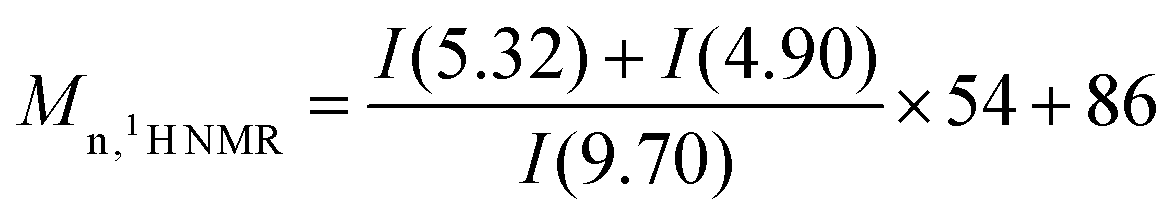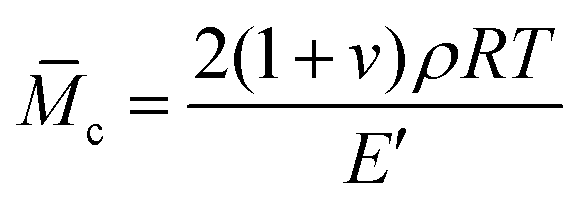DOI:
10.1039/C9PY01826C
(Paper)
Polym. Chem., 2020,
11, 1348-1355
Tuning the mechanical and dynamic properties of imine bond crosslinked elastomeric vitrimers by manipulating the crosslinking degree†
Received
2nd December 2019
, Accepted 29th December 2019
First published on 30th December 2019
Abstract
Vitrimers are a class of covalently crosslinked networks that act as a traditional thermoset at low temperatures while they can flow and act as a viscoelastic liquid at high temperatures through bond exchange reactions. The state-of-the-art approach to tune the mechanical and dynamic properties of vitrimers is to explore new exchangeable chemistries and alter catalyst systems and stoichiometric ratios of the starting materials. Herein, we initiate an alternative approach toward this goal. In particular, aldehyde group-terminated polybutadiene rubber (APB) with different molecular weights were prepared and subsequently crosslinked by tris(2-aminoethyl)amine through the formation of imine bond linkages. The modulus and ultimate strength of the imine bond crosslinked networks are consistently enhanced with the decrease of the molecular weight of APB precursors. In addition, APB with a higher molecular weight, i.e. a lower crosslinking degree, leads to a faster relaxation rate and a higher activation energy for network rearrangement. We envision that this work provides a methodology to tune the mechanical and dynamic properties of vitrimer materials by altering the precursor molecular weight and network crosslinking degree.
Introduction
Covalent adaptable polymer networks contain dynamic covalent bonds that can rearrange the network topology under external stimuli, which provides a trade-off between the malleability and covalently crosslinked architecture of polymers.1–4 Recently, Leibler and co-workers pioneered the concept of vitrimers, which are capable of altering their topologies through associative exchange reactions.5–9 Benefiting from the associative exchange mechanism, the network connectivity is maintained because the number of crosslinks remains invariable during network rearrangement; as a result the network is insoluble at all temperatures. In striking contrast to thermoplastic whose viscosity changes abruptly near the glass transition temperature, the viscosity of vitrimers exhibits a gradual Arrhenius-like variation with temperature, allowing them to be recycled and making them malleable in the solid state without the need for precise temperature control or a complicated mold.10,11 Due to these attractive properties, vitrimers have found promising applications in recyclable thermosetting polymers,12,13 mouldable liquid-crystalline elastomers,14 reconfigurable shape memory polymers,15,16 recycling carbon fibers from thermosetting binders,17,18 electronic skins19,20 and healable materials.21,22
To tune the thermomechanical performance and dynamic properties of vitrimers, three elegant routes have been established. The first approach is to explore new exchangeable chemistries to expand the galleries of vitrimer materials. Thus far, various exchangeable chemistries including disulfide exchange,23,24 boronic ester exchange,25,26 transalkylation,13,27 imine chemistry,28–30 olefin metathesis,31 transamination of vinylogous urethanes32,33 and siloxane equilibration34 have been explored in the context of vitrimers. For example, highly dynamic boronic ester bonds can endow the networks with self-healing ability at room temperature.21 Transamination exchange reactions can undergo two bond exchange mechanisms in fluorinated vinylogous urethane vitrimers, conferring the networks with a novel dual temperature response.32 The second approach is to alter the catalyst system in specific vitrimers. For example, Leibler et al. reported that the transesterification reaction rate of β-hydroxyl esters could be tuned by varying the amount and nature of the catalyst, thus leading to a tunable topology freezing transition temperature.11,35 Du Prez et al. reported that the kinetics of the transamination of vinylogous urethanes could be accelerated by acidic additives while it could be suppressed by basic additives, offering the possibility of manipulating the viscoelastic properties of vitrimers in a wide range.36 The third approach is to change the stoichiometric ratios among the starting materials.33,37 For instance, Zhang et al. demonstrated that the vitrimer network prepared by reacting epoxy and anhydride at a stoichiometric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 exhibited a higher modulus and a faster stress relaxation at elevated temperatures.38 However, to the best of our knowledge, the effects of the precursor molecular weight and network crosslinking degree on the final properties of vitrimers have rarely been reported and remain to be explored.
