Subtle backbone modifications control the interpenetration of dibenzosuberone-based coordination cages†
Thorben R. Schulte,
Marcel Krick,
Carmen I. Asche,
Sabrina Freye and
Guido H. Clever*
Institute of Inorganic Chemistry, Georg-August University Göttingen, Tammannstr. 4, 37077 Göttingen, Germany. E-mail: gclever@gwdg.de
First published on 18th June 2014
Abstract
In order to gain a better understanding of the formation of interpenetrated double-cages [Pd4L8] from banana-shaped ligands and square-planar metal cations, our previously reported dibenzosuberone-based ligand was synthetically modified. We show that the formation of double-cages tolerates a wide range of suberone backbone modifications, as long as the ketone functionality is preserved. Reduction of this group to a CH2-bridge allowed us to form a monomeric [Pd2L4] cage, instead. PM6 and DFT calculations were performed to study the role of the carbonyl group in the formation of double-cages yielding results that are in good agreement with the experiments.
Introduction
The rational synthesis of self-assembled supramolecular architectures plays a key role in understanding the complex structures found in nature and spurs the development of artificial nanostructures with various functions.1 Among the plethora of discrete self-assemblies that have been realized today, coordination cages are of particular interest.2–6 The defined inner cavity of such cages has been utilized for the selective recognition of a wide variety of guest molecules,7–9 the stabilization of reactive intermediates10–12 and the catalysis of chemical reactions.13,14 Furthermore, implemented functionalities such as light-switchable15 and redox-active16,17 backbones, paramagnetic metals18,19 as well as endohedral anchor groups20 have enriched the chemistry of coordination cages, recently.We have reported about a series of interpenetrated coordination cages21–23 based on square-planar coordinated Pd(II) cations and banana-shaped bis-monodentate pyridyl ligands24 with dibenzosuberone25 and phenothiazine17,26,27 backbones. In these structures, two [Pd2L4] cages28 dimerize as subunits to give a discrete interpenetrated [Pd4L8] double-cage comprising three pockets in which anionic guest