
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular through-space versus through-bond charge transfer in new donor-carbazole-acceptor type luminophores†
Jakub
Drapala
 a,
Anna
Sadocha
b,
Michał K.
Cyranski
b,
Malgorzata
Zagorska
a,
Adam
Pron
*a and
Irena
Kulszewicz-Bajer
*a
a,
Anna
Sadocha
b,
Michał K.
Cyranski
b,
Malgorzata
Zagorska
a,
Adam
Pron
*a and
Irena
Kulszewicz-Bajer
*a
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: adam.pron@pw.edu.pl; irena.bajer@pw.edu.pl
bFaculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
First published on 28th January 2025
Abstract
Five new donor–acceptor luminophores consisting of an acridan or phenoxazine donor and a benzonitrile (1–3) or benzothiadiazole (4, 5) acceptor connected via a carbazole linker are reported. All the synthesized compounds were electrochemically active and could be oxidized in either a two-step (1–4) or three-step (5) process consisting of electron abstraction from the donor substituent and central carbazole unit. The electrochemically determined ionization potential (IP) values depended on the type of the donor used, and the acridan-substituted carbazole derivative (compound 1) exhibited the highest IP value of 5.27 eV. Alternatively, the IP values of phenoxazine-substituted carbazoles (2–5) were lower by about 0.2 eV. Compounds 4 and 5, which were the derivatives containing benzothiadiazole acceptors, were electrochemically reduced in a quasi-reversible one-electron process, yielding the absolute value of electron affinity (|EA|) of 2.98 eV for 4. It was found that the |EA| of 5 increased to 3.12 eV as a result of the extended conjugation of its acceptor unit. The synthesized compounds can also be considered potential TADF luminophores as they exploited the so-called “through space charge transfer” (TSCT) effect. This was owing to their molecular architecture imposed by the rigid carbazole bridge, which enabled appropriate mutual orientation of the donor and acceptor units. They exhibited fluorescence in the blue-orange spectral range (480–653 nm), which was strongly dependent on the solvent polarity. The spectra recorded for molecular dispersions of 1–5 in Zeonex showed a significant increase in PLQY of 43% for 1 and 94% for 5. TDDFT calculations suggested that the observed emissions for compounds 1–4 stemmed from local electron density redistribution upon excitation. The donating nature of the bridge (carbazole) gave rise to an appreciable through-bond charge transfer. However, in case of 5, the emission process could have predominantly resulted from stronger through-space interactions. These calculation results fully corroborated with the obtained crystallographic data, indicating significantly closer vicinity of the donor and acceptor units in 5 as compared to the remaining compounds (1–4).
Introduction
Donor–acceptor (D–A) type organic compounds exhibiting thermally activated delayed fluorescence (TADF) have been commonly exploited as effective electroluminophores in organic light-emitting diodes (OLEDs). The significant increase in the external quantum efficiency (EQE) achieved by OLEDs through exploiting TADF luminophores is related to the harvesting of triplet excitons, which are then transformed into singlets, which are capable of undergoing the radiative process. Efficient reverse intersystem crossing (RISC) between triplet T1 and singlet S1 states can occur only in cases when the energy difference ΔE (S1–T1) is sufficiently small (<0.1 eV); as a consequence, the process can be thermally activated. Moreover, it has been shown that the RISC process is accelerated for molecules in which vibronic coupling between 3LE and 1CT states takes place.1–4 Compounds exhibiting the TADF effect must contain donor and acceptor units in a highly twisted structure, which ensures the location of HOMO and LUMO orbitals on different parts of the molecule, leading in effect to a small value of ΔE (S1–T1). A large number of TADF-type luminophores have been synthesized to date. Accordingly, the values of EQE obtained for new TADF materials used as emitters in OLEDs increased significantly, exceeding 30% in some cases.5–9 Conventional TADF compounds consist of donor and acceptor units linked via a π-conjugated bridge. Thus, the interaction between D and A units has to be considered as a through bond charge transfer (TBCT). Owing to the spatial separation of their frontier orbitals, these compounds frequently suffer from weak oscillator strength, low photoluminescent quantum yield (PLQY) and low RISC transition rate.10Recently, a new approach in the design of TADF-type emitters has been proposed, namely, the concept exploiting a through-space charge transfer (TSCT). Luminophores of this type should contain not orthogonally but cofacially aligned D and A units, wherein the CT states arise from the spatial interactions of frontier orbitals. In this respect, TSCT compounds are similar to exciplexes but with defined positions of the D and A moieties. While designing the chemical structure of TSCT compounds, special emphasis should be placed on the type of the bridge linking the D and A units as well as the nature of the donor and acceptor moieties. Moreover, special attention should be paid to minimize the distance between the D and A units.11 To ensure proper location of D and A units, xanthene,12,13 fluorenone,14 triptycene15 or acenaphthene16 bridges were used, which facilitate the face-to-face orientation. It should be noted that molecules containing a xanthene bridge are of U-shape; thus, the D and A units can be cofacially aligned. However, the spacing between the D and A units linked to 9,9-dimethylxanthene bridge at 4,5 positions exceeds 3.5 Å, resulting in inefficient interactions of frontier orbitals and, as a consequence, small PLQY.12 A decreased donor–acceptor distance below 3 Å measured in xanthene compounds substituted at 8, 9 positions results in high PLQY values, frequently exceeding 90%.13
Fluorenone and fluorene were also successfully used in the design and synthesis of TSCT emitters. Chemical fixation of the rigid spiro-structure of a benzophenone–fluorene acceptor and different donors led to TSCT luminophores exhibiting high PLQY due to structural stiffening, which suppressed non-radiative decay losses.14 Fluorenone was used to form spiroacridine donors, which were linked to 1,3,5-triazine or dicyanopyrazinophenanthrene acceptors, leading to π-stacked sandwiched D–A–D structures exhibiting good luminescent properties.17,18
There exist reports on carbazole as a bridging moiety in TSCT luminophores.19–21 However, due to its chemical nature, carbazole can be considered as a conjugated bridge or an additional donor in D-carbazole-A type compounds. Thus, both “through space” (TSCT) and/or through-bond (TBCT) interactions can be expected in these luminophores. Monkman et al.19 demonstrated that the type of interactions depended specifically on the electron donating/electron accepting strengths of the donors and acceptors linked to the carbazole bridge. For a strong donor and a strong acceptor, TSCT interactions dominated, whereas for a weak donor and a strong acceptor, both TSCT and TBCT were revealed. Moreover, the authors pointed out that only the through-space interaction gave rise to a fast RISC process and efficient TADF. Detailed consideration of the effect of donor and acceptor nature on the luminescent properties led to the conclusion that enhanced molecular rigidity of both moieties results in an increase in the π–π interactions and suppression of non-radiative decay, which was spectroscopically manifested by high PLQY values.20,21
In the work presented here, we focus on new carbazole-bridged donor–acceptor compounds containing phenoxazine or 9,9-dimethyl-9,10-dihydroacridine donors attached to the carbazole bridge via a phenylene linker. Benzonitrile or benzothiadiazole were used as acceptor units. The selection of this specific family of D-bridge-A compounds requires justification. In our previous papers, we demonstrated that phenoxazine, when combined with acridone or quinacridone, yields efficient TADF emitters,22 which can be used in the fabrication of light emitting diodes (LEDs) exhibiting high values of external quantum efficiency (EQE),23 which in selected cases exceeds 20%.24 Based on these findings, we were tempted to verify whether D-bridge-A compounds with phenoxazine donors and two different acceptors can be potential candidates for new TSCT luminophores. Indeed, these new compounds turned out very interesting not only from the point of view of their photophysics but also due to their reversible electrochemistry in some ambipolar-type compounds. As far as their luminescent properties are concerned, the main goal was to determine how the modification of the donor-bridge-acceptor structure, in terms of the chemical nature of D and A groups as well as positional isomerism, influences intramolecular through-bond and/or through-space interactions in the studied compounds. The presented study is comprehensive since it starts from the design and synthesis of new potential TSCT molecules; the research is then extended to the electrochemical determination of their redox properties, followed by detailed photophysical investigations in different environments. Crystallographic data provide information on the distance between the donor and acceptor units as well as on their mutual orientation, which are vital for the TCST and TBCT processes. Finally, the obtained experimental data are compared with the results of quantum chemical (TDDFT) calculations.
