
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sixty years of electrochemical optical spectroscopy: a retrospective
Chao-Yu
Li
 ab and
Zhong-Qun
Tian
*a
ab and
Zhong-Qun
Tian
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: zqtian@xmu.edu.cn
bSchool of Materials Science and Engineering, Tongji University, Shanghai, 201804, China
First published on 29th February 2024
Abstract
Sixty years ago, Reddy, Devanatan, and Bockris performed the first in situ electrochemical ellipsometry experiment, which ushered in a new era in the study of electrochemistry, using optical spectroscopy. After six decades of development, electrochemical optical spectroscopy, particularly electrochemical vibrational spectroscopy, has advanced from a phase of immaturity with few methods and limited applications to a phase of maturity with excellent substrate generality and significantly improved resolutions. Here, we divide the development of electrochemical optical spectroscopy into four phases, focusing on the proof-of-concept of different electrochemical optical spectroscopy studies, the emergence of plasmonic enhancement-based electrochemical optical spectroscopic (in particular vibrational spectroscopic) methods, the realization of electrochemical vibrational spectroscopy on well-defined surfaces, and the efforts to achieve operando spectroelectrochemical applications. Finally, we discuss the future development trend of electrochemical optical spectroscopy, as well as examples of new methodology and research paradigms for operando spectroelectrochemistry.
 Chao-Yu Li | Chao-Yu Li is a professor at School of Materials Science and Engineering, Tongji University, China. He obtained his PhD degree in physical chemistry from Xiamen University in 2016. And then, he carried out postdoctoral research at Massachusetts Institute of Technology and Emory University in the U.S.A, respectively. He joined Tongji University in 2023, and his main research interests include spectroelectrochemistry, electrochemical energy storage and conversion, plasmonics, and single-molecule spectroscopy, etc. |
 Zhong-Qun Tian | Zhong-Qun Tian is a full professor of chemistry at Xiamen University. He obtained his BSc degree from Xiamen University in 1982 and completed his PhD under the supervision of Prof. Martin Fleischmann at University of Southampton in 1987. Prof. Tian is a member of the Chinese Academy of Sciences, a Fellow of the Royal Society of Chemistry and the International Society of Electrochemistry, and served as President of the International Society of Electrochemistry from 2019 to 2020. His main research interests include SERS, spectroelectrochemistry, nanochemistry, plasmonics, and catassembly, etc. |
1. Introduction
Since the nineteenth century, the electrochemical research method has been developed mainly by using electric signals as excitation and detection means, i.e., to study the structure and reaction mechanism at electrode/electrolyte interfaces, with precise measurement of current, potential and charge (e.g., chronoamperometry, chronopotentiometry, cyclic voltammetry, AC impedance, etc.).1–4 The electrochemical methods developed with the advances in electronics have now achieved a very high detection sensitivity, capable of detecting a change in a sub-monolayer of atoms or molecules (i.e., the adsorption/desorption of a sub-monolayer of molecules on a single crystal electrode surface).1–4 However, the traditional electrochemical methods have their intrinsic limitations; for instance, the electrical signal is incapable of chemically recognizing specific molecules and revealing the bonding information and fine molecular structure of the individuals. Accordingly, to accurately identify various species on the electrode, and to explain the mechanism of electrochemical reaction, it is imperative to integrate other advanced technologies into the modern electrochemical methods, thus offering a great opportunity to apply optical spectroscopy in electrochemical studies5.After sixty years of development, the main frame of the electrochemical optical spectroscopy has been established, moving forward from the early phase of proof-of-concept of spectroscopic methods to the phase of making practical contributions to the field of electrochemistry.6 In particular, since 2010, the operando spectroscopic technique has attracted considerable attention because it allows for the real-time studies of the correlations between electrochemical performance with chemical and structural changes inside electrochemical devices under real working conditions, and this methodology, which advances the object of study from the previous ideal electrochemical interface to the practical interphase between the electrode and electrolyte (namely, “from the interface to the interphase”), has become an important application in spectroelectrochemistry. Therefore, it is necessary to review the history and different phases of the development of electrochemical optical spectroscopy (in this review, optical spectroscopy refers to the spectroscopy studies in the spectral wavelength range from ultraviolet to IR), and to discuss the new methodology and research paradigm for operando spectroelectrochemistry.
Here, as shown in Table 1, we divide the development into four phases, focusing on the proof-of-concept of different electrochemical optical spectroscopy studies (phase I), the emergence of plasmonic enhancement-based electrochemical vibrational spectroscopic methods (phase II), the realization of electrochemical vibrational spectroscopy on well-defined surfaces (phase III), and the development of operando electrochemical vibrational spectroscopy (phase IV).
| Development Phase | Spectroscopic technique | Year | Spectroscopic mode | Electrochemical system | Features and notes |
|---|---|---|---|---|---|
| Abbreviations: EC-IR, electrochemical infrared spectroscopy; EC-SHG, electrochemical second hormonic generation; EC-SFG, electrochemical sum frequency generation; EC-SERS, electrochemical surface-enhanced Raman spectroscopy; EC-SEIRAS, electrochemical surface-enhanced infrared absorption spectroscopy; TERS, tip-enhanced Raman spectroscopy; TMs, transition metals; TM-coated Au, transition metal-coated Au; EC-SHINERS, electrochemical shell-isolated nanoparticle-enhanced Raman spectroscopy; SHIN, shell-isolated nanoparticle; EC-nano FTIR, electrochemical Fourier transform infrared nano-spectroscopy; AQ-COOH, 2-anthraquinonecarboxylic acid; pNDMA, para-nitrosodimethylaniline; 4-PBT, (4′-(pyridin-4-yl)biphenyl-4-yl)methanethiol; SPP, surface plasmon polariton; SPM, scanning probe microscopy; TAS, Te2As3Se5. Both operando fiber-based EC-Raman and EC-IR refer to operando spectroscopic experiments in batteries. | |||||
| Phase I (proof-of-concept of electrochemical optical spectroscopies) | EC-ellipsometry7 | 1963 | Normal | Anodic formation of Hg2Cl2 films on Hg electrodes | First in situ electrochemical optical spectroscopy. Due to the limited detection sensitivity of instrument, the film electrode with a certain thickness containing a significant number of analytes is highly desirable and decisive. |
| EC-UV-Vis8 | 1964 | Normal | Electro-redox of ferrocyanide and chronopotentiometric electrooxidation of o-tolidine | First in situ spectroscopic study of electrochemical product in solution phase. The analytes need to absorb light in UV-Vis wavelength. | |
| EC-IR9 | 1966 | ATR-based | Electroreductions of 8-quinolinol and tetramethylbenzidine free radicals. | First in situ spectroelectrochemistry using vibrational spectroscopy, in which Ge was used simultaneously as a working electrode and a waveguide for multi-internal reflection. | |
| EC-SHG10 | 1967 | Normal | Electrified Si and Ag electrodes. | First in situ nonlinear spectroscopy at the electrochemical interface. | |
| EC-Raman11 | 1973 | Normal | Electrochemical deposition of Hg2Cl2, Hg2Br2, and HgO. | Hg2Cl2, Hg2Br2, and HgO, the same systems studied by Bockris in the first EC-ellipsometry, are strong Raman scatters, which facilitate the normal Raman measurement (external reflection) and also the optimization of the optic configuration and cell. | |
| EC-IR using external reflection12,13 | 1980 | Normal | Electrooxidation of thianthrene and the adsorption of indole and H2O on Pt electrodes. | An external-reflection mode was used for the first time to acquire IR absorption signals of molecules adsorbed at the electrode surface. | |
| EC-SFG14 | 1990 | Normal | Adsorption of CO and CN− on the Pt electrode surface. | First in situ nonlinear spectroscopy with molecular vibrational information at the electrochemical interface. | |
| Phase II (plasmonic enhancement-based electrochemical vibrational spectroscopies) | EC-SERS15,16 | 1974-1977 | Nanostructure-based | Adsorption of pyridine on an electrochemically roughened Ag electrode surface | The discovery of SERS, in which the surface enhancement effect enables the high-quality Raman spectroscopic measurement of (sub)monolayer of molecules adsorbed on the electrode surface. |
| EC-SERS on TMs17–20 | 1987 | Nanostructure-based | EC-SERS on different TM layers (i.e., Fe,20 Ni,17 Co,17 Pt,18 Pd,18 and Pb,19etc.) deposited on Au or Ag substrates | A strategy of “borrowing” high SERS activity from highly SERS-active Au or Ag substrates to probe the Raman signal on SERS-weakly or non-SERS-active TM surfaces. | |
| EC-SERS with self-assemble monodisperse colloids21 | 1995 | Nanostructure-based | EC-SERS on monodisperse Au or Ag nanoparticles assembled on organosilane-polymer-modified solid substrate | For the first time, the monodisperse high-SERS active nanoparticles were regularly arranged on the substrate for desirable SERS activity with good stability and reproducibility. | |
| EC-SEIRAS22–25 | Mid-1990s | Nanostructure-and ATR-based | Adsorption of AQ-COOH,22 water,23 water-sulfate,24 and pyridine25 on thin Ag or Au film electrodes. | Molecules on evaporated thin metal films exhibit enormously strong IR absorption; however, the enhancement factor of SEIRAS is modest compared to that of SERS. The vacuum evaporation rate or electrochemical deposition rate to prepare the metal film electrode are crucial to the enhancement. | |
| Phase III (electrochemical vibrational spectroscopies on well-defined surfaces) | EC-IR on single-crystal electrode26–28 | Late 1980s | ATR-based | Adsorption of CO26,27 and hydrogen28 at Pt single-crystal electrodes | The external-reflection spectroscopic mode promotes the vibrational spectroscopy on a well-defined electrode surface. SPP induced by ATR configuration enhance the Raman signal on atomically flat electrode surfaces. |
| EC-Raman on single-crystal electrodes29,30 | 1991;29 199830 | ATR-based | Adsorption of pNDMA at Ag single-crystal electrodes;29 adsorption of pyridine at Cu single-crystal electrodes30 | ||
| EC-SFG on single-crystal electrode31,32 | 1994 | Normal | Adsorption of hydrogen31 and cyanide32 at Pt single-crystal electrodes | ||
| EC-SHINERS33 | 2010 | Nanostructure-based | Adsorption of hydrogen at Pt single-crystal electrodes | The ultra-thin, pinhole-free dielectric shell of SHIN plays the key role. | |
| EC-TERS34,35 | 2015 | Tip-based | Protonation and deprotonation of 4-PBT on Au single-crystal electrodes;34 electrochemical redox of Nile blue on an ITO electrode.35 | The electrochemistry, plasmon-enhanced spectroscopy, and SPM with high spatial resolution are combined to enable vibrational spectroscopy on well-defined electrodes. | |
| EC-nano FTIR36 | 2019 | Tip-based | Potential-dependent aggregations of sulfate and ammonium at the graphene-electrolyte interface. | ||
| Phase IV (operando electrochemical vibrational spectroscopies) | Operando EC-IR37 and EC-Raman38,39 | Early 2010s | Normal | EC-IR of the adsorbed CO on the Pt membrane electrode in an operating fuel cell;37 EC-Raman of water distribution in Nafion membranes at various hydration states in operating fuel cells.38,39 | The design of cells compatible with operando spectroscopic experiments is essential, and the normal external-reflection spectroscopic mode is suitable under real working conditions. Normally, the exterior of the cell was changed to a transparent window, and the electrode component requires a hole to allow for the optic access. |
| Operando fiber-based EC-Raman40 | 2016 | Fiber-based | Operando study of electrolyte distribution inside a laminate cell. | Without using a transparent optic window in place of the battery exterior, optic fibers can be used as small probes embedded directly into the practical or commercial batteries for non-destructive analysis of the electrolytes during cycling. Normally, silicon and TAS are used as fiber materials for Raman and IR, respectively. | |
| Operando fiber-based EC-IR41 | 2022 | Fiber-based | Electrolyte analysis in a commercial 18650 jelly roll and a Swagelok cell. | ||
2. Four phases of development of electrochemical optical spectroscopy
2.1. Phase I
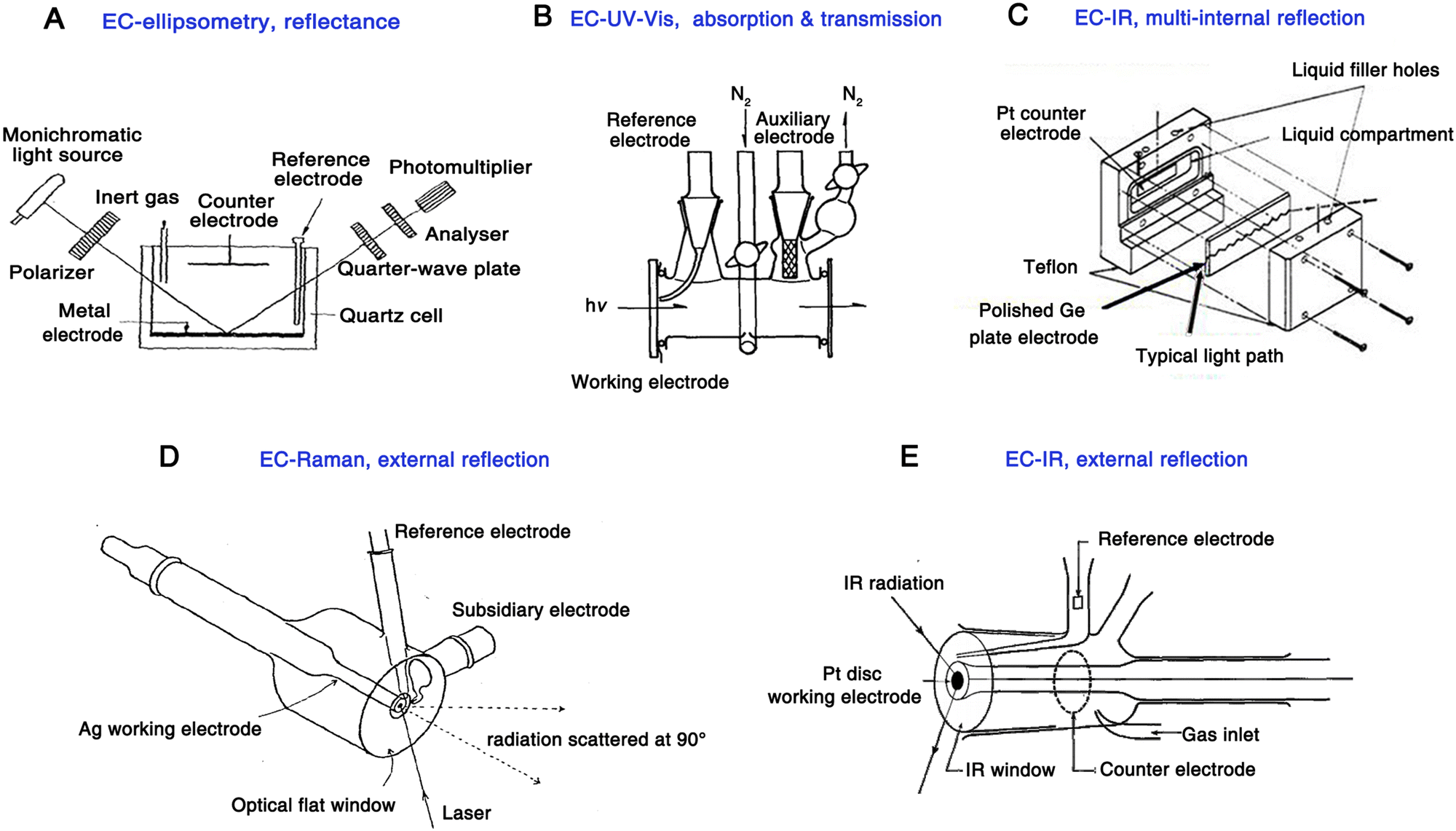 | ||
| Fig. 1 Schematics of the electrochemical cell for the pioneering work of EC-ellipsometry (A), EC-UV-Vis (B), EC-IR (multi-internal reflection) (C), EC-Raman (D), EC-IR (external reflection) (E), respectively. Fig. 1A is reproduced with permission from ref. 7. Copyright 1963 Elsevier B.V. Fig. 1B is reproduced with permission from ref. 43. Copyright 1968. Elsevier B.V. Fig. 1C is reproduced with permission from ref. 9. Copyright 1966 American Chemical Society. Fig. 1D is reproduced with permission from ref. 15. Copyright 1974 Elsevier B.V. Fig. 1E is reproduced with permission from ref. 44. Copyright 1984 Elsevier B.V. | ||
It is important to note that due to the limited detection sensitivity of the instrument during the early stages of spectroelectrochemistry, a calomel film electrode with a certain thickness played a crucial role in validating the proof-of-concept of the new method because it contains a considerable number of molecules, which greatly facilitated the acquisition of optical signals on electrode.
In 1964, Kuwana et al. used a simple but classic optical spectroscopic technique, UV-Vis spectroscopy, in the electrochemical system for the first time.8 They used a common transmission optical configuration for UV-Vis measurement and a transparent tin dioxide as the working electrode (Fig. 1B). In this work, the electro-redox of ferrocyanide in a KCl solution and the chronopotentiometric electrooxidation of o-tolidine were recorded. As a milestone in the early stages of spectroelectrochemistry, this was the first time when the electrochemical reaction product in the solution phase was studied using a spectroscopic technique. It is worth noting that, in UV-Vis measurements, this method is mainly used for analytes that can absorb excitation wavelengths of light.
In 1966, a meaningful electrochemical infrared (EC-IR) experiment was conducted by Mark and Pons,9 in which a polished semiconducting Ge plate was simultaneously used as a working electrode and a waveguide, enabling a multiple-reflection configuration of IR light (Fig. 1C). This is the first report of in situ electrochemical vibrational spectroscopy; however, it is worth noting that Ge is not a universal electrode material, and other electrode materials commonly used in the electrochemistry field, such as Au, Pt, and Pd, cannot be applied in the attenuated total reflectance (ATR)-IR mode, thus limiting the generality of this method at that time.
In 1973, Fleischmann et al. achieved an breakthrough in spectroelectrochemistry, that is, the application of Raman spectroscopy in the electrochemical study for the first time.11 A prominent advantage of Raman spectroscopy compared to IR absorption is that Raman spectra can be recorded by using lasers in the visible wavelength region. Therefore, the absorption of incident light by the aqueous solution and the glass optical window is negligible. As a result, it is more convenient to use a generalized electrochemical cell for Raman spectroscopy measurements, for example, the incident light can be directed to the electrode surface through a glass optical flat and the aqueous electrolyte (the electrochemical cell is shown in Fig. 1D15). This is the first time that an external reflection excitation-collection mode has been used in electrochemical vibrational spectroscopy, which is more suitable for the use of a common rod-like metal electrode in spectroelectrochemistry and thus more attractive to the electrochemistry community.
It should be noted that, in the absence of the resonance effect and surface enhancement, the differential Raman cross-section is generally smaller than 10−29 cm2 sr−1, which is approximately 10 orders of magnitude lower than IR absorption, resulting in an extremely weak Raman signal.50 Hence, back in 1972 when the authors discussed the possibility of obtaining Raman spectra from the electrode surface, the mercury/calomel system was recommended as calomel has an exceptionally large Raman scattering cross-section (i.e., a strong Raman scatter).44 At the same time, to generate a large-area mercury surface, thin films of Hg2Cl2 were formed on mercury droplets that had been electrodeposited onto a Pt disc foil11. As a result, a characteristic Hg–Hg stretch band at 168 cm−1 was observed, which disappeared upon shifting the potential to a more cathodic state, indicating that the signal originated from the electrode surface layer. Furthermore, similar observations were obtained with the Hg2Br2 and HgO system, suggesting for the first time that Raman scattering could be employed for in situ spectroelectrochemical studies.11,44
It is of significance that this mercury oxide and halide system is the same system which Bockris et al. studied ten years ago for the first in situ electrochemical ellipsometry measurement (1963),7 in which the excellent optical scatters facilitate the collection of spectroelectrochemical signals, the optimization of optical path, and the design of electrochemical Raman (EC-Raman) spectroscopic cells. However, given the larger number of molecules in the mercury oxide and halide system, it was still highly desirable to realize investigations of (sub-)monolayer adsorption (reaction) species on the electrode surface, which are more significant and more challenging. In practical terms, this means that the research object needs to be advanced from the film electrode studied in the early work to more important interfaces, such as the electrode/electrolyte interface, which requires a much higher detection sensitivity.
