
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermally activated structural phase transitions and processes in metal–organic frameworks
Celia
Castillo-Blas
 *a,
Ashleigh M.
Chester
a,
David A.
Keen
b and
Thomas D.
Bennett
a
*a,
Ashleigh M.
Chester
a,
David A.
Keen
b and
Thomas D.
Bennett
a
aDepartment of Materials Science and Metallurgy, University of Cambridge, 27 Charles Babbage Road, Cambridge, CB30FS, UK. E-mail: cc2078@cam.ac.uk
bISIS Facility, Rutherford Appleton Laboratory, Harwell Campus, OX11 0DE, Didcot, Oxfordshire, UK
First published on 1st March 2024
Abstract
The structural knowledge of metal–organic frameworks is crucial to the understanding and development of new efficient materials for industrial implementation. This review classifies and discusses recent advanced literature reports on phase transitions that occur during thermal treatments on metal–organic frameworks and their characterisation. Thermally activated phase transitions and procceses are classified according to the temperaturatures at which they occur: high temperature (reversible and non-reversible) and low temperature. In addition, theoretical calculations and modelling approaches employed to better understand these structural phase transitions are also reviewed.
 Celia Castillo-Blas | Dr Celia Castillo-Blas obtained her PhD degree in 2019 at the Institute of Materials Science of Madrid (ICMM-CSIC) under the supervision of Dr Felipe Gándara and Prof Ángeles Monge. In 2019, she moved to Universidad Autónoma de Madrid (Spain) for a postdoctoral position working at the group of Dr Ana Platero-Prats. Currently, she is a postdoctoral research associate at the group of Prof Thomas Bennett at the University of Cambridge and her research is focused on the preparation and characterisation of MOFs-inorganic glasses composites. She has also been a visiting researcher at the University of Jena. |
 Ashleigh M. Chester | Ashleigh M. Chester obtained her BSc degree in Chemistry from the University of Warwick in 2018 and completed her MSc at the University of St. Andrews in 2020. She is currently a PhD student at the Department of Materials Science and Metallurgy (University of Cambridge), working on the development of novel hybrid materials comprising inorganic glasses and metal-organic frameworks (MOFs). |
 David A. Keen | Professor David A. Keen was awarded his PhD at the University of Oxford in 1990, studying the structure of disordered materials by neutron scattering. He has worked on the local structure arrangements of condensed matter and has developed total scattering (pair distribution function, PDF) as well as experimental and reverse Monte Carlo computational methods to probe the relationship between structural disorder and the resulting physical properties of materials. He is currently a visiting professor at the Physics Department in Oxford University and research scientist at the ISIS Neutron Scattering Facility at the Rutherford Appleton Laboratory (Oxfordshire, UK). |
 Thomas D. Bennett | Prof Thomas Bennett obtained his PhD from the University of Cambridge in 2012, working under the supervision of Professor Anthony Cheetham FRS on the physical properties of hybrid frameworks. Currently, he is an Assistant Professor at the Department of Materials Science and Metallurgy (University of Cambridge), where his group focuses on hybrid melt-quenched glasses, stimuli-responsive framework behaviour and glass-based composites. He is also currently vice-chair of the international MOF advisory committee and has been a visiting researcher at the University of Canterbury New Zeland Te Whare Wānanga o Waitaha, University of Kyoto and the Wuhan University of Technology. |
1. Introduction
Metal–organic frameworks (MOFs) are a relatively new class of hybrid materials which have gained great attention in the field of the materials science for more than three decades, thanks to their versatility and their interesting and modulating properties. MOFs are formed by the self-assembly of an inorganic cluster, also denoted as a secondary building unit (SBU), and an organic linker to form porous periodic networks with different topologies.1 Owing to their remarkable porosity, MOFs have multiple desirable applications such as gas storage or separation,2 drug delivery systems,3 heterogeneous catalysis,4 and sensing,5 among others. As such, the structure–property relationship of these materials is crucial to the development of new materials for industrial implementation in these applications.Structural phase transitions can be triggered by different stimuli such as pressure,6 photochemical,7 guest adsorption/desorption,8 magnetism9 and most importantly, temperature.10
Temperature is a key factor for inducing solid-state transformations in all materials and compounds. However, despite the great scope of the available literature, most studies focus only on the thermal stability of materials at high temperatures.11 The lack of information on temperature-induced transformations is surprising given that many MOF applications are related to a better understanding of their response at different temperatures.
Several excellent papers have reviewed the thermal stability of MOFs,11 reversible single crystal-to-single crystal studies,8 MOF glasses,12,13 and MOF-derived materials obtained upon heating (carbons,14 metal oxides, metal chalcogenides, metal carbides and metal and metal oxide composites).15–18
Here we review the different nature of thermally activated, structural phase transitions and processes in MOFs and classify them according to how MOFs transform under different thermal treatments (Fig. 1). Moreover, we describe in detail the characterisation techniques and approaches to theoretical calculations used to classify these transitions.
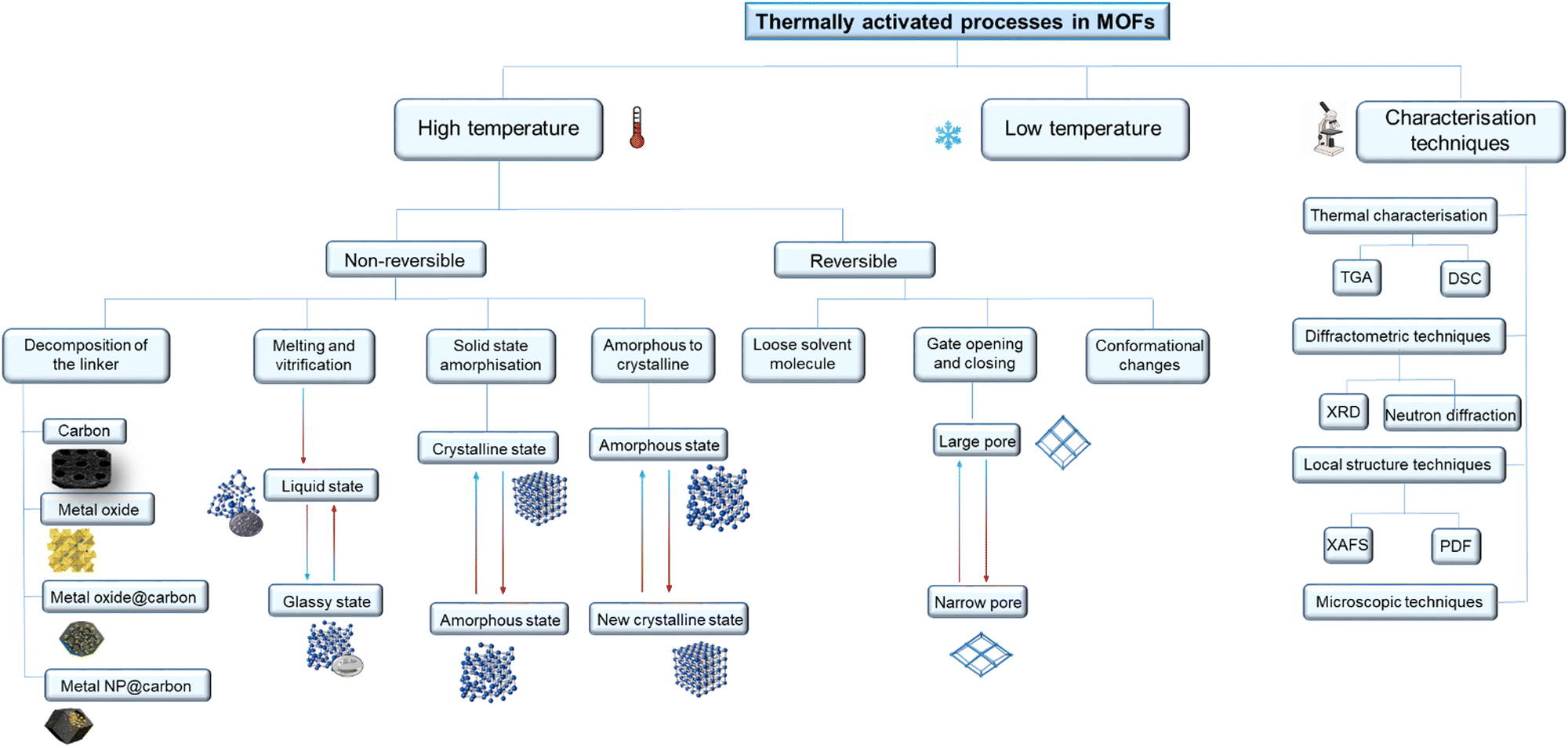 | ||
| Fig. 1 Schematic representation of the potential processes and structural phase transitions that can occur by applying thermal treatment to a MOF material. They have been classified into high temperature (non-reversible or reversible) or low temperature processes and transitions. Multiple characterisation techniques to understand these phase transitions and processes are also listed. | ||
In Section 2, the review focuses on non-reversible phase transitions induced upon heating, including partial or complete decomposition of the linker, and conformational or phase changes.
Section 3 reviews the reversible structural phase transitions that occur upon heating. The phenomenon of breathing, specifically related to flexible MOFs, is described in depth. Additionally, we describe the loss and recovery of solvent molecules that induce conformational or metal-coordination environment changes.
In Section 4, we concentrate on structural changes that occur when a material is cooled; these changes are less studied but relevant as most gas uptake isotherms are recorded at low temperatures under liquid N2 or using ice baths.
Section 5 summarises the characterisation techniques used to identify and monitor the structural phase transitions described above. These techniques include thermal characterisation, diffractometric, total scattering, microscopic and spectroscopic techniques.
Finally, Section 6 gives an account of the different theoretical calculations and modelling approaches employed to improve understanding of these structural phase transitions, as well as the thermodynamic implications to consider in these processes.
2. Non-reversible high temperature phase transitions
The thermal stability at high temperature of MOFs is often studied by thermal gravimetric analysis (TGA) as it provides information on the stability of the MOF upon heating. However, although it can reveal some phase transition temperatures, it does not provide insight on the induced structural changes and thus other techniques must be also utilised.Depending on the conditions used during the heating process, different structural transformations may occur. The control of heating rate, dwell temperature, heating time, gas atmosphere and flow rate are crucial to obtain desired structures and to induce the desired phase transitions.
2.1. Decomposition of the linker
MOFs are sensitive materials upon heating; MOF linkers and modulators frequently decompose above the decomposition temperature (Td) of the material, obtained from TGA. When this occurs, MOFs can form new materials with different chemical or structural characteristics in a non-reversible way, depending on the gas flow rate and the thermal treatment applied.This approach has been widely used to prepare different MOF-derived nanostructured materials such as carbons, metal oxides, metal oxides@carbon and metal particles@carbon materials, among others (Fig. 2). These types of materials have received much interest because they are often obtained via the formation of hollow or hierarchical structures.19 They are particularly suitable for electrochemical applications because MOFs have tuneable chemical compositions that can be designed at the molecular level. By judicious design of the parent MOF, advantageous properties, such as controlled porosity and high specific surface areas, can be translated to the MOF-derived material. Desired MOF-derived materials can be also obtained through a cautiously selected one-step thermal process from the parent MOF. This approach is widely applicable for their practical and industrial implementation.
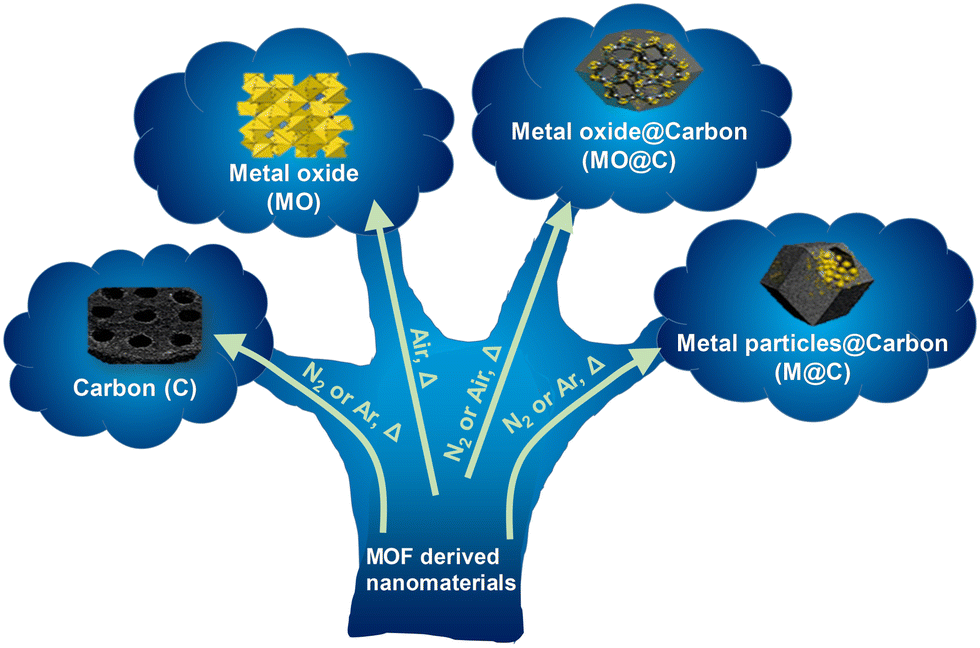 | ||
| Fig. 2 Schematic depiction showing the different MOF-derived nanocomposites and materials that can be obtained upon heating. | ||
Carbons can be synthesised from MOFs by a thermal process of carbonisation, using different synthetic approaches, with or without post-synthetic treatment. Carbonisation is a pyrolytic exothermic reaction where the organic linker partially decomposes and graphitises; the metal nodes are reduced from metal oxides to metal nanoparticles. Despite MOF carbonisation having been monitored using multiple characterisation techniques including powder X-ray diffraction, thermal gravimetric analysis or infrared spectroscopy, it is a very complex process involving multiple reactions, and its mechanism still remains unknown. Typically, carbonisation reactions are carried out in a reducing atmosphere, using N2, Ar, He or even H2 as the gas flow. Depending on the temperature, as well as the metal contained in the MOF, the metal can be reduced into nanoparticles and evaporated at a high temperature (1173 K). With these conditions, the reactions involved in this process might be, but not exclusively, dehydrogenation, condensation, hydrogen transfer and isomerization for the aliphatic chains and the posterior graphitisation at higher temperatures.52 These reactions often generate large amounts of CO2 and H2O gas, and they are associated with the partial decomposition of the linker and the loss of heteroatoms, as oxygen, nitrogen, hydrogen and phosphorus, among others.
The direct carbonisation of MOFs, involving just one thermal treatment step, often generates carbons with a low surface area. Usually this can be improved by a post-synthetic treatment, e.g. adding potassium hydroxide at high temperature and removing the potential impurities by acid washing. As a result, higher surface area carbons are obtained (>2000 m2 g−1).53 Furthermore, the adjustment of various parameters during carbonisation, such as heating rate, the nature of the MOF linker, and carbonisation time, has also been shown to be critical in determining the properties of the resulting carbonaceous materials.
An integral part of the structure of MOFs are metal-species in their SBUs. However, these metallic species must be removed to obtain high surface area carbon materials. A promising approach using Zn-based MOFs has been well-established to efficiently produce carbonaceous materials from MOFs.54 In this method, the MOF precursor undergoes an initial decomposition through heating in an inert atmosphere, leading to the transformation of zinc secondary building units (Zn-SBUs) into zinc oxide. Subsequently, the generated oxide is reduced to elemental zinc at 1173 K and then vaporised at this temperature, resulting in the formation of highly porous carbon material with tailored properties.
In a related example, a meticulous two-step carbonisation procedure was applied to isoreticular MOFs (IRMOFs) at temperatures of 823 K and 1173 K. This process was carefully monitored using X-ray diffraction and optical spectra (Fig. 3). Initially, the IRMOFs underwent transformation into a MOF-carbon composite at a relatively lower temperature (<1023 K), marked by the degradation of the primary MOF structure. Subsequently, as the temperature increased, a transition to a pure carbonaceous material occurred at 1173 K. During this transformation, zinc species originally present as ZnO were reduced to elemental zinc, which was subsequently vaporised. This series of reactions resulted in the production of a pure carbonaceous product, boasting an impressive surface area of 3447 m2 g−1.37
 | ||
| Fig. 3 Schematic depiction of the structural changes of IRMOF-1 with heat treatment to yield MDC-1.37 | ||
Alternatively, when a lower carbonisation temperature is applied, metals are retained in the carbonised samples. This also occurs in MOFs with SBUs consisting of metals with higher boiling points, such as iron, cobalt, copper or nickel. Conversely, these MOFs generate very porous carbon products after a post-acid wash treatment.55
In addition, the dimensionality of the MOF precursor can be retained in the final carbon products, culminating in possible materials with dimensionality ranging from zero-dimensional (0D) to three-dimensional (3D). An example of a 0D carbon material was prepared from ZIF-8 nanoparticles. These particles were heated to 773 K, producing carbon nanodots with an average particle size of 2.2 nm.24 Carbonaceous one-dimensional (1D) materials, such as carbon nanotubes, can also be obtained from MOFs with metal species such as Fe, Co, and Ni.56 In the high-temperature treatment, metallic nanoparticles were initially formed. Then, they catalysed the growth of carbon nanotubes on their surfaces.57 One such example was the pyrolysis without the addition of a catalyst or carrier gas at 773 K for 20 hours of [Ni3(BTC)2]·12H2O (BTC = benzene-1,3,5-tricarboxylate) in a tube furnace, which resulted in multi-walled carbon nanotubes (MWCNT).51 Utilising a different approach, two-dimensional (2D) carbon materials can also be synthesised from MOFs.58 For instance, a notable example involved the production of graphene-like carbon nanosheets by subjecting a composite of ZIF-7 and glucose to a carbonisation process in an Ar atmosphere at a temperature of 1223 K for five hours.42 ZIFs, zeolitic imidazolate frameworks, are a subclass of MOFs composed of divalent metal cation in a tetrahedral coordination environment linked through imidazolate ligands. Another example described the synthesis of a N-doped graphene particle analogue with a high nitrogen content (17.72%) from the pyrolysis of ZIF-8 particles for lithium storage applications.59 2D carbon nanosheets have been also prepared from an Al-MOF for use in Li–Se batteries.49 These examples demonstrate the structural versatility of MOF-derived carbon products; several additional examples of carbon-derived materials from MOFs are also summarised in Table 1. The thermal treatment used to prepare them, as well as their applications, are also given.
| MOF precursor | Product | Thermal treatment | Application | Ref. |
|---|---|---|---|---|
| ZIF-8 | NPC | 1173 K, N2 | ORR | 23 |
| ZIF-8 | CND-0D | 773 K, N2 | 24 | |
| ZIF-8 | N-PC | 1073 K, N2 | Sensing | 25 |
| ZIF-8 | NPC | 673 K, N2 | Adsorption of ethopabate | 26 |
| ZIF-8, sulfur powder | S-PC | 1323 K, NH3 | Lithium–sulphur batteries | 27 |
| ZIF-8 | N-PC | 973 K, N2 | Cancer therapy | 28 |
| ZIF-8, melamine | N-PC | 1223 K, N2 | Catalysis | 29 |
| ZIF-8 | PC | 1173 K, N2 | Batteries | 30 |
| ZIF-8 | N-PC | 1273 K, N2 | Water remediation | 31 |
| Zn-MOF | PC | 1173 K, Ar | Supercapacitor | 32 |
| Zn-MOF | PC | 1073 K, N2 | Energy storage | 33 |
| MOF-5 | PC | 1273 K, Ar | Water remediation | 34 |
| MOF-5 | PC | N2 | Catalysis | 35 |
| MOF-5 | PC | 1173 K | Capacitor | 36 |
| MOF-5 | PC | 1073 K, Ar | Gas storage | 37 |
| MOF-5 | PC | 823 K | Lithium batteries | 38 |
| MOF-74 (Zn/Fe) | PC | 1273 K, N2 | Water remediation | 39 |
| MOF-74 (Mg) | PC | 923 K, N2 | Capacitive deonisation | 40 |
| Mg-MOF | PC | 973 K, N2 | Capacitive deonisation | 41 |
| ZIF-7, glucose | NPC | 1223 K (5 h), Ar | 42 | |
| Zn-MOF | PC-2D | 1173 K, Ar | Supercapacitor | 43 |
| ZIF-67 | PC | 1073 K, Inert gas | Capacitor | 44 |
| MIL-88 (Fe) | PC | 1173 K, N2 | Capacitive deonisation | 45 |
| MIL-53(Fe) | PC | 1073 K, N2 | Water remediation | 46 |
| PCN-224, 222 | N-PC | 1273 K, N2 | CO2 reduction | 47 |
| UiO-66 | PC | 1073 K, N2 | Catalysis | 48 |
| Al-MOF | PC-2D | 1073 K, Ar | Batteries | 49 |
| ZIF-8 | HPC | 1273 K, N2 | 50 | |
| Ni3(BTC)2 | MWCNT | 51 |
Hollow carbon structures can also be produced from MOFs. The external-templating approach is conducted to create these materials with superior surface areas and loading capacities. This consists of coating a sacrificial or removable template, such as silica or polystyrene, with the desired MOF. In an example reported in 2014, hollow porous carbon (HPC) spheres were prepared by the pyrolysis of polystyrene@ZIF-8 core–shell microspheres.50
These MOF-derived metal oxides have been widely studied in different electrochemical applications such as lithium–air batteries,21,61–63 oxygen reduction reaction (ORR), carbon dioxide reduction, supercapacitors, water splitting,64 and others. They are also used as catalysts.