Research on this family of luminophores opens up new perspectives. So far, only in one of five studied compounds, i.e., the one containing benzothiazole disubstituted with thienyl rings as the acceptor, significant impact of the TSCT phenomenon on its luminescent properties has been found. However, the totality of the obtained results seems to indicate that further increase in the TSCT contribution at the expense of TBCT can be achieved by varying the acceptor strength and its geometry. Taking into account a significant number of potentially suitable acceptors, this approach seems promising. Replacing the carbazole linker by a shorter bridge, e.g., 1,8-disubstituted naphthalene, should also be considered as another rational step. Substitution in positions 1 and 8 with donor and acceptor moieties, respectively, could, in principle, yield a U-shape molecule characterized by a short stacking distance between the donor and acceptor. In this configuration, the TSCT mechanism should dominate over TBCT.
Results and discussion
Two series of D-carbazole-A compounds were prepared. The first series contained benzonitrile as an acceptor, differing in the type of the donor (phenoxazine or 9,9-dimethyl-9,10-dihydroacridine) and the positions of D, A substitution to the carbazole bridge. The second series contained the benzothiadiazole acceptor bearing one or two thiophene rings and phenoxazine as a donor. All the studied compounds are depicted in Chart 1. Thus, compound 1 contained 9,10-dihydro-9,9-dimethylacridine as a donor and benzonitrile as an acceptor. Other derivatives contained phenoxazine donor. In the case of 2 and 3, the positions of donor and acceptor were changed to estimate the impact of their positions on the molecular geometries and the distribution of electron density in both molecules. Compounds 4 and 5 contained 4-thienobenzothiadiazole and 4,7-dithienobenzothiadiazole attached to the carbazole core, respectively. | ||
| Chart 1 Structural formulae of the studied D-carbazole-A derivatives. | ||
Synthesis
The synthetic pathways leading to the studied derivatives are presented in Scheme 1 (detailed description of the synthetic procedures can be found in the ESI†). All compounds were synthesized using palladium-catalysed coupling reactions. Compounds 1, 2, 4 and 5 were prepared in the reaction of the adequate carbazole derivative with a suitable acceptor derivative. Thus, in the synthesis of 1 and 2, 1-bromo-3,6-di-t-butylcarbazole was reacted with 4-cyanophenylboronic acid pinacol ester in the presence of Pd(PPh3)4 catalyst. Then, N-substitution of the carbazole core with N-(4′-bromophenyl)-9,10-dihydro-9,9-dimethylacridine or N-(4′-bromophenyl)phenoxazine in the presence of Pd2(dba)3/t-Bu3P catalyst (reaction yields of 65% and 44%) led to compounds 1 and 2, respectively. The syntheses of 4 and 5 were performed in a similar manner. However, in these cases, carbazole substituted at position 1 with boronic acid pinacol ester was used as a substrate. The reactions of carbazole derivative with 4-bromo-7-thienobenzothiadiazole or 4-(5′-bromothieno)-7-thienobenzothiadiazole led to the corresponding intermediate products. Finally, compounds 4 and 5 were obtained in the reaction with N-(4′-bromophenyl)phenoxazine with yields of 88% and 76%, respectively. The synthetic route to compound 3 consisted of N-substitution of the carbazole core with 1-bromobenzonitrile in the first step. Then, this intermediate was brominated and substituted with phenoxazine derivative to give compound 3 with 60% yield. | ||
| Scheme 1 Synthetic routes to the studied derivatives D-carbazole-A: (a) compounds 1 and 2, (b) compounds 4 and 5, and (c) compound 3. | ||
Electrochemical studies
The studied compounds contain units that are known to undergo reversible oxidation (carbazole, phenoxazine, acridan) or reversible reduction (benzothiadiazole). These features are also reflected in the electrochemical studies of 1–5. Cyclic voltammograms (CV) of 1, 2 and 3 are presented in Fig. 1, whereas those of 4 and 5 are given in Fig. 2. The corresponding differential pulse voltammograms (DPV) can be found in the ESI† (Fig. S3.1 and S3.2). Redox potentials determined from cyclic voltammetry data are collected in Table 1. In the voltammograms of 1–3 registered at positive potentials (vs. Fc/Fc+), two consecutive 1e− oxidations are observed, revealed by two cathodic peaks of approximately the same surface area. The peak at lower potentials can be ascribed to the oxidation of the donor unit to a radical cation. 1 is the only compound containing 9,9-dimethyl-9,10-dihydroacridine, a weaker donor compared to the phenoxazine moiety in 2 and 3. As a result, its first oxidation peak is shifted towards higher potentials (0.58 V) as compared to the cases of 2 and 3, whose phenoxazine unit is oxidized at 0.37 and 0.34 V (vs. Fc/Fc+), respectively. Similar oxidation potentials are featured for other 9,9-dimethyl-9,10-dihydroacridine or phenoxazine derivatives reported previously.22,25–27 The second oxidation peak registered in the CVs can undoubtedly be attributed to the oxidation of the carbazole core. Its potential is the same for 1–3 (ca. 0.98 V vs. Fc/Fc+). Evidently, the carbazole bridge oxidation is essentially independent of the type of donors used and their substitution position, suggesting that the electron densities of both donor and bridging segments of the molecule do not overlap. Similar effect was observed by Maggiore et al. in the case of benzonitrile derivatives substituted with phenoxazine and carbazole.281–3, which contain cyanophenylene acceptor, are however very difficult to reduce to their radical anionic form, at least under the conditions used in this research for CV studies. Polarizations down to −2.0 V did not result in the appearance of any reduction-related cathodic peak. | ||