In 1980, a significant work was conducted by Bewick et al.,12,13 in which an external-reflection mode was used for the first time to acquire IR absorption signals at the electrode surface. Based on this configuration, a commonly used Pt disc-shaped electrode served as a working electrode to study the hydrogen and water adsorption on the Pt surface under potential modulations (Fig. 1E and 2A). Notably, to avoid the significant absorption of IR light by the electrolyte, a thin-layer electrolyte between the electrode surface and window was required. At the same time, the authors employed a continuous potential modulation method to improve the signal-to-noise ratio. This method was called electrochemically modulated infrared spectroscopy (EMIRS),51 which is the first electrochemical IR technique of using an electrode potential difference strategy.52
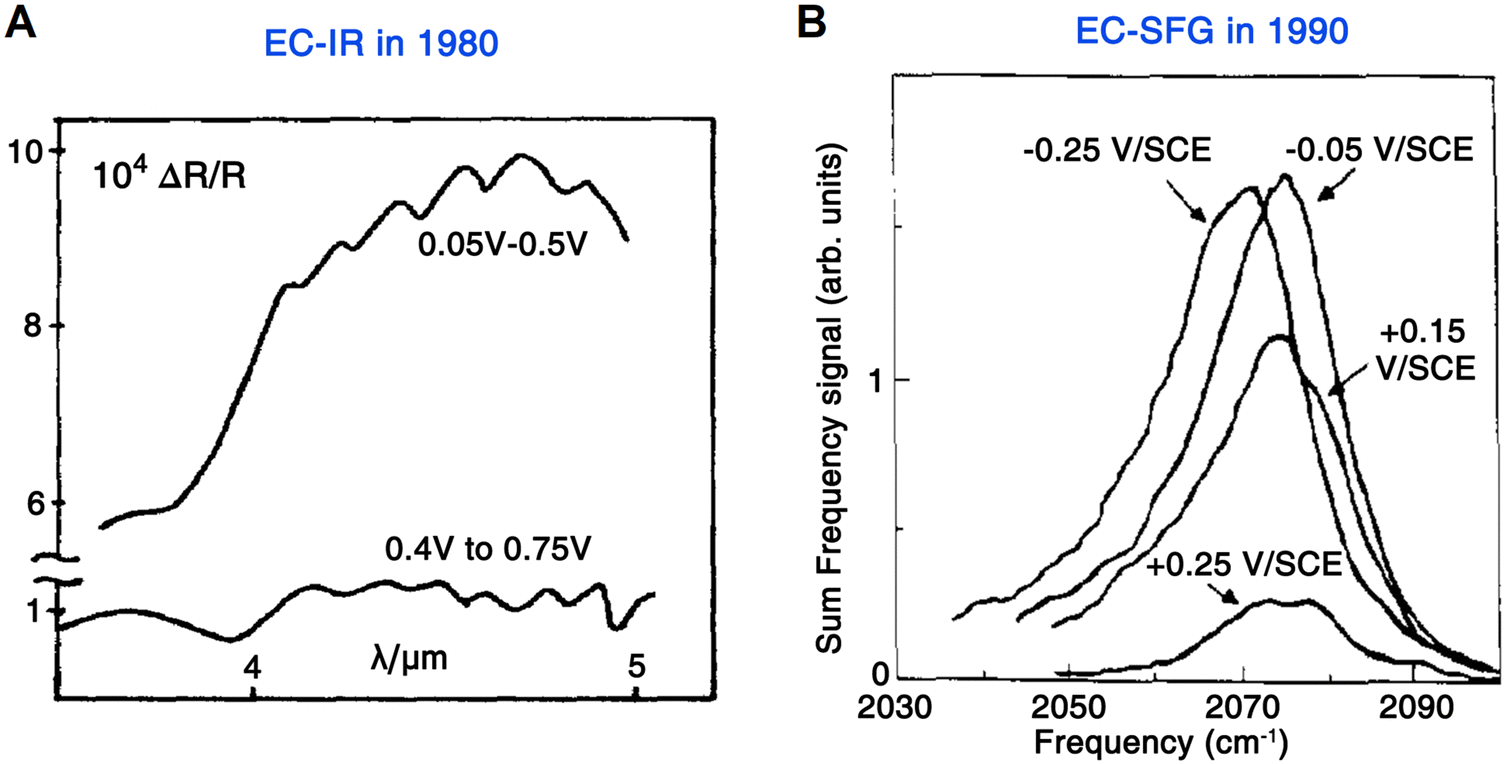 | ||
| Fig. 2 (A) First EC-IR measurement using an external-reflection mode by Bewick et al. in 1980, in which the spectra were recoded from the Pt/1 M H2SO4 interface under potential modulations. (B) First EC-SFG spectroscopy study by Tadjeddine et al. in 1990. These SFG spectra were obtained from CO adsorbed on a Pt electrode. Fig. 2A is reproduced with permission from ref. 12. Copyright 1980 Elsevier Ltd. Fig. 2B is reproduced with permission from ref. 14. Copyright 1990 Elsevier B.V. | ||
In the meantime, with short laser pulses, nonlinear optics started to play an important role in spectroelectrochemistry. Compared with IR and Raman spectroscopic methods, nonlinear spectroscopis methods, such as sum frequency generation (SFG) and second harmonic generation (SHG) are characterized by their inherent surface specificity and sensitivity.47 In 1967, Lee et al. reported the first electrochemical SHG measurements, in which the second-harmonic reflected light was in situ detected on electrified silicon and silver surfaces in KCl solutions.10 SFG (ωSFG = ω1 + ω2) is an extension of the SHG process (ω1 = ω2 = ω, ωSHG = 2ω), and in addition to having the high sensitivity of the SHG technique, SFG is also capable of probing the characteristic vibrational transitions of molecules.47
SFG spectroscopy has several distinct advantages over Raman scattering and IR absorption spectroscopy: (1) under the electric dipole approximation, SFG is only allowed in media with a broken centrosymmetry, making it an inherently surface specific spectroscopic technique; (2) at the interface of a molecule adsorbed on a metal, SFG can probe the response of the adsorbed molecule, electronic excitations of the metal, and the interaction of molecule adsorption modifying the density of states at the interface; (3) in cases involving short pulsed lasers, SFG is able to investigate the vibrational dynamics at the interface with a time resolution from nanosecond (ns) to less than 100 femtosecond (fs) and (4) the surface structure can be studied through anisotropy experiments, among other techniques.47,53–56
In 1987, in order to obtain the fingerprint vibrational spectroscopic information at the interface, Shen et al. developed IR-visible SFG spectroscopy, where one incident light source is a tunable IR laser and the other incident light source is a visible laser with a fixed wavelength, i.e., the IR pulse spatially and temporally overlaps with a visible pulse for the SFG signal.57–59 Three years later (1990), Guyot-Sionnest and Tadjeddine applied SFG spectroscopy to electrode–electrolyte interfaces for the first time.14 Because an IR beam is used as one of the excitations, a similar issue to that in EC-IR spectroscopy, i.e., the strong adsorption of IR light by electrolyte, should be tackled. Therefore, a thin-layer cell similar to the one commonly used in EC-IR spectroscopy was adopted in electrochemical sum frequency generation (EC-SFG) measurements to study the adsorption of CO, CN−, and SCN− anions on Pt electrodes. As shown in Fig. 2B, no background interference from the electrolyte or Pt electrode was observed in the SFG signal of the CO stretching vibration (i.e., a well-defined resonance of vibration over an unobservable contribution from metal), demonstrating the characteristic surface sensitivity of EC-SFG, which is crucial and invaluable for studying adsorbed species on the electrode surface.
Electrochemical vibrational spectroscopy, including EC-IR, EC-Raman, and EC-SFG, among others, is characterized by its ability to provide fingerprint vibrational information from the electrochemical interface or bulk phases. Furthermore, the significant advancements in nanoscience since the 1990s have led to an explosive growth of electrochemical vibrational spectroscopic research studies, which has promoted the important new development phases of spectroelectrochemistry. A search was conducted through Web of Science® using the key words ‘electrochemical Raman’, ‘electrochemical IR’, ‘electrochemical sum frequency generation’, ‘electrochemical UV-Vis’, ‘electrochemical second harmonic generation’, and ‘electrochemical ellipsometry’, respectively. As revealed in Fig. 3, in recent years, the number of research articles on electrochemical vibrational spectroscopic methods (including Raman, IR, and SFG) is nearly 6000 per year, which is almost 4 times of the total number of articles on EC-ellipsometry, EC-SHG, and EC-UV-Vis. Therefore, the significant and rapid growth of electrochemical vibrational spectroscopy have notably contributed to phases II to IV in the development of electrochemical optical spectroscopy.
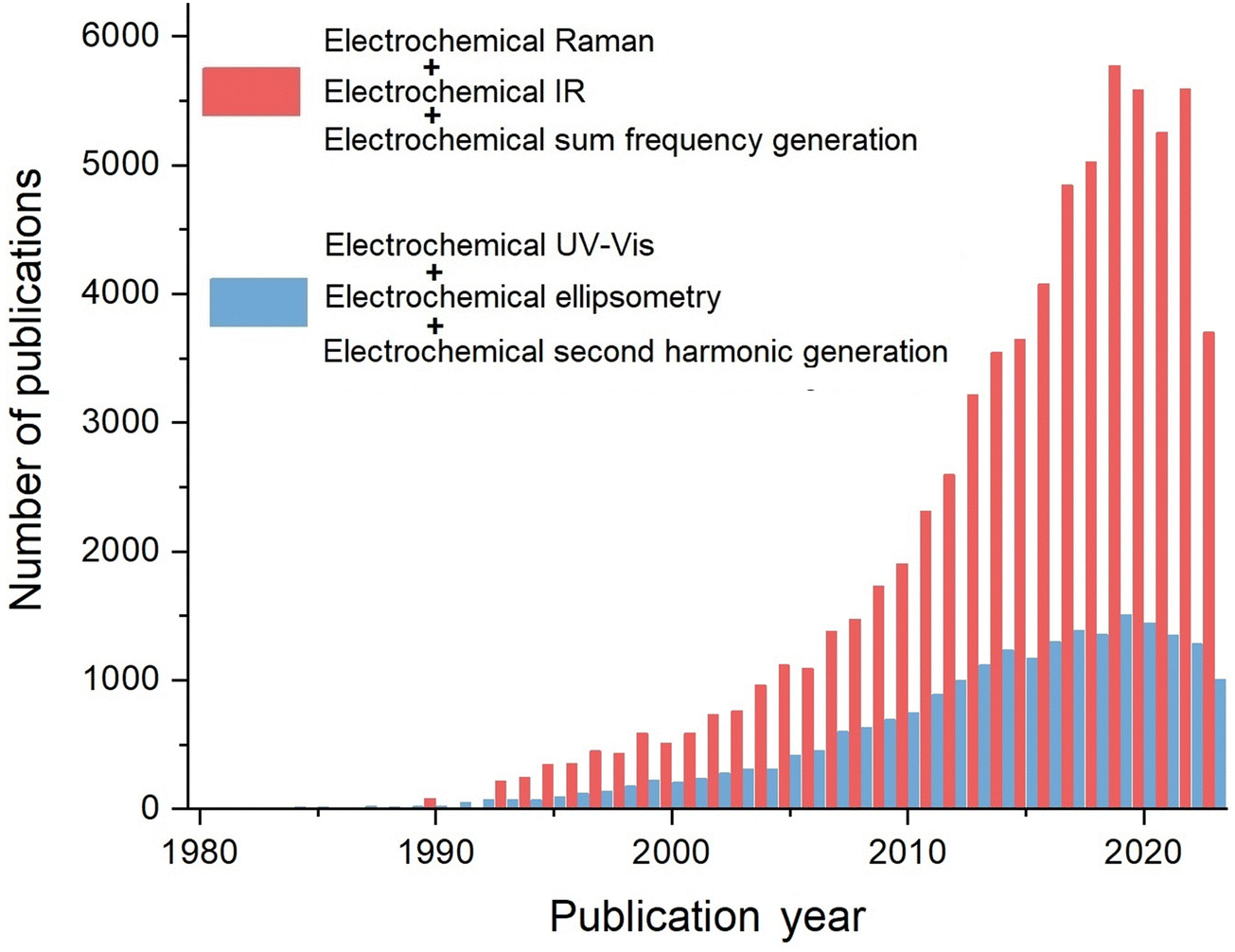 | ||
| Fig. 3 The total number of articles on electrochemical vibrational spectroscopy methods using the key words “electrochemical Raman”, “electrochemical IR”, and “electrochemical sum frequency generation”, and the total number of articles on other electrochemical optical spectroscopies using the keywords “electrochemical UV-Vis”, “electrochemical ellipsometry”, and “electrochemical second harmonic generation”, respectively. These data are obtained through Web of Science®. | ||
Herein, it is reasonable to present a comprehensive overview of the development of electrochemical optical spectroscopy methods, but with an emphasis on vibrational spectroscopy in light of historical and future development trends.
2.2. Phase II
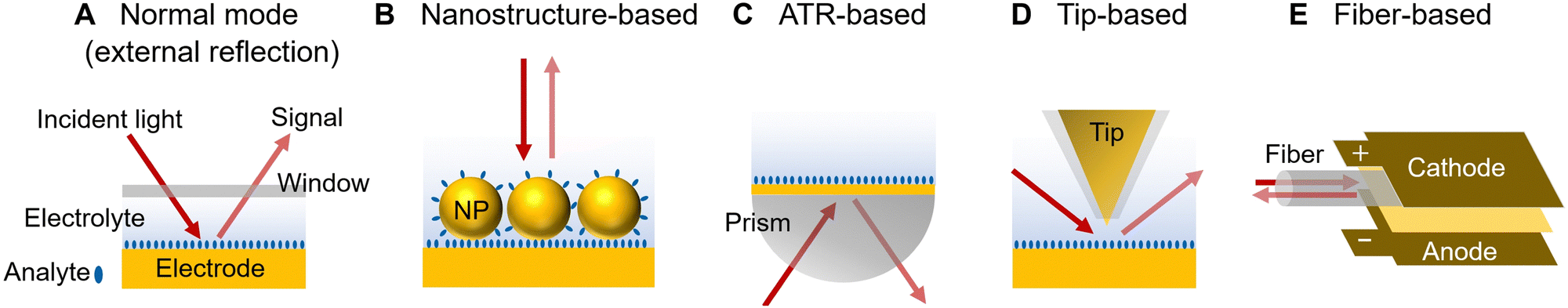 | ||
| Fig. 4 Different electrochemical vibrational spectroscopic modes. (A) Normal mode (external reflection). (B) Nanostructure-based mode. NP is an abbreviation for nanoparticle. (C) ATR-based mode. (D) Tip-based mode. (E) Fiber-based mode. | ||
In 1974, in order to overcome the extremely low detection sensitivity for surface adsorbed species in Raman spectroscopy, Fleischmann, Hendra, and McQuillan came up with a strategy to significantly increase the number of surface molecules, in which pyridine (Py) was chosen because of its very large Raman cross-section.15 They used potential-controlled oxidation and reduction cycles (ORC) to increase the surface area of a silver electrode in an aqueous 0.1 M KCl electrolyte containing 0.05 M Py. As shown in Fig. 5, by using a front-reflection cell (see Fig. 1D for the schematic of cell), the Raman spectra obtained on the electrochemically roughened silver electrode exhibit unexpectedly high quality, and the distinct potential dependence suggested that the Raman signal originates from the electrode surface-adsorbed species. In fact, the preliminary result of this work was briefly introduced during a general discussion at the conference of 56th Faraday Discussions in 1973,42 but it was formally published in 1974.15
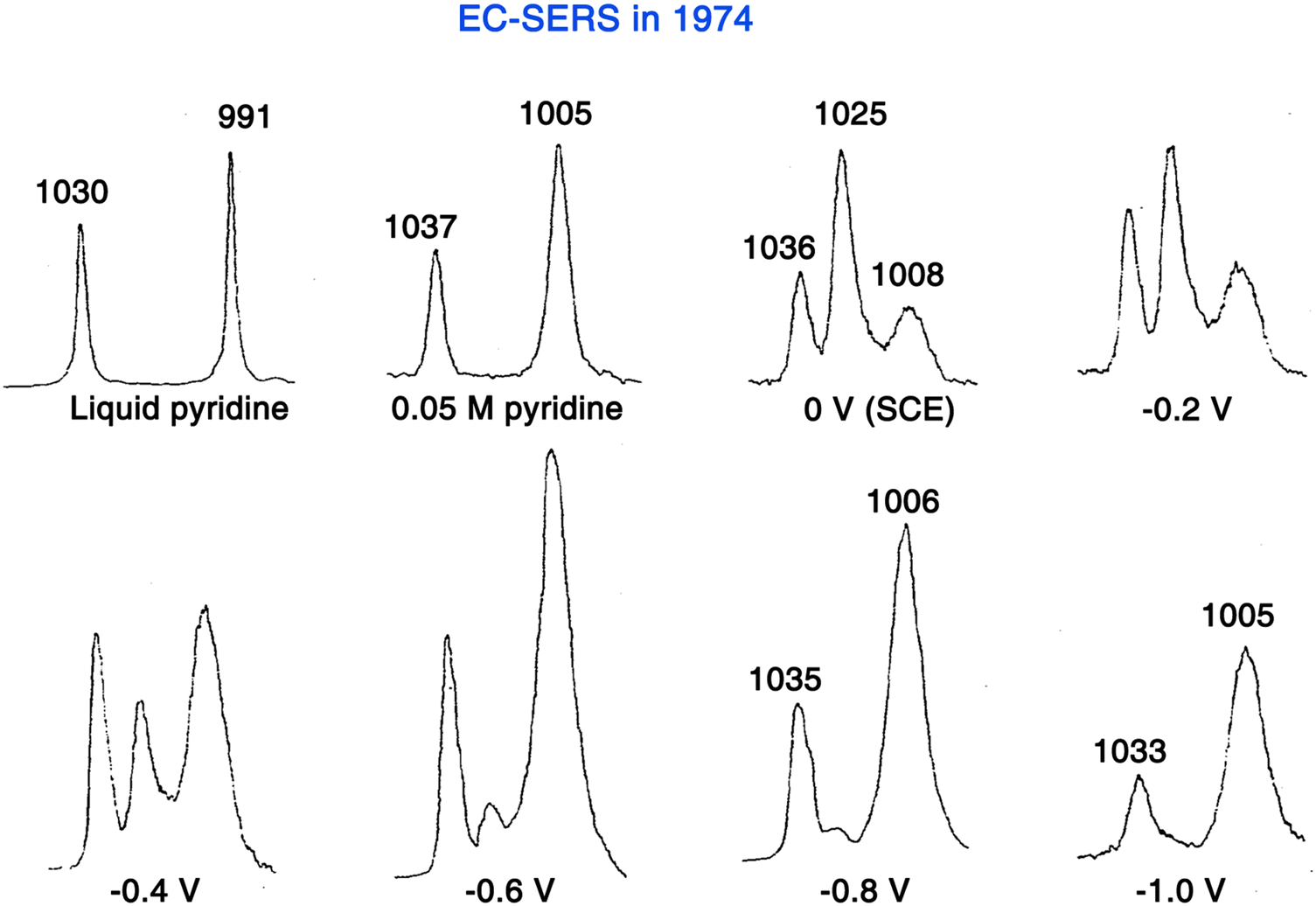 | ||
| Fig. 5 First SERS experiment by Fleischmann et al. in 1974, in which the adsorption of pyridine was studied on a roughened Ag electrode surface. Reproduced with permission from ref. 15. Copyright 1974 Elsevier B.V. | ||
In retrospect, although Fleischmann and co-authors were unaware of the SERS effect at the time, this work was the first SERS measurement, i.e., EC-SERS (Fig. 5). They initially believed that the electrochemical roughening largely increased the electrode surface area and thus the number of surface adsorbed molecules, resulting in the acquisition of strong Raman signals from the surface adsorbed Py. With careful calculations and experimental verifications, Van Duyne and Jeanmaire found that the major contribution to the strong Raman signal intensity of adsorbed Py species is an anomalous enhancement of 5 to 6 orders of magnitude compared to those predicted from the scattering cross-section for Py in bulk.16 However, their paper went through a long and exhaustive reviewing procedure and was eventually published in 1977.16 In the same year, Albrecht and Creighton independently reported a similar result.62 These pioneers presented strong evidence to demonstrate that the enormously strong surface Raman signals must be caused by a genuine enhancement of the Raman scattering efficiency itself. This Raman enhancement effect was later named SERS.63,64
The discovery of SERS created a sensation in the fields of surface science and spectroscopy;50,65 after comprehensively studying the SERS effect through experiments and theorical calculations, people realized the enhancement is largely owing to the excitation of surface plasmon resonance (SPR).66–70 In 1978, Moskovits proposed that the Raman enhancement in SERS is caused by the resonances of conduction electrons on the surface of roughened metal.70 Moreover, he predicted that the Raman enhancement could be realized on Au, Ag, and Cu colloids. In 1979, Creighton et al. conducted the first study of SERS using Ag and Au colloids, which were synthesized through wet-chemical methods.69
Looking back at the history of SERS, the enthusiasm in the fields of surface science and electrochemistry was greatly motivated after the discovery of SERS, but soon wore off in the late 1970s. The main reasons are the limitations on the substrate material and morphology of SERS.71 First, the generality of materials is limited to a few “free-electron like” metals, such as Au, Ag, and Cu.72 However, the application of SERS on other electrode materials important to electrochemistry, such as weakly SERS-active or SERS-inactive group VIIIB transition metals (TMs), was difficult.
