
Aliphatic C–H azidation through a peroxydisulfate induced radical pathway†
Xin
Li
a and
Zhang-Jie
Shi
*ab
aBeijing National Laboratory of Molecular Sciences and Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: zshi@pku.edu.cn
bState Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 18th August 2016
Abstract
A persulfate induced radical process to transform aliphatic tertiary C–H bonds into organic azides under transition-metal-free conditions has been developed. This method exhibits good chemo- and regioselectivity. A wide range of functional groups, including esters, halogens, azides, and dipeptides, are all tolerated well through the radical pathway, thus expanding the potential application of this method.
The selective functionalization of isolated, aliphatic C–H bonds is a long-standing goal in organic chemistry.1 Over the past few years, a number of strategies toward direct functionalization of selected aliphatic C–H bonds have been successfully established. Among them, C–H functionalization via a radical pathway is the earliest investigated and regarded as one of the more powerful tools in organic synthesis.2–6 In most of those cases, pre-installed functional groups were first activated to generate heteroatom-centered radicals. Subsequent intramolecular hydrogen atom abstraction afforded carbon-centered radicals, which further coupled with other radicals to give the final products. Alternatively, the intermolecular hydrogen atom abstraction from substrates to appropriate radical abstractors also produced carbon-centered radicals directly from C–H bonds, thus avoiding pre-functionalization.7,8 Just recently, we reported a regioselective aliphatic C–H bond oxidation using persulfate salts as the oxidant.9 In the reported process, the phthalimide group assisted two-step SET oxidation between substrates and sulfate radicals transformed the isolated aliphatic C–H bonds into carbocations, which further coupled with O-based nucleophiles to produce C–O bond formation products (Scheme 1, route 1). We speculated that, once the carbon-centered radicals were formed, they could be captured by appropriate reagents to forge other kinds of C–X bonds, thus preventing the subsequent oxidation step and meanwhile affording new products through direct C–H functionalization (Scheme 1, route 2).
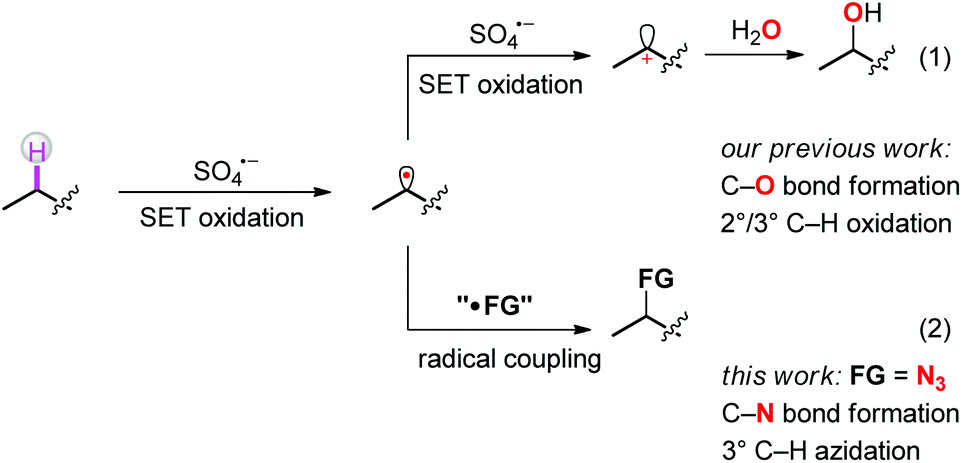 | ||
| Scheme 1 C–H functionalization via a peroxydisulfate induced radical pathway. | ||