molecules were found to bind in an allosteric fashion.25,27,29 We further showed, that the size of the guest in the central pocket controls the guest selectivity of the outer two pockets through a mechanical relay mechanism.29,31 In one of the latter examples, we studied a derivative of our prototypical dibenzosuberone backbone having a further aryl substituent covalently attached to the oxygenated carbon centre of the central seven-membered ring.30 We showed, that this structural modification of the ligand had a substantial influence on the cage assembly and anion binding properties of the system.
Here, we deliver further data on the impact of synthetic backbone modifications of the dibenzosuberone scaffold on the formation of interpenetrated coordination cages. While we show that certain derivatizations of the backbone and donor-sites do not interfere with cage dimerization in acetonitrile, we also demonstrate that another, surprisingly simple modification does indeed prohibit double-cage formation in this solvent. The monomeric species [Pd2L4] is formed in this case. Alternatively, dimerization can be suppressed by using DMSO as a solvent.
Results and discussion
Scheme 1 illustrates the synthetic ligand modifications discussed in this manuscript. The three new ligand derivatives L2–4 are depicted next to the already studied parental ligand L1. All ligand were derived from the common precursor dibenzosuberone 1 which is commercially available. After selective bromination to give 2, ligands L1 and L2 were obtained by Sonogashira or Suzuki cross-coupling reactions, respectively. Oxidative dehydrogenation of compound 2 using PCl5 in POCl332 yielded derivative 3 which was converted into ligand L3 by a Sonogashira reaction using 3-ethynylpyridine. Reduction of intermediate 3 with Al(Oi-Pr)3 at 300 °C32 gave dibenzo-cycloheptatriene derivative 4 which was reacted accordingly to give ligand L4. All three new ligands were employed in the formation of self-assembled coordination cages according to Scheme 1b. Therefore, the ligands were reacted with the palladium source [Pd(CH3CN)4](BF4)2 in a polar solvent such as DMSO or acetonitrile to give either a monomeric cage [Pd2L4] or a dimeric product [Pd4L8]. The details of the outcomes of these assembly reactions are discussed in the following paragraphs.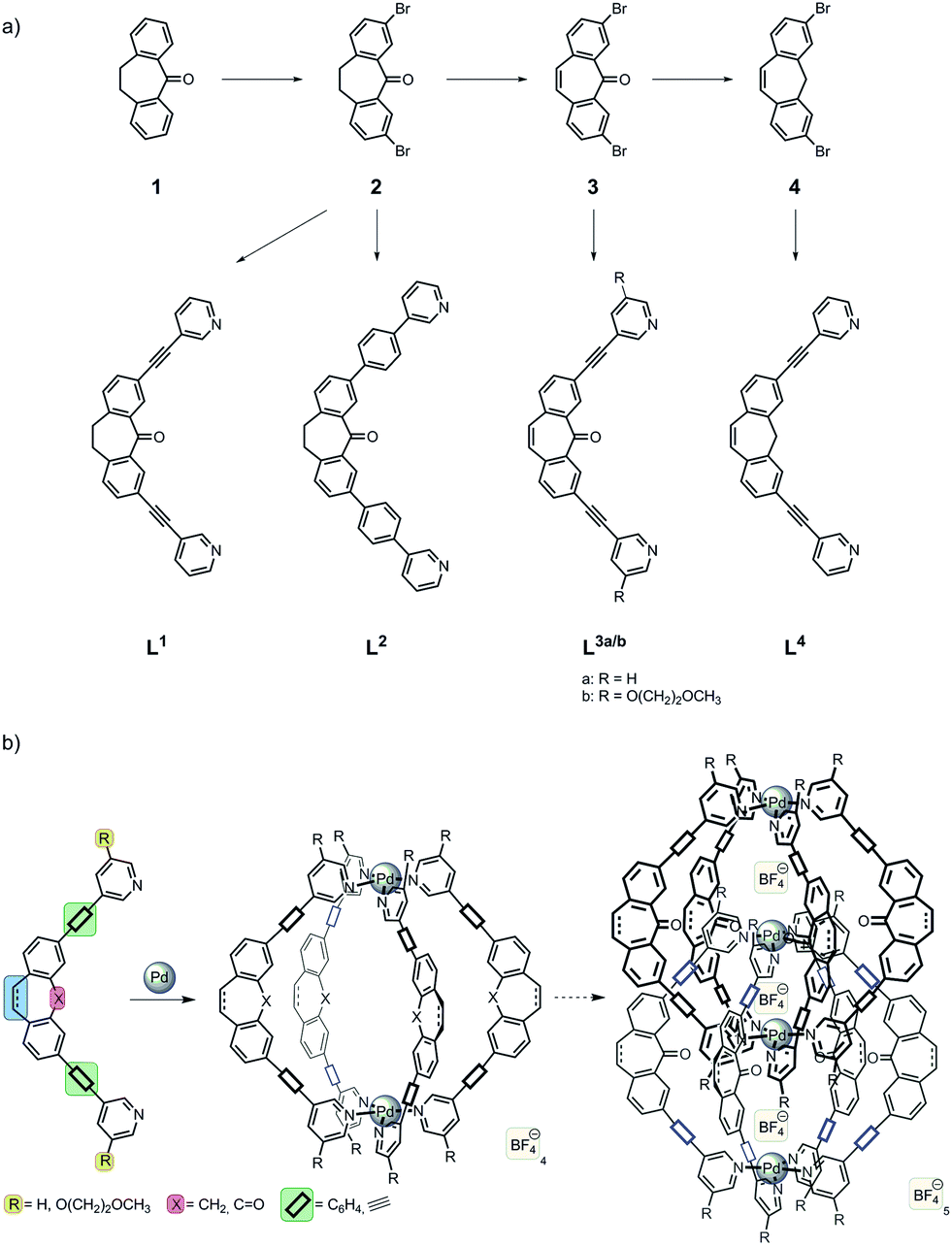 | ||