Therefore, a strategy of “borrowing” high SERS activity from highly SERS-active Au or Ag substrates was initially proposed by Van Duyne to study the adsorbates on semiconductor electrode surfaces, i.e., n-GaAS(100), in 1983.73 But in this method, the analytes were able to adsorb on the Ag surface as well, which may result in mixed signals from molecules adsorbed on both the semiconductor and Ag. In 1987, the Fleischmann group17,20 and Weaver group18,19 independently developed the “borrowing” strategy by coating a layer of different TMs, such as Fe,20 Ni,17 Co,17 Pt,18 Pd,18 Pb,19etc. onto the surface of Au or Ag substrates. It is worth noting that the thickness of the coating layer of TMs should be kept as thin as possible to prevent the decay of the electromagnetic field transmitting through the layer, thus “borrowing” stronger plasmonic enhancement from the Au core. In 2002, utilizing the under potential deposition (UPD) and redox replacement methods, the well-controlled ultra-thin (as thin as a single atomic layer) and pinhole-free Pt-group metal layer-coated Au nanoparticle-dispersed electrodes were developed by Weaver et al., enabling a strong SERS signal on weakly SERS-active TM surfaces by “borrowing” the enhancement from the highly SERS-active Au nanoparticle core.74
In 2004, Tian et al. proposed an alternative “borrowing” SERS activity strategy through which monodispersed Au core-TM shell nanoparticles (Au@TM) can be directly prepared via wet-chemical methods.75–77 In these methods, the material of TM shells can be changed from Pt to Pd, Ni, Rh, Ru, and Co, respectively; meanwhile, the monodispersed Au@TM nanoparticles can be conveniently drop cast onto the electrode surface, thereby facilitating the generalized spectroelectrochemical study.71
The another major limitation of SERS is that SERS was predominantly used to study the molecules adsorbed on the nanostructure-based ill-defined substrate surfaces, such as roughened electrodes15–20,73 or metal colloids;69,78 however, it was challenging to carry out EC-SERS on well-defined or well-ordered electrode surfaces. Fueled by the development in nanoscience, there has been a surge in activity in EC-SERS since the 1990s. One of the most significant works for preparing SERS substrates in a well-controlled manner was proposed by Natan et al. in 1995.21 In this work, highly SERS-active monodisperse Au or Ag nanoparticles were assembled on organosilane-polymer-modified solid substrates. Furthermore, conducting materials, such as indium-doped SnO2 and Pt could be used as the substrate to immobilize the colloids, which provided a new way for applications in spectroelectrochemistry.
An alternative approach to prepare large-area and well-ordered SERS substrates is nanosphere lithography (NSL), which has been predominantly developed by the Van Duyne group67,79,80 and Bartlett group81,82 since the early 2000s. In a representative procedure of the NSL method, monodispersed silica or polystyrene (PS) nanospheres are first self-assembled on the surface of a conductive substrate to obtain a monolayer or multilayers of a two-dimensional (2D) colloidal crystal mask, and then it is used as a template for the preparation of SERS-active metal films over nanosphere (MFON) electrodes80 or periodic particle arrays (e.g., metal nanotriangles).83
In 1982, Suëtaka et al. carried out a detailed study of the SEIRAS effect, where the IR absorption enhancement of fingerprint vibration bands below 2000 cm−1 from p-nitrobenzoic acid (e.g., NO2 symmetric stretching at 1345 cm−1) was examined.85 They found that the absorption enhancement on a ∼5 nm thick Ag film evaporated onto a Ge prism was about an order of magnitude.85 In this work, a thin Ag film with an island structure is necessary for the absorption enhancement, and the Ag thickness plays a dominating role in the enhancement mechanism.85,86
Osawa et al.22,87–90 and Suëtaka et al.85,86 delved deeply into the mechanism of SEIRAS in the 1980s to 1990s. It is worth noting that SEIRAS did not receive much attention due to limited enhancement in 1980s, and since the 1990s, it has received renewed interest owing to its applications,87 and in particular, many efforts have been devoted to the preparation and characterization of SEIRAS-related nanoparticles or nanostructures.61,91,92 Nowadays, the SEIRAS effect is applicable to a variety of surface adsorbed species on various metal films, such as Ag, Au, Cu, Pt, Pd, In, Ni, Al, etc.52,61,93
After the pioneering work, SEIRAS has been developed to promote the application of IR spectroscopy to the electrochemical interface, leading to a notable growth of EC-SEIRAS studies since the mid-1990s.22–25,87 Significantly, along with the SEIRAS effect, the application of the ATR mode greatly facilitates the EC-IR measurement on the metal film electrodes, and the Osawa group pioneered the ATR-SEIRAS studies of molecular adsorption and reactions at the electrode–electrolyte interface in the 1990s (a representative ATR-cell is shown in Fig. 6A).23–25,61,87 To realize high-performance electrochemical ATR-SEIRAS, it is crucial to prepare suitable thin metal films on the optical window. Advanced by nanotechnology, the ATR-SEIRAS mode has been revived due to the development of fabrication techniques with which different kinds of metal materials, such as Ag, Au, Cu, Pt, Pd, In, Ni, and Al,52,61,93 can be more easily deposited in the form of thin films on window materials through which IR light propagates. Interestingly, as shown in Fig. 6B, vacuum evaporated gold films with ordered nano-island structures can provide suitable SEIRAS enhancement while still exhibiting the electrochemical response of Au(111) single crystal electrodes.23–25
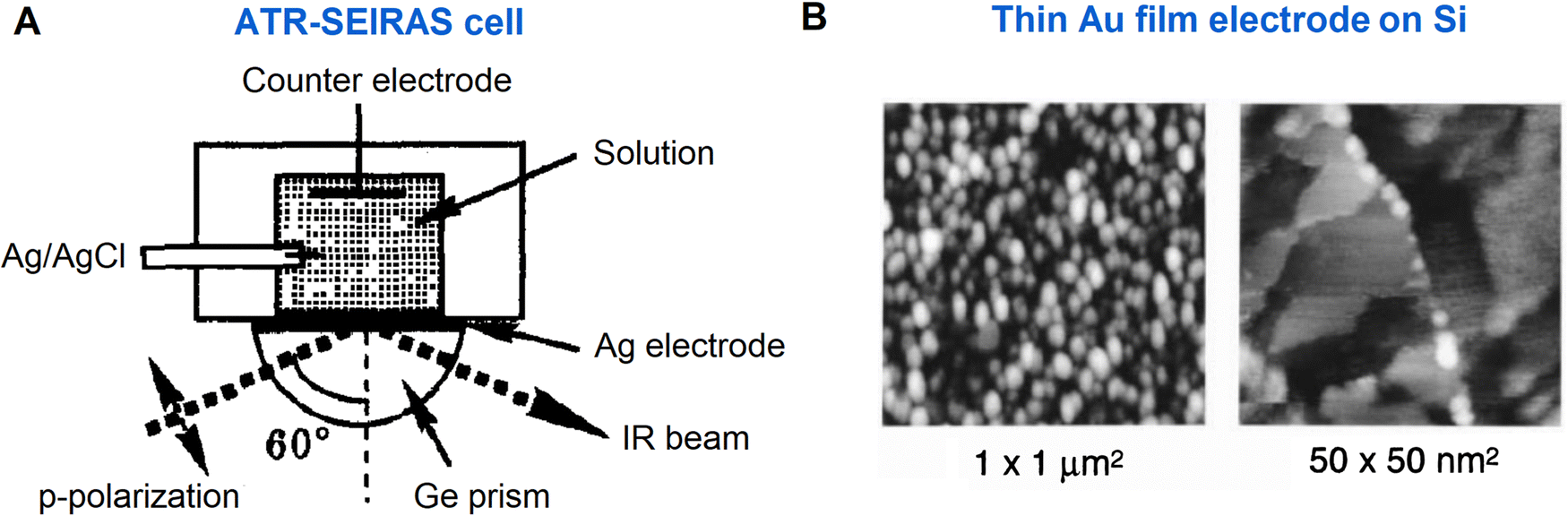 | ||
| Fig. 6 (A) Schematic of an ATR-SEIRAS cell. (B) STM images of a thin Au(111) film electrode for EC-SEIRAS experiments, where the electrode was obtained by vacuum-evaporation on a Si prism. Fig. 6A is reproduced with permission from ref. 22. Copyright 1993 Elsevier B.V. Fig. 6B is reproduced with permission from ref. 25. Copyright 1998 American Chemical Society. | ||
In addition to the SEIRAS effect, the presence of thin metal films introduced another interesting phenomenon at the electrochemical interface. An abnormal infrared effect (AIRE) was observed in the IR study of molecules adsorbed on the electrode surface in the presence of metal films, primarily by Sun et al.94,95 in the late 1990s. The IR spectra of CO and SCN− adsorbed on thin Pt, Pd, and Rh films deposited on glassy carbon (GC) electrodes showed an inversion in the sign of IR bands as well as an IR absorption enhancement. The AIRE was directly related to the reflection of IR light from the metal thin film electrode; however, it was not limited to the SPR-active coinage metals (i.e., Au, Ag, and Cu) and was not sensitive to surface roughness, which is not similar to the SEIRAS effect.94,95
Based on the surface enhancement effect, the vibrational spectroscopic signal of surface-adsorbed molecules can be enhanced up to several orders of magnitude (e.g., a Raman signal enhancement greater than 106-fold in EC-SERS), enabling the in situ vibrational detection of a (sub)monolayer of molecules on the surface.45,50 Thanks to the pioneering work and the excellent surface detection sensitivity, electrochemical surface-enhanced spectroscopy has become a significant experimental subfield in spectroelectrochemistry.5,45,61,101,102
2.3. Phase III
In spectroelectrochemistry, the use of structurally well-defined electrode surfaces, such as an atomically flat single-crystal electrode surface, can help to determine the coverage, arrangement, and orientation of adsorbed molecules, as well as the electrode surface state and optic field, respectively. Therefore, the substrates with well-defined surfaces greatly facilitate the systematic research studies on the electrochemical mechanism and its correlation with theory. In phase III of the development of electrochemical optical spectroscopy, as advanced by the rapid development of nanoscience since the 1990s, considerable efforts had been devoted to the realization of spectroelectrochemical measurements on well-defined surfaces.However, as compared to IR and SFG spectroscopy, Raman spectroscopic measurement on well-defined surfaces is more challenging due to the intrinsically extremely low optical cross-section. As shown in Fig. 4B, although the SERS effect greatly enhances the Raman signal of surface-adsorbed species, the electrode surface needs to be roughened or nanostructured to excite the localized surface plasmon resonance (LSPR) for high SERS activity.50 As a result, the Raman signal responses are inhomogeneously distributed over the electrode surface with ill-defined morphology. Furthermore, spatially-resolved SERS measurements on nanostructured electrode surfaces revealed that the plasmonic “hotspots” with large electromagnetic field enhancement can even provide site-specific redox potentials.105 Therefore, spectroelectrochemical studies on atomically flat single-crystal surfaces are important in surface electrochemistry, but highly challenging for conventional SERS. This intrinsic difficulty has led to a gradual decrease in the tidal wave of SERS research since the late 1980s.106 Different from the nanostructure-based SERS, an alternative way to enable the SERS effect on a smooth metal surface is to excite the surface plasmon polariton (SPP) through an ATR configuration (Fig. 4C), including the Otto configuration and Kretschmann configuration.60 In 1991, based on a Kretchmann configuration, the EC-Raman measurement was conducted on a well-defined Ag(111) film electrode epitaxially grown on mica.29 On the other hand, with an ATR-Otto configuration, the facet-dependent adsorption of pyridine was studied on three low-index Cu single-crystal electrode surfaces, i.e., Cu(111), (100), and (110), respectively.30 But there are still several limitations: first, the signal is still very weak, and a few systems with very strong Raman signals, e.g., para-nitrosodimethylaniline29 and pyridine,30 can be studied; second, related optical system configurations and electrochemical cells are complicated.
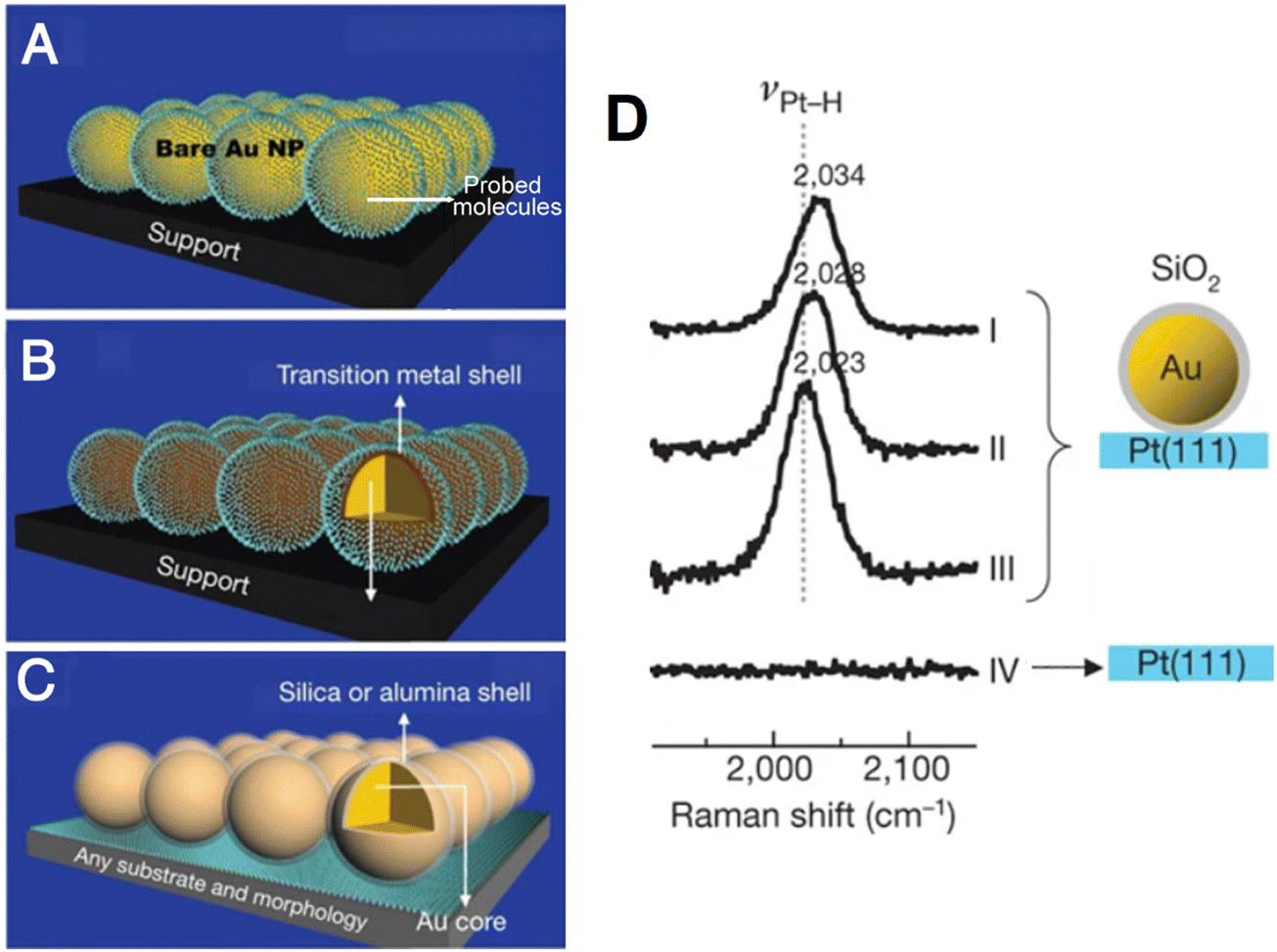 | ||
| Fig. 7 (A) and (B) Schematic of conventional SERS using Au nanoparticles (A) and transition metal shell coated Au nanoparticles (B). (C) Schematic of SHINERS. (D) Potential-dependent SHINERS spectra of hydrogen adsorbed on Pt(111) electrode surfaces. Reproduced with permission from ref. 33. Copyright 2010 Macmillan Publishers Limited. | ||
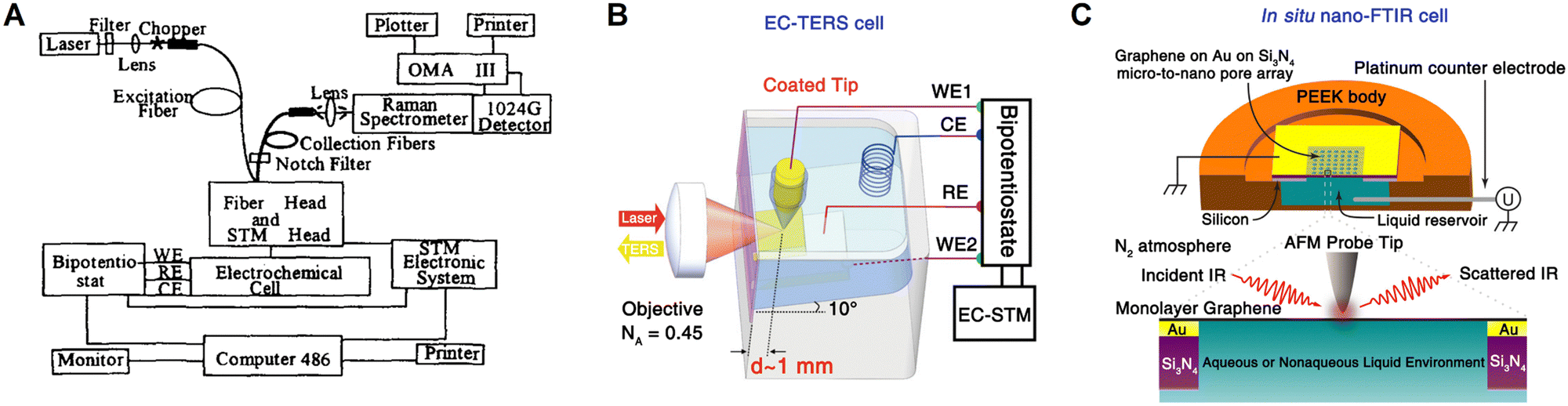 | ||
| Fig. 8 Schematics of the combined base of simultaneous STM and Raman spectroscopy (A), in situ cell of EC-TERS (B) and nano-FTIR (C), respectively. (A) is reproduced with permission from ref. 110. Copyright 1996 Elsevier. (B) is reproduced with permission from ref. 34. Copyright 2015 American Chemical Society. (C) is reproduced with permission from ref. 36. Copyright 2019 American Chemical Society. | ||
Based on SPM, tip-enhanced Raman spectroscopy (TERS) was independently invented by several groups in 2000,112–115 which can provide a Raman spectroscopic imaging resolution at a nanometer-scale.106 However, it is highly difficult to combine TERS and electrochemistry in the limited space of a SPM system. In 2015, Ren et al.34 and Van Duyne et al.35 independently reported electrochemical TERS (EC-TERS), ushering in a new era of nanospectroscopic research at electrochemical interfaces. On a Au(111) single-crystal surface, EC-TERS can reveal the potential-dependent protonation and deprotonation of the adsorbed aromatic molecules (a typical cell for EC-TERS is shown in Fig. 8B),34 and with the further improvement in sensitivity, this technique is currently able to probe a chemical reaction in solution with a spatial resolution of around 5 nm.116
On the other hand, the combination of SEIRAS effect and scanning near-field optical microscope has advanced nanoscale IR spectroscopy on a well-defined surface.117 In 2019, based on AFM, the first in situ electrochemical Fourier transform infrared nanospectroscopy (nano-FTIR, Fig. 8C) was realized on a graphene electrode surface, where the potential-dependent behavior of SO42− and NH4+ ions at the graphene/electrolyte interface was studied using nano-FTIR spectra.36 Furthermore, the combination of nano-FTIR and ATR-FTIR methods allows for the probing of subsurface depths at the nanoscale and microscale, respectively. This was achieved by applying a custom-built solid polymer electrolyte cell with a single layer of graphene as the working electrode, which is IR transparent.118 As a result, it was capable of simultaneously in situ studying both the electrochemical interface together with the bulk phase of solid polymer electrolyte.