The structure of the resulting oxide is intricately shaped by a combination of factors, including the original composition of the MOF, the specific temperature and the duration of the thermal treatment applied. For MOF precursors containing cobalt, manganese, or iron, the calcination process often forms spinel oxides as the prevailing outcome. However, non-spinel oxides, such as CoO, Fe2O3, and Mn2O3 can be also obtained by controlling temperature, gas flow, time heated and heating rate. Other examples indicate the calcination of Zn, Cu, Ni and Ti-based MOFs leads to MgO, ZnO, CuO, NiO, TiO2 and ZrO2 as the products.
To date, a great number of porous metal oxide nanomaterials have been derived from MOF precursors, including cupric oxide (CuO),65 cobalt oxide (Co3O4),66 iron oxide (Fe2O3),67 magnesium oxide (MgO),68 manganese oxide (Mn2O3),69 nickel oxide (NiO),70 titanium dioxide (TiO2),71 and zinc oxide (ZnO).72 This approach has also been expanded to mixed transition metal oxides, such as ferrites (CoFe2O4 and Mn1.8Fe1.2O4),73 cobaltites (ZnCo2O4, ZnxCo3−xO4) and manganites (ZnMn2O4), including four-metal oxides with the spinel structure (Zn3−x−y−zMnxCoyCazO4). These have been employed for the ORR and reverse water–gas shift reaction (carbon dioxide reduction).74,75 Several representative examples are summarised in Table 2, where the MOF precursor, thermal treatment and applications are listed.
| MOF precursor | Metal oxide | Thermal treatment | Application | Ref. |
|---|---|---|---|---|
| MOF-199 | CuO | Air, 723 K, 2 h | Capacitor | 65 |
| Mg-aph-MOF | MgO | Air, 773 K, 12 h | CO2 uptake | 68 |
| Mn-BTC | Mn2O3 | Air, 723 K, 2 h | Li-battery | 69 |
| Fe-MIL-88B | Fe2O3 | Air, 657 K | Magnetic material | 76 |
| Fe-MIL-88B | Fe3O4 | N2 973 K | Magnetic material | 76 |
| Ni-BTC | NiO | Air, 973 K, 1 h | Li-battery | 77 |
| Ni-BTA | NiO | Air, 773 K, 2 h | Li-battery | 70 |
| Co-BDC | Co3O4 | Air, 723 K, 2 h | Supercapacitor | 78 |
| (Co3(NDC)3(DMF)4) | Co3O4 | Air, 873 K, 1 h | Li-battery | 79 |
| MIL-125 (Ti) | TiO2 | Air, 673 K, 4 h | Li-battery | 71 |
| ZnMn2–ptcda | ZnMn2O4 | Air, 723 K, 1 h | 80 | |
| Ni–Fe–PB | NiFe2O4 | Air, 973 K, 6 h | Li-battery | 81 |
| Ni/Fe MIL-88 | Ni0.62Fe2.38O4 | Air, 773 K | Li-battery | 82 |
| ZIF-67 | NixCo3−xO4 | Air, 623 K, 1.5 h | OER | 83 |
| Zn1−x−y−zMnxCoyCaz(hfipbb) | Zn3−x−y−zMnxCoyCazO4 | 1073 K, 24 h | ORR, CO2 reduction | 74,75 |
| ZIF-67 | Hollow Co3O4 | 533 K, air, 3 h | OER | 84 |
| [Cu3(btc)2]n | Cu2O/CuO | Air, 623 K | Li-battery | 85 |
| Fe2Ni MIL-88/Fe MIL-88 | NiFe2O4/Fe2O3 | Air, 723 K, 6 h | Li-battery | 86 |
| ZIF-67@NiCoLDH | Co3O4/NiCo2O4 | 623 K, 2 h | OER, CO2 methanation | 87 |
| Co3[Fe(CN)6]2@Ni3[Co(CN)6]2 | Fe2O3@NiCo2O4 | Air, 723 K, 6 h | Li-battery | 88,89 |
During the process of synthesising metal oxides, meticulous control of various parameters holds significant importance. These include factors such as surface area, chemical composition, particle size and shape, as well as the oxidation state. A recent example showcased the control of composition using MOF-derived methods, where nanoplate assemblies of ZnMn2O4 from a bimetallic MOF, ZnMn2–ptcda (ptcda = perylene-3,4,9,10-tetracarboxylic dianhydride) were created through heat treatment.80 Temperature control was critical for obtaining the desired spinel oxide. Additionally, a mixed valence metal oxide hollow structure (CuO/CuO2) has been synthesised by thermolysis at 523 K from [Cu3(BTC)2]n.
In another example, Co3O4 particles, with a size of about 25 nm, but a low surface area of 5.3 m2 g−1, were obtained from a Co-MOF.79 A method for selectively converting MIL-88B (Materials from the Lavoisier Institute) into either Fe2O3 or Fe3O4 has been demonstrated. When MIL-88B is heated in the presence of air, it transforms directly into hematite (Fe2O3). However, employing a two-step process involving calcination followed by heating in an inert atmosphere yields magnetite (Fe3O4). By using MIL-88(Fe) as a sacrificial template, spindle-like mesoporous Fe2O3 structures with a surface area of 75 m2 g−1 were produced. This approach involved a two-step thermolysis, which included heating in a N2 environment, followed by calcination in air.76
Many MOFs have been designed with core–shell structures for different uses, but creating core–shell metal oxide nanostructures from MOFs has proven more challenging. These challenges notwithstanding, derived core–shell metal oxides have gained increasing attention because of their excellent electrochemical performance and catalytic properties as they combine the electrochemical properties of both oxides. As an illustrative example, a two-step methodology has been employed to fabricate Co3O4@NiCo2O4 double-shelled nanocages, featuring distinct shell compositions. This innovative approach employs MOF particles as precursors. In this work, ZIF-67 particles were dispersed within a Ni(NO3)2 solution, giving rise to the formation of ZIF@Ni–Co yolk-shelled structures (Fig. 4).87 A subsequent annealing treatment converted the ZIF-67 cores and Ni–Co shells into Co3O4 (inner core) and NiCo2O4 (external shell), respectively. Another representative example is the thermal conversion of Co3[Fe(CN)6]2@Ni3[Co(CN)6]2 nanotubes to porous Fe2O3@NiCo2O4 nanocages as anode materials for Li-batteries.88
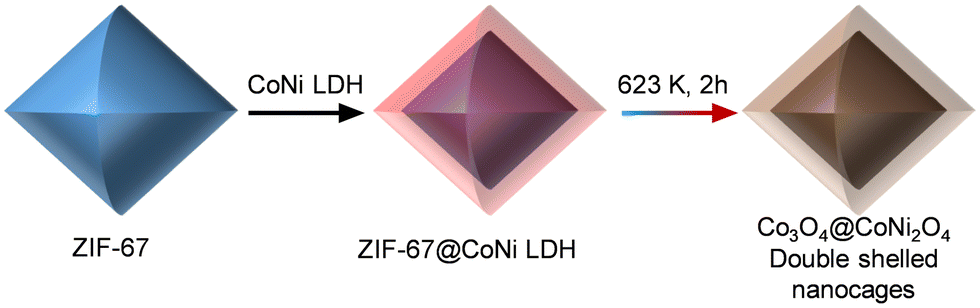 | ||
| Fig. 4 Schematic illustration of the formation process of Co3O4@NiCo2O4 double-shelled nanocages.87 | ||
Hollow metal oxide polyhedra can be also obtained using the external-templating approach. For this approach, a core–shell structure with ZIF-67 over micelles was prepared with a surfactant, alkyltrimethylammonium bromide (CnTAB; n = 12, 14 or 16), in aqueous media and calcined at 533 K for three hours.84
Traditional synthetic approaches of MO@C composites often lead to poor control over the metal oxide particle sizes because of agglomeration, which alters their electrode material performance. However, researchers can optimise thermolysis by heating MOFs, resulting in highly porous composites, MO@C and M@C (metal nanoparticles decorated carbon composites), because of the high surface area carbon produced in the process. This in turn mitigates particle agglomeration.
In one example, hybrid Co3O4@C porous nanowire arrays were prepared from a Co-naphthalenedicarboxylate MOF precursor with a layered crystalline structure. They were directly grown on copper foil using a low-temperature (353 K) hydrothermal process. Then, the components in the MOF were transformed into Co3O4 and carbon through carbonisation in a nitrogen atmosphere. This process produced nanowires with internal pores (Fig. 5). The resulting material had a high surface area of 251 m2 g−1 and a substantial carbon content of 52.1 wt%.92
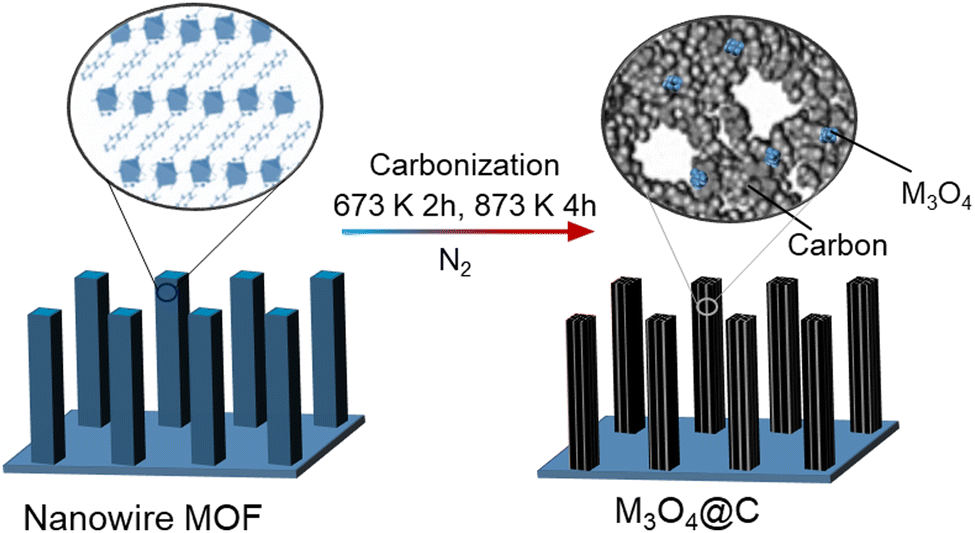 | ||
| Fig. 5 Schematic illustration for the fabrication of M3O4–C nanowire arrays.92 Inspired from ref. 92. | ||
A key study about the limits on obtaining metal, metal oxides and mixed metal–metal oxides particles concluded that the reduction potential of the coordination metal plays a critical role in determining the resultant product. The decarbonisation process generated metallic species through thermolysis when the reduction potential surpassed −0.27 V. However, at lower reduction potential values than −0.27 V, thermolysis produced a metal oxide as the resulting product.93 The study involved several MOFS, such as MOF-4, HKUST-1, Zn-ADA-1, MnHFMOF-D, MOF-CJ4, Cd-MOF-1, etc.
A MOF pyrolysis approach to prepare NP@C (carbon embedded nanoparticles) materials has also gained attention. Cobalt-based MOFs have been widely explored using this methodology to obtain Co@C–N (carbon–nitrogen embedded cobalt nanoparticles) and Co@C (carbon embedded cobalt nanoparticles) (Table 3).94
| MOF precursor | M@C/M@C–N | Thermal treatment | Application | Ref. |
|---|---|---|---|---|
| ZIF-67 | Co@C–N | 1073 K, 8 h, Ar | Oxidation of alcohols to esters | 94 |
| ZIF-67 | Co@C–N | 873–1173 K, 10 h, Ar | Catalysis | 96 |
| ZIF-67 | Co–CoO@C–N | 773–1073 K, 3 h, under N2 | Nitroarene hydrogenation | 97,98 |
| ZIF-67 | Co@CN | 1173 K, 2 h, N2 | ORR | 99 |
| Co9(btc)6(tpt)2(H2O)15 | Co@C–N | Ar 20 mL min−1, 773–1173 K | Oxydative amidation of aldehydes | 100 |
Co[(1,4-bdc)(ted)0.5]·2DMF·0.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) H2O H2O |
Co@C–N | 773–1073 K 8–15 h, Ar | Hydrogenation of aliphatic and aromatic | 101 |
| ZIF-8 | Zn@C–N | 973–1073 K, 2 h, heating rate 5 K min−1, N2 60 mL min−1 | CO2 cycloaddition with epoxides | 102 |
| MUV-3: [Fe(mIm)2] | Fe@C | 973 K, 3.5 h, under N2 | OER | 103 |
| Basolite F-300 | Fe@C | 773 K 8 h N2 | Fischer–Tropsch catalysis | 95 |
| Fe/Co-MOF | Fe/CoOx@C | 973–1173, 0.5 h, | OER | 104 |
Gascon et al. demonstrated an innovative approach, exemplifying excellent dispersion of iron nano-particles within a porous carbon matrix.95 This methodology involved the impregnation of the Fe-precursor (Basolite F-300) with a carbon source, followed by pyrolysis, as illustrated in Fig. 6.
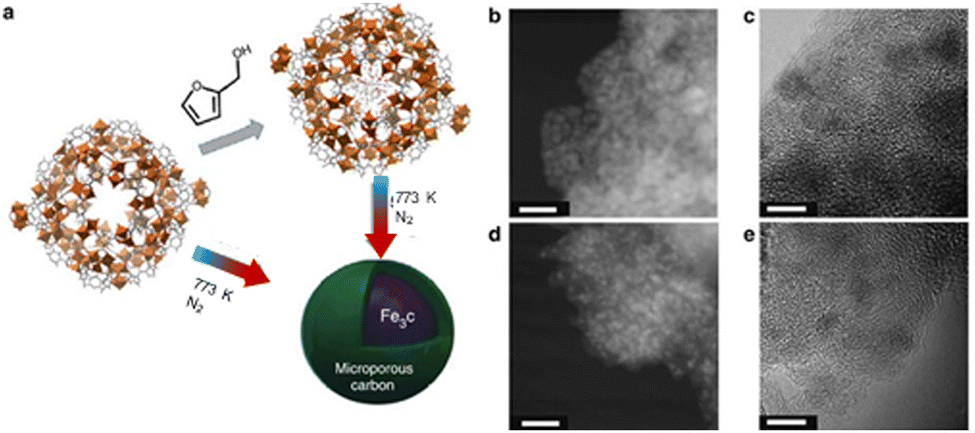 | ||
| Fig. 6 (a) MOF mediated synthesis strategy for the Basolite F-300 material. (b) High-angle annular dark-field scanning electron (HAADF STEM) images of Fe@C (scale bar, 20nm). (c) High-resolution (HRTEM) micrographs of 38-Fe@C (scale bar, 5 nm). (d) HAADF STEM of 25-Fe@C (scale bar, 20 nm) HRTEM image of 25-Fe@C (scale bar, 5nm). Adapted from ref. 95 with permission from Springer Nature, copyright 2015. | ||
Hollow M@carbon composites can be also prepared using a similar thermal treatment. These hollow materials have larger surface areas, superior loading capacities and lower densities than their counterparts. This makes them highly desirable materials for electrochemical and catalytic applications.
2.2. Melting and vitrification behaviour
Traditionally, the MOF field has focused almost entirely on the crystalline domain. However, order–disorder and solid–liquid-glass phase transitions upon heating are commonly observed and the resulting phases have great importance in materials science.105 Some of their properties are beneficial for material processing and provide unique features for their industrial implementation.106,107The solid to liquid transition is reached upon heating above the melting point of the material. This transformation is reversible and the mechanism can be explained for simple systems following the Lindemann criterion, which proposes that melting occurs when the root-square of the vibration amplitude of particles in crystals exceeds a critical threshold proportion of the interparticle distance.108,109 It is also crucial that the material possesses a decomposition temperature (Td) higher than the melting temperature (Tm), to avoid the total or partial decomposition of the material during melting. These temperatures vary according to the nature of the solid material (covalent, organic, inorganic), the applied pressure and the surrounding environment (argon, air, nitrogen, etc).110 These criteria mean that melting has rarely been observed because of the relatively low thermal stability of the MOF and coordination polymer (CP) materials.11
Different families of hybrid materials, such as hybrid-organic inorganic perovskites (HOIPs), CPs and porous organic cages (POCs) have shown melting upon heating. Usually, these materials have melting temperatures around 400–500 K.111 For example, dicyanamide-based HOIPs and their derivatives melt at 379–535 K.112 This melting temperature can easily be varied by changing the ammonium cation located at the perovskite A sites, as well as the B site metal cation.
Despite the reduction of intrinsic porosity of the crystalline MOF after a liquid phase transition, opportunities have been identified for the unique behaviour of dense and processable melt-quenched glass phases. Glassy MOFs are mouldable and grain boundary-free, both properties may facilitate the fabrication of homogeneous membranes for gas separation, solid electrolytes, optical materials and composite hybrids. However, until now, examples of porous 2D and 3D CPs or MOFs that melt remains scarce.
However, glassy MOFs can be synthesised using other routes including mechanical vitrification113 or direct synthesis.114,115 These avoid the issues associated with the thermal decomposition of MOFs, but the most conventional route to glassy MOFs formation remains melt-quenching.13
From a molecular-kinetics point of view, the equilibrium liquid to non-crystalline solid transition may be considered as a fourth order phase transition, different to gas, liquid and solid states.116 A melt-quenched glass, also known as a ‘frozen-liquid’, is a disordered material that lacks the periodicity of crystals, but behaves mechanically like solids. They are formed by cooling a liquid fast enough to avoid crystallisation (Fig. 7).117 They can be distinguished from other amorphous solids by the presence of a glass transition temperature (Tg). Tg is a temperature or temperature range over which a reversible transition between a glassy state and a more viscoelastic state occurs.
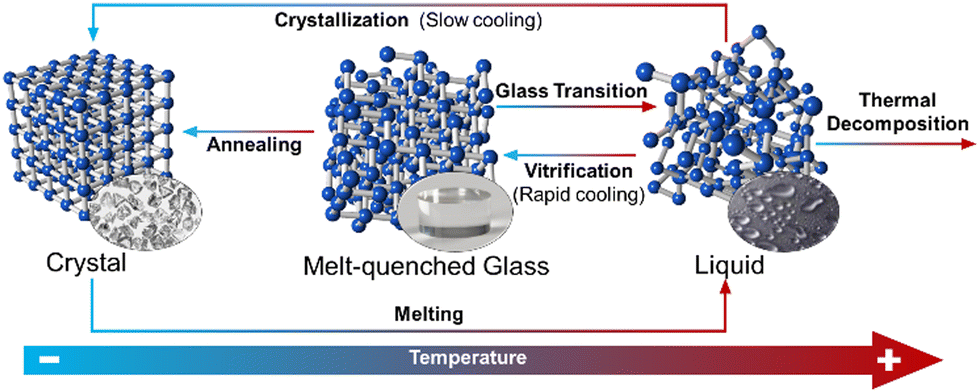 | ||
| Fig. 7 Schematic depiction of the processes for achieving melt-quenched CP/MOF glasses. | ||
The first MOF which demonstrated the ability to melt and form a glass after quenching was ZIF-4.118 ZIF-4, Zn(Im)2 (Im− = imidazolate), is a material with a cag network topology possessing one of the most complex behaviours upon heating in MOF-based materials.119 ZIF-4 amorphises at 603 K before recrystallising into a denser phase denoted ZIF-zni at 673 K.120 This denser ZIF-zni phase melts at 863 K, and then decomposes at a slightly higher temperature (873 K).121 The melted liquid can be quenched by rapid cooling down before decomposition occurs, producing a ZIF glass with a Tg of 565 K.118 However, the melting point of ZIF-zni is very high and exhaustive efforts have been explored to minimise this Tm value, whilst maintaining the porosity of the ZIF-4 structure and preventing decomposition of the material upon heating. The addition of other linkers into the ZIF-zni structure drastically changes the thermal behaviour of its structure. ZIF-61, [Zn(Im)1.35(mIm)0.65] (mIm = 2-methylimidazolate), decomposes upon heating without passing through the liquid state. On the other hand, trace amounts of 5-aminobenzimidazolate (abIm) into the ZIF-zni structure creates a new material, [Zn(Im)1.995(abIm)0.005], which melts at 842 K (a lower temperature than that of pristine ZIF-4 zni) with no evidence of decomposition to zinc oxide.122
The most well-known approach for reducing Tm is partial substitution of the imidazolate ligand by another imidazole-derived linker, such as benzylimidazolate (bIm−) in the structure. This led to the idea of exploring the thermal behaviour of ZIF-62, [Zn(Im)2−x(bIm)x] (Im− = imidazolate, x = 0.1–0.3). It was found that the Tgs of ZIF-62 (in the range of 571–593 K) can be tuned by changing the Im/bIm ratio.123 Isostructural ZIF analogues with cag topology have also been prepared with cobalt, which exhibit even lower Tg values (533–563 K). Additionally, multivariate-MOFs (MTV-MOFs) have Tgs which can be modified using mixtures of different linkers and metals.124
Moreover, the addition of other benzylimidazolate linkers with different functionalities has also been explored to investigate their thermal behaviour, improve their mechanical performance and add new properties. For example, by adding 2-aminobenzylimidazole as a linker, an amino functionality was introduced to create a new ZIF, ZIF-UC-6, with Tg and Tm values of 589 and 618 K, respectively.125 Interestingly, the surface of this glass can be post-synthetically modified, which changes its hydrophobicity. In a different example, the substitution of benzylimidazolate with purinate (pur−) in the ZIF-62 structure led to increased CO2 uptake capacity for both the resulting crystalline and glass ZIF-UC-7, Zn(Im)1.75(pur)0.25.126
Recently, imidazole was substituted in ZIF-4 by an imidazole containing cyano groups, forming ZIF-4-CNx materials, with the following compositions: [Zn(Im)2−x(CNIm)x] CNIm− = 4-cyanoimidazolate, (0.04 ≤ x ≤ 0.39) and [Zn(Im)2−x(dCNIm)x], dCNIm− = 4,5-dicyanoimidazolate, (0.10 ≤ x ≤ 0.28).127 This family of materials melts and forms melt-quenched glasses with a range of Tgs between 527 K and 249 K that can be easily varied controlling the Im/CNIm ratio.
Apart from Zn and Co, other metals can also be investigated. Recently, a very interesting iron non-porous CP [Fe3(Im)6(Him)2]128 was studied upon heating. This CP exhibits a mass loss associated with the removal of the terminal imidazoles under N2 atmosphere over 556 K. This mass loss is associated with a structural phase transition to a crystalline phase denoted MUV-24 [Fe(Im)2] with a lla topology.129 Further heating under N2 flow results in another structural transition to form a zni polymorph above 690 K. This polymorph is able to melt above 755 K and form a melt-quenched glass after rapid cooling. Additionally, changing the conditions upon heating produces another phase transition from MUV-24 (lla) to MUV-24 with a coi topology.
Although the ZIF family encompasses the most representative examples of melting materials, other coordination polymers (CPs) with different dimensionalities can also melt, where several can form a glass without recrystallisation upon rapid cooling down (Table 4).
| MOF/CP precursor | T m/K | T g/K | Ref. |
|---|---|---|---|
| ZIF-zni: [Zn(Im)2] | 836 | 565 | 118,121 |
| ZIF-4-CNx: [Zn(Im)2−x(CNIm)x] (x = 0.04–0.39) | 549–543 | 127 | |
| ZIF-4-dCNx: Zn(Im)2−x(dCNim)x (x = 0.10–0.28) | 545–527 | 127 | |
| ZIF-62: [Zn(Im)2−x(bIm)x] (x = 0.05–0.35) | 645–714 | 571–593 | 121,123,145 |
| ZIF-62 (Co): [Co(Im)2−x(bIm)x] (x = 0.10–0.30) | 659–705 | 533–263 | 121,145 |
| [Co0.2Zn0.8(Im)1.95(bIm)0.025(5-ClbIm)0.025] | 583 | 561 | 146 |
| ZIF-UC-1: [Zn(Im)2−x−y(bIm)x(mbIm)y] | 691–706 | 578–589 | 124 |
| ZIF-UC-2: [Zn(Im)1.90(6-Cl-5-FbIm)0.10] | 679 | 523 | 147 |
| ZIF-UC-3: [Zn(Im)1.75(5-Cl-2-mbIm)0.25] | 663 | 609 | 147 |
| ZIF-UC-4: [Zn(Im)1.63(5-FbIm)0.37] | 694 | 563 | 147 |
| ZIF-UC-5: [Zn(Im)1.69(5-ClbIm)0.31] | 705 | 593 | 147 |
| ZIF-UC-6: [Zn(Im)1.82(abIm)0.18] | 618 | 589 | 125 |
| ZIF-UC-7: [Zn(Im)1.75(pur)0.25] | 591 | 546 | 126 |
| TIF-4: [Zn(Im)1.5(mbIm)0.5] | 713 | 623 | 121 |
| ZIF-zni-NH2: [Zn(Im)1.995(abIm)0.005] | 842 | 616 | 122 |
| MUV-24: [Fe(Im)2] | 755 | 463 | 129 |
| [M(L1)2] (M = Zn, Co, Mn, Cd) | NA | 421–504 | 130 |
| [M(L1X)2] (L2X = L2, L2F, L2Cl, L2Br, L1NH2) | NA | 393–562 | 130 |
| [Co(L3)2] | NA | 498 | 130 |
| [Co(L4)2] | NA | 553 | 130 |
| [Co(L5)2] | NA | 526 | 130 |
| [Co(L6)2] | NA | 529 | 130 |
| [Co(byp)]fum | NA | 396 | 131 |
| [Co(byp)]bdc | NA | 393 | 131 |
| [Co(bpee)]bdc | NA | 393 | 131 |
| [Ag(pL2)(CF3SO3)]·2C6H6 | 544 | 434 | 148 |
| [Ag(mL1)(CF3SO3)]·2C6H6 | 442 | 341 | 148 |
| [Cu2(SCN)3(C2bpy)] | 460 | 341 | 149 |
| [Cu2(SCN)3(C4bpy)] | 411 | 332 | 149 |
| [Cu8(SCN)12(Phbpy)4] | 490 | 344 | 149 |
| [Cu(SCN)2(3-Pybpy)] | 476 | 345 | 149 |
Most meltable CPs and MOFs contain imidazolate and triazolate linkers combined with metal cations of the first transition series, most of them containing Zn2+. Very recently, Horike et al. have observed for the first time the vitrification phase transition of a coordination polymer containing carboxylate linkers via the decoordination of the water molecules from the network structure before recrystallisation to porous glasses.130 This has also been demonstrated in coordination polymers containing terephthalate linkers.131 These unprecedented results promise new and exciting avenues to obtain hybrid glasses (Table 4).
The reduced porosity question of these hybrid glasses is still unresolved, but several approaches have shown exciting results in boosting porosity in these materials.132 Some of them include the fabrication of membranes and composites comprising hybrid glasses. Alternatively, MOF-glass membranes, prepared via crystallite-MOF dispersion on a porous ceramic alumina support and posterior melting upon heating, have displayed excellent gas separation performances.133 In addition, a ZIF-62 glass foam self-supported membrane has been also fabricated through the thermal decomposition of a MOF. This material has a larger number of pores and shows a great permeance of CH4 compare to other ZIF-glasses.134
Another interesting approach is the incorporation of crystalline MOFs into a MOF-glass matrix to form composites.135 Moreover, glass matrices have demonstrated the ability to retain open-pore structural phases, increasing gas uptake.136 Recently, Knebel et al. have reported a synthetic approach to increase the CO2 diffusion in ZIF-62 glass by controlling the melting and tempering parameters, resulting in changes of the pore channel structure in the ångström-range.137 Another promising approach, still not applied in the synthesis of MOF glasses, is the use of small molecules such as carbonates, that decompose during the melting and tempering of the glass. This method has shown promise with introducing pores in inorganic glasses.138
As a result of the exciting properties of these glassy MOFs, this emerging field in materials science has gained great attention. These transformative materials can offer realistic solutions in luminescence, gas separation, catalysis, biomedical and optical applications with promising industrial implementation. Thanks to their intrinsic transparency and isotropic homogeneity, MOF glasses have demonstrated properties favourable to luminescence applications, especially if they contain lanthanides or luminophore ligands.139,140 Moreover, ZIF-62 and their derivatives exhibit high transmittance (∼90%)141 and exciting nonlinear optical response for potential use in photonics.142 Additionally, gas separation membranes containing MOF glasses have demonstrated promising results in the separation of gas mixtures, offering a more industrial applicable approach.133,143,144 Furthermore, imidazolate and purinate derivatives are biocompatible, which could show promise in biomedical applications such as drug delivery and scaffolds.
2.3. Solid state amorphisation
Temperature induced crystalline to amorphous transformations have been studied in detail owing to their impact on hybrid glasses in materials science. This thermal process does not involve partial or total decomposition of the linkers, and the MOF's chemical composition is retained. In 2010 the first example of an amorphous ZIF formed by thermal treatment was reported for Zn-ZIF-4 (cag topology) without any intermediate melting step.120 Later, Zn-based ZIF-1, ZIF-3 and Co-ZIF-4 with topologies crb, dft and cag, respectively, also amorphised upon heating.150 This effective amorphisation upon heating, without decomposition, has been linked to their topologies. Topologies such as cag, BCT or DFT are able to form amorphous materials without decomposition, whilst ZIF-8, ZIF-9, ZIF-11, ZIF-14, and ZIF-βqtz with SOD, SOD, RHO, ANA and qtz topologies respectively, decompose whilst undergoing an amorphous phase transition. Conversely, and very recently, a SOD topology ZIF (the same topology as ZIF-8), known as CdIF-1, has exhibited amorphisation without decomposition. After amorphisation, an improvement in diffusion and adsorption selectivities of n-C4H10/i-C4H10 was observed, where it was suggested that the longer Cd–N bonds are associated with a more flexible framework, culminating in these improved properties.151An exciting example of these transitions is the temperature-induced amorphisation of a Co-MOF (SCNU-Z6), [Co0.7HL] (HL = 2,6-bis-(4-imidazol-1-yl-phenyl)-4-[4-(2H-tetrazol-5-yl)-phenyl]-pyridine).152 This aMOF was formed by the removal of the solvent molecules in the framework over 368 K and exhibited excellent iodine capture capacity.
2.4. Amorphous to crystalline
Many studies have focused on the amorphisation of crystalline MOFs. However, despite their scarcity in the literature, amorphous-to-crystalline transformations have also been reported in MOFs. Glasses and amorphous materials are metastable, which means that they are able to transform into more stable structures and topologies with the application of high temperature over time. One of the most known MOF examples of this phenomenon is ZIF-8.Mechanically amorphised ZIF-8 has exhibited the formation of a new, crystalline quartz topology upon heating under argon flow (Fig. 8).153 This opens up the possibility of exploring the thermal behaviour of other amMOFs upon heating to obtain new desired crystalline phases.
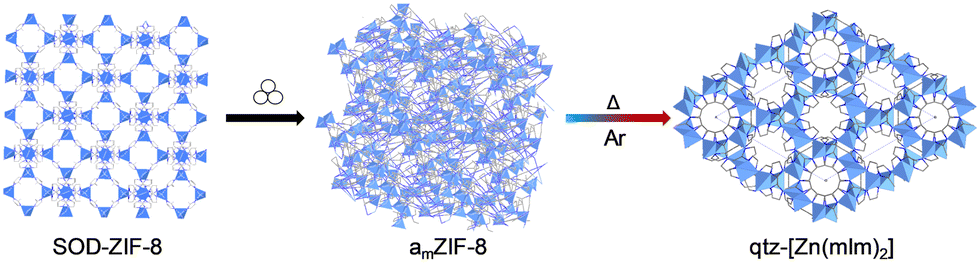 | ||
| Fig. 8 Schematic depiction of the synthetic approach to obtain a new qtz-topology from a mechanically amorphised ZIF.153 Reproduced from ref. 153 with permission from Royal Chemical Society, copyright 2022. | ||
3. Reversible high temperature phase transitions
3.1. Metal-coordination environment desolvation and guest molecules
One important characteristic of MOFs is their intrinsic porosity and their ability to host guest molecules. However, such adsorbed molecules can also be released upon heating, leading to a change in the structural conformation of the host-structure. Most guest molecules are located in the cavities of the MOF. However, there are examples where several molecules are also coordinated to the SBU, which are especially prevalent in non-porous materials and coordination polymers. It is often the case that these guest molecules interact with the host network in a supramolecular fashion.154,155 Therefore, elimination of these coordinated and/or uncoordinated solvent molecules may result in different structural transformations with variable significance, which may be reversible or non-reversible in nature (Fig. 9).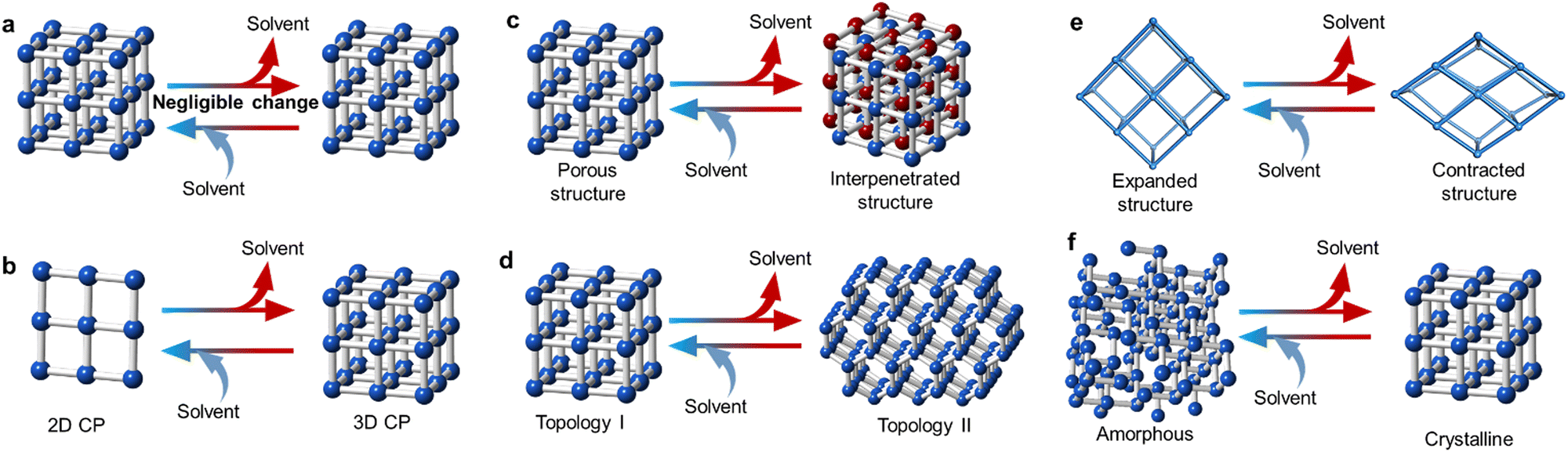 | ||
| Fig. 9 Schematic depiction of the potential changes after the loss of solvent molecule. (a) Negligible changes after solvent loss. This means only changes in the coordination environment of the metal cluster occur without changes to the whole structure. (b) Increase in the dimensionality of the structure after solvent loss. (c) Creation of an interpenetrated structure. (d) Topological change after solvent loss. (e) Contraction or expansion of the structure. This behaviour is commonly known as breathing. (f) Amorphisation or recrystallisation after solvent loss. Red and blue arrows indicate heating and cooling processes, respectively. | ||
In some cases, the exchange of coordinated solvent molecules leads to a drastic transformation across the entire framework. However, in some cases the loss of the solvent molecule results only in a slight structural distortion in the crystalline network, despite a large change in local coordination. This has been observed as colour changes in several cases. For example, a 3D magnetic MOF [KCo7(OH)3(ip)6(H2O)4]·12H2O (H2ip = isophthalic acid) underwent a reversible structural transformation to [KCo7(OH)3(ip)6] when heated to 393 K, accompanied by a colour change from pink to purple. Upon the removal of the solvent molecules during heating, a change in the coordination geometry was observed, but without noticeable crystal lattice variations.156 When the dehydrated purple crystals formed after the heat treatment were exposed to air for a few days, they reverted to their original red colour. Although both structures were similar, the metal coordination environments in the dehydrated compound were very different.
Some of these transitions can increase or decrease the dimensionality and the reorganising of the network. One remarkable example of this was reported by Kitagawa et al.157 Here, a porous 2D coordination polymer was reversibly transformed into a 3D structure by dehydration after heating at 353 K for four hours. The material, {[Cu2(TCI)(OH)(H2O)3]·1.5H2O}n, TCI = (tris(2-carboxyethyl)isocyanurate), contained a TCI flexible organic linker and a SBU consisting of a CuII ion. This material transforms into a 3D framework upon guest removal, caused by the sliding of the 2D sheets and contraction of the spaces between the layers. Here, versatile coordination geometry of the CuII ion and ligand flexibility played a key role in the reversible structural transformation, which was accompanied by a change in optical and magnetic properties. Another interesting example reported by Kondo et al. showed a structural change from a 0D CP {[Co(bpy)2(CH3CN)2(H2O)]·2(OTf)} (bpy = 4,4′-bipyridine, OTf = trifluoromethanesulfonate) into a 1D CP through the irreversible loss of the acetonitrile solvent after air exposure.158 Moreover, the 1D {[Co(bpy)2(OTf)2(H2O)2]·(bpy)} reversibility transformed into a 2D porous material, ([Co(bpy)2(OTf)2]), through loss of water molecules after a thermal treatment at 423 K under vacuum. Dimensionality changes can also be reached through solvent exchange. For example, a 2D Zn-CP exhibited a transformation into a 3D-MOF after exposure to dichloromethane at room temperature, improving the framework stability, porosity and hydrogen uptake.159
Interpenetrated structures can also be affected by the removal of coordinated solvent molecules. One such example was MOF-123, [Zn7O2(NBD)5(DMF)2] (NBD = 2-nitrobenzene-1,4-dicarboxylate), which contains DMF molecules occupying the channels.160 By heating to 593 K, MOF-123 undergoes a structural transformation, resulting in a doubled interpenetrated [Zn7O2(NBD)5] material, known as MOF-246. This transformation was reversible and was associated with a 50% reduction in the thickness of the crystal after loss of the DMF molecules.
Porous structures have the capacity to accommodate a diverse range of molecules within their channels. Within the lattice, solvent molecules can engage in interactions with the host material through supramolecular bonding. Interestingly, when these solvent molecules are either lost or adsorbed, it leads to only minor alterations in the overall structure and connections within the coordination network. The chosen method for the removal of solvent molecules can cause different types of structural changes. An example of this was shown by a Mn-based MOF, {[Mn(L)(X)](X)x}n, where L = and X is the guest molecule = pyridine, water, dimethylformamide, 2-picoline, 4-picoline, aniline, benzonitrile or 2,6-lutidine. Depending on the guest molecule in the pore, different coordination modes of the metal centre were observed, generating materials with various space groups.161 Another remarkable example is the dehydration of [Cu(iba)2]·2H2O (Hiba = 4-(1H-imidazol-1-yl)) benzoic acid which results in different porous polymorphs depending on the dehydration method.162 When the crystal was heated at 433 K for 12 hours, the product CP retained the original topology. However, when exposed to air at room temperature for two months, a 2D complex, [Cu(iba)2], was formed instead. Kinetic and/or thermodynamic processes are thought to control the different routes.
In most flexible porous materials, the reversible loss of the solvent serves as a distinctive feature, inducing a disruption within the host structure without the breakage or formation of new chemical bonds. This unique characteristic highlights the adaptability of these materials to environmental changes. Despite the absence of bond rearrangements, the intricate interplay of multi-variable coordination geometries exhibited by metal ions, the dynamic nature of metal–ligand bonds, and the arrangement of supramolecular packing contribute significantly to structural transformations. As an example, tetragonal [Ag6Cl(atz)4]OH·6H2O (Hatz = 3-amino-1,2,4,-triazole) can be dehydrated reversibly to obtain [Ag6Cl(atz)4]OH·xH2O.163 Here, the host framework structure has a large distortion from the original one, but the topology of the [Ag3(atz)2] network is retained with similar structural parameters. Both structures exhibited a six-fold interpenetration. Another example showed a dynamic MOF, CCIQS-1, exhibit different phase transitions as a result of solvation and desolvation processes.164 Desolvation of the as-synthesised phase shows a phase transition from Cmc21 to P212121, accompanied by a contraction of the unit cell and the opening of the hydrophilic channels. This process was selectively reversible by soaking the material in different solvents, such as DMF, THF, 1-hexene or mixtures of DMF and toluene.
Another interesting behaviour observed in the loss of solvent molecules is the structural memory effect. For example, Du et al. reported a reversible crystal-to-amorphous phase transition of a sponge-like MOF, [Mn(Me-ip)(DMF)]n, (Me-ipH2 = 5-methylisophthalic acid) to an amorphous phase, in which DMF molecules were released upon heating above 453 K. The crystallinity of this phase was recovered after 48-hour immersion in DMF.165 This material exhibits a crystalline-amorphous-crystalline behaviour recovering its crystalline structure from an amorphised structure. This memory effect can also be delayed, which decreases the crystallite-size whilst maintaining a mesostable open-pore phase when the solvent is removed. The close-pore phase is recovered after a thermal treatment. A remarkable example is [Cu2(bdc)2(bpy)]n, a porous framework reported by Kitagawa et al. where 50 nm crystallites in their dried form maintained the open-pore phase.166 However, >100 nm particles contained a mixture of phases (closed and open-pore phase) and crystallites with sizes over 300 nm exhibited a pristine closed-pore phase.
Guest loss in MOFs upon heating can also be used in sensing. For example, multiple MOFs have been investigated as volatile organic compounds (VOCs) colorimetric sensors thanks to their colour change after the VOC is removed.167,168
An interesting example is a luminescent cationic MOF, [{Zn(L)(MeOH)2}(NO3)2·xG]n (where L is 4,4′-ethylenedianiline and 2-pyridine-carboxaldehyde, and G are disordered guest molecules).169 The structure of this MOF changed drastically, in a reversible way, with the loss of methanol or water molecules, which led to changes in the shapes and sizes of the pores. In addition, the anion in this MOF can be easily exchanged with anions (ClO4−, SCN−, N3− and N(CN)2−), which enabled tuning of their luminescent properties, accompanied by expansion or contraction of the unit cell. A similar behaviour was observed in a layered Zn-MOF with nitrate anions located in the interlayer channels. In this example, anions could be exchanged, generating three different topologies depending on the nature of the guest anion.170
3.2. Gate opening and closing in flexible MOFs.
Typically, upon heating, materials expand in three dimensions because of the increased vibrations of their constituent atoms.171 In addition to positive expansion, unusual structural arrangements can lead to unexpected thermal expansion behaviours, such as negative (contraction) or zero. Several MOFs show a reversible change in their unit cell parameters, whilst maintaining their composition after thermal treatment. They are known as thermoresponsive MOFs.10 Thanks to their flexibility, MOFs often exhibit these anomalous behaviours.172 For example, MOF-5,173 IR-MOFs174 and HKUST-1,175 have shown remarkable experimental and theoretical negative thermal expansion (NTE). Moreover, MOF-339 has the largest theoretical negative thermal expansion value reported to date.176 However, this material is not accessible experimentally because of its low mechanical stability. Another example of anomalous behaviour is the anisotropic thermal expansion demonstrated by HMOF-1.177 In this example, the a cell parameter increased, b decreased and the c parameter remained unchanged upon heating. Additionally, a Zn-based MOF, [[Zn(napht)2(OH)2]n·nCH3OH], exhibited a large anisotropic thermal expansion of its network at 370 K.178 More specifically, this anisotropic behaviour involved positive thermal expansion (PTE) along the c axis accompanied by biaxial NTE along the a and b axes. A remarkable example reported by Kaskel et al. is (Cu)DUT-69, [Cu2(BBCDC)]Xn (X = solvent), which showed a change in the material's thermal behaviour from positive to negative thermal expansion, depending on the presence of solvent guest molecules.179 This behaviour could be tuned depending on the transition metal selected.Flexible MOF systems can be categorised according to the type of metal-node and how their coordination chemistry with the linker molecules controls the framework dynamics.
One type of distortion that has been observed in flexible MOFs has been termed breathing behaviour. This phenomenon is associated with reversible structural transitions of MOFs, where atoms are substantially displaced in the framework and the unit cell volume changes.10 These materials are able to transform between closed or narrow pore (np) to open or large pore (lp) conformations (Fig. 10). Unit cell parameters and crystallographic space groups of the distinct phases (narrow pore and large pore) are frequently different. Pioneering groups in this field include those of G. Ferey and S. Kitagawa, who have expanded this concept since 2004.180
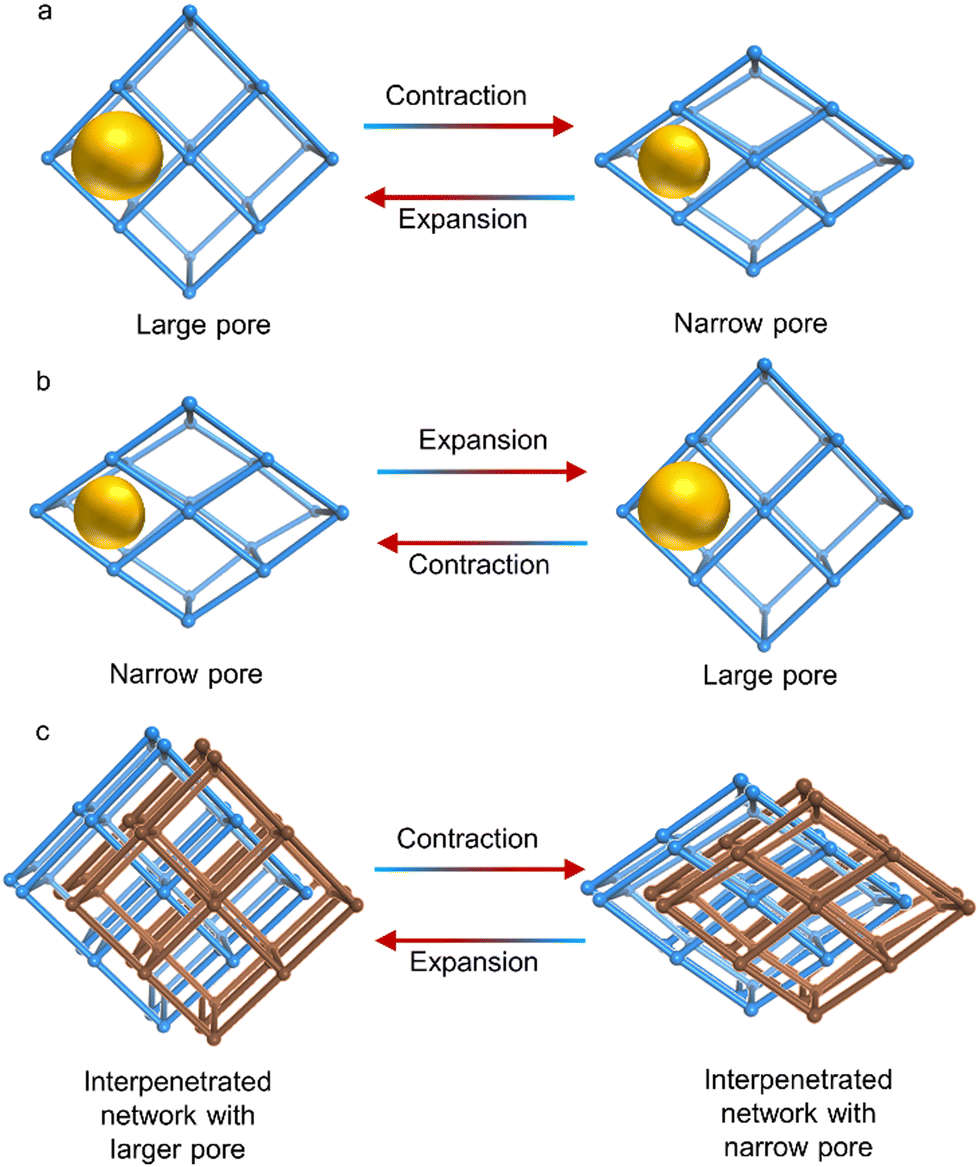 | ||
| Fig. 10 Schematic depiction of gate opening and closing processes. (a). Contraction of the unit cell upon heating. (b) Expansion of the cell upon heating. (c). Contraction of the cell upon heating with an interpenetrated structure. Yellow spheres indicate the pore size and red and blue arrows show heating and cooling processes, respectively. | ||
Similarly, various research groups have developed new, highly breathable frameworks. These frameworks can be adjusted in terms of their flexibility, rigidity, and how much they can expand and contract by using specific solvent molecules within their lattice structures.156,181 One framework, namely [Zn3(OH)2(btca)2]·DMF·4H2O (H2btca = benzotriazole-5-carboxylic acid),182 exists in a large pore phase, which then transforms into a small pore phase, [Zn3(OH)2(btca)2], at 713 K for 30 minutes. Two intermediates were identified at 493 K, [Zn3(OH)2(btca)2]·DMF·0.5H2O and [Zn3(OH)2(btca)2]·2H2O. Another example is Co(BDP)·2DEF·H2O, with DEF molecules present in the channels.183 Upon heating to 443 K, desolvation of these molecules occurred, generating a substantially different structure. The original structure was regenerated by exposing the desolvated one to DEF. These findings align with a flexing behaviour similar to an accordion, which alternately narrows and widens the channel pores. This behaviour has been previously shown in similar frameworks linked by terephthalate.184
One of the most remarkable examples of breathing was reported by C. Serre et al. in 2008.185 They observed that expansion upon solvent adsorption occurs without apparent bond breaking, in a series of isoreticular chromium and iron dicarboxilates with formula [M3IIIO(H2O)2X(fum)3]·guest (M = Fe, Cr; X = F, Cl, acetate), labelled MIL-88A to D [fum = fumarate (88A), terephthalate (1,4-BDC) (88B), 2,6-naphthalenedicarboxylate (NDC) (88C), and 4-4′-biphenyldicarboxylate (BPDC) (88D)]. The as-synthesised forms were able to accept solvents to produce the open form. However, heating and desolvating directly produced the dry form.
Apart from the expansion and contraction behaviour and the deformation of the coordination framework, sliding between closely packed layers or networks caused by guest exchange, dehydration, and sorption can also change the structure of a porous material.186 In certain cases, this process leads to a change in the packing mode. One of the first examples of this behaviour was a laminated framework, [Fe(pydc)(4,4′-bipy)]·H2O (H2pydc = 2,5-dicarboxypyridine, 4,4′-bipy = 4,4′-bipyridine).187 Here, authors observed structural contraction and reversible sliding of the layers in the material even though the space group and lattice parameters were maintained after heating. An interesting example of expansion in the unit cell and channel size was observed in the partially interpenetrated NOTT-202 material ([Me2NH2]1.75[In(C40H22O8)]1.75·12(C3H7NO)·10H2O).188 Guest removal of the material led to a structural phase transition with a change of symmetry from orthorhombic to monoclinic and a cell volume expansion of 11%.
Structural variation caused by the presence/absence of solvent molecules is not the only change that can occur with MOFs, changes in physicochemical properties such as colour can also take place. An illustrative example is the robust MOF [Ni(cyclam)(bpydc)]·5H2O (cyclam = 1,4,8,11-tetraazacyclotetradecane, bpydc2− = 2,2′-bipyridil-5,5′-dicarboxylate), which underwent dehydration concomitant with a colour change from yellow to pink when heated to 423 K.189 When the dehydrated crystal, [Ni(cyclam)(bpydc)], was subjected to air or water vapour for several minutes, the crystal returned to its original yellow colour. Notably, both processes occured while maintaining crystallinity, indicating preservation of the crystal structure with only a minor variation in the unit cell parameters. This suggests that exposure to air or water vapour leads to reversible changes in the crystal.