| Fig. 1 Cyclic voltammograms of 1 (a), 2 (b) and 3 (c). Concentration of the studied compounds: 1 × 10 3 M, electrolyte: 0.1 M Bu4NPF6 solution in DCM; scan rate: 50 mV s−1. | ||
 | ||
| Fig. 2 Cyclic voltammograms of 4 (a) and 5 (b). Concentration of the studied compounds: 1 × 103 M, electrolyte: 0.1 M Bu4NPF6 solution in DCM; scan rate: 50 mV s−1. | ||
| Compound | E(0/−1) | E(−1/0) | E(0/1) | E(1/2) | E(2/3) | E(2/1) | E(1/0) |
|---|---|---|---|---|---|---|---|
| 1 | 0.58 | 0.99 | 0.84 | 0.49 | |||
| 2 | 0.37 | 0.98 | 0.89 | 0.29 | |||
| 3 | 0.34 | 0.98 | 0.89 | 0.26 | |||
| 4 | −1.94 | −1.84 | 0.35 | 0.95 | 0.83 | 0.27 | |
| 5 | −1.78 | −1.70 | 0.36 | 0.77 | 0.94 | 0.63 | 0.28 |
Fig. 2 shows CVs of 4 and 5. At positive potentials, 4 undergoes two-step oxidation, differing from the oxidation of 1–3 only by more pronounced irreversibility of the carbazole oxidation process at 0.95 V vs. Fc/Fc+. In the CV of 5, an additional anodic peak appears at 0.77 V vs. Fc/Fc+, which, based on the literature data,29 can be attributed to the oxidation of the dithienobenzothiadiazole acceptor unit. Thus, in this case, all three units constituting the molecule are active in the oxidation processes. Polarization to negative potentials (vs. Fc/Fc+) gives rise to an anodic peak associated with quasi-reversible one-electron reduction of the benzothiadiazole unit to a radical anion.30–32 It should be noted that 5 is reduced at significantly higher potentials (−1.78 V vs. Fc/Fc+) as compared to 4 (−1.94 V vs. Fc/Fc+). This can be rationalized by more extended conjugation of the acceptor unit, in 5 which facilitates the delocalization of the surplus negative charge imposed upon one-electron reduction.
The electrochemical data derived from CVs were used for the calculation of the ionization potential (IP), electron affinity (EA) and electrochemical band gap (Eelg) of the investigated compounds (see Table 2). The obtained IP values are very similar for compounds 2–5 (in the range of 5.04–5.06 eV), which contain the same donor (phenoxazine). 1 containing 9,9-dimethyl-9,10-dihydroacridine, i.e., a donor somehow weaker than phenoxazine, shows a higher value of IP = 5.27 eV. The electron affinity of 5 (|EA| = 3.12 eV) is lower than that determined for 4 (|EA| = 2.98 eV). This result shows that the incorporation of an additional thiophene ring between the carbazole bridge and benzothiadiazole decreases the LUMO level as well as the electrochemical band gap (Eelg). These results are in good agreement with those calculated by DFT; especially for the EA values, the calculated IP values are somehow overvalued (see ESI,† Table S6.1). The electrochemical band gaps of 4 and 5 are 2.06 eV and 1.93 eV, respectively. These rather low values, together with the quasi-reversibility of the first oxidation and first reduction processes, imply ambipolar character of these compounds.
| Compound | E onsetred [V] | E onsetox [V] | EA [eV] | IP [eV] | E elg [eV] |
|---|---|---|---|---|---|
| IP and EA were calculated following a procedure: IP = |e|[Eonsetox + 4.8] [eV], EA = e[Eonsetred + 4.8] [eV] | |||||
| 1 | 0.47 | 5.27 | |||
| 2 | 0.26 | 5.06 | |||
| 3 | 0.24 | 5.04 | |||
| 4 | −1.82 | 0.24 | −2.98 | 5.04 | 2.06 |
| 5 | −1.68 | 0.25 | −3.12 | 5.05 | 1.93 |
Crystal structures
To get a deeper insight into the molecular shape and molecular interactions in the solid state, X-ray diffraction studies on single crystals of 1–5 were performed. One of the t-butyl groups in 2 and 3 is disordered over two or three positions, respectively, whereas the thiophene ring in 4 and 5 is disordered over two positions (for experimental details, see ESI†). Fig. 3 presents the molecular structures of all systems (for more details, see Fig. S4.1 in ESI†). Both donors and acceptors are strongly twisted with respect to the carbazole moiety. The twist angles between the carbazole and neighboring rings at central nitrogen (φ angle) or at the outer ring (θ angle) are given in Table 3. They can tolerate significant variations and range from 49.7° (for 5) to 62.5° (for 4) and from 41.9° (for 2) to 67.0° (for 4). These changes may result mostly from steric interactions (due to close proximity between substituents) and crystal packing. Electronic effects, due to intramolecular electron transfer, might also be considered, but in all cases, the phenoxazine or 9,9-dimethyl-9,10-dihydroacridine moieties adopt highly twisted or even perpendicular conformation, with respect to the phenylene ring. The twist is in general high and ranges from 72.3° (for 5) to 88.9° (for 2) (ϕ angle, see Table 3), which impedes efficient electron transfer. In turn, the intermolecular interactions may play a more important role. This can be exemplified by comparing 1 and 2. Replacing phenoxazine by 9,9-dimethyl-9,10-dihydroacridine leads to a change of the benzonitrile group twist by 11.0°. This is mostly caused by the crystal packing. All systems crystallize in centrosymmetric space groups: P21/n (1), P21/c (2) or P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (3, 4, 5); however, only 5 creates an architecture where the molecules may form a π-stacked structure. The neighboring phenoxazine moieties are situated in an anti-parallel way, as is shown in Fig. 4. The interplanar, separating distance is rather small and equals 3.453 Å. Despite this favorable arrangement, the interacting fragments are somewhat shifted. This shift can be quantified by the distance between their centroids, which equals 3.794 Å. This impacts the efficiency of the π–π interaction, which is reduced but still significant. The crystal packing of 1–4 but also 5 is predominantly stabilized by a set of weak CH⋯N or CH⋯π intermolecular interactions, which are observed in the unit cell at a distance of 2.7–2.9 Å in both cases and by weak van der Waals forces (see Fig. S.4.2–S4.6 in the ESI† for more detail).