Furthermore, scanning electrochemical microscopy (SECM) is a unique method to investigate the nanoscale information of structure–activity on electrode surfaces, in which an ultramicroelectrode (UME) tip is positioned near the substrate surface, which drives a redox reaction that records the diffusion-limited currents generated during scanning of the substrate surface, thus offering information on the surface morphology and local interfacial electrochemical activity.119 Combining SECM with optical spectroscopy enables the simultaneous in situ studies of the electrochemical processes and molecular vibrational signatures in local regions,120,121i.e., to study local surface modifications with self-assembled monolayers122 and the surface pH perturbations induced by the UME tip-driven hydrogen evolution reaction (HER).123
2.4. Phase IV
Unlike the concepts of ex situ and in situ, which were proposed in early times and have been applied ever since, the term operando originates from the Latin gerund, and it was introduced in 2002 as a tentative name for “real” reaction in situ spectroscopy.124 Both the terms in situ and operando originated from heterogeneous catalysis research,125 while the operando methodology was first used for the combination of in situ spectroscopy characterization and simultaneous evaluation of both the structure and activity of catalysts under working conditions.124,126 Figuratively speaking, ex situ techniques study a dead fish out of water (or a sliced fish), in situ techniques study a live fish restricted to a small fishbowl, while operando techniques can comprehensively study a fish swimming freely in the ocean. In contrast to ex situ techniques normally examining the sample outside the electrochemical working environment, in situ and operando techniques can study the surface and phase changes in the electrode bulk, and the electrochemically generated reaction intermediate. Normally, in situ measurements are suitable for the fundamental study on a well-defined or model electrode surface, whereas operando characterization emphasizes on the measurements in a practical/commercial device or an architecture highly resembling the practical one.101,127–129
3. Advances in in situ and operando applications
Nowadays, with a growing impact on electrochemical fields as diverse as electrochemical energy storage and conversion, electrolysis, electroplating, corrosion, electrochemical synthesis, biological electron-transfer processes, etc., in situ or operando characterization methods with desirable detection sensitivity and resolutions to understand the structure and dynamics of electrode–electrolyte interfaces/interphases have become increasingly significant.5,52,61,101 In the following, we will focus on the recent advances in in situ and operando applications of electrochemical vibrational spectroscopy.3.1 Electrical double layer
Probing the composition and structure of the electrical double layer is highly significant in electrochemical studies. Interfacial water has an important impact on the physical and chemical properties of the electrical double layer, and the EC-Raman investigation of interfacial water was first carried out by Fleischmann et al. in 1981.133 However, the conventional Raman spectroscopy is incapable of probing interfacial water on single-crystal electrodes with atomically flat surfaces, which limits the understanding of the atomic structure of interfacial water. As a key figure of merit, SHINs can be used as Raman signal amplifiers to obtain vibrational spectra on the surface of single-crystal electrodes without interference from the bulk electrolyte (Fig. 9A–C show the working principle and surface Raman signal enhancement in SHINERS during the study of interfacial water134).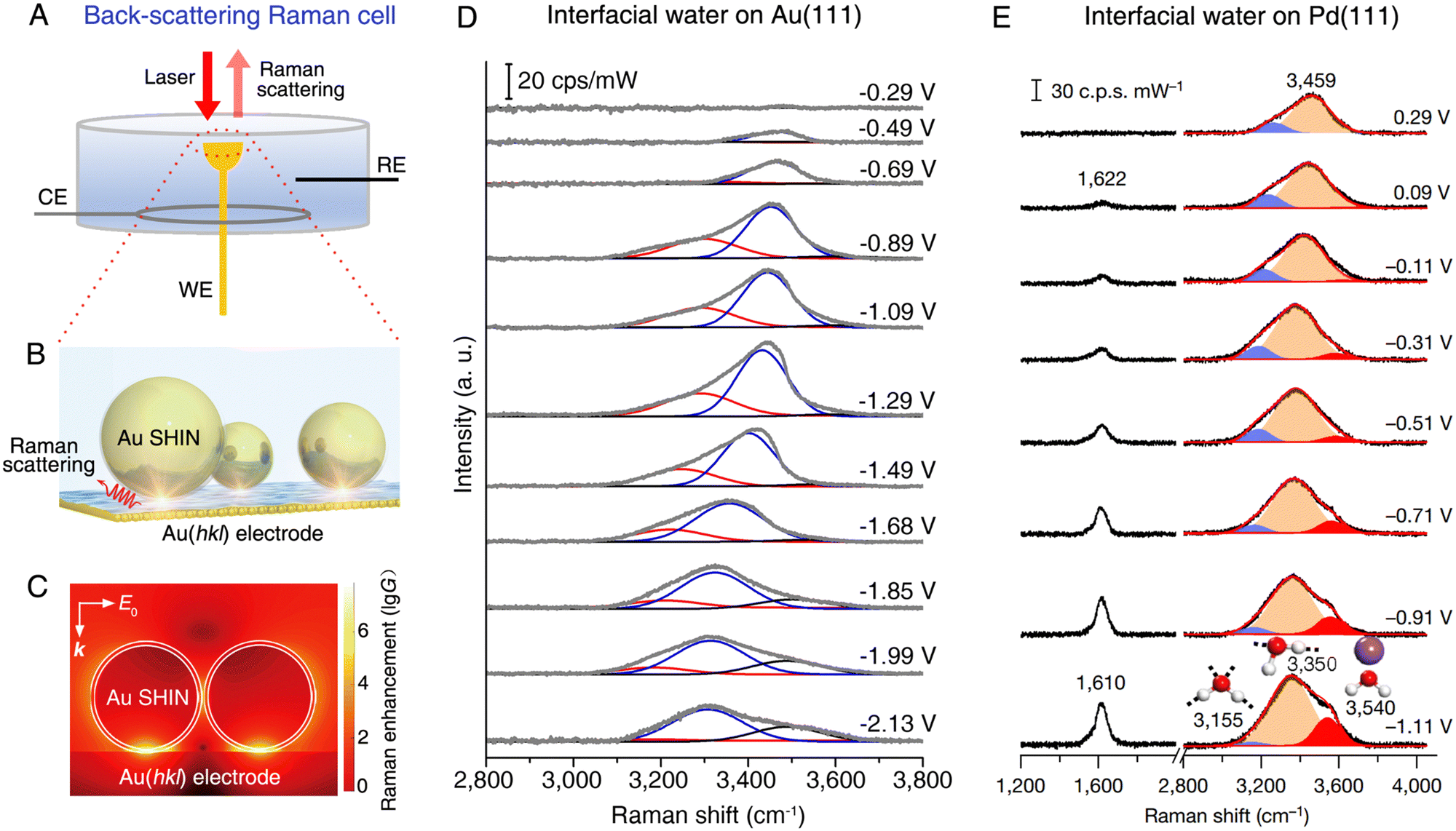 | ||
| Fig. 9 (A) Schematic of a back-scattering Raman cell for the SHINERS method on a single-crystal electrode. (B) Schematic of EC-SHINERS for interfacial water measurement. (C) Finite-element-method simulation of Raman signal enhancement on a Au electrode surface in EC-SHINERS. (D) In situ EC-SHINERS spectra of interfacial water on the Au(111) electrode in a 0.1 M Na2SO4 solution. The potential is referenced to potential of zero charge. (E) In situ EC-SHINERS spectra of interfacial water on the Pd(111) electrode in a 0.1 M NaClO4 solution (pH 11). The Pd(111) electrode is obtained by coating a Pd monolayer onto a Au(111) electrode surface and the potential is referenced to a reversible hydrogen electrode. (B)–(D) reproduced with permission from ref. 134. Copyright 2019 Springer Nature. (E), Reproduced with permission from ref. 135. Copyright 2021 Springer Nature. | ||
As shown in Fig. 9D and E, the SHINERS method is suitable for the in situ probing of the adsorption of interfacial water on Au(111)134,136 and Pd(111)135 electrodes, respectively. SHINERS spectra of interfacial water, particularly the OH stretching modes in the spectral range from 3000 to 3800 cm−1, show distinct potential dependence on both Au(111) (Fig. 9D) and Pd(111) (Fig. 9E) electrodes, indicating the changes in the structure of interfacial water, i.e., the orientation of interfacial water and related hydrogen-bonding networks. On the Au(111) electrode, the Stark tuning rate can be derived from the Raman frequency shift of the interfacial water under bias conditions, with which it is capable of revealing two structural transitions of interfacial water, i.e., from a “parallel” structure to a “one H-down” structure and then to a “two-H-down” structure, as the electrode potential was negatively scanned from potential of zero charge (PZC) toward the hydrogen evolution reaction (HER) region.134 On the surface of the Pd(111) electrode, the interpretations on the OH stretching mode indicate that interfacial water consists of hydrated cations and hydrogen-bonded water. Meanwhile, the interfacial water structurally transformed from a randomly distributed state to an ordered one in the HER region owing to the cation cooperation and the potential changes.135
Vibrational IR-visible SFG spectroscopy is a unique spectroscopic method for studying water molecules at the interface, where the OH frequency and intensity reveal the hydrogen bonding strength and ordering of water molecules,137 enabling the sensitive nonlinear spectroelectrochemical study of the structures of interfacial water and the electrode double layer.14,53,102,138–142 As shown in Fig. 10A and B, Benderskii et al. investigated the IR-visible SFG signal of D2O on a monolayer graphene electrode surface, and they found a pronounced asymmetry in the response of OD stretching to the positive and negative potentials.140 To detect the charge-neutral point on the graphene electrode, the minimum of potential-dependent G-band Raman frequency of graphene was measured and it was found at ∼ +0.1 V (vs. Ag/AgCl). For the spectra obtained at potentials more negative than −1.6 V, only a relatively narrow peak at 2697 cm−1 was found, which was assigned to the free OD stretching mode of D2O in the topmost monolayer (Fig. 10A). However, when the potential was set to positive values above −1.0 V, only the characteristic broad hydrogen-bonded bands from ∼2300 to ∼2700 cm−1 were observed and the narrow free-OD band disappeared.
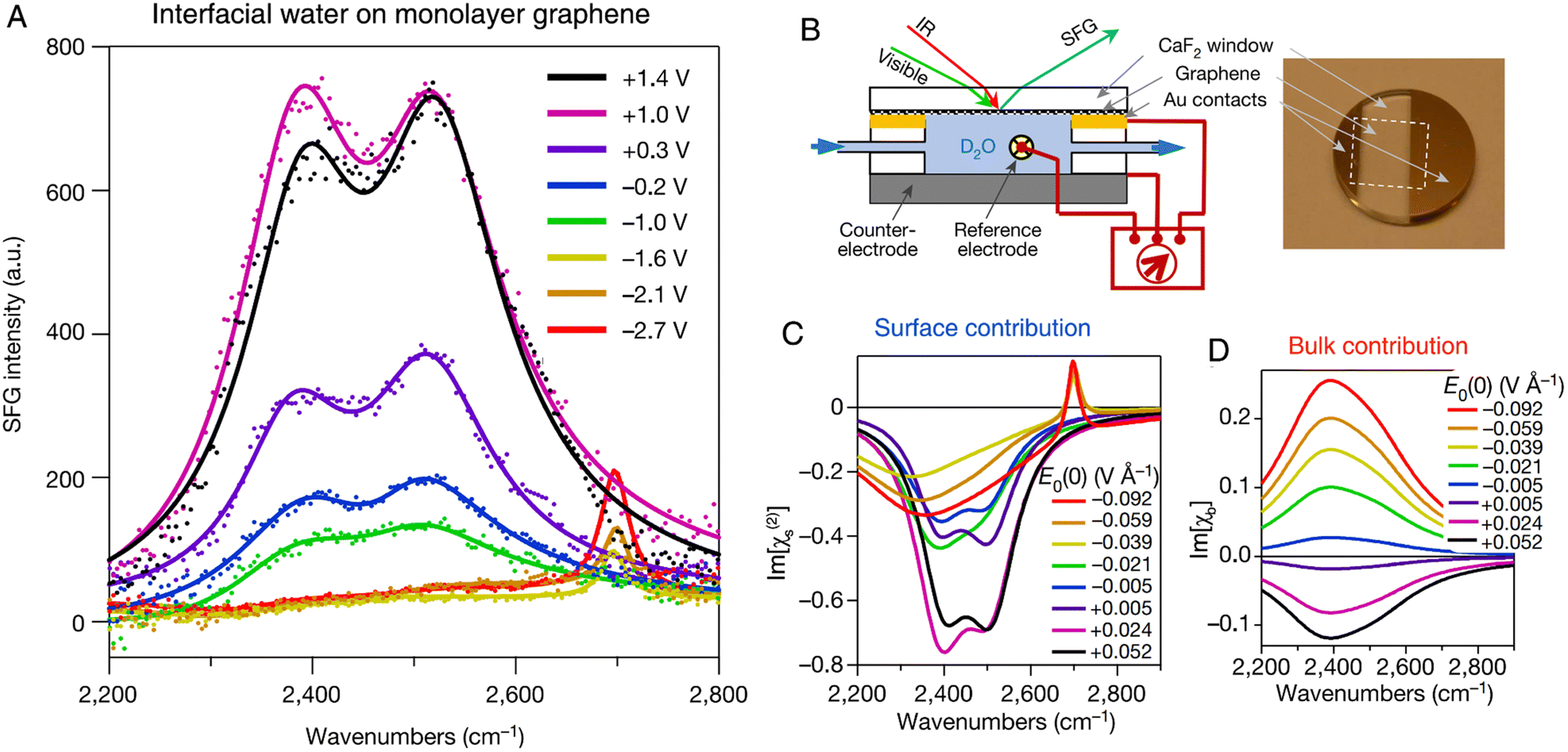 | ||
| Fig. 10 (A) In situ SFG spectra of D2O at the graphene electrode (no electrolyte is added to D2O). The potential is referenced to a Ag/AgCl electrode. (B) Schematic of an electrochemical cell and the corresponding photograph of a graphene electrode for SFG experiments. (C) and (D) The extracted surface (C) and bulk (D) contribution from SFG spectra as a function of the applied field. Reproduced with permission from ref. 140. Copyright 2021 Springer Nature. | ||
SFG is nominally a second-order nonlinear process (the second-order susceptibility tensor of the medium can be expressed as χ(2)), but the static electric field at the charged graphene–D2O interface gave rise to a third-order contribution (i.e., the χ(3) term). In contrast to the χ(2) term, which originates from only a few monolayers of water on surface, the χ(3) term may contain the contribution from the region through which the static electric field penetrates, i.e., the Debye screening length (in the order of 1 μm in this work). By comparing the imaginary parts of the surface (Im[χ(2)], Fig. 10C) and bulk (Im[χb], Fig. 10D) contributions from the spectra, the extracted potential-dependent Im[χ(2)] does not follow the linear response as that of Im[χb], exhibiting significant asymmetry at positive and negative potentials. The unusual nonlinear response of interfacial water to the applied electric field suggested that treating interfacial water as a simple linear dielectric medium should be carefully considered and further examined.
3.2. Electrocatalysis
Vibrational spectroscopy (such as IR and Raman spectroscopy) exhibits an energy resolution of around two orders of magnitudes higher than conventional electrochemical techniques; therefore, it can be used to carefully study the molecular structure of adsorbed molecules or oxidation (reduction) of molecules, enabling a detailed analysis of the reaction mechanism in electrocatalysis. For instance, in the spectroelectrochemical study of electrocatalytic reactions, IR absorption spectroscopy has been combined with cyclic voltammetry (CV) in the in situ investigation of direct oxidation of methanol on the Pt electrode.143 In the CVs of both the positive and negative potential sweeps of the polycrystalline Pt electrode, the dominant and shoulder peaks were almost merged. However, in the corresponding IR absorption spectra with a spectral resolution at 4 cm−1, the linearly and bridged-bonded CO molecules were clearly differentiated at the beginning of potential scan. It was clearly demonstrated that vibrational spectroscopy is beneficial for a higher energy resolution to identify the adsorbed species as compared to the conventional electrochemical technique.143 With this intrinsic advantage, IR spectroscopy has received great attention in the study of significant electrochemical CO2 reduction reactions (CO2RR), i.e., the CO2RR on Cu electrocatalysts, where Cu is one excellent electrocatalyst material for CO2RR and also a suitable substrate for high SEIRAS activity.61,144 Based on in situ ATR-SEIRAS, Shao et al. studied the role of a bicarbonate-based electrolyte, a widely used aqueous electrolyte, in the CO2RR on a Cu thin film electrode supported on a Au substrate.145 As the potential was scanned from +0.3 toward −1.3 V in CO2-saturated KHCO3, they observed several distinct vibrational features in the spectral region from 1300 to 2400 cm−1, where the depletion of CO2 (i.e., the IR absorption band at ∼2340 cm−1) and increase of adsorbed CO (i.e., the broad band at ∼2080 cm−1) become visible at ∼ −0.4 V vs. RHE, close to the onset of the reduction current in the linear scanning voltammogram at ∼−0.3 V (Fig. 11A and B). However, they found that the surface-adsorbed CO species could be detected in the absence of CO2 purge, i.e., the bands around 2020 and 2070 cm−1 originating from the different adsorption configuration of CO on Cu were still observable in the control experiment in Ar-saturated 0.1 M KHCO3 (Fig. 11C). This observation indicated that bicarbonate anions were the main CO2 source in the CO2RR process on the Cu electrode. Furthermore, they carried out an isotopic experiment by purging the unlabeled CO2 to a labeled KH13CO3 electrolyte and measured the time-dependent SEIRAS spectra at −0.6 V. As shown in Fig. 11D, a depletion of 13CO2 in the solution was observed at 2277 cm−1 in the first 3s, followed by the simultaneous appearance of adsorbed 13CO at ∼1990 cm−1, which offered direct spectroscopic evidence that the CO2 in equilibrium with CO3− was the main CO2 source.145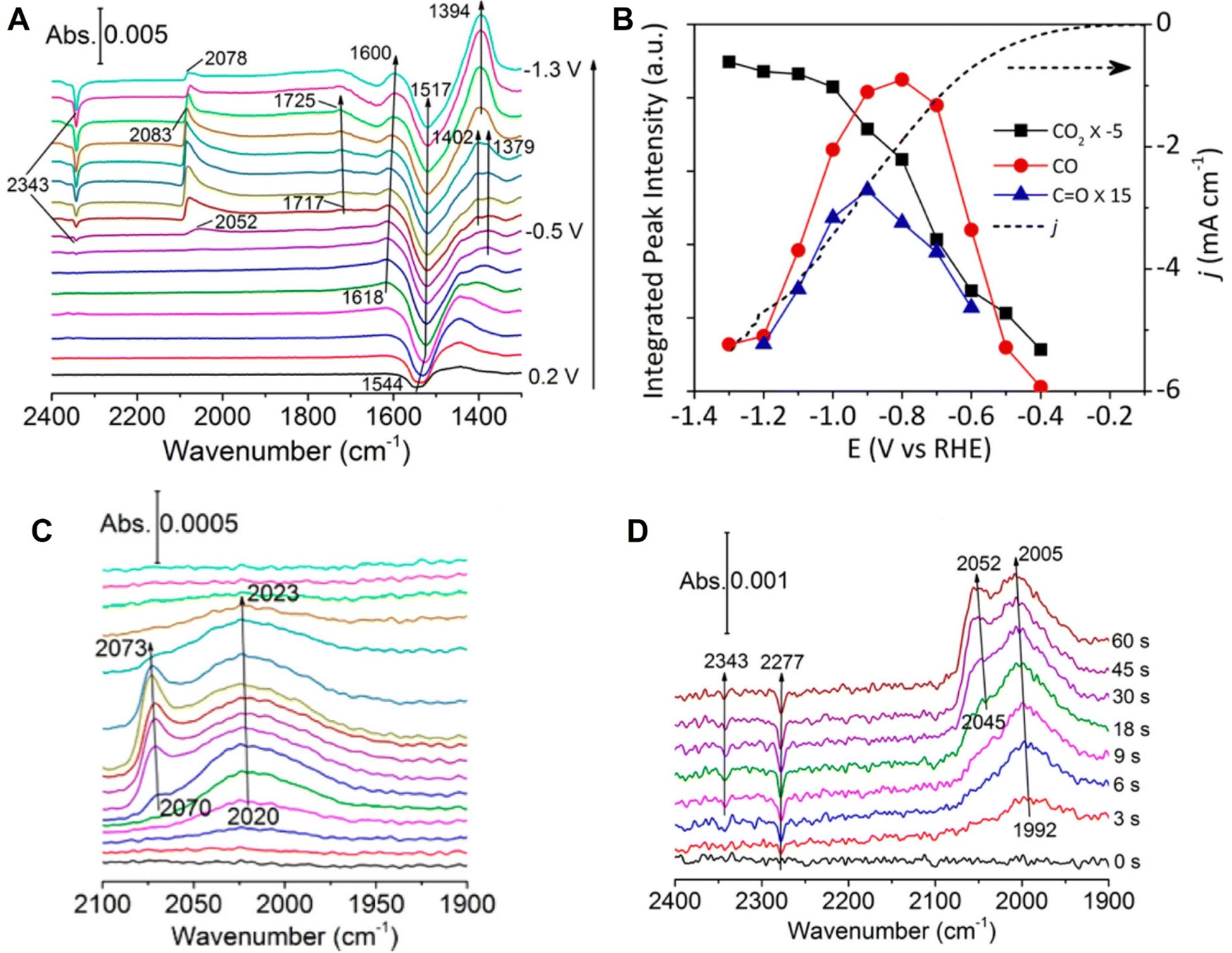 | ||
| Fig. 11 (A) In situ ATR-SEIRAS spectra on a Cu thin film electrode in a CO2-saturated 0.1 MKHCO3 electrolyte. (B) Corresponding cyclic voltammogram curve (dotted line) and peak intensities in (A). (C) ATR-SEIRAS spectra measured in an Ar-saturated 0.1 MKHCO3 electrolyte. (D) Time-dependent ATR-SEIRAS on the Cu thin film electrode after stepping from 0.2 to −0.6 V vs. RHE in a CO2 purged 0.1 M KH13CO3 electrolyte. In (A) and (C), spectrum obtained at 0.3 V was used as a reference. In (D), the reference spectrum was obtained at 0.2 V. Reproduced with permission from ref. 145. Copyright 2017 American Chemical Society. | ||
The understanding of the relationships between the surface structure and catalytic activity can greatly benefit the design and synthesis of high-performance electrocatalysts. Pt electrocatalysts play a significant role in the oxygen reduction reaction (ORR); however, the related Raman spectroscopic studies are limited to nanostructured Pt electrode surfaces; therefore they are unable to investigate the structure–activity relationships on structurally well-defined single-crystal electrodes.71 To unravel the reaction mechanism of ORR on a single-crystal Pt(hkl) electrode surface, Li et al. used in situ EC-SHINERS to identify the intermediates involved in the ORR in both acidic environments, in which they found different pathway mechanisms of the ORR process on Pt(111), Pt(100), and Pt(110) electrode surfaces.146 As shown in Fig. 12, they spread SHINs (pinhole-free Au@SiO2 nanoparticles) onto the Pt single-crystal electrode surface to enhance the Raman signal from the interfacial region (the related experimental configuration is similar to that illustrated in Fig. 9). In a 0.1 M HClO4 solution, in situ EC-SHINERS spectra were recorded on the Pt(111) electrode from 1.1 to 0.5 V vs. RHE, in which a Raman peak of 732 cm−1 appeared at 0.8 V (Fig. 12A). Based on the deuterium isotopic substitution experiment and density functional theory (DFT) calculations, this peak was assigned to the O–O stretching mode of HO2* species on Pt(111). In addition, as revealed in Fig. 12B, the Raman peak corresponding to HO2* species appeared at a potential very close to the potential at which the ORR process achieved the limiting diffusion current (∼0.7 V), indicating a crucial role of HO2* species in the ORR under acid conditions. In sharp contrast, the important HO2* species was absent on Pt(100) and Pt(110) electrode surfaces; however, two Raman peaks at 1030 and 1080 cm−1 were observed, which were attributed to the symmetric stretching mode of ClO3 in HClO4 and Pt-OH bending mode OH* species. Apparently, the EC-SHINERS method provided direct spectroscopic evidence of the different ORR mechanisms on three Pt(hkl) electrodes in an acidic solution, that is, the ORR on the Pt(111) surface by the generation of HO2*, while on Pt(100) and Pt(110) electrode surfaces by the formation of OH*.
