
Synthesis of trifluoromethylthiolated azacalix[1]arene[3]pyridines from the Cu(II)-mediated direct trifluoromethylthiolation reaction of arenes via reactive arylcopper(III) intermediates†
Fei
Wang
a,
Liang
Zhao
b,
Jingsong
You
a and
Mei-Xiang
Wang
*b
aKey Laboratory of Green Chemistry and Technology (MOE), College of Chemistry, Sichuan University, Chengdu 610064, China
bKey Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: wangmx@mail.tsinghua.edu.cn
First published on 20th May 2016
Abstract
Using azacalix[1]arene[3]pyridines and their high valent organocopper(II) and (III) complexes as probes, arene C–H bond trifluoromethylthiolation was investigated. While arylcopper(II) compounds appeared almost inert, arylcopper(III) compounds reacted efficiently with a nucleophilic trifluoromethylthiolating reagent. Under mild conditions, azacalix[1]arene[3]pyridines underwent the regioselective Cu(ClO4)2-mediated one-pot trifluoromethylthiolation reaction with Me4NSCF3 to afford low-rim functionalized macrocycles.
Introduction
Heteracalixaromatics1,2 are a new generation of macrocycles in supramolecular chemistry. As revealed by us1 and others,2,3 one of the pronounced structural features of heteracalixaromatics is self fine-tunability of the macrocyclic conformation, cavity and electronic properties because different bridging heteroatoms are able to form various conjugation systems with their adjacent aromatic rings. As a consequence, heteracalixaromatics behave as smart macrocyclic host molecules, exhibiting versatile molecular recognition properties towards charged and electron neutral guests.1–6 For example, azacalix[4]pyridines adopt highly symmetric saddle-shaped conformations to interact with transition metal ions via dipole–ion interactions,4 the partially protonated azacalix[4]pyridines form distorted saddle-shaped conformations to bind to anions mainly through the hydrogen bond interactions.5 Dichloro-substituted oxacalix[2]arene[2]triazine, on the other hand, forms a range of V-shaped clefts of varied sizes in which two triazine rings act as a pair of clippers to complex anions of different geometries and volumes by means of cooperative anion–π and lone-pair electron–π interactions.6,7Heteracalixaromatics are synthesized conveniently by means of the stepwise fragment coupling strategy and a one-pot reaction protocol from readily available starting materials.1–3 The post-macrocyclization chemical manipulations provide an alternative route to tailor-made functional heteracalixaromatics. One of the noteworthy examples is the Cu(II)-catalyzed and mediated arene C–H bond functionalization of azacalix[1]arene[3]pyridines with a range of carbon and heteroatom nucleophiles, which enables the generation of diverse functional macrocycles.8 It has been revealed that the reaction proceeds through a key electrophilic metalation step to afford structurally well-defined arylcopper(II). Oxidation of arylcopper(II) complexes by free copper(II) species leads to arylcopper(III) intermediates which undergo cross-coupling reactions with nucleophiles.9 It should also be noted that other structurally well-defined arylcopper(III) compounds have been reported by Hedman, Hodgson and Llobet10 from the reaction of arene-embedded aza-crowns and Cu(ClO4)2. Ribas et al.11 have proposed recently a five-step mechanism to account for the formation of aryl–Cu(III) from the Cu(II) salt-mediated arene C–H activation. The rate-limiting step is believed to be a C–H bond cleavage via a concerted proton-coupled electron transfer (PCET) process.
Aryl trifluoromethyl sulfides have been attracting considerable attention because they possess unique chemical, physical and biological activities due to the extremely high lipophilicity of the trifluoromethylthio group. Aryl trifluoromethyl sulfide compounds have found applications in agrochemical and materials science. The synthesis of aryl trifluoromethyl sulfides is predominantly executed by either direct trifluoromethylthiolation of prefunctionalized aromatic substrates or trifluoromethylation of aromatic thiols and derivatives.12–16 Nucleophilic aromatic substitution of aryl halides with various trifluoromethanethiolate sources such as CuSCF3 constitutes a conventional method to construct aryl trifluoromethyl sulfides.12,15 Noticeably, with the advent of easy-to-use trifluoromethylthiolating reagents, direct trifluoromethylthiolation has gained increasing popularity.13–15 For example, electrophilic N-trifluoromethylthiosaccharin has been reported to trifluoromethylthiolate a variety of electron-rich aromatic compounds when chlorotrimethylsilane or triflic acid is used as an activator.17 Catalyzed by transition metals, trifluoromethanesulfenates,18N-trifluoromethylthiol phthalimide,19,20 S8/CF3SiMe3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 and Me4NSCF322 are able to react with arylboronic acids to form trifluoromethylthiolated arenes. Transition metals such as palladium23 and nickel24 also catalyze the cross coupling reaction between pre-functionalized arenes and AgSCF3 and Me4NSCF3, resulting in the formation of Caryl–SCF3 bonds. It is particularly worth noting that, by applying a directing group strategy, Daugulis25a reported in 2012 a Cu(OAc)2-promoted trifluoromethylthiolation of aryl C–H bonds with CF3SSCF3. It is probably the only example of copper-catalyzed arene trifluoromethylthiolation involving a C–H bond activation process.25b In contrast to the rapid development of synthetic methods, the mechanism of transition metal-catalyzed and -promoted trifluoromethylthiolation of aromatic compounds remains elusive.12–16 The formidable challenges in mechanistic elucidation include, if not all, the isolation or observation of structurally well-defined organometallic intermediates.
21 and Me4NSCF322 are able to react with arylboronic acids to form trifluoromethylthiolated arenes. Transition metals such as palladium23 and nickel24 also catalyze the cross coupling reaction between pre-functionalized arenes and AgSCF3 and Me4NSCF3, resulting in the formation of Caryl–SCF3 bonds. It is particularly worth noting that, by applying a directing group strategy, Daugulis25a reported in 2012 a Cu(OAc)2-promoted trifluoromethylthiolation of aryl C–H bonds with CF3SSCF3. It is probably the only example of copper-catalyzed arene trifluoromethylthiolation involving a C–H bond activation process.25b In contrast to the rapid development of synthetic methods, the mechanism of transition metal-catalyzed and -promoted trifluoromethylthiolation of aromatic compounds remains elusive.12–16 The formidable challenges in mechanistic elucidation include, if not all, the isolation or observation of structurally well-defined organometallic intermediates.
As a continuation of our research project on heteracalixaromatics,1 we endeavored to construct trifluoromethylthiolated azacalixpyridine macrocycles that may possess unique properties in selective extraction of transition metals. Importantly, having had isolated arylcopper(II) and arylcopper(III) complexes in hand,9 we envisioned that azacalix[1]arene[3]pyridines would act as a unique molecular platform, permitting an in-depth study of copper salt-mediated arene C–H bond trifluoromethylthiolation. We report herein a simple and convenient one-pot synthesis of trifluoromethylthiolated azacalix[1]arene[3]pyridines. We demonstrate for the first time that Cu(II)-mediated arene C–H bond trifluoromethylthiolation proceeds through an organometallic reaction mechanism via an arylcopper(III) intermediate.