| Scheme 1 (a) Synthesis of ligands L1–4 from the common precursor dibenzosuberone 1; (b) schematic illustration of the influence of the ligand modifications on the cage assembly. | ||
Exchange of alkyne for 1,4-phenylene linkers
The exchange of the alkyne linkers of the previously studied ligand L1 for 1,4-phenylene linkers yielded a ligand L2 that is slightly longer in terms of its N–N distance (17.7 Å) than the parental structure (14.9 Å). In DMSO as a solvent, the reaction of ligand L2 with the Pd salt leads to the formation of a monomeric cage species [Pd2L24] as can be deduced from the signal shifting (but not splitting) in the 1H NMR spectra (Fig. 1a) and the occurrence of signals for the species [Pd2L24]4+ and [Pd2L24 + BF4]3+ in the ESI mass spectrum (Fig. 1b).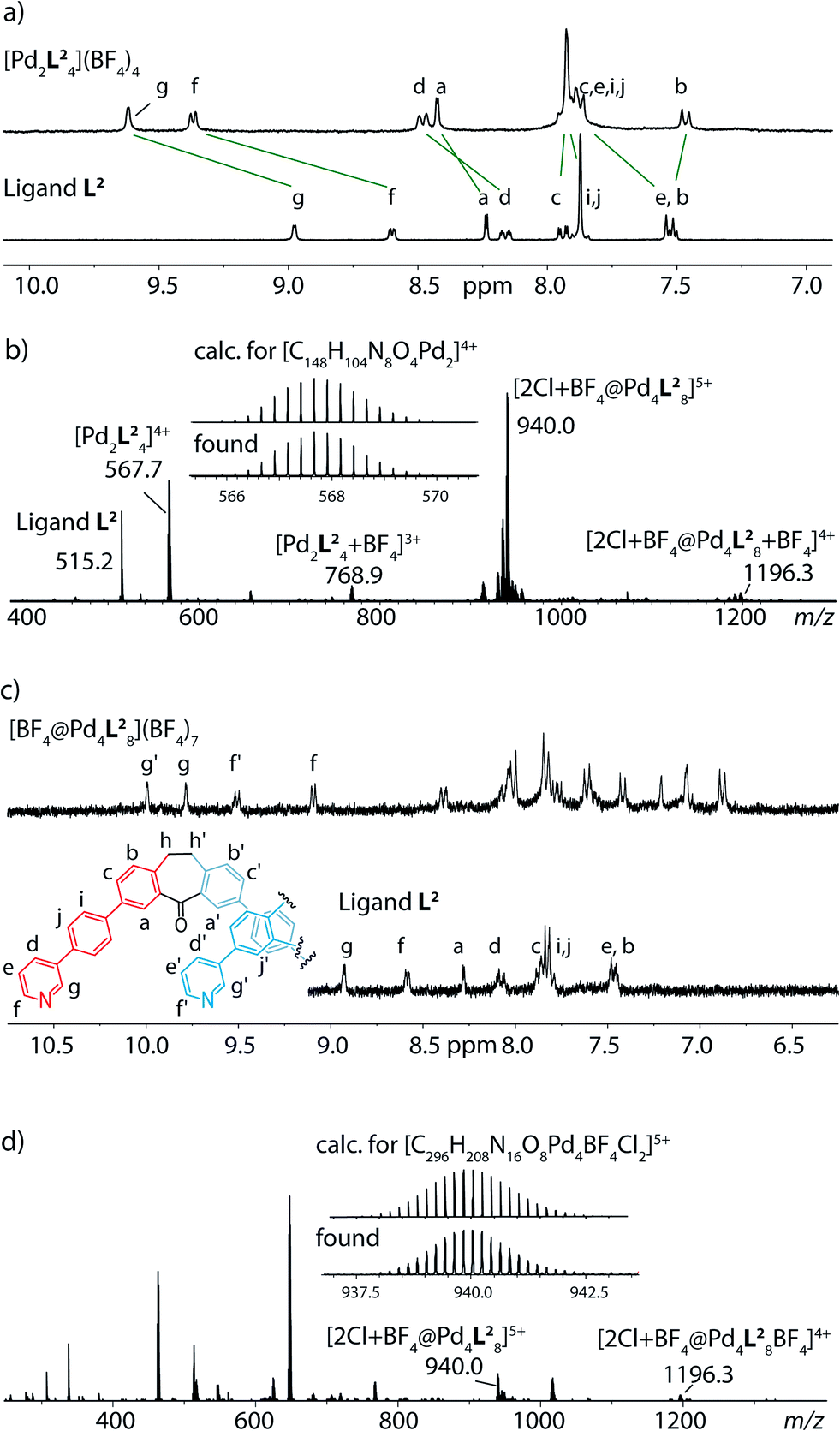 | ||
| Fig. 1 (a) 1H NMR spectrum and (b) ESI mass spectrum showing the formation of monomeric cage [Pd2L24] in DMSO. The spectra in (c and d) support the formation of the interpenetrated double-cage [Pd4L28] in acetonitrile, instead. Signals assignable to double-cages in spectrum (b) most probably result from a solvent change prior to the mass spectrometric measurement which induces the interpenetration reaction. As reported before, chloride-bound species result from machine contaminations and support the tremendous halide binding affinity of these systems. | ||
Nevertheless, ligand L2 smoothly reacts to an interpenetrated double-cage [Pd4L28] when acetonitrile is used as a solvent, as shown by the previously described effect of signal splitting in the NMR spectra (Fig. 1c) and the observation of signals for the dimeric species [2Cl + BF4@Pd4L28]5+ and [2Cl + BF4@Pd4L28 + BF4]4+ in the mass spectrum (Fig. 1d). This finding is in accordance with our previous observations using acetonitrile (or acetone) as a solvent. The fact that DMSO leads to the formation of monomeric cages, instead, may arise from different entropic contributions of releasing the larger DMSO or the smaller acetonitrile solvent molecules from the interior of the monomeric cages upon dimer formation.