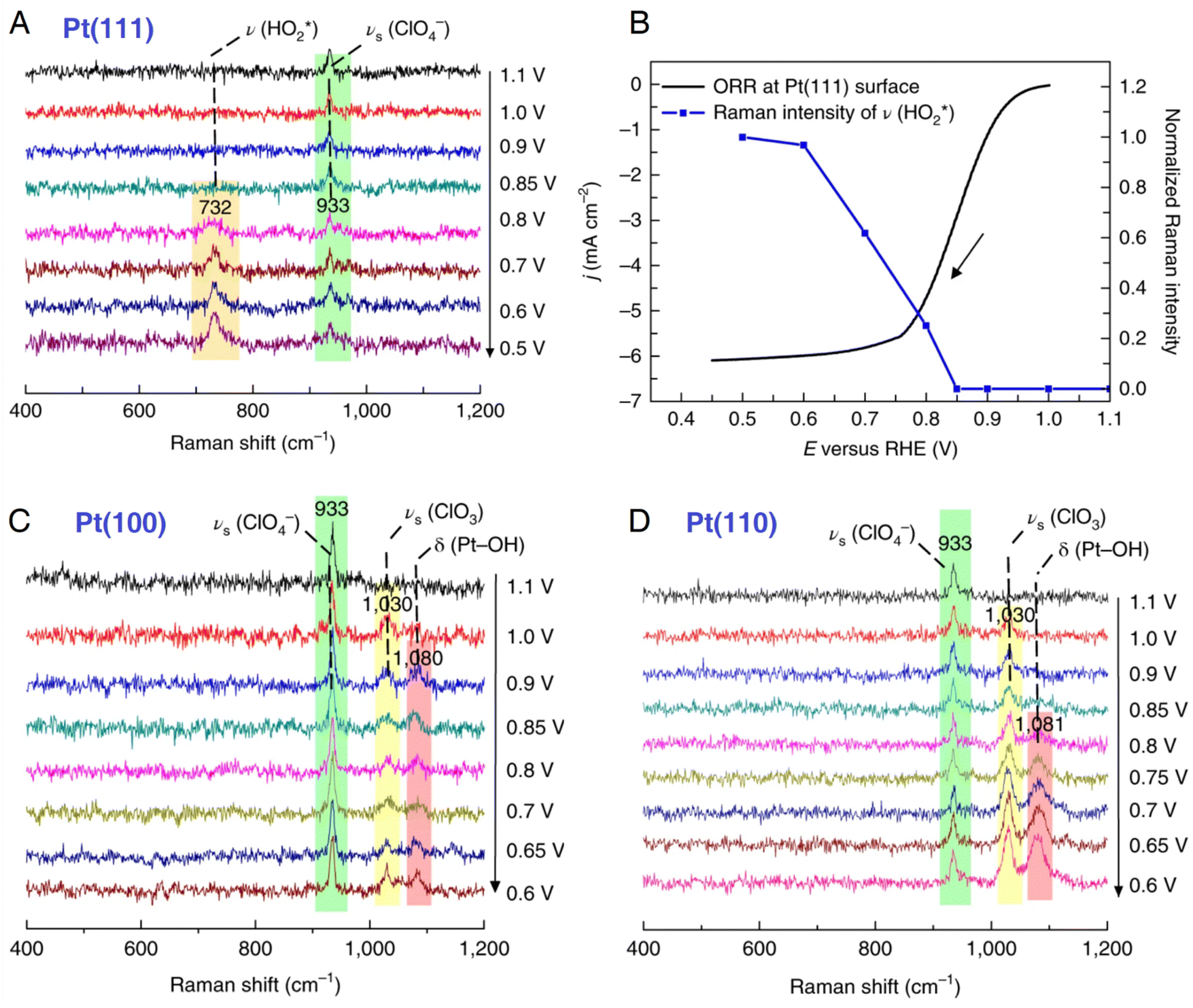 | ||
| Fig. 12 (A) In situ EC-SHINERS spectra on the Pt(111) electrode in O2-saturated 0.1 M HClO4. (B) Potential-dependent Raman intensity of HO2* species on Pt(111) versus ORR current. ORR current was obtained in a O2-saturated 0.1 M HClO4 solution using a rotating disk electrode Pt(111) electrode with a rotation rate of 1600 rpm. and a scan rate of 50 mV s−1. (C) and (D) In situ EC-SHINERS spectra on Pt(100) (C) and Pt(110) (D) electrodes in O2-saturatred 0.1 M HClO4. Reproduced with permission from ref. 146. Copyright 2018 Springer Nature. | ||
3.3 Rechargeable batteries
Recently, operando characterization methods have received great attention due to their ability to probe the structural, chemical, and mechanical changes in electrochemical energy devices under their real working conditions.101,127,147,148 In rechargeable batteries, operando methods benefit largely from the collaboratively combination of diverse characterization techniques to understand the complex interrelated phenomena, such as the relationships between structural transformation and chemical reaction and performance of device.149 As a representative example shown in Fig. 13, Zhou et al. carried out an operando spectroelectrochemical study of the anionic redox process in Li-ion batteries by correlating vibrational Raman spectroscopy and differential electrochemical mass spectrometry (DEMS).150 The exploration of oxygen-related anionic redox (peroxo-like (O2)n− species) activity in Li-ion batteries opens up a new way for boosting the capacity limit of electrode materials and thus has attracted much interest, but it also calls for advanced operando characterization techniques to probe the underlying nature of the anionic redox activity.151,152 Vibrational Raman spectroscopy is a powerful characterization method of probing oxygen-related species at electrochemical interfaces.146,153–155 As shown in Fig. 13A, a Li-rich Ni/Co-free O3-type Li0.6[Li0.2Mn0.8]O2 was used as the cathode, and assembled into an operando Raman cell with Li foil serving as both the reference and counter electrode. During the first and second charge–discharge cycles, a vibrational feature appeared in the region from 795 to 845 cm−1 that was attributed to the O–O stretching mode of peroxo species (Fig. 13B), indicating reversible redox properties. However, the peak position shifted toward a higher wavenumber in the second cycle, which suggested a longer O–O distance of peroxo species that would cause the generation of superoxo species (the Raman peak at 1104 cm−1 at the end of the first cycle) and gaseous O2 (a O2 evolution rate of 254 μmol g−1 as quantified by DEMS). Furthermore, the electrochemical decomposition of electrolyte at high voltage and the nucleophilic attack on propylene carbonate by superoxo species could be detected by operando Raman spectroscopy, as indicated by the appearance of a Raman peak around 1080 cm−1 revealing the formation of Li2CO3. Therefore, by combining operando Raman spectroscopy and DEMS, relatively reversible oxygen-related redox and electrolyte decomposition processes were monitored in real-time, allowing a better understanding of anionic redox activity in high-capacity electrode materials.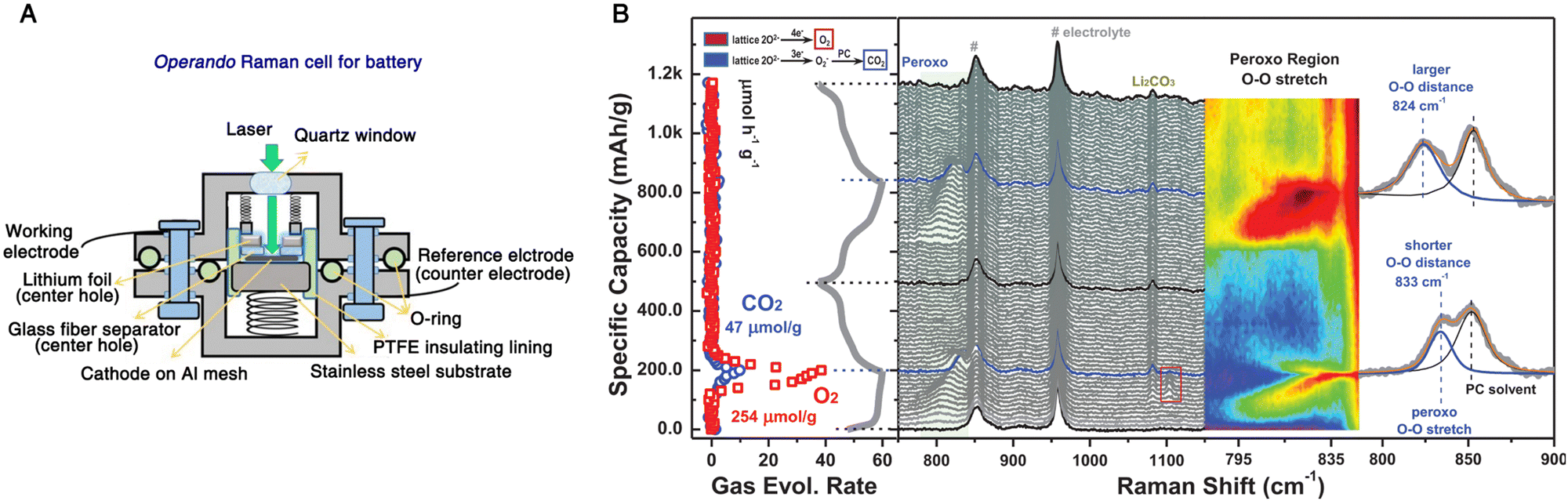 | ||
| Fig. 13 (A) Schematic of an operando Raman cell for Li-ion batteries. (B) Operando Raman spectra and DEMS of the gas evolution rate during the first and second cycle of a Li0.6[Li0.2Mn0.8]O2 cathode in an electrolyte consisting of 1 M LiClO4 in propylene carbonate. The peaks marked by the number sign at ∼850 and 920 cm−1 originate from the electrolyte. Fig. 13A is reproduced with permission from ref. 153. Copyright 2018 the Royal Society of Chemistry. Fig. 13B is reproduced with permission from ref. 150. Copyright 2020 John Wiley and Sons. | ||
Probing the chemical composition and structures of electrode-electrolyte interfaces (EEIs) or solid-electrolyte interphases (SEIs) is one of the most significant applications of electrochemical vibrational spectroscopy in Li-ion chemistry. Although the thickness of EEIs is only on the nanometer scale, its formation and evolution play a crucial role in achieving a Li-ion battery with high performance and a long cycle life.156 By coupling with electrochemical operation, optical spectroscopy methods with high surface sensitivity, such as nanostructure- and ATR-based vibrational spectroscopy56,129 and surface plasmon resonance spectroscopy,157,158 provide prominent opportunities for in situ/operando tracking of the chemical and structural evolution of electrode/electrolyte interfaces or interphases. Shao-Horn et al. used in situ FTIR spectroscopy to study the formation of EEIs, that is, the oxidation of carbonate-based electrolyte on a Ni-rich LiNi0.8Co0.1Mn0.1O2 (NMC811) electrode surface.159 Usually, a thin film electrode directly deposited on a prism is preferred for ATR-IR measurements, but it is challenging to prepare a NMC thin film electrode in this configuration. Therefore, in this work, a composite NMC/glassy fiber was used as a working electrode (positive electrode) that was placed downward, facing the Pt thin film supported by a CaF2 prism; thereby the reaction intermediates generated from the NMC electrode could be detected through IR absorption in an internal reflection geometry (Fig. 14A). As shown by the in situ FTIR difference spectra on an NMC811 electrode charged to 4.4 V in ethylene carbonate with 1.5 M LiPF6 (Fig. 14B), several bands appeared in the spectral region from ∼1750 to 1830 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching region) at 3.8 V. These peaks were assigned to vinylene carbonate (∼1830 cm−1), ethylene carbonate (∼1800 cm−1), Li+-associated ethylene carbonate (∼1770 cm−1), and oligomers with ethylene carbonate-like rings (∼1760 cm−1), respectively. It is noted that vinylene carbonate is one of decomposed products of ethylene carbonate, that is, vinylene carbonate results from the removal of two hydrogens from ethylene carbonate, forming a CC bond in the ring. Meanwhile, the oligomer possibly resulted from the opening and polymerization of ethylene carbonate. By potentiostatically holding at 4.4 V for a period of time (0 to 60 min), no obvious changes in the peak intensities were observed (Fig. 14C). In contrast, in the time-dependent measurement at open circuit potential (OCP) following the holding experiment at 4.4 V, the peak intensities of ethylene carbonate, vinylene carbonate, and oligomers decreased quickly, indicating these species diffused away from the EEI at OCP (Fig. 14D). However, a deconvoluted peak at 1813 cm−1 hardly changed with increasing time, which was assigned to dehydrogenated ethylene carbonate. This observation suggested that dehydrogenated ethylene carbonate adsorbed on the oxide surface of the NMC electrode, possibly on the surface oxygen through a C–Osurface bond generated during the dehydrogenation reaction of ethylene carbonate. As NMC811 was galvanostatically charged to 4.4 V, potentiostatically held at 4.4 V, and rested at OCP, the potential-dependent intensity trends of peaks were consistent with the corresponding voltage profile (Fig. 14E), showing that in situ FTIR spectroscopy is sufficient in revealing the chemical and structural changes in the EEI, thus giving insights into the strategies to develop stable and high-performance Li-ion batteries.
O stretching region) at 3.8 V. These peaks were assigned to vinylene carbonate (∼1830 cm−1), ethylene carbonate (∼1800 cm−1), Li+-associated ethylene carbonate (∼1770 cm−1), and oligomers with ethylene carbonate-like rings (∼1760 cm−1), respectively. It is noted that vinylene carbonate is one of decomposed products of ethylene carbonate, that is, vinylene carbonate results from the removal of two hydrogens from ethylene carbonate, forming a CC bond in the ring. Meanwhile, the oligomer possibly resulted from the opening and polymerization of ethylene carbonate. By potentiostatically holding at 4.4 V for a period of time (0 to 60 min), no obvious changes in the peak intensities were observed (Fig. 14C). In contrast, in the time-dependent measurement at open circuit potential (OCP) following the holding experiment at 4.4 V, the peak intensities of ethylene carbonate, vinylene carbonate, and oligomers decreased quickly, indicating these species diffused away from the EEI at OCP (Fig. 14D). However, a deconvoluted peak at 1813 cm−1 hardly changed with increasing time, which was assigned to dehydrogenated ethylene carbonate. This observation suggested that dehydrogenated ethylene carbonate adsorbed on the oxide surface of the NMC electrode, possibly on the surface oxygen through a C–Osurface bond generated during the dehydrogenation reaction of ethylene carbonate. As NMC811 was galvanostatically charged to 4.4 V, potentiostatically held at 4.4 V, and rested at OCP, the potential-dependent intensity trends of peaks were consistent with the corresponding voltage profile (Fig. 14E), showing that in situ FTIR spectroscopy is sufficient in revealing the chemical and structural changes in the EEI, thus giving insights into the strategies to develop stable and high-performance Li-ion batteries.