(3, 4, 5); however, only 5 creates an architecture where the molecules may form a π-stacked structure. The neighboring phenoxazine moieties are situated in an anti-parallel way, as is shown in Fig. 4. The interplanar, separating distance is rather small and equals 3.453 Å. Despite this favorable arrangement, the interacting fragments are somewhat shifted. This shift can be quantified by the distance between their centroids, which equals 3.794 Å. This impacts the efficiency of the π–π interaction, which is reduced but still significant. The crystal packing of 1–4 but also 5 is predominantly stabilized by a set of weak CH⋯N or CH⋯π intermolecular interactions, which are observed in the unit cell at a distance of 2.7–2.9 Å in both cases and by weak van der Waals forces (see Fig. S.4.2–S4.6 in the ESI† for more detail).
 | ||
| Fig. 3 Molecular structures of 1–5. In the case of disorder, the parts with bigger occupancies are shown. The figures with all details (disorder and ADPs) are given in (ESI†). | ||
| φ [°] | θ [°] | ϕ [°] | |
|---|---|---|---|
| 1 | 58.7 | 52.9 | 83.7 |
| 2 | 55.4 | 41.9 | 88.9 |
| 3 | 58.3 | 56.7 | 77.3 |
| 4 | 62.5 | 67.0 | 88.3 |
| 5 | 49.7 | 50.9 | 72.3 |
 | ||
| Fig. 4 Stacking interaction with separating distance in 5. | ||
To summarize, taking into account the molecular shapes of the studied molecules in the solid state as well as their crystal packing, only 5 seems to adopt the geometry allowing for a significant contribution of TSCT in its emission processes.
Photophysical properties
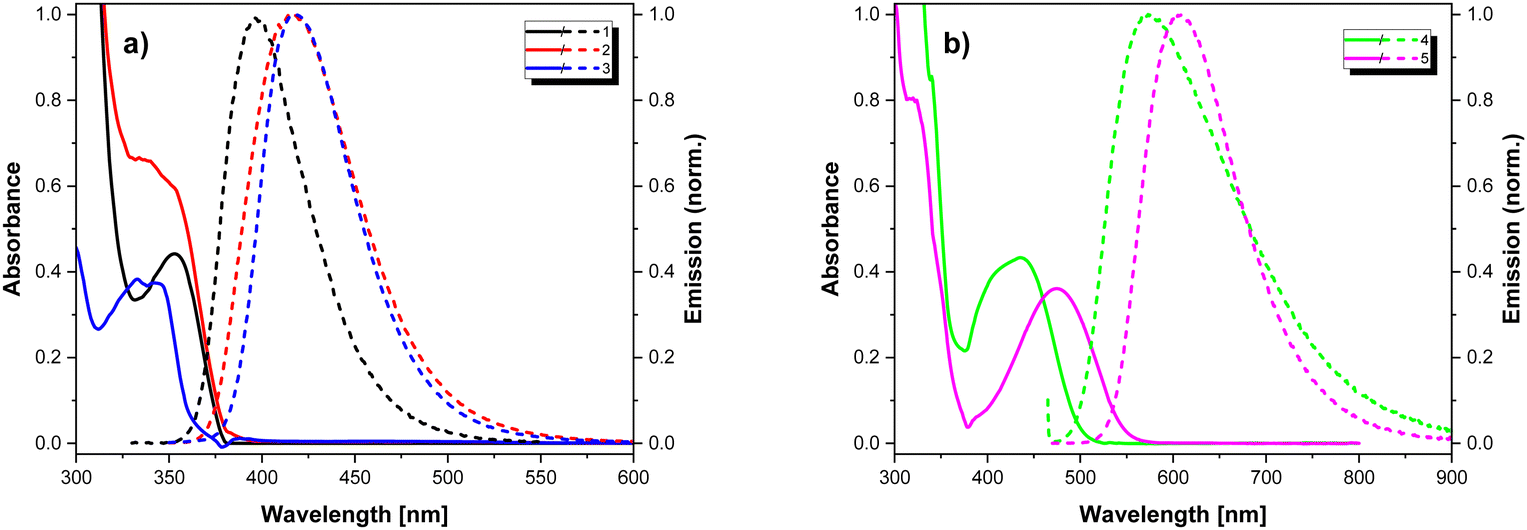
The studied compounds contain chromophore groups of different types, which, when combined in one molecule, may give rise to a rather complex spectroscopic behaviour. Thus, detailed spectroscopic investigations were performed in different environments, including solvents of different polarity, frozen solvents and solid Zeonex matrix.Fig. 5 shows the absorption spectra of 1–5 measured for solutions in DCM and the emission ones recorded in PhMe. The corresponding optical parameters are collected in Table 4. Intense bands present in the absorption spectra at ca. 300 nm can be attributed to the π–π* transition in the donor segments, while lower energy bands arise from the presence of the electron-accepting units. In particular, compounds 4 and 5 exhibit well-resolved low-energy bands at 423 and 467 nm, respectively, which can have CT character. The emission spectra of all compounds display a single band with no visible vibronic structure. The compounds exhibit low to moderate photoluminescence quantum yields with the highest values observed for 1 and 5 (21% and 24%, respectively). The measured PL lifetimes values are typical of fluorescence and range from 4.8 to 12.2 ns.