A new phenomenon that has emerged in MOF-composites is the ability to lock the wide-pore or large-pore phase of the MOF, even after complete desolvation. ZIF-7, [Zn(bIm)2] (bIm = benzimidazolate), exhibits a phase transition between wide-pore (ZIF-7-I) and narrow-pore (ZIF-7-II) as a result of desolvation. ZIF-7-I was locked by a mixed-matrix membrane containing ZIF-7-I nanocrystals and rigid polyimides. Retaining this wide-pore structure increased the selectivity and permeability for O2/N2 separation.190
Crystal engineering can also be used to control breathing processes. A recent study showed that an isostructural MIL-53 MOF, NU-2002 with formula [Al(OH)(BPDCA)]n (BPDCA = bicyclo[1.1.1]pentane-1,3-dicarboxylic acid), displayed increased efficiency in separating hexane isomers. This was achieved by increasing the linker dimensionality by using a 3D linker, BPDCA, instead of a 2D linker, terephthalic acid. This in turn controlled the structural breathing of the MOF.191
3.3. Conformational changes
The alterations in MOF structure triggered by temperature fluctuations can result in either polymerisation or depolymerisation processes, involving the breaking or formation of bonds and prompting movements within the lattice. Additionally, this rearrangement has the potential to influence the dimensionality of the material, meaning it can impact the spatial arrangement or configuration of the compound in terms of its three-dimensional structure. One example reported by Zhou et al. showed how a hydrated phase, [Cu(tzc)(dpp)]n·2H2O, transformed into an anhydrous phase [Cu(tzc)(dpp)]nvia the monohydrate intermediate phase [Cu(tzc)(dpp)]n·H2O.192 Upon heating to 423 K, the [Cu(tzc)(dpp)]n phase transformed into a new orthorhombic polymorph which was then transformed into a monoclinic phase upon cooling at 100 K (Fig. 11). These polymorphs exhibited different magnetic behaviour.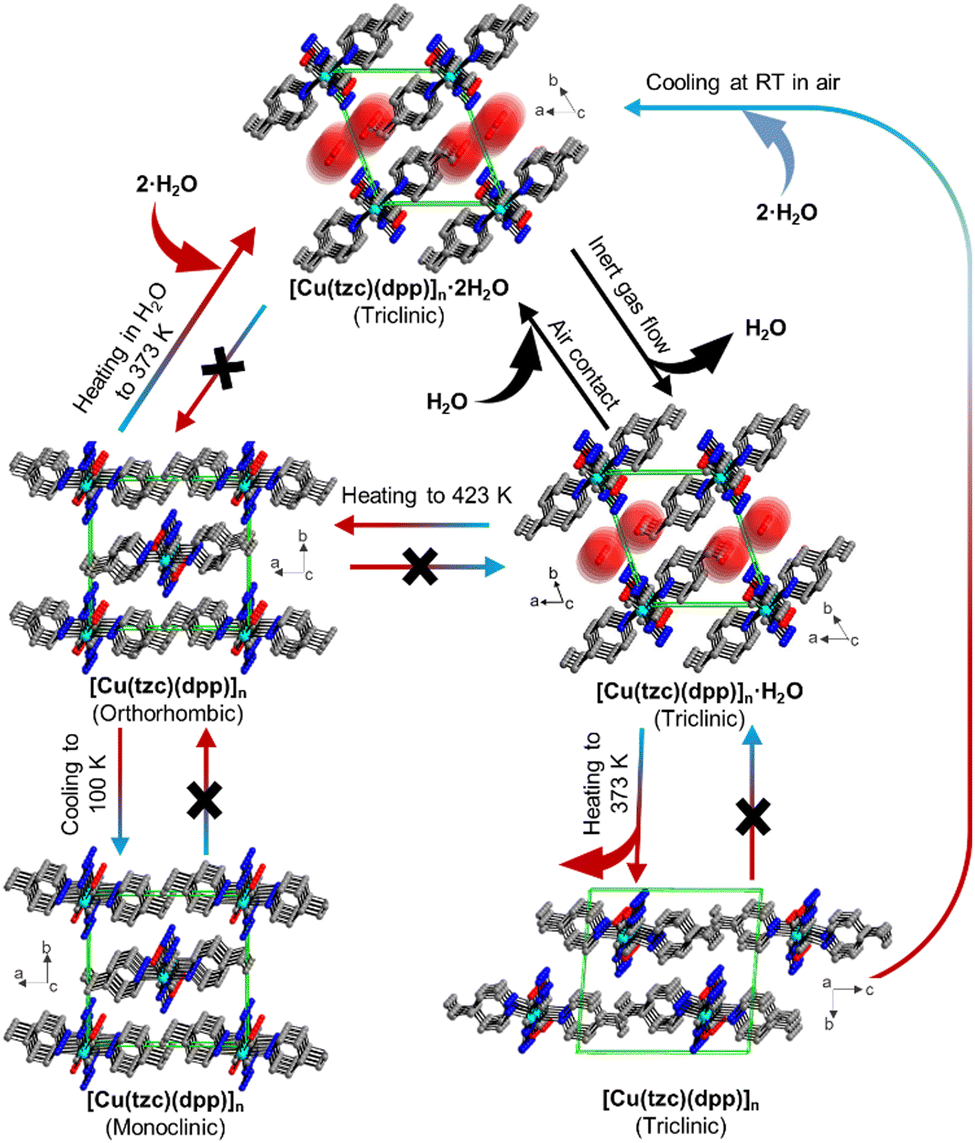 | ||
| Fig. 11 Crystal structures of the hydrates [Cu(tzc)(dpp)]n·2H2O, [Cu(tzc)(dpp)]n·H2O and their polymorphic anhydrate modifications [Cu(tzc)(dpp)]n viewed as stacked chains in a single unit cell. Arrows and labels indicate the directions and conditions, respectively, for phase transitions occurring between these phases.192 (tzc: tetrazolate-5-carboxylate, dpp: 1,3-di(4-pyridyl)propane). | ||
Thallapally et al. showed that a heterometallic MOF, {[Yb6Cu12(OH)4(PyC)12(H2O)36]·(NO3)14·xS}n (where H2PyC = 4-Pyrazolecarboxylic acid, S = unassigned free solvent molecules), transformed into a different phase, {[Yb4O(H2O)4Cu8(OH)8/3(PyC)8(HCOO)4]·(NO3)10/3·xS}n, upon heating above 393 K.193 The channel sizes and topologies of the two phases were identical, but structural changes were observed in the metal nodes by single-crystal X-ray diffraction (SCXRD). These changes were also probed by ab initio molecular dynamics simulations.
4. Low temperature phase transitions
Examples of phase transitions in MOFs occurring at room temperature or below are less common than those at high temperatures. A possible explanation for this is that thermal stability is usually studied at higher temperatures to identify the decomposition temperature of the material and to monitor the potential loss of solvent molecules. As such, many potential structural phase transitions occurring at lower temperatures may be present, yet unobserved. This is surprising considering that one of the most promising applications of MOFs is gas capture and separation, where isotherms of N2, H2, CO2, and others, are collected below room temperature, where structural transitions could affect the MOF's capture and separation ability.In most cases, structural phase transitions caused by cooling below room temperature are reversible. An interesting example is the structural study of PCN-526, [Cd8C96H48Cl5N40O6].194 Here, the phase transition occurred at 110 K. The authors demonstrated that during this phase transition, the one-dimensional channel of the MOF was distorted from a square to rectangular shape, whilst maintaining its crystallinity. Additionally, the mechanism for the transformation was linked to the modification of the occupancy or species of metal ions in the cavities.
Another illustrative example is a MOF with a rod-shaped SBU, [Zn0.59Co0.41(hfipbb)] [(hfipbb = 4,4′-(hexafluoroisopropylidene)-bis(benzoic acid))], which exhibited a doubling of cell volume at low temperature.195 According to variable temperature single crystal X-ray diffraction experiments, the lattice parameter c doubled when cooling to 150 K from room temperature as a result of a phase transition. At room temperature, data showed the presence of only one tetrahedral metal site, with a mixed Zn/Co occupancy. A phase transition was first observed at 150 K, with the corresponding doubling of the c parameter. After the phase transition, two independent crystallographic metal positions appeared in the SBU, exhibiting a tetrahedral-distorted coordination, denoted as Td1 and Td2 in Fig. 12. Analysis of the difference Fourier maps revealed a large residual electron density around the metal positions, which were assigned as additional metal sites with partial occupancy (Td1b and Td2b), arising from the ongoing atomic rearrangement. However, the presence of residual electron density indicated that the transformation was not complete until 50 K. At this temperature, the crystal structure was equivalent to one with the composition Zn0.21Co0.79(hfipbb), with both octahedral and tetrahedral metal sites. These results suggested that an increment in the cobalt content in the SBU had a stabilising effect on the octahedral site.
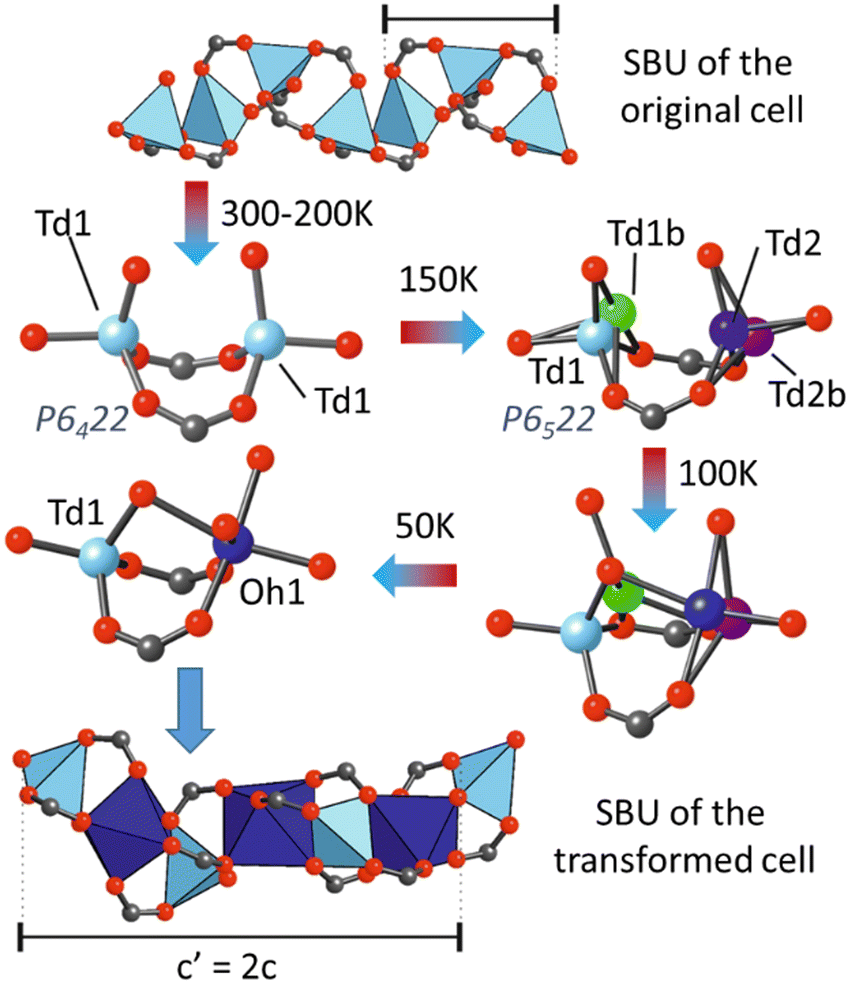 | ||
| Fig. 12 Schematic depiction of the structural transition occurring in the [Zn0.79Co0.21(hfipbb)] material upon cooling. The inorganic SBU might adapt to include octahedrally coordinated cations, which results in a unit cell transformation with a doubled c parameter.195 Image modified with permission of Science. | ||
Another example, [Ag6Cl(atz)4]OH·6H2O (Hatz = 3-amino-1,2,4,-triazole) with a tetragonal phase can be transformed into an orthorhombic phase at 103 K.163 The amino groups in both cases were two-fold disordered. When the orthorhombic phase was heated to 293 K, it slowly transformed back to tetragonal.
A similar, reversible structural transformation occurs in DUT-7, Zn4O((S)-L)3(DMF)10(H2O)3.5 ((S)-L = 2,2′-spirobiindane-5,5′-dicarboxylic acid), upon cooling to 100 K.196 During this transformation, a notable change occured where one of the zinc atoms within the Zn4O cluster transitioned from a tetrahedral to an octahedral environment. This shift was caused by the coordination of two additional solvent molecules. This demonstrates cluster flexibility, a phenomenon that can provide fresh perspectives on the catalytic activity of MOFs.
5. Characterisation of phase transitions
5.1. Thermal characterisation
Thermogravimetric analysis (TGA) is a widely applied characterisation technique used for the thermal analysis of MOFs because of its availability and straightforward analysis.11 It records mass loss of the sample upon heating at a specific heating rate under a specific atmosphere. TGA data usually provide useful information to calculate the composition, defect concentration and the solvent molecules present in a MOF's pores.197 It also provides the decomposition temperature (Td) of MOFs and can be used to calculate the composition of a material after heating. This is particularly useful for the preparation of MOF-derived materials described in the non-reversible phase transitions section.However, TGA has some limitations, such as low resolution for the majority of the available data because of the large temperature intervals (50 or 100 K). In addition, clearly identifying the difference between the plateau of the TGA curve and the decomposition curve is sometimes challenging, especially when solvent loss is involved. This makes it difficult to identify the different decomposition steps. The TGA curve is also sensitive to experimental factors such as heating rate, heating atmosphere and the pre-treatments carried out. This causes substantial differences in Td values for the same material,198 which means that experimental details should be reported alongside Td. The idea of thermal stability in general is somewhat misrepresented, where it can be argued that classifying a material as thermally stable at a given temperature requires isothermal experiments to recreate actual working conditions.
As such, TGA data are not enough to fully characterise the thermal properties of MOFs; TGA analysis should be accompanied with differential scanning calorimetry (DSC) and a crystallographic characterisation technique, such as X-ray or neutron diffraction.
Differential scanning calorimetry has become, in addition to TGA, a universal standard tool for the characterisation of phase transitions, owing to its ultrahigh sensitivity to small energy fluctuations. A typical DSC experiment consists of heating a sample dynamically or isothermally by applying heat flow under a specific atmosphere. Both the sample and a reference are monitored as a function of time and temperature.199 The calorimeter measures the heat/energy absorbed (endothermic reaction) or released (exothermic reaction) by the sample when it is subjected to a specific temperature path (Fig. 13).200
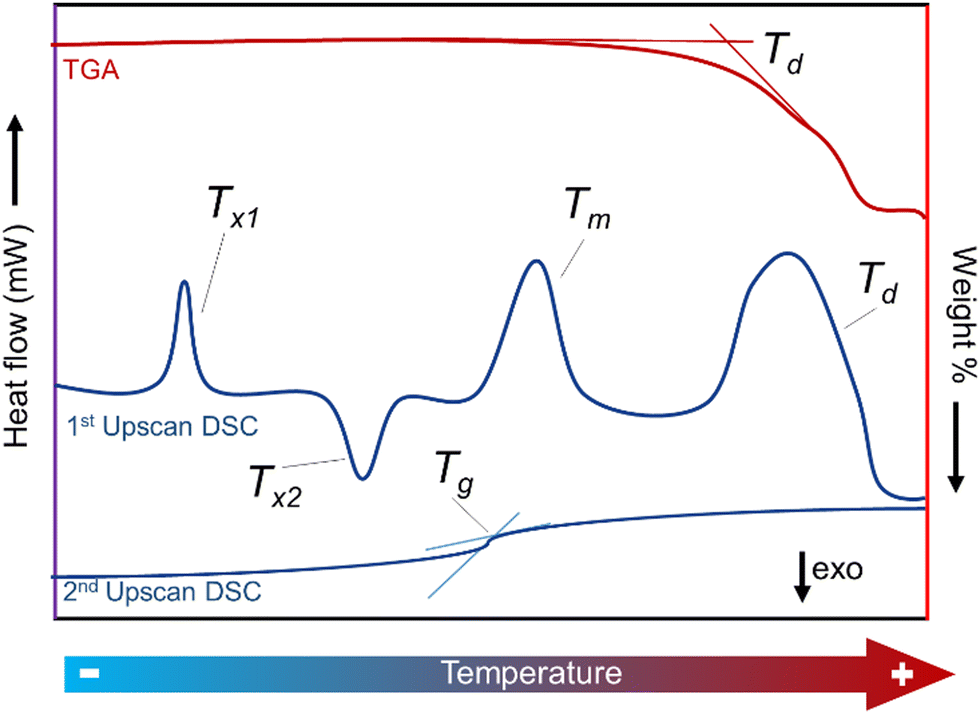 | ||
| Fig. 13 DSC depiction (Heat flow vs. Temperature) and thermogravimetric analysis of a meltable crystalline MOF. TGA is represented as a red line showing a weight loss upon heating, indicating the decomposition temperature (Td). DSC 1st and 2nd upscans are depicted in the graph as blue curves. 1st upscan shows multiple processes upon heating. Firstly, an endothermic recrystallisation (structural phase transition) denoted as Tx1. The second feature shows an exothermic phase transition (Tx2). Melting and decomposition processes also appear as endothermic features. A Tg appears in the 2nd DSC upscan of a crystalline MOF, after the material melted and formed a glassy phase during heating in the 1st upscan. | ||
This information is also particularly important for differentiating between glassy and amorphous MOFs, where glassy MOFs have a Tg, and amorphous MOFs do not. It is also useful for identifying melting processes and the melting temperature of a material.
5.2. X-ray and neutron diffraction
Several spectroscopic techniques have been developed to provide structural information of materials, crystallography is still the most important and straightforward method to visualise 3D MOF structures and their structural changes.Microcrystalline powders are generally more readily available than single crystals. However, due to extensive overlap of diffraction peaks, powder X-ray diffraction (PXRD) patterns offer comparatively less structural information with less accuracy compared to SCXRD experiments. Additionally, ab initio determination of unknown and complex crystal structures from PXRD data remains a substantial, and sometimes impossible, challenge. In contrast, solving structures from SCXRD data has become a routine and reliable method.201 SCXRD patterns measured using common in-house X-ray sources can provide precise atomic coordinates, bond lengths, bond angles, as well as information on atomic thermal displacements and occupancies. Such detailed structural insights are generally unavailable from PXRD, even when measured using highly monochromatised and intense synchrotron radiation. The inherent limitations of PXRD underscore the continued importance and utility of SCXRD for obtaining accurate and comprehensive crystallographic data.
However, it is not always possible to obtain single crystals with MOF materials. Although PXRD is more challenging for structure solution, it is possible to solve structures using multiple crystallographic approaches such as direct methods202 or charge-flipping methods.203
In addition, neutron diffraction experiments can also be employed to monitor structural phase transitions and processes, despite the lack of examples in MOFs. Although most organic linkers contain hydrogen atoms, which contribute to the diffraction pattern by increasing the background, this technique may be useful to study the contribution of multiple phases at variable temperatures (VT). An example is the VT study of a bimetallic MOF, [Zn1−xCox(hfipbb)] containing two different metals. The presence of two different phases with different metal sequences in their SBUs were observed through Rietveld refinement of the VT-neutron diffraction data.195
One of the most remarkable studies shows the polymorphism of ZIF-4 through the collection of multiple measurements at variable pressure and temperature. With this information, a pressure–temperature phase diagram was prepared (Fig. 14).204 These types of experiments can be extended to different MOFs to construct similar phase diagrams, and to explore unknown structures in operando mode, which will have a great impact on their industrial implementation.
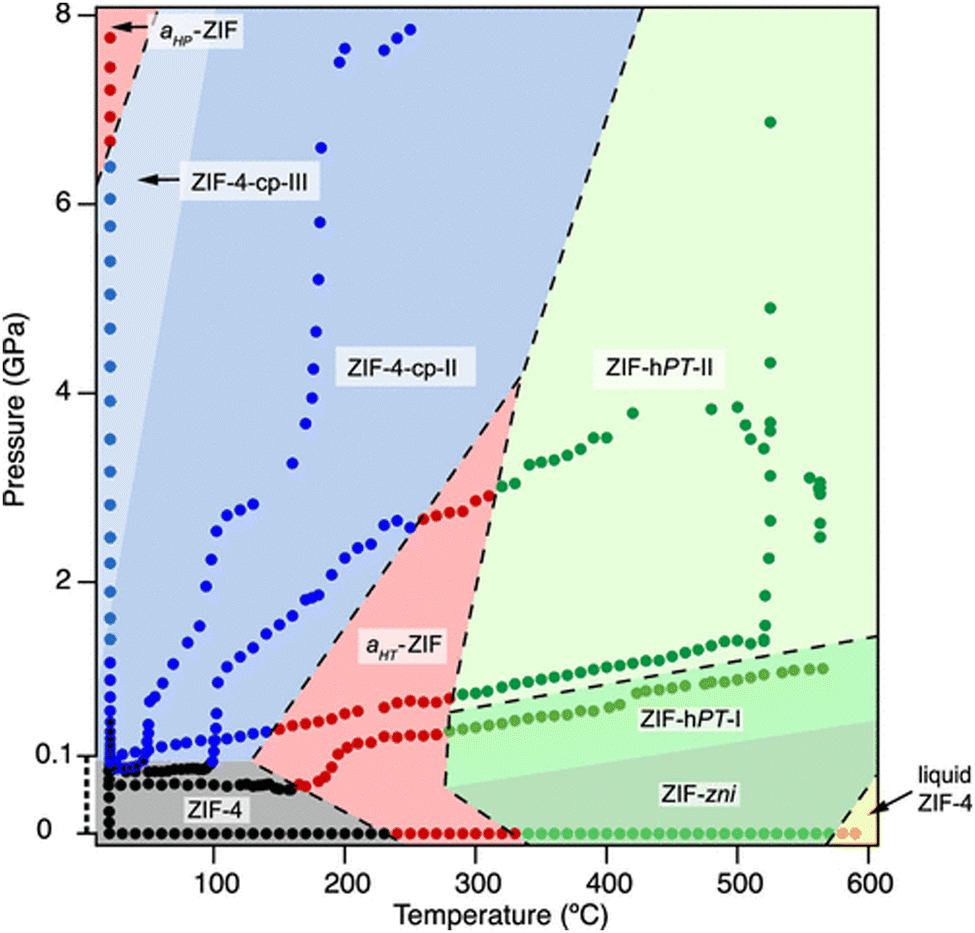 | ||
| Fig. 14 Pressure–temperature phase diagram of ZIF-4. The pressure range from 0 to 0.1 GPa has been magnified for better visibility and is thus not to scale. Experimental points are represented as circle symbols, and they are coloured according to the phases observed in situ (ZIF-4 cag: black, ZIF-4cp-II: blue, amorphous ZIF: red, ZIF-zni: green, ZIF-liquid: yellow). Coloured outlines of phase boundaries are drawn as guides to the eye. Dashed lines indicate irreversible, reconstructive transition.204 Reproduced from ref. 204 with permission from American Chemical Society, copyright 2019. | ||
5.3. Characterisation techniques of local structure
Total X-ray and neutron scattering experiments to extract the pair distribution function (PDF) have gained attention in recent years to analyse defective and highly disordered MOF materials. This technique analyses both Bragg and diffuse scattering signals simultaneously and is thus ideal to analyse MOFs.PDF gives information in real space instead of reciprocal space, as diffraction. This gives key information on the local structure of both crystalline and amorphous MOFs.205
This technique is particularly important for the characterisation of MOF glasses, which lack long-range order, and it allows comparison of the local structure of a crystalline MOF with its glassy analogue. Moreover, multivariate and linear regression analysis of PDF data provides information about the atom–atom interactions at the interface of MOF-composites.206,207 In addition, in situ PDF experiments are an effective way to monitor melting and vitrification processes, providing insights into changes in the local structure upon heating.
These in situ experiments also unveil local distortions. One interesting example was reported by Platero-Prats et al., in which NU-100 and UiO-66 were studied by VT-PDF measurements.208 Structural transitions of the Zr6- and Hf6-nodes in both structures, only visible when observing their local structures by VT-PDF, were deciphered (Fig. 15). These phase transition are important to understand as they may affect the catalytic activity of these materials.
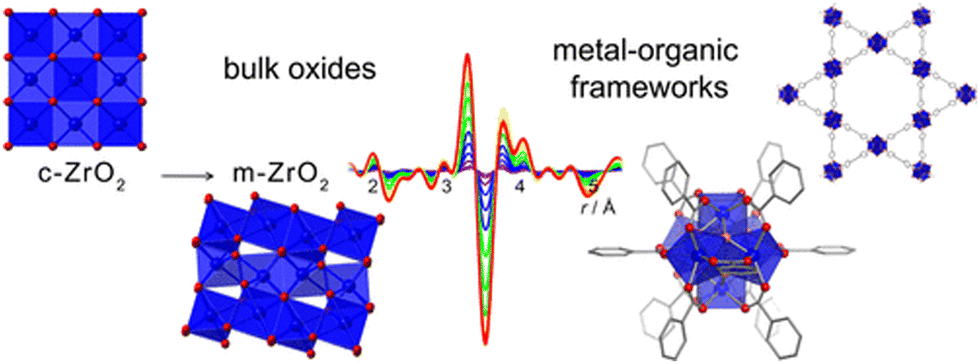 | ||
| Fig. 15 Schematic depiction of the metal-node phase transition of NU-1000 and UiO-66 structures upon heating.208 Reproduced from ref. 208 with permission from American Chemical Society, copyright 2017. | ||
The structural dynamics of the local structure of MOFs can also be monitored by spectroscopic techniques, such as X-ray absorption spectroscopy (XAS). One example of utilising this technique was a study on a Ca-MOF, Ca(BDC)(DMF)(H2O), which has the same topology of MIL-53. Loss of the DMF molecules on the surface of the MOF crystallites occurred before the decoordination of the solvent. This study suggested that the DMF was already partially decoordinated at 423 K in different domains, in agreement with NMR analyses. The phase transition was completed at 673 K, also confirmed by XRD Rietveld refinement.209
5.4. Microscopic techniques
Transmission electron microscopy (TEM) has been employed by MOF researchers to study dynamic processes of MOFs because of recent developments in camera and detector technology, which allows diffraction studies to be performed using a very small amount of sample, with minimal beam damage.210Development of microscopic techniques has allowed researchers to study dynamic processes in different soft materials, such as phase transitions and self-assemblies.211,212 However, examples of MOF studies are still rare because of the experimental limitations observing the spatiotemporal evolution of their structures.
Microscopic in situ experiments using variable temperature liquid-cell transmission electron microscopy (VT-LCTEM) aim to solve this issue. This promising approach has been used to study the growth mechanisms of ZIF-8.213 In another example, VT-LCTEM, combined with HRTEM, enabled the observation of a phase transition from a microporous MOF, scu-NU-906, to a mesoporous MOF, csq-NU-1008, [Zr6O4(OH)8(OH2)4(TCPB-Br2)2] (TCPB-Br2 = 1,4-dibromo-2,3,5,6-tetrakis(4-carboxyphenyl)benzene) (Fig. 16).214 Moreover, in situ TEM has been recently used to construct phase diagrams of many Zr-MOFs, providing information on phase, crystallinity, crystallite size and morphology.215
 | ||
| Fig. 16 TEM images during the reaction of the phase transition transformation over time by the addition of formic acid to NU-906. (a) and (b) NU-906, (c) and (d) NU-1008 isolated after two hours (e) and (f) NU-1008 isolated after two days. Insets: Fast Fourier transform (FFT) of the image inside the red square, cropped at the predominant lattice fringes, and enlarged image inside the white square.214 Adapted from ref. 214 with permission from the American Chemical Society, copyright 2020. | ||
Another in situ approach is ultrafast transmission electron microscopy (UTEM), using dark-field imaging. This promising methodology has been employed to solve tantalum disulphide charge-ordered phases.216
Temperature control when using microscopes is still a challenge, but these microscopic approaches will be able to monitor thermoactivated phase transitions in an image, and also in a diffraction mode, in the near future.
6. Theoretical calculations and modelling
Theoretical calculations and modelling methods have been extensively used in recent years to complement experimental work and to help researchers better understand material structures and properties.Density-functional theory (DFT) is probably the most extended computational quantum mechanical modelling approach used in materials science to investigate structural configurations of materials. DFT methods have been used in MOF chemistry to calculate diffusion activation energies,217 elucidate gas adsorption sites218 and to understand mechanical219 and luminescent properties.220
Structural determination of amorphous structures at the microscopic or bulk scale is always challenging. Despite the tremendous efforts using total scattering and different microscopic, diffractometric and spectroscopic techniques, often only information at the local scale is obtained.
To address this challenge, multiple computational methods have been applied to model these highly disordered structures. Several strategies have been employed to generate potential models. For example, several strategies involve simulating a phase transition from the parent crystalline MOF to an amorphous one using molecular dynamics (MD). Some ab initio MDs have been successfully used to simulate the melting of crystalline MOFs to their liquid state,221,222 and to model the corresponding melt-quenched glasses.223 However, the formation of most amorphous MOFs requires the simulation of bond breaking and reformation, which considerably increases the computational cost of this method.224 Therefore, reactive force fields (ReaxFF) have been also proposed as an alternative approach, a compromise between chemical accuracy and computational cost. This method, which has connection-dependent terms, enables the simulation of breaking-reformation processes in atomic bonds.225
However, glass-MOF models obtained through ReaxFF have been found to be remarkably different from the ab initio counterparts, in both local and medium scale, as well as in the bulk properties.226
A rare example of a mixed-linker metal–organic framework incorporating a semi-rigid ditopic diacetylene ligand is UMON-44, {[Zn(L1)(L2)](DMF)2}n (L1: 1,6-bis(1-imidazolyl)-2,4-hexadiyne, L2: isophthalate).227 This flexible structure revealed two phase transitions upon heating, and the presence of large positive and negative thermal expansions. Computational methods effectively differentiated between two polymorphs of UMON-44 by precisely replicating their essential structural and spectroscopic features. This was done by extrapolating free energies at finite temperatures and checking the specific temperature a phase became more stable than the preceding one.
Breathing behaviour mechanisms have also been studied by MD within artificial periodic boundary conditions to simulate crystalline materials using small unit cells and non-defective MOFs.228 For example, the method of breathing of the MIL-53 material has been exhaustively analysed in the last years. Original studies of the breathing mechanism suggested that the phase transition occured in a cooperative manner through Monte Carlo simulations, where the transformation was layer-by layer.229 However, more recent studies strongly contest this. Body dispersion calculations within a random-phase approximation suggest that even at 0 K, a small fraction of the large-pore phase is still present.230
More realistic force-field-based MD have been recently used, which enable large-scale cells. These results suggest this phenomenon has its origin in spatial disorder as interfacial defects, as a consequence of thermal treatments, generating unknown phases coexisting with the known crystalline ones.228 This was also confirmed using non-periodic nanocrystallite models in other MOFs, such as DMOF-1 and DUT-128.231
7. Conclusions and perspectives
Multiple thermally activated processes, which may be reversible or irreversible, can occur upon heating a MOF.Typically, non-reversible processes are related to the total or partial decomposition of the linker. The control of the thermal and atmospheric parameters during heating are crucial to obtain the desired MOF-derived nanostructures. These derived materials are widely employed for electrochemical and catalytic applications.
Other non-reversible processes such as structural phase transformations have been classified and described in detail. Several MOFs and 3D CPs can melt and form a liquid state, with the ability to form melt-quenched glasses after rapid cooling. Despite the scarcity of melt-quenched MOF glasses, this new branch of the MOF field has received great attention thanks to the promising mechanical and optical properties of MOF glasses. Less common transitions upon heating, such as crystalline to amorphous (non-glassy) and amorphous (non-glassy) to crystalline are also possible.
Reversible structural phase transitions are mostly related to the loss of solvent molecules. For example, the phenomenon of breathing has received great interest in the MOF-field because of the different porosities (and thus guest sorption behaviours) that can be reached upon heating.
Other conformational changes can also appear upon heating in a reversible way, with or without decoordination of a solvent molecule.
Low temperature structural phase transitions were also reviewed here. Even though reports are still rare at present, they may have a fundamental impact in gas sorption and magnetic studies.
Characterisation of phase transitions are crucial to understand their nature and also to identify the initial and the transformed structure. In this regard, in situ experiments will become crucial to monitor phase transitions.
Multiple techniques must be employed to identify different phase transitions. Thermal analysis through TGA and DSC allows differentiation between amorphous and glass phases. Additionally, diffractometric techniques give structural information through single crystal or powder analysis.
Advances in electron microscopy will also allow further in-depth monitoring of phase transitions in MOFs. This developing field will have a tremendous impact on the identification and monitoring of phase transitions in diffraction and image mode.
These advances in characterisation and increased focus on the understanding of diverse structural transitions in MOFs will have significant effects tremendous impact on the MOF field because of the link between structural variation and transitions and the MOFs physical and chemical properties.
Abbreviations
| 3-Pybpy | [3,1′:4′,4′′terpyridin]-1′-ium |
| 5-Cl-2-mbIm | 5-Chloro-2-methylbenzimidazolate |
| 5-FbIm | 5-Fluorobenzimidazolate |
| 5-ClbIm | 5-Chlorobenzimidazolate |
| 6-Cl-5-FbIm | 6-Chloro-5-fluorobenzimidazolate |
| BBCDC | Biphenyl bis(carbazole dicarboxylate) |
| bIm | Benzenimidazolate |
| bpee | trans-1,2-bis(4-pyridyl)ethylene |
| bpy | 4,4′-Bipyridine |
| C2bpy | 1-Ethyl-[4,4′-bipyridin]-1-ium |
| C4bpy | 1-Butyl-[4,4′-bipyridin]-1-ium |
| dCNIm | 4,5-Dicyanoimidazolate |
| DEF | Diethylformamide |
| DFT | Density functional theory |
| DMF | Dimethylformamide |
| DSC | Differential scanning calorimetry |
| FFT | Fast Fourier transform |
| fum | fumarate |
| H2hfipbb | 4,4′-(Hexafluoroisopropylidene)bis(benzoic acid) |
| H2BDC | Benzenedycarboxylic acid |
| HL | 2,6-Bis-(4-imidazol-1-yl-phenyl)-4-[4-(2H-tetrazol-5-yl)-phenyl]-pyridine |
| H2PyC | 4-Pyrazolecarboxylic acid |
| H2TTPP | 5,10,15,20-Tetrakis[4-(2H-tetrazol-5-yl)phenyl]porphyrin |
| H3BTC | Benzenetricarboxylic acid |
| Hatz | 3-Amino-1,2,4,-triazole |
| HMOF | Hinged metal–organic framework |
| HPC | Hollow porous carbon |
| Im | Imidazolate |
| L1 | Isonicotinate |
| L2 | Pyridine-3-carboxylate |
| L2Br | 3-Bromoisonicotinate |
| L2Cl | 3-Chloroisonicotinate |
| L2F | 3-Fluoroisonicotinate |
| L2NH2 | 3-Aminoisonicotinate |
| L3 | (E)-3-(Pyridin-4-yl)acrylate |
| L4 | 4-(Pyridin-4-yl)benzoate |
| L5 | (E)-4-(2-(Pyridin-4-yl)vinyl)benzoate |
| L6 | 4-(1H-imidazol-1-yl)benzoate |
| mIm | 2-Methylimidazolate |
| mL1 | 1,3,5-Tris((3-cyanophenylethynyl)benzene) |
| MD | Molecular dynamics |
| MOF | Metal–organic framework |
| MUV | Materials of the University of Valencia |
| napht | 4-(1H-naphtho[2,3-d]imidazol-1-yl)benzoic acid |
| NBD | 2-Nitrobenzene-1,4-dicarboxylate |
| pL2 | 1,3,5-Tris(4-ethynylbenzonitrile)benzene |
| Phbpy | 1-Phenyl-[4,4′-bipyridin]-1-ium |
| Pur | Purinate |
| Pair distribution function | |
| PXRD | Powder X-ray diffraction |
| SEM | Scanning electron microscopy |
| ted | Triethylenediamine |
| TEM | Transmission electron microscopy |
| TGA | Thermal gravimetric analysis |
| TCPB-Br2 | 1,4-Dibromo-2,3,5,6-tetrakis(4-carboxyphenyl)benzene |
| UTEM | Ultrafast transmission electron microscopy |
| ZIF | Zeolitic imidazolate framework |
Author contributions
C. C. B. conceptualised this review. All authors contributed with useful discussions. C. C. B. prepared the manuscript with inputs of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. C. B., A. M. C. and T. D. B. thank Leverhulme Trust Research Project Grant (RPG-2020-005). T. D. B. also thanks the Royal Society for both a University Research Fellowship (URF\R\211013) and a research grant (RGS\R2\212221).Notes and references
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
.