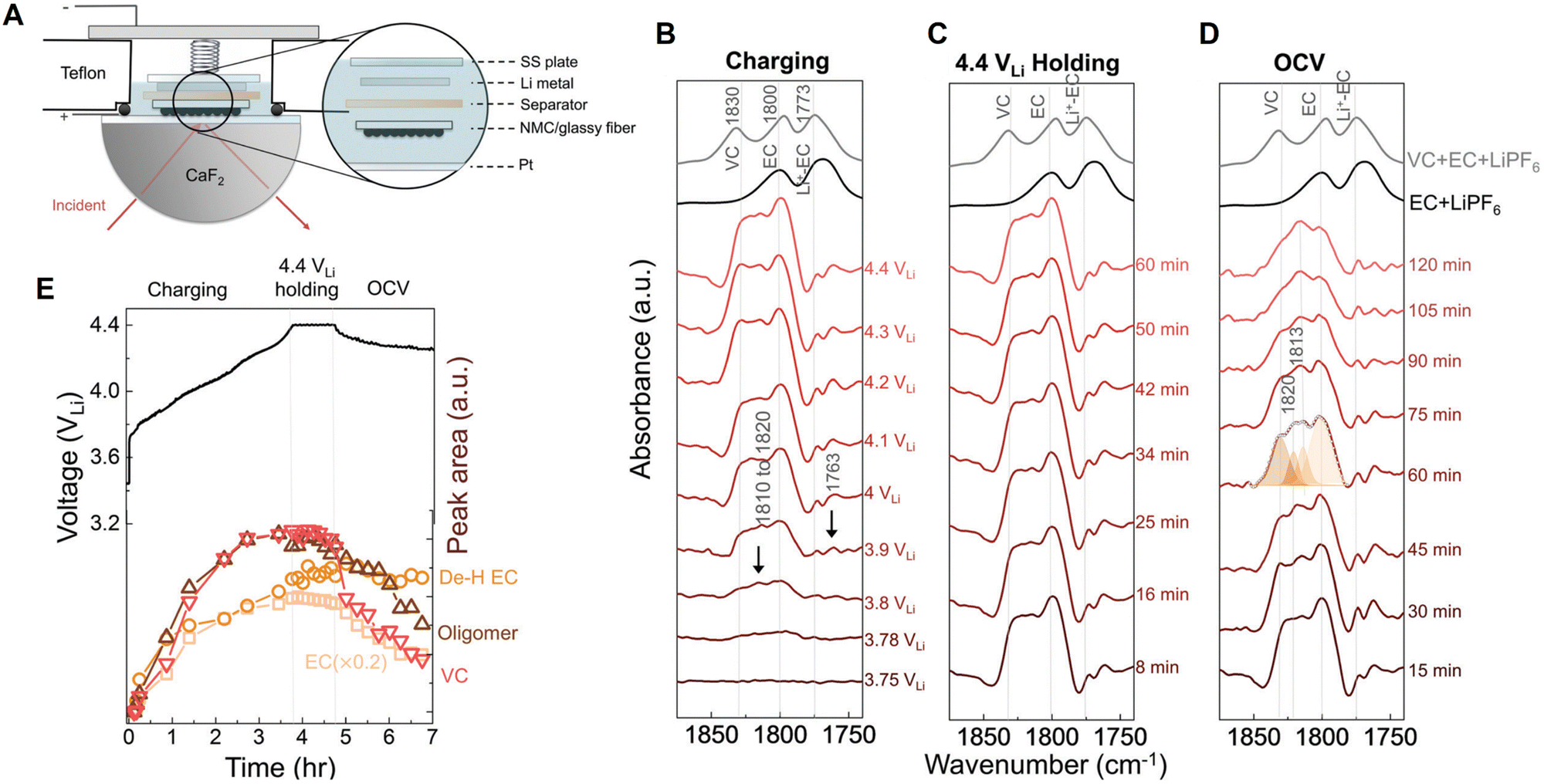 | ||
| Fig. 14 (A) Schematic of an in situ FTIR cell. (B)–(D) In situ FTIR difference spectra on the LiNi0.8Co0.1Mn0.1O2 electrode during charging to 4.4 V (B), potentiostatic holding at 4.4 V (C), resting at OCV (D) in ethylene carbonate with 1.5 M LiPF6. (E) Corresponding electrochemical curve of the LiNi0.8Co0.1Mn0.1O2 electrode galvanostatically charged to 4.4 V, held at 4.4 V, and then rested at OCV; and deconvoluted IR intensity of peaks at 1800 cm−1 (ethylene carbonate), ∼1813 cm−1 (dehydrogenated ethylene carbonate, De–H EC), ∼1820 cm−1 (oligomers), and 1830 cm−1 (vinylene carbonate), respectively. OCV is an abbreviation for open circuit voltage, and the potential was referred to Li+/Li. Reproduced with permission from ref. 159. Copyright 2020 the Royal Society of Chemistry. | ||
Solid-state batteries are anticipated to be an alternative to conventional lithium-ion batteries since they replace the flammable liquid electrolyte with a solid electrolyte, allowing for tighter packing of battery components, while also providing safety advantages.160,161 However, as the active electrode materials are normally opaque, the light beam is unable to penetrate through the closely packed solid layers, making it difficult to perform operando spectroscopic experiments in a traditional manner, e.g., focusing the incident beam perpendicularly through the liquid electrolyte bulk to the electrode surface. As shown in Fig. 15, in the spectroelectrochemical study of a solid-state lithium–sulfur (Li–S) battery, the operando Raman cell with an open side was placed inside an inert argon environment to enable the spectroscopic detection of each battery component from the cross-sectional direction.162 As revealed by the operando Raman spectra, sulfur (S8, of which the Raman peaks are at 158, 220, and 473 cm−1, respectively) underwent gradual reduction but was not completely reduced during the discharge process, while an intermediate species, that is, Li2S2 (438 cm−1) appeared at the half charge state (2.359 V) and then disappeared at the full charge state (2.789 V), indicating that Li2S was first oxidized to Li2S2, and then to S8 during the charging process.162 With a side opening cell, this method is suitable for the characterization of a solid-state electrolyte in operando, and thus to understand the emerging solid–solid electrochemical interfaces/interphases.
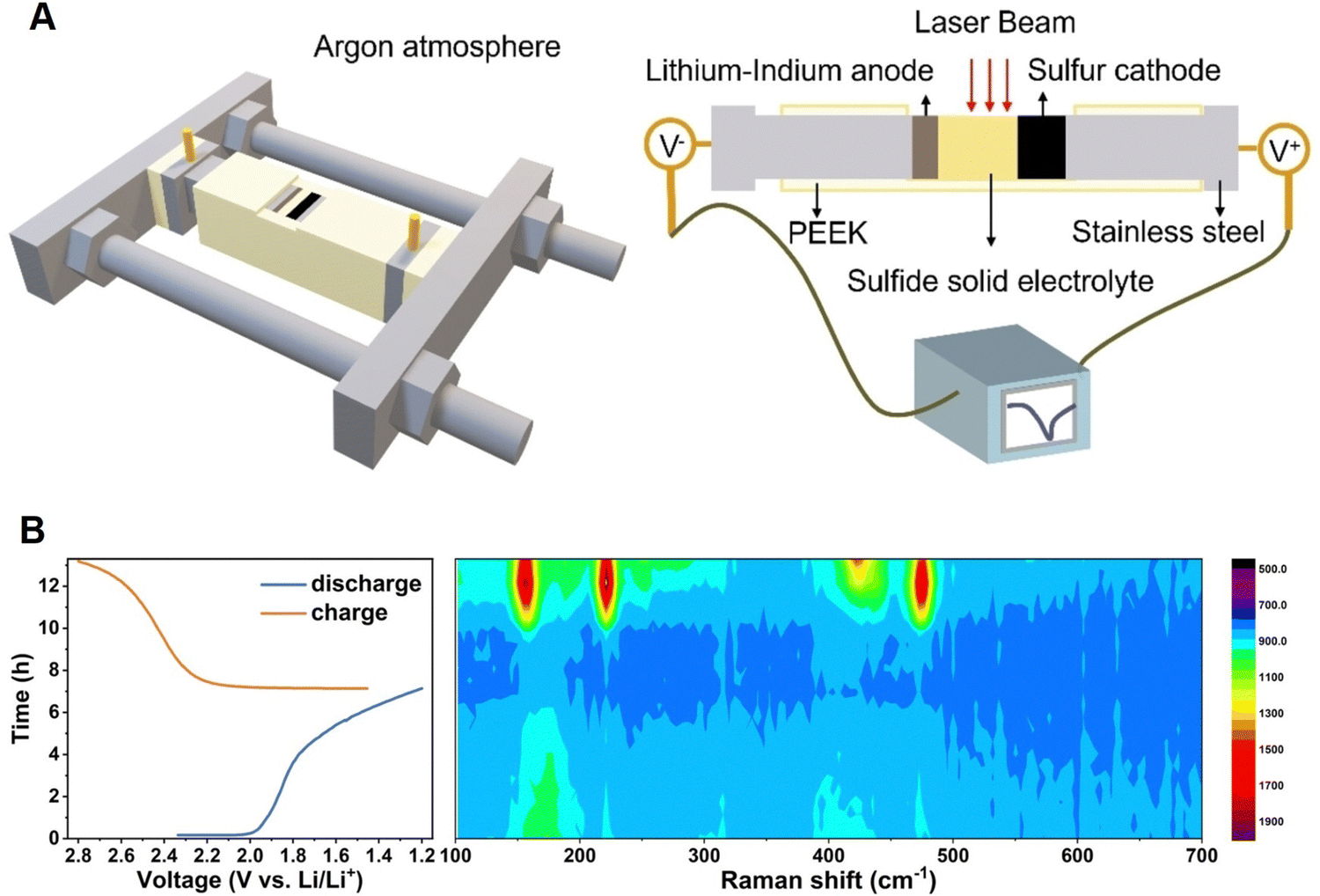 | ||
| Fig. 15 (A) Schematic of an operando Raman spectroscopic cell for solid-state batteries and the corresponding components of the setup. (B) Time–voltage profile of a solid-state Li–S battery and the corresponding operando Raman spectra contour. Reproduced with permission from ref. 162. Copyright 2023 Wiley-VCH GmbH. | ||
The development of electrochemical vibrational spectroscopy offers great opportunities for conducting in situ and operando studies in important areas such as fundamental electrochemistry, electrocatalysis, rechargeable batteries, etc. Each electrochemical vibrational spectroscopy technique (i.e., EC-IR, EC-Raman, and EC-SFG) has advantages and limitations, which are briefly summarized in Table 2, and can be selected according to the practical requirements and applications.
| Vibrational spectroscopy | IR | SFG | Raman |
|---|---|---|---|
| a With a normal external reflection mode. b With nanostructure-, ATR-, or tip-based modes, the surface selectivity is high. c With a nanostructure-based mode, the detection sensitivity is up to a single-molecule level.45 | |||
| Spectroscopic mode | Normal mode; | Normal mode; | Normal mode; |
| Nanostructure-based mode; | Nanostructure-based mode; | Nanostructure-based mode; | |
| ATR-based mode; | ATR-based mode | ATR-based mode; | |
| Tip-based mode; | Tip-based mode; | ||
| Fiber-based mode | Fiber-based mode | ||
| Detection sensitivity | High | High | Lowa; |
| Very highc | |||
| Surface selectivity | Lowa; | Intrinsically high | Lowa; |
| Highb | Highb | ||
| Applications | Electric double layer;12,13,23,24,163–166 | Electric double layer;102,138–142 | Electric double layer;133–136,167,168 |
| Electrocatalysis;143,145,169–174 | Electrocatalysis;175–178 | Electrocatalysis;146,179–184 | |
| Rechargeable battery118,159,185,186 | Rechargeable battery187–189 | Rechargeable battery154,190–193 | |
| Advantages | Sensitive to *CO species, due to the high IR absorption cross-section of C–O bond; feasible to probe both the SEI and the electrolyte layer close to the surface; no interference by fluorescence background, etc. | Suitable for measurement on well-defined surface; intrinsic high surface selectivity and sensitivity in probing the orientation of surface species, e.g., molecular catalysts in heterogeneous electrocatalysis, etc. | Laser with a wavelength in visible region is feasible for operando experiments; no interference by H2O and CO2; suitable for the detection in low-frequency regions, e.g., the libration mode of water (<800 cm−1), metal–carbon and/or metal–oxygen bonds, etc. |
| Challenges | Strong IR adsorption by H2O and CO2; possible interference from a window or prism (e.g., the interference in a low-frequency spectral region);influence of thin-layer electrolyte on electrochemistry, etc. | Strong IR adsorption by H2O and CO2; possible interference from a window or prism (e.g., the interference in a low-frequency spectral region); influence of a thin-layer electrolyte on electrochemical responses; smooth surface is desired, etc. | The cross-section of Raman scattering is very low; the surface-enhancement effect limits the generalities of surface morphology and electrode material; possible interference by a fluorescence background, etc. |
4. Summary and outlook
4.1. Development trends
The application of in situ optical spectroscopy in electrochemistry was carried out six decades ago, with a significant growth since the 1990s, driven by advancements in nanoscience. The development of electrochemical optical spectroscopy, in particular vibrational spectroscopy, has made incalculable contribution not only to the progress of electrochemistry, but also to physical and chemical science and technology. It is foreseeable that electrochemical optical spectroscopy will be further innovated in both theory and experiments with the development of new spectroscopic techniques, instrumentation, nanotechnology, data-driven methods, etc.The main development direction of electrochemical optical spectroscopy must be consistent with that of electrochemistry. Currently, electrochemical energy is of paramount importance in electrochemistry; therefore, it is desirable to further develop a new strategy, methodology, and research paradigm with respect to the different key scientific and technical problems in electrochemical energy studies.
For practical applications of electrochemical optical spectroscopy, the energy conversion and storage systems are in strong demand and facing the challenges of new systems with complex structures, raising unprecedented requirements for the capability of obtaining more reliable and useful information of various electrochemical interfaces under working conditions, i.e., developing operando spectroscopic methods with high detection sensitivity, along with high temporal and spatial resolutions. Furthermore, there are still great difficulties in operando studies under low and high temperature working conditions, i.e., probing the emerging wide-temperature batteries for application under extreme climate/operation conditions.194,195 It is imperative to develop new spectroscopic methods for characterization in such complex and extreme environments.
To discuss these significant explorations, examples illustratin the new strategies, methodologies, and research paradigm in electrochemical optical spectroscopy are presented below.
4.2. New strategies, methodologies, and research paradigms
It is worth noting that SERS-active substrate materials have been expanded beyond “free-electron like” metals to the semiconductor/dielectric substrates198 as well as graphene materials,199 opening up new avenues for EC-SERS applications. In particular, the roles of such SERS-active substrates are manifold, e.g., graphene can simultaneously act as a Raman probe to reveal the spectroelectrochemical features of carbon materials.200–202
As shown in Fig. 16A–F, Yamanaka et al. placed eight ultra-fine fiber Raman probes inside a practical laminate lithium-ion battery at different positions to monitor the electrolyte concentration variations.130 The probe consists of two silica-based optical fibers, i.e., one excitation fiber for laser light and one collection fiber for Raman signals, where the total cross-sectional area of the probe is around 30 μm × 60 μm.40,130 Usually, the Raman spectroscopic measurement offers an averaged information of electrolyte from a battery. However, based on the ultra-fine spectroscopic probes at different locations inside the battery, the concentration gradient of electrolyte in the plane parallel to the electrode surfaces was observed and the corresponding changes during cycles were monitored in operando. Hence, this measurement will be beneficial for the studies of ion migration and related position-dependent reactions inside a battery.
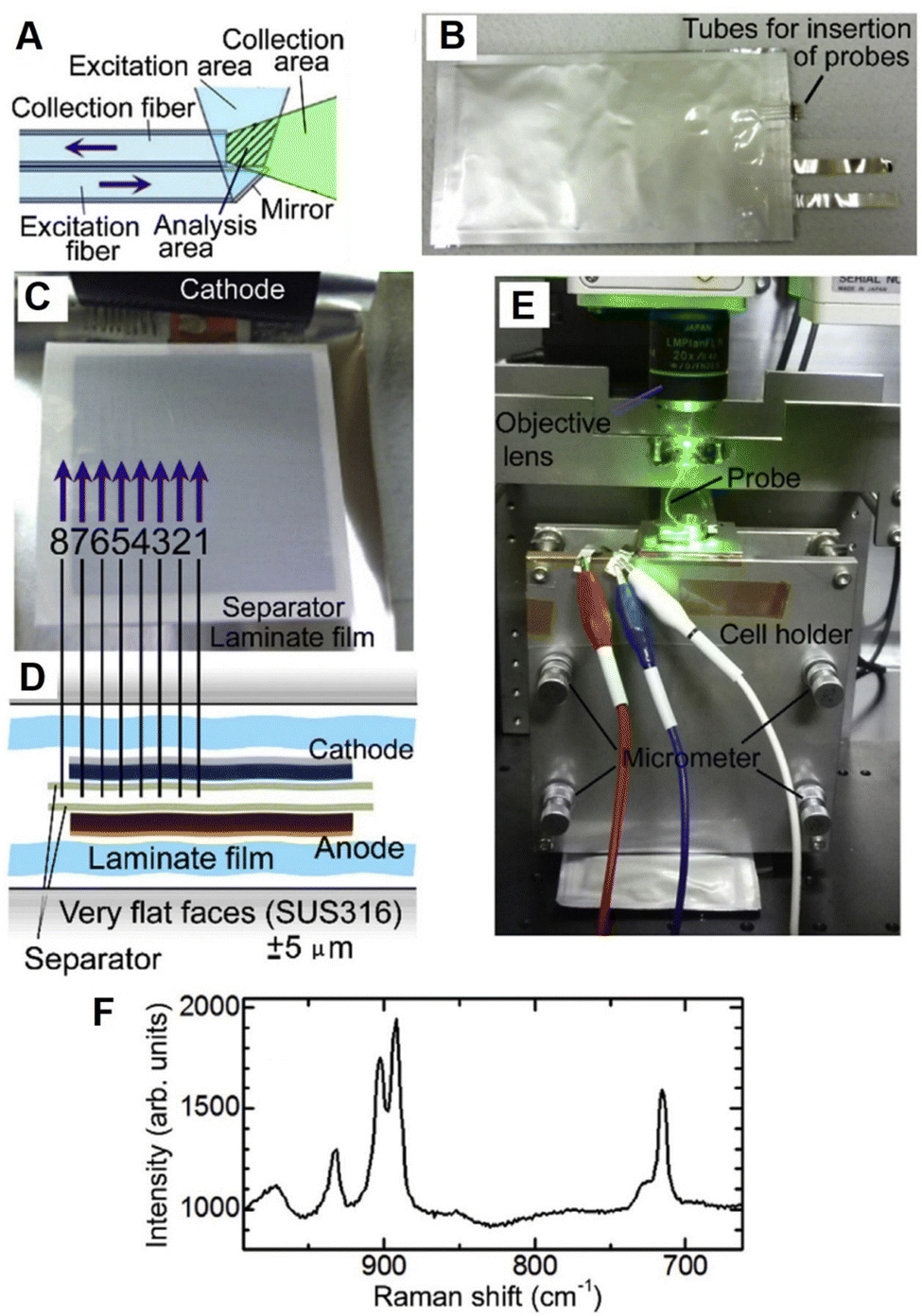 | ||
| Fig. 16 (A)–(F) Operando Raman spectroscopic measurement inside a laminate cell using microprobes. (A) Schematic of probe consisting of one excitation optical fiber and one collection fiber. (B) Photograph of a laminate cell inserted with eight microprobes. (C) Inside and (D) cross-section view of the laminate cell with the probes. (E) Photograph of setup of the operando fiber-based Raman spectroscopic measurement. (F) Raman spectrum of electrolyte inside the laminate cell. Reproduced with permission from ref. 130. Copyright 2017 Elsevier B.V. | ||
Since silica-based fiber is typically limited to the spectral range from 0.8 to 2 μm, to enable to fiber-based spectroelectrochemistry in the IR range, chalcogenide glass fiber can be employed instead.41 As shown by the schematic in Fig. 17A, Gervillié-Mouravieff et al. used a Te2AS3Se5 (TAS) fiber to conduct operando fiber-based IR evanescent wave spectroscopy (FEWS) in the commercial 18650 Na-ion battery.41 The TAS fiber was passed through a pre-drilled 18650 jelly roll cell (the hole was then closed by with epoxy) to monitor the electrolyte decomposition reaction. Operando IR-FEWS spectra clearly reveal the dynamics of electrolyte species, e.g., dimethyl carbonate (DMC) and PF6−, and identify the decomposition reaction intermediates/products, e.g., alkyl carbonate and methoxy species, during the charge and discharge processes, respectively (Fig. 17B and C). This fiber-based EC-IR approach provides an opportunity to rapidly diagnose the dynamics of electrolyte in a battery, which can be expected to offer additional important molecular insights into the different components of an electrochemical device under real working conditions.