Desaturation of the backbone ethylene bridge
Next, we examined modifications concerning the core region of the dibenzosuberone-derived ligands. Therefore, the seven-membered ring was desaturated at its ethylene bridge yielding ligand L3a. Reaction with [Pd(CH3CN)4](BF4)2 in acetonitrile first gave monomeric species [Pd2L3a4] as a kinetic intermediate as was shown by an 1H NMR measurement conducted briefly after mixing the ligand with the metal source (Fig. 2a) which is in good accordance with our previously reported observations using ligand L1.25 When the reaction was allowed to proceed further, this monomeric species was consumed in the formation of a follow-up product that precipitated out of solution. We suspected that this compound was the interpenetrated double-cage, but unfortunately, it turned out to be of such low solubility, that its characterization by NMR spectroscopy and ESI mass spectrometry failed. In order to elucidate the nature of this follow-up project, we decided to synthesize ligand derivative L3b which contains two solubilizing methoxy–ethoxy chains attached to both pyridine rings. When ligand L3b was used as a reactant in the cage formation, no precipitate was formed, even after prolonged heating of the solution. Indeed, the thermodynamic product in this case proofed to be the interpenetrated cage [3BF4@Pd4L3b8] according to the characteristic signal pattern in its 1H NMR spectrum and the occurrence of signals for the charged species [3BF4@Pd4L3b8]5+, [3BF4@Pd4L3b8 + BF4]4+ and [3BF4@Pd4L3b8 + 2BF4]3+ in the ESI mass spectrum. It is worth mentioning, that this is also the first demonstration that banana-shaped bis-pyridyl ligands carrying an extra substituent at both pyridine rings are able to form double-cages, since molecular modeling studies indicate that the steric situation around the inner Pd(pyridine)4-planes is substantially crowded. Nevertheless, a double-cage is formed using ligand L3b, presumably because the attached chains are facing no hindrance when protruding between the flanking backbones of the interpenetrating cage subunits.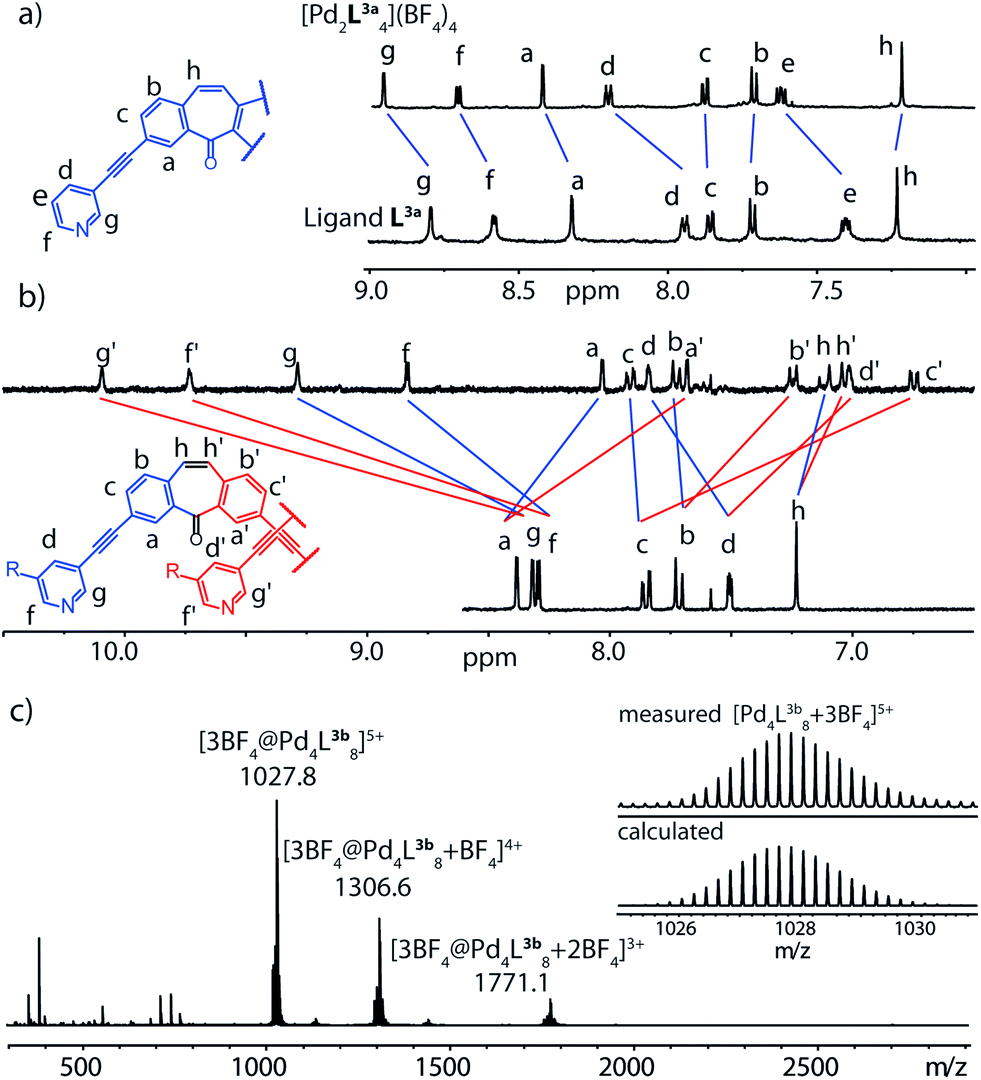 | ||
| Fig. 2 (a) 1H NMR spectroscopic monitoring of (a) the formation of a monomeric cage [Pd2L3a4] as an intermediate before the onset of precipitation and (b) the formation of an interpenetrated double-cage [Pd4L3b8] based on a ligand derivative with higher solubility. (c) Also the ESI mass spectrum supports dimerization in this case. | ||
Deoxygenation of the backbone carbonyl group
In a further synthetic step, we decided to remove the inward pointing carbonyl group of the ligand by a reductive protocol and obtained ligand L4. To our surprise, the reaction of this ligand derivative with Pd(II) cations in acetonitrile led to the formation of a monomeric coordination cage [Pd2L44] as indicated by the lack of a signal splitting in the 1H NMR spectrum and the observation of signals for the species [Pd2L44]4+, [Pd2L44 + BF4]3+ and [Pd2L44 + 2BF4]2+ in the mass spectrum of the reaction product (Fig. 3a and b). | ||
| Fig. 3 (a) 1H NMR and (b) ESI mass spectrum supporting the formation of a monomeric cage [Pd2L44] from ligand L4 and Pd(II) cations in acetonitrile. | ||
In order to gain further insights into the reasons behind the formation of a monomeric cage product in this case, we performed a series of electronic structure calculations. First, we calculated the energies of the formal homodesmic dimerization of two monomeric cages {[Pd2L1,3,44](BF4)2}2+ to give double-cages {[Pd4L1,3,48](BF4)4}4+ under conservation of the total number of counter anions on the left and right side of the equation. Therefore, geometries were optimized on semiempiric PM6 level of theory and dimerization energies ΔEdim (PM6 as well as DFT B3LYP/LANL2DZ) were calculated according to the formula given in Fig. 4a (for details see the ESI†). In agreement with the experimental results, it was found that the formation of double-cages based on ligand L4 is strongly disfavored as compared to double-cage formation from ligand L1 (PM6: +235.8 kJ mol−1, DFT: +271.7 kJ mol−1) as well as compared to double-cage formation from ligand L3 (PM6: +108.9 kJ mol−1, DFT: +281.6 kJ mol−1). As one plausible factor contributing to the stabilization of the double-cages [Pd4L18] and [Pd4L38] we postulate an attractive interaction between the carbonyl oxygen substituents and the polarized ring planes of the inner Pd(pyridine)4 moieties in the interpenetrated double-cages. Since ligand L4 does not possess this carbonyl oxygen atom, this tentative contribution to the double-cage stabilization is lost. Fig. 4b details this situation for one of the two equivalent inner Pd(pyridine)4-planes taken from the single crystal X-ray structure of double-cage [Pd4L18].25 The semi-space filling representation clearly shows that the carbonyl oxygen substituents of the four interpenetrating ligand backbones are perfectly complementing the gaps between the four pyridine rings of the Pd(pyridine)4-propeller. Each oxygen atom is showing distances of 2.53 and 2.64 Å to the next pyridine ring planes and a distance of 4.29 Å to the palladium cation. In order to check if this arrangement contributes an attractive term to the overall energy of dimerization, we performed a rigid potential energy scan of the fragment