:0.5 exhibited a higher modulus and a faster stress relaxation at elevated temperatures.38 However, to the best of our knowledge, the effects of the precursor molecular weight and network crosslinking degree on the final properties of vitrimers have rarely been reported and remain to be explored.
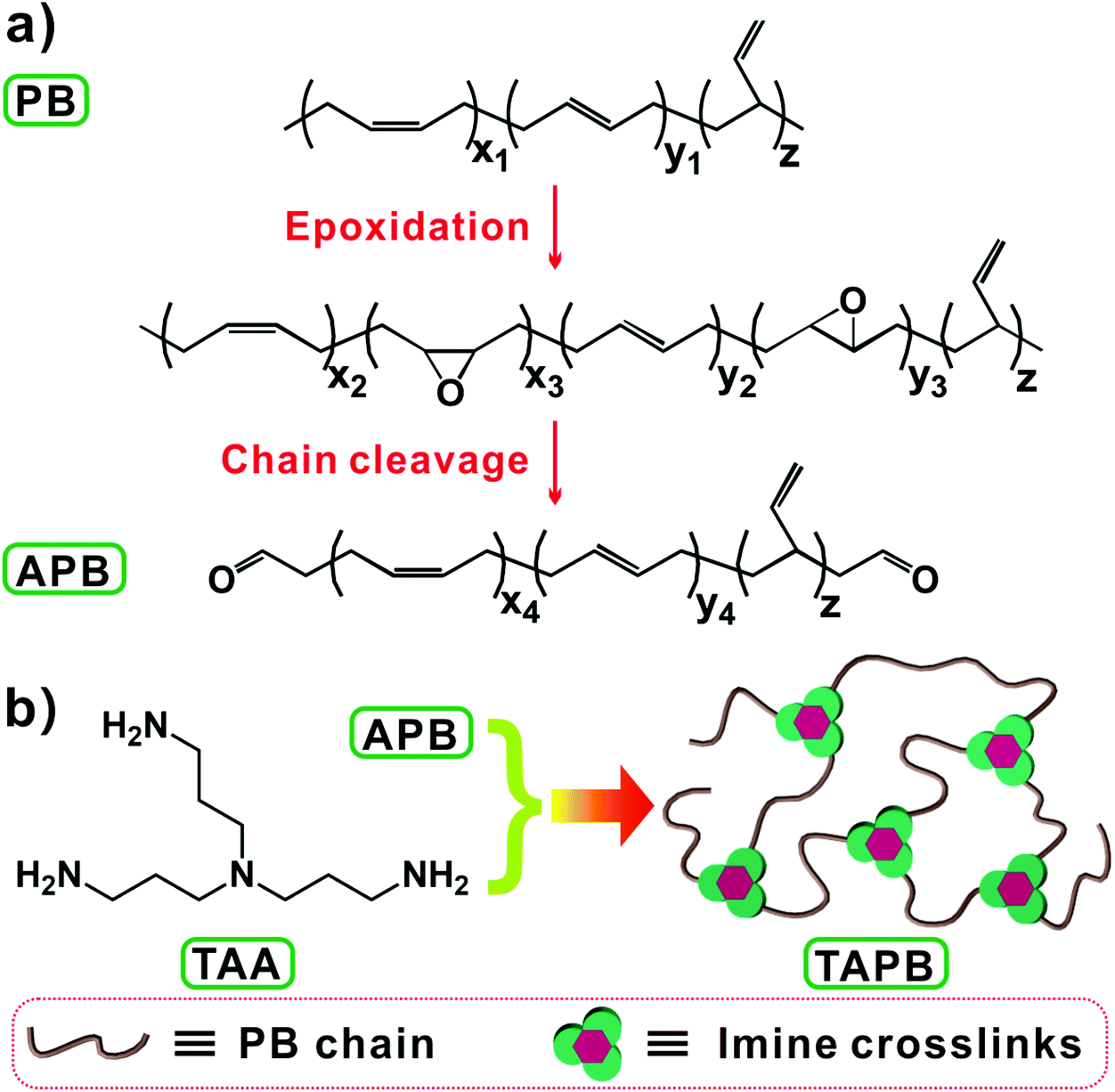
Imine bonds that are derived from the condensation reaction between aldehyde and amino groups are types of readily accessible and catalyst-free dynamic covalent bonds.28,39 Recently, Zhang et al. synthesized a series of malleable polyimine thermosets from terephthaldehyde and multifunctional amines, and the mechanical properties and moisture sensitivity could be manipulated through the judicious selection of monomers.18,28,40 In this work, we prepared imine bond-crosslinked elastomeric vitrimers by crosslinking aldehyde group-terminated polybutadiene (APB) with tris(2-aminoethyl)amine (TAA), whose mechanical performance and dynamic properties could be tuned by changing the molecular weight of APB. To this end, APB with different molecular weights were firstly prepared through a one-pot method by combining epoxidation and chain-cleavage reactions of polybutadiene (Scheme 1a), and then the resulting APB was crosslinked by TAA through imine bond crosslinks (Scheme 1b). The effects of the APB molecular weight and network crosslinking density on the mechanical properties and bond exchange reaction kinetics of the networks were studied.