- X. Zhao, Y. Wang, D.-S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef PubMed
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed
- I. E. Collings and A. L. Goodwin, J. Appl. Phys., 2019, 126, 181101 CrossRef
- Z. Xiao, H. F. Drake, G. S. Day, J. E. Kuszynski, H. Lin, H. Xie, P. Cai, M. R. Ryder and H.-C. Zhou, Cell Rep. Phys. Sci., 2022, 3, 101074 CrossRef CAS
- E. Fernandez-Bartolome, A. Martinez-Martinez, E. Resines-Urien, L. Piñeiro-Lopez and J. S. Costa, Coord. Chem. Rev., 2022, 452, 214281 CrossRef CAS
- G. Mínguez Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533–557 RSC
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC
- C. Healy, K. M. Patil, B. H. Wilson, L. Hermanspahn, N. C. Harvey-Reid, B. I. Howard, C. Kleinjan, J. Kolien, F. Payet, S. G. Telfer, P. E. Kruger and T. D. Bennett, Coord. Chem. Rev., 2020, 419, 213388 CrossRef CAS
- T. D. Bennett and S. Horike, Nat. Rev. Mater., 2018, 3, 431–440 CrossRef
- N. Ma and S. Horike, Chem. Rev., 2022, 122, 4163–4203 CrossRef CAS PubMed
- J. Ding, Y. Tang, S. Zheng, S. Zhang, H. Xue, Q. Kong and H. Pang, Nano Res., 2022, 15, 6793–6818 CrossRef CAS
- K.-Y. Zou and Z.-X. Li, Chem. – Eur. J., 2018, 24, 6506–6518 CrossRef CAS PubMed
- S. Yang, L. Peng, S. Bulut and W. L. Queen, Chem. – Eur. J., 2019, 25, 2161–2178 CrossRef CAS PubMed
- X. F. Lu, Y. Fang, D. Luan and X. W. D. Lou, Nano Lett., 2021, 21, 1555–1565 CrossRef CAS PubMed
- B. Li, L. Hong, C. Jing, X. Yue, H. Huang, Q. Jiang and J. Tang, Microporous Mesoporous Mater., 2024, 365, 112836 CrossRef CAS
- Z.-X. Cai, Z.-L. Wang, J. Kim and Y. Yamauchi, Adv. Mater., 2019, 31, 1804903 CrossRef PubMed
- B. You, N. Jiang, M. Sheng, W. S. Drisdell, J. Yano and Y. Sun, ACS Catal., 2015, 5, 7068–7076 CrossRef CAS
- X.-C. Xie, K.-J. Huang and X. Wu, J. Mater. Chem. A, 2018, 6, 6754–6771 RSC
- L. Zhang, H. Bin Wu and X. W. (David) Lou, J. Am. Chem. Soc., 2013, 135, 10664–10672 CrossRef CAS PubMed
- L. Zhang, Z. Su, F. Jiang, L. Yang, J. Qian, Y. Zhou, W. Li and M. Hong, Nanoscale, 2014, 6, 6590–6602 RSC
- A. J. Amali, H. Hoshino, C. Wu, M. Ando and Q. Xu, Chem. – Eur. J., 2014, 20, 8279–8282 CrossRef CAS PubMed
- H. Xu, S. Zhou, L. Xiao, H. Wang, S. Li and Q. Yuan, J. Mater. Chem. C, 2015, 3, 291–297 RSC
- H. Hu, G. Ruan, X. Jiang, H. Pan, Z. Wu and Y. Huang, New J. Chem., 2022, 46, 8224–8231 RSC
- X. Li, Q. Sun, J. Liu, B. Xiao, R. Li and X. Sun, J. Power Sources, 2016, 302, 174–179 CrossRef CAS
- X. Pan, L. Bai, H. Wang, Q. Wu, H. Wang, S. Liu, B. Xu, X. Shi and H. Liu, Adv. Mater., 2018, 30, 1800180 CrossRef PubMed
- X. Li, J. Zhang and W. Li, J. Ind. Eng. Chem., 2016, 44, 146–154 CrossRef CAS
- J. Xu, J. Wang, L. Ge, J. Sun, W. Ma, M. Ren, X. Cai, W. Liu and J. Yao, J. Colloid Interface Sci., 2022, 610, 98–105 CrossRef CAS PubMed
- Z. Zhang, Y. Chen, P. Wang, Z. Wang, C. Zuo, W. Chen and T. Ao, J. Hazard. Mater., 2022, 423, 127103 CrossRef CAS PubMed
- X.-M. Cao, Z.-J. Sun, S.-Y. Zhao, B. Wang and Z.-B. Han, Mater. Chem. Front., 2018, 2, 1692–1699 RSC
- N. Liu, X. Liu and J. Pan, J. Colloid Interface Sci., 2022, 606, 1364–1373 CrossRef CAS PubMed
- S. Li, X. Zhang and Y. Huang, J. Hazard. Mater., 2017, 321, 711–719 CrossRef CAS PubMed
- S. Cheng, N. Shang, C. Feng, S. Gao, C. Wang and Z. Wang, Catal. Commun., 2017, 89, 91–95 CrossRef CAS
- L. Chang, J. Li, X. Duan and W. Liu, Electrochim. Acta, 2015, 176, 956–964 CrossRef CAS
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, Chem. Mater., 2012, 24, 464–470 CrossRef CAS
- S. J. Yang, S. Nam, T. Kim, J. H. Im, H. Jung, J. H. Kang, S. Wi, B. Park and C. R. Park, J. Am. Chem. Soc., 2013, 135, 7394–7397 CrossRef CAS PubMed
- M. del Rio, J. C. Grimalt Escarabajal, G. Turnes Palomino and C. Palomino Cabello, Chem. Eng. J., 2022, 428, 131147 CrossRef CAS
- C.-P. Li, Y.-Q. Wu, F.-Y. Zhang, L.-X. Gao, D.-Q. Zhang and Z.-X. An, Sep. Purif. Technol., 2021, 277, 119618 CrossRef CAS
- T. Hussain, P. Nie, B. Hu, X. Shang, J. Yang and J. Liu, J. Mater. Sci., 2021, 56, 10282–10292 CrossRef CAS
- W. Xia, B. Qiu, D. Xia and R. Zou, Sci. Rep., 2013, 3, 1935 CrossRef PubMed
- J. Hwang, R. Yan, M. Oschatz and B. V. K. J. Schmidt, J. Mater. Chem. A, 2018, 6, 23521–23530 RSC
- N. L. Torad, R. R. Salunkhe, Y. Li, H. Hamoudi, M. Imura, Y. Sakka, C.-C. Hu and Y. Yamauchi, Chem. – Eur. J., 2014, 20, 7895–7900 CrossRef CAS PubMed
- X. Xu, J. Li, M. Wang, Y. Liu, T. Lu and L. Pan, ChemElectroChem, 2016, 3, 993–998 CrossRef CAS
- T. Van Tran, D. T. C. Nguyen, H. T. N. Le, T. T. K. Tu, N. D. Le, K. T. Lim, L. G. Bach and T. D. Nguyen, J. Environ. Chem. Eng., 2019, 7, 102881 CrossRef
- Y. Hou, X.-J. Hu, H.-Y. Tong, Y.-B. Huang and R. Cao, Inorg. Chem. Commun., 2020, 114, 107825 CrossRef CAS
- S. Cheng, N. Shang, X. Zhou, C. Feng, S. Gao, C. Wang and Z. Wang, New J. Chem., 2017, 41, 9857–9865 RSC
- W. Jin, H.-J. Li, J. Zou, S. Inguva, Q. Zhang, S. Zeng, G. Xu and X. Zeng, Mater. Lett., 2019, 252, 211–214 CrossRef CAS
- H. J. Lee, S. Choi and M. Oh, Chem. Commun., 2014, 50, 4492–4495 RSC
- L. Chen, J. Bai, C. Wang, Y. Pan, M. Scheer and X. You, Chem. Commun., 2008, 1581–1583 RSC
- X. Zhang, T. Kitao, D. Piga, R. Hongu, S. Bracco, A. Comotti, P. Sozzani and T. Uemura, Chem. Sci., 2020, 11, 10844–10849 RSC
- Q. Wang, W. Xia, W. Guo, L. An, D. Xia and R. Zou, Chem. – Asian J., 2013, 8, 1879–1885 CrossRef CAS PubMed
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed
- M. Hu, J. Reboul, S. Furukawa, N. L. Torad, Q. Ji, P. Srinivasu, K. Ariga, S. Kitagawa and Y. Yamauchi, J. Am. Chem. Soc., 2012, 134, 2864–2867 CrossRef CAS PubMed
- W. Hu, M. Zheng, B. Xu, Y. Wei, W. Zhu, Q. Li and H. Pang, J. Mater. Chem. A, 2021, 9, 3880–3917 RSC
- C. Wang, Y. V. Kaneti, Y. Bando, J. Lin, C. Liu, J. Li and Y. Yamauchi, Mater. Horiz., 2018, 5, 394–407 RSC
- X. Xu, R. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988–4991 CrossRef CAS PubMed
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed
- R. R. Salunkhe, Y. V. Kaneti and Y. Yamauchi, ACS Nano, 2017, 11, 5293–5308 CrossRef CAS PubMed
- X. Tan, Y. Wu, X. Lin, A. Zeb, X. Xu, Y. Luo and J. Liu, Inorg. Chem. Front., 2020, 7, 4939–4955 RSC
- W. Zhou, Y. Tang, X. Zhang, S. Zhang, H. Xue and H. Pang, Coord. Chem. Rev., 2023, 477, 214949 CrossRef CAS
- Y. Li, Y. Xu, W. Yang, W. Shen, H. Xue and H. Pang, Small, 2018, 14, 1704435 CrossRef PubMed
- M. Ali, E. Pervaiz, T. Noor, O. Rabi, R. Zahra and M. Yang, Int. J. Energy Res., 2021, 45, 1190–1226 CrossRef CAS
- T.-T. Li, J. Qian and Y.-Q. Zheng, RSC Adv., 2016, 6, 77358–77365 RSC
- C. Li, T. Chen, W. Xu, X. Lou, L. Pan, Q. Chen and B. Hu, J. Mater. Chem. A, 2015, 3, 5585–5591 RSC
- L. Zhang, H. Bin Wu, S. Madhavi, H. H. Hng and X. W. (David) Lou, J. Am. Chem. Soc., 2012, 134, 17388–17391 CrossRef CAS PubMed
- T. K. Kim, K. J. Lee, J. Y. Cheon, J. H. Lee, S. H. Joo and H. R. Moon, J. Am. Chem. Soc., 2013, 135, 8940–8946 CrossRef CAS PubMed
- F. Zheng, S. Xu, Z. Yin, Y. Zhang and L. Lu, RSC Adv., 2016, 6, 93532–93538 RSC
- S. Kong, R. Dai, H. Li, W. Sun and Y. Wang, ACS Sustainable Chem. Eng., 2015, 3, 1830–1838 CrossRef CAS
- Z. Xiu, M. H. Alfaruqi, J. Gim, J. Song, S. Kim, T. V. Thi, P. T. Duong, J. P. Baboo, V. Mathew and J. Kim, Chem. Commun., 2015, 51, 12274–12277 RSC
- X. Yang, L. Qiu and X. Luo, RSC Adv., 2018, 8, 4890–4894 RSC
- F. Zheng, D. Zhu, X. Shi and Q. Chen, J. Mater. Chem. A, 2015, 3, 2815–2824 RSC
- C. Castillo-Blas, N. López-Salas, M. C. Gutiérrez, I. Puente-Orench, E. Gutiérrez-Puebla, M. L. Ferrer, M. Á. Monge and F. Gándara, J. Am. Chem. Soc., 2019, 141, 1766–1774 CrossRef CAS PubMed
- C. Castillo-Blas, C. Álvarez-Galván, I. Puente-Orench, A. García-Sánchez, F. E. Oropeza, E. Gutiérrez-Puebla, Á. Monge, V. A. de la Peña-O’Shea and F. Gándara, Nano Res., 2021, 14, 493–500 CrossRef CAS
- W. Cho, S. Park and M. Oh, Chem. Commun., 2011, 47, 4138–4140 RSC
- H. Pang, B. Guan, W. Sun and Y. Wang, Electrochim. Acta, 2016, 213, 351–357 CrossRef CAS
- F. Zhang, L. Hao, L. Zhang and X. Zhang, Int. J. Electrochem. Sci., 2011, 6, 2943–2954 CrossRef CAS
- B. Liu, X. Zhang, H. Shioyama, T. Mukai, T. Sakai and Q. Xu, J. Power Sources, 2010, 195, 857–861 CrossRef CAS
- J. Zhao, F. Wang, P. Su, M. Li, J. Chen, Q. Yang and C. Li, J. Mater. Chem., 2012, 22, 13328–13333 RSC
- H. Yu, H. Fan, B. Yadian, H. Tan, W. Liu, H. H. Hng, Y. Huang and Q. Yan, ACS Appl. Mater. Interfaces, 2015, 7, 26751–26757 CrossRef CAS PubMed
- Y. Xia, B. Wang, G. Wang, X. Liu and H. Wang, ChemElectroChem, 2016, 3, 299–308 CrossRef CAS
- R. P. Antony, A. K. Satpati, K. Bhattacharyya and B. N. Jagatap, Adv. Mater. Interfaces, 2016, 3, 1600632 CrossRef
- Y. Chuan Tan and H. Chun Zeng, Chem. Commun., 2016, 52, 11591–11594 RSC
- L. Hu, Y. Huang, F. Zhang and Q. Chen, Nanoscale, 2013, 5, 4186–4190 RSC
- G. Huang, F. Zhang, L. Zhang, X. Du, J. Wang and L. Wang, J. Mater. Chem. A, 2014, 2, 8048–8053 RSC
- H. Hu, B. Guan, B. Xia and X. W. (David) Lou, J. Am. Chem. Soc., 2015, 137, 5590–5595 CrossRef CAS PubMed
- G. Huang, L. Zhang, F. Zhang and L. Wang, Nanoscale, 2014, 6, 5509–5515 RSC
- M. Ni, Y. Zhu, C. Guo, D.-L. Chen, J. Ning, Y. Zhong and Y. Hu, ACS Catal., 2023, 13, 2502–2512 CrossRef CAS
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 RSC
- K. Shen, X. Chen, J. Chen and Y. Li, ACS Catal., 2016, 6, 5887–5903 CrossRef CAS
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed
- R. Das, P. Pachfule, R. Banerjee and P. Poddar, Nanoscale, 2012, 4, 591–599 RSC
- W. Zhong, H. Liu, C. Bai, S. Liao and Y. Li, ACS Catal., 2015, 5, 1850–1856 CrossRef CAS
- V. P. Santos, T. A. Wezendonk, J. J. D. Jaén, A. I. Dugulan, M. A. Nasalevich, H.-U. Islam, A. Chojecki, S. Sartipi, X. Sun, A. A. Hakeem, A. C. J. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Nat. Commun., 2015, 6, 6451 CrossRef CAS PubMed
- X. Wang and Y. Li, J. Mol. Catal. A: Chem., 2016, 420, 56–65 CrossRef CAS
- X. Ma, Y.-X. Zhou, H. Liu, Y. Li and H.-L. Jiang, Chem. Commun., 2016, 52, 7719–7722 RSC
- Y.-X. Zhou, Y.-Z. Chen, L. Cao, J. Lu and H.-L. Jiang, Chem. Commun., 2015, 51, 8292–8295 RSC
- Y.-Z. Chen, C. Wang, Z.-Y. Wu, Y. Xiong, Q. Xu, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed
- J. Long, K. Shen and Y. Li, ACS Catal., 2017, 7, 275–284 CrossRef CAS
- J. Long, Y. Zhou and Y. Li, Chem. Commun., 2015, 51, 2331–2334 RSC
- M. Ding, S. Chen, X.-Q. Liu, L.-B. Sun, J. Lu and H.-L. Jiang, ChemSusChem, 2017, 10, 1898–1903 CrossRef CAS PubMed
- J. López-Cabrelles, J. Romero, G. Abellán, M. Giménez-Marqués, M. Palomino, S. Valencia, F. Rey and G. Mínguez Espallargas, J. Am. Chem. Soc., 2019, 141, 7173–7180 CrossRef PubMed
- W. Chen, Y. Zhang, G. Chen, R. Huang, Y. Zhou, Y. Wu, Y. Hu and K. (Ken) Ostrikov, J. Mater. Chem. A, 2019, 7, 3090–3100 RSC
- C. A. Angell, Science, 1995, 267, 1924–1935 CrossRef CAS PubMed
- J. Ren, H. W. Langmi, B. C. North and M. Mathe, Int. J. Energy Res., 2015, 39, 607–620 CrossRef CAS
- S. M. Collins, K. E. MacArthur, L. Longley, R. Tovey, M. Benning, C. B. Schönlieb, T. D. Bennett and P. A. Midgley, APL Mater., 2019, 7, 091111 CrossRef
- C. Chakravarty, P. G. Debenedetti and F. H. Stillinger, J. Chem. Phys., 2007, 126, 204508 CrossRef PubMed
- S. A. Khrapak, Phys. Rev. Res., 2020, 2, 12040 CrossRef CAS
- A. Seeger, D. Freitag, F. Freidel and G. Luft, Thermochim. Acta, 2004, 424, 175–181 CrossRef CAS
- B. K. Shaw, A. R. Hughes, M. Ducamp, S. Moss, A. Debnath, A. F. Sapnik, M. F. Thorne, L. N. McHugh, A. Pugliese, D. S. Keeble, P. Chater, J. M. Bermudez-Garcia, X. Moya, S. K. Saha, D. A. Keen, F.-X. Coudert, F. Blanc and T. D. Bennett, Nat. Chem., 2021, 13, 778–785 CrossRef CAS PubMed
- B. K. Shaw, C. Castillo-Blas, M. F. Thorne, M. L. Ríos Gómez, T. Forrest, M. D. Lopez, P. A. Chater, L. N. McHugh, D. A. Keen and T. D. Bennett, Chem. Sci., 2022, 13, 2033–2042 RSC
- M. F. Thorne, A. F. Sapnik, L. N. McHugh, A. M. Bumstead, C. Castillo-Blas, D. S. Keeble, M. Diaz Lopez, P. A. Chater, D. A. Keen and T. D. Bennett, Chem. Commun., 2021, 57, 9272–9275 RSC
- Y. Zhao, S.-Y. Lee, N. Becknell, O. M. Yaghi and C. A. Angell, J. Am. Chem. Soc., 2016, 138, 10818–10821 CrossRef CAS PubMed
- W. Xu, N. Hanikel, K. A. Lomachenko, C. Atzori, A. Lund, H. Lyu, Z. Zhou, C. A. Angell and O. M. Yaghi, Angew. Chem., Int. Ed., 2023, e202300003 CAS
- V. I. Dimitrov, J. Non-Cryst. Solids, 2005, 351, 2394–2402 CrossRef CAS
- P. G. Debenedetti and F. H. Stillinger, Nature, 2001, 410, 259–267 CrossRef CAS PubMed
- T. D. Bennett, J.-C. Tan, Y. Yue, E. Baxter, C. Ducati, N. J. Terrill, H. H.-M. Yeung, Z. Zhou, W. Chen, S. Henke, A. K. Cheetham and G. N. Greaves, Nat. Commun., 2015, 6, 8079 CrossRef CAS PubMed
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed
- T. D. Bennett, A. L. Goodwin, M. T. Dove, D. A. Keen, M. G. Tucker, E. R. Barney, A. K. Soper, E. G. Bithell, J.-C. Tan and A. K. Cheetham, Phys. Rev. Lett., 2010, 104, 115503 CrossRef PubMed
- T. D. Bennett, Y. Yue, P. Li, A. Qiao, H. Tao, N. G. Greaves, T. Richards, G. I. Lampronti, S. A. T. Redfern, F. Blanc, O. K. Farha, J. T. Hupp, A. K. Cheetham and D. A. Keen, J. Am. Chem. Soc., 2016, 138, 3484–3492 CrossRef CAS PubMed
- A. M. Bumstead, M. F. Thorne, A. F. Sapnik, C. Castillo-Blas, G. I. Lampronti and T. D. Bennett, Dalton Trans., 2022, 51, 13636–13645 RSC
- L. Frentzel-Beyme, M. Kloß, P. Kolodzeiski, R. Pallach and S. Henke, J. Am. Chem. Soc., 2019, 141, 12362–12371 CrossRef CAS PubMed
- M. L. Ríos Gómez, G. I. Lampronti, Y. Yang, J. C. Mauro and T. D. Bennett, Dalton Trans., 2020, 49, 850–857 RSC
- A. M. Bumstead, I. Pakamorė, K. D. Richards, M. F. Thorne, S. S. Boyadjieva, C. Castillo-Blas, L. N. McHugh, A. F. Sapnik, D. S. Keeble, D. A. Keen, R. C. Evans, R. S. Forgan and T. D. Bennett, Chem. Mater., 2022, 34, 2187–2196 CrossRef CAS PubMed
- A. M. Bumstead, C. Castillo-Blas, I. Pakamorė, M. F. Thorne, A. F. Sapnik, A. M. Chester, G. Robertson, D. J. M. Irving, P. A. Chater, D. A. Keen, R. S. Forgan and T. D. Bennett, Chem. Commun., 2023, 59, 732–735 RSC
- J. Song, L. Frentzel-Beyme, R. Pallach, P. Kolodzeiski, A. Koutsianos, W.-L. Xue, R. Schmid and S. Henke, J. Am. Chem. Soc., 2023, 145, 9273–9284 CrossRef CAS PubMed