shown in Fig. 4b by varying all four oxygen–palladium distances between 3.29 and 6.29 Å in steps of 0.25 Å using two different dispersion corrected DFT functionals implemented in Gaussian '09 together with two different Ahlrich basis sets (DFT M06-2X/def2-svp, ωB97XD/def2-svp and ωB97XD/def2-tzvp). All three calculations show a minimum at a Pd–O distance of about 4.2 Å which is in good agreement with the distance extracted from the X-ray structure of [Pd4L18]. It is worth mentioning, that this minimum was only produced when using dispersion corrected DFT functionals; the popular B3LYP functional did not yield a minimum in the potential energy surface scan, regardless of the size of the basis set. The average energy gain per carbonyl group was 0.016 Hartree (42.0 kJ mol−1) which would add up eightfold to give a stabilizing contribution of 336 kJ mol−1 for the formation of double-cage [Pd4L18] from ligand L1. Fig. 4(c and d) show, that a similar oxygen–pyridine contact can be postulated for double-cage [Pd4L38] (based on the PM6 calculated structure) but not for a hypothetical double-cage [Pd4L48] which lacks the carbonyl groups. Interestingly, the calculated energy contribution of 336 kJ mol−1 stemming from the eight oxygen–pyridine interactions in [Pd4L18] compares quite well with the 271.7 kJ mol−1 stabilization of this double-cage as compared to the hypothetical, oxygen-free double-cage [Pd4L48] that was the result of the B3LYP/LANL2DZ//PM6 geometry optimizations mentioned above (although the latter calculations were performed without dispersion correction).
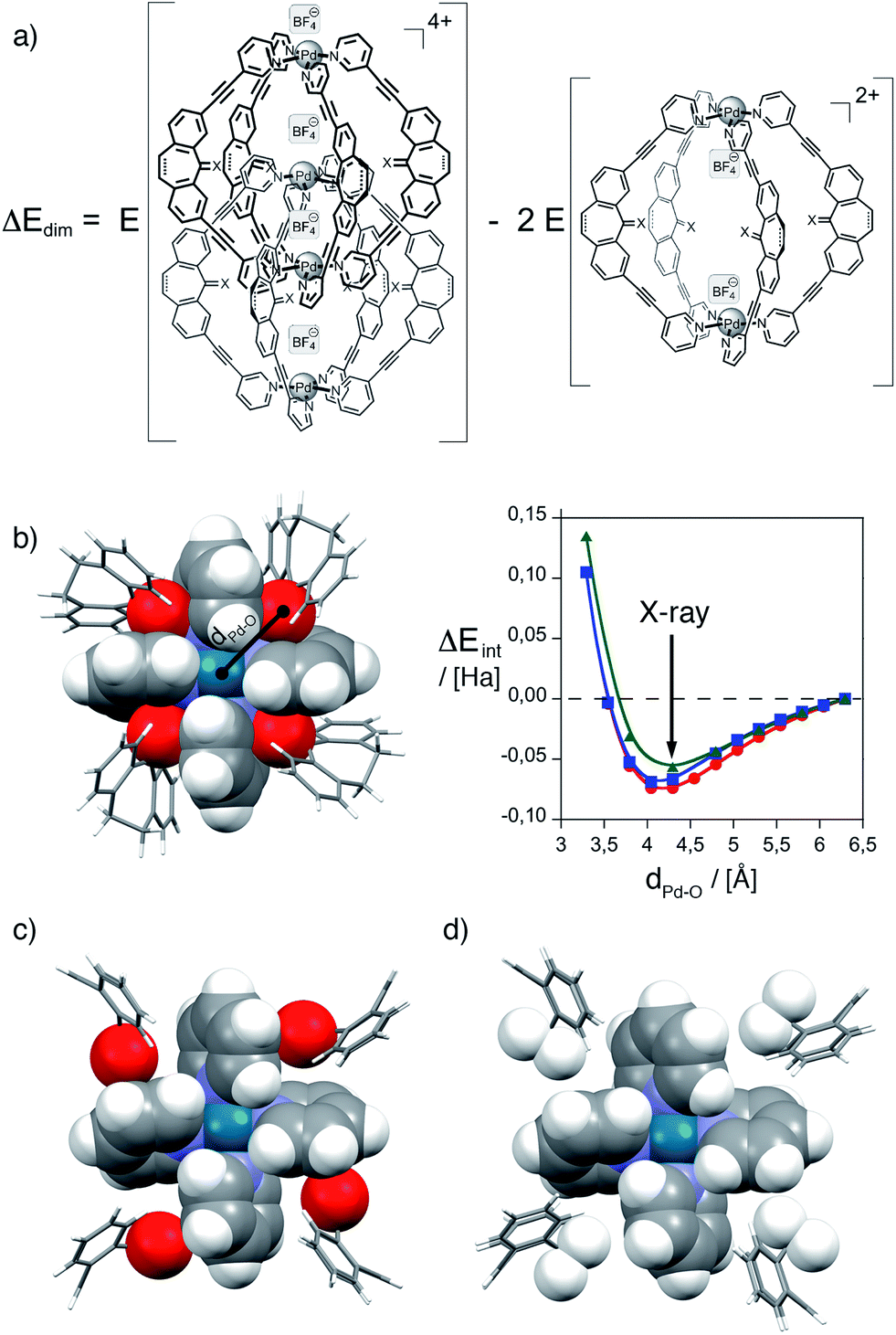 | ||
| Fig. 4 Electronic structure calculation results. (a) Homodesmic reaction scheme for the calculation of the dimerization energy Edim for the mono- and double-cages based on ligands L1, L3 and L4. (b) Inner Pd(pyridine)4-plane of double-cage [Pd4L18] and result of the potential energy scan for the interpenetration energy Eint under variation of the palladium–oxygen distance dPd–O (red circles: ωB97XD/def2-svp, blue squares: M06-2X/def2-svp, green triangles: ωB97XD/def2-tzvp). (c) Inner Pd(pyridine)4-plane of [Pd4L38]. (d) Inner Pd(pyridine)4-plane of [Pd4L48]. The Pd(pyridine)4-planes and the central substituents of the interpenetrating ligands are drawn in space filling mode, the remaining structure in stick representation (carbon: grey, hydrogen: white, nitrogen: blue, oxygen: red, palladium: turquois). | ||
In case of the dibenzosuberone-derived cages, the interaction between the carbonyl groups and Pd(pyridine)4 planes seems to play a crucial role for the stabilization of the double-cage structure. It is interesting to note, however, that other Pd4L8 double-cage structures reported by us17,30 and Kuroda et al.22 do not have carbonyl groups in equivalent positions.