 | ||
| Fig. 5 UV-vis (solid lines) (in DCM) and fluorescence (dashed lines) (in toluene) spectra (a) 1–3 and (b) 4, 5. | ||
| λ abs [nm] DCM | λ em [nm] DCM | PLQY DCM | λ em [nm] PhMe | PLQY PhMeaerated | PL lifetime [ns] PhMeaerated | |
|---|---|---|---|---|---|---|
| a PLQY standards Quinine sulfate in 0.05 M H2SO4. b Coumarin 153 in EtOH. c Fluorescein in 0.1 M NaOH. d Samples excited at the lowest energy band maximum. | ||||||
| 1 | 353, 279 | 480 | 0.10a | 439 | 0.21a | 8.9 |
| 2 | 340 (sh),300, 277 | 542 | 0.02a | 472 | 0.02a | 12.2 |
| 3 | 334, 298, 278 | 521 | 0.04a | 449 | 0.04a | 4.8 |
| 4 | 423, 320, 296 | 606 | 0.00b | 573 | 0.01b | 11.0 |
| 5 | 467, 318, 300 | 653 | 0.00c | 605 | 0.24c | 10.4 |
In the series of compounds bearing benzonitrile electron-accepting moiety (1–3), the highest bathochromic shift is observed for 2, indicating stronger conjugation in the π-bond system, confirmed by the smallest twist angles φ and θ calculated for this compound (see Table 3). In the case of compounds containing benzothiadiazole acceptors (4, 5), 5 emits at longer wavelength, which manifests more extended conjugation in its acceptor unit. Switching from a nonpolar solvent (PhMe) to a polar one (DCM) results in a bathochromic shift of the emission maxima together with the broadening of emission bands with a significant drop in the PLQY values (see Table 4). These changes point out the polarised nature of the excited state (see Fig. S5.3, ESI†). The spectra of 1–3 and 5 do not show any dependence on the excitation wavelength. On the contrary, in the case of 4, the character of the spectra is strongly dependent on the excitation energy. While exciting the acceptor part of 4 (longer wavelengths), its emission spectrum exhibits a single well-defined band. Upon excitation with shorter wavelengths (within the common spectral range of the donor and acceptor part of the molecule), a contribution from a lower energy band is observed and the emission maximum shows a bathochromic shift (from 573 nm to 635 nm, see Fig. 6). This finding suggests energy transfer from the donor to the acceptor part of the molecule and the formation of a charge-transfer excited state.
 | ||
| Fig. 6 Emission spectra of 4 in PhMe upon excitation with different wavelengths. | ||
In order to further investigate the nature of 1–5 compounds’ emission, additional measurements in the deaerated PhMe were performed. In the cases of 1–3 and 5, the measurements yield spectra of identical shape but higher PLQY as compared to those recorded in the same but aerated solvent. Only the spectrum of 4 shows a small bathochromic shift upon PhMe deaeration (slightly higher contribution of a possible charge transfer state). The observed increased emission is accompanied by a considerable increase of the PL lifetime (Fig. S5.4, ESI†). Both parameters increase by a similar factor (see Table 5). The observed differences vanish upon consecutive aerating of the studied deaerated solutions. These findings indicate engagement of the triplet excited state in the process of emissive deexcitation.
| PLQY PhMedegassed | PLQY PhMedegassed/aeratedd | PL lifetime [ns] PhMedegassed | PL lifetime PhMedegassed/aerated | |
|---|---|---|---|---|
| a PLQY standards Quinine sulfate in 0.05 M H2SO4. b Coumarin 153 in EtOH. c Fluorescein in 0.1 M NaOH. d Calculated as the ratio of the integrals of the emission spectra with respect to the solution absorbance at the excitation wavelength. | ||||
| 1 | 0.33a | 1.61 | 14.3 | 1.61 |
| 2 | 0.06a | 2.69 | 31.9 | 2.61 |
| 3 | 0.05a | 1.26 | 6.1 | 1.27 |
| 4 | 0.01b | 1.44 | 21.0 | 1.91 |
| 5 | 0.37c | 1.58 | 15.8 | 1.52 |
In order to further elucidate the effects of environmental factors on the emissive processes of the studied compounds, the photophysical properties of their molecular blends in Zeonex (1% wt concentration) as well as in frozen PhMe solutions (at 77 K) were determined. The results are collected in Fig. 7, 8 and Table 6.
 | ||
| Fig. 7 UV-vis (solid lines) and PL (dashed lines) spectra of the molecular dispersions of 1–5 (1 wt% concentration) in Zeonex (a) 1–3, (b) 4 and 5. | ||
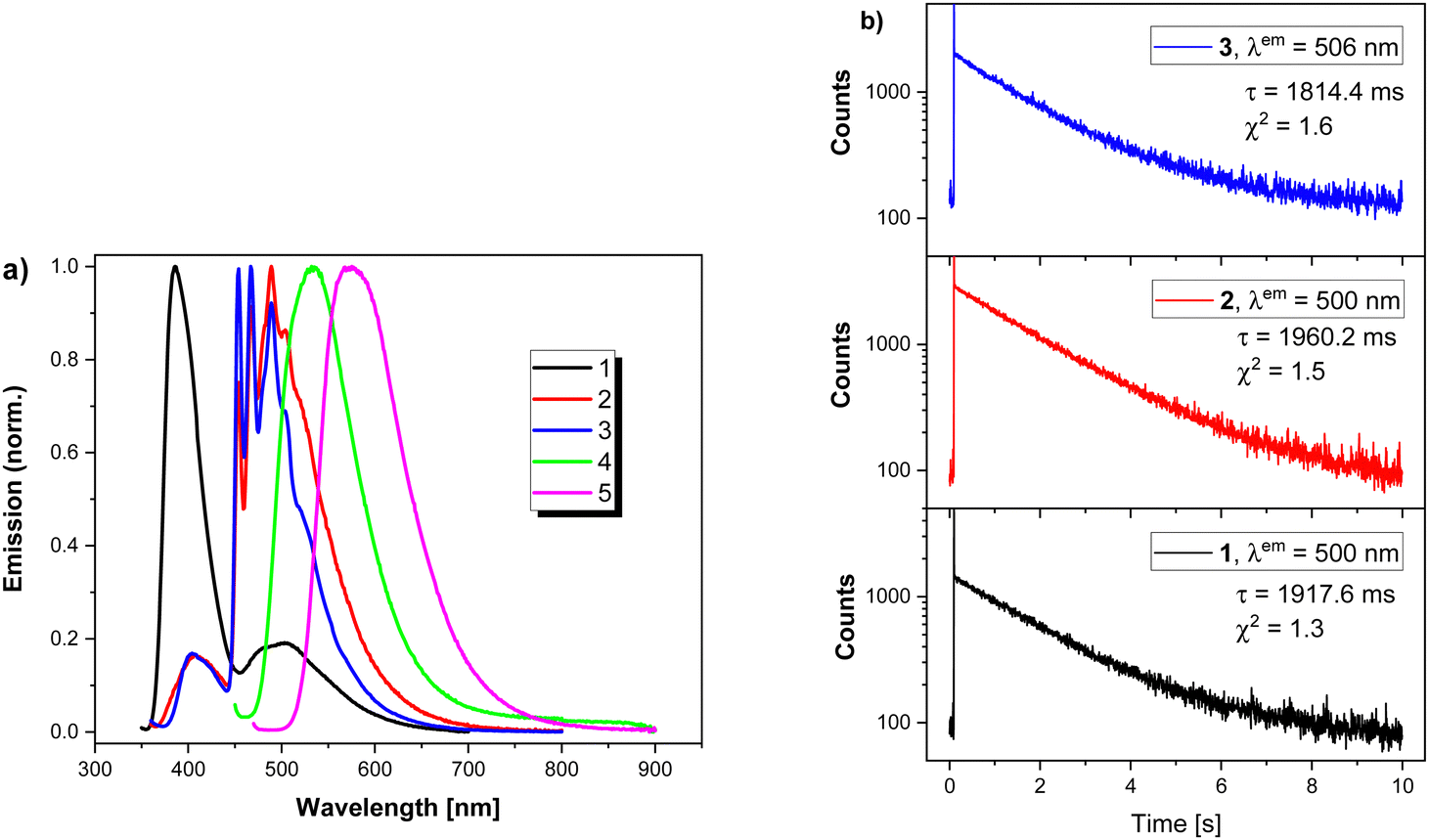 | ||
| Fig. 8 (a) PL spectra of frozen PhMe solutions of 1–5 at 77 K. (b) Phosphorescence decay of 1–3. | ||
| λ abs [nm] | λ em [nm] | PLQY Zeonex | Fluorescence lifetime [ns] | χ 2 | |
|---|---|---|---|---|---|
| a Integrating sphere at 330 nm. b Integrating sphere at 415 nm excitation wavelength. c Monoexponential fit. d Biexponential fit. | |||||
| 1 | 353 | 396 | 0.43a | 5.8 | 2.8 |
| 2 | ca. 340 (sh), 302 | 418 | 0.05a | 6.9c | 12.6c |
| 3.6 (40%), 9.8 (60%)d | 1.2d | ||||
| 3 | 343, 333, 298 | 417 | 0.07a | 3.1c | 4.9c |
| 2.5 (78%), 5.9 (22%)d | 1.2d | ||||
| 4 | 436, 324 | 545 | 0.18b | 22.3c | 9.8c |
| 12.2 (46%), 33.4 (54%)d | 1.5d | ||||
| 5 | 474, 320, 301 | 589 | 0.94b | 8.9 | 1.6 |
Expectedly, the character of the absorption spectra were retained. On the other hand, the rigid and non-polar environment of the Zeonex matrix caused a hypsochromic shift and narrowing of the emission spectra, as compared to those measured at RT in PhMe solutions. Geometrical restrictions imposed by a rigid environment resulted in an increase of the PLQY and almost in all cases shortened the PL lifetimes. In three cases (2, 3 and 4), the fluorescence decay profiles did not fit well into a single exponential decay model. This finding suggests either a case of mixed local and charge-transfer emission or presence of two emitting species. The latter variant related to 4 can be justified by the existence of two rotational isomers (rotation around Ccarbazole–Cbenzothiadiazole bond). For 2 and 3, two conformations of the phenoxazine moiety (axial or equatorial) can be envisioned.33 In this regard, single exponential decay of 1 is probably a result of low conformational geometrical distribution. The monoexponential decay of 5 can be related to pure emission from the electron-accepting unit. Conjugation extended by the carbazole unit in 5 results in slightly red-shifted emission in PhME and DCM. On the other hand, a subtle blue-shift is observed in the Zeonex matrix (see Fig. S5.5, ESI† for comparison). The CT emission may be more pronounced in the case of 4. Molecular blend of 4 in Zeonex exhibits the highest value of fluorescence lifetime. Moreover, the PL lifetime of 4 in Zeonex is higher than that measured for PhMe solution.
The PL studies in frozen PhMe solutions indicated clear differences in the emission properties at low temperatures. The emission spectrum of 1 shows two separate bands, namely, a high energy (386 nm) band ascribed to fluorescence and a low energy (506 nm) band due to phosphorescence. 4 and 5 exhibit similar behaviour in frozen PhMe solutions and in Zeonex matrix. Again, double exponential decay is observed for 4. 2 and 3 constitute much more complex cases. Both compounds exhibit strongly structured emission spectra in PhMe at 77 K. Similar to 1, the high energy band corresponds to fluorescence. Strongly structured emission within the 450–700 nm spectral range has several origins. On a shorter time scale (200 μs measurement window), double exponential decay is observed with luminescence lifetimes of approximately 1 μs and 8 μs whose contributions are highly dependent on the selected emission wavelength. Upon increasing the measurement window up to 10 s, clear long-lived phosphorescence is observed (Fig. 8).
Quantum chemical calculations
The performed computations involved the optimization of the studied compounds structures in their ground (S0) and first singlet excited state (S1). Optimized geometries showed the expected separation of HOMO and LUMO orbitals on the electron donating and electron accepting parts of the molecule (see Table S6.2, ESI†).In order to further investigate the nature of the electronic transitions in the studied molecules, theoretical absorption and emission spectra were calculated. TDDFT computations on S0 geometry indicate that the lowest energy excitations exhibit strongly mixed character. In order to get a clear view of the excitations of interest, natural transition orbitals analysis was performed.
In case of 1, the lowest energy transition is characterised by a considerable oscillator strength (fS0→S1 = 0.1425) and corresponds to the electron density transfer from the carbazole unit to the electron-accepting benzonitrile unit (see Table S6.3, ESI†). HOMO → LUMO transition does not have any contribution to this excitation. Interestingly, in the absorption spectrum, there are two low intensity excitations (fS0→S2 = 0.0003 and fS0→S3 = 0.0016) of similar energy. Both transitions consist of electron density transfer from the donor (acridan) to the acceptor part (benzonitrile) of the molecule.
Compounds 2 and 3 display a similar pattern of electronic transitions structure. Two lowest energy excitations correspond to the weak electron density transfer (f = 0.0004–0.0019) from the donor part of the molecule (phenoxazine) to its orthogonally placed π-spacer (phenylene ring) with a slight contribution of electron donation to the benzonitrile unit. The latter is the most pronounced in the case of S0 → S2 transition in compound 3. This is revealed by considerable dispersion of the LUNTO orbital between phenoxazine, phenylene and benzonitrile units. Similar to compound 1, first intense electronic transitions (S0 → S3 with f = 0.1395 and f = 0.1293 for 2 and 3, respectively) involve electron density transfer from carbazole to benzonitrile.
The second series of the compounds exhibit some analogies to the first one. Although the experimental absorption spectra of 4 and 5 contain well-separated low-energy absorption bands, computations reveal the presence of low intensity excitations alongside intense, rather local transitions in the acceptor segment (from thiophene and carbazole to benzothiadiazole). These low intensity bands are pure HOMO–LUMO transitions. Compound 5 can form three conformers differing in the orientation of the electron-accepting unit with respect to the phenoxazine moiety (see Fig. 9).
 | ||
| Fig. 9 Conformers of 5 considered for the computations. | ||
In the case of 4 and two conformers of 5 (5c1 and 5c2), the calculated lowest energy transitions of high intensity correspond to S0 → S2 (with f = 0.2349 and f = 0.4036 for 4 and 5c1, respectively). They can be attributed to the electron density transfer from phenoxazine to benzothiadiazole. In contrast, 5c3 exhibits inverse order (but identical in character) of the two lowest electronic transitions (Fig. S6.2, ESI†).
TDDFT calculations performed on optimized S1 geometries yield mirror images of the HONTO and LUNTO orbitals with respect to the lowest intense absorption transitions (see Table S6.4, ESI†). In most cases, the calculated Stokes’ shifts closely reflect the experimental values (Table 7). These results suggest that the observed emission in 1–4 stems from local electron density redistribution upon excitation. Relatively high discrepancies are observed for compound 5. These can be a direct result of either conformational equilibrium or stronger through-space interactions of the donor and acceptor.
| Calculated | Experimental | |||||
|---|---|---|---|---|---|---|
| E abs [eV] | E em [eV] | Stokes shift [eV] | E abs [eV] | E em [eV] | Stokes shift [eV] | |
| a Calculated from the first intense excitation. b Maximum estimated from the deconvolution of the absorption spectrum. | ||||||
| 1 | 4.0384 | 3.3769 | 0.6615 | 3.51 | 2.82 | 0.69 |
| 2 | 4.0403a | 3.0441 | 0.9962 | 3.60b | 2.63 | 0.98 |
| 3 | 4.1240a | 3.1727 | 0.9513 | 3.71 | 2.76 | 0.95 |
| 4 | 3.1355 | 2.3231 | 0.8124 | 2.93 | 2.16 | 0.77 |
| 5c1 | 2.9221 | 2.1108 | 0.8113 | 2.65 | 2.05 | 0.61 |
| 5c2 | 2.9143 | 2.1673 | 0.7470 | |||
| 5c3 | 2.8463 | 1.9632 | 0.8831 | |||
In order to explain the observed increase in PLQY and PL lifetime upon deaerating the investigated solutions of 1–5, additional TDDFT computations for the S1 geometry were performed. The results are collected in Fig. S6.1-2 (ESI†). For compounds 1–3, the energy diagrams show the presence of several triplet excited states of lower energy than that of the molecule in the relaxed S1 state. Singlet–triplet gaps are of the order of 0.01 eV. Such proximity of the triplet excited states may significantly influence the deexcitation process. In this view, the observed increase in PLQY and PL lifetimes upon deaerating may possibly result from the presence of previously oxygen-quenched deexcitation channels involving molecules in the triplet excited state. Low singlet–triplet gap enables effective intersystem crossing as well as reverse intersystem crossing. This could justify the observed rather slight elongation of the PL lifetime.
Compound 4 does not show a triplet excited state with low S1–T gap, while the case of 5 is more complex. Clear dependence of the energetic structure on the conformation of the molecule is visible. Conformers 5c1 and 5c2 show high singlet–triplet gap. On the other hand, 5c3 exhibits a very low S–T gap (0.0056 eV). This may possibly originate from close distance between the benzothiadiazole and phenoxazine units. The distances between centroids of phenyl rings within benzothiadiazole and phenoxazine are 3.629 Å, 4.532 Å and 3.404 Å for 5c1, 5c2 and 5c3, respectively. These results support claims that through-space interactions between the electron donating and electron accepting units in the studied compounds influence radiative de-excitation of the molecules. However, the geometry of 5 in the crystalline phase deviates significantly from the three conformers optimized in vacuum. Notably, the distance between the donor and acceptor parts of the molecule exceeds 5 Å (see ESI,† Fig. S4.6). This discrepancy is explained by a closer examination of the molecule's stacking arrangement (Fig. 4). In the crystal, the phenoxazine moiety of one molecule interweaves between the donor and acceptor parts of a neighboring molecule, causing substantial distortion of the BTD moiety from the plane of the carbazole linker. This observation highlights the high flexibility of the BTD moiety relative to the rest of the molecule and underscores the importance of considering multiple conformers when analyzing the spectroscopic properties.
Conclusions
To summarize, we synthesized five donor-carbazole-acceptor derivatives containing benzonitrile or benzothiadiazole as acceptors and acridan or phenoxazine as donors. All studied compounds are electrochemically active, exhibiting two-step (compounds 1–4) or three-step (compound 5) quasi-reversible oxidation. Compounds 4 and 5 are in addition ambipolar in nature since they could be quasi-reversibly reduced to the radical anion state in a 1e process. The investigated derivatives exhibit fluorescence in the blue-orange spectral range, strongly dependent on the polarity of the solvents used. PLQY values measured in a rigid matrix (Zeonex) are significantly improved as compared to those determined for toluene or DCM solutions. This improvement can have its origin in the confinement of intermolecular interactions. TDDFT calculations suggest that the observed emission of 1–4 stems from the local electron density redistribution upon excitation. A partial overlap of HONTO and LUNTO suggests some involvement of a through-bond charge transfer. In the case of 5, the emission process may result from stronger through-space charge transfer interactions due to a relatively small distance between the donor and acceptor units.Data availability
The data supporting this article have been included as part of the ESI.† The crystal structures were deposited at the Cambridge Crystallographic Data Centre with the following numbers: CCDC 2401006–2401010. These data can be obtained free of charge from www.ccdc.cam.ac.uk/structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre, Poland (NCN, Grant Opus no. 2022/45/B/ST5/02120). The X-ray structures were determined at the Czochralski Laboratory of Advanced Crystal Engineering (Faculty of Chemistry, University of Warsaw).References
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS.
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, e1603282 CrossRef PubMed.
- P. L. dos Santos, M. K. Etherington and A. P. Monkman, J. Mater. Chem. C, 2018, 6, 4842–4853 RSC.
- T. L. Wu, M. J. Huang, C. C. Lin, P. Y. Huang, T. Y. Chou, R. W. Chen-Cheng, H. W. Lin, R. S. Liu and C. H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS.
- T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS.
- Y. Zou, J. Hu, M. Yu, J. Miao, Z. Xie, Y. Qiu, X. Cao and C. Yang, Adv. Mater., 2022, 34, 2201442 CrossRef CAS.
- Y.-C. Cheng, X.-C. Fan, F. Huang, X. Xiong, J. Yu, K. Wang, C.-S. Lee and X.-H. Zhang, Angew. Chem., Int. Ed., 2022, 61, e20221257 Search PubMed.
- K. R. Naveen, H. I. Yang and J. H. Kwon, Commun. Chem., 2022, 5, 149 CrossRef CAS PubMed.
- M. Godumala, S. Choi, M. J. Cho and D. H. Choi, J. Mater. Chem. C, 2019, 7, 2172 RSC.
- A. Batra, G. Kladnik, H. Vázquez, J. Meisner, L. Floreano, C. Nuckolls, D. Cvetko, A. Morgante and L. Venkataraman, Nat. Commun., 2012, 3, 1086 CrossRef PubMed.
- H. Tsujimoto, D. G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894–4900 CrossRef CAS.
- T. Huang, Q. Wang, G. Meng, L. Duan and D. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200059 CrossRef CAS.
- Y. Song, M. Tian, R. Yu and L. He, ACS Appl. Mater. Interfaces, 2021, 13, 60269–60278 CrossRef CAS.
- J.-R. Mistry, S. Montanaro and I. A. Wright, Mater. Adv., 2023, 4, 787–803 RSC.
- S. Kumar, L. Gomes Franca, K. Stavrou, E. Crovini, D. B. Cordes, A. M. Z. Slawin, A. P. Monkman and E. Zysman-Colman, J. Phys. Chem. Lett., 2021, 12, 2820–2830 CrossRef CAS PubMed.
- X.-Q. Wang, S.-Y. Yang, Q.-S. Tian, C. Zhong, Y.-K. Qu, Y.-J. Yu, Z.-Q. Jiang and L.-S. Liao, Angew. Chem., Int. Ed., 2021, 60, 5213–5219 CrossRef CAS PubMed.
- Y.-J. Yu, M. Song, X.-Y. Meng, Y.-K. Qu, X.-Q. Wang, L. Chen, S.-Y. Yang, D.-Y. Zhou, Z.-Q. Jiang and L.-S. Liao, Org. Lett., 2023, 25, 6024–6028 CrossRef CAS.
- H. Miranda-Salinas, Y.-T. Hung, Y.-S. Chen, D. Luo, H.-C. Kao, C.-H. Chang, K.-T. Wong and A. Monkman, J. Mater. Chem. C, 2021, 9, 8819–8833 RSC.
- C. Wu, W. Liu, K. Li, G. Cheng, J. Xiong, T. Teng, C.-M. Che and C. Yang, Angew. Chem., Int. Ed., 2021, 133, 4040–4044 CrossRef.
- X.-F. Song, C. Jiang, N. Li, J. Miao, K. Li and C. Yang, Chem. Sci., 2023, 14, 12246–12254 RSC.
- I. Kulszewicz-Bajer, M. Zagorska, M. Banasiewicz, P. A. Gunka, P. Toman, B. Kozankiewicz, G. Wiosna-Salyga and A. Pron, Phys. Chem. Chem. Phys., 2020, 22, 8522–8534 RSC.
- I. Kulszewicz-Bajer, M. Guzauskas, M. Makowska-Janusik, M. Zagorska, M. Mahmoudi, J. V. Grazulevicius, A. Pron and D. Volyniuk, J. Mater. Chem. C, 2022, 10, 12377–12391 RSC.
- M. Guzauskas, D. Volyniuk, I. Kulszewicz-Bajer, M. Mahmoudi, A. Lazauskas, V. Jasinskas, V. Gulbinas, A. Pron and J. V. Grazulevicius, Adv. Opt. Mater., 2023, 2301059 CrossRef CAS.
- Y. Liu, Q. Chen, Y. Tong and Y. Ma, Macromolecules, 2020, 53, 7053–7062 CrossRef CAS.
- F.-Y. Hao, Y.-Z. Shi, K. Wang, X.-C. Fan, L. Wu, J. Ye, C.-J. Zheng, Y.-Q. Li, X.-M. Oua and X.-H. Zhang, J. Mater. Chem. C, 2020, 8, 10416–10421 RSC.
- Y. Yan, R. Walser-Kuntz and M. S. Sanford, ACS Mater. Lett., 2022, 4, 733–739 CrossRef CAS.
- A. Maggiore, Y. Qu, R. Guillot, P. Pander, M. Vasylieva, P. Data, F. B. Dias, P. Audebert, G. Clavier and F. Miomandre, J. Phys. Chem. B, 2022, 126, 2740–2753 CrossRef CAS PubMed.
- P. Ledwon, N. Thomson, E. Angioni, N. J. Findlay, P. J. Skabara and W. Domagala, RSC Adv., 2015, 5, 77303–77315 RSC.
- M. Shen, J. R. Lopez, J. Huang, Q. Liu, X. H. Zhu and A. J. Bard, J. Am. Chem. Soc., 2010, 132, 13453–13461 CrossRef CAS PubMed.
- P. Ledwon, P. Zassowski, T. Jarosz, M. Lapkowski, P. Wagner, V. Cherpak and P. Stakhira, J. Mater. Chem. C, 2016, 4, 2219–2227 RSC.
- N. A. Pushkarevsky, E. A. Chulanova, L. A. Shundrin, A. I. Smolentsev, G. E. Salnikov, E. A. Pritchina, A. M. Genaev, I. G. Irtegova, I. Y. Bagryanskaya, S. N. Konchenko, N. P. Gritsan, J. Beckmann and A. V. Zibarev, Chem. – Eur. J., 2019, 25, 806–816 CrossRef CAS.
- M. Hodée, J. Massue, S. Achelle, A. Fihey, D. Tondelier, G. Ulrich, F. Robin-le-Guen and C. Katan, Phys. Chem. Chem. Phys., 2023, 25, 32699–32708 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2401006–2401010. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc04884a |
| This journal is © The Royal Society of Chemistry 2025 |