
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of mono-, di-, and triaminobismuthanes and observation of C–C coupling of aromatic systems with bismuth(III) chloride†
Christian
Hering-Junghans
ab,
Axel
Schulz
*ac,
Max
Thomas
a and
Alexander
Villinger
a
aInstitut für Chemie, Universität Rostock, Albert-Einstein-Str. 3a, 18059 Rostock, Germany. E-mail: axel.schulz@uni-rostock.de
bDepartment of Chemistry, University of Alberta, 11227 Saskatchewan Dr., T62 2G2 Edmonton, Alberta, Canada
cAbteilung Materialdesign, Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
First published on 19th February 2016
Abstract
The reaction of lithium N-trimethylsilyl-amides of the type RN(SiMe3)Li with bismuth(III) chloride yielded mono-, di- or triaminobismuthanes depending on the sterical demand of the anilide ligand R and the used stoichiometry. For the bulky Mes* substituent the reaction with BiCl3 resulted in the formation of a C–C coupling product as the main product besides a small amount of the expected Mes*N(SiMe3)BiCl2.
Introduction
C–C coupling reactions of aromatic systems in the presence of bismuth(III) chloride have been known since 2002, when Hanna and coworkers described the reaction of LiOAr (Ar = 2,6-di-tert-butylphenyl) with BiCl3.1 A complex mixture of organic compounds along with a black precipitate was observed by the authors. Beside HOAr, the main products of this reaction were a diol (1) and a dione (2) as illustrated in Scheme 1,1,2 and their formation can be rationalized by a mechanism involving radical intermediates.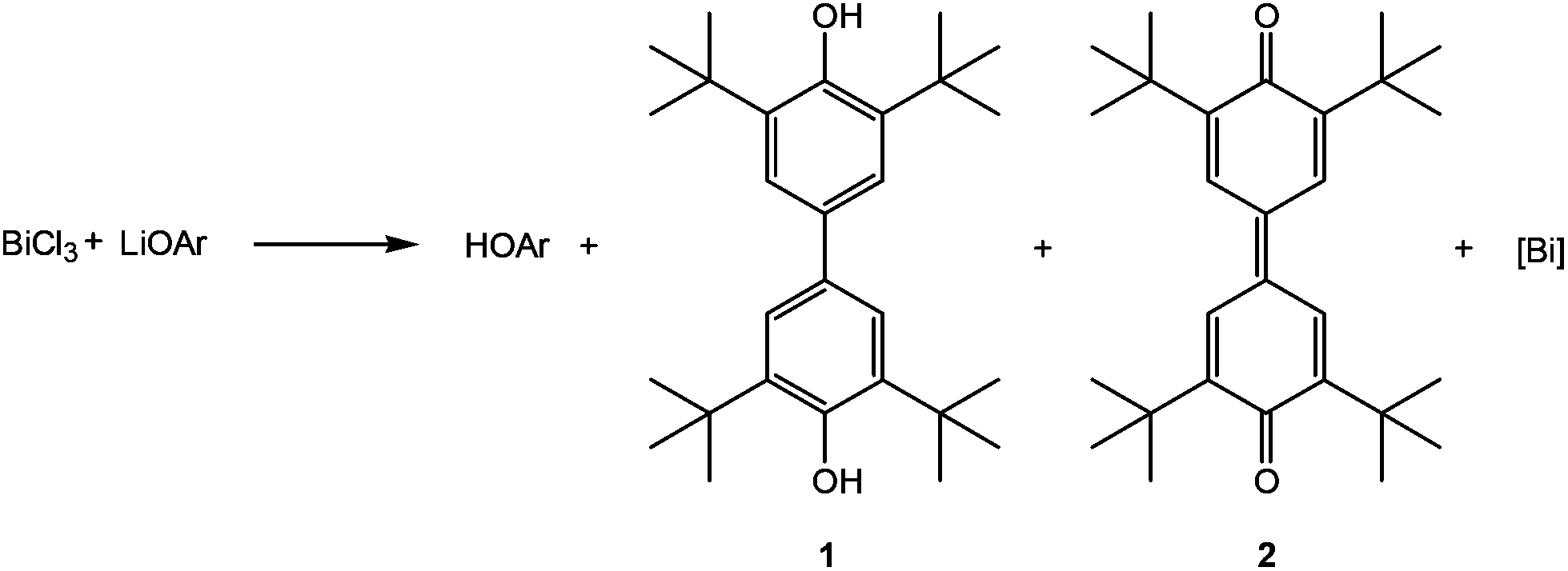 | ||
| Scheme 1 Reaction of LiOAr with BiCl3 by Hanna et al. (Ar = 2,6-di-tert-butylphenyl). | ||
With the aid of EPR spectroscopy Hanna and coworkers could experimentally detect Bi(II) radicals in the reaction mixture.1 In 2011 the group of Evans reported the preparation of a formal Bi(II) species with an NCN pincer-type ligand and a CH-activated ArO-group (Ar = 2,6-ditertbutylphenyl) binding to bismuth through the 4-position (3, Scheme 2).2 In contrast the reaction of the BiCl2-precursor with KOAr′ (Ar′ = Dmp, 2,6-dimethylphenyl; Dipp, 2,6-diisopropylphenyl) substituents yielded the corresponding oxidobismuthanes, a C–H activation of the para-position could not be observed. In 2014, Evans et al. presented a catalytic cycle based on their pincer complex 3 (Scheme 2).3
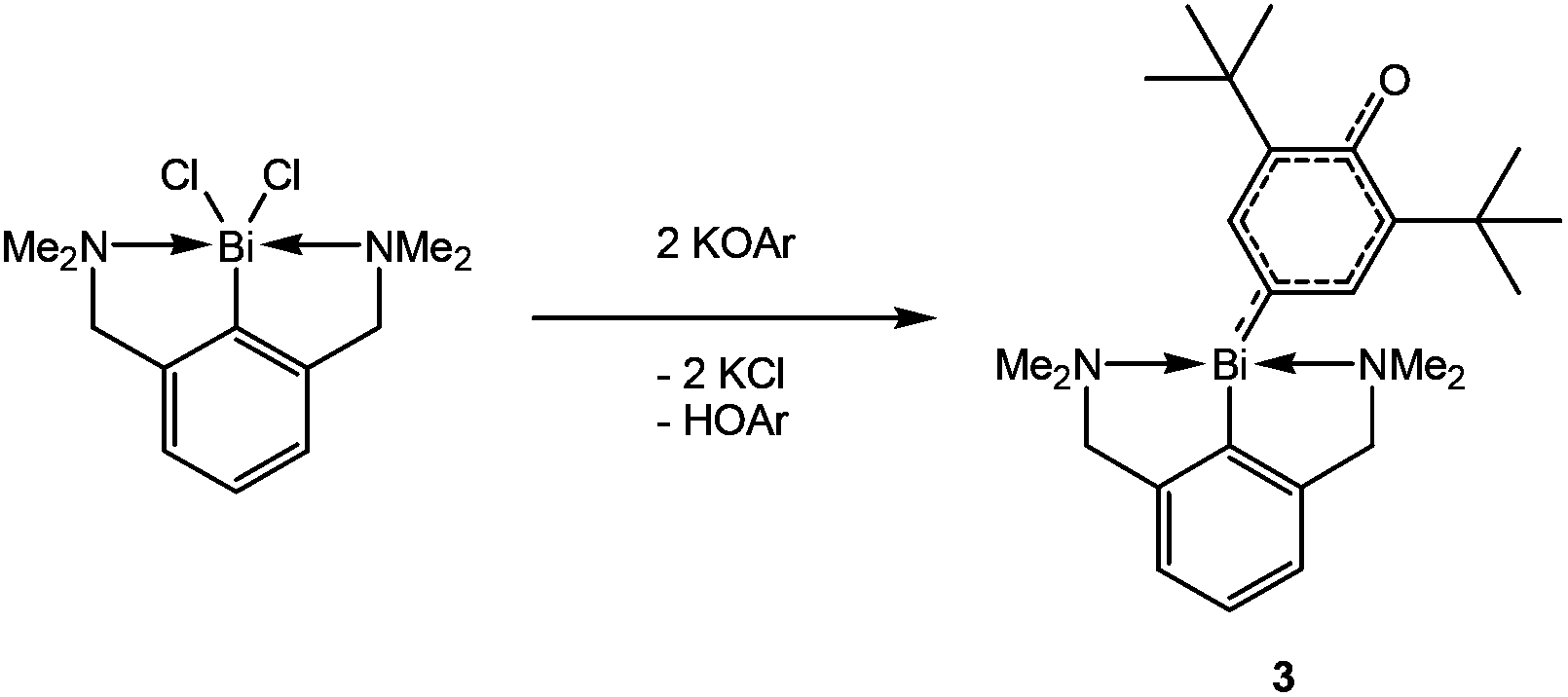 | ||
| Scheme 2 Synthesis of 3 by Evans et al. (Ar = 2,6-di-tert-butylphenyl). | ||
With this complex in hand, the group of Evans was able to introduce a carboxyl group using 2,6-di-tert-butylphenolat and CO2 as starting materials. The carboxyl group in this reaction could also be replaced by a NO functionality.4 Just recently, a stable Bi(II)-radical was reported by the Coles group, making this a vital field of research.5 To the best of our knowledge, there are no similar reactions of amides (4, R = aryl in Scheme 3) reported. Herein, we report on the systematic study of silylated bulky mono-, di- and triaminobismuthanes of the type RN(SiMe3)BiCl2, [RN(SiMe3)]2BiCl and [RN(SiMe3)]3Bi. There is only scarce information on substituted aminobismuthanes6 which are valuable starting materials for several transformations in main group chemistry such as the generation of cyclo-dibismadiazanes [R′Bi(μ-NR)]2,7–9 bismuthenium cations,10–14 or Bi–N precursors.15–23 For example, Bi(NMe2)3 is used in CVD (chemical vapour deposition) processes to deposit bismuth nitrides as suggested by Errington and Norman.23 The isolation of (silylated) aminobismuthanes is complicated since often coincident formation of monoaminodichloro-, diaminochloro-, and triaminobismuthanes can be observed (Scheme 3). Additionally, Me3SiCl-elimination can result in the formation of iminobismuthanes or their respective dimers, the dibismadiazanes, can be obtained depending on the bulkiness of the substituents.24 Hence, it is vital to find specific synthetic protocols to generate specific aminobismuthanes, iminobismuthanes or cyclo-dibismadiazane, exclusively. This can be achieved by the utilization of Me3Si-substituted bulky amido groups N(SiMe3)R (Scheme 3) and by the use of lithium amides rather than amines.
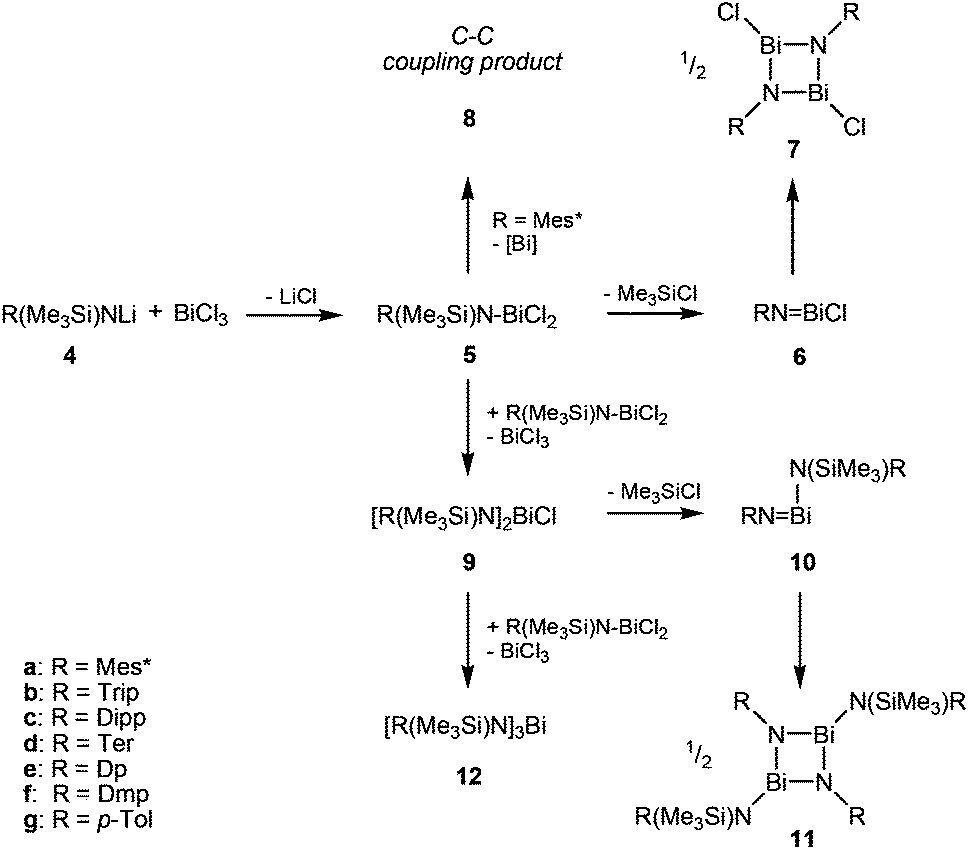 | ||
| Scheme 3 Product distribution in the synthesis of mono-, di- and triamino bismuthanes. | ||
Results and discussion
Syntheses
In a series of experiments we studied the reaction of BiCl3 with 1, 2 and 3 equivalents of Li[N(SiMe3)R] (R = Mes* (4a), Trip (4b), Dipp (4c), Ter (4d), Dp (4e), Dmp (4f), p-Tol (4g); Mes* = 2,4,6-tri-tert-butylphenyl, Trip = 2,4,6-triisopropylphenyl, Dipp = 2,6-diisopropylphenyl, Ter = 2,6-bis(2,4,6-trimethylphenyl)-phenyl, Dp = 3,4-dimethylphenyl, Dmp = 2,6-dimethylphenyl, p-Tol = 4-methylphenyl) at ambient temperature. The lithium amides 4 were readily obtained in the reaction of the respective N-trimethylsilyl-aniline derivatives with n-BuLi, whereas in most cases 4 can be generated in situ prior to the reaction with BiCl3.Astonishingly, treatment of BiCl3 with equimolar amounts of 4 resulted, depending on the steric strain, in a complex mixture of products as illustrated in Scheme 3 (cf. steric strain can be evaluated using the maximal cone angle at 1.45 Å:25 251° Mes*, 222° Trip, 223° Dipp, 232° Ter, 158° Dp, 199° Dmp, 158° p-Tol).
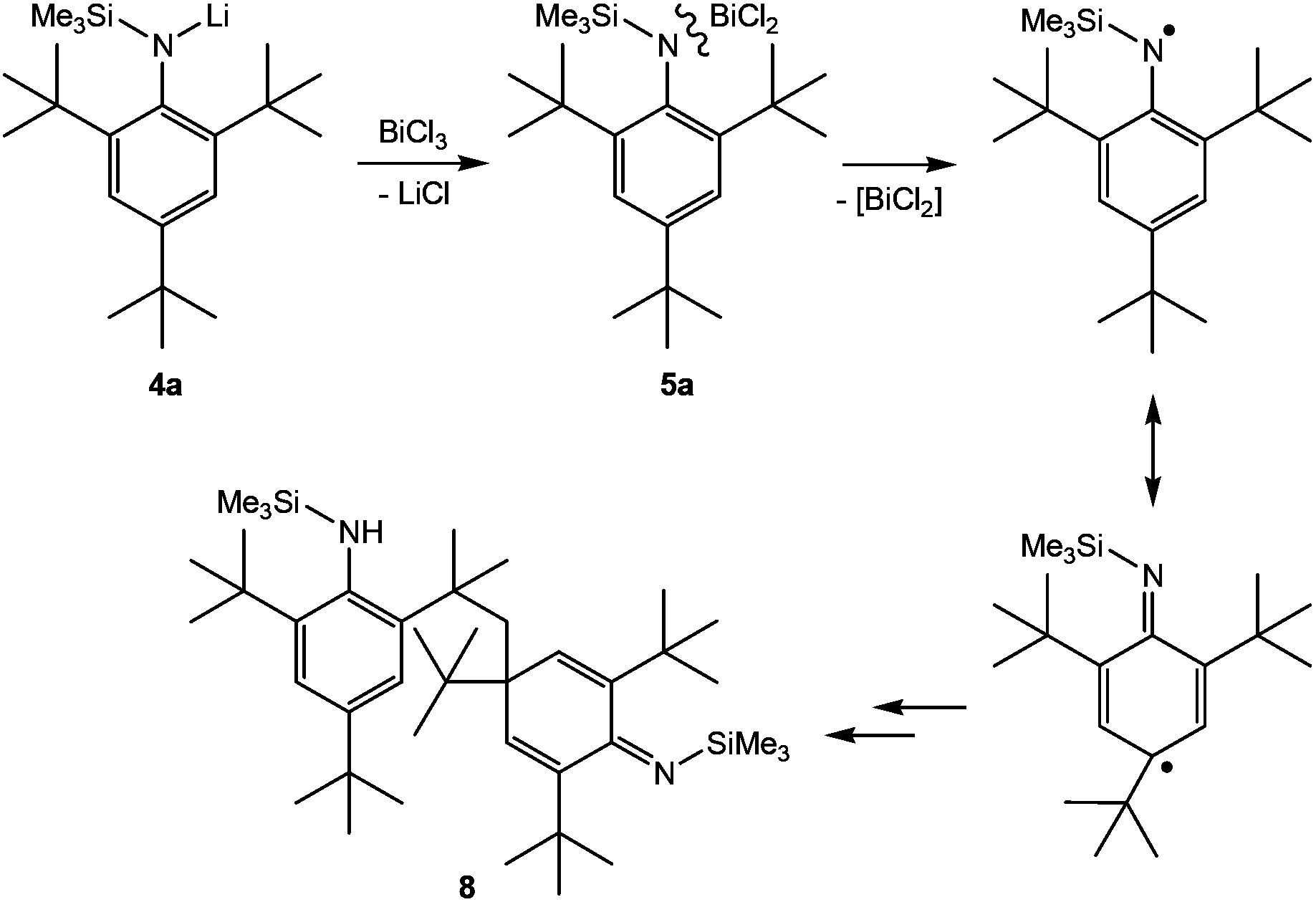 | ||
| Scheme 4 Formation of 5a and postulated reaction to the coupling product 8 in analogy to the suggested reaction pathway by Hanna et al. | ||
Upon addition of a solution of Mes*N(SiMe3)Li at room temperature to a suspension of BiCl3 first the formation of Mes*N(SiMe3)BiCl2 (5a) was observed as indicated by 1H NMR data. The yellow solution darkens upon complete addition of 4a and eventually gave the aforementioned black suspension. Presumably, decomposition is initiated by homolytic dissociation of the N–Bi bond affording, after a complex rearrangement, C–C coupling product 8 besides a black mixture of bismuth containing compounds (for example BiCl3 thf could be identified) as illustrated in Scheme 4. It needs to be emphasized that the formation of 8 under this reaction conditions is reproducible and usually yields 8 in isolated yields of 40–50% with respect to 4a (Fig. 1). When the addition of 4a to BiCl3 was carried out at low temperatures (−80 °C), we were successful to isolate 5a in small amounts besides 8 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 ratio) upon slow warming to −20 °C and fractional crystallization. Hence, this experimentally proves that the initial formation of 5a represents the first reaction step as discussed before. The presence of 5a was unequivocally confirmed by X-ray diffraction (Fig. 2). Crystals of 5a 1.5 toluene were obtained from recrystallization in toluene (Fig. 2). Using isolated and recrystallized 4a rather than generating it in situ (in the reaction with BiCl3 in diethyl ether at −80 °C) suppressed the formation of the C–C coupling product, however, not entirely. It was possible to obtain 5a as the main product in using this protocol. Using freshly sublimed BiCl3 also helped to increase the amount of 5a formed and suppressed formation of 8. The influence of freshly sublimed BiCl3 was already discussed by Evans et al. in the synthesis of Bi[N(SiM3)2]3.26 In the reaction with 4a the as supplied bismuth(III)-chloride tends to pronounce the formation of 8. 5a is stable in the solid state with respect to the formation of 8. In dichloromethane solution the formation of a black precipitate indicated a partial decomposition within 120 h at room temperature, but no decomposition products could be identified in the 1H NMR spectrum, which suggest decomposition into NMR-silent or insoluble products. Interestingly, Burford and co-workers reported the formation of (Mes*NH)3Bi in the reaction of three equivalents of the corresponding lithium amide with one equivalent BiCl3.27 Obviously, substitution of H by Me3Si prevents a higher degree of substitution at the bismuth.
:9 ratio) upon slow warming to −20 °C and fractional crystallization. Hence, this experimentally proves that the initial formation of 5a represents the first reaction step as discussed before. The presence of 5a was unequivocally confirmed by X-ray diffraction (Fig. 2). Crystals of 5a 1.5 toluene were obtained from recrystallization in toluene (Fig. 2). Using isolated and recrystallized 4a rather than generating it in situ (in the reaction with BiCl3 in diethyl ether at −80 °C) suppressed the formation of the C–C coupling product, however, not entirely. It was possible to obtain 5a as the main product in using this protocol. Using freshly sublimed BiCl3 also helped to increase the amount of 5a formed and suppressed formation of 8. The influence of freshly sublimed BiCl3 was already discussed by Evans et al. in the synthesis of Bi[N(SiM3)2]3.26 In the reaction with 4a the as supplied bismuth(III)-chloride tends to pronounce the formation of 8. 5a is stable in the solid state with respect to the formation of 8. In dichloromethane solution the formation of a black precipitate indicated a partial decomposition within 120 h at room temperature, but no decomposition products could be identified in the 1H NMR spectrum, which suggest decomposition into NMR-silent or insoluble products. Interestingly, Burford and co-workers reported the formation of (Mes*NH)3Bi in the reaction of three equivalents of the corresponding lithium amide with one equivalent BiCl3.27 Obviously, substitution of H by Me3Si prevents a higher degree of substitution at the bismuth.
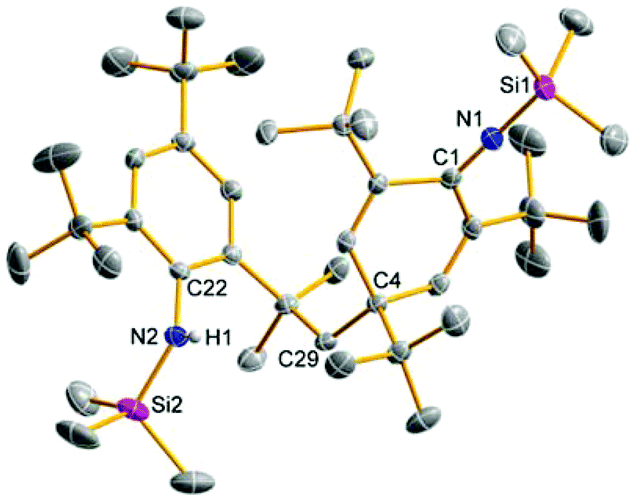 | ||
| Fig. 1 ORTEP drawing of the molecular structure of 8. Thermal ellipsoids with 50% probability at 173 K (hydrogen atoms omitted for clarity except from N–H). Selected distances (Å) and angles (°): Si1–N1 1.689(2), Si2–N2 1.732(2), N1–C1 1.268(2), N2–C22 1.438(2), C29–C4 1.565(3), Si1–N1–C1 175.9(2), N2–Si2–C22 135.2(2). | ||
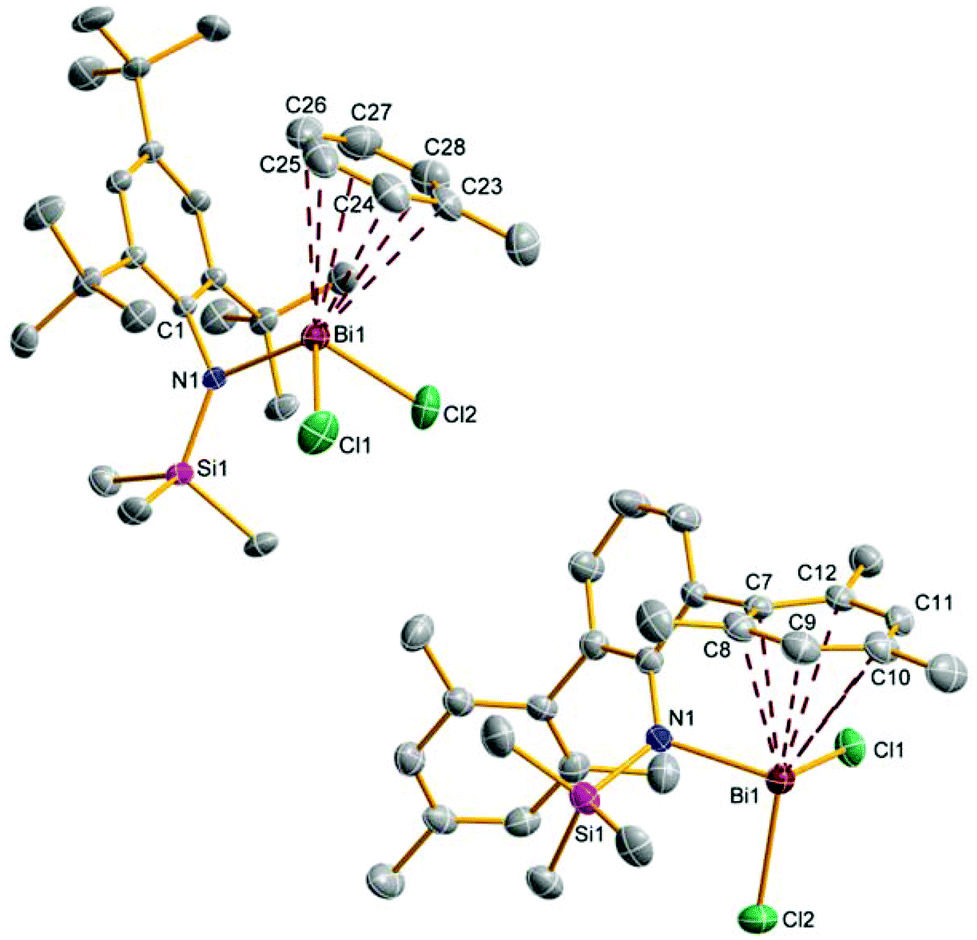 | ||
| Fig. 2 ORTEP drawing of the molecular structure of 5a (left) and 5d (right). Thermal ellipsoids with 50% probability at 173 K (hydrogen atoms omitted for clarity). Selected distances (Å) and angles (°): 5a: Bi1–N1 2.146(1), Bi1–Cl1 2.4771(5), Bi1–Cl2 2.4920(5), N1–Si1 1.747(2), Bi1–C23 3.340(2), Bi1–C24 3.376(2), Bi1–C25 3.474(3), Bi1–C26 3.517(2), Bi1–C27 3.472(3), Bi1–C28 3.386(3), Cl1–Bi1–Cl2 89.86(2), Cl2–Bi1–N1 103.57(3), N1–Bi1–Cl1 99.82(3), Bi1–N1–C1 101.49(8); 5d: Bi1–N1 2.176(3), Bi1–Cl1 2.485(1), Bi1–Cl2 2.517(1), Bi1–C7 2.940(4), Bi1–C8 3.223(4), Bi1–C9 3.524(5), Bi1–C10 3.637(4), Bi1–C11 3.484(4), Bi1–C12 3.191(4), N1–Bi1–Cl1 100.31(8), N1–Bi1–Cl2 94.09(8), Cl1–Bi1–Cl2 87.64(3), Bi1–N1–C1 118.3(2). | ||
We were interested in the generality of this C–C coupling reaction, prompting us to study differently substituted N-trimethylsilyl-amides, however, C–C coupling processes were only observed for the very bulky Mes* substituent.
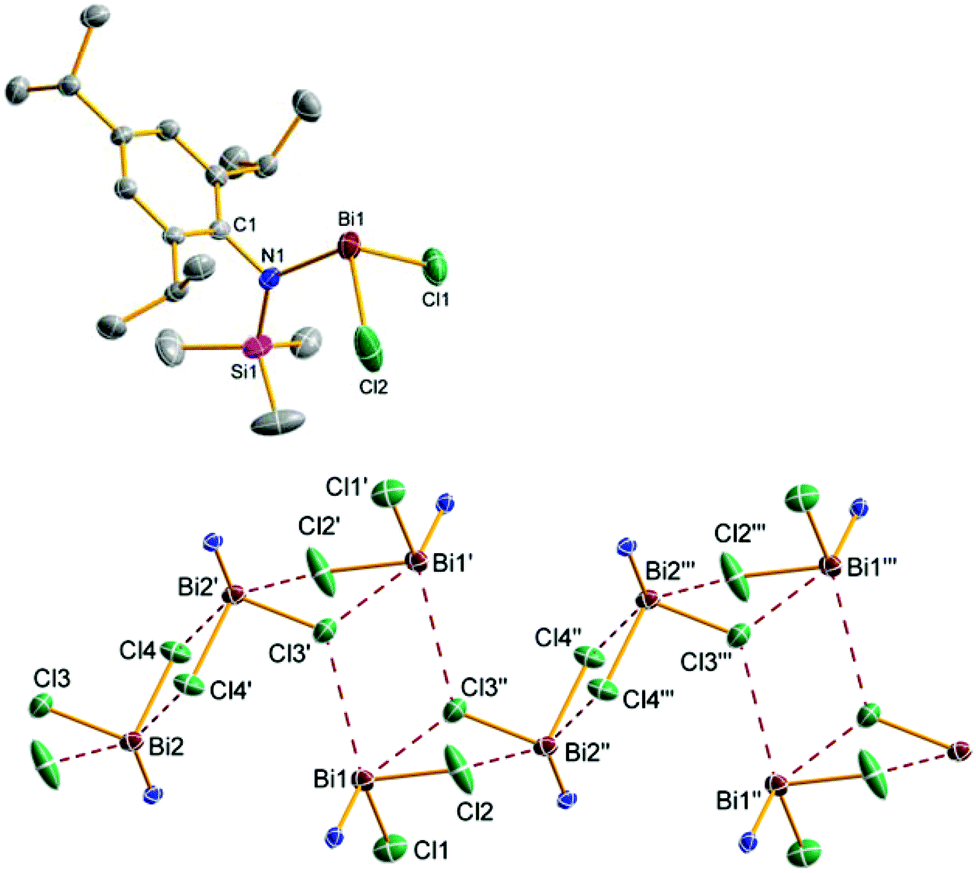 | ||
| Fig. 3 ORTEP drawing of the molecular structure of 5b (top, only one independent molecule is shown) and representation of the chain-like structure in the crystal of 5b (bottom, view along b-axis). Thermal ellipsoids with 50% probability at 173 K (hydrogen atoms omitted for clarity). Selected distances (Å) and angles (°): Bi1–Cl1 2.449(1), Bi1–Cl2 2.482(1), Bi1–Cl3 2.4946(9), Bi2–Cl4 2.5063(9), Bi1–N1 2.111(3), Bi2–N2 2.133(3), N1–Bi1–Cl1 97.84(7), Cl1–Bi1–Cl2 93.85(4), Cl2–Bi1–N1 97.05(7); long range distances: Bi2′–Cl4 3.593(1), Bi2′–Cl2′ 3.359(1), Bi1′–Cl3′ 3.755(1), Bi1–Cl3′ 3.761(1). | ||
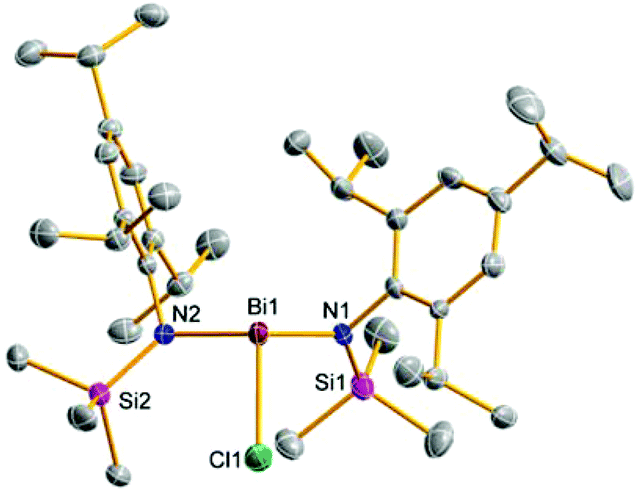 | ||
| Fig. 4 ORTEP drawing of the molecular structure of 9b. Thermal ellipsoids with 50% probability at 173 K (hydrogen atoms omitted for clarity). Selected distances (Å) and angles (°): Bi1–Cl1 2.4862(7), Bi1–N1 2.140(2), Bi1–N2 2.179(2), N1–Bi1–Cl1 97.35(5), Cl1–Bi1–N2 95.83(5), N2–Bi1–N1 108.50(7). | ||
:1 stoichiometry resulted in the formation of mono-substitution product DippN(SiMe3)BiCl2 (5c), which could be crystalized beside [DippN(SiMe3)]2BiCl (9c). Unfortunately, even isolated crystals of 5c were contaminated with 9c and thus could not be fully characterized. Interestingly, reaction of two or three equivalents 4c with bismuth(III)-chloride afforded DippN(SiMe3)H as the main product. In analogy to this reaction Roesky and co-workers demonstrated that the reaction of two or three equivalents of DippN(H)Li with BiCl3 led to a four-membered heterocycle of the type [DippNBiN(H)Dipp]2, the dimerization product of the intermediately formed iminobismuthane, similar to ring 11 (Scheme 3).28 Burford et al. reported a similar reaction but added t-BuN(H)Li as additional base to the reaction mixture of DippN(H)Li and BiCl3. This reaction also afforded [DippNBiN(H)Dipp]2 but in higher yields and with an easier isolation of the four-membered ring system, because the byproduct t-BuNH2 is considerably easier to remove than DippNH2.24
:1 mixture of 5e and 9e were obtained (Fig. 5). Generally, both the double as well as the triple substitution product can either be formed by reaction of 5e with 4e and 9e with 4e, respectively, or as depicted in Scheme 3 by BiCl3 elimination from 5e and 9e respectively. As shown in Fig. 5, in the solid state structure of 5e/9e BiCl3 moieties are preformed, so it can be assumed that in solution dimeric or even oligomeric units are present which react under BiCl3 elimination to give the triaminobismuthane 12e. Crystals of 5e/9e are only stable in the solid state, in solution they readily release BiCl3 affording 12e (Fig. 6), which is also the main product, when three equivalents of 4e are reacted with BiCl3.
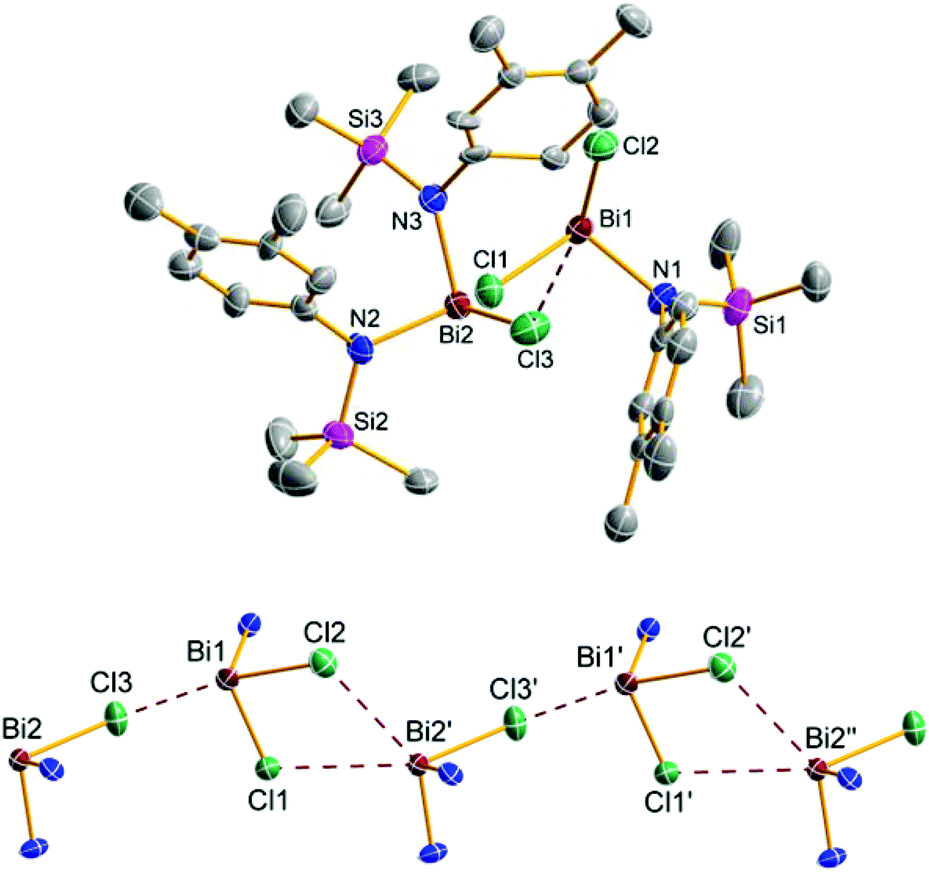 | ||
| Fig. 5 ORTEP drawing of the molecular structure of 5e/9e (top) and representation of the chain-like structure in the crystal of 5e/9e (bottom, view along a-axis). Thermal ellipsoids with 50% probability at 173 K (hydrogen atoms omitted for clarity). Selected distances (Å) and angles (°): Bi1–Cl1 2.4702(2), Bi1–Cl2 2.550(2), Bi2–Cl3 2.581(2), Bi1–N1 2.116(6), Cl2–Bi1–Cl3 171.000(6), Cl1–Bi1–Cl3 83.360(7); long range distances: Bi1–Cl3 3.057(2), Bi2′–Cl1 3.516(2), Bi2′–Cl2 3.644(2), Bi1–Bi2 4.8481(3). | ||
 | ||
| Fig. 6 ORTEP drawing of the molecular structure of 12g (top) and 12e (bottom). Thermal ellipsoids with 50% probability at 173 K (hydrogen atoms omitted for clarity). Selected distances (Å) and angles (°): 12g: Bi1–N1 2.150(3), Bi1–N2 2.162(3), Bi1–N3 2.154(3), N1–Bi1–N2 98.6(1), N2–Bi1–N3 98.6(1), N3–Bi1–N1 98.0(1); 12e: Bi1–N1 2.159(3), Bi1–N2 2.155(3), Bi1–N3 2.155(3), N1–Bi1–N2 98.5(1), N2–Bi1–N3 97.9(1), N3–Bi1–N1 98.7(1). | ||
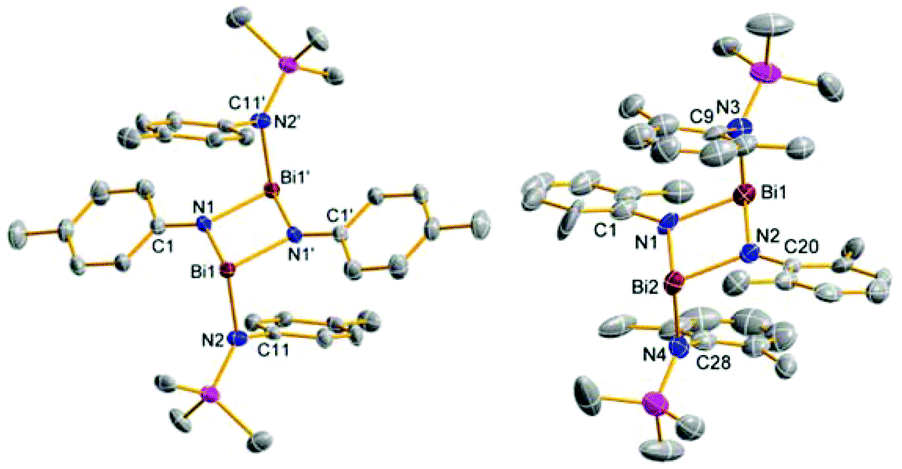 | ||
| Fig. 7 ORTEP drawing of the molecular structure of 11g (left) and 11f (right). Thermal ellipsoids with 50% probability at 173 K (hydrogen atoms omitted for clarity). Selected distances (Å) and angles (°): 11g: Bi1–N1 2.129(2), Bi1–N1′ 2.147(2), Bi1–N2 2.165(2), N1–Bi1–N2 95.29(8), N2–Bi1–N1′ 96.61(8), N1′–Bi1–N1 75.62(8), Bi1–N1–C1 127.0(2), C1–N1–Bi1′ 127.3(2), Bi1′–N1–Bi1 104.38(8), Bi1–N1–C1–C2 155.7(2); 11f: Bi1–N1 2.16(1), Bi1–N2 2.164(7), Bi1–N3 2.176(9), N2–Bi2 2.154(9), Bi2–N1 2.142(7), Bi2–N4 2.175(9), N1–Bi1–N2 77.1(3), N2–Bi1–N3 99.5(3), N3–Bi1–N1 101.7(3), N1–Bi2–N2 77.7(3), N2–Bi2–N4 102.1(3), N4–Bi2–N1 98.7(3), Bi1–N2–C20 127.9(7), C20–N2–Bi2 129.7(6), Bi2–N2–Bi1 102.3(4), Bi1–N2–C20–C21 142.6(8). | ||
:1 stoichiometry led to a mixture of products. From this mixture we were able to isolate the triple substitution product [p-TolN(SiMe3)]3Bi (12g) as pale yellow crystals suitable for X-ray structure elucidation (Fig. 6). However, utilization of an excess of base 4g (2 or 3 equivalents) resulted in the formation of 12g (80%) when freshly sublimed BiCl3 was added. Interestingly, when BiCl3 was used as delivered by Alfa Aesar traces of four-membered heterocycle 11g were observed. Red crystals of 11g suitable for X-ray structure determination were isolated from such an experiment.
X-ray elucidation
X-ray quality crystals were selected in Fomblin YR-1800 perfluoroether (Alfa Aesar) at ambient temperature. The samples were cooled to 173(2) K during measurement. Selected structural data are listed in Table 1, the molecular structures are shown in Fig. 1–7.| Bi–Cl | Bi–N | Cl–Bi–Cl | Cl–Bi–N | N–Bi–N | |
|---|---|---|---|---|---|
| a Exocyclic Bi–N distance. b Nring–Bi–Nexocyclic. c Ring adopts C1 symmetry. d Ring adopts Ci symmetry. | |||||
| 5a | 2.4771(5) | 2.146(1) | 89.9(2) | 99.82(3) | — |
| 2.4920(5) | — | — | 103.57(3) | — | |
| 5b | 2.449(1) | 2.111(3) | 93.85(4) | 97.05(7) | — |
| 2.483(1) | — | — | 97.84(7) | — | |
| 9b | 2.4862(7) | 2.140(2) | — | 95.83(5) | 108.51(7) |
| — | 2.178(2) | — | 97.35(5) | — | |
| 5c | 2.4746(9) | 2.125(3) | 94.90(3) | 92.50(7) | — |
| 2.5124(9) | — | — | 95.99(7) | — | |
| 2.4687(8) | 2.128(2) | 84.24(3) | 101.95(7) | — | |
| 2.6409(8) | — | — | 94.48(7) | — | |
| 2.4631(8) | 2.142(2) | 92.16(3) | 93.19(7) | — | |
| 2.6580(8) | — | — | 96.16(7) | — | |
| 2.4369(9) | 2.126(3) | 97.86(3) | 95.89(8) | — | |
| 2.515(1) | — | — | 94.58(7) | — | |
| 9c | 2.497(2) | 2.170(3) | — | 96.48(9) | 112.5(2) |
| — | 2.147(3) | — | 98.6(1) | — | |
| 2.477(1) | 2.182(3) | — | 95.17(8) | 110.7(1) | |
| — | 2.148(3) | — | 96.74(8) | — | |
| 5d | 2.485(1) | 2.176(3) | 87.64(3) | 94.09(8) | — |
| 2.517(1) | — | — | 100.31(8) | — | |
| 5e in | 2.550(2) | 2.116(6) | 89.60(7) | 94.7(1) | — |
| 5e/9e | 2.470(2) | — | — | 95.2(2) | — |
| 9e in | 2.581(2) | 2.137(6) | — | 94.6(2) | 95.7(2) |
| 5e/9e | — | 2.141(6) | — | 97.9(2) | — |
| 12e | — | 2.159(3) | — | — | 98.7(1) |
| — | 2.155(3) | — | — | 97.9(1) | |
| — | 2.155(3) | — | — | 98.5(1) | |
| 11f | — | 2.176(9)a | — | — | 99.5(3)b |
| — | 2.175(9)a | — | — | 102.1(3)b | |
| — | 2.16(1) | — | — | 98.7(3)b | |
| — | 2.142(7) | — | — | 101.7(3)b | |
| — | 2.154(9) | — | — | 77.1(3) | |
| — | 2.164(7) | — | — | 77.7(3) | |
| 11g | — | 2.165(2)a | — | — | 96.61(8)b |
| — | 2.129(2) | — | — | 75.62(8) | |
| — | 2.147(2) | — | — | 95.29(8) | |
| 12g | — | 2.150(3) | — | — | 98.5(1) |
| — | 2.162(3) | — | — | 98.6(1) | |
| — | 2.154(3) | — | — | 98.0(1) | |
As expected the molecular structures of all bismuth species feature a strongly distorted trigonal pyramidal coordination environment about the Bi center. Despite the pyramidal Bi centers, the four-membered rings in 11g and 11f are planar with both exocyclic amino groups in a trans orientation (deviation from planarity 11f: 0.0°, 11g: <2°). As can be seen from the data in Table 1, the molecular structures of all considered species display a fairly sharp distribution of Bi–N distances between 2.111–2.182 Å in accord with a typical polar Bi–N single bond (cf. ∑rcov(Bi–N) = 2.22 Å;29 2.16–2.17 Å in [DipNBiN(H)Dip]2]).24,28
The Bi–Cl bond lengths are in the range between 2.4369 to 2.658 Å, which is also indicative single bonds (∑rcov(Bi–Cl) = 2.50 Å).29 With respect to the angles around the bismuth center, the following trend was observed: <(Cl–Bi–Cl) (84.2–97.9°) ≪ (Cl–Bi–N) (92.5–103.6°) ≪ (N–Bi–N) (95.7–112.5°) which can mainly be attributed to the bulky Me3Si and phenyl substituents attached to the N atoms.
The most prominent structural features are secondary inter- and intramolecular interactions stabilizing the Lewis acidic bismuth center. Three different types of such interactions were observed: (i) intramolecular π-arene⋯Bi contacts, (ii) intermolecular π-arene⋯Bi contacts and (iii) chlorine atoms as bridges between different Bi centers. Strong secondary interactions of type (i) (Menshutkin type π complexes)30 with one phenyl group of the terphenyl substituent were observed for 5d (Fig. 2) as indicated by very short Bi⋯Carene distances (2.940–3.637 Å) which are well within the range of van der Waals radii (∑rvdW(C⋯Bi) = 3.77 Å).31
Interestingly, this strong interaction led to a considerable distortion of the highly flexible terphenyl substituent as indicated by e.g. the deviation from planarity of the involved phenyl ring (largest C–C–C–C torsion angle 9.3°). In 5a an intermolecular Menshutkin type π complex with one co-crystallized toluene solvent molecule is found with Bi⋯Carene distances between 3.340–3.517 Å.
As depicted in Fig. 3 and 5, a chain-like structure arises for 5b and 5e/9e due to weak Cl⋯Bi interactions between adjacent molecules. In 5b different Cl⋯Bi interactions can be discussed as indicated by the following four long range distances: Bi2′–Cl4 3.593(1), Bi2′–Cl2′ 3.359(1), Bi1′–Cl3′ 3.755(1), and Bi1–Cl3′ 3.761(1) Å. These rather long Bi⋯Cl distances (cf. ∑rvdW(Bi⋯Cl) = 3.81 Å)31 are significantly longer than the intramolecular Bi–Cl bonds (2.449–2.506 Å, ∑rcov(Bi–Cl) = 2.50 Å)29 in accord with a polar Bi–Cl single bond (see above).
To the best of our knowledge compound 5e/9e is the first example of co-crystallized mono and double substituted aminochloropnictanes. Both species are linked by three Cl⋯Bi interactions (Bi1–Cl3 3.057(2), Bi2′–Cl1 3.516(2), and Bi2′–Cl2 3.644(2) Å, Fig. 5) leading to the formation one-dimensional chains along the a-axis with alternating 5e and 9e molecules in the solid state.
Conclusions
Depending on the steric demand of the aryl group and the used stoichiometry the reaction of RN(SiMe3)Li with BiCl3 leads to mono, double or even triple substitution at the bismuth atom. With bulky groups attached to the amide, preferentially the products of mono substitution (5) are found (Scheme 3), while triple substitution is mainly observed for smaller groups. The aminodichlorobismuthanes 5 with smaller R groups readily undergo BiCl3 elimination reactions to form diaminochlorobismuthanes (9) or even triaminobismuthanes (12). Instead of triaminobismuthanes also cyclo-dibismadiazanes 11 can be formed when an excess of amide with a less bulky group is used and in solution both compounds 11 and 12 can even co-exist as well as in the solid state. For bulky groups, which are not able to form triple substitution products, the reaction of a huge excess of amide with BiCl3 leads to the formation of the amine RN(SiMe3)H. We want to stress that all reaction conditions (stoichiometry, solvent, reaction temperature, the use of isolated lithium amide or in situ generated and use of sublimed or unsublimed BiCl3) have a strong influence on the formed products and their distribution in mixtures. Even small changes to the reaction conditions can lead to very different results.In the solid state the Lewis acidic bismuth atoms in these compounds can be stabilized by secondary interactions with aromatic solvents or intramolecularly, such as in the terphenyl substituted species. Another way to stabilize bismuth centers was established by bridging Cl atoms as found in some of the mono- and dichloro bismuthanes.
C–C coupling reactions rather than substitution was only observed for the supermesityl substituted derivative, which forms in the first step the mono-substitution product, that readily undergoes a C–C coupling process, presumably by a radical mechanism.
Acknowledgements
We are indebted to Dr Dirk Michalik for NMR measurements.Notes and references
- T. A. Hanna, A. L. Rieger, P. H. Rieger and X. Wang, Inorg. Chem., 2002, 41, 3590–3592 CrossRef CAS PubMed.
- I. J. Casely, J. W. Ziller, M. Fang, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 5244–5247 CrossRef CAS PubMed.
- D. R. Kindra and W. J. Evans, Dalton Trans., 2014, 43, 3052–3054 RSC.
- D. R. Kindra, I. J. Casely, J. W. Ziller and W. J. Evans, Chem. – Eur. J., 2014, 20, 15242–15247 CrossRef CAS PubMed.
- R. J. Schwamm, J. R. Harmer, M. Lein, C. M. Fitchett, S. Granville and M. P. Coles, Angew. Chem., Int. Ed., 2015, 54, 10630–10633 CrossRef CAS PubMed.
- H. Suzuki and Y. Matano, Organobismuth Chemistry, Elsevier Science B. V., Amsterdam, 2001 Search PubMed.
- G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815–7880 CrossRef CAS PubMed.
- (a) M. S. Balakrishna, D. J. Eisler and T. Chivers, Chem. Soc. Rev., 2007, 36, 650–664 RSC; (b) G. G. Briand, T. Chivers and M. Parvez, Can. J. Chem., 2003, 81, 169–174 CrossRef CAS.
- D. Michalik, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2010, 49, 7575–7477 CrossRef CAS PubMed.
- M. Veith, B. Bertsch and V. Huch, Z. Anorg. Allg. Chem., 1988, 559, 73–88 CrossRef CAS.
- W. Baumann, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2008, 47, 9530–9532 CrossRef CAS PubMed.
- M. Lehmann, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2012, 51, 8087–8091 CrossRef CAS PubMed.
- R. J. Schwamm, B. M. Day, M. P. Coles and C. M. Fitchett, Inorg. Chem., 2014, 53, 3778–3787 CrossRef CAS PubMed.
- C. Hering-Junghans, M. Thomas, A. Villinger and A. Schulz, Chem. – Eur. J., 2015, 21, 6713–6717 CrossRef CAS PubMed.
- O. J. Scherer, P. Hornig and M. Schmidt, J. Organomet. Chem., 1966, 6, 259–264 CrossRef CAS.
- M. Veith and B. Bertsch, Z. Anorg. Allg. Chem., 1988, 557, 7–22 CrossRef CAS.
- A. J. Edwards, M. A. Beswick, J. R. Galsworthy, M. A. Paver, P. R. Raithby, M.-A. Rennie, C. A. Russell, K. L. Verhorevoort and D. S. Wright, Inorg. Chim. Acta, 1996, 248, 9–14 CrossRef CAS.
- B. Nekoueishahraki, P. P. Samuel, H. W. Roesky, D. Stern, J. Matussek and D. Stalke, Organometallics, 2012, 31, 6697–6703 CrossRef CAS.
- (a) B. M. Day and M. P. Coles, Organometallics, 2013, 32, 4270–4278 CrossRef CAS; (b) R. J. Schwamm, M. P. Coles and C. M. Fitchett, Organometallics, 2015, 34, 2500–2507 CrossRef CAS.
- M. Brym, C. M. Forsyth, C. Jones, P. C. Junk, R. P. Rose, A. Stasch and D. R. Turner, Dalton Trans., 2007, 3282–3288 RSC.
- D. Dange, A. Davey, J. A. B. Abdalla, S. Aldridge and C. Jones, Chem. Commun., 2015, 51, 7128–7131 RSC.
- B. N. Diel, T. L. Hubler and W. G. Ambacher, Heteroat. Chem., 1999, 10, 423–429 CrossRef CAS.
- W. Clegg, N. A. Compton, R. J. Errington, G. A. Fisher, M. E. Green, D. C. A. Hockless and N. C. Norman, Inorg. Chem., 1991, 30, 4680–4683 CrossRef CAS.
- N. Burford, T. S. Cameron, K.-C. Lam, D. J. LeBlanc, C. L. B. Macdonald, A. D. Phillips, A. L. Rheingold, L. Stark and D. Walsh, Can. J. Chem., 2001, 79, 342–348 CrossRef CAS.
- A. Schulz, Z. Anorg. Allg. Chem., 2014, 640, 2183–2192 CrossRef CAS.
- W. J. Evans, D. B. Rego and J. W. Ziller, Inorg. Chim. Acta, 2007, 360, 1349–1353 CrossRef CAS.
- N. Burford, C. L. B. Macdonald, K. N. Robertson and T. S. Cameron, Inorg. Chem., 1996, 35, 4013–4016 CrossRef CAS PubMed.
- U. Wirringa, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Inorg. Chem., 1994, 33, 4607–4608 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- H. Schmidbaur and A. Schier, Organometallics, 2008, 27, 2361–2395 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, full characterization of all compounds. CCDC 1447892–1447910. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00229c |
| This journal is © The Royal Society of Chemistry 2016 |