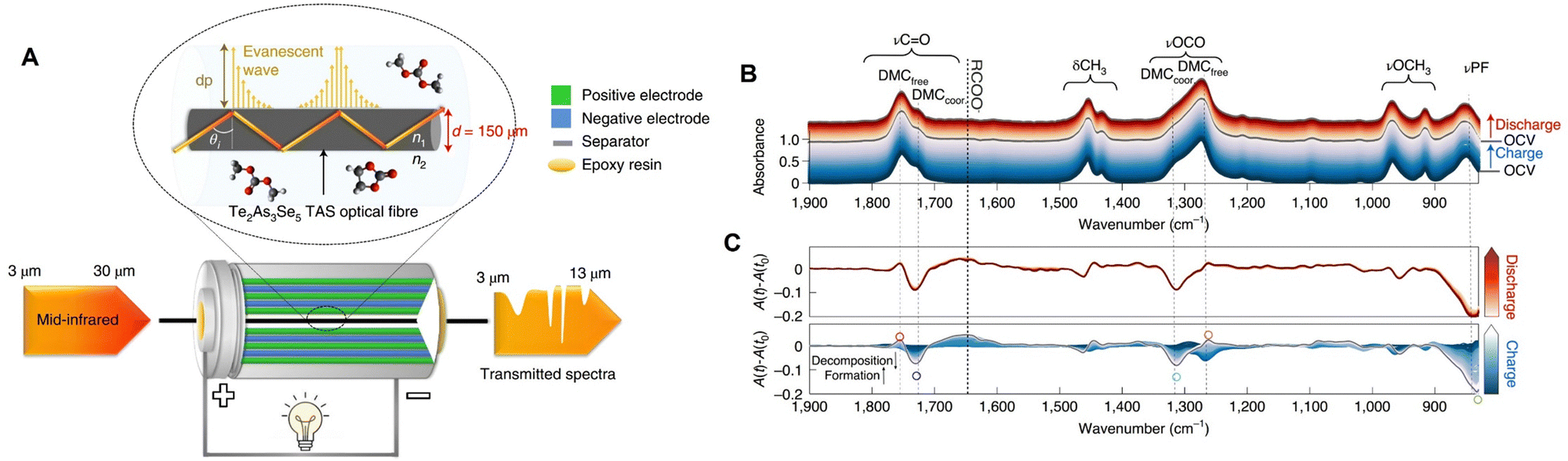 | ||
| Fig. 17 (A) Schematic of operando IR fiber evanescent wave spectroscopy (FEWS) using mid-IR light in the range from 830 to 5000 cm−1 passing through the Te2As3Se5 (TAS) fiber core (a diameter of 150 μm). The TAS fiber is located in the central void of a commercial 18650 jelly roll, allowing IR-FEWS measurements of the electrolyte under operando conditions. (B) Operando IR-FEWS spectra obtained during the first charge–discharge cycle. The spectra in charge were colored in blue, while those in discharge were colored in red. (C) The relative IR absorbance changes (A(t) − A(t0)) obtained in charge (bottom) and discharge (top). Reproduced with permission from ref. 41. Copyright 2022 Springer Nature. | ||
Rather than just monitoring the state of electrochemical devices, we can anticipate more important advances in operando spectroelectrochemical techniques. The synergistic combination of operando characterization, real-time analysis of information, and autonomous adjustment of the working condition of devices is a new research paradigm to be developed. In particular, an implantable diagnostic tool is desirable to collect the operando information of a device and then to immediately communicate with the outside world.203 As inspired by the fiber optical diagnosis as we discussed above, an optical fiber can be inserted inside a battery to collect the operando information, such as operando Raman40,130,131 or IR41 spectra, and transmit the these signals simultaneously, and therefore, we can anticipate an important progress toward a new research paradigm of AI-driven operando spectroelectrochemistry in the coming years.
Optical spectroscopic instruments are strongly dependent on the power of the light source and the sensitivity of the detector. It is well known that the Raman scattering phenomenon was discovered in 1928, but it was rapidly developed and widely applied until the invention of lasers in 1960.45 Therefore, the laser technology has been indispensable to the development and popularization of Raman spectroscopy. In particular, in operando research studies, there are enormous demands in the laser power, efficiency, and wavelength tuning range of incident light source. In this regard, the powerful free electron laser (FEL) technology is capable of fully tunable wavelength, high-quality light beam, high-level polarization, high peak brightness, and high average power, etc.209 Therefore, the application of FEL will be definitely beneficial for progress of spectroelectrochemistry.6
Compared to vibrational spectroscopy, EC-UV-Vis spectroscopy has advantages in sensitivity and quantitative analytical capability for the study of electrolyte bulk; it has widely contributed to the in situ characterization of electrogenerated species and study of redox processes.213,214 Thus, EC-UV-Vis is suitable for the quantitative analysis of large number of intermediates during electrochemical synthesis.
The reversible chemical/electrochemical interconversion between quinone and hydroquinone has been widely recognized in electrochemical synthesis and organic redox flow batteries.215,216 Recently, it was used in a nickel-catalyzed cross-electrophile coupling (XEC) reaction to synthesize pharmaceutical intermediates on a hectogram scale (100 g scale).215 In this work, nickel-catalyzed XEC reaction was supported by a quinone-mediated H2 anode under non-aqueous conditions. UV-Vis spectroscopy was employed to in situ investigate the interplay between thermal catalytic hydrogenation of sodium anthraquinone-2-sulfonate (AQS) and the electrochemical oxidation of the corresponding anthrahydroquinone (AQSH2) in real time. As shown in Fig. 18, the redox speciation of AQS (with an absorption band at ∼330 nm) and AQSH2 (with an adsorption band at ∼380 nm) species in the anolyte reservoir was studied using UV-Vis spectroscopy during various processes, such as hydrogenation-only or electrolysis-only loop, or both loops operating simultaneously. This spectroelectrochemical method demonstrates significant potential in the real-time analysis for the development of highly-efficient, large-scale synthetic electrochemistry.
 | ||
| Fig. 18 (A) Schematic of mediated H2 anode by paring the thermal hydrogenation of AQS and the electrochemical oxidation of AQSH2 in real time. (B) In situ UV-Vis spectra of the redox speciation of AQS and AQSH2 species in the anolyte reservoir for various processes, such as hydrogenation-only or electrolysis-only loop, or both loops operating simultaneously. Reproduced with permission from ref. 215. Copyright 2023 Springer Nature. | ||
After sixty years of development and growth, electrochemical optical spectroscopy has become one of the most important branches of spectroelectrochemistry. Based on the promising development trend, we are optimistic that in the near future, the emergence of new breakthroughs in spectroelectrochemistry will bring incalculable impact not only to the progress of electrochemistry, but also on surface/interface science and the wider fields of materials science and technology.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grants no. 22372121, 92372108, 21991130, and 22032004) and the Fundamental Research Funds for the Central Universities of China.Notes and references
- J. O. 'M. Bockris and K. Khan, Surface Electrochemistry, Plenum, New York, 1993 Search PubMed.
- H. D. Abruña, Electrochemical Interfaces—Modern Techniques for in-situ Interface Characterization, VCH, Berlin, 1991 Search PubMed.
- J. Lipkowski and P. N. Ross, Adsorption at Electrode Surface, VCH, New York, 1992 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2000 Search PubMed.
- In-situ Spectroscopic Studies of Adsorption at the Electrode and Electrocatalysis, ed. S.-G. Sun, P. A. Christensen and A. Wieckowski, Elsevier Science B.V., Amsterdam, 2007 Search PubMed.
- Z.-Q. Tian, et al., Spectro-electrochemistry, Chemical Industry Press Co., Ltd., Beijing, 2021 Search PubMed.
- A. K. N. Reddy, M. A. V. Devanathan and J. O. 'M. Bockris, J. Electroanal. Chem., 1963, 6, 61–67 CAS.
- T. Kuwana, R. K. Darlington and D. W. Leedy, Anal. Chem., 1964, 36, 2023–2025 CrossRef CAS.
- H. B. Mark and B. S. Pons, Anal. Chem., 1966, 38, 119–121 CrossRef CAS.
- C. H. Lee, R. K. Chang and N. Bloembergen, Phys. Rev. Lett., 1967, 18, 167–170 CrossRef CAS.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, J. Chem. Soc., Chem. Commun., 1973, 80–81 RSC.
- A. Bewick, K. Kunimatsu and B. Stanley Pons, Electrochim. Acta, 1980, 25, 465–468 CrossRef CAS.
- A. Bewick and K. Kunimatsu, Surf. Sci., 1980, 101, 131–138 CrossRef CAS.
- P. Guyot-Sionnest and A. Tadjeddine, Chem. Phys. Lett., 1990, 172, 341–345 CrossRef CAS.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- M. Fleischmann, Z. Q. Tian and L. J. Li, J. Electroanal. Chem. Interfacial Electrochem., 1987, 217, 397–410 CrossRef CAS.
- L. W. H. Leung and M. J. Weaver, J. Am. Chem. Soc., 1987, 109, 5113–5119 CrossRef CAS.
- L.-W. H. Leung and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1987, 217, 367–384 CrossRef CAS.
- G. Mengoli, M. M. Musiani, M. Fleischman, B. Mao and Z. Q. Tian, Electrochim. Acta, 1987, 32, 1239–1245 CrossRef CAS.
- R. G. Freeman, K. C. Grabar, K. J. Allison, R. M. Bright, J. A. Davis, A. P. Guthrie, M. B. Hommer, M. A. Jackson, P. C. Smith, D. G. Walter and M. J. Natan, Science, 1995, 267, 1629–1632 CrossRef CAS PubMed.
- M. Osawa, K.-i Ataka, K. Yoshii and T. Yotsuyanagi, J. Electron Spectros. Relat. Phenomena, 1993, 64–65, 371–379 CrossRef CAS.
- K.-i Ataka, T. Yotsuyanagi and M. Osawa, J. Phys. Chem., 1996, 100, 10664–10672 CrossRef CAS.
- K.-i Ataka and M. Osawa, Langmuir, 1998, 14, 951–959 CrossRef CAS.
- W.-B. Cai, L.-J. Wan, H. Noda, Y. Hibino, K. Ataka and M. Osawa, Langmuir, 1998, 14, 6992–6998 CrossRef CAS.
- F. Kitamura, M. Takeda, M. Takahashi and M. Ito, Chem. Phys. Lett., 1987, 142, 318–322 CrossRef CAS.
- F. Kitamura, M. Takahashi and M. Ito, J. Phys. Chem., 1988, 92, 3320–3323 CrossRef CAS.
- R. J. Nichols and A. Bewick, J. Electroanal. Chem. Interfacial Electrochem., 1988, 243, 445–453 CrossRef CAS.
- S. P. Byahut and T. E. Furtak, Electrochim. Acta, 1991, 36, 1879–1882 CrossRef CAS.
- A. Bruckbauer and A. Otto, J. Raman Spectrosc., 1998, 29, 665–672 CrossRef CAS.
- A. Peremans and A. Tadjeddine, Phys. Rev. Lett., 1994, 73, 3010–3013 CrossRef CAS PubMed.
- W. Daum, K. A. Friedrich, C. Klünker, D. Knabben, U. Stimming and H. Ibach, Appl. Phys. A, 1994, 59, 553–562 CrossRef.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- Z.-C. Zeng, S.-C. Huang, D.-Y. Wu, L.-Y. Meng, M.-H. Li, T.-X. Huang, J.-H. Zhong, X. Wang, Z.-L. Yang and B. Ren, J. Am. Chem. Soc., 2015, 137, 11928–11931 CrossRef CAS PubMed.
- D. Kurouski, M. Mattei and R. P. Van Duyne, Nano Lett., 2015, 15, 7956–7962 CrossRef CAS PubMed.
- Y.-H. Lu, J. M. Larson, A. Baskin, X. Zhao, P. D. Ashby, D. Prendergast, H. A. Bechtel, R. Kostecki and M. Salmeron, Nano Lett., 2019, 19, 5388–5393 CrossRef CAS PubMed.
- I. Kendrick, D. Kumari, A. Yakaboski, N. Dimakis and E. S. Smotkin, J. Am. Chem. Soc., 2010, 132, 17611–17616 CrossRef CAS PubMed.
- M. Hara, J. Inukai, K. Miyatake, H. Uchida and M. Watanabe, Electrochim. Acta, 2011, 58, 449–455 CrossRef CAS.
- P. Huguet, A. Morin, G. Gebel, S. Deabate, A. K. Sutor and Z. Peng, Electrochem. Commun., 2011, 13, 418–422 CrossRef CAS.
- T. Yamanaka, H. Nakagawa, M. Ochida, S. Tsubouchi, Y. Domi, T. Doi, T. Abe and Z. Ogumi, J. Phys. Chem. C, 2016, 120, 2585–2591 CrossRef CAS.
- C. Gervillié-Mouravieff, C. Boussard-Plédel, J. Huang, C. Leau, L. A. Blanquer, M. B. Yahia, M. L. Doublet, S. T. Boles, X. H. Zhang, J. L. Adam and J. M. Tarascon, Nat. Energy, 2022, 7, 1157–1169 CrossRef.
- I. Bergman, B. E. Conway, A. Bewick, T. Kuwana, J. D. E. McIntyre, C. L. Gardner, E. J. Casey, M. A. Barrett, H. Angerstein-Kozlowska, D. M. Kolb, M. Keddam, A. J. McQuillan, J. S. Clarke, A. T. Kuhn and W. J. Orville-Thomas, Faraday Spec. Discuss. Chem. Soc., 1973, 56, 152–170 RSC.
- J. W. Strojek and T. Kuwana, J. Electroanal. Chem., 1968, 16(4), 471–483 CrossRef CAS.
- P. J. Hendra, Int. J. Vib. Spect., 2000, 4, 1 Search PubMed.
- Z.-Q. Tian and B. Ren, Encyclopedia of Electrochemistry, Wiley-VCH, 2007, ch. 6, pp. 572–659 Search PubMed.
- M. R. Anderson and C. D. Taylor, Encyclopedia of Analytical Chemistry, 2007 Search PubMed.
- Y. R. Shen, Nature, 1989, 337, 519–525 CrossRef CAS.
- G. A. Somorjai and Y. Li, Introduction to surface chemistry and catalysis, John Wiley & Sons, 2010 Search PubMed.
- Z.-Q. Tian and B. Ren, Annu. Rev. Phys. Chem., 2004, 55, 197–229 CrossRef CAS PubMed.
- Z.-Q. Tian, B. Ren and D.-Y. Wu, J. Phys. Chem. B, 2002, 106, 9463–9483 CrossRef CAS.
- A. Bewick, K. Kunimatsu, B. S. Pons and J. W. Russell, J. Electroanal. Chem. Interfacial Electrochem., 1984, 160, 47–61 CrossRef CAS.
- Z.-Q. Tian and B. Ren, Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, 2006 Search PubMed.
- A. Tadjeddine and F. Vidal, In-situ Spectroscopic Studies of Adsorption at the Electrode and Electrocatalysis, ed. S.-G. Sun, P. A. Christensen and A. Wieckowski, Elsevier Science B.V., Amsterdam, 2007, pp. 273–298 Search PubMed.
- A. McClelland and Z. Chen, Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd., 2008 Search PubMed.
- S. Ye and K. Uosaki, in Encyclopedia of Electrochemistry, ed. A. J. Bard and M. Stratmann, Wiley-VCH Verlag GmbH, Weinheim, 2007, vol. 10, pp. 513–553 Search PubMed.
- A. Ge, K.-i Inoue and S. Ye, J. Chem. Phys., 2020, 153, 170902 CrossRef CAS PubMed.
- J. H. Hunt, P. Guyot-Sionnest and Y. R. Shen, Chem. Phys. Lett., 1987, 133, 189–192 CrossRef CAS.
- X. D. Zhu, H. Suhr and Y. R. Shen, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 3047–3050 CrossRef CAS PubMed.
- P. Guyot-Sionnest, J. H. Hunt and Y. R. Shen, Phys. Rev. Lett., 1987, 59, 1597–1600 CrossRef CAS PubMed.
- J.-F. Li, Y.-J. Zhang, S.-Y. Ding, R. Panneerselvam and Z.-Q. Tian, Chem. Rev., 2017, 117, 5002–5069 CrossRef CAS PubMed.
- M. Osawa, in Near-Field Optics and Surface Plasmon Polaritons, ed. S. Kawata, Springer Berlin Heidelberg, Berlin, Heidelberg, 2001, pp. 163–187 Search PubMed.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- R. P. Van Duyne, in Chemical and Biochemical Applications of Lasers, ed. C. B. Moore, Academic Press, 1979, pp. 101–185 Search PubMed.
- R. K. Chang and T. E. Furtak, Surface Enhanced Raman Scattering, Plenum Press, New York, 1982 Search PubMed.
- M. Moskovits, J. Raman Spectrosc., 2005, 36, 485–496 CrossRef CAS.
- E. C. Le Ru and P. G. Etchegoin, in Principles of Surface-Enhanced Raman Spectroscopy, ed. E. C. Le Ru and P. G. Etchegoin, Elsevier, Amsterdam, 2009, pp. 121–183 Search PubMed.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed.
- S. A. Maier, Plasmonics: fundamentals and applications, Springer, New York, 2007 Search PubMed.
- J. A. Creighton, C. G. Blatchford and M. G. Albrecht, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 790–798 RSC.
- M. Moskovits, J. Chem. Phys, 1978, 69, 4159–4161 CrossRef CAS.
- Z.-Q. Tian, B. Ren, J.-F. Li and Z.-L. Yang, Chem. Commun., 2007, 3514–3534 RSC.
- S.-Y. Ding, J. Yi, J.-F. Li, B. Ren, D.-Y. Wu, R. Panneerselvam and Z.-Q. Tian, Nat. Rev. Mater., 2016, 1, 16021 CrossRef CAS.
- R. P. Van Duyne and J. P. Haushalter, J. Phys. Chem., 1983, 87, 2999–3003 CrossRef CAS.
- S. Park, P. Yang, P. Corredor and M. J. Weaver, J. Am. Chem. Soc., 2002, 124, 2428–2429 CrossRef CAS PubMed.
- L. Lu, G. Sun, H. Zhang, H. Wang, S. Xi, J. Hu, Z. Tian and R. Chen, J. Mater. Chem., 2004, 14, 1005–1009 RSC.
- J.-W. Hu, Y. Zhang, J.-F. Li, Z. Liu, B. Ren, S.-G. Sun, Z.-Q. Tian and T. Lian, Chem. Phys. Lett., 2005, 408, 354–359 CrossRef CAS.
- J.-F. Li, Z.-L. Yang, B. Ren, G.-K. Liu, P.-P. Fang, Y.-X. Jiang, D.-Y. Wu and Z.-Q. Tian, Langmuir, 2006, 22, 10372–10379 CrossRef CAS PubMed.
- C. G. Blatchford, J. R. Campbell and J. A. Creighton, Surf. Sci., 1982, 120, 435–455 CrossRef CAS.
- C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2001, 105, 5599–5611 CrossRef CAS.
- L. A. Dick, A. D. McFarland, C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2002, 106, 853–860 CrossRef CAS.
- P. N. Bartlett, J. J. Baumberg, S. Coyle and M. E. Abdelsalam, Faraday Discuss., 2004, 125, 117–132 RSC.
- S. Mahajan, M. Abdelsalam, Y. Suguwara, S. Cintra, A. Russell, J. Baumberg and P. Bartlett, Phys. Chem. Chem. Phys., 2007, 9, 104–109 RSC.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. V. Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- A. Hartstein, J. R. Kirtley and J. C. Tsang, Phys. Rev. Lett., 1980, 45, 201–204 CrossRef CAS.
- A. Hatta, T. Ohshima and W. Suëtaka, Appl. Phys. A, 1982, 29, 71–75 CrossRef.
- A. Hatta, Y. Suzuki and W. Suëtaka, Appl. Phys. A, 1984, 35, 135–140 CrossRef.
- M. Osawa, Bull. Chem. Soc. Jpn, 1997, 70, 2861–2880 CrossRef CAS.
- M. Osawa, M. Kuramitsu, A. Hatta, W. Suëtaka and H. Seki, Surf. Sci., 1986, 175, L787–L793 CrossRef CAS.
- M. Osawa, K.-I. Ataka, K. Yoshii and Y. Nishikawa, Appl. Spectrosc., 1993, 47, 1497–1502 CrossRef CAS.
- Y. Nishikawa, T. Nagasawa, K. Fujiwara and M. Osawa, Vib. Spectrosc., 1993, 6, 43–53 CrossRef CAS.
- P. A. Christensen, in Encyclopedia of Electrochemistry, 2007 Search PubMed.
- H.-L. Wang, E.-M. You, R. Panneerselvam, S.-Y. Ding and Z.-Q. Tian, Light Sci. Appl., 2021, 10, 161 CrossRef CAS PubMed.
- M. Osawa, in In-situ Spectroscopic Studies of Adsorption at the Electrode and Electrocatalysis, ed. S.-G. Sun, P. A. Christensen and A. Wieckowski, Elsevier Science B.V., Amsterdam, 2007, pp. 209–246 Search PubMed.
- G.-Q. Lu, S.-G. Sun, S.-P. Chen and L.-R. Cai, J. Electroanal. Chem., 1997, 421, 19–23 CrossRef CAS.
- G.-Q. Lu, S.-G. Sun, L.-R. Cai, S.-P. Chen, Z.-W. Tian and K.-K. Shiu, Langmuir, 2000, 16, 778–786 CrossRef CAS.
- E. V. Alieva, L. A. Kuzik and V. A. Yakovlev, Chem. Phys. Lett., 1998, 292, 542–546 CrossRef CAS.
- E. W. M. van der Ham, Q. H. F. Vrehen, E. R. Eliel, V. A. Yakovlev, E. V. Alieva, L. A. Kuzik, J. E. Petrov, V. A. Sychugov and A. F. G. van der Meer, J. Opt. Soc. Am. B, 1999, 16, 1146–1152 CrossRef CAS.
- S. Baldelli, A. S. Eppler, E. Anderson, Y.-R. Shen and G. A. Somorjai, J. Chem. Phys., 2000, 113, 5432–5438 CrossRef CAS.
- T. Kawai, D. J. Neivandt and P. B. Davies, J. Am. Chem. Soc., 2000, 122, 12031–12032 CrossRef CAS.
- Y. He, H. Ren, E.-M. You, P. M. Radjenovic, S.-G. Sun, Z.-Q. Tian, J.-F. Li and Z. Wang, Phys. Rev. Lett., 2020, 125, 047401 CrossRef CAS PubMed.
- Y. Yang, Y. Xiong, R. Zeng, X. Lu, M. Krumov, X. Huang, W. Xu, H. Wang, F. J. DiSalvo, J. D. Brock, D. A. Muller and H. D. Abruña, ACS Catal., 2021, 11, 1136–1178 CrossRef CAS.
- W.-T. Liu and Y. R. Shen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1293–1297 CrossRef CAS PubMed.
- C. Korzeniewski, in In-situ Spectroscopic Studies of Adsorption at the Electrode and Electrocatalysis, ed. S.-G. Sun, P. A. Christensen and A. Wieckowski, Elsevier Science B.V., Amsterdam, 2007, pp. 179–208 Search PubMed.
- N. Furuya, S. Motto and K. Kunimatsu, J. Electroanal. Chem. Interfacial Electrochem., 1988, 239, 347–360 CrossRef CAS.
- A. J. Wilson and K. A. Willets, Nano Lett., 2014, 14, 939–945 CrossRef CAS PubMed.
- C.-Y. Li, J. Yi, R. Hu, J.-F. Li and Z.-Q. Tian, in Encyclopedia of Nanomaterials, ed. Y. Yin, Y. Lu and Y. Xia, Elsevier, Oxford, 1st edn, 2023, vol. 1, pp. 511–535 Search PubMed.
- D. M. Kolb, Angew. Chem., Int. Ed., 2001, 40, 1162–1181 CrossRef CAS PubMed.
- O. M. Magnussen, Chem. Rev., 2002, 102, 679–726 CrossRef CAS PubMed.
- A. A. Gewirth and B. K. Niece, Chem. Rev., 1997, 97, 1129–1162 CrossRef CAS PubMed.
- Z. Q. Tian, W. H. Li, B. Ren, B. W. Mao, J. G. Chen, J. Q. Mu, X. D. Zhuo and D. Wang, J. Electroanal. Chem., 1996, 401, 247–251 CrossRef.
- Z.-Q. Tian, W.-H. Li, J.-Q. Mu, B.-W. Mao, J.-G. Chen, X.-D. Zhuo, W. Zheng, D. Wang and E.-R. Yan, Acta Phys. Chim., 1994, 10, 1062–1065 CAS.
- M. S. Anderson, Appl. Phys. Lett., 2000, 76, 3130–3132 CrossRef CAS.
- N. Hayazawa, Y. Inouye, Z. Sekkat and S. Kawata, Opt. Commun., 2000, 183, 333–336 CrossRef CAS.
- R. M. Stöckle, Y. D. Suh, V. Deckert and R. Zenobi, Chem. Phys. Lett., 2000, 318, 131–136 CrossRef.
- B. Pettinger, G. Picardi, R. Schuster and G. Ertl, Electrochemistry, 2000, 68, 942–949 CrossRef CAS.
- S.-C. Huang, X. Wang, Q.-Q. Zhao, J.-F. Zhu, C.-W. Li, Y.-H. He, S. Hu, M. M. Sartin, S. Yan and B. Ren, Nat. Commun., 2020, 11, 4211 CrossRef CAS PubMed.
- J. J. Schwartz, D. S. Jakob and A. Centrone, Chem. Soc. Rev., 2022, 51, 5248–5267 RSC.
- X. He, J. M. Larson, H. A. Bechtel and R. Kostecki, Nat. Commun., 2022, 13, 1398 CrossRef CAS PubMed.
- S. Amemiya, A. J. Bard, F.-R. F. Fan, M. V. Mirkin and P. R. Unwin, Annu. Rev. Anal. Chem., 2008, 1, 95–131 CrossRef CAS PubMed.
- C. L. Brosseau, A. Colina, J. V. Perales-Rondon, A. J. Wilson, P. B. Joshi, B. Ren and X. Wang, Nat. Rev. Methods Primers, 2023, 3, 79 CrossRef CAS.
- W. Schuhmann, D. Öhl and D. M. Morales, in Springer Handbook of Advanced Catalyst Characterization, ed. I. E. Wachs and M. A. Bañares, Springer International Publishing, Cham, 2023, pp. 189–211 Search PubMed.
- J. Clausmeyer, M. Nebel, S. Grützke, Y. U. Kayran and W. Schuhmann, ChemPlusChem, 2018, 83, 414–417 CrossRef CAS PubMed.
- K. O. Hatfield, M. T. Gole, N. B. Schorr, C. J. Murphy and J. Rodríguez-López, Anal. Chem., 2021, 93, 7792–7796 CrossRef CAS PubMed.
- M. A. Bañares and I. E. Wachs, J. Raman Spectrosc., 2002, 33, 359–380 CrossRef.
- A. Gurlo and R. Riedel, Angew. Chem., Int. Ed., 2007, 46, 3826–3848 CrossRef CAS PubMed.
- B. M. Weckhuysen, Chem. Commun., 2002, 97–110 RSC.
- A. M. Tripathi, W.-N. Su and B. J. Hwang, Chem. Soc. Rev., 2018, 47, 736–851 RSC.
- J. Tan, D. Liu, X. Xu and L. Mai, Nanoscale, 2017, 9, 19001–19016 RSC.
- L. Meyer, N. Saqib and J. Porter, J. Electrochem. Soc., 2021, 168, 090561 CrossRef.
- T. Yamanaka, H. Nakagawa, S. Tsubouchi, Y. Domi, T. Doi, T. Abe and Z. Ogumi, J. Power Sources, 2017, 359, 435–440 CrossRef CAS.
- E. Miele, W. M. Dose, I. Manyakin, M. H. Frosz, Z. Ruff, M. F. L. De Volder, C. P. Grey, J. J. Baumberg and T. G. Euser, Nat. Commun., 2022, 13, 1651 CrossRef CAS PubMed.
- T. Lombardo, M. Duquesnoy, H. El-Bouysidy, F. Årén, A. Gallo-Bueno, P. B. Jørgensen, A. Bhowmik, A. Demortière, E. Ayerbe, F. Alcaide, M. Reynaud, J. Carrasco, A. Grimaud, C. Zhang, T. Vegge, P. Johansson and A. A. Franco, Chem. Rev., 2022, 122, 10899–10969 CrossRef CAS PubMed.
- M. Fleischmann, P. J. Hendra, I. R. Hill and M. E. Pemble, J. Electroanal. Chem. Interfacial Electrochem., 1981, 117, 243–255 CrossRef CAS.
- C.-Y. Li, J.-B. Le, S. Chen, Z.-L. Yang, J.-F. Li, J. Cheng and Z.-Q. Tian, Nat. Mater., 2019, 18, 697–701 CrossRef CAS PubMed.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- C.-Y. Li, M. Chen, S. Liu, X. Lu, J. Meng, J. Yan, H. D. Abruña, G. Feng and T. Lian, Nat. Commun., 2022, 13, 5330 CrossRef CAS PubMed.
- G. Gonella, E. H. G. Backus, Y. Nagata, D. J. Bonthuis, P. Loche, A. Schlaich, R. R. Netz, A. Kühnle, I. T. McCrum, M. T. M. Koper, M. Wolf, B. Winter, G. Meijer, R. K. Campen and M. Bonn, Nat. Rev. Chem., 2021, 5, 466–485 CrossRef CAS PubMed.
- Y. Tong, I. Y. Zhang and R. K. Campen, Nat. Commun., 2018, 9, 1313 CrossRef PubMed.
- Y. Tong, F. Lapointe, M. Thämer, M. Wolf and R. K. Campen, Angew. Chem., Int. Ed., 2017, 56, 4211–4214 CrossRef CAS PubMed.
- A. Montenegro, C. Dutta, M. Mammetkuliev, H. Shi, B. Hou, D. Bhattacharyya, B. Zhao, S. B. Cronin and A. V. Benderskii, Nature, 2021, 594, 62–65 CrossRef CAS PubMed.
- Z. D. Schultz, S. K. Shaw and A. A. Gewirth, J. Am. Chem. Soc., 2005, 127, 15916–15922 CrossRef CAS PubMed.
- S. Nihonyanagi, S. Ye, K. Uosaki, L. Dreesen, C. Humbert, P. Thiry and A. Peremans, Surf. Sci., 2004, 573, 11–16 CrossRef CAS.
- Y. X. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, J. Am. Chem. Soc., 2003, 125, 3680–3681 CrossRef CAS PubMed.
- Y. Hori, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer New York, New York, NY, 2008, pp. 89–189 Search PubMed.
- S. Zhu, B. Jiang, W.-B. Cai and M. Shao, J. Am. Chem. Soc., 2017, 139, 15664–15667 CrossRef CAS PubMed.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, Nat. Energy, 2019, 4, 60–67 CrossRef CAS.
- J. Li and J. Gong, Energy Environ. Sci., 2020, 13, 3748–3779 RSC.
- J. Pu, C. Zhong, J. Liu, Z. Wang and D. Chao, Energy Environ. Sci., 2021, 14, 3872–3911 RSC.
- D. Atkins, E. Capria, K. Edström, T. Famprikis, A. Grimaud, Q. Jacquet, M. Johnson, A. Matic, P. Norby, H. Reichert, J.-P. Rueff, C. Villevieille, M. Wagemaker and S. Lyonnard, Adv. Energy Mater., 2022, 12, 2102694 CrossRef CAS.
- X. Cao, H. Li, Y. Qiao, M. Jia, X. Li, J. Cabana and H. Zhou, Adv. Mater., 2021, 33, 2004280 CrossRef CAS PubMed.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J. M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- B. Qiu, M. Zhang, Y. Xia, Z. Liu and Y. S. Meng, Chem. Mater., 2017, 29, 908–915 CrossRef CAS.
- Y. Qiao, S. Guo, K. Zhu, P. Liu, X. Li, K. Jiang, C.-J. Sun, M. Chen and H. Zhou, Energy Environ. Sci., 2018, 11, 299–305 RSC.
- Y. Qiao, J. Yi, S. Wu, Y. Liu, S. Yang, P. He and H. Zhou, Joule, 2017, 1, 359–370 CrossRef CAS.
- C.-Y. Li, J.-C. Dong, X. Jin, S. Chen, R. Panneerselvam, A. V. Rudnev, Z.-L. Yang, J.-F. Li, T. Wandlowski and Z.-Q. Tian, J. Am. Chem. Soc., 2015, 137, 7648–7651 CrossRef CAS PubMed.
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H.-H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS PubMed.
- X. Shan, U. Patel, S. Wang, R. Iglesias and N. Tao, Science, 2010, 327, 1363–1366 CrossRef CAS PubMed.
- M. Kitta, K. Murai, K. Yoshii and H. Sano, J. Am. Chem. Soc., 2021, 143, 11160–11170 CrossRef CAS PubMed.
- Y. Zhang, Y. Katayama, R. Tatara, L. Giordano, Y. Yu, D. Fraggedakis, J. G. Sun, F. Maglia, R. Jung, M. Z. Bazant and Y. Shao-Horn, Energy Environ. Sci., 2020, 13, 183–199 RSC.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- M. B. Dixit, J.-S. Park, P. Kenesei, J. Almer and K. B. Hatzell, Energy Environ. Sci., 2021, 14, 4672–4711 RSC.
- D. Cao, X. Sun, F. Li, S.-M. Bak, T. Ji, M. Geiwitz, K. S. Burch, Y. Du, G. Yang and H. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202302363 CrossRef CAS PubMed.
- A. Yamakata and M. Osawa, J. Am. Chem. Soc., 2009, 131, 6892–6893 CrossRef CAS PubMed.
- Y.-J. Zhang, Z.-F. Su, J.-F. Li and J. Lipkowski, J. Phys. Chem. C, 2020, 124, 13240–13248 CrossRef CAS.
- A. E. Russell, A. S. Lin and W. E. O'Grady, J. Chem. Soc. Faraday Trans., 1993, 89, 195–198 RSC.
- M. A. Habib and J. O. Bockris, Langmuir, 1986, 2, 388–392 CrossRef CAS.
- Z. Q. Tian, Y. X. Chen, B. W. Mao, C. Z. Li, J. Wang and Z. F. Liu, Chem. Phys. Lett., 1995, 240, 224–229 CrossRef CAS.
- Y.-X. Chen and A. Otto, J. Raman Spectrosc., 2005, 36, 736–747 CrossRef CAS.
- A. Cuesta, G. Cabello, C. Gutiérrez and M. Osawa, Phys. Chem. Chem. Phys., 2011, 13, 20091–20095 RSC.
- C. T. Williams, C. G. Takoudis and M. J. Weaver, J. Phys. Chem. B, 1998, 102, 406–416 CrossRef CAS.
- X. Chang, S. Vijay, Y. Zhao, N. J. Oliveira, K. Chan and B. Xu, Nat. Commun., 2022, 13, 2656 CrossRef CAS PubMed.
- M.-H. Shao, P. Liu and R. R. Adzic, J. Am. Chem. Soc., 2006, 128, 7408–7409 CrossRef CAS PubMed.
- Z. Su, M. Karaskiewicz, J. Rogalski, R. Bilewicz and J. Lipkowski, J. Electroanal. Chem., 2020, 875, 113820 CrossRef CAS.
- I. A. Cechanaviciute, R. P. Antony, O. A. Krysiak, T. Quast, S. Dieckhöfer, S. Saddeler, P. Telaar, Y.-T. Chen, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2023, 62, e202218493 CrossRef CAS PubMed.
- R. L. Behrens, A. Lagutchev, D. D. Dlott and A. Wieckowski, J. Electroanal. Chem., 2010, 649, 32–36 CrossRef CAS.
- N. García Rey and D. D. Dlott, J. Phys. Chem. C, 2015, 119, 20892–20899 CrossRef.
- M. L. Clark, A. Ge, P. E. Videla, B. Rudshteyn, C. J. Miller, J. Song, V. S. Batista, T. Lian and C. P. Kubiak, J. Am. Chem. Soc., 2018, 140, 17643–17655 CrossRef CAS PubMed.
- F. Vidal, B. Busson, C. Six, O. Pluchery and A. Tadjeddine, Surf. Sci., 2002, 502–503, 485–489 CrossRef CAS.
- B. Ren, X. Q. Li, C. X. She, D. Y. Wu and Z. Q. Tian, Electrochim. Acta, 2000, 46, 193–205 CrossRef CAS.
- H. Yang, Y. Yang and S. Zou, J. Phys. Chem. C, 2007, 111, 19058–19065 CrossRef CAS.
- Y. Zhao, X.-G. Zhang, N. Bodappa, W.-M. Yang, Q. Liang, P. M. Radjenovica, Y.-H. Wang, Y.-J. Zhang, J.-C. Dong, Z.-Q. Tian and J.-F. Li, Energy Environ. Sci., 2022, 15, 3968–3977 RSC.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS.
- T. A. Galloway and L. J. Hardwick, J. Phys. Chem. Lett., 2016, 7, 2119–2124 CrossRef CAS PubMed.
- Y.-H. Wang, J.-B. Le, W.-Q. Li, J. Wei, P. M. Radjenovic, H. Zhang, X.-S. Zhou, J. Cheng, Z.-Q. Tian and J.-F. Li, Angew. Chem., Int. Ed., 2019, 58, 16062–16066 CrossRef CAS PubMed.
- J.-T. Li, S.-R. Chen, F.-S. Ke, G.-Z. Wei, L. Huang and S.-G. Sun, J. Electroanal. Chem., 2010, 649, 171–176 CrossRef CAS.
- S.-I. Pyun and Y.-G. Ryu, J. Electroanal. Chem., 1998, 455, 11–17 CrossRef CAS.
- Q. Peng, J. Chen, H. Ji, A. Morita and S. Ye, J. Am. Chem. Soc., 2018, 140, 15568–15571 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- A. Ge, D. Zhou, K.-I. Inoue, Y. Chen and S. Ye, J. Phys. Chem. C, 2020, 124, 17538–17547 CrossRef CAS.
- C.-Y. Li, Y. Yu, C. Wang, Y. Zhang, S.-Y. Zheng, J.-F. Li, F. Maglia, R. Jung, Z.-Q. Tian and Y. Shao-Horn, J. Phys. Chem. C, 2020, 124, 4024–4031 CrossRef CAS.
- T. A. Galloway, L. Cabo-Fernandez, I. M. Aldous, F. Braga and L. J. Hardwick, Faraday Discuss., 2017, 205, 469–490 RSC.
- S. Hy, Felix, Y.-H. Chen, J.-Y. Liu, J. Rick and B.-J. Hwang, J. Power Sources, 2014, 256, 324–328 CrossRef CAS.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J.-M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X.-Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- Y. Feng, L. Zhou, H. Ma, Z. Wu, Q. Zhao, H. Li, K. Zhang and J. Chen, Energy Environ. Sci., 2022, 15, 1711–1759 RSC.
- S.-Y. Ding, E.-M. You, J. Yi, J.-F. Li and Z.-Q. Tian, Faraday Discuss., 2017, 205, 457–468 RSC.
- L. Deng, Y. Zhai, Y. Chen, N. Wang and Y. Huang, J. Phys. D: Appl. Phys., 2019, 52, 43LT01 CrossRef CAS.
- I. Alessandri and J. R. Lombardi, Chem. Rev., 2016, 116, 14921–14981 CrossRef CAS PubMed.
- X. Ling, L. Xie, Y. Fang, H. Xu, H. Zhang, J. Kong, M. S. Dresselhaus, J. Zhang and Z. Liu, Nano Lett., 2010, 10, 553–561 CrossRef CAS PubMed.
- M. Kalbac, H. Farhat, J. Kong, P. Janda, L. Kavan and M. S. Dresselhaus, Nano Lett., 2011, 11, 1957–1963 CrossRef CAS PubMed.
- L. Kavan and L. Dunsch, ChemPhysChem, 2007, 8, 974–998 CrossRef CAS PubMed.
- W. Xu, N. Mao and J. Zhang, Small, 2013, 9, 1206–1224 CrossRef CAS PubMed.
- C. P. Grey and J. M. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef PubMed.
- C. Shi, W. Zhang, R. L. Birke and J. R. Lombardi, J. Phys. Chem., 1990, 94, 4766–4769 CrossRef CAS.
- C. Shi, W. Zhang, J. R. Lombardi and R. L. Birke, J. Phys. Chem., 1992, 96, 10093–10096 CrossRef CAS.
- C. Zong, C.-J. Chen, M. Zhang, D.-Y. Wu and B. Ren, J. Am. Chem. Soc., 2015, 137, 11768–11774 CrossRef CAS PubMed.
- C. Fang, R. R. Frontiera, R. Tran and R. A. Mathies, Nature, 2009, 462, 200–204 CrossRef CAS PubMed.
- R. R. Frontiera, A.-I. Henry, N. L. Gruenke and R. P. Van Duyne, J. Phys. Chem. Lett., 2011, 2, 1199–1203 CrossRef CAS PubMed.
- K.-J. Kim, Z. Huang and R. Lindberg, Synchrotron Radiation and Free-Electron Lasers: Principles of Coherent X-Ray Generation, Cambridge University Press, Cambridge, 2017 Search PubMed.
- F. Gomollón-Bel, Chem. Int., 2023, 45, 14–22 Search PubMed.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef PubMed.
- D. Zhang, R. Wang, X. Wang and Y. Gogotsi, Nat. Energy, 2023, 8, 567–576 CrossRef CAS.
- K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Nat. Rev. Chem., 2017, 1, 0039 CrossRef CAS.
- J. Twilton, M. R. Johnson, V. Sidana, M. C. Franke, C. Bottecchia, D. Lehnherr, F. Lévesque, S. M. M. Knapp, L. Wang, J. B. Gerken, C. M. Hong, T. P. Vickery, M. D. Weisel, N. A. Strotman, D. J. Weix, T. W. Root and S. S. Stahl, Nature, 2023, 623, 71–76 CrossRef CAS PubMed.
- D. G. Kwabi, Y. Ji and M. J. Aziz, Chem. Rev., 2020, 120, 6467–6489 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |