
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble helicene possessing B–N dative bonds built on 1,4-dihydropyrrolo[3,2-b]pyrrole core†
Wojciech D.
Petrykowski
 ab,
Nicolas
Vanthuyne
c,
Carmelo
Naim
d,
Francesco
Bertocchi
e,
Yevgen M.
Poronik
a,
Arkadiusz
Ciesielski
b,
Michał K.
Cyrański
*b,
Francesca
Terenziani
*e,
Denis
Jacquemin
*df and
Daniel T.
Gryko
*a
ab,
Nicolas
Vanthuyne
c,
Carmelo
Naim
d,
Francesco
Bertocchi
e,
Yevgen M.
Poronik
a,
Arkadiusz
Ciesielski
b,
Michał K.
Cyrański
*b,
Francesca
Terenziani
*e,
Denis
Jacquemin
*df and
Daniel T.
Gryko
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44-52, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl
bFaculty of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland. E-mail: mkc@chem.uw.edu.pl
cAix Marseille Univ, CNRS, Centrale Med, FSCM, Marseille, France
dNantes Université, CNRS, CEISAM, UMR6230, F-44000, Nantes, France. E-mail: Denis.Jacquemin@univ-nantes.fr
eDepartment of Chemistry, Life Sciences and Environmental Sustainability, University of Parma, 43124 Parma, Italy. E-mail: francesca.terenziani@unipr.it
fInstitut Universitaire de France (IUF), F-75005, Paris, France
First published on 2nd April 2025
Abstract
Just four steps are required to transform 2-nitrobenzaldehyde into centrosymmetric, quadrupolar N,B-doped nanographenes possessing two nitrogen–boron dative bonds. A convergent fragment coupling strategy allowed rapid access to key intermediates bearing the 1,4-dihydropyrrolo[3,2-b]pyrrole core. 2,6-Di-tert-butylpyridine turned out to be the best base for the formation of B←N bonds. This synthetic strategy can be extended to encompass double helicenes possessing two [7]helicene units bearing four five-membered rings. The size of the peripheral arm influences the reaction output: in the case of replacing benzene with dibenzothiophene, the yield decreases from 75% to 16%. Interestingly only two enantiomers and not meso form are formed in the latter case. The obtained double helicene containing 14 fused rings, exhibits green emission characterized by reasonable fluorescence quantum yield reaching 0.38. This dye has average circularly polarized luminescence brightness (BCPL) of about 15 M−1 cm−1. The analysis of the electronic structure of the dyes with quantum chemical methods reveals highly-delocalized excited states with the core of the dye acting as a electron-donating moiety.
Introduction
The incorporation of boron–nitrogen bonds into the structure of polycyclic aromatic hydrocarbons (PAHs) offers polarization of π-systems, which leads to the modulation of their photophysical properties. The extraordinary advances in the chemistry of azaborines have been fueled by the isosteric character of the N–B bond versus the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.1–10 In contrast, the approach to aromatic architectures with B←N dative bonds has a much shorter history which started from a key paper by Yamaguchi in 2006.11 The presence of four-coordinated boron atoms in such dyes represents an entirely new handle for tuning the optoelectronic properties.12,13 During the last decade, Jäkle and co-workers described several polycyclic structures possessing two B←N bonds e.g. dye 1 (Fig. 1).14–17 Extensive efforts have been devoted to advancing this chemistry by Pischel and Nakamura in 2020, who reported a bis-boronated green light-emitter 2, which possessed a nearly quantitative fluorescence quantum yield (Φfl) (Fig. 1).18,19 Similarly, in 2021, we reported a polarized B←N-containing chromophore 3 with an electron-donating 1,4-dihydropyrrolo[3,2-b]pyrrole system incorporated into its central part to obtain derivatives that show efficient green and red light emission.20 However, the chemical diversity of these structures cannot cover the fact that until recently the vast majority of functional dyes possessing B←N bonds had a planar π-system.
