank
b and
Thomas J. J.
Müller
*a
aInstitut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany. E-mail: ThomasJJ.Mueller@hhu.de
bInstitut für Anorganische Chemie und Strukturchemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany
First published on 19th May 2016
Abstract
N-Boc protected 1-aryl propargyl carbamates and acid chlorides can be readily transformed in a one-pot fashion by a coupling–isomerization–elimination (CIE) sequence into 4-substituted 5-(2-oxoethyl) oxazol-2(3H)-ones in moderate to good yield.
Introduction
Oxazol-2(3H)-one is an interesting derivative of the parent oxazole. While the aromatic structure is formally negated by the presence of an amide carbonyl group, the amide resonance retains it (Fig. 1).1 As a consequence, the N-proton is strongly acidified (pKa = 15.0) in comparison to the saturated oxazolidin-2-one (pKa = 20.9) or the structurally related open-chain ethyl carbamate (pKa = 24.6).2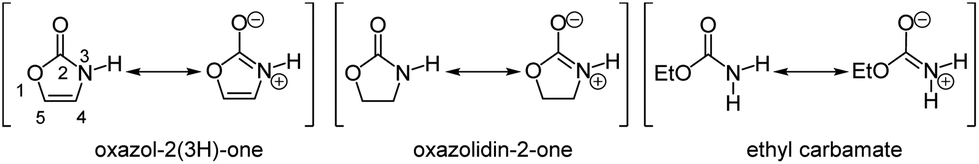 | ||
| Fig. 1 Oxazol-2(3H)-one, oxazolidin-2-one, and carbamate resonance structures. | ||
The structural motif of oxazol-2(3H)-one is not only present in muscazone (A), a toxic, psychoactive ingredient of amanita muscaria,3i.e. fly agaric, but also in highly active herbicides against broadleaf and narrowleaf weeds plaguing rice fields, such as compound B, which does not affect the growth of the crop (Fig. 2).4
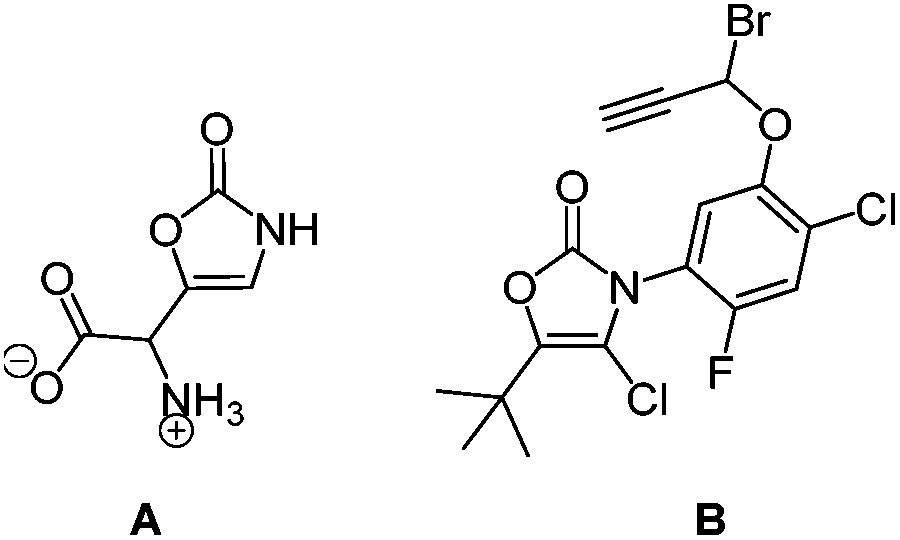 | ||
| Fig. 2 Muscazone (A), an oxazol-2(3H)-one natural product, and a highly potent herbicide (B). | ||
Chiral oxazolidin-2-ones have found broad application as Evans’ auxiliaries in enantioselective syntheses,5 such as asymmetric aldol additions, Diels–Alder reactions, 1,3-dipolar cycloadditions, and radical reactions,6 and as protected 1,2-amino alcohols in the synthesis of biologically active compounds.7 Furthermore they have reached particular attention as building blocks in natural product synthesis.8 Oxazolidinones and oxazolones can also be found in the core of a series of antimicrobial active ingredients.9 For instance, the antibiotic linezolid (zyvoxid® by Pfizer) was found to be active against multiresistant Gram positive bacteria (Fig. 3).10 Interestingly, linezolid does not act as a classical peptide transferase inhibitor but rather binds to the 23S portion of the 50S subunit of tRNA and affects an early stage of translation.11 Combretoxazolones are oxazolinones that were recognized as analogues of combretastatin A-4 (Fig. 3), a biologically active constituent of several anticancer agents. Similar activity found against the same cancer cell lines is attributed to the configurational fixation of the Z-configured double bond in the heterocyclic congeners (Fig. 3).12
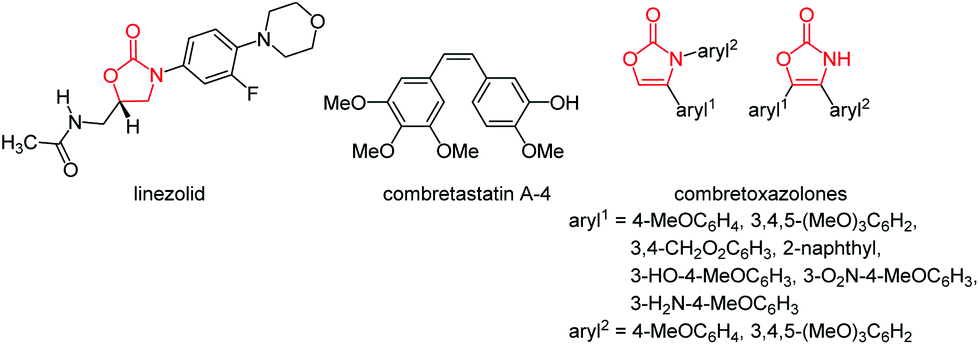 | ||
| Fig. 3 Oxazolidinone linezolid (left) and two cytotoxic combretoxazolones (right), analogues of anticancer active component combretastatin A-4 (middle). | ||
Finally, oxazol-2-ones in their own right have become valuable synthetic building blocks for multiple transformations.13 From N-acyl oxazol-2-ones14 over activation and displacements with oxygen, nitrogen, and carbon nucleophiles to give oxazoles,15 they span the range to Diels–Alder reactions with inverse electron demand, since oxazol-2-ones can be regarded as electron rich dienophiles.16
The classical syntheses of oxazoles predominantly rely on carbonyl condensation and some famous name reactions have paved the way to this important class of five-membered heterocycles.17,18 Recently, we reported an efficient three-component amidation–coupling–cyclization–isomerization (ACCI) of specifically 2,5-disubstituted oxazoles, which proceeds via an intramolecular Michael-type addition after the activating formation of an ynone by Sonogashira coupling (Scheme 1).19 By employing the heterocyclization concept and by addressing the methylene carbonyl functionality in the product for a Fischer indole synthesis a novel three-component synthesis of deep-blue luminescent 5-(3-indolyl)oxazoles could be successfully disclosed.20 Later a related strategy was reported by Wachenfeldt and co-workers who took advantage of a solvent-free amidation of N-benzyl propargylamines under microwave irradiation furnishing trisubstituted oxazoles with concomitant, aromatizing cleavage of the benzyl group in good to excellent yield.21 Although classical syntheses22 and recent contributions on Au(I)-,23–26 Pd(II)-,27 and Lewis acid28 and ionic liquid29 catalyzed cycloisomerization syntheses of structurally related oxazolidinones have been reported, the catalyzed, diversity-oriented formation of oxazol-2-ones has remained unexplored.