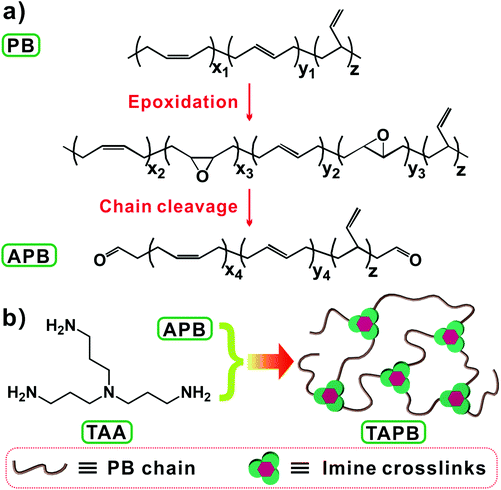 |
| | Scheme 1 (a) One-pot synthesis of APB. (b) Preparation of TAPB by reacting APB with TAA. | |
Experimental
Materials
Commercially available cis-1,4-polybutadiene rubber (PB, trade name: BR 9000, vinyl content = 2.1 wt%) was purchased from Beijing Yanshan Petrochemical Co. Ltd, Sinopec, China. 3-Chloroperbenzoic acid (mCPBA, 75%), periodic acid (H5IO6, 99%), tris(2-aminoethyl)amine (TAA, 97%), n-butylamine (BA, 99%) and 2,6-di-tert-butyl-4-methylphenol (BHT, AR) were obtained from Sigma-Aldrich. Sodium bicarbonate, Celite, tetrahydrofuran, cyclohexane, methanol and toluene were purchased from Tianjin Fuyu Fine Chemical Co. Ltd.
Synthesis of APB
The synthetic procedure of APB is shown in Scheme 1a, according to the previously reported literature.41 Briefly, 5 g of PB was dissolved in 100 mL of cyclohexane, into which a mCPBA solution in tetrahydrofuran (25 mL) with a varying molar ratio of the mCPBA/butadiene unit was added dropwise within 0.5 h. The epoxidation reaction was carried out at room temperature for 1 h. Afterwards, a H5IO6 solution (the molar number of H5IO6 is equal to that of mCPBA) in tetrahydrofuran (25 mL) was added into the above solution to initiate the chain-cleavage reaction for 2 h at room temperature, followed by the addition of sodium bicarbonate for neutralization. The resulting solution was then filtered through Celite to remove insoluble iodic acid, and the filtrate was precipitated and washed with methanol. Finally, the antioxidant agent BHT was added, and the product was vacuum dried at 40 °C overnight to obtain APB-x, in which x represents the percentage of the molar ratio of the mCPBA/butadiene unit (Table 1).
Table 1 Molar ratio of the mCPBA/butadiene unit, molecular weights of APB determined by 1H NMR and GPC, and molar number of aldehyde groups per 100 g of APB
| |
mCPBA/butadiene unit (%) |
M
n (g mol−1) |
Aldehyde group molar number per 100 g APBa (mmol) |
|
1H NMR |
GPC |
|
Molar number of aldehyde groups was calculated using the molecular weight measured by 1H NMR and a functionality of 2.
|
| APB-0.5 |
0.5 |
37134 |
15917 |
5.39 |
| APB-0.55 |
0.55 |
18510 |
11658 |
10.80 |
| APB-0.65 |
0.65 |
16010 |
11229 |
12.49 |
| APB-1.2 |
1.20 |
10710 |
5227 |
18.67 |
| APB-2.0 |
2.00 |
5067 |
3301 |
39.47 |
Synthesis of imine bond crosslinked APB (TAPB)
12 g of APB was dissolved in 75 mL of toluene. After the solution was stirred in an ice-bath for 1 h, a stoichiometric amount of TAA in 15 mL of toluene was added, followed by vigorous stirring in an ice-bath for 5 min. The mixture was immediately transferred into a Teflon mold and the solvent was allowed to evaporate at room temperature. After that, the obtained film was left at 50 °C for 6 h and 90 °C for 6 h. In the context, TAPB-x represents TAA crosslinked APB-x.
Characterization
Proton nuclear magnetic resonance spectra (1H NMR) were recorded on a Bruker AVANCE III 600 MHz spectrometer. Fourier transform infrared (FTIR) spectroscopy was performed on a Bruker Vertex 70 FTIR spectrometer. The molecular weight was determined by gel permeation chromatography (GPC) using a Waters 2414 series system in tetrahydrofuran and calibrated with the polystyrene standard. Thermal gravimetric analysis (TGA) was performed on a TA Q50 thermogravimetric analyzer under a N2 atmosphere at 10 °C min−1.
Sol fraction, swelling ratio, and crosslinking density were measured based on equilibrium swelling tests by immersing the samples in toluene.42 The detailed procedure is described in the ESI.†
Uniaxial tensile test and cyclic tensile test were conducted on dogbone-shaped samples (ca. 60 mm × 4 mm × 0.5 mm) with a gauge length of 40 mm using a U-CAN UT-2060 instrument at room temperature with an extension rate of 500 mm min−1. At least four specimens were measured for the uniaxial tensile test, and the average value was calculated. The reprocessability was conducted by grinding the samples into a fine powder using an Ultracentrifugal Grinder FM200 machine, and then the powder was screened with 70 mesh sieves, followed by compression molding at 160 °C for 40 min.
Creep tests, cyclic strain-recovery tests and stress relaxation experiments were conducted on TA Q800 DMA apparatus. For the creep experiment, a nominal stress of 0.15 MPa was applied on the sample after 15 min equilibration at 150 °C. For the cyclic strain-recovery test, the stress was alternated between 0.1 MPa for 30 min and 0 MPa for 5 min in each cycle. For the stress relaxation test, after equilibrating at desired temperatures for 15 min, the sample was stretched to a constant strain of 2% in the linear viscoelastic region and the stress decay was monitored over time.
Results and discussion
Covalent crosslinking of APB with TAA
To prepare APB, the double bonds in the PB backbone were partially epoxidized and then cleaved at the exact epoxidized sites. As shown in the FTIR spectrum of PB in Fig. 1a, the absorptions at 1653, 965, 730 and 912 cm−1 are due to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in cis-1,4 and bending vibrations of trans-1,4, cis-1,4 and 1,2-vinyl, respectively. Considering the epoxidation selectivity of the double bonds where 1,2-vinyl groups are hardly epoxidized by mCPBA,41,43 the FTIR absorption peak of 1,2-vinyl at 912 cm−1 is chosen as an internal reference. Compared with PB, a new absorption peak appears at 1729 cm−1 in the FTIR spectrum of APB-2.0, which originates from the stretching vibrations of CO in the aldehyde groups.41 With the increase of mCPBA loading, the absorption related to the aldehyde groups is consistently intensified (Fig. S1†), indicating a higher aldehyde group content. Fig. 1b and S2† depict the 1H NMR spectra of PB and APB. When compared to PB, new resonances at 9.70 and 2.27–2.44 ppm appear in the 1H NMR spectrum of APB, which are ascribed to the protons in the aldehyde groups and methylene protons connected to the aldehyde groups, respectively. Because the epoxidation and chain-cleavage reactions are controllable and highly efficient,41 APB with different molecular weights can be readily obtained by varying the epoxidation degree. According to the integral area in 1H NMR spectra, the molecular weights (Mn) of APB are calculated based on eqn (1), where I(5.32), I(4.90) and I(9.70) are the integral area of peaks related to the signals of 1,4-isomer, 1,2-vinyl and aldehyde groups, respectively. As expected, the molecular weights of APB are consistently decreased with an increase in the mCPBA loading (Table 1). Moreover, the molecular weights measured by GPC also show a similar tendency (Table 1, Fig. S3†), although the values are not identical to those obtained from 1H NMR due to different measurement methods.
C in cis-1,4 and bending vibrations of trans-1,4, cis-1,4 and 1,2-vinyl, respectively. Considering the epoxidation selectivity of the double bonds where 1,2-vinyl groups are hardly epoxidized by mCPBA,41,43 the FTIR absorption peak of 1,2-vinyl at 912 cm−1 is chosen as an internal reference. Compared with PB, a new absorption peak appears at 1729 cm−1 in the FTIR spectrum of APB-2.0, which originates from the stretching vibrations of CO in the aldehyde groups.41 With the increase of mCPBA loading, the absorption related to the aldehyde groups is consistently intensified (Fig. S1†), indicating a higher aldehyde group content. Fig. 1b and S2† depict the 1H NMR spectra of PB and APB. When compared to PB, new resonances at 9.70 and 2.27–2.44 ppm appear in the 1H NMR spectrum of APB, which are ascribed to the protons in the aldehyde groups and methylene protons connected to the aldehyde groups, respectively. Because the epoxidation and chain-cleavage reactions are controllable and highly efficient,41 APB with different molecular weights can be readily obtained by varying the epoxidation degree. According to the integral area in 1H NMR spectra, the molecular weights (Mn) of APB are calculated based on eqn (1), where I(5.32), I(4.90) and I(9.70) are the integral area of peaks related to the signals of 1,4-isomer, 1,2-vinyl and aldehyde groups, respectively. As expected, the molecular weights of APB are consistently decreased with an increase in the mCPBA loading (Table 1). Moreover, the molecular weights measured by GPC also show a similar tendency (Table 1, Fig. S3†), although the values are not identical to those obtained from 1H NMR due to different measurement methods.| |  | (1) |
 |
| | Fig. 1 (a) FTIR spectra of PB, APB-2.0 and TAPB-2.0. (b) 1H NMR spectra of PB and APB-2.0. | |
The imine bond crosslinked TAPB was synthesized by reacting the as-prepared APB with TAA, which is explicitly confirmed by the FTIR results. Compared with APB, the absorption peak for aldehyde groups at 1729 cm−1 is almost disappeared and a new absorption peak ascribed to the stretching vibration of CN at 1595 cm−1 is observed in the FTIR spectra of TAPB (Fig. 1a and S4†). These observations indicate the formation of imine bonds through the condensation reaction between the aldehyde and amino groups. It should be noted that small amounts of free amino and aldehyde moieties are still present in the networks (Fig. S4†), which may be because the high viscosity of the network limits further reaction at the later curing stage. The resulting TAPB samples can't be dissolved in their good solvents such as toluene, which is an indicator of the formation of covalently crosslinked networks. After immersing the samples in toluene at room temperature for 3 days to achieve swelling equilibrium, the sol fraction and swelling ratio of TAPB are consistently decreased with the decrease of the Mn of APB (Fig. 2a and Table S1†). This is because a lower molecular weight of APB implies a denser network structure, as evidenced by the increased crosslinking density with decreasing molecular weight (Fig. 2a and Table S1†). Furthermore, the average molecular weight between two adjacent crosslinks (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) c) in TAPB is calculated according to eqn (2):
c) in TAPB is calculated according to eqn (2):
| |  | (2) |
where
ν is Poisson's ratio (here it is 0.5 for rubber),
ρ is the density of TAPB,
R is the universal gas constant and
E′ is the storage modulus at 25 °C obtained from DMA measurements. As expected, the
c value of TAPB is increased linearly with the molecular weight of APB (
Fig. 2b and Table S1
†).
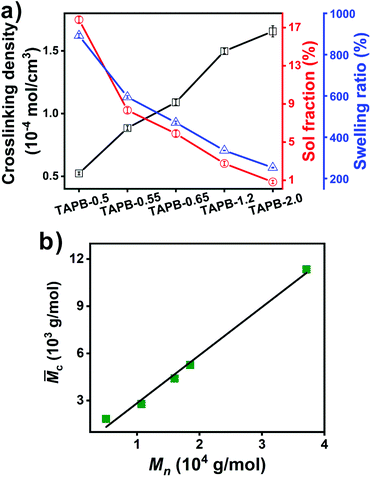 |
| | Fig. 2 (a) Crosslinking density, sol fraction and swelling ratio of TAPB. (b) Relationship between c of TAPB and Mn (determined by 1H NMR) of APB. | |
Mechanical properties and dynamic properties of TAPB
The typical stress–strain curves of TAPB are illustrated in Fig. 3, and the detailed mechanical properties are tabulated in Table S2.† TAPB samples have Young's modulus of 0.24–3.06 MPa, an ultimate stress of 0.42–1.16 MPa and a breaking strain of 53–451%. The samples can be restored to their original shape after being stretched to a large deformation and exhibit little hysteresis loss in the loading–unloading cycle (Fig. S5†), showing good elasticity due to covalently crosslinked elastomers. It is evident that the mechanical properties can be easily tuned by manipulating the molecular weight of APB. Specifically, with the decrease of Mn, the Young's modulus and ultimate stress are consistently improved, while the breaking strain is gradually decreased (Fig. 3). This is because a lower Mn represents a shorter flexible chain and a higher crosslinking density in the network, which results in a more constrained and rigid network.