Conclusions
In this paper we have examined some factors leading to the formation of monomeric and interpenetrated dimeric coordination cages from bis-monodentate ligands and square-planar metal cations with a focus on synthetic modifications of the ligand structure.Previously, we have reported the double-cage forming ligand L1 in which the pyrdiyl-donors are attached to the dibenzosuberone backbone via alkyne linkers.25 We also showed that a shorter ligand (lacking the alkyne spacers) is not able to form interpenetrated double-cages. Here, on the other hand, we show that the elongation of the ligand by exchanging the alkynes with 1,4-phenylene bridges does not interfere with the formation of double-cages.
The dibenzosuberone backbone structure can be modified with the following implications for the double-cage formation: oxidation of the ethylene bridge to a double bond is tolerated as is the attachment of chain substituents to the meta-position of the pyridine rings. Reduction of the carbonyl group to a CH2 group removes the ligand's ability to form interpenetrated dimers and the assembly stops at the stage of the monomeric cages. Monomeric cages are also obtained when DMSO instead of acetonitrile is used as a solvent for the self-assembly.
Currently, we are trying to transfer the learned assembly and interpenetration principles to ligands with other backbone structures in order to gain further control over the monomer/dimer equilibrium and the selectivity for anion binding.
Acknowledgements
We thank the Fonds der Chemischen Industrie and the DFG (CL 489/2-1) for generous support. We thank Dr Michael John and Dr Holm Frauendorf for their help in obtaining NMR spectroscopic and mass spectrometric data, respectively.Notes and references
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2009 Search PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825 CrossRef CAS PubMed.
- M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161 RSC.
- T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476 RSC.
- D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136 CrossRef CAS PubMed.
- N. Gimeno and R. Vilar, Coord. Chem. Rev., 2006, 250, 3161 CrossRef CAS PubMed.
- S. O. Kang, J. M. Llinares, V. W. Day and K. Bowman-James, Chem. Soc. Rev., 2010, 39, 3980 RSC.
- J. L. Sessler, P. A. Gale and W.-S. Cho, Anion receptor chemistry, Royal Society of Chemistry, Cambridge, 2006 Search PubMed.
- D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2006, 45, 745 CrossRef CAS PubMed.