- S. J. Rettig, A. Storr, D. A. Summers, R. C. Thompson and J. Trotter, J. Am. Chem. Soc., 1997, 119, 8675–8680 CrossRef CAS
- L. León-Alcaide, R. S. Christensen, D. A. Keen, J. L. Jordá, I. Brotons-Alcázar, A. Forment-Aliaga and G. Mínguez Espallargas, J. Am. Chem. Soc., 2023, 145, 11258–11264 CrossRef PubMed
- Y.-S. Wei, Z. Fan, C. Luo and S. Horike, Nat. Synth., 2024, 3, 214–223 CrossRef
- Z. Fan, Y.-S. Wei, C. Das, K. Kanamori, H. Yamada, K. Ohara and S. Horike, Chem. Commun., 2023, 59, 14317–14320 RSC
- L. N. McHugh and T. D. Bennett, J. Mater. Chem. A, 2022, 10, 19552–19559 RSC
- Y. Wang, H. Jin, Q. Ma, K. Mo, H. Mao, A. Feldhoff, X. Cao, Y. Li, F. Pan and Z. Jiang, Angew. Chem., Int. Ed., 2020, 132, 4395–4399 CrossRef
- Z. Yang, Y. Belmabkhout, L. N. McHugh, D. Ao, Y. Sun, S. Li, Z. Qiao, T. D. Bennett, M. D. Guiver and C. Zhong, Nat. Mater., 2023, 22, 888–894 CrossRef CAS PubMed
- S. Li, S. Yu, S. M. Collins, D. N. Johnstone, C. W. Ashling, A. F. Sapnik, P. A. Chater, D. S. Keeble, L. N. McHugh, P. A. Midgley, D. A. Keen and T. D. Bennett, Chem. Sci., 2020, 11, 9910–9918 RSC
- C. W. Ashling, D. N. Johnstone, R. N. Widmer, J. Hou, S. M. Collins, A. F. Sapnik, A. M. Bumstead, P. A. Midgley, P. A. Chater, D. A. Keen and T. D. Bennett, J. Am. Chem. Soc., 2019, 141, 15641–15648 CrossRef CAS PubMed
- O. Smirnova, S. Hwang, R. Sajzew, L. Ge, A. Reupert, V. Nozari, S. Savani, C. Chmelik, M. R. Reithofer, L. Wondraczek, J. Kärger and A. Knebel, Nat. Mater., 2024, 23, 262–270 CrossRef CAS PubMed
- M. T. Islam, L. Macri-Pellizzeri, V. Sottile and I. Ahmed, Biomater. Sci., 2021, 9, 1826–1844 RSC
- Y. Hirai, T. Nakanishi, Y. Kitagawa, K. Fushimi, T. Seki, H. Ito, H. Fueno, K. Tanaka, T. Satoh and Y. Hasegawa, Inorg. Chem., 2015, 54, 4364–4370 CrossRef CAS PubMed
- E. T. Spielberg, E. Edengeiser, B. Mallick, M. Havenith and A.-V. Mudring, Chem. – Eur. J., 2014, 20, 5338–5345 CrossRef CAS PubMed
- A. Qiao, H. Tao, M. P. Carson, S. W. Aldrich, L. M. Thirion, T. D. Bennett, J. C. Mauro and Y. Yue, Opt. Lett., 2019, 44, 1623–1625 CrossRef CAS PubMed
- M. A. Ali, X. Liu, Y. Li, J. Ren and J. Qiu, Inorg. Chem., 2020, 59, 8380–8386 CrossRef CAS PubMed
- C. Ma, N. Li, D. Li, Z. Gu, Z. Qiao and C. Zhong, J. Membr. Sci., 2023, 683, 121873 CrossRef CAS
- Y. Zhang, Y. Wang, H. Xia, P. Gao, Y. Cao, H. Jin and Y. Li, Chem. Commun., 2022, 58, 9548–9551 RSC
- L. Frentzel-Beyme, M. Kloß, R. Pallach, S. Salamon, H. Moldenhauer, J. Landers, H. Wende, J. Debus and S. Henke, J. Mater. Chem. A, 2019, 7, 985–990 RSC
- A. M. Bumstead, M. F. Thorne and T. D. Bennett, Faraday Discuss., 2021, 225, 210–225 RSC
- J. Hou, M. L. Ríos Gómez, A. Krajnc, A. McCaul, S. Li, A. M. Bumstead, A. F. Sapnik, Z. Deng, R. Lin, P. A. Chater, D. S. Keeble, D. A. Keen, D. Appadoo, B. Chan, V. Chen, G. Mali and T. D. Bennett, J. Am. Chem. Soc., 2020, 142, 3880–3890 CrossRef CAS PubMed
- C. Das, T. Ogawa and S. Horike, Chem. Commun., 2020, 56, 8980–8983 RSC
- S. S. Nagarkar, H. Kurasho, N. T. Duong, Y. Nishiyama, S. Kitagawa and S. Horike, Chem. Commun., 2019, 55, 5455–5458 RSC
- T. D. Bennett, D. A. Keen, J.-C. Tan, E. R. Barney, A. L. Goodwin and A. K. Cheetham, Angew. Chem., Int. Ed., 2011, 50, 3067–3071 CrossRef CAS PubMed
- S. Park and H.-K. Jeong, J. Mater. Chem. A, 2022, 10, 4992–4998 RSC
- Y. Feng, M.-Y. Zou, H.-C. Hu, W.-H. Li, S.-L. Cai, W.-G. Zhang and S.-R. Zheng, Chem. Commun., 2022, 58, 5013–5016 RSC
- M. F. Thorne, C. Castillo-Blas, L. N. McHugh, A. M. Bumstead, G. Robertson, A. F. Sapnik, C. S. Coates, F. N. Sayed, C. P. Grey, D. A. Keen, M. Etter, I. da Silva, K. Užarević and T. D. Bennett, Chem. Commun., 2022, 58(11949), 11952 Search PubMed
- P.-Q. Liao, D.-D. Zhou, A.-X. Zhu, L. Jiang, R.-B. Lin, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2012, 134, 17380–17383 CrossRef CAS PubMed
- J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2008, 130, 6010–6017 CrossRef CAS PubMed
- X.-N. Cheng, W.-X. Zhang, Y.-Y. Lin, Y.-Z. Zheng and X.-M. Chen, Adv. Mater., 2007, 19, 1494–1498 CrossRef CAS
- S. K. Ghosh, W. Kaneko, D. Kiriya, M. Ohba and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 8843–8847 CrossRef CAS PubMed
- A. Kondo, T. Nakagawa, H. Kajiro, A. Chinen, Y. Hattori, F. Okino, T. Ohba, K. Kaneko and H. Kanoh, Inorg. Chem., 2010, 49, 9247–9252 CrossRef CAS PubMed
- L. Wen, P. Cheng and W. Lin, Chem. Commun., 2012, 48, 2846–2848 RSC
- S. B. Choi, H. Furukawa, H. J. Nam, D.-Y. Jung, Y. H. Jhon, A. Walton, D. Book, M. O’Keeffe, O. M. Yaghi and J. Kim, Angew. Chem., Int. Ed., 2012, 51, 8791–8795 CrossRef CAS PubMed
- M. C. Das and P. K. Bharadwaj, Chem. – Eur. J., 2010, 16, 5070–5077 CrossRef CAS PubMed
- X.-D. Chen, X.-H. Zhao, M. Chen and M. Du, Chem. – Eur. J., 2009, 15, 12974–12977 CrossRef CAS PubMed
- J.-P. Zhang, Y.-Y. Lin, W.-X. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2005, 127, 14162–14163 CrossRef CAS PubMed
- M. de J. Velásquez-Hernández, V. B. López-Cervantes, E. Martínez-Ahumada, M. Tu, U. Hernández-Balderas, D. Martínez-Otero, D. R. Williams, V. Martis, E. Sánchez-González, J.-S. Chang, J. S. Lee, J. Balmaseda, R. Ameloot, I. A. Ibarra and V. Jancik, Chem. Mater., 2022, 34, 669–677 CrossRef
- C.-B. Tian, R.-P. Chen, C. He, W.-J. Li, Q. Wei, X.-D. Zhang and S.-W. Du, Chem. Commun., 2014, 50, 1915–1917 RSC
- Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov, T. Tsuruoka, S. Isoda, W. Kosaka, O. Sakata and S. Kitagawa, Science, 2013, 339, 193–196 CrossRef CAS PubMed
- Z. Hu, C. Tao, H. Liu, X. Zou, H. Zhu and J. Wang, J. Mater. Chem. A, 2014, 2, 14222–14227 RSC
- H. Chen, Z. You, X. Wang, Q. Qiu, Y. Ying and Y. Wang, Chem. Eng. J., 2022, 446, 137098 CrossRef CAS
- B. Manna, A. K. Chaudhari, B. Joarder, A. Karmakar and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 998–1002 CrossRef CAS PubMed
- R. Guo, Y. Liu, Z. Wang, H. Wang and H. Liu, CrystEngComm, 2023, 25, 4157–4166 RSC
- V. A. Drebushchak, J. Therm. Anal. Calorim., 2020, 142, 1097–1113 CrossRef CAS
- S. R. G. Balestra, R. Bueno-Perez, S. Hamad, D. Dubbeldam, A. R. Ruiz-Salvador and S. Calero, Chem. Mater., 2016, 28, 8296–8304 CrossRef CAS PubMed
- N. Lock, Y. Wu, M. Christensen, L. J. Cameron, V. K. Peterson, A. J. Bridgeman, C. J. Kepert and B. B. Iversen, J. Phys. Chem. C, 2010, 114, 16181–16186 CrossRef CAS
- D. Dubbeldam, K. S. Walton, D. E. Ellis and R. Q. Snurr, Angew. Chem., Int. Ed., 2007, 46, 4496–4499 CrossRef CAS PubMed
- Y. Wu, A. Kobayashi, G. J. Halder, V. K. Peterson, K. W. Chapman, N. Lock, P. D. Southon and C. J. Kepert, Angew. Chem., Int. Ed., 2008, 47, 8929–8932 CrossRef CAS PubMed
- J. D. Evans, J. P. Dürholt, S. Kaskel and R. Schmid, J. Mater. Chem. A, 2019, 7, 24019–24026 RSC
- L. D. DeVries, P. M. Barron, E. P. Hurley, C. Hu and W. Choe, J. Am. Chem. Soc., 2011, 133, 14848–14851 CrossRef CAS PubMed
- I. Grobler, V. J. Smith, P. M. Bhatt, S. A. Herbert and L. J. Barbour, J. Am. Chem. Soc., 2013, 135, 6411–6414 CrossRef CAS PubMed
- B. Garai, V. Bon, A. Efimova, M. Gerlach, I. Senkovska and S. Kaskel, J. Mater. Chem. A, 2020, 8, 20420–20428 RSC
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed
- A. Demessence and J. R. Long, Chem. – Eur. J., 2010, 16, 5902–5908 CrossRef CAS PubMed
- J. Xiao, Y. Wu, M. Li, B.-Y. Liu, X.-C. Huang and D. Li, Chem. – Eur. J., 2013, 19, 1891–1895 CrossRef CAS PubMed
- H. J. Choi, M. Dincă and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848–7850 CrossRef CAS PubMed
- P. L. Llewellyn, S. Bourrelly, C. Serre, Y. Filinchuk and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 7751–7754 CrossRef CAS PubMed
- C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CrossRef CAS PubMed
- K. Biradha, Y. Hongo and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3395–3398 CrossRef CAS PubMed
- M.-H. Zeng, X.-L. Feng and X.-M. Chen, Dalton Trans., 2004, 2217–2223 RSC
- S. Yang, X. Lin, W. Lewis, M. Suyetin, E. Bichoutskaia, J. E. Parker, C. C. Tang, D. R. Allan, P. J. Rizkallah, P. Hubberstey, N. R. Champness, K. Mark Thomas, A. J. Blake and M. Schröder, Nat. Mater., 2012, 11, 710–716 CrossRef CAS PubMed
- E. Y. Lee and M. P. Suh, Angew. Chem., Int. Ed., 2004, 43, 2798–2801 CrossRef CAS PubMed
- L. Xiang, D. Liu, H. Jin, L.-W. Xu, C. Wang, S. Xu, Y. Pan and Y. Li, Mater. Horiz., 2020, 7, 223–228 RSC
- C. S. Smoljan, Z. Li, H. Xie, C. J. Setter, K. B. Idrees, F. A. Son, F. Formalik, S. Shafaie, T. Islamoglu, L. K. Macreadie, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2023, 145, 6434–6441 CrossRef CAS PubMed
- M. Wriedt, A. A. Yakovenko, G. J. Halder, A. V. Prosvirin, K. R. Dunbar and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 4040–4050 CrossRef CAS PubMed
- Y. Han, M. A. Sinnwell, S. J. Teat, M. L. Sushko, M. E. Bowden, Q. R. S. Miller, H. T. Schaef, L. Liu, Z. Nie, J. Liu and P. K. Thallapally, Adv. Sci., 2019, 6, 1802056 CrossRef PubMed
- D. Liu, T.-F. Liu, Y.-P. Chen, L. Zou, D. Feng, K. Wang, Q. Zhang, S. Yuan, C. Zhong and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 7740–7746 CrossRef CAS PubMed
- C. Castillo-Blas, V. A. de la Peña-O’Shea, I. Puente-Orench, J. Romerode Paz, R. Sáez-Puche, E. Gutiérrez-Puebla, F. Gándara and Á. Monge, Sci. Adv., 2017, 3, e1700773 CrossRef PubMed
- K. Gedrich, I. Senkovska, I. A. Baburin, U. Mueller, O. Trapp and S. Kaskel, Inorg. Chem., 2010, 49, 4440–4446 CrossRef CAS PubMed
- I. A. Lázaro, Eur. J. Inorg. Chem., 2020, 4284–4294 CrossRef
- J. B. James and Y. S. Lin, J. Phys. Chem. C, 2016, 120, 14015–14026 CrossRef CAS
-
H. J. Höhne, G. W. H. Hemminger and W. F. Flammersheim, Differential Scanning Calorimetry, Springer, 2nd edn, 2003 Search PubMed
- Q. Zheng, Y. Zhang, M. Montazerian, O. Gulbiten, J. C. Mauro, E. D. Zanotto and Y. Yue, Chem. Rev., 2019, 119, 7848–7939 CrossRef CAS PubMed
- F. Gándara and T. D. Bennett, IUCrJ, 2014, 1, 563–570 CrossRef PubMed
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
- F. Gándara, F. J. Uribe-Romo, D. K. Britt, H. Furukawa, L. Lei, R. Cheng, X. Duan, M. O’Keeffe and O. M. Yaghi, Chem. – Eur. J., 2012, 18, 10595–10601 CrossRef PubMed
- R. N. Widmer, G. I. Lampronti, S. Chibani, C. W. Wilson, S. Anzellini, S. Farsang, A. K. Kleppe, N. P. M. Casati, S. G. MacLeod, S. A. T. Redfern, F.-X. Coudert and T. D. Bennett, J. Am. Chem. Soc., 2019, 141, 9330–9337 CrossRef CAS PubMed
- C. Castillo-Blas, J. M. Moreno, I. Romero-Muñiz and A. E. Platero-Prats, Nanoscale, 2020, 12, 15577–15587 RSC
- C. Castillo-Blas, A. M. Chester, R. P. Cosquer, A. F. Sapnik, L. Corti, R. Sajzew, B. Poletto-Rodrigues, G. P. Robertson, D. J. M. Irving, L. N. McHugh, L. Wondraczek, F. Blanc, D. A. Keen and T. D. Bennett, J. Am. Chem. Soc., 2023, 145, 22913–22924 CrossRef CAS PubMed
- A. M. Chester, C. Castillo-Blas, R. Sajzew, B. P. Rodrigues, R. Mas-Balleste, A. Moya, J. E. Snelson, S. M. Collins, A. F. Sapnik, G. P. Robertson, D. J. M. Irving, L. Wondraczek, D. A. Keen and T. D. Bennett, Chem. Sci., 2023, 14, 11737–11748 RSC
- A. E. Platero-Prats, A. Mavrandonakis, L. C. Gallington, Y. Liu, J. T. Hupp, O. K. Farha, C. J. Cramer and K. W. Chapman, J. Am. Chem. Soc., 2016, 138, 4178–4185 CrossRef CAS PubMed
- M. Mazaj, G. Mali, M. Rangus, E. Žunkovič, V. Kaučič and N. Zabukovec Logar, J. Phys. Chem. C, 2013, 117, 7552–7564 CrossRef CAS
- X. Gong, K. Gnanasekaran, Z. Chen, L. Robison, M. C. Wasson, K. C. Bentz, S. M. Cohen, O. K. Farha and N. C. Gianneschi, J. Am. Chem. Soc., 2020, 142, 17224–17235 CrossRef CAS PubMed
- Z. Lyu, L. Yao, W. Chen, F. C. Kalutantirige and Q. Chen, Chem. Rev., 2023, 123(7), 4051–4145 CrossRef CAS PubMed
- A. Rizvi, J. T. Mulvey, B. P. Carpenter, R. Talosig and J. P. Patterson, Chem. Rev., 2021, 121, 14232–14280 CrossRef CAS PubMed
- J. P. Patterson, P. Abellan, M. S. Denny, C. Park, N. D. Browning, S. M. Cohen, J. E. Evans and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137, 7322–7328 CrossRef CAS PubMed
- J. Lyu, X. Gong, S.-J. Lee, K. Gnanasekaran, X. Zhang, M. C. Wasson, X. Wang, P. Bai, X. Guo, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 4609–4615 CrossRef CAS PubMed
- X. Gong, K. Gnanasekaran, K. Ma, C. J. Forman, X. Wang, S. Su, O. K. Farha and N. C. Gianneschi, J. Am. Chem. Soc., 2022, 144, 6674–6680 CrossRef CAS PubMed
- D. Thomas, D. Till and R. Claus, Science, 2021, 371, 371–374 CrossRef PubMed
- T. Watanabe, S. Keskin, S. Nair and D. S. Sholl, Phys. Chem. Chem. Phys., 2009, 11, 11389–11394 RSC
- K. Sillar, A. Hofmann and J. Sauer, J. Am. Chem. Soc., 2009, 131, 4143–4150 CrossRef CAS PubMed
- C. L. Hobday, R. J. Marshall, C. F. Murphie, J. Sotelo, T. Richards, D. R. Allan, T. Düren, F.-X. Coudert, R. S. Forgan, C. A. Morrison, S. A. Moggach and T. D. Bennett, Angew. Chem., Int. Ed., 2016, 55, 2401–2405 CrossRef CAS PubMed
- M. Ji, X. Lan, Z. Han, C. Hao and J. Qiu, Inorg. Chem., 2012, 51, 12389–12394 CrossRef CAS PubMed
- R. Gaillac, P. Pullumbi and F. X. Coudert, J. Phys. Chem. C, 2018, 122, 6730–6736 CrossRef CAS
- R. Gaillac, P. Pullumbi, K. A. Beyer, K. W. Chapman, D. A. Keen, T. D. Bennett and F.-X. Coudert, Nat. Mater., 2017, 16, 1149–1154 CrossRef CAS PubMed
- R. Gaillac, P. Pullumbi, T. D. Bennett and F.-X. Coudert, Chem. Mater., 2020, 32, 8004–8011 CrossRef CAS
- A. U. Ortiz, A. Boutin, A. H. Fuchs and F.-X. Coudert, J. Phys. Chem. Lett., 2013, 4, 1861–1865 CrossRef CAS PubMed
- T. P. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. M. Aktulga, T. Verstraelen, A. Grama and A. C. T. van Duin, npj Comput. Mater., 2016, 2, 15011 CrossRef CAS
- N. Castel and F.-X. Coudert, J. Phys. Chem. C, 2022, 126, 19532–19541 CrossRef CAS
- A. Duarte Rodrigues, K. Fahsi, X. Dumail, N. Masquelez, A. van der Lee, S. Mallet-Ladeira, R. Sibille, J.-S. Filhol and S. G. Dutremez, Chem. – Eur. J., 2018, 24, 1586–1605 CrossRef CAS PubMed
- S. M. J. Rogge, M. Waroquier and V. Van Speybroeck, Nat. Commun., 2019, 10, 4842 CrossRef PubMed
- C. Triguero, F.-X. Coudert, A. Boutin, A. H. Fuchs and A. V. Neimark, J. Phys. Chem. Lett., 2011, 2, 2033–2037 CrossRef CAS
- J. Wieme, K. Lejaeghere, G. Kresse and V. Van Speybroeck, Nat. Commun., 2018, 9, 4899 CrossRef CAS PubMed
- L. Schaper and R. Schmid, Commun. Chem., 2023, 6, 233 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |