DOI:
10.1039/C6QO00321D
(Research Article)
Org. Chem. Front., 2016,
3, 1331-1336
Towards taxane analogues synthesis by dienyne ring closing metathesis†‡
Received
1st July 2016
, Accepted 17th August 2016
First published on 18th August 2016
Abstract
Herein we report the facile construction of taxadiene analogues by ring closing metathesis of dienynes built on a cyclohexenone with a natural configuration at C1 and its further transformation to incorporate most of the key functional groups of taxol.
Introduction
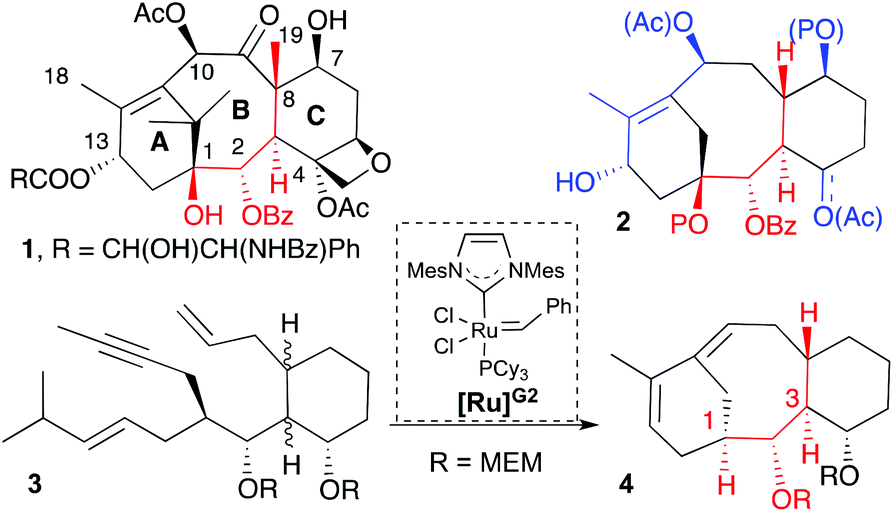
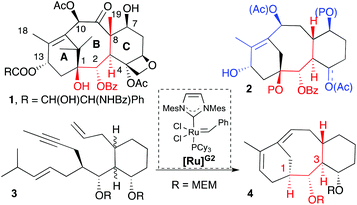
Taxol (1, Scheme 1) is a natural product widely prescribed for the treatment of various types of cancer, for example, ovarian, breast, lung and liver.1 The high demand for this drug has stimulated intensive research that aims to find easy-to-access and thus inexpensive sources of supply.2 Despite the health improvement of patients with cancer, harmful side effects, drug resistance development, and poor water solubility3 are problems that call for further research to find less structurally complex and easy to prepare analogues that show improved physical and anticancer properties. On the other hand, from the perspective of synthetic chemists, the taxol structure represents a significant challenge because of the large number of stereogenic centres, as well as its highly congested and densely oxygenated tetracyclic structure.1e To date, multiple approaches have been taken to the synthesis of taxanes where the fused bicyclic [5.3.1] system imposes a major challenge due to the presence of the eight-membered ring that is neither enthalpically nor entropically favorable.4 Furthermore the presence of a double bond at a carbon bridge-head provides additional strain to the structure.4,1e Inspired by the biosynthetic pathway of 1, in which the tandem cyclization of geranylgeranyl diphosphate, a polyenic precursor, generates the carbocyclic core,5 we designed a one step synthesis for the [5.3.1] bicyclic system of taxanes based on the cascade ring-closing dienyne metathesis (RCDEYM, Scheme 1).6 We believe that this approach fulfils the idea of rapid access to potential anticancer candidates. Previously we used this synthetic methodology to combine steroid and taxane structures to make taxosteroids.7 Additionally, we observed that the cyclization of a dienyne built on a cyclohexyl scaffold with the same relative configuration in C1, C2, C3 and C8 as the natural product did not cyclize, but its diastereomer with the opposite configuration in C1 did (Scheme 1).7 In this scenario we envisioned that the replacement of the hydrogen atom at C1 by a hydroxyl group (Scheme 1, compound 2) would favor a preorganized conformation reactive towards the RCDEYM reaction. Our precedent work with taxosteroid reactivity7 together with the reported work by Prunet's group utilizing our synthetic approach to synthesize a taxane with the hydroxyl group at C1 supported this hypothesis.8 Inspired by the biosynthesis of terpene natural products, herein we report the rapid access to highly functionalized taxane analogues (16,17,18-trinortaxanes) by a cascade RCDEYM process reminiscent of the cyclase phase,9 to forge the [5.3.1] bicyclic system and further functionalization of the resulting taxadiene analogues just as it occurs in the oxidase phase of natural products.10
 |
| | Scheme 1 Chemical structures of taxol (1) and highly oxidated taxoid 2, and previously reported RCDEYM for the rapid access to the [5.3.1] bicyclic system (A and B) of taxanes. | |
Results and discussion
Taxane skeleton construction
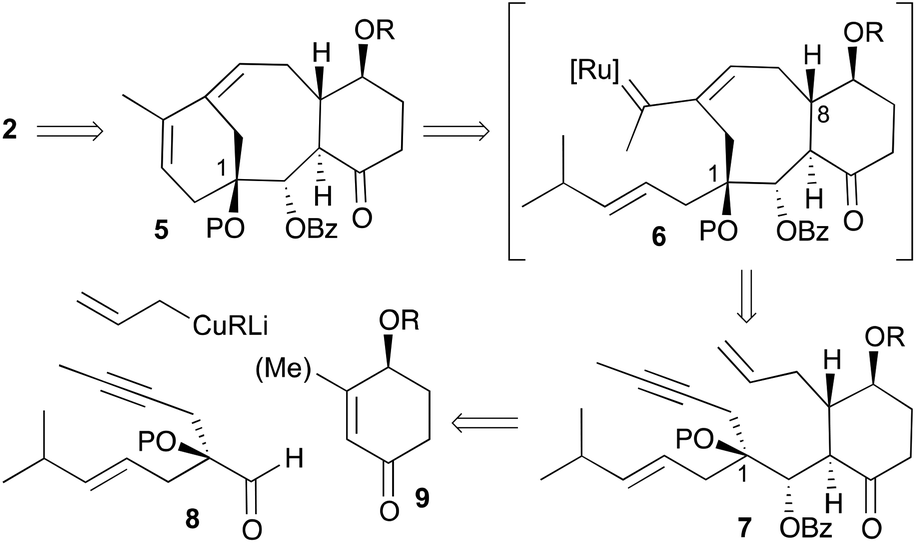
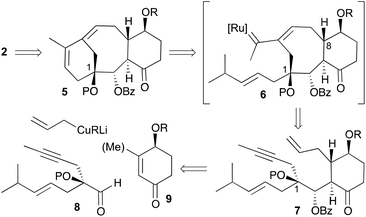
As mentioned earlier, our synthetic approach took advantage of the tandem ring-closing metathesis of acyclic dienynes (Scheme 2).11,12 Having in mind the biosynthesis of terpene-natural products,5 we envisioned a strategy (Scheme 2) where taxane analogues 2 could be easily accessed by a two-phase process: a tandem RCDEYM reaction of dienynes 7 (cyclase phase)9 and a latter functionalization of the double bonds of 16,17,18-trinortaxadienes 5 (oxidase phase).10 We reasoned that the cascade metathesis on dienyne 7 would start at the least substituted double bond to generate a metal–carbene intermediate that would subsequently react with the alkyne to give the metal alkylidene intermediate 6.13 Intermediate 6 would undergo a ring closing metathesis with the remaining double bond to give rise to the six-membered ring A of taxadiene 5. Dienyne 7 was predicted to arise from the conjugate addition of allyl cuprate to cyclohexenone 9 followed by trapping of the resulting enolate with aldehyde 8 that carries the enyne moiety and the hydroxyl group on carbon C1. The use of 3-methylcyclohexenones could allow the incorporation of the methyl group at C8 (Me-19), although this goal was not addressed in the present work.
 |
| | Scheme 2 Retrosynthetic analysis for the synthesis of taxane analogue 2 based on the cascade ring-closing metathesis of dienyne 7. | |
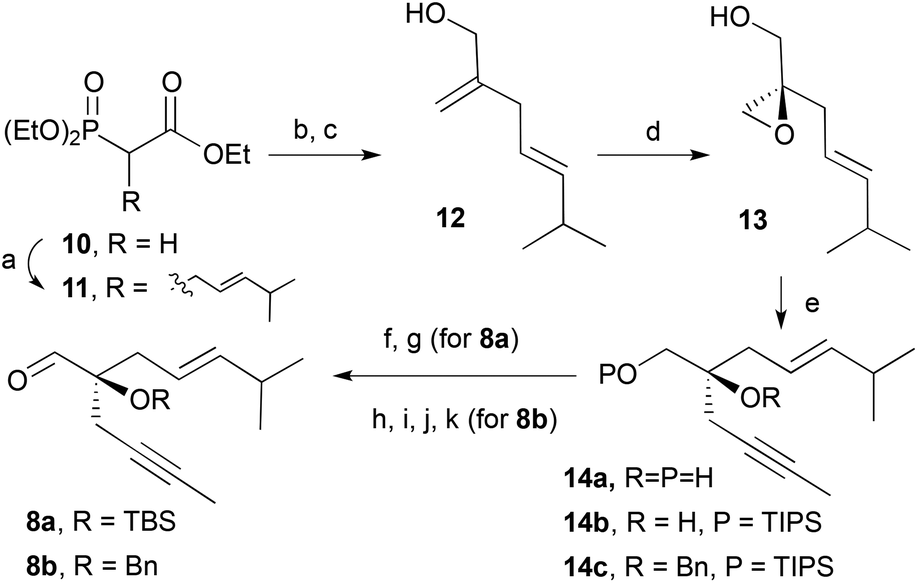
As outlined in the retrosynthetic scheme, we planned to set the configuration of C1 at an early stage to control the diastereoselective formation of the other chiral centres. To this end, we reasoned that the Sharpless epoxidation14 of allylic alcohol 12 (Scheme 3) would be the most reliable method to induce the required chirality. The synthesis of aldehyde 8 started with the alkylation of triethyl phosphonoacetate (10) with (E)-4-methyl-1-bromopent-2-ene,15 Horner–Wadsworth–Emmons16 reaction of the resulting product with formaldehyde followed by reduction with DIBAL-H of the ester afforded the allylic alcohol 12 in good yield. With compound 12 in hand, we were in a position to install the C–O bond at C1 in an asymmetric fashion. Thus Sharpless epoxidation14 of allylic alcohol 12 using (−)-diisopropyl tartrate [(−)-DIPT]16 as the chiral ligand produced epoxy-alcohol 13 with the natural product configuration (1S) in 93% yield and 90% enantiomeric excess. Addition of epoxide 13 in THF to a solution of 1-propynyllithium at −78 °C afforded diol 14a in excellent yield.17 Finally, Swern oxidation18 followed by protection of the tertiary alcohol by a tert-butyldimethylsilyl ether provided aldehyde 8a. Aiming to evaluate the effects of the steric congestion on the metathesis reaction, the benzyl-protected aldehyde (8b) was also prepared. Direct benzylation of the unprotected aldehyde did not proceed as expected, for this reason we resorted to a four step approach in which the primary alcohol was protected by a triisopropylsilyl ether (14b). Treatment of 14b with sodium hydride followed by benzyl bromide and tetrabutylammonium iodide provided the fully protected diol 14c. The use of other silyl groups (TBS or TMS) to protect the primary alcohol provided, under a variety of benzylation conditions, large amounts of the product derived from the silyl migration to the tertiary alcohol.17 Finally, the silyl group was removed under acidic conditions and the resulting alcohol was oxidized under Swern conditions to afford aldehyde 8b in 50% yield from diol 14a.