As we know, organic azides are versatile building blocks of great synthetic value.10 Not only are they readily converted into many other compounds such as amines, amides, imines, heterocycles, and nitrene intermediates, but they also play an irreplaceable role in “click” chemistry.11 However, except for the early elegant work on weak aliphatic C–H bond azidation,12 the direct azidation of isolated, “inactivated” aliphatic C–H bonds is still challenging, and only a few examples of isolated C–H azidation with transition-metal catalysts or under transition-metal-free conditions have been reported.13–15 In view of the current progress and based on our recent developments in C–H oxidation chemistry, we decided to pursue the coupling between carbon-centered radicals and azidation reagents as the model reaction to fulfill our above-mentioned design (Scheme 1, route 2). When we were preparing this manuscript, Tang and coworkers reported a beautiful example to proceed the C–H azidation in the presence of a weak base.16
Our investigation began with the evaluation of several azidation reagents for the coupling reactions. At first, N-Phth-i-hexylamine 1a, sodium persulfate, and different kinds of potential azidation reagents were heated at 50 °C in acetonitrile/water mixed solvents for 4 hours (Table 1, entries 1–6). It was found that sodium azide, which was successfully applied in the Mn-catalyzed azidation system,14b gave no azidation product under the present reaction conditions (entry 1). TMSN3 also failed to promote the desired reaction (entry 2). The target azidation products were obtained in moderate yields when sulfonyl azides were employed as azidation reagents (entries 3–6). Among them, 3-PySO2N3 gave the most promising result (entry 3). Subsequent screening of radical initiators showed that potassium persulfate was the best choice for the reaction (entry 7). Further optimizations revealed that an obviously improved yield was achieved by elevating the temperature to 90 °C and increasing the persulfate loading to three equivalents (entry 11). However, other attempts such as elevating the temperature further, increasing the persulfate and 3-PySO2N3 loading, prolonging the reaction time, and adding base or transition-metal salts all gave negative effects (entries 10 and 12–16). It should be stated that the reaction atmosphere was crucial for the outcome of the reaction. The reaction proceeded efficiently under argon while failing in air (entry 17).
|
|
||||
|---|---|---|---|---|
| Entry | M2S2O8 (equiv.) | [N3] (equiv.) | Condition | Yieldb (%) |
| a 1a (0.10 mmol), initiator, azide reagent, water (0.5 mL), acetonitrile (1.0 mL), Ar, in a sealed tube. b GC yields. c Isolated yield. d K2CO3 (1.0 equiv.) was added. e Fe(OAc)2 (0.1 equiv.) was added. f The reaction was performed under air. Phth = phthalyol. TPS = 2,4,6-triisopropyl-phenylsulfonyl. | ||||
| 1 | Na2S2O8 (2.0) | NaN3 (2.0) | 50 °C, 4 h | nd |
| 2 | Na2S2O8 (2.0) | TMSN3 (2.0) | 50 °C, 4 h | nd |
| 3 | Na2S2O8 (2.0) | 3-PySO2N3 (2.0) | 50 °C, 4 h | 45 |
| 4 | Na2S2O8 (2.0) | C12H25SO2N3 (2.0) | 50 °C, 4 h | 33 |
| 5 | Na2S2O8 (2.0) | TPS-N3 (2.0) | 50 °C, 4 h | 23 |
| 6 | Na2S2O8 (2.0) | TsN3 (2.0) | 50 °C, 4 h | 37 |
| 7 | K2S2O8 (2.0) | 3-PySO2N3 (2.0) | 50 °C, 4 h | 54 |
| 8 | (NH4)2S2O8 (2.0) | 3-PySO2N3 (2.0) | 50 °C, 4 h | 42 |
| 9 | K2S2O8 (2.0) | 3-PySO2N3 (2.0) | 90 °C, 4 h | 75 |
| 10 | K2S2O8 (2.0) | 3-PySO2N3 (2.0) | 100 °C, 4 h | 72 |
| 11 | K2S2O8 (3.0) | 3-PySO2N3 (2.0) | 90 °C, 4 h | 87 (71c) |
| 12 | K2S2O8 (4.0) | 3-PySO2N3 (2.0) | 90 °C, 4 h | 78 |
| 13 | K2S2O8 (3.0) | 3-PySO2N3 (3.0) | 90 °C, 4 h | 88 |
| 14 | K2S2O8 (3.0) | 3-PySO2N3 (2.0) | 90 °C, 6 h | 80 |
| 15d | K2S2O8 (3.0) | 3-PySO2N3 (2.0) | 90 °C, 4 h | 42 |
| 16e | K2S2O8 (3.0) | 3-PySO2N3 (2.0) | 90 °C, 4 h | 79 |
| 17f | K2S2O8 (3.0) | 3-PySO2N3 (2.0) | 90 °C, 4 h | nd |
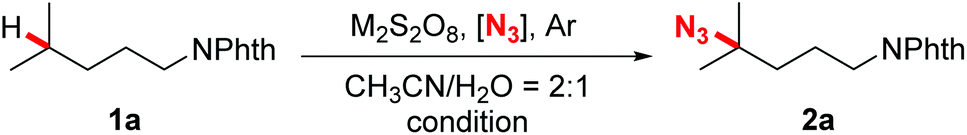
With the optimized reaction conditions in hand, we next evaluated the substrate scope and the selectivity of the reaction. As illustrated in Scheme 2, a wide variety of N-Phth-amines with different alkyl side chains were transformed into azide compounds with good yields and excellent selectivity. These data showed that the length of the alkyl side chains did not affect the efficiency of the reaction very much (2a–2d), while suitable electronic features were essential for both the chemo- and regioselectivity of this reaction. Because tertiary C–H bonds are more electron-rich than secondary and primary ones, this azidation reaction exhibited an overwhelming bias toward tertiary C–H bonds no matter where they are located (2b–2n). When secondary C–H bonds and tertiary C–H bonds were at the same distance from an electron-withdrawing group (PhthN–), the azidation took place only at the more electron-rich tertiary C–H bonds, leaving the secondary ones intact (2i, 2j and 2m). Moreover, due to the inductive effects resulting in electronic distinctions between two inequivalent tertiary C–H bonds, this reaction also showed a good remote regioselectivity. The azidation preferred to occur at more electron-rich, remote tertiary C–H bonds, giving remote functionalized products as the major regioisomers (2j–2l). Besides linear alkyl side chains, the C–H azidation also proceeded well on substrates with cyclic carbon backbones. For example, methylcyclohexylamines exhibited good reactivity toward sulfonyl azide and gave satisfactory yields (2m and 2n). In contrast with the tertiary C–H bonds, secondary bonds exhibited rather lower reactivity and poorer regioselectivity toward azidation under the standard reaction conditions, affording a mixture of regioisomers in modest yield (2o).
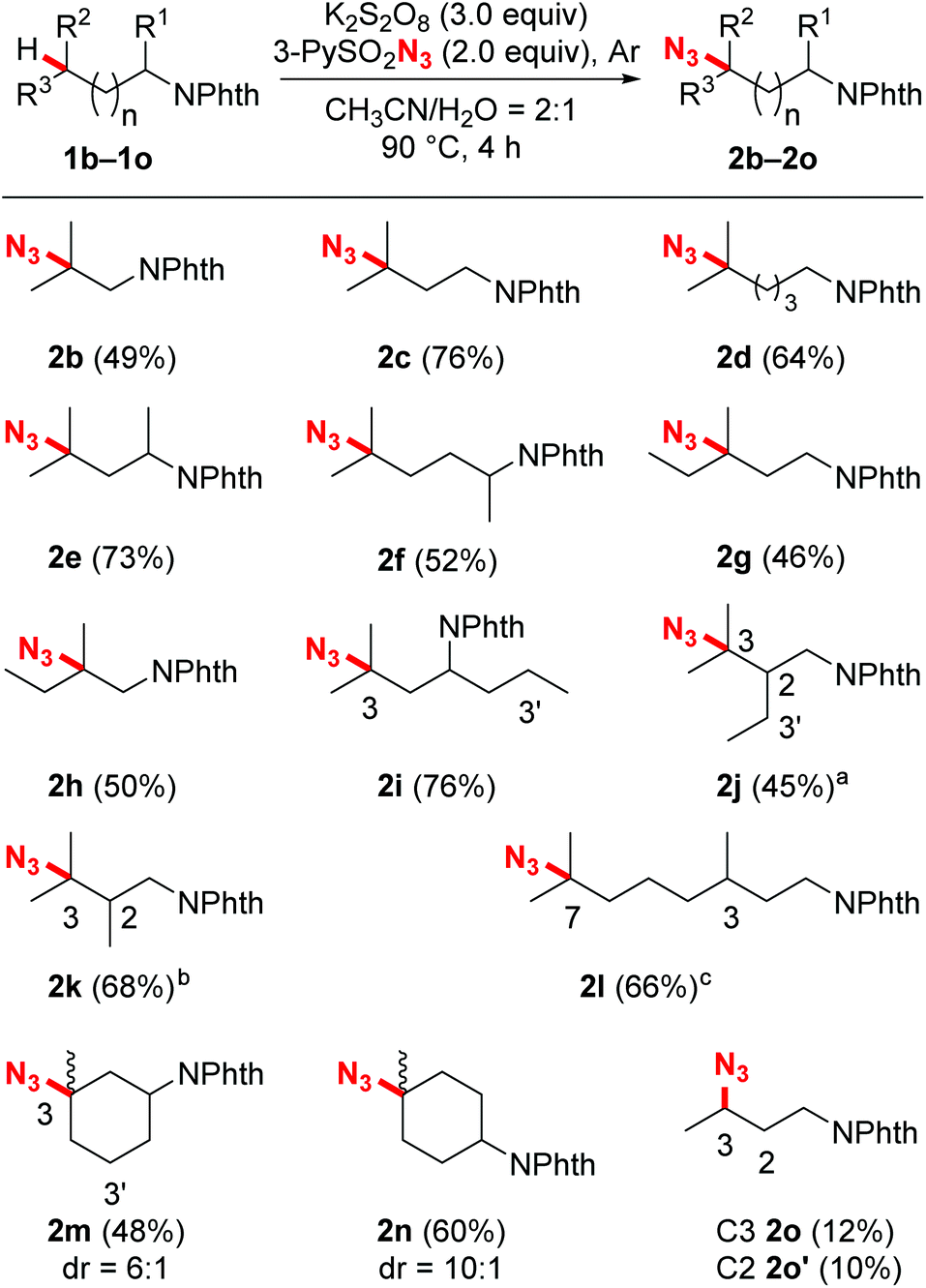 | ||
Scheme 2 Substrate scope for the C–H azidation. Reaction conditions: N-Phth-protected amines (0.10 mmol), K2S2O8 (0.30 mmol), 3-PySO2N3 (0.20 mmol), water (0.5 mL), acetonitrile (1.0 mL), Ar, 90 °C, 4 h. Isolated yields. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C3:C2 > 20:1.bC3:C2 = 14:1. cC7:C3 = 3:1. C3:C2 > 20:1.bC3:C2 = 14:1. cC7:C3 = 3:1. | ||
After accomplishing the reactivity and selectivity studies, the functional group compatibility was investigated. As shown in Scheme 3, a broad array of functional groups were tolerated very well in this C–H azidation reaction. For instance, amino acid esters (2p and 2q), as well as protected alcohol groups such as benzoate and mesylate (2r–2t) all coupled with 3-PySO2N3 readily, affording moderate to good yields. It should be mentioned that halogens, specifically iodine, which are variably tolerated in radical processes, all survived well under the reaction conditions. Good efficiency and yields were obtained in the cases of chloro- and bromo-substituted substrates (2u and 2v). Although an iodo-substituted substrate reacted with 3-PySO2N3 sluggishly under the standard conditions, a 49% yield with 47% starting material recovered showed that a good mass balance was obtained (2w). Besides, a pre-installed azide group was also compatible with the reaction, producing the di-azide compound 2x. This azidation protocol could also be extended to the modification of synthetic peptides (2y and 2z). The electron-rich, remote tertiary C–H bond selectivity was maintained very well, and good yields were obtained. When there were two inequivalent tertiary C–H bonds existing in a dipeptide, for example, N-Phth-Leu-Val-OMe, the more remote and electron-rich tertiary C–H bond in the leucine residue was tagged with an azide group preferentially (2z).
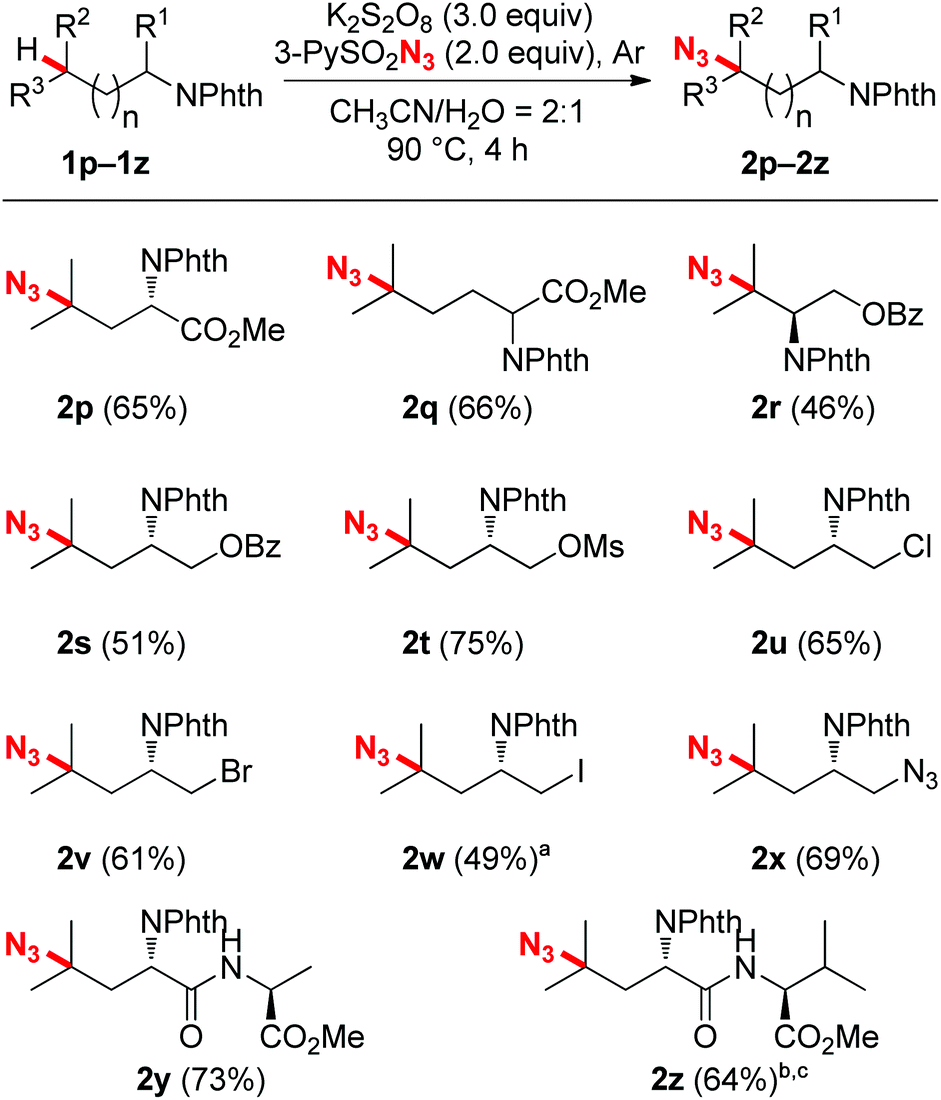 | ||
| Scheme 3 Functional group compatibility for the C–H azidation. Reaction conditions: N-Phth-protected amino compounds (0.10 mmol), K2S2O8 (0.30 mmol), 3-PySO2N3 (0.20 mmol), water (0.5 mL), acetonitrile (1.0 mL), Ar, 90 °C, 4 h. Isolated yields. a47% of starting material was recovered. b1.5 equiv. 3-PySO2N3 was used.cDi-azidation product was isolated in 12% yield. | ||
A plausible mechanism is outlined in Scheme 4. At first, sulfate radical anions are generated by the pyrolysis of persulfate salts. The sulfate radicals abstract the hydrogen atoms from the most electron-rich positions of the substrates to afford carbon radicals II. Subsequently, the radicals II attack the terminal nitrogen atoms of sulfonyl azides to form intermediates III,17 which then decompose into the final azidation products IV and sulfonyl radicals V. The latter are further oxidized by remaining sulfate radicals to sulfonyl cations and subsequently react with O-based nucleophiles such as water to form pyridine-3-sulfonic acids.18 As shown in Scheme 1, once the carbon radicals II form, there are two competing reaction pathways, i.e. radical trapping by the sulfonyl azides and further oxidation by the persulfates. The 1H-NMR figures of crude products clearly showed that under the standard reaction conditions, the azidation products were obtained overwhelmingly more than the oxidation products,19 indicating that the radical trapping reaction is more faster than the second oxidation.
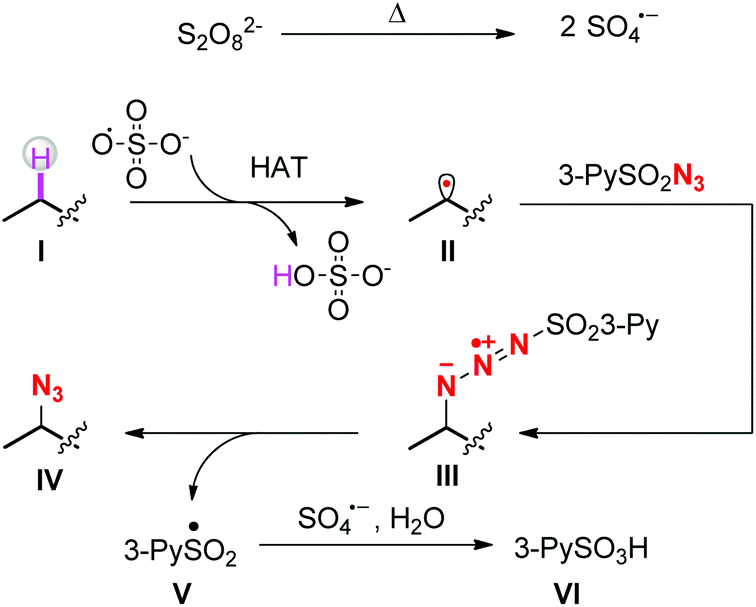 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In conclusion, an efficient and practical C–H azidation method has been developed. This radical process proceeded in an aqueous solvent without a transition-metal catalyst, exhibiting good remote tertiary C–H bond selectivity and broad functional group compatibility. Tertiary C–H bonds in a series of substrates, including dipeptides, can be easily transformed into organic azides in good yields. Many functional groups, including halogens, can be tolerated under the radical conditions.Acknowledgements
We gratefully acknowledge the financial support from the National Basic Research Program of China (973 Program) (Grant No. 2015CB856600) and the National Nature Science Foundation of China (Grant No. 21332001).Notes and references
- (a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (c) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (d) Y. Ishihara and P. S. Baran, Synlett, 2010, 1733 CAS; (e) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed; (f) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (g) D. Y.-K. Chen and S. W. Youn, Chem. – Eur. J., 2012, 18, 9452 CrossRef CAS PubMed; (h) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (i) J. J. Li, C–H Bond Activation in Organic Synthesis, CRC Press, 2015 Search PubMed.
- For the Hofmann–Löffler–Freytag reaction, see: (a) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1883, 16, 558 CrossRef; (b) K. Löffler and C. Freytag, Ber. Dtsch. Chem. Ges., 1909, 42, 3427 CrossRef; (c) M. E. Wolff, Chem. Rev., 1963, 63, 55 CrossRef CAS; (d) E. J. Corey and W. R. Hertler, J. Am. Chem. Soc., 1958, 80, 2903 CrossRef CAS; (e) P. Buchschacher, J. Kalvoda, D. Arigoni and O. Jeger, J. Am. Chem. Soc., 1958, 80, 2905 CrossRef CAS.
- For the Barton reaction, see: (a) D. H. R. Barton, J. M. Beaton, L. E. Geller and M. M. Pechet, J. Am. Chem. Soc., 1960, 82, 2640 CrossRef CAS; (b) D. H. R. Barton, J. M. Beaton, L. E. Geller and M. M. Pechet, J. Am. Chem. Soc., 1961, 83, 4076 CrossRef CAS; (c) G. Majetich and K. Wheless, Tetrahedron, 1995, 51, 7095 CrossRef CAS; (d) Ž. Čeković, Tetrahedron, 2003, 59, 8073 CrossRef.
- For the Norrish-Yang reaction, see: P. Wessig, Stereoselective Photocyclization of Ketones (Norrish–Yang Reaction), in Handbook of C–H Transformations, ed. G. Dyker, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005, vol. 2, pp. 569–579 and references therein Search PubMed.
- For selected reviews, see: (a) M. Nechab, S. Mondal and M. P. Bertrand, Chem. – Eur. J., 2014, 20, 16034 CrossRef CAS PubMed; (b) S. Chiba and H. Chen, Org. Biomol. Chem., 2014, 12, 4051 RSC; (c) T. Taniguchi, D. Hirose and T. Hashimoto, Synlett, 2014, 2531 CrossRef CAS.