 |
| | Fig. 3 Typical stress–strain curves of TAPB. | |
Due to the dynamic nature of imine bonds, TAPB samples are expected to be capable of rearranging their topologies via imine exchange at elevated temperatures. As shown in Fig. 4a, the samples show a sudden increase in strains upon loading at 30 °C, and the strains remain invariable with time while recovering to their original length immediately after unloading, exhibiting a typical feature of covalently crosslinked networks. However, when the temperature is elevated to 150 °C, the samples show an obvious viscous behavior after thermal expansion and elastic deformation. Such a discrepancy in the viscoelasticity at 30 and 150 °C can be interpreted by the fact that network topology rearrangement controlled by the imine exchange is frozen at low temperatures while it is activated at high temperatures.
 |
| | Fig. 4 (a) Cyclic strain/recovery profiles of TAPB-0.55, TAPB-0.65 and TAPB-1.2 at 30 and 150 °C with a stress of 0.1 MPa. (b) Creep experiments for TAPB with a constant stress of 0.15 MPa at 150 °C. (c) Stress relaxation curves for TAPB-0.65 at different temperatures under a constant strain of 2%. (d) Fitting of τ to the Arrhenius law for TAPB. | |
Fig. 4b shows the creep curves of TAPB at 150 °C. After a sudden increase in strain due to the elastic response, the strains for all samples are increased linearly with time, demonstrating that the networks can flow at high temperatures. This malleability is induced by imine exchange at high temperatures, which enables the rearrangement of the network topology to adapt to the external force. The relaxation time can be calculated according the following equations:44
where
η is the viscosity (Pa s),
σ is the stress (Pa),
ε′ is the strain rate (s
−1), and
E is the Young's modulus at 150 °C. Here,
σ is 0.15 MPa for all the samples,
ε′ is the slope of the creep curves, and
E is determined as the slope of the stress–strain curves (0.5–5% strain) measured by DMA Q800 at 150 °C. All these parameters are listed in
Table 2. It can be seen that the relaxation time
τ is prolonged with decreasing molecular weight (
i.e., increasing crosslinking density). This is because more imine exchange is required to achieve network topology rearrangement in the samples having a higher crosslinking density.
44
Table 2 Strain rate, Young's modulus at 150 °C, relaxation time and activation energy of TAPB
| Samples |
ε′ (10−2 min−1) |
E (MPa) |
τ (10−2 min) |
E
a (kJ mol−1) |
| TAPB-0.5 |
1624.0 |
0.42 |
2.2 |
85.6 |
| TAPB-0.55 |
245.7 |
1.13 |
5.4 |
76.5 |
| TAPB-0.65 |
55.5 |
1.50 |
18.0 |
70.7 |
| TAPB-1.2 |
13.8 |
2.52 |
43.1 |
59.0 |
| TAPB-2.0 |
3.8 |
4.04 |
97.7 |
49.2 |
Fig. 4c and S6† show the relaxation curves of TAPB at different temperatures. Apparently, the relaxation rate is promoted by elevating the temperature, because the relaxation process is actually controlled by thermo-activated imine exchange which is accelerated with temperature. The relaxation time (τ) determined as the time required to reach a relaxation of 37% (1/e) of the initial stress exhibits an Arrhenius-like temperature dependence (Fig. 4d), indicating the associative pathway of imine exchange.28 The activation energy (Ea) of TAPB is calculated according to eqn (5) and tabulated in Table 2, which is within the previously reported Ea values for imine exchange reactions (48–157 kJ mol−1).28,30,45–47 Counterintuitively, the Ea values for TAPB are consistently increased with the decrease of the crosslinking density. Generally, a higher crosslinking density may result in a higher energy barrier for network rearrangement due to the restricted mobility of the polymer chains.37,48 Herein, unlike previously reported vitrimers that were synthesized from small molecules, TAPB is prepared using macromolecular APB as precursors and the network contains a rather lower fraction of imine bonds. In addition, the segment mobility almost does not change with the crosslinking density, as evidenced by the identical glass transition temperature among the TAPB samples (Table S1†). Therefore, it is reasonable to conclude that the concentration of imine bonds may dominate the network arrangement rather than chain mobility. Specifically, the samples with a lower crosslinking density indicate a lower concentration of imine bonds, which reduces the chance of the imine bonds approaching and colliding with each other to achieve effective exchange, and thus a higher energy is required to realize network rearrangement. It should be noted that creep and relaxation measurements were conducted after 15 min thermal equilibrium at 150 °C, during which the residual amino and aldehyde moieties were capable of fully reacting. This notion is evidenced by the disappearance of the FTIR absorptions related to the amino and aldehyde groups in the samples after being maintained at 150 °C for 15 min (Fig. S7†). Therefore, the residual free amino and aldehyde groups in the networks will not influence the dynamics of the networks.
| |  | (5) |
It is generally accepted that reshaping or recycling a permanently crosslinked polymeric material is inherently difficult. Herein, TAPB containing dynamic imine linkages can alter network topologies through associative imine exchange and exhibit gradual viscosity vibrations with temperature, enabling them to be reshaped in the solid state. As a proof of concept, the strip samples can be reshaped into complex shapes by bending or twisting them and allowing them to relax stress at 160 °C for 1 h (Fig. 5a). This process doesn't need precise temperature control and complicated mold, which provides significant feasibility to process geometrically complex objects.
 |
| | Fig. 5 (a) Image presentation of malleability for TAPB-1.2. (b) Recycling of TAPB-0.65 by hot pressing. (c) Recovery ratio of the mechanical properties for TAPB samples. (d) Typical stress–strain curves of TAPB-0.65 after multiple cycles of recycling. The scale bar represents 1 cm. | |
To demonstrate the reprocessibility, TAPB samples after the tensile test are ground into a fine power and then subjected to compression molding at 160 °C for 40 min. As shown in Fig. 5b, a new consolidated and smooth sample is obtained. The recycled samples can recuperate most of their original mechanical properties (Fig. 5c and S8†), and the stress–strain curves of TAPB-0.65 are almost overlapped after multiple cycles of recycling (Fig. 5d), revealing good recyclability of the TAPB samples. It should be mentioned that the recovery ratio of TAPB-1.2 and TAPB-2.0 is slightly higher than 100%, which may be because the reaction of the residual amino and aldehyde groups during the remolding process increases the crosslinking density. According to TGA tests (Fig. S9†), the onset degradation temperature of TAPB is above 300 °C and their weights remain almost invariable after keeping at 200 °C for 2 h, indicating that the TAPB samples are thermally stable during reprocessability.
The above-mentioned recycling process relies on the imine exchange reactions in TAPB networks in the solid state. Considering that imine bonds can undergo exchange reactions with free amine groups (as shown in Fig. 6a),49 we set out to recycle TAPB by adding monofunctional amines to degrade the crosslinked network. As shown in Fig. 6b, taking TAPB-0.5 as an example, it can only be swollen in toluene. When monofunctional amines (such as low boiling point n-butylamine) are added into the solution, the swollen TAPB-0.5 is completely collapsed and dissolved after 3 days at room temperature. This is because the networks are de-crosslinked through the exchange reactions between the imine crosslinks and n-butylamine. Afterwards, the solution mixture can be precipitated in methanol to obtain n-butylamine modified linear APB. By reintroducing TAA, the n-butylamine can be replaced by TAA and then evaporated during the successive curing process, and ultimately new crosslinked samples can be retrieved (Fig. 6c).
 |
| | Fig. 6 (a) Exchange reaction between imine bonds with free amino groups. (b) Schematic diagram of chemical recycling for TAPB using n-butylamine. (c) TAPB-0.5 just immersed in toluene (left). (i) TAPB-0.5 was immersed in toluene for 3 days. (ii) n-Butylamine was added and the solution was left for another 3 days at room temperature. (iii) The mixture solution was precipitated in methanol and dried. (iv) Conceptual recycling by adding TAA into the degraded product and curing. | |
Conclusions
In summary, we reported a methodology to manipulate the mechanical and dynamic properties of vitrimers by varying the precursor molecular weight and network crosslinking degree. APB with different molecular weights were firstly prepared through chemical epoxidation and chain-cleavage reactions of PB and were then crosslinked by TAA through imine bond crosslinks. The network crosslinking density is increased with the decrease of the molecular weight of the APB precursor. As the crosslinking density increases, the modulus and strength of TAPB are consistently improved while the breaking strain is gradually decreased due to a more constrained and rigid network. The samples having a higher crosslinking density exhibit a slower relaxation rate because more imine exchange is required to achieve network topology rearrangement. Interestingly, the energy barrier for the network rearrangement is increased with the decrease of the crosslinking density, as a low concentration of imine bonds reduces the chance of the imine bonds approaching and colliding with each other to achieve effective exchange. All the samples are malleable and can recuperate most of their original mechanical properties after recycling. Additionally, chemical recycling of the samples can be realized by de-crosslinking the imine bond crosslinks using monofunctional amines through imine exchange. Overall, this work provides an alternative method to tune the mechanical and dynamic properties of vitrimer materials by altering the precursor molecular weight and network crosslinking degree.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by the China National Funds for Distinguished Young Scientists (51825303) and the National Natural Science Foundation of China (51790503, 51703064 and 51673065).