- M. Kawano, Y. Kobayashi, T. Ozeki and M. Fujita, J. Am. Chem. Soc., 2006, 128, 6558 CrossRef CAS PubMed.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed.
- U. Brinker and J.-L. Mieusset, Molecular encapsulation: Organic reactions in constrained systems, Wiley, Chichester, West Sussex, UK, 2010 Search PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319 CrossRef CAS PubMed.
- S. Bivaud, J.-Y. Balandier, M. Chas, M. Allain, S. Goeb and M. Sallé, J. Am. Chem. Soc., 2012, 134, 11968 CrossRef CAS PubMed.
- M. Frank, J. Hey, I. Balcioglu, Y.-S. Chen, D. Stalke, T. Suenobu, S. Fukuzumi, H. Frauendorf and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 10102 CrossRef CAS PubMed.
- Y. Liu, C. Hu, A. Comotti and M. D. Ward, Science, 2011, 333, 436 CrossRef CAS PubMed.
- M. B. Duriska, S. M. Neville, B. Moubaraki, J. D. Cashion, G. J. Halder, K. W. Chapman, C. Balde, J.-F. Létard, K. S. Murray, C. J. Kepert and S. R. Batten, Angew. Chem., 2009, 121, 2587 CrossRef.
- M. C. Young, A. M. Johnson, A. S. Gamboa and R. J. Hooley, Chem. Commun., 2013, 49, 1627 RSC.
- M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52 CrossRef CAS PubMed.
- M. Fukuda, R. Sekiya and R. Kuroda, Angew. Chem., 2008, 120, 718 CrossRef.
- A. Westcott, J. Fisher, L. P. Harding, P. Rizkallah and M. J. Hardie, J. Am. Chem. Soc., 2008, 130, 2950 CrossRef CAS PubMed.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848 RSC.
- S. Freye, J. Hey, A. Torras-Galán, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191 CrossRef CAS PubMed.
- M. Frank, L. Krause, R. Herbst-Irmer, D. Stalke and G. H. Clever, Dalton Trans., 2014, 43, 4587 RSC.
- M. Frank, J. M. Dieterich, S. Freye, R. A. Mata and G. H. Clever, Dalton Trans., 2013, 42, 15906 RSC.
- Several other groups have reported coordination cages of the general formula [M2L4]4+ where M is a square-planar coordinated metal ion such as PdII or PtII or octahedrally coordinated metal fragments such as CoX2 and L are bis-pyridyl ligands bridging the two metal centers. See for example: (a) D. A. McMorran and P. J. Steel, Angew. Chem., 1998, 110, 3495 (Angew. Chem., Int. Ed., 1998, 37, 3295) CrossRef; (b) D. K. Chand, K. Biradha and M. Fujita, Chem. Commun., 2001, 1652 RSC; (c) C. Su, Y. P. Cai, C. Chen, M. D. Smith, W. Kaim and H. C. zur Loye, J. Am. Chem. Soc., 2003, 125, 8595 CrossRef CAS PubMed; (d) N. L. S. Yue, D. J. Eisler, M. C. Jennings and R. J. Puddephatt, Inorg. Chem., 2004, 43, 7671 CrossRef CAS PubMed; (e) H. Amouri, L. Mimassi, M. N. Rager, B. E. Mann, C. Guyard-Duhayon and L. Raehm, Angew. Chem., Int. Ed., 2005, 44, 4543 CrossRef CAS PubMed; (f) G. H. Clever, S. Tashiro and M. Shionoya, Angew. Chem., Int. Ed., 2009, 48, 7010 CrossRef CAS PubMed; (g) P. Liao, B. W. Langloss, A. M. Johnson, E. R. Knudsen, F. S. Tham, R. R. Julian and R. J. Hooley, Chem. Commun., 2010, 46, 4932 RSC; (h) J. D. Crowley and E. L. Gavey, Dalton Trans., 2010, 39, 4035 RSC; (i) N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438 CrossRef CAS PubMed; (j) J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778 RSC.
- S. Freye, D. M. Engelhard, M. John and G. H. Clever, Chem.–Eur. J., 2013, 19, 2114 CrossRef CAS PubMed.
- S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476 CrossRef CAS PubMed.
- J. M. Dieterich, G. H. Clever and R. A. Mata, Phys. Chem. Chem. Phys., 2012, 14, 12746 RSC.
- Y. Wei and C.-T. Chen, J. Am. Chem. Soc., 2007, 129, 7478 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Ligand and cage synthetic data, further computational details. See DOI: 10.1039/c4ra04679j |
| This journal is © The Royal Society of Chemistry 2014 |