
C(sp3)–H bond functionalization by sequential hydride transfer/cyclization: electronic effect and steric effect controlled regioselectivity
Liang
Wang
and
Jian
Xiao
*
College of Chemistry and Pharmaceutical Sciences, Qingdao Agricultural University, 266109, Qingdao, China. E-mail: chemjianxiao@163.com
First published on 3rd March 2016
Abstract
The electronic effect and steric effect have a dramatic impact on the fascinating cascade [1,n]-hydride transfer/cyclization. The regioselectivity of two potential hydrogen donors could be perfectly tuned to construct different skeletons.
 Liang Wang | Liang Wang received his PhD degree from the University of Heidelberg in 2011 working under the supervision of Prof. Dr Dirk Menche, after which he worked as a postdoctoral fellow at the University of Basel with Prof. Dr Andreas Pfaltz (2012–2013). In 2014 he took the position of professor at Qingdao Agricultural University, China. His research interests include C–H functionalization and cascade reactions. |
 Jian Xiao | Jian Xiao was born in Shandong province in 1981 and received his PhD degree from Nanyang Technological University under the guidance of Prof. Teck-Peng Loh in 2009. After one more year as a postdoc research fellow in the same group, he joined the Dalian Institute of Chemical Physics, Chinese Academy of Sciences as an Associate Professor (2010–2012). In 2012, he was appointed as a full professor at Qingdao Agricultural University. In 2013–2014, he worked as a research fellow in the Scripps Research Institute with Prof. Carlos F. Barbas III. His research interest includes a new synthetic methodology involving organocatalysis and asymmetric catalysis, focusing on organocatalytic asymmetric C–H functionalization and cycloaddition reactions. |
Although cascade [1,n]-hydride transfer/cyclization was identified more than one hundred years ago,1 this internal redox process was largely overlooked until the beginning of this century. Inspired by the pioneering work from the groups of Sames,2 Seidel,3 and Akiyama,4 recent decades have witnessed a strong renaissance in this area as an intriguing avenue for C(sp3)–H bond functionalization.
It is well known that cleavage of the C(sp3)–H bond is the rate-determining step in C(sp3)–H bond functionalization and the hydride transfer dramatically relies on the substituent effects. The presence of heteroatoms, such as nitrogen and oxygen, will facilitate the hydride migration due to the following three factors. Firstly, heteroatoms with high electronegativities will polarize the C–X bond, causing the weakening of the C(sp3)–H bond. Secondly, the hyperconjugation effect of the σ*C–H orbital with a neighbouring heteroatom lone pair or π-orbital promotes the hydride shift (Fig. 1).5 This effect not only weakens the C(sp3)–H bond but also increases the negative charge density of the hydrogen atom. Thirdly, the carbocationic intermediate generated upon hydride migration can be stabilized by adjacent heteroatoms, aromatic or alkyl substituents via p–p conjugation, π–p conjugation or σ–p hyperconjugation, respectively. Consequently, the rate of C(sp3)–H bond cleavage is closely associated with the stability of the cationic intermediate, thus any factor which can stabilize this intermediate will dramatically promote this process, while the groups destabilizing it will retard the proximal C(sp3)–H bond cleavage. Notably, without the electronic assistance, highly electrophilic hydride acceptors and harsh conditions are always required to get decent yields.
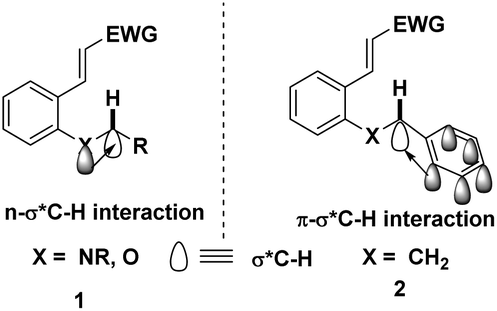 | ||
| Fig. 1 The electronic assistance from the heteroatom and the aryl group. | ||
The electronic effect also has a drastic influence on the regioselectivity. When two potential hydrides, Hα and Hβ, were available in substrate 1, Hα would migrate to create the more stable iminium intermediate 2 preferentially over migration of Hβ to furnish intermediate 4.6 The subsequent barrier-less and swift intramolecular nucleophilic attack on the iminium subunit resulted in the cyclized product 3, even though the electrophilic moiety was more sterically hindered (Scheme 1a).7 Similarly, if two methylene moieties served as potential hydride donors, the methylene substituted with an electron-rich group would be more reactive (Scheme 1b).8 Consequently, the electron-donating substituents significantly facilitated its proximal C(sp3)–H bond cleavage. In this case, the regioselectivity was dominated by the electronic effect instead of the steric effect of hydride donors.
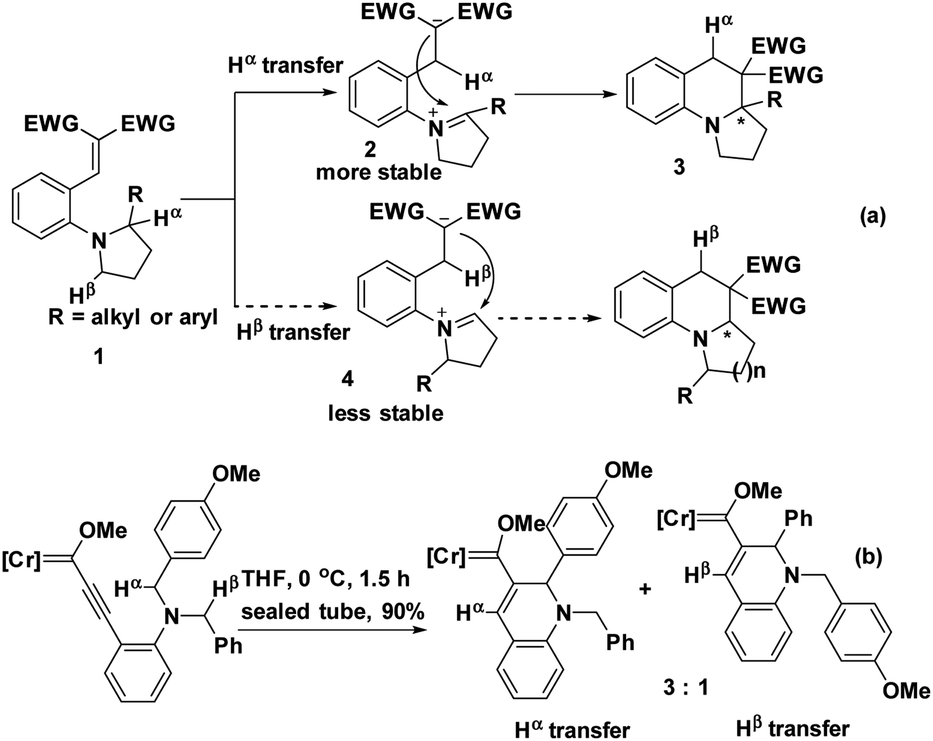 | ||
| Scheme 1 The electronic effect on the regioselectivity. | ||
In addition to the electronic effect, this cascade reaction can also be strongly impacted by the distance between hydride donors and acceptors, and any factor rendering them closer to each other will drastically promote this process. Barluenga and Ballesteros et al. demonstrated that even a comparatively inert non-benzylic secondary C(sp3)–H bond could be functionalized directly with a vicinal gold(III)-activated alkyne serving as a hydride acceptor, without electronic assistance from a heteroatom (Scheme 2a).9 Akiyama et al. also observed an intriguing ortho-substituent acceleration effect in the cascade [1,5]-hydride transfer/cyclization reaction (Scheme 2b).4a
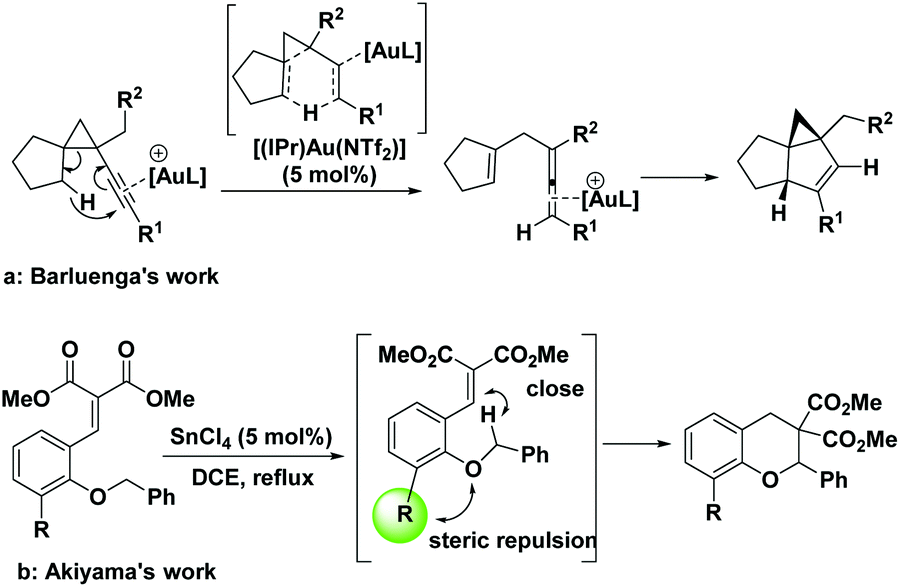 | ||
| Scheme 2 The steric effect on the cascade hydride transfer/cyclization. | ||
The precise regioselectivity-controlling effect of initial C(sp3)–H functionalization was elegantly demonstrated by Akiyama and coworkers, who recently reported a fascinating Yb(OTf)3-catalyzed double C(sp3)–H bond functionalization of benzylamine derivative 5 through a sequential hydride shift/cyclization process (Scheme 3).4e Both the electronic-effect and steric effect were exploited to precisely manipulate the sequence of two possible hydride migrations in Akiyama's work. In this reaction, the initial hydride shift ([1,4]- or [1,6]-hydride shift) was completely controlled by the steric effect of the α substituent of trifluoromethyl ketone. By tuning the R group of the ketone, the sequence of functionalization of two potential hydrogen donors was totally reversed. A bicyclo[3.2.2]nonane skeleton 6 was provided from substrate 5a (R = H) through a sequential [1,6]- and [1,5]-hydride shift process. In sharp contrast, when a benzyl group was substituted at the α position of the trifluoroacetyl group, a [1,4]- and [1,5]-hydride shift occurred successively in substrate 5b, resulting in the formation of a fused bicyclic product 7. Surprisingly, in the absence of a bulky R group, the sterically disfavored 7-membered ring 8 was preferentially generated rather than the sterically favored 5-membered ring 9. DFT calculation and theoretical studies revealed that the resonance stabilization in the benzylidene carbonyl moiety and the steric repulsion of the α-substituent were the key points driving the reaction course.
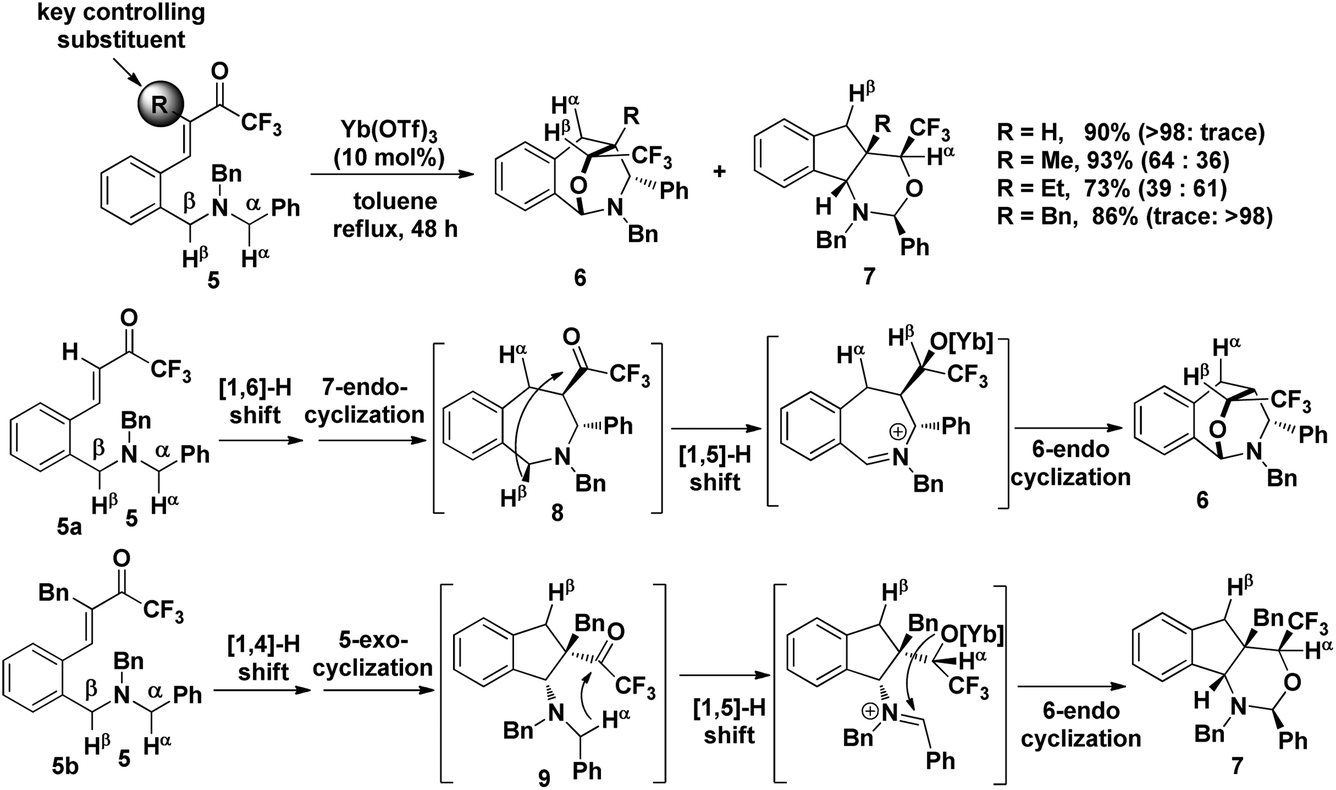 | ||
| Scheme 3 Double C(sp3)–H bond functionalization of the benzylamine derivative. | ||
In addition to the rationalization proposed by Akiyama et al., the regioselectivity can be interpreted in an intuitive way (Scheme 4). In substrate 5a, Cα–Hα is linked with a normal phenyl group, while Cβ–Hβ is substituted with the electron-deficient phenyl group, and the σ*C–H orbital of the Cα–Hα bond is impacted to a larger extent than that of the Cβ–Hβ bond through hyperconjugation, rendering the Cα–Hα bond much easier to cleave. Consequently, Hα will migrate preferentially to furnish intermediate I which undergoes a low-barrier and swift cyclization to afford a 7-membered intermediate 8, instead of the formation of a 5-membered 10. Remarkably, a sterically disfavored 7-membered ring was formed rather than a sterically favored 5-membered ring. Once again, the regioselectivity is thoroughly governed by the electronic effect rather than the steric effect.
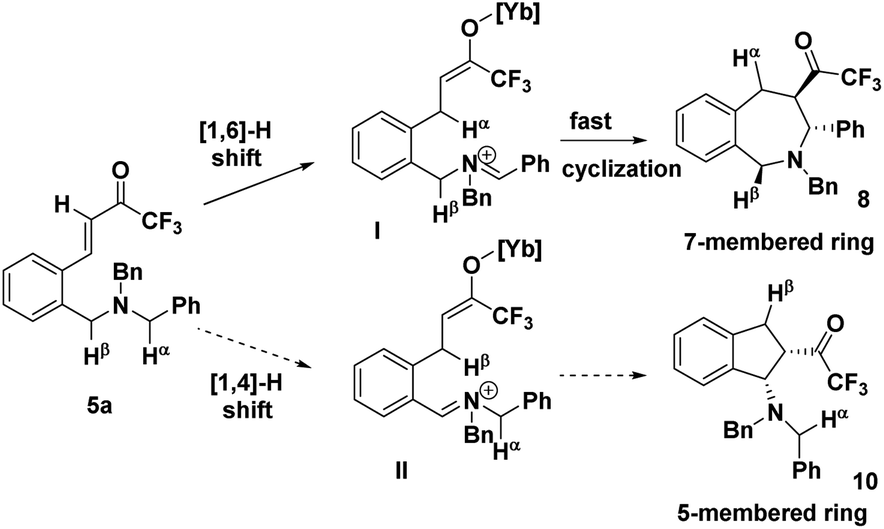 | ||
| Scheme 4 Electronic effect controlled regioselectivity. | ||
When the bulky benzyl group was substituted at the position α to a trifluoroacetyl group as in substrate 5b, the situation is thoroughly reversed. The sterically demanding benzyl group would induce a large steric repulsion between the hydrogen acceptor (α and β carbon of the ketone) and the methylene on which Hα is located, pushing the sterically hindered Hα moiety away, leading to the conformer 11. As the reactivity of the hydrogen donor is quite sensitive to the distance between the donor and the acceptor, the vicinal Hβ would shift preferentially, followed by a fast intramolecular nucleophilic attack to afford the intermediate 9 (Scheme 5). In this case, the steric effect dominated the sequence of hydride migration rather than the electronic effect.
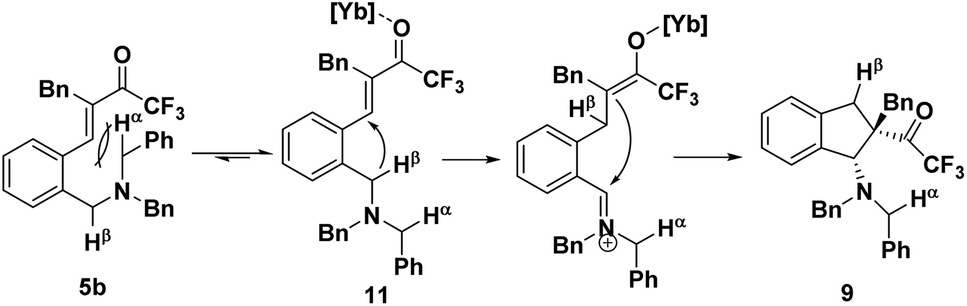 | ||
| Scheme 5 Steric effect controlled regioselectivity. | ||
In summary, we have highlighted the key role of the electronic effect and steric effect in controlling the regioselectivity of [1,n]-hydride transfer/cyclization. The origin of this phenomenon has been rationalized and summarized. The recent double cascade [1,n]-hydride transfer/cyclization elegantly demonstrated the great power of this strategy, in which the regioselectivity of two potential hydrogen donors could be perfectly tuned with the aid of the “two hands”, the electronic effect and the steric effect, to construct structurally diverse molecules in an atom-economical manner. The chemistry of [1,n]-hydride transfer/cyclization is of high significance, which, however, is still under development. We believe that this useful strategy has a brilliant future and will inspire more application for construction of complex architectures.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (no. 21102142) and the Talents of High Level Scientific Research Foundation (no. 6631112323 and 6631115015) of Qingdao Agricultural University.References
- (a) M. C. Haibach and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5010 CrossRef CAS PubMed; (b) B. Peng and N. Maulide, Chem. – Eur. J., 2013, 19, 13274 CrossRef CAS PubMed; (c) L. Wang and J. Xiao, Adv. Synth. Catal., 2014, 356, 1137 CrossRef CAS; (d) P. Mátyus, O. Éliás, P. Tapolcsányi, Á. Polonka-Bálint and B. Halász-Dajka, Synthesis, 2006, 2625 CrossRef.
- (a) S. J. Pastine, K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2005, 127, 12180 CrossRef CAS PubMed; (b) K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2009, 131, 402 CrossRef CAS PubMed; (c) P. A. Vadola and D. Sames, J. Am. Chem. Soc., 2009, 131, 16525 CrossRef CAS PubMed; (d) S. J. Pastine and D. Sames, Org. Lett., 2005, 7, 5429 CrossRef CAS PubMed; (e) K. M. McQuaid, J. Z. Long and D. Sames, Org. Lett., 2009, 11, 2972 CrossRef CAS PubMed; (f) P. A. Vadola, I. Carrera and D. Sames, J. Org. Chem., 2012, 77, 6689 CrossRef CAS PubMed.
- (a) S. Murarka, C. Zhang, M. D. Konieczynska and D. Seidel, Org. Lett., 2009, 11, 129 CrossRef CAS PubMed; (b) S. Murarka, I. Deb, C. Zhang and D. Seidel, J. Am. Chem. Soc., 2009, 131, 13226 CrossRef CAS PubMed; (c) C. Zhang, C. K. De, R. Mal and D. Seidel, J. Am. Chem. Soc., 2008, 130, 416 CrossRef CAS PubMed; (d) M. C. Haibach, I. Deb, C. K. De and D. Seidel, J. Am. Chem. Soc., 2011, 133, 2100 CrossRef CAS PubMed.
- (a) K. Mori, T. Kawasaki, S. Sueoka and T. Akiyama, Org. Lett., 2010, 12, 1732 CrossRef CAS PubMed; (b) K. Mori, S. Sueoka and T. Akiyama, J. Am. Chem. Soc., 2011, 133, 2424 CrossRef CAS PubMed; (c) K. Mori, K. Ehara, K. Kurihara and T. Akiyama, J. Am. Chem. Soc., 2011, 133, 6166 CrossRef CAS PubMed; (d) K. Mori, T. Kawasaki and T. Akiyama, Org. Lett., 2012, 14, 1436 CrossRef CAS PubMed; (e) K. Mori, K. Kurihara, S. Yabe, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2014, 136, 3744 CrossRef CAS PubMed; (f) K. Mori, N. Umehara and T. Akiyama, Adv. Synth. Catal., 2015, 357, 901 CrossRef CAS.
- (a) P. Brunet and J. D. Wuest, J. Org. Chem., 1996, 61, 2020 CrossRef CAS; (b) M. Alajarin, B. Bonillo, M. Ortin, P. Sanchez-Andrada and A. Vidal, Org. Lett., 2006, 8, 5645 CrossRef CAS PubMed; (c) K. Mori, S. Sueoka and T. Akiyama, Chem. Lett., 2011, 40, 1386 CrossRef CAS.
- (a) L. Zhang, L. Chen, J. Lv, J.-P. Cheng and S. Luo, Chem. – Asian J., 2012, 7, 2569 CrossRef CAS PubMed; (b) L. Chen, L. Zhang, J. Lv, J. Cheng and S. Luo, Chem. – Eur. J., 2012, 18, 8891 CrossRef CAS PubMed.
- (a) V. Ojea and J. M. Quintela, Heterocycles, 1993, 36, 1337 CrossRef CAS; (b) W. H. N. Nijhuis, W. Verboom, A. A. El-Fadl, S. Harkema and D. N. Reinhoudt, J. Org. Chem., 1989, 54, 199 CrossRef CAS.
- J. Barluenga, M. Fananas-Mastral, F. Aznar and C. Valdes, Angew. Chem., Int. Ed., 2008, 47, 6594 CrossRef CAS PubMed.
- J. Barluenga, R. Sigueiro, R. Vicente, A. Ballesteros, M. Tomas and M. A. Rodriguez, Angew. Chem., Int. Ed., 2012, 51, 10377 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2016 |