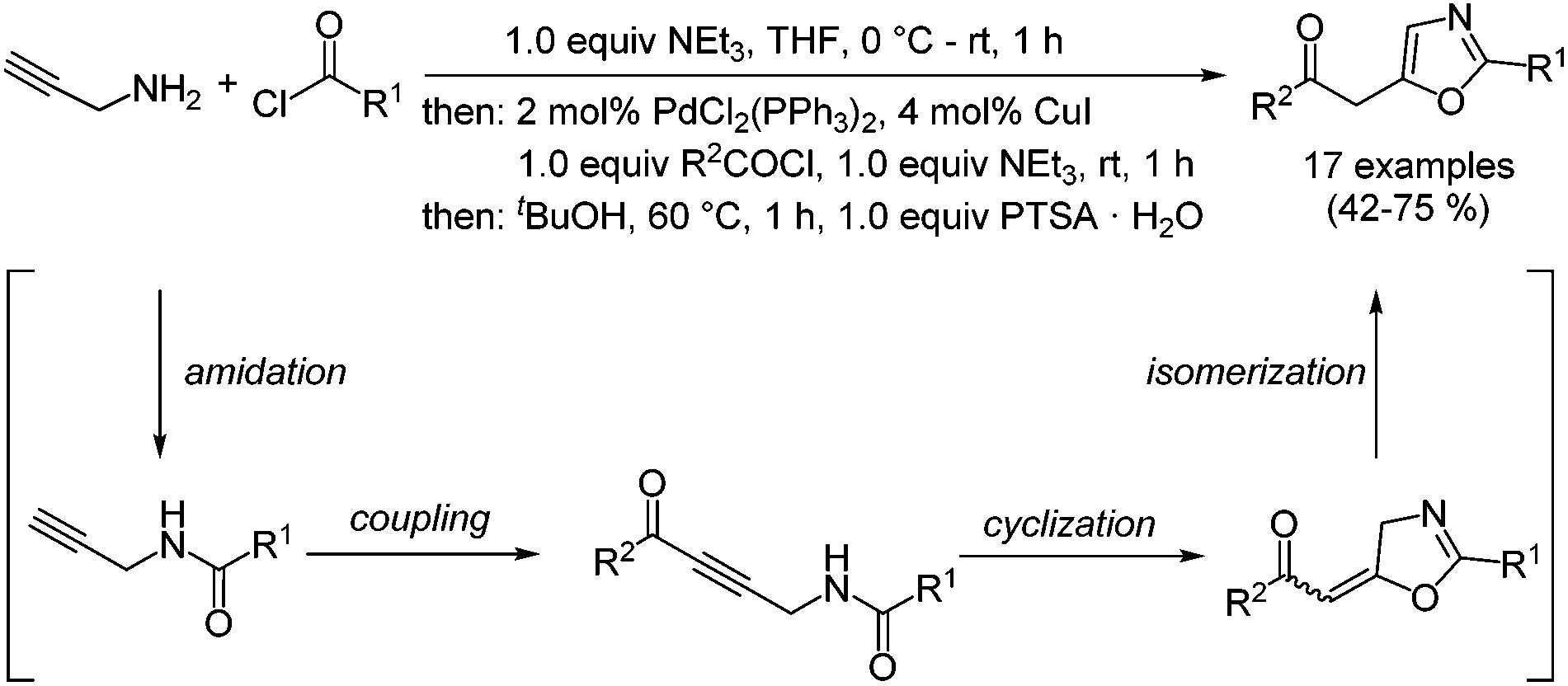 | ||
| Scheme 1 One-pot amidation–coupling–cyclization–isomerization (ACCI) synthesis of 4-substituted 5-(2-oxoethyl) oxazol-2(3H)-ones. | ||
In the course of our studies on three-component coupling–addition–cyclocondensation synthesis30 of N-Boc 2-substituted 4-iodopyrroles from acid chlorides and N-Boc protected propargyl carbamates,31 we discovered by serendipity that oxazol-2-ones were formed when 1-aryl substituted N-Boc protected propargyl carbamates were used as starting materials (Scheme 2). Important to notice is that oxazol-2-ones were already obtained upon aqueous workup of the ynone intermediate, indicating that sodium iodide was not involved in the cyclization. In addition, trifluoroacetic acid could also catalyze the cyclization–elimination reaction. Here, we report the optimization of this unprecedented formation of oxazol-2-ones and the methodological development of their synthesis by coupling–isomerization–elimination (CIE).
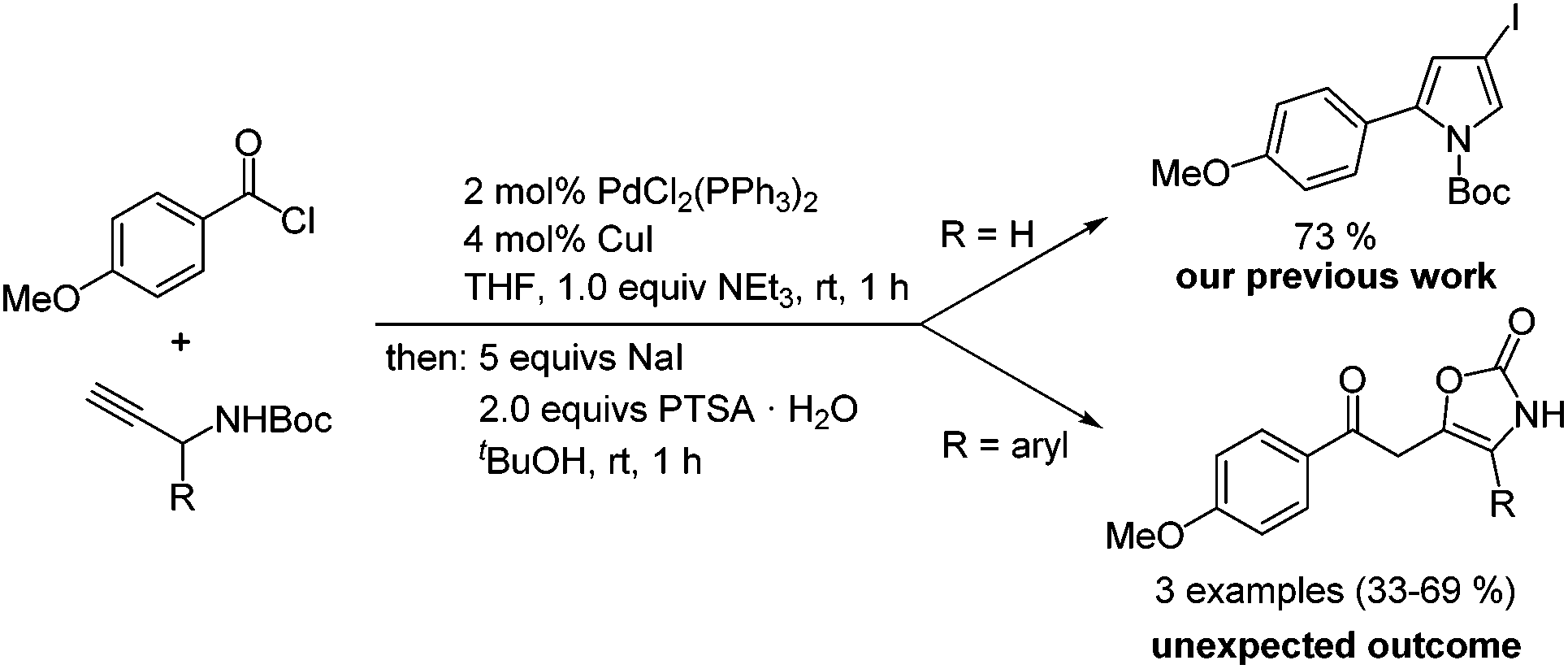 | ||
| Scheme 2 Competing formation of oxazol-2-ones in the three-component coupling–addition–cyclocondensation synthesis of N-Boc 2-substituted 4-iodopyrroles. | ||
Results and discussion
Encouraged by the serendipitous finding of oxazol-2-ones (Scheme 2) we first set out to optimize the overall sequence with respect to catalytic acid, solvent, reaction temperature, and time. As a model reaction 4-methoxybenzoylchloride (1a) and tert-butyl(1-phenylprop-2-yn-1-yl) carbamate (2a) were chosen as reaction partners. For ynone formation the standard conditions were employed (Scheme 3).32 However, we were never able to isolate the corresponding ynone with this peculiar substitution pattern (vide infra). First, the effect of various Brønsted acids on the isomerization–elimination was investigated at room temperature for 1 h (Table 1).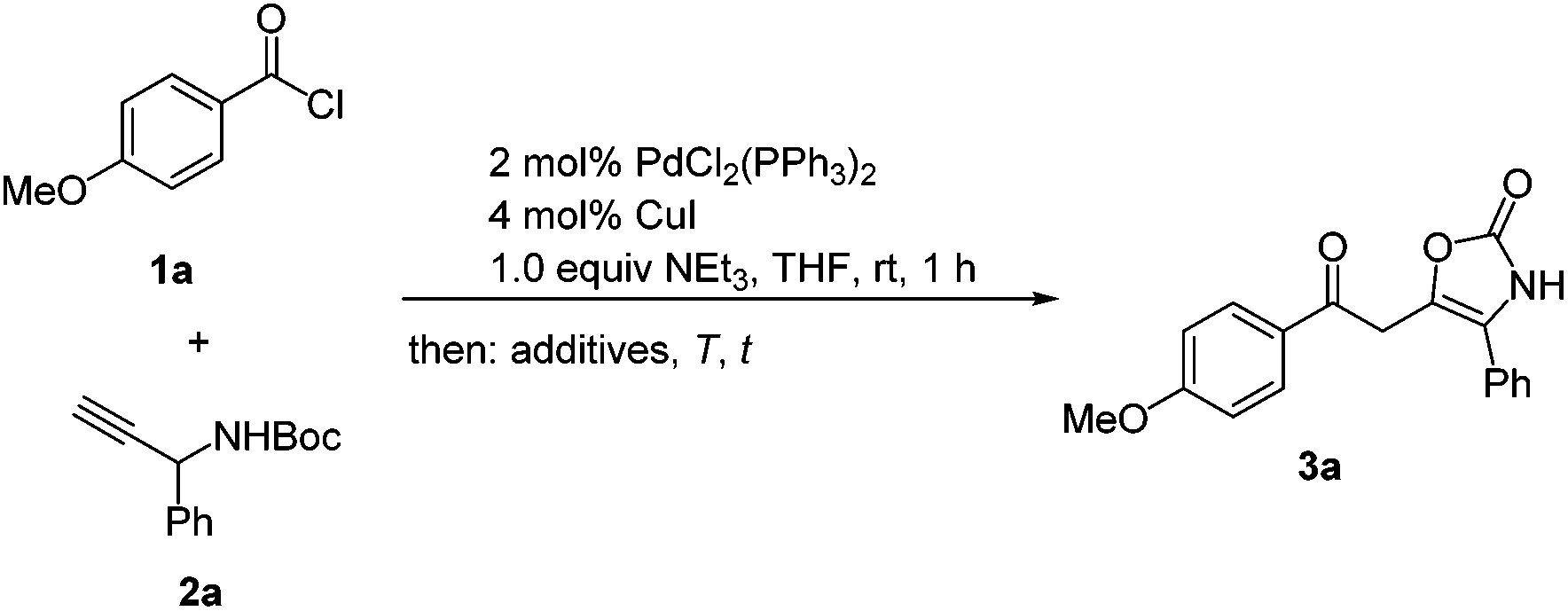 | ||
| Scheme 3 Optimization of the coupling–isomerization–elimination synthesis (CIE) of the 2-oxoethyl oxazol-2(3H)-one 3a. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| Entry | Acidb | Oxazol-2(3H)-one 3ac |
|---|---|---|
| a All reactions were performed on a 1.0 mmol scale at concentrations c0(1a) = c0(2a) = 0.2 m, the isomerization–elimination step was performed at room temp for 1 h. b 2.0 equiv. of acid were added in the isomerization–elimination step. c Isolated yields after chromatography on silica gel. d 37% aqueous solution. e Impure product. | ||
| 1 | PTSA·H2O, tBuOH | 72% |
| 2 | PTSA·H2O | 70% |
| 3 | CF3CO2H | 70% |
| 4 | CH3SO3H | 59% |
| 5 | HCld | 37% |
| 6 | CH3CO2H | 40%e |
| 7 | H2O | 33% |
It could be shown that the addition of tBuOH as a co-solvent as under 4-iodopyrrole formation conditions was not required for PTSA monohydrate (pKa ≈ 0.7) as an acid (Table 1, entries 1 and 2). A slightly increased acidity by changing the acid component to trifluoroacetic acid (pKa ≈ 0.2) did not increase the yield (Table 1, entry 3), whereas stronger acids, such as methane sulfonic acid (pKa ≈ −1.9) and concentrated hydrochloric acid, even diminished the yields of oxazolone 3a (Table 1, entries 4 and 5). Weaker acetic acid and water were not efficient proton sources for the isomerization–elimination (Table 1, entries 6 and 7).
Further on, the solvent, reaction temperature, and reaction time were screened maintaining PTSA monohydrate as a catalyst for the product forming step (Table 2).
a
| Entry | Solvent | Temperature T [°C] | Time t [min] | Oxazol-2(3H)-one 3ab |
|---|---|---|---|---|
| a The reactions were performed on a 1.0 mmol scale at concentrations c0(1a) = c0(2a) = 0.2 m, the coupling step was performed at room temp for 1 h and the isomerization–elimination step was performed at room temp for 1 h and 2.0 equiv. of PTSA·H2O were added in the isomerization–elimination step. b Isolated yields after chromatography on silica gel. c c 0(1a) = c0(2a) = 0.4 m. d 1.0 equiv. of NEt3 (99%), first dried with KOH pellets then distilled from Na/benzophenone. e 1.0 equiv. of NEt3 (99%), dried with KOH pellets. f 2.0 equiv. of NEt3 (99%), dried with KOH pellets. g 2.0 equiv. of NEt3 (99%), first dried with KOH pellets then distilled from Na/benzophenone and addition of 3.0 equiv. of PTSA·H2O in the isomerization–elimination step. | ||||
| 1c | THFd | 23 | 60 | 66% |
| 2c | THF | 0–4 | 60 | 68% |
| 3c | THFd | 23 | 120 | 65% |
| 4e | THF | 23 | 60 | 68% |
| 5f | THF | 23 | 60 | 65% |
| 6e | THF | 50 | 60 | 58% |
| 7 | THF | 23 | 30 | 72% |
| 8g | THF | 23 | 30 | 65% |
| 9f | THF | 23 | 30 | 61% |
| 10f | THF | 23 | 15 | 55% |
| 11f | THF | 23 | 10 | 55% |
| 12f | 1,4-dioxane | 23 | 30 | 52% |
| 13c | CH2Cl2 | 23 | 60 | 57% |
As a solvent best suitable for both the coupling and the isomerization–elimination steps THF gave higher yields (Table 2, entries 1–11) than 1,4-dioxane and dichloromethane (Table 2, entries 12 and 13). Performing the coupling step with carefully dried triethylamine (distillation from Na/benzophenone) as a base and thereby minimizing the water concentration gave comparable yields (Table 2, entries 1–3) to simply dried triethylamine (storage with KOH pellets) (Table 2, entries 4–6). For the sake of practicability the employment of peculiarly dried triethylamine is therefore not necessary. Complete conversion can be achieved after 30 min at room temperature (entry 7) whereas the addition of 2 equiv. of triethylamine does not improve the yield (entries 8 and 9). Further reduction of the reaction time results in lower yields (entries 10 and 11). Increasing the reaction temperature to 50 °C gave already lower yields (Table 2, entry 6). In summary, the optimal conditions of the coupling–isomerization–elimination sequence are given under entry 7. With these conditions in hand, the methodological scope and limitation study was performed with various acid chlorides 1 and 1-aryl substituted N-Boc protected propargyl carbamates 225,33,34 to give 4-substituted 5-(2-oxoethyl) oxazol-2(3H)-ones 3 in moderate to good yields (Scheme 4, Table 3).
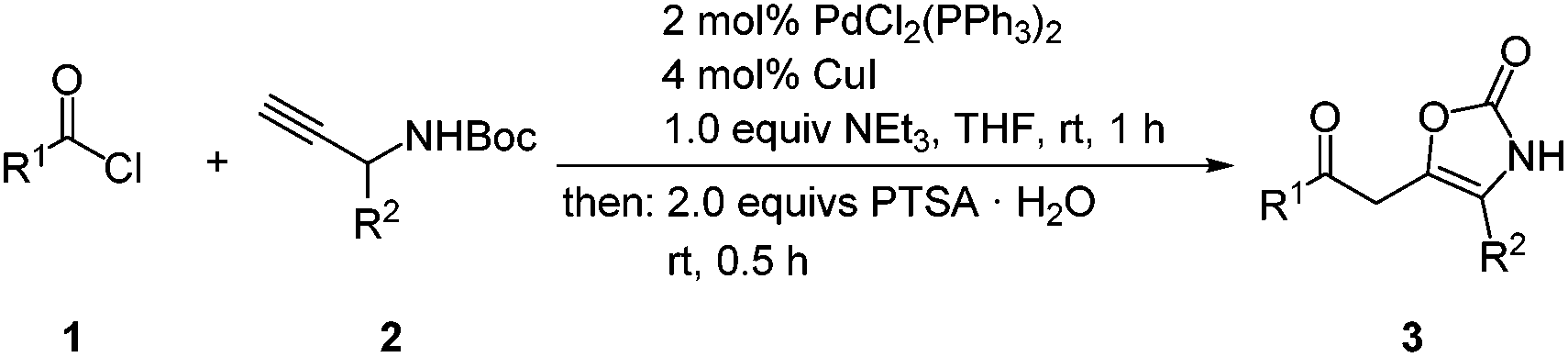 | ||
| Scheme 4 General procedure for the coupling–isomerization–elimination synthesis of oxazole-2(3H)-ones 3. | ||
a
| Entry | Acid chloride 1 | N-Boc 1-aryl-propargyl carbamate 2 | 4-Substituted 5-(2-oxoethyl) oxazol-2(3H)-one 3 (yield)b |
|---|---|---|---|
| a The reactions were performed at a 1 mmol scale with concentrations c0(1a) = c0(2a) = 0.4 m, the coupling step was performed at room temp for 1 h and the isomerization–elimination step was performed at room temp for 0.5 h. b Isolated yields after chromatography on silica gel. c The reaction was performed at 0.5 mmol scale. d The coupling was performed for 1.5 h (monitored by TLC). e The coupling was performed in 3.5 h (monitored by TLC). f The coupling was performed for 2.0 h (monitored by TLC). g The isomerization–elimination step was performed for 1 h (monitored by TLC). h The coupling was performed for 24 h (monitored by TLC). | |||
| 1 | R1 = p-MeOC6H4 (1a) | R2 = Ph (2a) | 3a (73%) |
| 2 | R1 = m-MeOC6H4 (1b) | 2a | 3b (70%) |
| 3c,d | R1 = o-MeOC6H4 (1c) | 2a | 3c (81%) |
| 4 | R1 = p-MeC6H4 (1d) | 2a | 3d (47%) |
| 5 | R1 = m-MeC6H4 (1e) | 2a | 3e (49%) |
| 6c,e | R1 = o-MeC6H4 (1f) | 2a | 3f (55%) |
| 7 | R1 = p-PhC6H4 (1g) | 2a | 3g (61%) |
| 8 | R1 = Ph (1h) | 2a | 3h (46%) |
| 9 | R1 = p-FC6H4 (1i) | 2a | 3i (55%) |
| 10 | R1 = m-FC6H4 (1j) | 2a | 3j (71%) |
| 11 | R1 = o-FC6H4 (1k) | 2a | 3k (57%) |
| 12f | R1 = 3,5-F2C6H3 (1l) | 2a | 3l (30%) |
| 13c | R1 = p-BrC6H4 (1m) | 2a | 3m (66%) |
| 14 | R1 = 2-thienyl (1n) | 2a | 3n (62%) |
| 15c,g | R1 = iPr (1o) | 2a | 3o (54%) |
| 16c,h | R1 = cyclopropyl (1p) | 2a | 3p (24%) |
| 17 | R1 = 1-adamantyl (1q) | 2a | 3q (27%) |
| 18c | 1a | R2 = p-MeOC6H4 (2b) | 3r (41%) |
| 19 | 1a | R2 = p-MeC6H4 (2c) | 3s (37%) |
| 20 | 1a | R2 = p-ClC6H4 (2d) | 3t (55%) |
| 21g | 1a | R2 = o-ClC6H4 (2e) | 3u (10%) |
| 22 | 1a | R2 = 2-thienyl (2f) | 3v (43%) |
| 23 | 1n | 2c | 3w (60%) |
The structures of the 5-substituted 2-oxoethyl oxazol-2(3H)-ones 3 were unambiguously assigned by 1H and 13C NMR and IR spectroscopy, mass spectrometry, and combustion analysis or high resolution mass spectrometry. Most characteristically, the methylene protons between the ketone and the oxazol-2(3H)-one appear as singlets at δ 3.90–3.95 for aliphatic substituents R1 and at δ 4.15–4.54 for (hetero)aryl substituents R1 in the 1H NMR spectra. The corresponding resonances of the methylene carbon nuclei can be found in the 13C NMR spectra between δ 35.0 and 40.3. The ketone carbon nuclei appear between δ 188.0 and 209.8 depending on the nature of the (hetero)aromatic or (cyclo)aliphatic substituent. The oxazolone carbonyl nuclei can be detected at higher field in a very narrow range around δ 155. In the proton spectra at lower field the heterocyclic NH-amide protons appear as broad signals between δ 11.0 and 11.3. In the IR spectra the two distinct carbonyl stretching vibrations can be assigned for the methylene ketones appearing between 1694 and 1661 cm−1 and for the oxazol-2-ones in a range from 1753 to 1736 cm−1 typical for oxazol-2(3H)-one.35 The structure was additionally corroborated by an X-ray structure analysis of 5-substituted 2-oxoethyl oxazol-2(3H)-one 3r indicating the formation of unsymmetrical dimers by amide hydrogen bonding in the solid state (Fig. 4).36
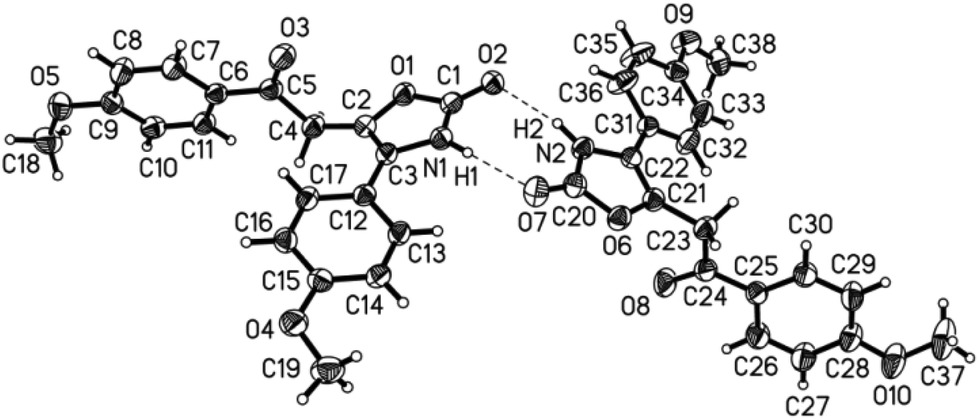 | ||
| Fig. 4 ORTEP plot of the molecular structure of the 4-substituted 5-(2-oxoethyl) oxazol-2(3H)-one 3r. | ||
The presented one-pot coupling–isomerization–elimination (CIE) synthesis of 4-substituted 5-(2-oxoethyl) oxazol-2(3H)-ones 3 can be performed with a broad range of (hetero)aroyl chlorides ranging from electron rich (Table 3, entries 1, 3, 4, 6, 7, 18–22) over electroneutral (Table 3, entries 5 and 8) to electron deficient (Table 3, entries 2, 9–13), heterocyclic (Table 3, entries 14 and 23) and even certain (cyclo)aliphatic acid chlorides (Table 3, entries 15–17). The substitution pattern of N-Boc protected 1-(hetero)aryl propargyl carbamates 2 also allows for the implementation of electron releasing and withdrawing substituents as well as heteroaromatic substituents, however, an ortho-substituent apparently hampers the cycloisomerization (Table 3, entry 21).
As a consequence of the elusiveness of the ynone coupling product and the subsequent acid mediated isomerization–elimination to furnish 4-substituted 5-(2-oxoethyl) oxazol-2(3H)-ones 3 we reasoned that in case of a strict absence of an acid the tert-butyl group will be kept intact and thereby the corresponding 2-tert-butoxy oxazole derivative should be accessible. Indeed, upon reacting p-methoxy benzoyl chloride (1a) and N-Boc protected 1-phenyl propargyl carbamate (2a) under modified Sonogashira coupling conditions32 and upon basic aqueous workup and chromatography on silica gel the 2-tert-butoxy oxazole 5 was obtained in 71% yield (Scheme 5).
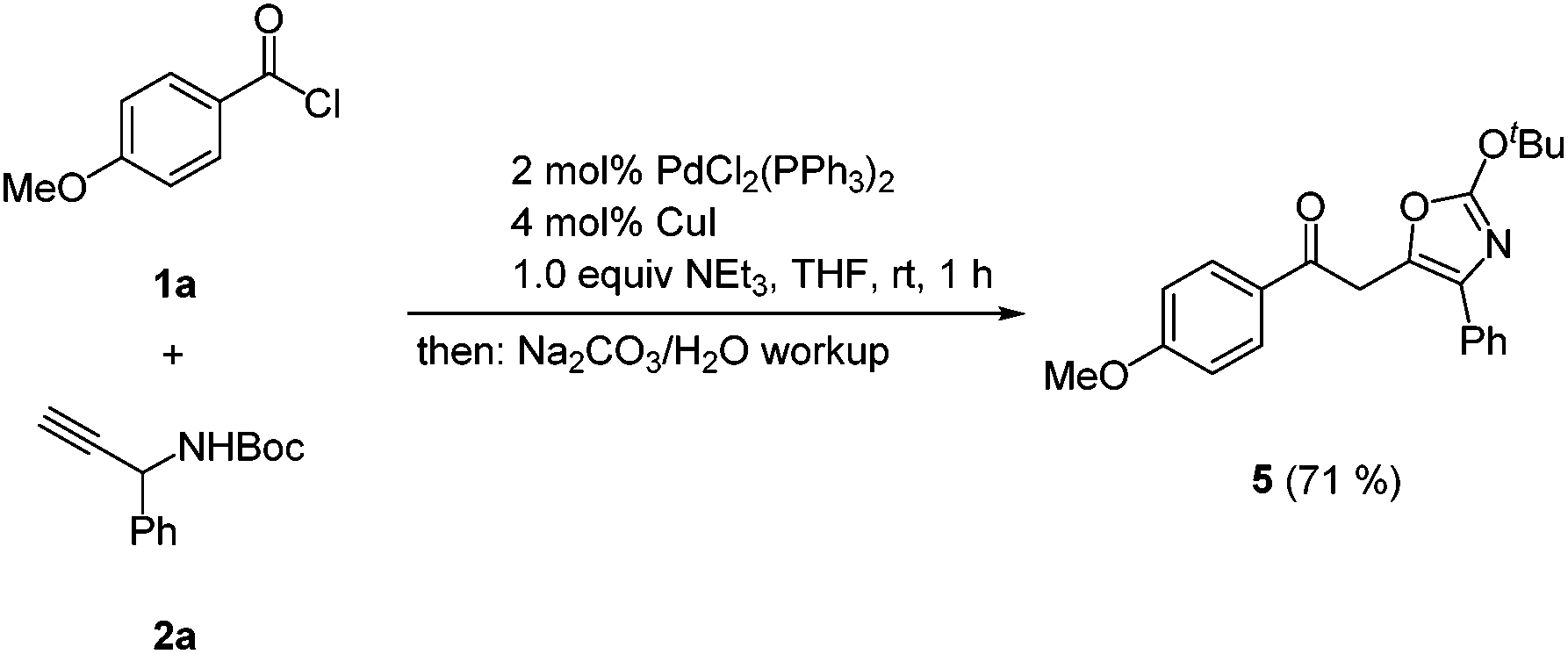 | ||
| Scheme 5 Coupling-cycloisomerization synthesis of the 4-substituted 5-(2-oxoethyl) 2-tert-butoxy oxazole 5. | ||
Mechanistically, this observation can be interpreted as a consequence of a steric interaction of the tert-butyl carbamate with the 1-aryl moiety which is present in the N-Boc substituted 1-aryl substituted propargyl carbamate. Thus, the oxygen atom of the carbamate immediately undergoes a Michael-type on the triple bond of the ynone that is formed by the cross-coupling reaction, resulting in the observed cyclization. In the absence of 1-aryl substitution the ynone becomes persistent to spontaneous Michael addition, as supported by the consecutive transformation to 4-iodo pyrroles.31 Hence, the acid in this novel one-pot CIE synthesis of 4-substituted 5-(2-oxoethyl) oxazol-2(3H)-ones cannot be attributed to the cycloisomerization, but rather to induce tert-butyl cleavage.
Conclusion
Here, we have unraveled and developed an unusual coupling–isomerization–elimination synthesis of 4-substituted 5-(2-oxoethyl) oxazol-2(3H)-ones, a subclass of side-chain functionalized oxazol-2(3H)-one derivatives. The substitution pattern at the two diversity points, i.e. the acid chloride and the N-Boc protected 1-aryl substituted propargyl carbamate is fairly broad and allows electronically diverse decoration of the title compound class by a straightforward one-pot protocol. The isolation of a 5-substituted 2-oxoethyl 2-tert-butoxy oxazole product as a consequence of strict absence of acid not only elucidates the overall mechanistic rationale but also sets the stage for further developing one-pot methodologies with this intermediate. Further studies directed to develop propargyl amide and carbamate based one-pot sequences initiated by cross-coupling are currently underway.Experimental
Synthetic procedures
| Entry | Acid chloride 1 [mg] (mmol) | N-Boc 1-arylpropargyl carbanate 2 [mg] (mmol) | 4-Substituted 5-(2-oxoethyl) oxazol-2(3H)-one 3 [mg] (%) |
|---|---|---|---|
| a The coupling was performed for 1.5 h (monitored by TLC). b The coupling was performed for 3.5 h (monitored by TLC). c The coupling was performed for 2.0 h (monitored by TLC). d The coupling was performed for 24 h (monitored by TLC). e The isomerization was performed for 1.0 h (monitored by TLC). | |||
| 1 | 176 (1.00) of 1a | 232 (1.00) of 2a | 224 (73) of 3a |
| 2 | 173 (1.00) of 1b | 232 (1.00) of 2a | 215 (70) of 3b |
| 3a | 88 (0.5) of 1c | 116 (0.50) of 2a | 125 (81) of 3c |
| 4 | 158 (1.00) of 1d | 232 (1.00) of 2a | 136 (47) of 3d |
| 5 | 158 (1.00) of 1e | 232 (1.00) of 2a | 145 (49) of 3e |
| 6b | 79 (0.50) of 1f | 116 (0.50) of 2a | 81 (55) of 3f |
| 7 | 216 (1.00) of 1g | 232 (1.00) of 2a | 217 (61) of 3g |
| 8 | 142 (1.00) of 1h | 232 (1.00) of 2a | 130 (46) of 3h |
| 9 | 165 (1.00) of 1i | 232 (1.00) of 2a | 163 (55) of 3i |
| 10 | 162 (1.00) of 1j | 232 (1.00) of 2a | 210 (71) of 3j |
| 11 | 162 (1.00) of 1k | 232 (1.00) of 2a | 169 (57) of 3k |
| 12c | 181 (1.00) of 1l | 232 (1.00) of 2a | 95 (30) of 3l |
| 13 | 112 (0.50) of 1m | 116 (0.50) of 2a | 118 (66) of 3m |
| 14 | 150 (1.00) of 1n | 232 (1.00) of 2a | 178 (62) of 3n |
| 15d | 54 (0.50) of 1o | 116 (0.50) of 2a | 67 (54) of 3o |
| 16d | 52 (0.50) of 1p | 116 (0.50) of 2a | 30 (24) of 3p |
| 17 | 203 (1.00) of 1q | 232 (1.00) of 2a | 93 (27) of 3q |
| 18 | 88 (0.50) of 1a | 131 (0.50) of 2b | 69 (41) of 3r |
| 19 | 176 (1.00) of 1a | 246 (1.00) of 2c | 119 (37) of 3s |
| 20 | 176 (1.00) of 1a | 266 (1.00) of 2d | 190 (55) of 3t |
| 21e | 176 (1.00) of 1a | 266 (1.00) of 2e | 34 (10) of 3u |
| 22 | 176 (1.00) of 1a | 237 (1.00) of 2f | 135 (43) of 3v |
| 23 | 150 (1.00) of 1n | 246 (1.00) of 2c | 179 (60) of 3w |
:1) as a pale brown solid.
R
f (petroleum ether/ethyl acetate 1:1) = 0.32, Mp 161–166 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 3.85 (s, 3 H), 4.41 (s, 2 H), 7.02–7.12 (m, 2 H), 7.32–7.50 (m, 5 H), 7.97–8.08 (m, 2 H), 11.15 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 35.5 (CH2), 55.7 (CH3), 114.1 (CH), 123.9 (Cquat), 126.0 (CH), 127.2 (Cquat), 128.4 (CH), 128.7 (Cquat), 129.0 (CH), 130.5 (Cquat), 130.9 (CH), 155.0 (Cquat), 163.7 (Cquat), 193.3 (Cquat). MS (EI), m/z: 309 (M+, 2), 135 (C8H7O2+, 100), 107 (C7H7O+, 8), 92 (C6H4O+, 5), 77 (C6H5+, 13). IR (ATR), ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) [cm−1]: 1736 (s), 1676 (m). Anal. calcd for C18H15NO4 (309.3): C 69.89, H 4.89, N 4.53; Found: C 69.73, H 4.84, N 4.44.
[cm−1]: 1736 (s), 1676 (m). Anal. calcd for C18H15NO4 (309.3): C 69.89, H 4.89, N 4.53; Found: C 69.73, H 4.84, N 4.44.
:1 to 1:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.12, Mp 151–154 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 3.80 (s, 3 H), 4.49 (s, 2 H), 7.16–7.30 (m, 1 H), 7.32–7.57 (m, 7 H), 7.58–7.69 (m, 1 H), 11.17 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 36.1 (CH2), 55.4 (CH3), 113.0 (CH), 119.9 (CH), 120.9 (CH), 124.1 (Cquat), 126.0 (CH), 127.1 (Cquat), 128.5 (CH), 129.0 (CH), 130.0 (CH), 130.2 (Cquat), 137.2 (Cquat), 155.0 (Cquat), 159.5 (Cquat), 194.9 (Cquat). MS (EI), m/z: 309 (M+, 11), 174 ((M − C8H7O2)+, 4), 135 (C8H7O2+, 100), 107 (C7H7O+, 15), 103 (11), 92 (C6H4O+, 6), 77 (C6H5+, 13). IR (ATR), [cm−1]: 1751 (s), 1684 (m). Anal. calcd for C18H15NO4 (309.3): C 69.89, H 4.89, N 4.53; Found: C 69.81, H 5.13, N 4.31.
:1) as a colorless solid.
R
f (petroleum ether/ethyl acetate 1:1) = 0.32, Mp 177–181 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 3.79 (s, 3 H), 4.33 (s, 2 H), 7.04 (dt, J = 7.4 Hz, J = 0.7 Hz, 1 H), 7.17 (d, J = 8.1 Hz, 1 H), 7.30–7.50 (m, 5 H), 7.52–7.64 (m, 2 H), 11.12 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 40.3 (CH2), 55.8 (CH3), 112.5 (CH), 120.6 (CH), 123.8 (Cquat), 126.0 (CH), 126.8 (Cquat), 127.2 (Cquat), 128.4 (CH), 129.0 (CH), 129.8 (CH), 130.4 (Cquat), 134.4 (CH), 155.0 (Cquat), 158.3 (Cquat), 196.6 (Cquat). MS (EI), m/z: 309 (M+, 2), 149 (10), 135 (C8H7O2+, 100), 105 (12), 77 (C6H5+, 23), 44 (12), 43 (25), 40 (60). IR (ATR), [cm−1]: 1744 (s), 1668 (m). Anal. calcd for C18H15NO4 (309.3): C 69.89, H 4.89, N 4.53; Found: C 69.88, H 4.87, N 4.32.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.14, Mp 187–193 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 2.39 (s, 3 H), 4.45 (s, 2 H), 7.30–7.50 (m, 7 H), 7.89–7.99 (m, 2 H), 11.15 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 21.2 (CH3), 35.7 (CH2), 124.0 (Cquat), 126.0 (CH), 127.1 (Cquat), 128.4 (CH), 128.6 (CH), 129.0 (CH), 129.4 (CH), 130.4 (Cquat), 133.3 (Cquat), 144.4 (Cquat), 155.0 (Cquat), 194.5 (Cquat). MS (EI), m/z: 293 (M+, 6), 174 ((M − C8H7O)+, 2), 119 (C8H7O+, 100), 91 (C7H7+, 18), 77 (C6H5+, 4). IR (ATR), [cm−1]: 1742 (s), 1684 (m). HRMS (ESI): m/z calcd for C18H16NO3 ((M + H)+): 294.1125; Found: 294.1119.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.16, Mp 149–160 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 2.39 (s, 3 H), 4.45 (s, 2 H), 7.30–7.50 (m, 7 H), 7.89–7.99 (m, 2 H), 11.15 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 20.9 (CH3), 35.9 (CH2), 124.0 (Cquat), 125.7 (CH), 126.0 (CH), 127.1 (Cquat), 128.4 (CH), 128.7 (CH), 128.8 (CH), 129.0 (CH), 130.3 (Cquat), 134.4 (CH), 135.8 (Cquat), 138.3 (Cquat), 155.0 (Cquat), 195.1 (Cquat). MS (EI), m/z: 293 (M+, 8), 174 ((M − C8H7O)+, 4), 119 (C8H7O+, 100), 103 (10), 91 (C7H7+, 21), 77 (C6H5+, 5). IR (ATR), [cm−1]: 1748 (s), 1686 (m). HRMS (ESI): m/z calcd for C18H16NO3 ((M + H)+): 294.1125; Found: 294.1124.
:1) as a colorless solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.43, Mp 169–172 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 2.38 (s, 3 H), 4.40 (s, 2 H), 7.20–7.58 (m, 8 H), 7.80–7.98 (m, 1 H), 11.15 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 20.8 (CH3), 38.5 (CH2), 124.1 (Cquat), 125.8 (CH), 126.0 (CH), 127.1 (Cquat), 128.4 (CH), 129.0 (CH), 129.2 (CH), 130.3 (Cquat), 131.8 (CH), 131.9 (CH), 136.5 (Cquat), 137.7 (Cquat), 154.9 (Cquat), 198.5 (Cquat). MS (EI), m/z: 293 (M+, 3), 174 ((M − C8H7O)+, 2), 119 (C8H7O+, 100), 116 (23), 115 (11), 105 (C7H5O+, 10), 103 (11), 91 (C7H7+, 30), 77 (C6H5+, 9), 43 (20), 40 (36). IR (ATR), [cm−1]: 1737 (s), 1692 (m), 1680 (m). Anal. calcd for C18H15NO3 (293.3): C 73.71, H 5.15, N 4.78; Found: C 73.49, H 5.28, N 4.64.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.14, Mp 189–201 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 4.54 (s, 2 H), 7.30–7.59 (m, 8 H), 7.71–7.81 (m, 2 H), 7.81–7.92 (m, 2 H), 8.04–8.20 (m, 2 H), 11.18 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 35.9 (CH2), 124.1 (Cquat), 126.0 (CH), 127.0 (CH), 127.07 (CH), 127.10 (Cquat), 128.4 (CH), 128.5 (CH), 129.0 (CH), 129.1 (CH), 129.2 (CH), 130.2 (Cquat), 134.6 (Cquat), 138.8 (Cquat), 145.1 (Cquat), 155.0 (Cquat), 194.6 (Cquat). MS (EI), m/z: 355 (M+, 3), 182 (14), 181 (C13H9O+, 100), 174 ((M − C13H9O)+, 1), 153 (C12H9+, 13), 152 (21), 77 (C6H5+, 3). IR (ATR), [cm−1]: 1746 (s), 1674 (m). Anal. calcd for C23H17NO3 (355.4): C 77.73, H 4.82, N 3.94; Found: C 77.53, H 4.87, N 3.91.
:1) as a colorless solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.16, Mp 189–194 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 4.51 (s, 2 H), 7.32–7.48 (m, 5 H), 7.51–7.61 (m, 2 H), 7.64–7.74 (m, 1 H), 7.97–8.10 (m, 2 H), 11.16 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 35.9 (CH2), 124.1 (Cquat), 126.0 (CH), 127.1 (Cquat), 128.41 (CH), 128.44 (CH), 128.8 (CH), 129.0 (CH), 130.2 (Cquat), 133.8 (CH), 135.8 (Cquat), 155.0 (Cquat), 195.0 (Cquat). MS (EI), m/z: 279 (M+, 9), 174 ((M − C7H5O)+, 9), 105 (C7H5O+, 100), 103 (17), 77 (C6H5+, 25). IR (ATR), [cm−1]: 1742 (s), 1690 (s). Anal. calcd for C17H13NO3 (279.3): C 73.11, H 4.69, N 5.02; Found: C 72.95, H 4.80, N 4.89.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.16, Mp 164–172 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 4.50 (s, 2 H), 7.26–7.55 (m, 7 H), 8.02–8.23 (m, 2 H), 11.16 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 35.9 (CH2), 115.9 (d, J = 22 Hz, CH), 124.1 (Cquat), 126.0 (CH), 127.1 (Cquat,), 128.4 (CH), 129.0 (CH), 130.1 (Cquat), 131.5 (d, J = 10 Hz, CH), 132.6 (d, J = 3 Hz, Cquat), 155.0 (Cquat), 165.4 (d, J = 253 Hz, Cquat), 193.7 (Cquat). MS (EI), m/z: 297 (M+, 8), 174 ((M − C7H4FO)+, 19), 123 (C7H4FO+, 100), 104 (C7H4O+, 9), 103 (18), 95 (C6H4F+, 16), 77 (C6H5+, 10). IR (ATR), [cm−1]: 1748 (s), 1688 (m). Anal. calcd for C17H12FNO3 (297.3): C 68.68, H 4.07, N 4.71; Found: C 68.65, H 4.21, N 4.57.
:1 to 1:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.15, Mp 185–192 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 4.53 (s, 2 H), 7.28–7.49 (m, 5 H), 7.49–7.68 (m, 2 H), 7.75–7.96 (m, 2 H), 11.17 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 36.1 (CH2), 115.1 (d, J = 22 Hz, CH), 120.7 (d, J = 21 Hz, CH), 124.2 (Cquat), 124.6 (d, J = 3 Hz, CH), 126.1 (CH), 127.0 (Cquat,), 128.5 (CH), 129.0 (CH), 129.9 (Cquat), 131.1 (d, J = 8 Hz, CH), 137.9 (d, J = 6 Hz, Cquat), 155.0 (Cquat), 162.2 (d, J = 245 Hz, Cquat), 194.2 (d, J = 2 Hz, Cquat). MS (EI), m/z: 297 (M+, 19), 175 (11), 174 ((M − C7H4FO)+, 100), 123 (C7H4FO+, 88), 104 (C7H4O+, 12), 103 (48), 95 (C6H4F+, 21), 77 (C6H5+, 12), 43 (10). IR (ATR), [cm−1]: 1753 (s), 1692 (m). Anal. calcd for C17H12FNO3 (297.3): C 68.68, H 4.07, N 4.71; Found: C 68.53, H 4.33, N 4.51.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.16, Mp 162–176 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 4.39 (d, J = 1.6 Hz, 2 H), 7.14–7.58 (m, 7 H), 7.64–7.79 (m, 1 H), 7.88 (td, J = 7.7 Hz, J = 1.7 Hz, 1 H), 11.17 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 39.6 (d, J = 7 Hz, CH2), 117.0 (d, J = 23 Hz, CH), 124.2 (Cquat), 124.7 (d, J = 12 Hz, Cquat), 124.9 (d, J = 3 Hz, CH), 126.1 (CH), 127.0 (Cquat), 128.5 (CH), 129.0 (CH), 129.7 (d, J = 2 Hz, Cquat), 130.5 (d, J = 2 Hz, CH), 135.6 (d, J = 9 Hz, CH), 155.0 (Cquat), 161.1 (d, J = 255 Hz, Cquat), 193.1 (d, J = 4 Hz, Cquat). MS (EI), m/z: 297 (M+, 13), 174 ((M − C7H4FO)+, 40), 123 (C7H4FO+, 100), 104 (C7H4O+, 10), 103 (27), 95 (C6H4F+, 11), 77 (C6H5+, 9). IR (ATR), [cm−1]: 1744 (s), 1686 (s). Anal. calcd for C17H12FNO3 (297.3): C 68.68, H 4.07, N 4.71; Found: C 68.47, H 4.28, N 4.47.
:1 to 1:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.21, Mp 185–197 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 4.56 (s, 2 H), 7.31–7.48 (m, 5 H), 7.62 (tt, J = 9.0 Hz, J = 2.3 Hz, 1 H), 7.68–7.79 (m, 2 H), 11.18 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 36.2 (CH2), 109.1 (t, J = 26 Hz, CH), 111.8 (dd, J = 18 Hz, J = 8 Hz, CH), 124.4 (Cquat), 126.1 (CH), 127.0 (Cquat), 128.5 (CH), 129.0 (CH), 129.5 (Cquat), 138.9 (t, J = 8 Hz, Cquat), 155.0 (Cquat), 162.5 (dd, J = 248 Hz, J = 12 Hz, Cquat), 193.2 (t, J = 2 Hz, Cquat). MS (EI), m/z: 315 (M+, 14), 277 ((M − F2)+, 11), 183 (11), 175 (14), 174 ((M − C7H3F2O)+, 100), 141 (C7H3F2O+, 69), 149 (12), 138 (20), 125 (16), 113 (40), 111 (25), 109 (13), 105 (C7H5O+, 45), 104 (26), 103 (59), 99 (14), 97 (30), 95 (17), 85 (26), 83 (24), 81 (15), 77 (C6H5+, 30), 71 (33), 69 (23), 57 (44), 55 (20), 43 (43). IR (ATR), [cm−1]: 1749 (s), 1726 (m), 1694 (m). Anal. calcd for C17H11F2NO3 (315.3): C 64.76, H 3.52, N 4.44; Found: C 64.47, H 3.78, N 4.23.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.16, Mp 198–201 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 4.50 (s, 2 H), 7.31–7.41 (m, 1 H), 7.41–7.49 (m, 4 H), 7.73–7.83 (m, 2 H), 7.92–8.02 (m, 2 H), 11.17 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 35.9 (CH2), 124.2 (Cquat), 126.0 (CH), 127.0 (Cquat), 128.0 (CH), 128.4 (Cquat), 129.0 (CH), 129.9 (Cquat), 130.4 (CH), 131.9 (CH), 134.8 (Cquat), 155.0 (Cquat), 194.4 (Cquat). MS (EI), m/z: 359 (M(81Br)+, 6), 357 (M(79Br)+, 6), 277 ((M − HBr)+, 15), 185 (C7H481BrO+, 97), 183 (C7H479BrO+, 100), 174 ((M − C7H4BrO)+, 37), 172 (30), 157 (C6H481Br+, 18), 155 (C6H479Br+, 18), 131 (10), 121 (19), 105 (33), 104 (24), 103 (48), 77 (C6H5+, 32), 76 (13), 69 (11), 43 (25). IR (ATR), [cm−1]: 1744 (s), 1690 (m). Anal. calcd for C17H12BrNO3 (358.2): C 57.00, H 3.38, N 3.91; Found: C 57.13, H 3.49, N 3.84.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.13, Mp 188–190 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 4.42 (s, 2 H), 7.05–7.32 (m, 1 H), 7.33–7.66 (m, 5 H), 7.88–8.44 (m, 2 H), 11.19 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 36.0 (CH2), 124.3 (Cquat), 126.1 (CH), 127.0 (Cquat), 128.6 (CH), 129.0 (CH), 129.1 (CH), 129.9 (Cquat), 134.8 (CH), 136.1 (CH), 142.6 (Cquat), 154.9 (Cquat), 188.0 (Cquat). MS (EI), m/z: 285 (M+, 15), 174 ((M − C5H3OS)+, 25), 111 (C5H3OS+, 100), 103 (24), 83 (C4H3S+, 4), 77 (C6H5+, 11). IR (ATR), [cm−1]: 1753 (s), 1661 (m). Anal. calcd for C15H11NO3S (285.3): C 63.14, H 3.89, N 4.91; Found: C 63.29, H 3.95, N 4.66.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.17, Mp 102–105 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 1.05 (d, J = 6.9 Hz, 6 H), 2.76 (sept, J = 6.9 Hz, 1 H), 3.91 (s, 2 H), 7.28–7.55 (m, 5 H), 11.11 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 17.8 (CH3), 37.2 (CH2), 39.8 (CH), 123.7 (Cquat), 125.9 (CH), 127.1 (Cquat), 128.4 (CH), 129.0 (CH), 130.3 (Cquat), 154.9 (Cquat), 209.2 (Cquat). MS (EI), m/z: 245 (M+, 23), 175 (39), 174 ((M − C4H7O)+, 88), 149 (29), 131 (15), 105 (13), 104 (35), 103 (100), 77 (C6H5+, 31), 71 (C4H7O+, 66), 69 (12), 57 (18), 55 (12), 44 (17), 43 (C3H7+, 81), 41 (18), 40 (68). IR (ATR), [cm−1]: 1748 (s), 1709 (m), 1682 (w). Anal. calcd for C14H15NO3 (245.3): C 68.56, H 6.16, N 5.71; Found: C 68.80, H 6.44, N 5.51.
:1) as a brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.13, Mp 144–148 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 0.77–1.04 (m, 4 H), 2.05–2.21 (m, 1 H), 3.95 (s, 2 H), 7.29–7.57 (m, 5 H), 11.14 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 10.9 (CH2), 20.1 (CH), 39.6 (CH2), 123.8 (Cquat), 126.0 (CH), 127.0 (Cquat), 128.4 (CH), 129.0 (CH), 130.1 (Cquat), 154.9 (Cquat), 205.4 (Cquat). MS (EI), m/z: 243 (M+, 17), 174 ((M − C4H5O)+, 36), 105 (31), 104 (16), 103 (46), 77 (C6H5+, 27), 69 (C4H5O+, 100), 57 (11), 43 (15), 41 (C3H5+, 25). IR (ATR), [cm−1]: 1744 (s), 1701 (s), 1687 (w). Anal. calcd for C14H13NO3 (243.3): C 69.12, H 5.39, N 5.76; Found: C 69.20, H 5.64, N 5.48.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.21, Mp 182–194 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 1.59–1.75 (m, 6 H), 1.76–1.88 (m, 6 H), 1.94–2.06 (m, 3 H), 3.90 (s, 2 H), 7.22–7.55 (m, 5 H), 11.07 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 27.3 (CH), 33.4 (CH2), 35.9 (CH2), 37.2 (CH2), 46.0 (Cquat), 123.8 (Cquat), 125.8 (CH), 127.2 (Cquat), 128.3 (CH), 128.9 (CH), 130.7 (Cquat), 154.9 (Cquat), 209.8 (Cquat). MS (EI), m/z: 337 (M+, 4), 163 (C11H15O+, 4), 136 (11), 135 (C10H15+, 100), 77 (C6H5+, 5). IR (ATR), [cm−1]: 1755 (s), 1705 (m). HRMS (ESI): m/z calcd For C21H24NO3 ((M + H)+): 338.1756; Found: 338.1753.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 1:1) = 0.26, Mp 162–169 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 3.76 (s, 3 H), 3.85 (s, 3 H), 4.35 (s, 2 H), 6.90–7.03 (m, 2 H), 7.03–7.12 (m, 2 H), 7.30–7.44 (m, 2 H), 7.94–8.09 (m, 2 H), 11.04 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 35.4 (CH2), 55.3 (CH3), 55.6 (CH3), 114.0 (CH), 114.5 (CH), 119.5 (Cquat), 123.7 (Cquat), 127.5 (CH), 128.7 (Cquat), 129.3 (Cquat), 130.9 (CH), 155.0 (Cquat), 159.3 (Cquat), 163.6 (Cquat), 193.4 (Cquat). MS (EI), m/z: 339 (M+, 4), 204 ((M − C8H7O2)+, 3), 135 (C8H7O2+, 100), 107 (C7H7O+, 6), 92 (C6H4O+, 4), 77 (C6H5+, 7). IR (ATR), [cm−1]: 1748 (s), 1683 (m). Anal. calcd for C19H17NO5 (339.3): C 67.25, H 5.05, N 4.13; Found: C 67.49, H 5.34, N 3.84.
:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.09, Mp 167–175 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 2.30 (s, 3 H), 3.85 (s, 3 H), 4.38 (s, 2 H), 6.99–7.15 (m,2 H), 7.16–7.29 (m, 2 H), 7.29–7.40 (m, 2 H), 7.93–8.10 (m, 2 H), 11.08 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 20.8 (CH3), 35.5 (CH2), 55.6 (CH3), 114.1 (CH), 123.9 (Cquat), 124.3 (Cquat), 125.9 (CH), 128.7 (Cquat), 129.5 (CH), 130.0 (Cquat), 130.9 (CH), 138.0 (Cquat), 155.0 (Cquat), 163.6 (Cquat), 193.4 (Cquat). MS (EI), m/z: 323 (M+, 4), 188 ((M − C8H7O2)+, 1), 136 (10), 135 (C8H7O2+, 100), 119 (10), 107 (C7H7O+, 4), 92 (C6H4O+, 4), 91 (5), 77 (C6H5+, 7). IR (ATR), [cm−1]: 1759 (s), 1670 (m). Anal. calcd for C19H17NO4 (323.3): C 70.58, H 5.30, N 4.33; Found: C 70.27, H 5.47, N 4.27.
:1) as a light yellow solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.10, Mp 186–195 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 3.85 (s, 3 H), 4.42 (s, 2 H), 7.07 (d, J = 8.9 Hz, 2 H), 7.28–7.67 (m, 4 H), 8.01 (d, J = 8.8 Hz, 2 H), 11.20 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 35.4 (CH2), 55.7 (CH3), 114.1 (CH), 123.1 (Cquat), 126.0 (Cquat), 127.7 (CH), 128.7 (Cquat), 129.0 (CH), 130.9 (CH), 131.1 (Cquat), 133.0 (Cquat), 154.8 (Cquat), 163.7 (Cquat), 193.2 (Cquat). MS (EI), m/z: 343 (M(35Cl)+, 1), 208 ((M(35Cl) − C8H7O)+, 1), 135 (C8H7O+, 100), 107 (C7H7O+, 4), 92 (C6H4O+, 4), 77 (C6H5+, 6). IR (ATR), [cm−1]: 1742 (s), 1682 (m). Anal. calcd for C18H14ClNO4 (343.8): C 62.89, H 4.10, N 4.07; Found: C 62.83, H 4.27, N 3.84.
:1) as a yellow solid.
R
f (petroleum ether/ethyl acetate 1:1) = 0.18, Mp 102–106 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 3.84 (s, 3 H), 4.15 (s, 2 H), 6.94–7.14 (m, 2 H), 7.36–7.55 (m, 3 H), 7.55–7.66 (m, 1 H), 7.85–8.03 (m, 2 H), 10.99 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 35.0 (CH2), 55.6 (CH3), 114.0 (CH), 121.3 (Cquat), 126.1 (Cquat), 127.6 (CH), 128.6 (Cquat), 130.0 (CH), 130.7 (CH), 131.2 (CH), 131.8 (CH), 132.0 (Cquat), 132.9 (Cquat), 154.9 (Cquat), 163.5 (Cquat), 193.0 (Cquat). MS (EI), m/z: 343 (M(35Cl)+, 1), 135 (C8H7O+, 100), 107 (C7H7O+, 6), 92 (C6H4O+, 6), 77 (C6H5+, 9), 58 (10). IR (ATR), [cm−1]: 1751 (s), 1684 (m). Anal. calcd for C18H14ClNO4 (343.8): C 62.89, H 4.10, N 4.07; Found: C 62.74, H 4.38, N 3.78.
:1 to 1:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.10, Mp 176–186 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 3.86 (s, 3 H), 4.44 (s, 2 H), 6.52–7.23 (m, 3 H), 7.30 (d, J = 2.4 Hz, 1 H), 7.57 (d, J = 4.6 Hz, 1 H), 8.03 (d, J = 8.5 Hz, 2 H), 11.32 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 35.7 (CH2), 55.7 (CH3), 114.1 (CH), 119.3 (Cquat), 125.6 (CH), 126.5 (CH), 127.8 (CH), 128.0 (Cquat), 128.6 (Cquat), 130.1 (Cquat), 130.8 (CH), 154.6 (Cquat), 163.7 (Cquat), 192.7 (Cquat). MS (EI), m/z: 315 (M+, 3), 180 ((M − C8H7O)+, 1), 135 (C8H7O+, 100), 107 (C7H7O+, 5), 92 (C6H4O+, 4), 77 (C6H5+, 8). IR (ATR), [cm−1]: 1751 (m), 1667 (w). Anal. calcd for C16H13NO4S (315.3): C 60.94, H 4.16, N 4.44; Found: C 60.95, H 4.37, N 4.29.
:1 to 1:1) as a light brown solid.
R
f (petroleum ether/ethyl acetate 2:1) = 0.14, Mp 178–192 °C. 1H NMR (DMSO-d6, 300 MHz), δ = 2.31 (s, 3 H), 4.39 (s, 2 H), 7.17–7.32 (m, 3 H), 7.32–7.42 (m, 2 H), 8.08 (d, J = 4.4 Hz, 2 H), 11.12 (s, 1 H). 13C NMR (DMSO-d6, 75 MHz), δ = 20.8 (CH3), 36.0 (CH2), 124.1 (Cquat), 124.2 (Cquat), 126.0 (CH), 129.0 (CH), 129.3 (Cquat), 129.5 (CH), 134.7 (CH), 136.0 (CH), 138.1 (Cquat), 142.6 (Cquat), 154.9 (Cquat), 188.0 (Cquat). MS (EI), m/z: 299 (M+, 31), 189 (13), 188 ((M − C5H3OS)+, 100), 119 (15), 118 (14), 117 (45), 115 (13), 111 (C5H3OS+, 100), 91 (C7H7+, 14). IR (ATR), [cm−1]: 1736 (s), 1657 (m). Anal. calcd for C16H13NO3S (299.3): C 64.20, H 4.38, N 4.68; Found: C 64.03, H 4.49, N 4.55.
Coupling-cycloisomerization synthesis of 2-oxoethyl 2-tert-butoxy oxazole 5 under basic conditions
In an oven-dried Schlenk tube with stir bar and screw cap PdCl2(PPh3)2 (14 mg, 0.02 mmol, 2 mol%) and CuI (8 mg, 0.04 mmol, 4 mol%) were placed under nitrogen. Then, dry THF (2.5(5.5 mL)) was added and the mixture was degassed with nitrogen for 5 min. The 4-methoxy benzoylchloride (4b) (176 mg, 1.00 mmol), N-Boc 1-phenylpropargyl carbamate (2a) (232 mg, 1.00 mmol), and dry triethylamine (0.14 mL, 1.00 mmol) were successively added and the mixture was stirred at room temp (water bath) for 1 h. The conversion was monitored by TLC. Then, aqueous saturated Na2CO3 solution (5 mL) was added and the aqueous layer was extracted with dichloromethane (3 × 5 mL). The combined organic phases were dried with anhydrous sodium sulfate and the solvents were removed under reduced pressure. The crude product was adsorbed on Celite© and chromatographed on silica gel (petroleum ether/ethyl acetate/triethylamine 100:10:1) to give 259 mg (71%) of analytically pure 2-(2-(tert-butoxy)-4-phenyloxazol-5-yl)-1-(4-methoxyphenyl)-ethanone (5) as an orange red solid.
R
f (petroleum ether/ethyl acetate 10:1) = 0.18. 1H NMR (CDCl3, 300 MHz), δ = 1.59 (s, 9 H), 3.87 (s, 3 H), 4.35 (s, 2 H), 6.88–7.00 (m, 2 H), 7.26–7.42 (m, 3 H), 7.61–7.71 (m, 2 H), 7.95–8.06 (m, 2 H). 13C NMR (CDCl3, 75 MHz), δ = 28.2 (CH3), 36.4 (CH2), 55.9 (CH3), 85.1 (Cquat), 114.3 (CH), 127.1 (CH), 127.9 (CH), 128.9 (CH), 129.5 (Cquat), 131.3 (CH), 132.6 (Cquat), 135.0 (Cquat), 136.7 (Cquat), 159.1 (Cquat), 164.2 (Cquat), 193.4 (Cquat). MS (EI), m/z: 365 (M+, 0.2), 350 ((M − CH3)+, 0.3), 309 ((M − C4H9 + H)+, 7), 136 (9), 135 (C8H7O2+, 100), 105 (C7H5O+, 6), 77 (C6H5+, 9), 57 (C4H9+, 14), 41 (16).
Acknowledgements
The authors cordially thank the Fonds der Chemischen Industrie for financial support.Notes and references
- G. V. Boyd, Sci. Synth., 2001, 11, 383 Search PubMed.
- X.-M. Zhang and F. G. Bordwell, J. Org. Chem., 1994, 59, 6456 CrossRef CAS.
- C. H. Eugster, Naturwissenschaften, 1968, 55, 305 CrossRef CAS PubMed.
- N. Kudo, M. Taniguchi, S. Furuta, K. Sato, T. Endo and T. Honma, J. Agric. Food Chem., 1998, 46, 5305 CrossRef CAS.
- D. A. Evans, Aldrichimica Acta, 1982, 15, 23 CAS.
- Asymmetric synthesis of nitrogen heterocycles, ed. J. Royer, Wiley-VCH Verlag, Weinheim, 2009, p. 235 Search PubMed.
- J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835 CrossRef PubMed.
- D. W. Knight, Oxazole and Its Derivatives, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chatopadhyay, Wiley-VCH, Weinheim, 2011, p. 403 Search PubMed.
- M. Daneshtalab, Top. Heterocycl. Chem., 2006, 2, 153 CAS.
- M. R. Barbachyn and C. W. Ford, Angew. Chem., Int. Ed., 2003, 42, 2010 CrossRef CAS PubMed.
- D. Shinabarger, Expert Opin. Invest. Drugs, 1999, 8, 1195 CrossRef CAS PubMed.
- N.-H. Nam, Y. Kim, Y.-J. You, D.-H. Hong, H.-M. Kim and B.-Z. Ahn, Bioorg. Med. Chem. Lett., 2001, 11, 3073 CrossRef CAS PubMed.
- For a review on the synthetic potential of oxazol-2-ones, see e.g. S. P. Fearnley, Curr. Org. Chem., 2004, 8, 1289 CrossRef CAS.
- T. Kunieda, Y. Abe, T. Higuchi and M. Hirobe, Tetrahedron Lett., 1981, 22, 1257 CrossRef CAS; T. Kunieda, T. Higuchi, Y. Abe and M. Hirobe, Tetrahedron, 1983, 39, 3253 CrossRef.
- R. Gompper and F. Effenberger, Chem. Ber., 1959, 1928 CrossRef CAS.
- J. A. Deyrup and H. L. Gingrich, Tetrahedron Lett., 1977, 3115 CrossRef CAS.
- G. V. Boyd, Sci. Synth., 2001, 11, 420 Search PubMed.
- For Blümlein-Lewy synthesis, see e.g. T. Eicher and S. Hauptmann, The Chemistry of Heterocycles, Wiley-VCH, Weinheim, 2nd edn, 2003, p. 127 Search PubMed. For Van Leusen synthesis and Robinson-Gabriel synthesis, see e.g. J. J. Li, Name Reactions, A Collection of Detailed Reaction Mechanisms, Springer-Verlag, Berlin, 3rd edn, 2006, pp. 506 and 601 Search PubMed.
- E. Merkul and T. J. J. Müller, Chem. Commun., 2006, 4817 RSC; E. Merkul, O. Grotkopp and T. J. J. Müller, Synthesis, 2009, 502 Search PubMed.
- O. Grotkopp, A. Ahmad, W. Frank and T. J. J. Müller, Org. Biomol. Chem., 2011, 9, 8130 CAS.
- H. V. Wachenfeldt, F. Paulsen, A. Sundin and D. Strand, Eur. J. Org. Chem., 2013, 4578 CrossRef.
- For a review on oxazolidinones, see e.g. M. E. Dyen and D. Swern, Chem. Rev., 1967, 67, 197 CrossRef CAS PubMed.
- R. Robles-Machin, J. Adrio and J. C. Carretero, J. Org. Chem., 2006, 71, 5023 CrossRef CAS PubMed.
- A. Buzas and F. Gagosz, Synlett, 2006, 2727 CAS.
- E.-S. Lee, H.-S. Yeom, J.-H. Hwang and S. Shin, Eur. J. Org. Chem., 2007, 3503 CrossRef CAS.
- D. Scarpi, S. Begliomini, C. Prandi, A. Oppedisano, A. Deagostino, E. Gómez-Bengoa, B. Fiser and E. G. Occhiato, Eur. J. Org. Chem., 2015, 3251 CrossRef CAS.
- B. Gabriele, P. Plastina, G. Salerno, R. Mancuso and M. Costa, Org. Lett., 2007, 9, 3319 CrossRef CAS PubMed.
- L. C. Wilkins, P. Wieneke, P. D. Newman, B. M. Kariuki, F. Rominger, A. S. K. Hashmi, M. M. Hansmann and R. L. Melen, Organometallics, 2015, 34, 5298 CrossRef CAS.
- J. Hu, J. Ma, Z. Zhang, Q. Zhu, H. Zhou, W. Lu and B. Han, Green Chem., 2015, 17, 1219 RSC.
- For related three-component syntheses of 3-halo furans, see: A. S. Karpov, E. Merkul, T. Oeser and T. J. J. Müller, Chem. Commun., 2005, 2581 RSC; A. S. Karpov, E. Merkul, T. Oeser and T. J. J. Müller, Eur. J. Org. Chem., 2006, 2991 CrossRef CAS.
- E. Merkul, C. Boersch, W. Frank and T. J. J. Müller, Org. Lett., 2009, 11, 2269 CrossRef CAS PubMed.
- A. S. Karpov and T. J. J. Müller, Org. Lett., 2003, 5, 3451 CrossRef CAS PubMed; D. M. D'Souza and T. J. J. Müller, Nat. Protoc., 2008, 3, 1660 CrossRef PubMed.
- According to synthetic procedures reported in T. Mecozzi and M. Petrini, J. Org. Chem., 1999, 64, 8970 CrossRef CAS PubMed.
- 1-Methyl substituted N-boc protected propargyl amide neither gave conclusive results nor selective product formation. Therefore, 1-alkyl substituted derivatives were omitted in this methodological study.
- K.-H. Scholz, H.-G. Heine and W. Hartmann, Liebigs Ann. Chem., 1976, 1319 CrossRef CAS.
- Crystallographic data (excluding structure factors) for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Centre: CCDC 1465496 (3r). See ESI.‡.
Footnotes |
| † Dedicated to Professor Dr Barry M. Trost on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: 1H and 13C NMR spectra and analytical data of compounds 2, and 1H and 13C NMR spectra of compounds 3, cif file of the X-ray structure analysis of compound 3r. CCDC 1465496. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00138f |
| This journal is © the Partner Organisations 2016 |