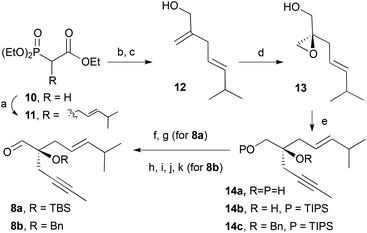 |
| | Scheme 3 Conditions: (a) i. NaH, THF, ii. (E)-1-bromo-4-methylpent-2-ene, 76%; (b) (H2CO)n, K2CO3, H2O, 80 °C; (c) DIBAL-H, THF, −78 °C, 92% (two steps); (d) (−)-DIPT, Ti(iPrO)4, TBHP, MS4 Å, CH2Cl2, 93% (90% ee); (e) 1-bromoprop-1-ene, nBuLi, THF, −78 °C–r.t., 94%; (f) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C, 73%; (g) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 68%; (h) TIPSCl, Et3N, DMAP, CH2Cl2, 76%; (g) NaH, BnBr, TBAI, 15-crown-5, THF, 81%; (h) HCl (aq.), MeOH, 97%; (i) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C, 83%. | |
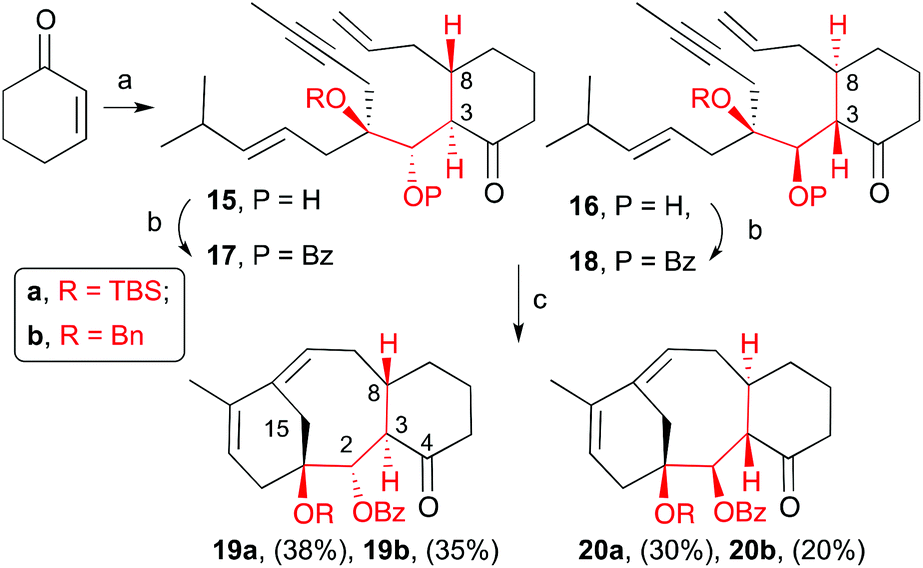
Once aldehydes 8a and 8b were synthesized, we set out to prepare the enyne precursors 17a,b and 18a,b (Scheme 4). Conjugate addition of allyl cuprate to cyclohexenone followed by aldol reaction of the resulting enolate with aldehyde 8a provided dienynes 15a and 16a as an inseparable mixture of two trans-addition isomers in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio and 75% yield. Similar results were obtained with aldehyde 8b to give 15b and 16b (5:3, 79%). The trans configuration (C3–C8) was assumed, and later confirmed, based on the known anti-aldol selectivity.19 The resulting mixture was treated with benzoyl triflate or a mixture of benzoyl chloride and silver triflate in the presence of pyridine to give benzoates 17a, 18a and 17b, 18b in good yields. Similar to the alcohol precursors, these benzoates were inseparable by column chromatography. With the dienyne precursors in hand, we were able to test our hypothesis of whether the hydroxyl protected group at C1 is capable of favouring the RCDEYM reaction of the dienyne possessing the same stereochemistry as the natural product. To our delight, we observed that when the benzoates, as a mixture of the two diastereomers (17a and 18a), were treated with 6–10 mol% of second generation Grubbs’ catalyst [Ru]G2 in refluxing dichloromethane provided two UV visible compounds that were identified as the tricyclic derivatives 19a and 20a (Scheme 4). Both compounds were easily separated by flash chromatography and obtained in 38% (19a) and 30% (20a) yields (68% overall yield). It is remarkable that dienynes 17a and 18a easily gave the RCDEYM reaction products for two reasons: in previous examples the presence of the ketone in C4 resulted in starting material decomposition and moreover dienyne 17a cyclizes in very good yield even though the deoxy-structure 3 (Scheme 1) with the same relative stereochemistry as 1 decomposed under the metathesis conditions.7 In addition, taxadiene 19a is the major product of the reaction. Similarly, treatment of the benzyl dienynes 17b and 18b with the [Ru]G2 in dichloromethane also provided the corresponding tricyclic products 19b and 20b in similar yields. The relative stereochemistry of taxadiene analogues was established by NOESY experiments and X-ray crystallography20 as shown in the appendix attached in the ESI and illustrated in ESI Fig. 1–3.‡
:2 ratio and 75% yield. Similar results were obtained with aldehyde 8b to give 15b and 16b (5:3, 79%). The trans configuration (C3–C8) was assumed, and later confirmed, based on the known anti-aldol selectivity.19 The resulting mixture was treated with benzoyl triflate or a mixture of benzoyl chloride and silver triflate in the presence of pyridine to give benzoates 17a, 18a and 17b, 18b in good yields. Similar to the alcohol precursors, these benzoates were inseparable by column chromatography. With the dienyne precursors in hand, we were able to test our hypothesis of whether the hydroxyl protected group at C1 is capable of favouring the RCDEYM reaction of the dienyne possessing the same stereochemistry as the natural product. To our delight, we observed that when the benzoates, as a mixture of the two diastereomers (17a and 18a), were treated with 6–10 mol% of second generation Grubbs’ catalyst [Ru]G2 in refluxing dichloromethane provided two UV visible compounds that were identified as the tricyclic derivatives 19a and 20a (Scheme 4). Both compounds were easily separated by flash chromatography and obtained in 38% (19a) and 30% (20a) yields (68% overall yield). It is remarkable that dienynes 17a and 18a easily gave the RCDEYM reaction products for two reasons: in previous examples the presence of the ketone in C4 resulted in starting material decomposition and moreover dienyne 17a cyclizes in very good yield even though the deoxy-structure 3 (Scheme 1) with the same relative stereochemistry as 1 decomposed under the metathesis conditions.7 In addition, taxadiene 19a is the major product of the reaction. Similarly, treatment of the benzyl dienynes 17b and 18b with the [Ru]G2 in dichloromethane also provided the corresponding tricyclic products 19b and 20b in similar yields. The relative stereochemistry of taxadiene analogues was established by NOESY experiments and X-ray crystallography20 as shown in the appendix attached in the ESI and illustrated in ESI Fig. 1–3.‡
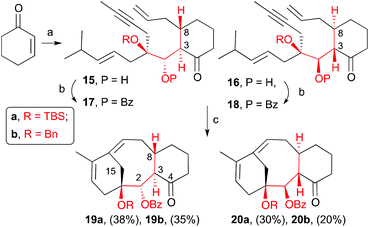 |
| | Scheme 4 Conditions: (a) i. allylMgBr, CuBr·DMS, LiCl, THF, −78 °C, ii. 8a/b, THF, r.t., 75% for 15a/16a (3:2 ratio); 79% for 15b/16b (5:3 ratio); (b) BzCl, TfOAg, py, THF, 60 °C, 77% for 17a/18a; 71% for 17b; 74% for 18b; (c) [Ru]G2, CH2Cl2, reflux, 68% for 19a/20a; 55% for 19b/20b. | |
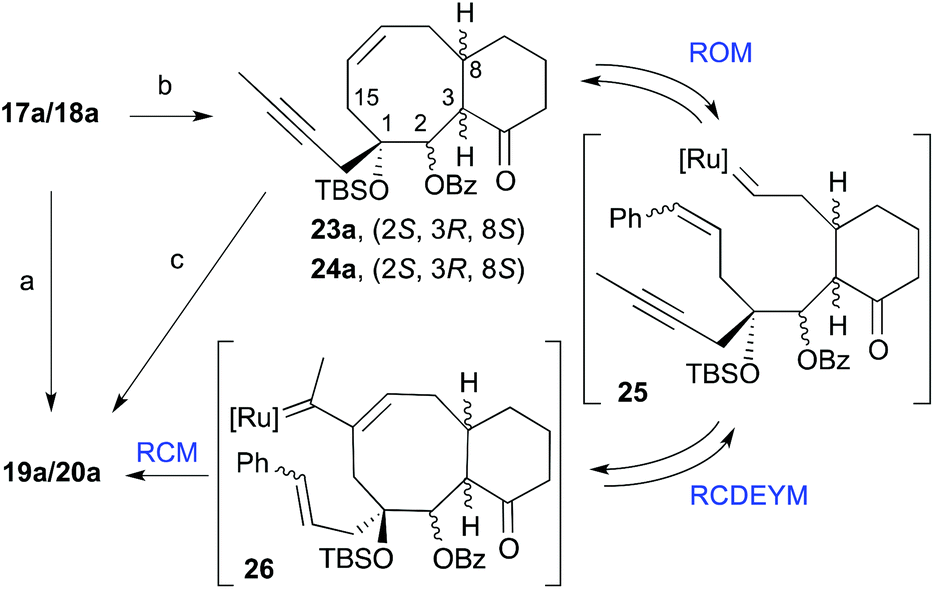
In an attempt to improve the yields of the cyclization we treated dienynes 17a and 18a with 15 mol% of the Grubbs catalyst [Ru]G2 in benzene at 40 °C (Scheme 5). Under these conditions the cyclooctene derivatives 23a and 24a were obtained as a result of the diene-ring closing metathesis of the alkenes. Interestingly, treatment of this cyclooctene mixture (23a/24a) with 15 mol% of the [Ru]G2 catalyst in refluxing benzene for 1 h gave the corresponding taxadienes (19a and 20a) in 64% yield. This observation is interesting from the mechanistic standpoint, because this implies that in benzene these dienynes most likely undergo the ring-closing diene metathesis (RCM) faster than RCDEYM. Therefore the sequence of events is as in Scheme 5. Most probably, under these conditions a fast RCM provided the cyclooctene ring. At a higher temperature, the ring opening metathesis (ROM) starts to compete with the RCM to initially generate the metal–carbene complexes 25. The subsequent reaction of 25 with the alkyne moiety (enyne cyclization, RCDEYM) forms the cyclooctene ring with concomitant formation of the metal alkylidene intermediates 26. Finally, ring-closing metathesis (RCM) of 26 with the last olefin moiety generates the six-membered taxane A-ring.
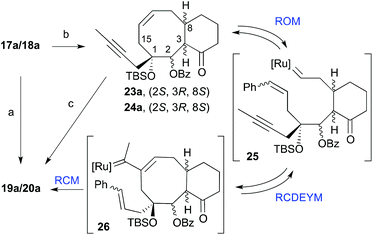 |
| | Scheme 5 Conditions: (a) [Ru]G2, CH2Cl2, reflux, 68%; (b) [Ru]G2, C6H6, 40 °C, 74%; (c) [Ru]G2, C6H6, reflux, 64%. | |
Synthesis of 4-tert-butylsilyloxy-taxadiene analogue 33a
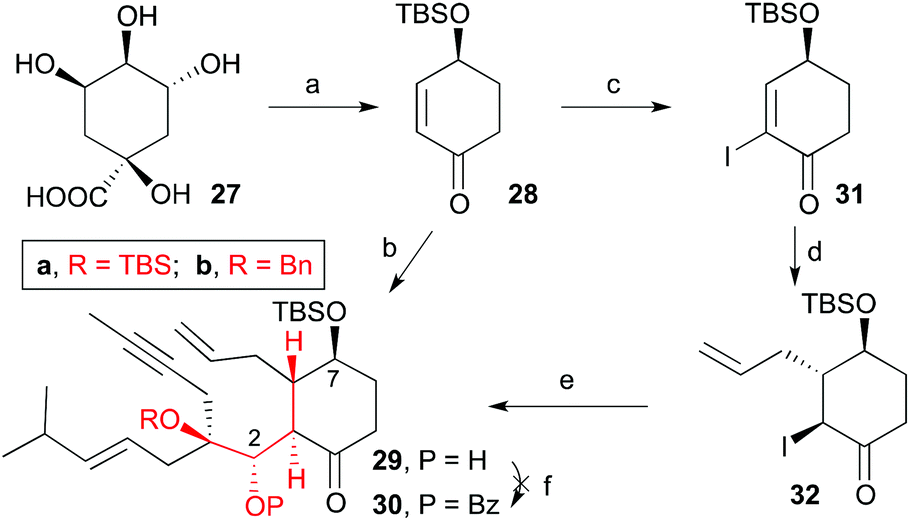
One of the drawbacks of this approach is that the cuprate addition to the cyclohexenone forms two enantiomeric enolates which upon reaction with aldehydes 8a,b, provides two diastereomers, 15a,b and 16a,b, with the incoming aldehyde trans to the allyl group (Scheme 4). Additionally, only one of each pair (15a,b) has the configuration of the natural product. Consequently a stereoselective cuprate addition was required to generate only one dienyne with relative and absolute stereochemistry matching that of taxol (1). To this end, we decided to install a bulky substituent (tert-butyldimethylsilyloxy) at C4 to direct the addition of the allyl group to the α,β-unsaturated ketone 28anti to the bulky group (Scheme 6). Moreover, this group at C4 corresponds with the hydroxyl group at C7 in the taxol skeleton. The pure enantiomer of the 4-(tert-butyldimethylsilyloxy)cyclohex-2-enone (28) was prepared from (−)-quinic acid (27) according to procedures reported in the literature (see ESI Scheme 1‡).21
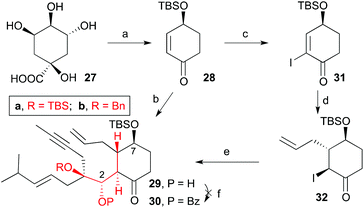 |
| | Scheme 6 Conditions: (a) see ESI Scheme 1;‡ (b) i. allylMgBr, CuBr·DMS, LiCl, THF, −78 °C; ii. 8a, THF, −78 °C, 20%; (c) I2, CCl4, py, 0 °C, 78%; (d) allylMgBr, CuBr·DMS, LiCl, THF, −78 °C, 76%; (e) i. EtMgBr, THF, 0 °C; ii. 8b, THF, 0 °C, 50%; (f) BzCl, TfOAg, py, THF, 60 °C. | |
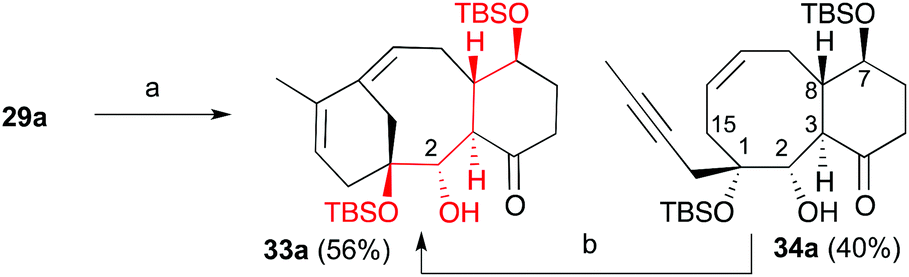
When we subjected ketone 28 and aldehyde 8a to the previously optimized 1,4-addition-aldol reaction conditions, the addition product 29a was obtained as the sole diastereomer, although in 20% overall yield (40% considering the recovered aldehyde). All attempts to optimize this transformation by changing the copper (CuCN, CuI, etc.) or propenyl-metal sources (Li or Mg), reaction times and temperatures were unsuccessful. Alternatively, we trapped the enolate as trimethylsilyl or acetyl derivatives and used the latter to generate the reactive enolate under more appropriate conditions. Unfortunately, no reaction was observed after treatment of these enolate derivatives with a variety of organometalic reagents (nBuLi, MeLi, EtMgBr) or Lewis acids (BF3, TiCl4 or SnCl4) in the presence of model aldehydes. To overcome this problem, ketone 28 was transformed into 2-iodocyclohexenone 31 by treatment with iodine in carbon tetrachloride and pyridine.22 Compound 31 was then transformed into α-iodoketone 32 by reaction with allylmagnesium bromide and copper bromide. Next, compound 32 was treated with ethylmagnesium bromide followed by addition of aldehyde 8b to afford hydroxyketone 29b in 50% yield as a single diastereoisomer.22 We tried to transform the alcohol at C2 into the benzoate, but the previously described conditions failed to give the desired product. Despite previous observations that the metathesis of dienynes bearing the hydroxyl group on C2 and the ketone group on C4 provides complex reaction mixtures,6,7 we decided to subject the dienyne 29a to the RCDEYM conditions (Scheme 7). The metathesis–cyclization of this compound using 10 mol% of the [Ru]G2 catalyst in refluxing dichloromethane provided the tricyclic derivative 33a in 56% yield, together with the cyclooctene derivative 34a (40%).6,7,23 It is worth noting that the cyclooctene intermediate 34a can be transformed into the taxane derivative 33a by further treatment with the [Ru]G2 catalyst. Taking into account this result, the ease of formation of the highly functionalized taxane skeleton by RCDEYM from acyclic dienynes anchored to a cyclohexane platform is remarkable.
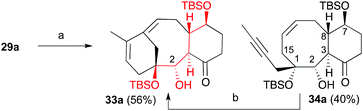 |
| | Scheme 7 Conditions: (a) [Ru]G2, CH2Cl2, 40 °C, 56%; (b) [Ru]G2, C6H6, 90 °C, 48%. | |
Taxane functionalization
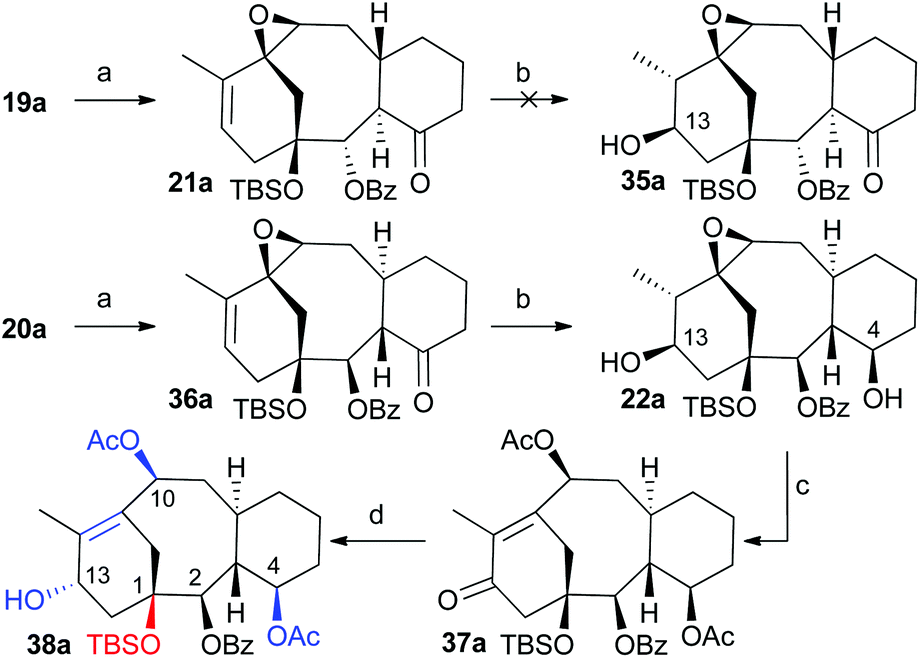
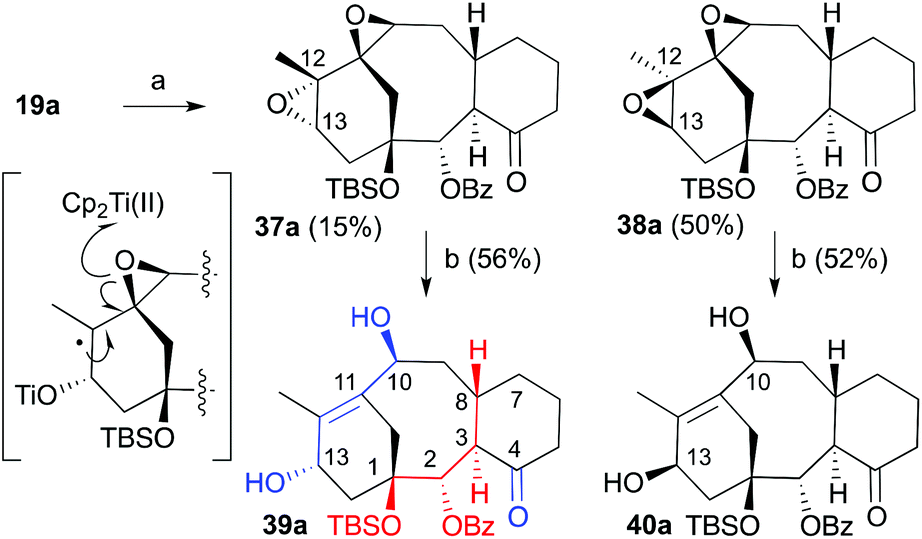
Once the cyclization methodology was established (cyclase phase) our next objective was the incorporation of additional functional groups (oxidase-like phase) on the carbocyclic system in an efficient way. To demonstrate the potential of these taxadiene analogues as a valuable source of taxane-like derivatives we further functionalize the tricyclic core to install the hydroxyl group on C13 and acetoxy group on C10 (Scheme 8). To this end, we made use of the methodology previously developed in our group.7 Therefore treatment of compounds 19a or 20a (Scheme 8) with one equivalent of m-chloroperbenzoic acid (MCPBA) at 0 °C gave the oxiranes 21a and 36a in 65% and 76% yields, respectively. In both cases, the oxygen addition took place, as expected, at the most strained and therefore more reactive C10–C11 double bond and through the least hindered exo-face. The X-ray diffraction of crystals of this compound allowed us to confirm the proposed structure (ESI Fig. 3a‡).20 Hydroboration–oxidation of natural configuration precursor 21a gave complex mixtures in which the expected alcohol 35a was not observed. All attempts to carry out this transformation under milder conditions were unsuccessful. On the other hand, the hydroboration–oxidation only took place on epoxide 36a to afford 22a in very good yield. The addition of the borane took place from the exo-face. In addition to the reaction of the double bond, under these conditions the ketone group was also reduced, with the diastereoselectivity dictated by the approach of borane from the least hindered α-face. The relative stereochemistry was assigned by X-ray crystallography (ESI Fig. 3b‡). As the resulting alcohol at C13 has the opposite configuration of the natural product, we corrected the stereochemistry by an oxidation–reduction procedure. The oxidation of 22a with PDC was followed by treatment of the resulting crude mixture with alumina and acetic anhydride in the presence of DMAP to give diacetylketone 37a in 75% yield. Interestingly, only the hydroxyl group at C13 was oxidized under these conditions while the hydroxyl group in C4 remained unaffected. Finally, Luche's reduction of the unsaturated ketone provided allylic alcohol 38a in almost quantitative yield. The resulting compound has the hydroxyl group in the endo position, which is the same as the natural product. Additionally, the obtained taxane precursor bears some of the key features of the natural product, such as the benzoate at C2, the protected hydroxyl group at C1, the acetates at C4 and C10 and the free hydroxyl group ready to incorporate the amino acid side chain.24
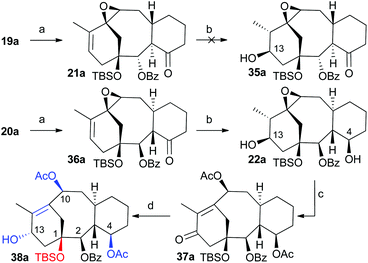 |
| | Scheme 8 Conditions: (a) MCPBA, CH2Cl2, 0 °C, 62% for 21a, 76% for 36a; (b) i. BH3·THF, CH2Cl2, 0 °C, ii. NaOH, H2O2, 76% for 22a; (c) i. PDC, CH2Cl2, ii. Al2O3, CH2Cl2, iii. Ac2O, DMAP, 74%; (d) NaBH4, CeCl3·7H2O, MeOH, 0 °C, 96%. | |
To resolve the observed problem with the reactivity of the isomer with the natural configuration, a different alternative was approached. For this purpose, we studied the incorporation of the 2-en-1,4-diol system presented on taxol through the previously developed ring opening of diepoxides through a titanium induced radical methodology.25 For this purpose the diene 19a was treated with MCPBA at 0 °C. Unfortunately a mixture of diepoxides 37a and 38a in a 1:3 ratio was obtained in 64% overall yield (Scheme 9). Both epoxides are differentiated in the relative orientation of epoxide at C12–C13. The major isomer was the one with the β-face configuration that was, in previous studies, the only isomer that underwent the double oxirane opening. This isomer is the precursor of a synthetic intermediate with the opposite configuration at C13.25 Mechanistically, the process was explained based on a titanium induced sequential epoxide opening of the dioxirane moiety.26 The observed lack of reactivity of the trans-diepoxide was justified at that time in terms of the steric hindrance provided by the tricyclic skeleton.25 The resulting diepoxides were subjected to the in situ generated titanocene(III) complex.26 Interestingly both of them underwent the double opening to provide diols 39a and 40a in 58 and 52% yield, respectively. Therefore, we have access to a highly advanced precursor taxane analogue with the configuration of the natural product although, at this moment, as the minor product. The main product in this strategy has the opposite configuration at C13 but the previously developed sequence based on the oxidation/reduction should allow inverting the configuration of this stereogenic center.
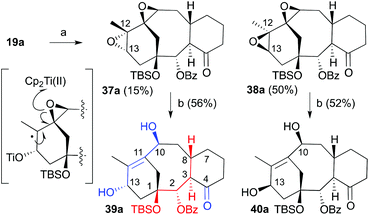 |
| | Scheme 9 Conditions: (a) MCPBA, CH2Cl2, 64%; (b) [Cp2TiCl2], Zn, THF, 0 °C. | |
Conclusions
In summary, we were able to install the stereogenic center at C1 with the same configuration of taxol (1) using the asymmetric Sharpless epoxidation. We showed that the protected hydroxyl group at C1 indeed favors the cascade ring-closing dienyne metathesis (RCDEYM) of dienynes 17a,b featuring a ketone at C4 and with the same stereochemistry at C1, C2, C3 and C8 of the natural product that previously has proven to be unreactive and without the need of additional tethering that pre-organizes the reactive unsaturations. Therefore, we were able to prepare the taxadiene analogues 19a,b, 20a,b and 33a furnished with the oxygenated functional groups at C1 and C2 along with the ketone at C4, and the two double bonds C10–C11 and C12–C13 that allow for further functionalization. It was observed that the cascade RCDEYM of dienyne 29a, carrying the seemingly labile groups ketone at C4 and unprotected alcohol at C2, takes place at unprecedented high yield. We demonstrated the potential of these taxadiene analogues as a source of various derivatives by further functionalization, the oxidase-like phase, of 19a to prepare the 16,17,18-nortaxane 39a furnished with the hydroxyl groups in C10, C13, the double bond C11–C12 and a carbonyl group at C4.
Acknowledgements
This work was supported by the Spanish Ministry of Economy and Competitivity (Mineco) and the ERDF (CTQ2010-15725, CTQ2013-43264-R and CTQ2014-51912-REDC), and by the Xunta de Galicia and the ERDF (GPC2013-039, EM2012/117 and EM2014/011). N. S. thanks Mineco for his FPU contract and S. P.-E. to the CONACYT for his fellowship (no. 162219). We also acknowledge the ORFEO-CINCA network.
Notes and references
-
(a)
M. Suffness, TAXOL®: Science and Applications, CRC Press, 1995 Search PubMed
 ;
(b)
H. Itokawa and K.-H. Lee, Taxus: The Genus Taxus, Taylor & Francis, 2003 Search PubMed ;
(c) V. A. Weger, J. H. Beijnen and J. H. M. Schellens, Anti-Cancer Drugs, 2014, 25, 488 CrossRef PubMed ;
(d) J. A. Yared and K. H. R. Tkaczuk, Drug Des., Dev. Ther., 2012, 6, 371 CAS ;
(e) Y.-F. Wang, Q.-W. Shi, M. Dong, H. Kiyota, Y.-C. Gu and B. Cong, Chem. Rev., 2011, 111, 7652 CrossRef CAS PubMed .
;
(b)
H. Itokawa and K.-H. Lee, Taxus: The Genus Taxus, Taylor & Francis, 2003 Search PubMed ;
(c) V. A. Weger, J. H. Beijnen and J. H. M. Schellens, Anti-Cancer Drugs, 2014, 25, 488 CrossRef PubMed ;
(d) J. A. Yared and K. H. R. Tkaczuk, Drug Des., Dev. Ther., 2012, 6, 371 CAS ;
(e) Y.-F. Wang, Q.-W. Shi, M. Dong, H. Kiyota, Y.-C. Gu and B. Cong, Chem. Rev., 2011, 111, 7652 CrossRef CAS PubMed .
-
(a) J. N. Denis, A. E. Greene, D. Guenard, F. Gueritte-Voegelein, L. Mangatal and P. Potier, J. Am. Chem. Soc., 1988, 110, 5917 CrossRef CAS ;
(b) I. Ojima, I. Habus, M. Zhao, M. Zucco, Y. H. Park, C. M. Sun and T. Brigaud, Tetrahedron, 1992, 48, 6985 CrossRef CAS .
-
(a) R. Krisna and L. D. Mayer, Eur. J. Pharm. Sci., 2000, 11, 265 CrossRef ;
(b) G. Arpino, S. De Placido and C. De Angelis, Anti-Cancer Drugs, 2015, 26, 117 CrossRef CAS PubMed ;
(c) Z. Liu, F. Zhang, G. Y. Koh, X. Dong, J. Hollingsworth, J. Zhang, P. S. Russo, P. Yang and R. W. Stout, Anti-Cancer Drugs, 2015, 26, 167 CrossRef CAS PubMed ;
(d) S. Koudelka and J. Turánek, J. Controlled Release, 2014, 163, 322 CrossRef PubMed ;
(e) P. Ma and R. J. Mumper, J. Nanomed. Nanotechnol., 2013, 4, 1 Search PubMed ;
(f) P. Zhao and D. Astruc, ChemMedChem, 2012, 7, 952 CrossRef CAS PubMed .
-
(a) K. C. Nicolaou, Z. Yang, J. J. Liu, H. Ueno, P. G. Nantermet, R. K. Guy, C. F. Claiborne, J. Renaud, E. A. Couladouros, K. Paulvannan and E. J. Sorensen, Nature, 1994, 367, 630 CrossRef CAS PubMed ;
(b) R. A. Holton, H. B. Kim, C. Somoza, F. Liang, R. J. Biediger, P. D. Boatman, M. Shindo, C. C. Smith and S. Kim, J. Am. Chem. Soc., 1994, 116, 1599 CrossRef CAS .
-
(a) S. Howat, B. Park, I. S. Oh, Y.-W. Jin, E.-K. Lee and G. J. Loake, New Biotechnol., 2014, 31, 242 CrossRef CAS PubMed ;
(b) R. Croteau, R. E. B. Ketchum, R. M. Long, R. Kaspera and M. R. Wildung, Phytochem. Rev., 2006, 5, 75 CrossRef CAS PubMed ;
(c) H. Li, T. Horiguchi, R. Croteau and R. M. Williams, Tetrahedron, 2008, 64, 6561 CrossRef CAS PubMed .
-
(a) R. García-Fandiño, E. M. Codesido, E. Sobrazo-Sánchez, L. Castedo and J. R. Granja, Org. Lett., 2004, 6, 193 CrossRef PubMed ;
(b) M. J. Aldegunde, R. García-Fandiño, L. Castedo and J. R. Granja, Chem. – Eur. J., 2007, 13, 5135 CrossRef CAS PubMed .
- M. J. Aldegunde, L. Castedo and J. R. Granja, Org. Lett., 2008, 10, 3789 CrossRef CAS PubMed .
-
(a) A. Letort, R. Aouzal, C. Ma, D.-L. Long and J. Prunet, Org. Lett., 2014, 16, 3300 CrossRef CAS PubMed ;
(b) C. Ma, A. Letort, R. Aouzal, A. Wilkes, G. Maiti, L. J. Farrugia, L. Ricard and J. Prunet, Chem. – Eur. J., 2016, 22, 6891 CrossRef CAS PubMed .
-
(a) P. D. Goldring, G. Pattenden and S. L. Rimmington, Tetrahedron, 2009, 65, 6670 CrossRef ;
(b) J. Petrignet, A. Boudhar, G. Blond and J. Suffert, Angew. Chem., Int. Ed., 2011, 50, 3285 CrossRef CAS PubMed ;
(c) A. Mendoza, Y. Ishibara and P. S. Baran, Nat. Chem., 2012, 4, 21 CrossRef CAS PubMed .
-
(a) N. C. Wilde, M. Isomura, A. Mendoza and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 4909 CrossRef CAS PubMed ;
(b) Y. Ishihara, A. Mendoza and P. S. Baran, Tetrahedron, 2013, 69, 5685 CrossRef CAS PubMed ;
(c) K. Chen and P. S. Baran, Nature, 2009, 459, 824 CrossRef CAS PubMed ;
(d) C. Yuan, Y. Jin, N. C. Wilde and P. S. Baran, Angew. Chem., Int. Ed., 2016, 55, 8420 CrossRef .
-
(a) A. Michaut and J. Rodriguez, Angew. Chem., Int. Ed., 2006, 45, 5740 CrossRef CAS PubMed ;
(b) M. E. Maier, Angew. Chem., Int. Ed., 2000, 39, 2073 CrossRef CAS .
-
(a) T. J. Katz and T. M. Sivavec, J. Am. Chem. Soc., 1985, 107, 737 CrossRef CAS ;
(b) S.-H. Kim, W. J. Zuercher, N. B. Bowden and R. H. Grubbs, J. Org. Chem., 1996, 61, 1073 CrossRef CAS ;
(c) W. J. Zuercher, M. Scholl and R. H. Grubbs, J. Org. Chem., 1998, 63, 4291 CrossRef CAS ;
(d) F.-D. Boyer, I. Hanna and L. Ricard, Org. Lett., 2004, 6, 1817 CrossRef CAS PubMed ;
(e) I. Dragutan, V. Dragutan, A. Demonceau and L. Delaude, Curr. Org. Chem., 2013, 17, 2678 CrossRef CAS .
-
(a) R. García-Fandiño, L. Castedo, J. R. Granja and D. J. Cárdenas, Dalton Trans., 2007, 2925 RSC ;
(b) F. Núñez-Zarur, X. Solans-Monfort, L. Rodríguez-Santiago and M. Sodupe, ACS Catal., 2013, 3, 206 CrossRef ;
(c) F. Núñez-Zarur, X. Solans-Monfort, L. Rodríguez-Santiago, R. Pleixats and M. Sodupe, Chem. – Eur. J., 2011, 17, 7506 CrossRef PubMed ;
(d) O. S. Lee, K. H. Kim, J. Kim, K. Kwon, T. Ok, H. Ihee, H.-Y. Lee and J.-H. Sohn, J. Org. Chem., 2013, 78, 8242 CrossRef CAS PubMed .
-
(a)
T. Katsuki and V. Martin, in Asymmetric epoxidation of allylic alcohols: The Katsuki-Sharpless epoxidation reactions en: Organic Reactions, ed. J. Ojima, Wiley-VCH, Weinheim, 1996, vol. 48 Search PubMed ;
(b)
D. Schinzer, in Asymmetric synthesis. The Sharpless epoxidation en: Organic Synthesis Highlights II, ed. H. Waldmann, Wiley-VCH, Weinheim, 1995 Search PubMed .
- A. van der Klei, R. de Jong, J. Lugtenburg and A. Tielens, Eur. J. Org. Chem., 2002, 3015 CrossRef CAS .
- J. M. Schomaker and B. Borham, Org. Biomol. Chem., 2004, 2, 621 CAS .
- D. Toussaint and J. Suffert, Org. Synth., 1999, 76, 214 CrossRef CAS .
- D. Evans, S. W. Kaldor, T. K. Jones, J. Clardy and T. J. Stout, J. Am. Chem. Soc., 1990, 112, 7001 CrossRef CAS .
- B. Schetter and R. Mharwald, Angew. Chem., Int. Ed., 2006, 45, 7506 CrossRef CAS PubMed .
-
(a) H. D. Flack, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1983, 39, 876 CrossRef ;
(b) H. D. Flack and G. Bernardinelli, J. Appl. Crystallogr., 2000, 33, 1143 CAS .
-
(a) B. M. Trost and A. G. Romero, J. Org. Chem., 1986, 51, 2332 CrossRef CAS ;
(b) Z. X. Wang, S. M. Miller and O. P. Anderson, J. Org. Chem., 1999, 64, 6443 CrossRef CAS ;
(c) J. E. Audia, R. Boisvert, A. D. Patten, A. Villalobos and S. J. Danishefsky, J. Org. Chem., 1989, 54, 3738 CrossRef CAS .
- D. A. William and Y. Kobayashi, J. Org. Chem., 2002, 67, 8771 CrossRef PubMed .
-
(a) L. Hyldtoft and R. Madsen, J. Am. Chem. Soc., 2000, 122, 8444 CrossRef CAS ;
(b) B. M. Kariuki, W. M. Owton, J. M. Percy, S. Pintat, C. A. Smith, N. S. Spencer, A. C. Thomas and M. Watson, Chem. Commun., 2002, 228 RSC .
- T. Ganesh, C. Yang, A. Norris, T. Glass, S. Bane, R. Ravindra, A. Banerjee, B. Metaferia, S. L. Thomas, P. Giannakakou, A. A. Alcaraz, A. S. Lakdawala, J. P. Snyder and D. G. I. Kingston, J. Med. Chem., 2007, 50, 713 CrossRef CAS PubMed .
- M. J. Aldegunde, L. Castedo and J. R. Granja, Chem. – Eur. J., 2009, 15, 4785 CrossRef CAS PubMed .
- K. Daasbjerg, H. Svith, S. Grimme, M. Gerenkamp, C. Mück-Lichtenfeld, A. Gansäuer, A. Barchuk and F. Keller, Angew. Chem., Int. Ed., 2006, 45, 2041 CrossRef CAS PubMed ; A. Gansäuer, A. Barchuk, F. Keller, M. Schmitt, S. Grimme, M. Gerenkamp, C. Mück-Lichtenfeld, K. Daasbjerg and H. Svith, J. Am. Chem. Soc., 2007, 129, 1359 CrossRef PubMed ; A. Gansäuer, C.-A. Fan, F. Keller and J. Keil, J. Am. Chem. Soc., 2007, 129, 3484 CrossRef PubMed .
Footnotes |
| † Dedicated to Professor Barry Trost on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1489402 and 1489403. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00321d |
|
| This journal is © the Partner Organisations 2016 |

 and
J. R.
Granja
and
J. R.
Granja