References
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- Y. Jin, Z. Lei, P. Taynton, S. Huang and W. Zhang, Matter, 2019, 1, 1456–1493 CrossRef.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS.
- W. K. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- H. Xiang, J. Yin, G. Lin, X. Liu, M. Rong and M. Zhang, Chem. Eng. J., 2019, 358, 878–890 CrossRef CAS.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- Z. Q. Pei, Y. Yang, Q. M. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2014, 13, 36–41 CrossRef CAS PubMed.
- N. Zheng, Z. Z. Fang, W. K. Zou, Q. Zhao and T. Xie, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS PubMed.
- Z. Pei, Y. Yang, Q. Chen, Y. Wei and Y. Ji, Adv. Mater., 2016, 28, 156–160 CrossRef CAS PubMed.
- K. Yu, Q. Shi, M. L. Dunn, T. Wang and H. J. Qi, Adv. Funct. Mater., 2016, 26, 6098–6106 CrossRef CAS.
- P. Taynton, H. G. Ni, C. P. Zhu, K. Yu, S. Loob, Y. H. Jin, H. J. Qi and W. Zhang, Adv. Mater., 2016, 28, 2904–2909 CrossRef CAS PubMed.
- Z. Zou, C. Zhu, Y. Li, X. Lei, W. Zhang and J. Xiao, Sci. Adv., 2018, 4, eaaq0508 CrossRef PubMed.
- H. Tran, V. R. Feig, K. Liu, Y. Zheng and Z. Bao, Macromolecules, 2019, 52, 3965–3974 CrossRef CAS.
- O. R. Cromwell, J. Chung and Z. B. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- H. P. Xiang, H. J. Qian, Z. Y. Lu, M. Z. Rong and M. Q. Zhang, Green Chem., 2015, 17, 4315–4325 RSC.
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. Y. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- Y. Chen, Z. Tang, X. Zhang, Y. Liu, S. Wu and B. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 24224–24231 CrossRef CAS PubMed.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS.
- P. Taynton, K. Yu, R. K. Shoemaker, Y. H. Jin, H. J. Qi and W. Zhang, Adv. Mater., 2014, 26, 3938–3942 CrossRef CAS PubMed.
- X. F. Lei, Y. H. Jin, H. L. Sun and W. Zhang, J. Mater. Chem. A, 2017, 5, 21140–21145 RSC.
- H. Zhang, D. Wang, W. X. Liu, P. C. Li, J. J. Liu, C. Y. Liu, J. W. Zhang, N. Zhao and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2011–2018 CrossRef CAS.
- Y. X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed.
- M. Guerre, C. Taplan, R. Nicolaÿ, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2018, 140, 13272–13284 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. D. Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- X. Wu, X. Yang, R. Yu, X. J. Zhao, Y. Zhang and W. Huang, J. Mater. Chem. A, 2018, 6, 10184–10188 RSC.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- W. Denissen, M. Droesbeke, R. Nicolay, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef PubMed.
- K. Yu, P. Taynton, W. Zhang, M. L. Dunn and H. J. Qi, RSC Adv., 2014, 4, 48682–48690 RSC.
- T. Liu, C. Hao, S. Zhang, X. N. Yang, L. W. Wang, J. R. Han, Y. Z. Li, J. N. Xin and J. W. Zhang, Macromolecules, 2018, 51, 5577–5585 CrossRef CAS.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- P. Taynton, C. Zhu, S. Loob, R. Shoemaker, J. Pritchard, Y. Jin and W. Zhang, Polym. Chem., 2016, 7, 7052–7056 RSC.
- Q. Zhou, S. Jie and B. G. Li, Ind. Eng. Chem. Res., 2014, 53, 17884–17893 CrossRef CAS.
- P. J. Flory and J. J. Rehner, J. Chem. Phys., 1943, 11, 512–520 CrossRef CAS.
- M. Aguiar, S. C. De Menezes and L. Akcelrud, Macromol. Chem. Phys., 1994, 195, 3937–3948 CrossRef CAS.
- Y. X. Lu, F. Tournilhac, L. Leibler and Z. B. Guan, J. Am. Chem. Soc., 2012, 134, 8424–8427 CrossRef CAS PubMed.
- S. Dhers, G. Vantomme and L. Avérous, Green Chem., 2019, 21, 1596–1601 RSC.
- H. Zheng, Q. Liu, X. Lei, Y. Chen, B. Zhang and Q. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2531–2538 CrossRef CAS.
- S. Wang, S. Ma, Q. Li, W. Yuan, B. Wang and J. Zhu, Macromolecules, 2018, 51, 8001–8012 CrossRef CAS.
- Y. Nishimura, J. Chung, H. Muradyan and Z. B. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01826c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*