Results and discussion
To understand the reactivity of high valent arylcopper(II) and arylcopper(III) species in trifluoromethylthiolation processes, we initiated our study by scrutinizing the reactions of isolated macrocyclic arylcopper(II) complex 1 and arylcopper(III) complex 2 with both electrophilic N-trifluoromethylthiosaccharin 317 and trifluoromethanesulfenate 418 and nucleophilic Me4NSCF35. When treated with equimolar 3 at ambient temperature in acetonitrile, arylcopper(II) 1a was converted into a mixture of products whereas arylcopper(III) complex 2a remained intact. Surprisingly, instead of the desired trifluoromethylthiolated benzene product 6a, double trifluoromethylthiolation took place exclusively on two proximal pyridine units to form products 7a, 7b and 7c in 11%, 7% and 13% yields, respectively (Fig. 1). Since the reaction between the parent azacalix[1]arene[3]pyridine 8a and 3 gave almost the same results (Fig. 1) (see chemical yields in parentheses), products 7a–c were most probably derived from direct trifluoromethylthiolation of 8a rather than organocopper(II) 1a. A preferential electrophilic substitution reaction on pyridine rings rather than on the benzene moiety concurs with our previous discovery.26 The 1,3-alternate conformation of azacalix[m]arene[n]pyridines (m = n = 2; m = 1, n = 3) results in the stronger conjugation between bridging nitrogen atoms and pyridine rings, leading to the enhancement of the electron density of pyridines.26 The less electrophilic trifluoromethylthiolating reagent trifluoromethanesulfenate 418 did not react with both arylcopper(II) and arylcopper(III) compounds 1a and 2a.
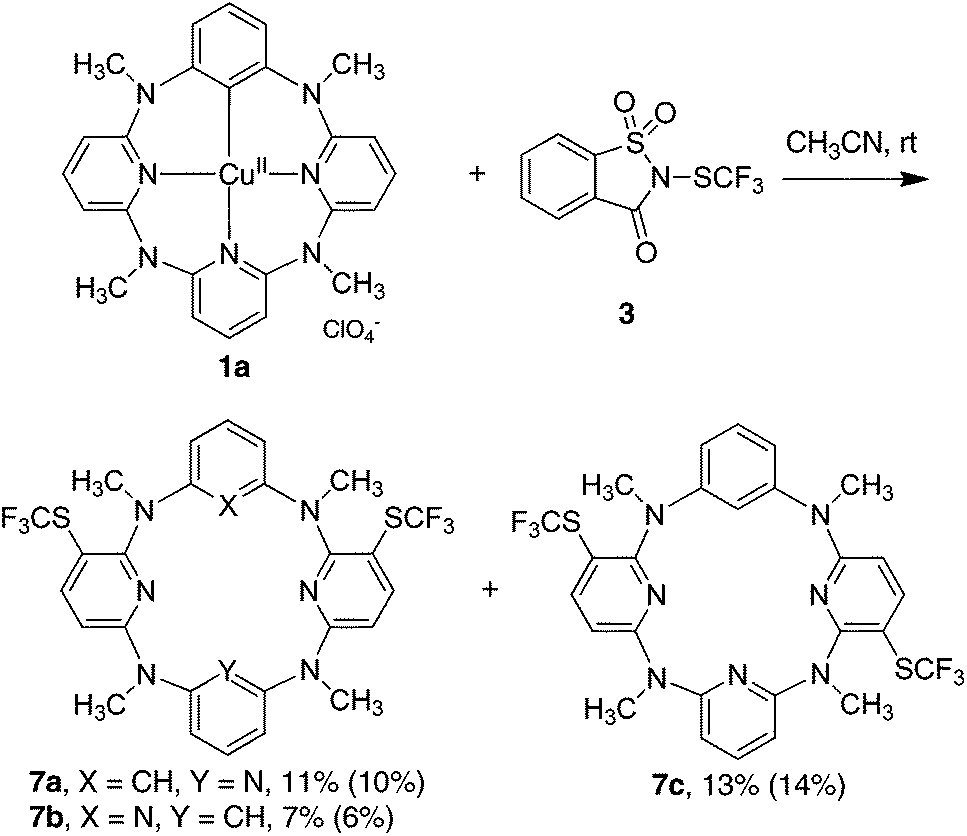 | ||
| Fig. 1 Reaction of phenylcopper(II) 1a with trifluoromethylthiolating reagent 3 (chemical yields in parentheses are from the reaction of parent azacalix[1]arene[3]pyridine 8a with 3). | ||
In stark contrast to electrophilic trifluoromethylthiolating reagents, nucleophilic Me4NSCF35 underwent the reaction smoothly with arylcopper(III) complex 2a at 80 °C in acetonitrile to afford trifluoromethylthiolated arene 6a in 55% yield. The increase of 5 led to the improvement of chemical yield. An excellent yield of 6a (92%) for instance was obtained when 1.5 equivalents of 5 were used in the reaction (Fig. 2). Even at room temperature the reaction gave 67% yield of 6a. The reaction was not influenced by the presence of a radical scavenger such as TEMPO, BHT or 1,1-diphenylethene, as the reaction afforded 6a in 84%, 88% or 86% yield, respectively. It is also important to note that, based on mass spectrometric analysis (see the ESI†), only copper(I) was generated from the reaction. It is indicative of a reductive elimination reaction. However, under the identical conditions, the reaction between arylcopper(II) complex 1a and 5 was very sluggish, and only a small amount of 6a (16%) was isolated along with the recovery of the parent azacalix[1]arene[3]pyridine 8a in 53% yield after work-up (Fig. 2). The outcomes indicate convincingly that both Caryl–Cu(II) and Caryl–Cu(III) bonds are inert towards electrophilic trifluoromethylthiolating reagents. The arylcopper(III) complex reacts highly efficiently with nucleophilic Me4NSCF3 whereas the arylcopper(II) analog is nearly inactive.
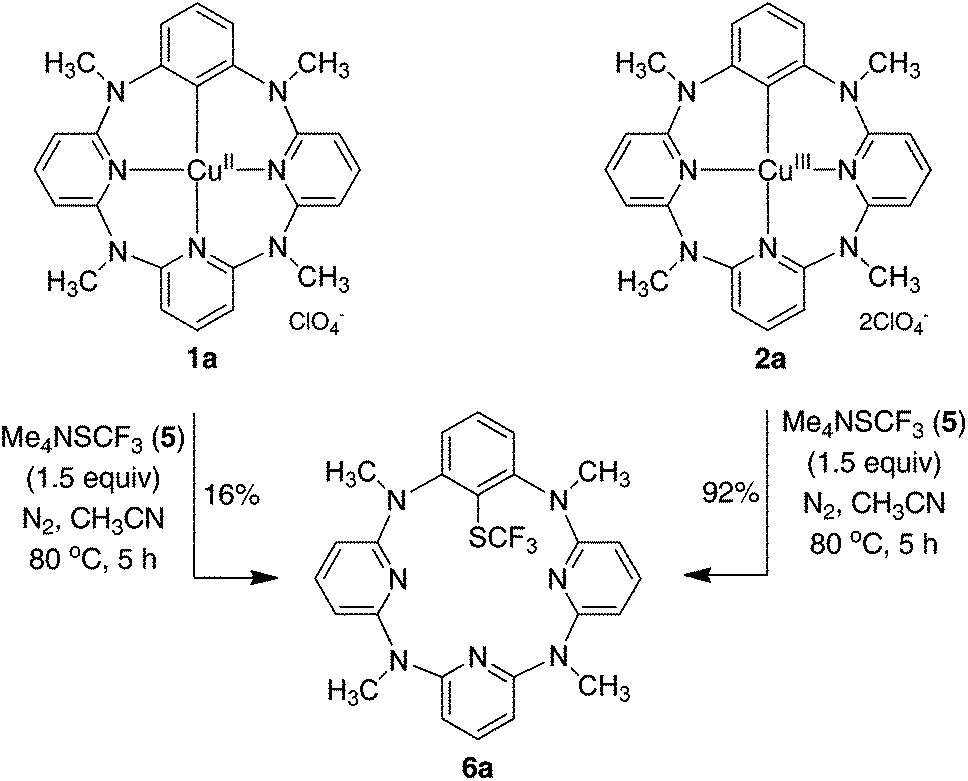 | ||
| Fig. 2 Reaction of phenylcopper(II) 1a and phenylcopper(III) 2a with trifluoromethylthiolating reagent 5. | ||
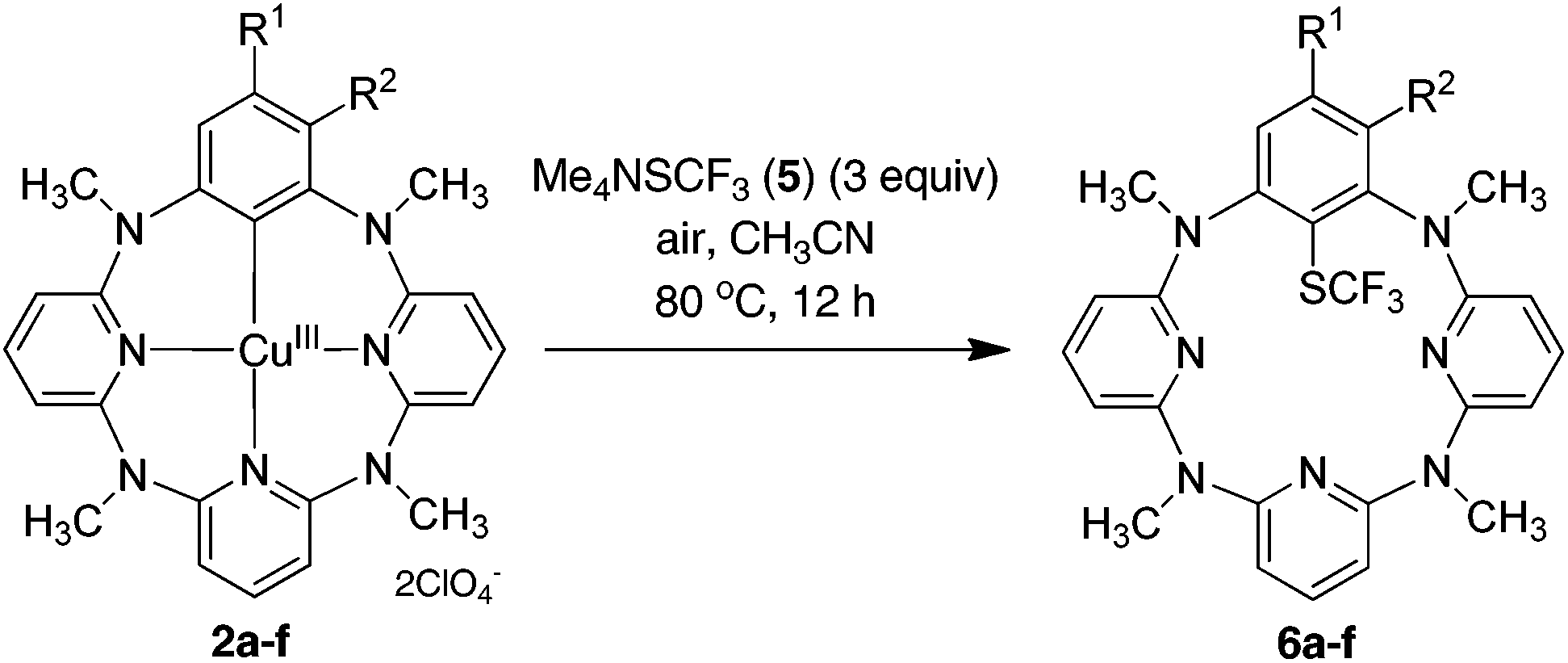
The reaction of arylcopper(III) compound 2a with 5 was carried out conveniently under aerobic conditions, albeit the yield of 6a was slightly decreased to 76%. Gratifyingly, the trifluoromethylthiolated product 6a was obtained in an excellent yield when the trifluoromethylthiolating reagent 5 was used in three equivalents. The trifluoromethylthiolation reaction was easily expanded to other arylcopper(III) substrates. As summarized in Table 1, all arylcopper(III) reactants 2a–f tested underwent efficient reaction with 5 to furnish the C–SCF3 bond forming products 6a–f in the yield ranging from 82% to 98%. It is obvious that the presence of an electron-withdrawing group on the benzene ring, irrespective of its substitution pattern, benefited the reaction. This has been evidenced by the higher chemical yields obtained from the reaction of 2a–d than 2e and 2f.
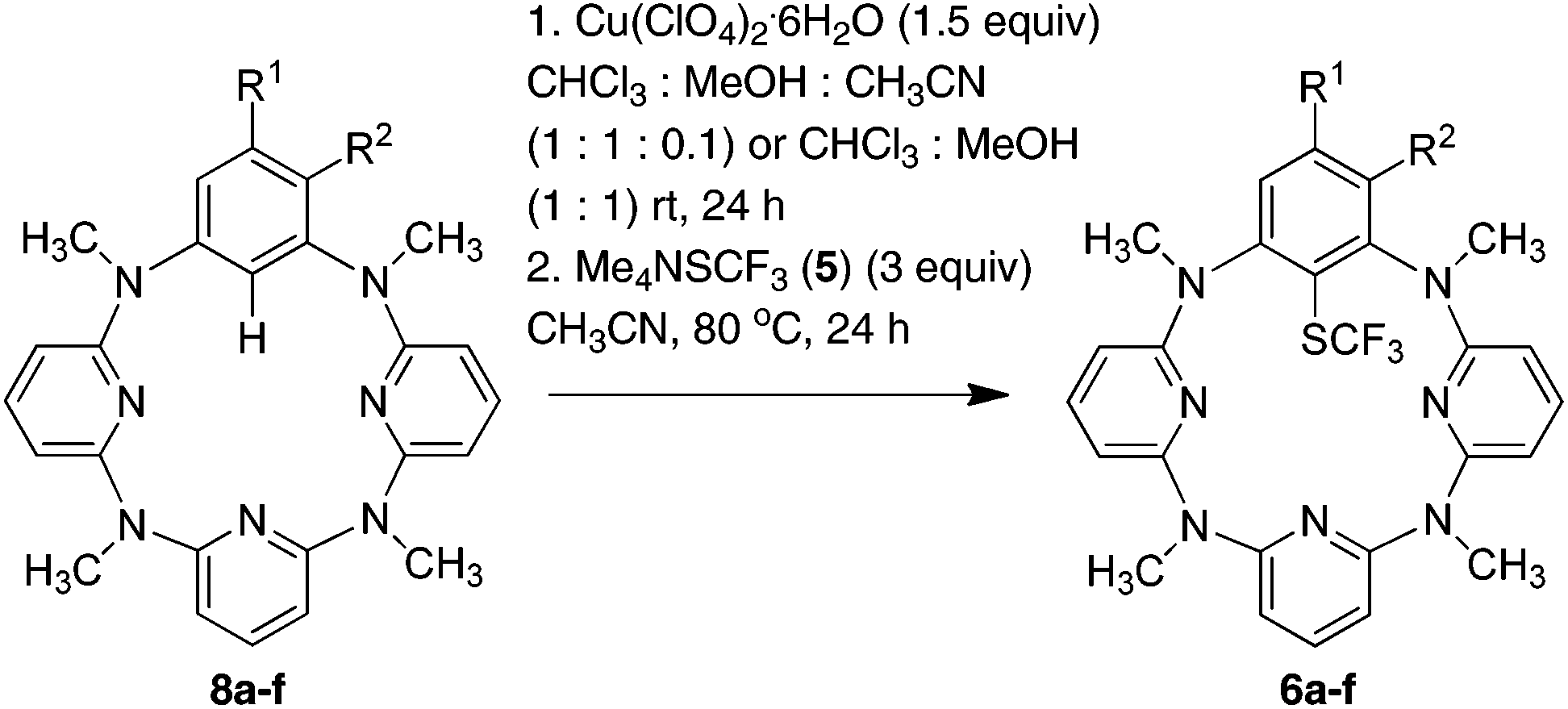
Encouraged by the facile reaction between arylcopper(III) complexes 2 with Me4NSCF35, we then explored the direct trifluoromethylthiolation reaction of arenes that are embedded in a macrocycle mediated by a copper(II) salt. Since the arylcopper(III) complex 2a is generated easily from the reaction of azacalix[1]arene[3]pyridine 8a with Cu(ClO4)2·6H2O through a direct electrophilic metalation and subsequent disproportionation cascade as we reported before,8,9 a one-pot reaction of 8a and 5 in the presence of Cu(ClO4)2·6H2O (1.5 equiv.) was attempted. Unfortunately, the reaction under the otherwise identical conditions gave only a small amount of product 6a (23%). The low efficiency of the reaction was most probably attributable to the decomposition of trifluoromethylthiolate 5 upon interaction with free copper ions.27 To circumvent this problem, we then conducted a step-wise copper(II)-mediated trifluoromethylthiolation of arene 8a in a one-pot fashion without isolation of arylcopper(III) species. Thus interaction of azacalix[1]arene[3]pyridine 8a with Cu(ClO4)2·6H2O in a mixture of chloroform and methanol at ambient temperature led to the formation of 6a. A solution of 5 (3 equiv.) in acetonitrile was added and the resulting mixture was kept at 80 °C for another 24 h to afford the trifluoromethylthiolated product 6a in 90% yield. Other azacalix[1]arene[3]pyridines 8b–f underwent a similar one-pot reaction with 5b mediated by the copper(II) salt, yielding the corresponding products 6b–f (Table 2). The slightly diminished chemical yields from the one-pot reaction in comparison with the reaction employing isolated arylcopper(III) complexes was mainly ascribed to the instability of trifluoromethylthiolating reagents under the one-pot reaction conditions27 along with the lower conversion of azacalix[1]arene[3]pyridines into the arylcopper(III) complexes in some cases.9 It may be worth noting that the anion of the arylcopper(III) complex did not affect the cross coupling reaction. This has been evidenced by the formation of 6a in 89%, 87% and 83% yield, respectively when Cu(BF4)2·6H2O, Cu(OTf)2 and Cu(NO3)2·3H2O were used instead of Cu(ClO4)2·6H2O. It should be pointed out that the one-pot reaction utilizing either one equivalent of Cu(ClO4)2·6H2O or less than two equivalents of Me4NSCF3 all resulted in the moderate yields of products 6a–f. The reaction of 8a with 5 using a sub-stoichiometric amount of copper(II) salt (50 mol%) only yielded 28% of 6a (see the ESI†).
The structures of products were characterized on the basis of spectroscopic data and microanalyses. To understand the conformation of trifluoromethylthiolated products, single crystals of 6a, 7b and 7c were cultivated and their molecular structures were determined by means of single crystal X-ray diffraction analysis. As illustrated in Fig. 3 and S2, S3,† trifluoromethylthiolated azacalix[1]arene[3]pyridine 6a adopts a nearly symmetric 1,3-alternate conformation while ditrifluoromethylthiolated azacalix[1]arene[3]pyridines 7b and 7c give heavily distorted 1,3-alternate conformers in the solid state. It is interesting to note that, all macrocyclic azacalix[1]arene[3]pyridine products give one set of distinct proton and carbon resonance signals in their 1H and 13C NMR spectra, respectively (see the ESI†). The outcomes suggest the existence of an equilibrium mixture of macrocyclic conformers that undergo very rapid interconversions at room temperature relative to the NMR time scale or of highly symmetric conformational structures in solution.28
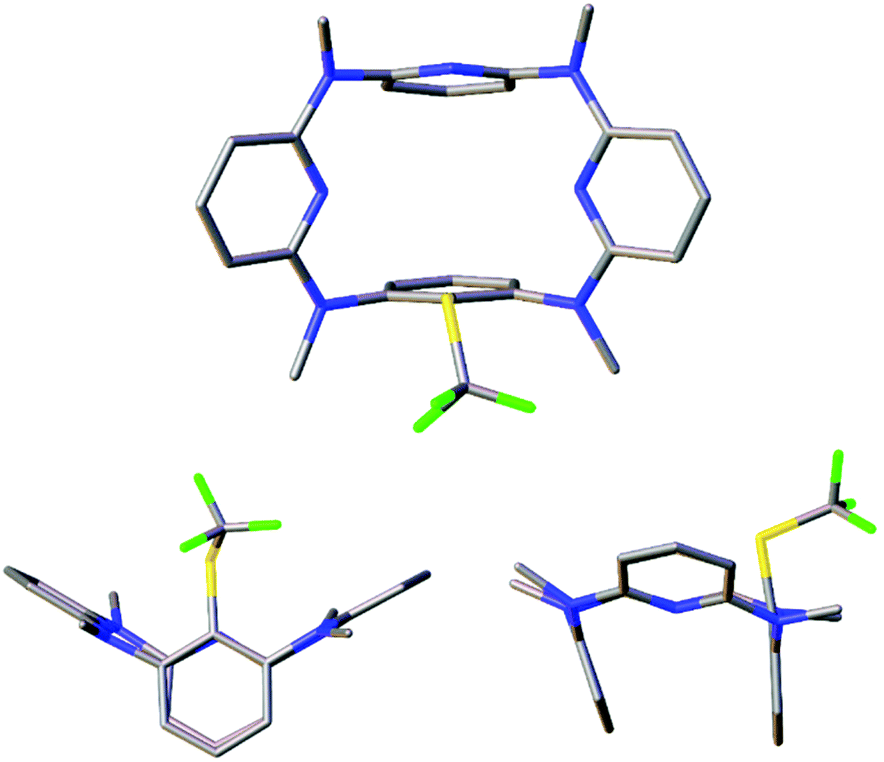 | ||
| Fig. 3 X-ray molecular structure of 6a with top (top) and side (bottom) views (CCDC 1474334). | ||
The aforementioned experimental evidence allows us to propose a reaction mechanism for trifluoromethylthiolation of arene compounds in question. As depicted in Fig. 4, the reaction between azacalix[1]arene[3]pyridines and copper(II) affords arylcopper(II) complexes which undergo a further oxidation reaction with free copper(II) to produce arylcopper(III) intermediates. The cross-coupling reaction with trifluoromethylthiolate yields aryl trifluoromethyl sulfides. It is worth noting that the azacalix[1]arene[3]pyridine macrocycle provides a unique multidentate ligand to stabilize high valent organocopper species, enabling the isolation of arylcopper(II) and arylcopper(III) species. Without a macrocyclic ligand, high valent arylcopper compounds cannot be isolated or even detected due to their reactive nature. The outcomes of the current study are reminiscent of other copper catalyzed and mediated C–H bond and even C–X bond trifluoromethylthiolations. It may imply that the copper catalyzed and mediated C–H bond and C–X bond trifluoromethylthiolations may probably proceed through high valent organocopper intermediates.29
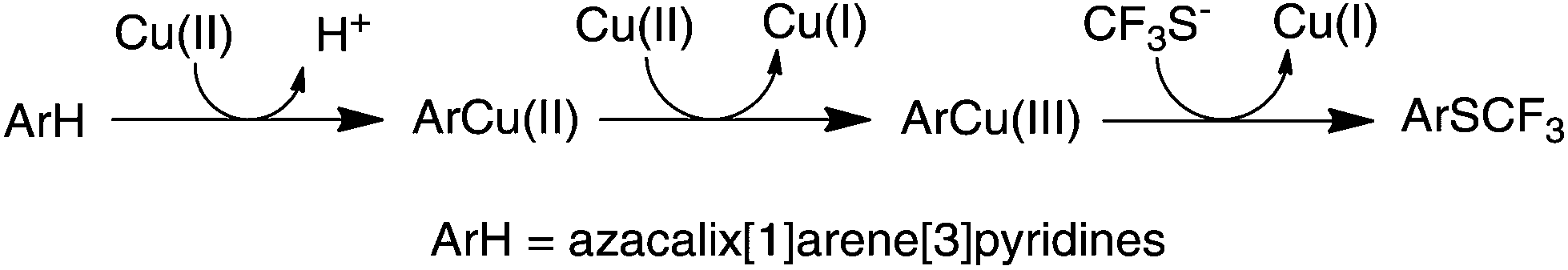 | ||
| Fig. 4 Reaction pathway for the Cu(II)-mediated arene C–H bond trifluoromethylthiolation with Me4NSCF3. | ||
Experimental
General procedure for the reaction of arylcopper(III) complexes 2a–f with Me4NSCF35: synthesis of trifluoromethylthiolated azacalix[1]arene[3]pyridines 6a–f
A mixture of arylcopper(III) complexes 2a–f (0.2 mmol) and Me4NSCF35 (105 mg, 0.6 mmol) in dry CH3CN (4 mL) was reacted at 80 °C for 12 h. A saturated aqueous solution of EDTA (2 mL) and brine (40 mL) was added, and the mixture was extracted with DCM (3 × 20 mL). The combined organic layer was dried with anhydrous Na2SO4. After removal of the solvent, the residue was chromatographed on a silica gel column (100–200 mesh) using a mixture of petroleum ether, DCM and ethyl acetate (12:2:1) as the mobile phase to give pure products 6a–f. The products are fully characterized by using spectroscopic data and elemental analysis data which are listed below.
6a (95 mg, 91% yield): white solid, mp 265–266 °C; 1H NMR (400 MHz, CDCl3) δ 7.45 (t, J = 8.0 Hz, 2H), 7.32 (t, J = 8.0 Hz, 1H), 7.08 (t, J = 7.8 Hz, 1H), 6.95 (d, J = 7.8 Hz, 2H), 6.53 (d, J = 7.8 Hz, 2H), 6.08 (d, J = 8.2 Hz, 2H), 6.05 (d, J = 8.2 Hz, 2H), 3.24 (s, 6H), 3.15 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −38.7 (s, 3F); 13C NMR (100 MHz, CDCl3) δ 158.8, 158.7, 157.2, 153.6, 139.1, 137.1, 132.6, 132.2 (q, J = 303.0 Hz), 127.5, 126.5, 120.3, 95.8, 95.2, 38.9, 36.4; IR (KBr, cm−1) ν 2907, 1598, 1580, 1568, 1477, 1422. HRMS (ESI-ion trap) calcd for C26H25F3N7S: [M + H]+ 524.1839. Found: 524.1833. Elemental analysis calcd (%) for C26H24F3N7S: C, 59.64; H, 4.62; N, 18.73. Found: C, 59.71; H, 4.71; N, 18.52. A high quality single crystal for X-ray diffraction analysis was obtained by diffusing n-hexane vapor into the solution of 6a in DCM.
6b (99 mg, 90% yield): greenish solid, mp 201–202 °C; 1H NMR (400 MHz, CDCl3) δ 7.47 (t, J = 8.0 Hz, 2H), 7.35 (t, J = 7.8 Hz, 1H), 7.23 (s, 2H), 6.62 (d, J = 7.8 Hz, 2H), 6.14 (d, J = 7.8 Hz, 2H), 6.09 (d, J = 7.8 Hz, 2H), 3.25 (s, 6H), 3.16 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −37.7 (s, 3F); 13C NMR (100 MHz, CDCl3) δ 158.8, 158.4, 157.0, 154.5, 139.5, 137.5, 134.0, 131.3, 129.5 (q, J = 308.0 Hz), 120.5, 117.7, 116.0, 96.9, 95.8, 38.5, 36.6; IR (KBr, cm−1) ν 2909, 2235, 1580, 1562, 1475, 1424. HRMS (ESI-ion trap) calcd for C27H24F3N8S: [M + H]+ 549.1791. Found: 549.1791. Elemental analysis calcd (%) for C27H23F3N8S: C, 59.11; H, 4.23; N, 20.43. Found: C, 58.85; H, 4.40; N, 20.13.
6c (99 mg, 89% yield): white solid, mp 206–207 °C; 1H NMR (400 MHz, CDCl3) δ 7.46 (t, J = 7.8 Hz, 2H), 7.34 (t, J = 7.6 Hz, 1H), 6.97 (s, 2H), 6.62 (d, J = 7.8 Hz, 2H), 6.13 (d, J = 8.2 Hz, 2H), 6.08 (d, J = 7.8 Hz, 2H), 3.27 (s, 6H), 3.15 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −38.5 (s, 3F); 13C NMR (100 MHz, CDCl3) δ 158.8, 158.6, 157.1, 154.4, 139.2, 137.7, 137.0, 129.8 (q, J = 308.0 Hz), 128.4, 125.6, 120.5, 96.3, 95.4, 38.6, 36.4; IR (KBr, cm−1) ν 2910, 1586, 1563, 1477, 1424. HRMS (ESI-ion trap) calcd for C26H24ClF3N7S: [M + H]+ 558.1449. Found: 558.1447. Elemental analysis calcd (%) for C26H23ClF3N7S: C, 55.96; H, 4.15; N, 17.57. Found: C, 55.85; H, 4.21; N, 17.54.
6d (109 mg, 98% yield): white solid, mp 240–241 °C; 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 7.8 Hz, 1H), 7.44 (d, J = 7.8 Hz, 1H), 7.40 (d, J = 8.2 Hz, 1H), 7.17 (t, J = 7.8 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H), 6.63 (d, J = 7.8 Hz, 1H), 6.55 (d, J = 7.8 Hz, 1H), 6.12–6.04 (m, 4H), 3.26 (s, 6H), 3.15 (s, 3H), 3.11 (s, 3H); 19F NMR (376 MHz, CDCl3) δ −37.9 (s, 3F); 13C NMR (100 MHz, CDCl3) δ 158.9, 158.7, 158.5, 157.1, 156.3, 152.1, 150.0, 139.3, 137.4, 133.7, 133.3, 129.9 (q, J = 308.0 Hz), 129.5, 129.0, 120.5, 120.3, 96.2, 96.1, 95.3, 94.5, 38.6, 37.0, 36.3, 36.2; IR (KBr, cm−1) ν 2909, 1583, 1561, 1476, 1423. HRMS (ESI-ion trap) calcd for C26H24ClF3N7S: [M + H]+ 558.1449. Found: 558.1446. Elemental analysis calcd (%) for C26H23ClF3N7S: C, 55.96; H, 4.15; N, 17.57. Found: C, 55.63; H, 4.16; N, 17.54.
6e (88 mg, 82% yield): white solid, mp 193–194 °C; 1H NMR (400 MHz, CDCl3) δ 7.45 (t, J = 8.0 Hz, 2H), 7.10 (t, J = 7.8 Hz, 1H), 6.79 (s, 2H), 6.55 (d, J = 7.8 Hz, 2H), 6.08 (d, J = 7.8 Hz, 2H), 6.05 (d, J = 8.2 Hz, 2H), 3.25 (s, 6H), 3.14 (s, 6H), 2.30 (s, 3H); 19F NMR (376 MHz, CDCl3) δ −38.9 (s, 3F); 13C NMR (100 MHz, CDCl3) δ 158.9, 158.7, 157.3, 153.2, 143.0, 139.1, 136.4, 130.2 (q, J = 308.0 Hz), 128.5, 122.8, 120.3, 95.9, 95.3, 38.9, 36.4, 21.3; IR (KBr, cm−1) ν 2911, 1581, 1563, 1478, 1423. HRMS (ESI-ion trap) calcd for C27H27F3N7S: [M + H]+ 538.1995. Found: 538.1992. Elemental analysis calcd (%) for C27H26F3N7S: C, 60.32; H, 4.87; N, 18.24. Found: C, 60.09; H, 4.84; N, 18.23.
6f (93 mg, 84% yield): white solid, mp 211–212 °C; 1H NMR (400 MHz, CDCl3) δ 7.45 (t, J = 8.0 Hz, 2H), 7.13 (t, J = 7.6 Hz, 1H), 6.56 (d, J = 7.8 Hz, 2H), 6.52 (s, 2H), 6.08 (d, J = 8.2 Hz, 2H), 6.05 (d, J = 7.8 Hz, 2H), 3.79 (s, 3H), 3.25 (s, 6H), 3.15 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −39.3 (s, 3F); 13C NMR (100 MHz, CDCl3) δ 163.1, 158.9, 158.8, 157.2, 154.7, 139.1, 136.5, 130.2 (q, J = 308.0 Hz), 120.4, 117.4, 113.8, 95.9, 95.2, 55.6, 38.7, 36.3; IR (KBr, cm−1) ν 2907, 1579, 1563, 1476, 1421. HRMS (ESI-ion trap) calcd for C27H27F3N7OS: [M + H]+ 554.1944. Found: 559.1944. Elemental analysis calcd (%) for C27H26F3N7OS: C, 58.58; H, 4.73; N, 17.71. Found: C, 58.40; H, 4.82; N, 17.56.
General procedure for the one-pot Cu(II)-mediated reaction of azacalix[1]arene[3]pyridines 8a–f with Me4NSCF35: synthesis of trifluoromethylthiolated azacalix[1]arene[3]pyridines 6a–f
A mixture of azacalix[1]arene[3]pyridines 8a–f (0.2 mmol) and Cu(ClO4)2·6H2O (111 mg, 0.3 mmol) in CHCl3 (4 mL), methanol (4 mL) and acetonitrile (0.4 mL) (for substrates 8b–f) or in CHCl3 (4 mL) and methanol (4 mL) (for substrate 8a) was reacted at room temperature under atmospheric conditions for 24 h. A solution of Me4NSCF35 (105 mg, 0.6 mmol) in CH3CN (3.6 mL) (for the reaction of 8b–f) or in CH3CN (4 mL) (for the reaction of 8a) was added, and the reaction mixture was continuously stirred at 80 °C for another 24 h. Following the same work-up procedure as that for the reaction 2a–f with 5, products 6a–f were obtained in 58–91% yields.Reaction of arylcopper(II) complex 1a with N-trifluoromethylthiosaccharin 3
Under nitrogen protection, a mixture of arylcopper(II) complex 1a (117 mg, 0.2 mmol) and N-trifluoromethylthiosaccharin 3 (57 mg, 0.2 mmol) in dry acetonitrile (4 mL) was continuously stirred at 25 °C for 24 h. A saturated aqueous solution of EDTA (2 mL) and brine (40 mL) was added, and the mixture was extracted with DCM (3 × 20 mL). The combined organic layer was dried with anhydrous Na2SO4. After removal of the solvent, the residue was chromatographed on a silica gel column (100–200 mesh) using a mixture of petroleum ether, DCM and ethyl acetate (12:6:1) as the mobile phase to give pure products 7a (14 mg, 11%), 7b (9 mg, 7%) and 7c (16 mg, 13%).
Reaction of azacalix[1]arene[3]pyridine 8a with N-trifluoromethylthiosaccharin 3
Under nitrogen protection, a mixture of 8a (424 mg, 1 mmol) and N-trifluoromethylthiosaccharin 3 (283 mg, 1 mmol) in dry acetonitrile (20 mL) was continuously stirred at 25 °C for 24 h. A saturated aqueous solution of EDTA (10 mL) and brine (200 mL) was added, and the mixture was extracted with DCM (3 × 80 mL). The combined organic layer was dried with anhydrous Na2SO4. After removal of the solvent, the residue was chromatographed on a silica gel column (200–300 mesh) using a mixture of petroleum ether, DCM and ethyl acetate (12:6:1) as the mobile phase to give pure products 7a (62 mg, 10%), 7b (38 mg, 6%) and 7c (87 mg, 14%).
7a: white solid, mp 235–236 °C; 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.2 Hz, 2H), 7.43 (t, J = 7.6 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 6.60 (d, J = 7.8 Hz, 2H), 6.56 (d, J = 7.8 Hz, 2H), 6.45 (d, J = 8.2 Hz, 2H), 5.60 (s, 1H), 3.31 (s, 6H), 3.27 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −43.5 (s, 6F); 13C NMR (100 MHz, CDCl3) δ 162.5, 160.0, 158.1, 150.8, 148.5, 139.4, 129.6 (q, J = 308.0 Hz), 129.4, 117.0, 116.6, 112.6, 108.7, 102.7, 40.8, 36.9; IR (KBr, cm−1) ν 2900, 1588, 1569, 1421, 1106. HRMS (ESI-ion trap) calcd for C27H24F6N7S2: [M + H]+ 624.1433. Found: 624.1436.
7b: white solid, mp 210–211 °C; 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 8.2 Hz, 2H), 7.16 (dt, J = 7.9, 2.6 Hz, 2H), 6.78 (dd, J = 7.8, 1.8 Hz, 2H), 6.62 (s, 1H), 6.38 (d, J = 8.2 Hz, 2H), 6.17 (d, J = 7.8 Hz, 2H), 3.24 (s, 6H), 3.23 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −43.6 (s, 6F); 13C NMR (100 MHz, CDCl3) δ 163.5, 160.0, 158.9, 148.9, 148.0, 137.9, 129.7 (q, J = 308.0 Hz), 129.6, 126.2, 123.5, 109.1, 104.5, 100.8, 39.1, 39.0; IR (KBr, cm−1) ν 2921, 1576, 1543, 1474, 1420, 1378, 1153, 1124, 1105. HRMS (ESI-ion trap) calcd for C27H24F6N7S2: [M + H]+ 624.1433. Found: 624.1433. A high quality single crystal for X-ray diffraction analysis was obtained by diffusing n-hexane vapor into the solution of 7b in DCM. For the X-ray molecular structure of 7b (CCDC 1474335), see the ESI.†
7c: white solid, mp 270–271 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 7.8 Hz, 2H), 7.41 (t, J = 8.0 Hz, 1H), 7.15 (t, J = 8.0 Hz, 1H), 6.66 (d, J = 6.9 Hz, 1H), 6.58 (d, J = 7.8 Hz, 1H), 6.46 (t, J = 8.0 Hz, 2H), 6.38 (d, J = 7.8 Hz, 2H), 5.86 (s, 1H), 3.22 (s, 3H), 3.20 (s, 3H), 3.18 (s, 3H), 3.18 (s, 3H); 19F NMR (376 MHz, CDCl3) δ −42.7 (s, 3F), −43.0 (s, 3F); 13C NMR (100 MHz, CDCl3) δ 161.9, 161.7, 161.6, 160.7, 159.6, 157.1, 150.8, 148.7, 148.0, 147.9, 139.5, 129.9 (q, J = 309.0 Hz), 129.8 (q, J = 309.0 Hz), 129.7, 118.1, 117.2, 115.5, 109.9, 108.3, 108.1, 107.1, 105.3, 104.3, 40.0, 39.3, 38.0, 37.5; IR (KBr, cm−1) ν 2911, 1603, 1565, 1479, 1423, 1373, 1161, 1099. HRMS (ESI-ion trap) calcd for C27H24F6N7S2: [M + H]+ 624.1433. Found: 624.1436. A high quality single crystal for X-ray diffraction analysis was obtained by diffusing n-hexane vapor into the solution of 7c in DCM. For the X-ray molecular structure of 7c (CCDC 1474336), see the ESI.†
Conclusions
In summary, we have achieved very convenient and straightforward copper(II)-mediated trifluoromethylthiolation of azacalix[1]arene[3]pyridine macrocycles using a readily available nucleophilic reagent Me4NSCF3 under mild conditions. By employing isolated organocopper(II) and organocopper(III) complexes, we have shown convincingly that direct arene C–H bond trifluoromethylthiolation proceeds through arylcopper(III) rather than arylcopper(II) reactive intermediates. The heteracalixaromatics along with the structurally well-defined organometallic complexes provide powerful molecular tools in the study of arene C–H bond functionalizations, permitting in-depth comprehension of the mechanistic aspects of transition metal catalysis. Applications of functionalized azacalix[1]arene[3]pyridines in molecular recognition and self-assembly are under investigation and results will be reported in due course.Acknowledgements
We thank the National Natural Science Foundation of China (21572111, 21132005, 21421064, 91427301) and Tsinghua University for financial Support.Notes and references
- (a) M.-X. Wang, X.-H. Zhang and Q.-Y. Zheng, Angew. Chem., Int. Ed., 2004, 43, 838 CrossRef CAS PubMed; (b) M.-X. Wang and H.-B. Yang, J. Am. Chem. Soc., 2004, 126, 15412 CrossRef CAS PubMed; (c) M.-X. Wang, Chem. Commun., 2008, 4541 RSC; (d) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182 CrossRef CAS PubMed.
- (a) W. Maes and W. Dehaen, Chem. Soc. Rev., 2008, 37, 2393 RSC; (b) H. Tsue, K. Ishibashi and R. Tamura, Top. Heterocycl. Chem., 2008, 17, 73 CAS; (c) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291 CrossRef CAS PubMed.
- For selected examples, see: (a) J. L. Katz and B. A. Tschaen, Org. Lett., 2010, 12, 4300 CrossRef CAS PubMed; (b) C.-F. Chen, Chem. Commun., 2011, 47, 1674 RSC; (c) W. van Rossom, M. Ovaere, L. van Meervelt, W. Dehaen and W. Maes, Org. Lett., 2009, 11, 1681 CrossRef CAS PubMed; (d) M.-L. Ma, X.-Y. Li and W. Ke, J. Am. Chem. Soc., 2009, 131, 8338 CrossRef CAS PubMed; (e) M. Xue and C.-F. Chen, Org. Lett., 2009, 11, 5294 CrossRef CAS PubMed; (f) H. Konishi, S. Hashimoto, T. Sakakibara, S. Matsubara, Y. Yasukawa, O. Morikawa and K. Kobayashi, Tetrahedron Lett., 2009, 50, 620 CrossRef CAS; (g) J.-M. Raimundo, Z. Chen and O. Siri, Chem. Commun., 2011, 47, 10410 RSC; (h) Y. Zhu, J. Yuan, Y. Li, M. Gao, L. Cao, J. Ding and A. Wu, Synlett, 2011, 52 Search PubMed; (i) C.-S. Zuo, O. Wiest and Y.-D. Wu, J. Phys. Org. Chem., 2011, 24, 1157 CrossRef CAS; (j) H. Tsue, H. Takahashi, K. Ishibashi, R. Inoue, S. Shimizu, D. Takahashi and R. Tamura, CrystEngComm, 2012, 14, 1021 RSC; (k) A. L. Vicente, J. M. Caio, J. Sardinha, C. Moiteiro, R. Delgado and V. Felix, Tetrahedron, 2012, 68, 670 CrossRef CAS; (l) C. Gargiulli, G. Gattuso, A. Notti, S. Pappalardo, M. F. Parisi and F. Puntoriero, Tetrahedron Lett., 2012, 53, 616 CrossRef CAS; (m) Y. Visitaev, I. Goldberg and A. Vigalok, Inorg. Chem., 2013, 52, 6779 CrossRef CAS PubMed; (n) C. Zhang, Z. Wang, S. Song, X. Meng, Y.-S. Zheng, X.-L. Yang and H.-B. Xu, J. Org. Chem., 2014, 79, 2729 CrossRef CAS PubMed; (o) J. W. Wackerly, M. Zhang, S. T. Nodder, S. M. Carlin and J. L. Katz, Org. Lett., 2014, 16, 2920 CrossRef CAS PubMed.
- (a) H.-Y. Gong, Q.-Y. Zheng, X.-H. Zhang, D.-X. Wang and M.-X. Wang, Org. Lett., 2006, 8, 4895 CrossRef CAS PubMed; (b) H.-Y. Gong, D.-X. Wang, Q.-Y. Zheng and M.-X. Wang, Tetrahedron, 2009, 65, 87 CrossRef CAS; (c) E.-X. Zhang, D.-X. Wang, Z.-T. Huang and M.-X. Wang, J. Org. Chem., 2009, 74, 8595 CrossRef CAS PubMed.
- H.-Y. Gong, X.-H. Zhang, D.-X. Wang, H.-W. Ma, Q.-Y. Zheng and M.-X. Wang, Chem. – Eur. J., 2006, 12, 9262 CrossRef CAS PubMed.
- (a) D.-X. Wang, Q.-Y. Zheng, Q.-Q. Wang and M.-X. Wang, Angew. Chem., Int. Ed., 2008, 47, 7485 CrossRef CAS PubMed; (b) D.-X. Wang and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 892 CrossRef CAS PubMed; (c) Y. Chen, D.-X. Wang, Z.-T. Huang and M.-X. Wang, Chem. Commun., 2011, 47, 8112 RSC; (d) S. Li, S.-X. Fa, Q.-Q. Wang, D.-X. Wang and M.-X. Wang, J. Org. Chem., 2012, 77, 1860 CrossRef CAS PubMed; (e) Q. He, Y.-F. Ao, Z.-T. Huang and D.-X. Wang, Angew. Chem., Int. Ed., 2015, 54, 11785 CrossRef CAS PubMed.
- J. Zhang, B. Zhou, Z.-R. Sun and X.-B. Wang, Phys. Chem. Chem. Phys., 2015, 17, 3131 RSC.
- (a) B. Yao, D.-X. Wang, Z.-T. Huang and M.-X. Wang, Chem. Commun., 2009, 2899 RSC; (b) Z.-L. Wang, L. Zhao and M.-X. Wang, Org. Lett., 2011, 13, 6560 CrossRef CAS PubMed; (c) Z.-L. Wang, L. Zhao and M.-X. Wang, Org. Lett., 2012, 14, 1472 CrossRef CAS PubMed; (d) Z.-L. Wang, L. Zhao and M.-X. Wang, Chem. Commun., 2012, 48, 9418 RSC; (e) B. Yao, Z.-L. Wang, H. Zhang, D.-X. Wang, L. Zhao and M.-X. Wang, J. Org. Chem., 2012, 77, 3336 CrossRef CAS PubMed; (f) H. Zhang, L. Zhao, D.-X. Wang and M.-X. Wang, Org. Lett., 2013, 15, 3836 CrossRef CAS PubMed; (g) B. Yao, Y. Liu, L. Zhao, D.-X. Wang and M.-X. Wang, J. Org. Chem., 2014, 79, 11139 CrossRef CAS PubMed.
- H. Zhang, B. Yao, L. Zhao, D.-X. Wang, B.-Q. Xu and M.-X. Wang, J. Am. Chem. Soc., 2014, 136, 6326 CrossRef CAS PubMed.
- X. Ribas, D. A. Jackon, B. Donnadieu, J. Mahia, T. Parella, R. Xifra, B. Hedman, K. O. Hodgson, A. Llobet and T. D. P. Stack, Angew. Chem., Int. Ed., 2002, 41, 2991 CrossRef CAS.
- X. Ribas, C. Calle, A. Poater, A. Casitas, L. Gómez, R. Xifra, T. Parella, J. Benet-Buchholz, A. Schweiger, G. Mitrikas, M. Solà, A. Llobet and T. D. P. Stack, J. Am. Chem. Soc., 2010, 132, 12299 CrossRef CAS PubMed.
- X.-H. Hu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef PubMed.
- L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed.
- X. Shao, C. Xu, L. Lu and Q. Shen, Acc. Chem. Res., 2015, 48, 1227 CrossRef CAS PubMed.
- H. Zheng, Y. Huang and Z. Weng, Tetrahedron Lett., 2016, 57, 1397 CrossRef CAS.
- (a) P. Chen and G. Liu, Synthesis, 2013, 2919 CAS; (b) G. Landelle, A. Panossian, S. Pazenok, J.-P. Vors and F. Leroux, Beilstein J. Org. Chem., 2013, 9, 2427 Search PubMed; (c) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415 CrossRef CAS.
- C.-F. Xu, B.-Q. Ma and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 9316 CrossRef CAS PubMed.
- (a) X.-X. Shao, X.-Q. Wang, T. Yang, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 3457 CrossRef CAS PubMed; (b) E. V. VInogradova, P. Müller and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 53, 3125 CrossRef CAS PubMed.
- R. Pluta, P. Nikolaienko and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 1650 CrossRef CAS PubMed.
- K. Kang, C.-F. Xu and Q. Shen, Org. Chem. Front., 2014, 1, 294 RSC.
- C. Chen, Y. Xie, L. Chu, R.-W. Wang, X. Zhang and F.-L. Qing, Angew. Chem., Int. Ed., 2012, 51, 2492 CrossRef CAS PubMed.
- C.-P. Zhang and D. A. Vicic, Chem. – Asian J., 2012, 7, 1756 CrossRef CAS PubMed.
- G. Teverovskiy, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 7312 CrossRef CAS PubMed.
- (a) C.-P. Zhang and D. A. Vicic, J. Am. Chem. Soc., 2012, 134, 183 CrossRef CAS PubMed; (b) G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164 CrossRef CAS PubMed.
- (a) L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237 CrossRef CAS PubMed; (b) For a similar example of trifluoromethylthiolation of arenes catalyzed by Pd, see: J. Xu, P. Chen, J. Ye and G. Liu, Acta Chim. Sin., 2015, 73, 1294 CrossRef CAS.
- (a) B. Yao, D.-X. Wang, H.-Y. Gong, Z.-T. Huang and M.-X. Wang, J. Org. Chem., 2009, 74, 5361 CrossRef CAS PubMed; (b) W.-S. Ren, L. Zhao and M.-X. Wang, J. Org. Chem., 2015, 80, 9272 CrossRef CAS PubMed.
- J. H. Clark and H. Smith, J. Fluorine Chem., 1993, 61, 223 CrossRef CAS.
- J.-T. Li, L.-X. Wang, D.-X. Wang, L. Zhao and M.-X. Wang, J. Org. Chem., 2014, 79, 2178 CrossRef CAS PubMed.
- (a) A. Weng, W. He, C. Chen, R. Lee, D. Tan, Z. Lai, D. Kong, Y. Yuan and K.-W. Huang, Angew. Chem., Int. Ed., 2013, 52, 1548 CrossRef PubMed; (b) J. Xu, X. Mu, P. Chen, J. Ye and G. Liu, Org. Lett., 2014, 16, 3942 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all products, and X-ray structures of 6a, 7b and 7c (CIFs). CCDC 1474334–1474336. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00161k |
| This journal is © the Partner Organisations 2016 |