- For selected examples, see: (a) K. Chen, J. M. Richter and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 7247 CrossRef CAS PubMed; (b) T. Liu, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 5871 CrossRef CAS PubMed; (c) M. A. Bigi, S. A. Reed and M. C. White, J. Am. Chem. Soc., 2012, 134, 9721 CrossRef CAS PubMed; (d) D. N. Zalatan and J. Du Bois, Synlett, 2009, 143 CAS; (e) T. Taniguchi, Y. Sugiura, T. Hatta, A. Yajima and H. Ishibashi, Chem. Commun., 2013, 49, 2198 RSC; (f) T. Hashimoto, D. Hirose and T. Taniguchi, Angew. Chem., Int. Ed., 2014, 53, 2730 CrossRef CAS PubMed; (g) J. Ozawa, M. Tashiro, J. Ni, K. Oisaki and M. Kanai, Chem. Sci., 2016, 7, 1904 RSC; (h) K. A. Hollister, E. S. Conner, M. L. Spell, K. Deveaux, L. Maneval, M. W. Beal and J. R. Ragains, Angew. Chem., Int. Ed., 2015, 54, 7837 CrossRef CAS PubMed; (i) X. Zhu, Y.-F. Wang, W. Ren, F.-L. Zhang and S. Chiba, Org. Lett., 2013, 15, 3214 CrossRef CAS PubMed; (j) Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894 CrossRef CAS PubMed; (k) P. Yu, J.-S. Lin, L. Li, S.-C. Zheng, Y.-P. Xiong, L.-C. Zhao, B. Tan and X.-Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 11890 CrossRef CAS PubMed; (l) C. G. Francisco, R. Freire, A. J. Herrera, I. Peréz-Martín and E. Suárez, Org. Lett., 2002, 4, 1959 CrossRef CAS PubMed; (m) E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591 CrossRef CAS PubMed; (n) H. Lu, H. Jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192 CrossRef CAS PubMed.
- For selected reviews, see: (a) F. Minisci, E. Vismara and F. Fontana, Heterocycles, 1989, 28, 489 CrossRef CAS; (b) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef CAS PubMed.
- For selected examples, see: (a) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS PubMed; (b) M. S. Chen and M. C. White, Science, 2010, 327, 566 CrossRef CAS PubMed; (c) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard, III and J. T. Groves, Science, 2012, 337, 1322 CrossRef CAS PubMed; (d) Q. Michaudel, D. Thevenet and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 2547 CrossRef CAS PubMed; (e) X. Li, H.-Y. Wang and Z.-J. Shi, New J. Chem., 2013, 37, 1704 RSC; (f) J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2014, 53, 1565 CrossRef PubMed; (g) S. Guo, X. Zhang and P. Tang, Angew. Chem., Int. Ed., 2015, 54, 4065 CrossRef CAS PubMed; (h) H. Wu, Z. Xiao, J. Wu, Y. Guo, J.-C. Xiao, C. Liu and Q.-Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 4070 CrossRef CAS; (i) X. Zhang, S. Guo and P. Tang, Org. Chem. Front., 2015, 2, 806 RSC; (j) W. Chen, H. Zheng, X. Pan, Z. Xie, X. Zan, B. Sun, L. Liu and H. Lou, Tetrahedron Lett., 2014, 55, 2879 CrossRef CAS; (k) J. K. Laha, K. S. S. Tummalapalli, A. Nair and N. Patel, J. Org. Chem., 2015, 80, 11351 CrossRef CAS PubMed; (l) J. Ma, W. Yi, G. Lu and C. Cai, Org. Biomol. Chem., 2015, 13, 2890 RSC.
- X. Li, X. Che, G.-H. Chen, J. Zhang, J.-L. Yan, Y.-F. Zhang, L.-S. Zhang, C.-P. Hsu, Y. Q. Gao and Z.-J. Shi, Org. Lett., 2016, 18, 1234 CrossRef CAS PubMed.
- (a) S. Bräse and K. Banert, Organic Azides: Syntheses and Applications, John Wiley & Sons, Ltd, Chichester, UK, 2010 Search PubMed; (b) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef PubMed; (c) E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297 CrossRef CAS; (d) D. Intrieri, P. Zardi, A. Caselli and E. Gallo, Chem. Commun., 2014, 50, 11440 RSC; (e) M. Minozzi, D. Nanni and P. Spagnolo, Chem. – Eur. J., 2009, 15, 7830 CrossRef CAS PubMed; (f) S. Chiba, Synlett, 2012, 21 CrossRef CAS; (g) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831 RSC; (h) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040 CrossRef CAS PubMed.
- (a) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905 CrossRef CAS PubMed; (b) M. Grammel and H. C. Hang, Nat. Chem. Biol., 2013, 9, 475 CrossRef CAS PubMed; (c) E. Lallana, R. Riguera and E. Fernandez-Megia, Angew. Chem., Int. Ed., 2011, 50, 8794 CrossRef CAS PubMed; (d) C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Bräse, Chem. Soc. Rev., 2011, 40, 4840 RSC.
- (a) P. Magnus and J. Lacour, J. Am. Chem. Soc., 1992, 114, 767 CrossRef CAS; (b) P. Magnus, J. Lacour and W. Weber, J. Am. Chem. Soc., 1993, 115, 9347 CrossRef CAS; (c) P. Magnus, C. Hulme and W. Weber, J. Am. Chem. Soc., 1994, 116, 4501 CrossRef CAS; (d) Y. Kita, H. Tohma, T. Takada, S. Mitoh, S. Fujita and M. Gyoten, Synlett, 1994, 427 CrossRef CAS; (e) P. Magnus, J. Lacour, P. A. Evans, M. B. Roe and C. Hulme, J. Am. Chem. Soc., 1996, 118, 3406 CrossRef CAS; (f) C. Viuf and M. Bols, Angew. Chem., Int. Ed., 2001, 40, 623 CrossRef CAS; (g) C. M. Pedersen, L. G. Marinescu and M. Bols, Org. Biomol. Chem., 2005, 3, 816 RSC; (h) T. Harschneck, S. Hummel, S. F. Kirsch and P. Klahn, Chem. – Eur. J., 2012, 18, 1187 CrossRef CAS PubMed; (i) Q.-H. Deng, T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2013, 135, 5356 CrossRef CAS PubMed; (j) M. V. Vita and J. Waser, Org. Lett., 2013, 15, 3246 CrossRef CAS PubMed; (k) H. Chen, W. Yang, W. Wu and H. Jiang, Org. Biomol. Chem., 2014, 12, 3340 RSC; (l) P. T. G. Rabet, G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2016, 18, 1646 CrossRef CAS PubMed.
- (a) M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 5290 CrossRef CAS PubMed; (b) X. Huang and J. T. Groves, ACS Catal., 2016, 6, 751 CrossRef CAS; (c) W. Song, S. I. Kozhushkov and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 6576 CrossRef CAS PubMed.
- (a) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600 CrossRef CAS PubMed; (b) X. Huang, T. M. Bergsten and J. T. Groves, J. Am. Chem. Soc., 2015, 137, 5300 CrossRef CAS PubMed; (c) Y. Wang, G.-X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679 RSC; (d) S. Kamijo, M. Watanabe, K. Kamijo, K. Tao and T. Murafuji, Synthesis, 2016, 115 Search PubMed.
- (a) A. P. Krasutsky, C. J. Kuehl and V. V. Zhdankin, Synlett, 1995, 1081 CrossRef CAS; (b) V. V. Zhdankin, A. P. Krasutsky, C. J. Kuehl, A. J. Simonsen, J. K. Woodward, B. Mismash and J. T. Bolz, J. Am. Chem. Soc., 1996, 118, 5192 CrossRef CAS.
- X. Zhang, H. Yang and P. Tang, Org. Lett., 2015, 17, 5828 CrossRef CAS PubMed.
- C. Ollivier and P. Renaud, J. Am. Chem. Soc., 2001, 123, 4717 CrossRef CAS PubMed.
- After the reaction, pyridine-3-sulfonic acid was detected by LC-MS.
- Azidation products/oxidation products >33/1, see the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00237d |
| This journal is © the Partner Organisations 2016 |