C bond.1–10 In contrast, the approach to aromatic architectures with B←N dative bonds has a much shorter history which started from a key paper by Yamaguchi in 2006.11 The presence of four-coordinated boron atoms in such dyes represents an entirely new handle for tuning the optoelectronic properties.12,13 During the last decade, Jäkle and co-workers described several polycyclic structures possessing two B←N bonds e.g. dye 1 (Fig. 1).14–17 Extensive efforts have been devoted to advancing this chemistry by Pischel and Nakamura in 2020, who reported a bis-boronated green light-emitter 2, which possessed a nearly quantitative fluorescence quantum yield (Φfl) (Fig. 1).18,19 Similarly, in 2021, we reported a polarized B←N-containing chromophore 3 with an electron-donating 1,4-dihydropyrrolo[3,2-b]pyrrole system incorporated into its central part to obtain derivatives that show efficient green and red light emission.20 However, the chemical diversity of these structures cannot cover the fact that until recently the vast majority of functional dyes possessing B←N bonds had a planar π-system.
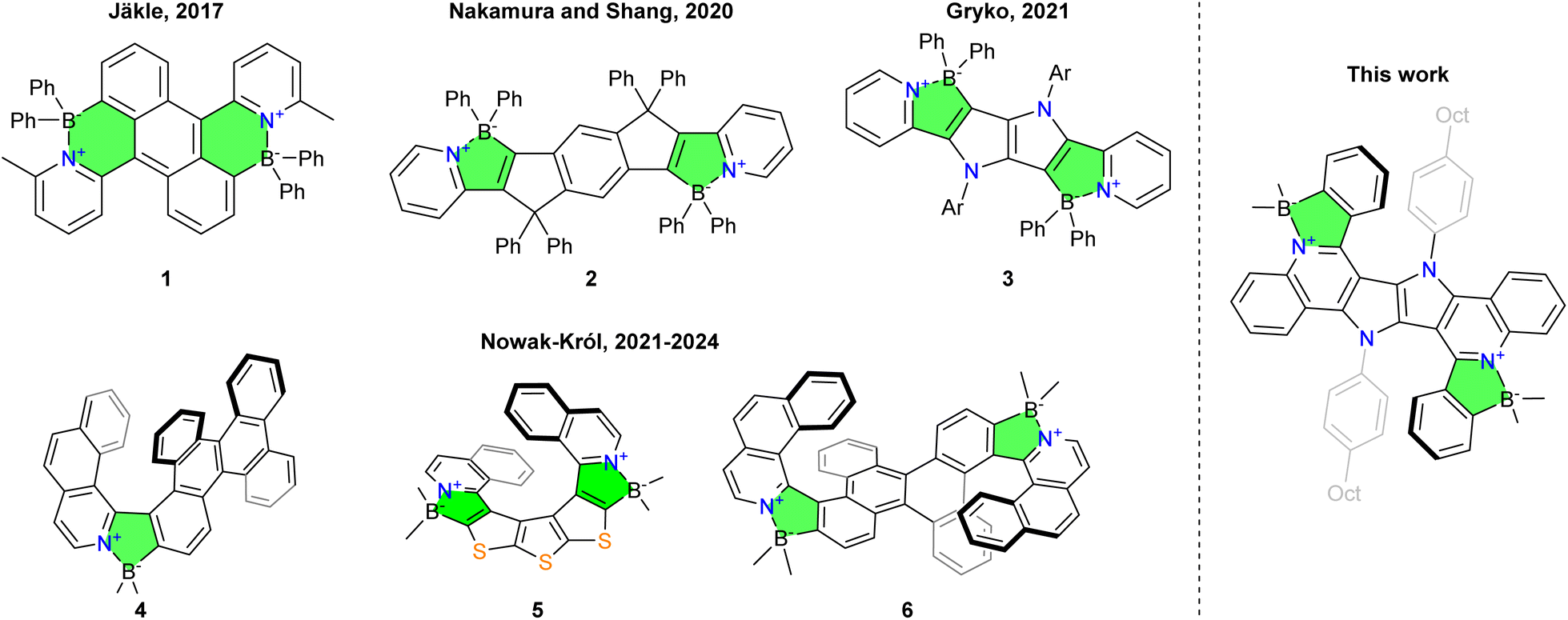 | ||
| Fig. 1 Chemical structures of selected examples of dyes bearing B←N dative bonds bridging aromatic systems. | ||
Within the quest of discovering organic chromophores possessing strong circularly polarized luminescence (CPL), attention has been focused on helicenes – inherently chiral polycyclic aromatic compounds composed of rings condensed in the ortho position in a way that they twist into a helix shape.21–24 The main arguments driving research on helicenes are possible applications of CPL which include OLEDs,25 fluorescence microscopy,26etc.22 It is obvious that materials other than archetypal benzene-only helicenes may offer greater chemical variability and more appreciable chiroptical properties.27–39 Among many heterocyclic helicenes, dyes possessing an azaborine moiety were reported in a few works.40–42
Intriguingly, as reported predominantly by Nowak-Król and co-workers, helicenes bearing B←N bond(s) possess appreciable optoelectronic properties. π-Expanded helicene 4 has a relatively large Φfl of 0.31 and |glum| of 2.2 × 10−3 with λem at 510 nm.43–45 Other examples include unusual helicene 5 containing three thiophene rings fused together,46 and the first double helicenes encompassing B←N bonds (e.g.6),47 possessing bright green fluorescence and reasonable |glum| values (Fig. 1). Clearly, a better understanding of the relationship between the structure of hetero-helicenes and their chiroptical properties requires explaining how the position and the type of heteroatoms affects electronic and magnetic dipole moments. This begs the question if these parameters could be modulated by incorporating multiple B←N bonds in a double helical architecture. To tackle these challenges, we target double helicenes possessing multiple B←N dative bonds at the periphery and another heterocyclic scaffold which exhibits quadrupolar symmetry.
Design and synthesis
In principle there are a few heterocyclic scaffolds suitable to serve as a central core in the construction of double helicenes. We have chosen 1,4-dihydropyrrolo[3,2-b]pyrrole (DHPP)48,49 as the central core because of the following combination of properties: (1) centrosymmetric, quadrupolar skeleton with near-C2h symmetry; (2) strong emission intensity, which remains almost constant regardless of structural modifications; (3) straightforward synthesis leading to the formation of a densely substituted scaffold which is amenable to facile functionalization.50Critically the incorporation of B←N dative bonds requires the presence of a pyridine moiety within the structure. In departure from reported pathways, we conceived a convergent fragment coupling strategy starting from a multicomponent reaction leading to the DHPP core, followed by closing two pyridine rings via a double Pictet–Spengler reaction allowing the installation of diverse aromatic scaffolds in the late stage. Thus, our synthetic design relies on the previously disclosed synthesis of ladder-type π-expanded DHPPs possessing quinoline units to build a basic centrosymmetric skeleton.51 In the final step, the obtained intermediates are subjected to the reaction with boron tribromide in the presence of a base (usually DIPEA) and then the bromine atoms are replaced with methyl groups upon treatment with trimethylaluminum, as established by Murakami.52
At the outset we have chosen dye 8, possessing simple phenyls at positions 9 and 13, as a model system, anticipating that the significant steric hindrance in the envisioned dyes would probably require optimization of the standard reaction conditions. Substrate 8 is easily accessible by the reduction/Pictet–Spengler sequence of DHPP 7 with benzaldehyde.51 As for the borylation reaction, standard conditions described by Murakami52 and optimized by Ingleson53 give extremely low conversion of 8 and only the mono-borylated product could be isolated. We noticed that increasing the temperature significantly boosts the reaction rate, but the best results are achieved by changing the base to 2,6-di-tert-butylpyridine (DBP). A more challenging substrate 10, obtained under slightly modified conditions of the aforementioned condensation, with bulkier naphthalene moieties, also undergoes electrophilic borylation to 11 with satisfactory yield thanks to the use of DBP. We underline that the syntheses of 9 and 11, shown in Scheme 1, does not require any column chromatography, as all the products and intermediates are purified by recrystallization. Notably, although dyes 9 and 11 are formally a double [5]helicene and a double [6]helicene respectively, their racemization barrier is too low to enable separation at RT, due to the presence of multiple five-membered rings.
 | ||
| Scheme 1 The syntheses of boron complexes 9, 11 and 13 from DHPP 7. 8, 9: Ar = phenyl, 10, 11: Ar = 1-naphthyl, 12, 13: Ar = 1-dibenzo[b,d]thienyl. | ||
Finally, we sought to further validate our strategy by the preparation of a double helicene which has a sufficient racemization barrier. With the optimized conditions in hands, the synthesis of double azaborahelicenes based on the DHPP skeleton with large steric hindrance, ensuring a stable configuration, becomes attainable. An aldehyde with an appropriate structure is required for the condensation step, in particular we chose dibenzo[b,d]thiophene-1-carbaldehyde, as it can be smoothly obtained in one step from a commercially available substrate. Its condensation with diamino-DHPP, derived from dye 7, gives 12 in low yield, albeit sufficient for the next step. The final borylation of 12 with BBr3/DBP delivers dye 13, a double helicene whose configuration is stable at room temperature (Fig. 2). Double helicene 13 was obtained as a racemic mixture, interestingly however that the meso-form was not detected. The relatively low yield of double helicene 13 (16%) prompted us to attempt to identify side-products which consists of only traces of the meso form among a few unstable substances. Both enantiomers of 13 were successfully isolated using HPLC on a chiral stationary phase with 99.5% enantiomeric excess (ee). Additional details can be found in the ESI.†
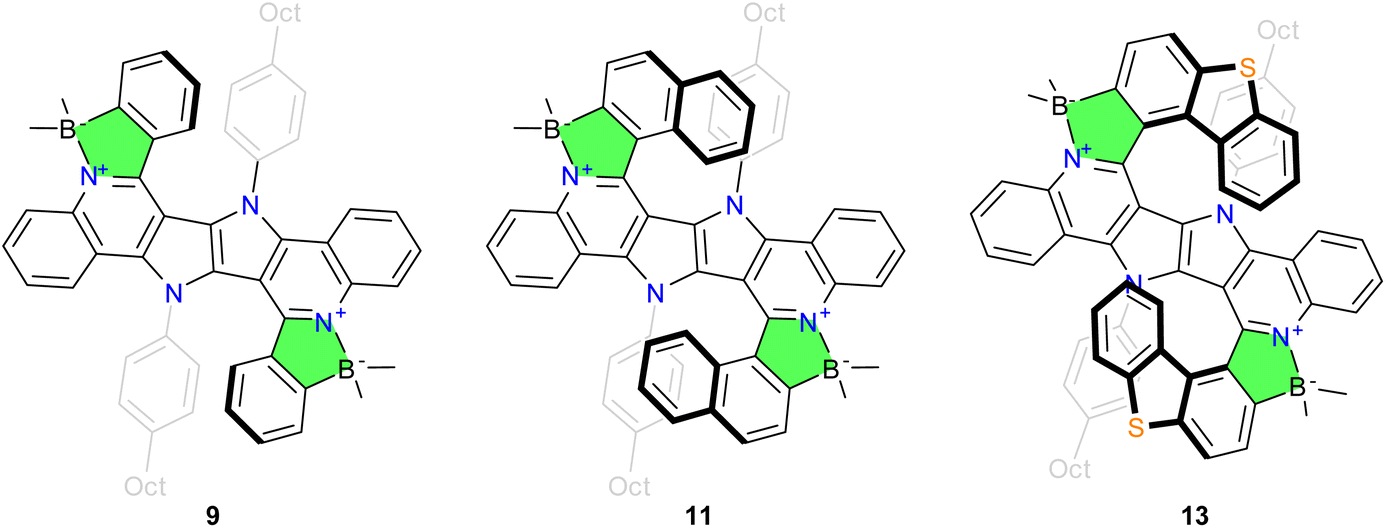 | ||
| Fig. 2 Chemical structures of the new dyes 9, 11 and 13. | ||
X-ray crystallography
Compound 11 forms large single crystals during purification, which are sufficient for X-ray structural analysis. The experiment confirms the expected structure of 11, with boron atoms incorporated into the five-membered rings (for another possibility see Fig. S6†). Molecules of 11 crystallize in a centrosymmetric P21/n space group with 4 molecules in the unit cell (Fig. S3†). Out of the two possible diastereoisomers (Ci and C2) only the chiral forms (C2) are present in the crystal. However, due to the location of the molecule in the general position of the unit cell and the disorder of the octyl substituents, deviations from ideal C2 symmetry occur. The side chains adopt three alternative positions, as shown in Fig. S4.† The core of the molecule is significantly bent. The angle formed by the outer rings (marked in yellow) is 37.78(7)°. The angles between the planes passing through the rings of the helicene fragments (marked in red and blue) are 38.14(7)° and 44.22(7)°, respectively (Fig. S5†).Single crystals of 13 was obtained by slow vapor diffusion of diethyl ether into concentrated solution of 13 in dichloromethane. The crystallographic experiment confirms the expected structure of 13 (see Fig. 3). This compound also crystallizes in P21/n space group with 4 molecules in the unit cell (Fig. S8†), similarly to 11, with a comparable degree of disorder in the octyl chains, which exist in two alternative positions (Fig. S9†). The helical part of the molecule is highly curved to the point that the peripheral rings are almost perpendicular to each other.
 | ||
| Fig. 3 Structure 13 with anisotropic displacement model at 50% probability level. For clarity, all hydrogen atoms are omitted. CCDC 2430266. | ||
Photophysical properties
Having three new helical π-expanded DHPPs 9, 11 and 13 in hand, we studied their photophysical properties (Fig. 4 and Table 1). The absorption spectra of all the new dyes look similar, with intense absorption bands around 340 nm along with a weaker absorption at 400–450 nm (typical of NB helicenes43–47), which is consistent with the computational data (vide infra). This correlates well with the structure being a hybrid of double helicenes and a ladder-type π-expanded heterocycle. Additionally, the increase of the chromophore length gives rise to only a moderate bathochromic shift of the absorption maxima. On the other hand, λabs values hardly change in different solvents, implying a relatively small change in dipole moment between the two electronic states. | ||
| Fig. 4 Absorption (solid line) and emission (dotted line) spectra of compounds 9 (top) and 13 (bottom) in toluene (blue), CH2Cl2 (green) and DMF (red). | ||
a
λ
abs/λem – absorption/emission wavelength, ε – molar absorption coefficient, Φfl – fluorescence quantum yield, Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) – Stokes shift. The values of λabs marked with an asterisk are estimated, as these maxima are not visible due to overlapping bands. – Stokes shift. The values of λabs marked with an asterisk are estimated, as these maxima are not visible due to overlapping bands.
|
|---|

|
The analysis of the fluorescence spectra shows that the emission of all compounds synthesized is more affected by the solvent polarity. The shape of the emission spectra in non-polar toluene is vibronically-resolved, which is consistent with low inhomogeneous broadening. Increasing the solvent polarity induces a progressive red-shift and broadens the emission band, suggesting a partial charge-transfer character of the emissive state. Dyes 9 and 11 show similar Stokes shift values in each solvent suggesting no specific effect of chromophore expansion on its emission. Lower values of Δ for 13 indicate the smallest geometry change upon internal relaxation following photoexcitation within the dyes synthesized.
All the new compounds have fluorescence quantum yields in the range of 0.23 to 0.39, whilst only slightly affected by the solvent polarity. In comparison to known NB helicenes 4–6, the new dyes exhibit similar spectral profiles, with intense short-wavelength and weak long-wavelength bands, however the Φfl values are comparable or higher.43–47 The Stokes shifts of 9, 11 and 13 are significantly higher than that of 3 and its derivatives, while Φfl values are approximately 2 times lower, which indicates that the new chromophore is less rigid than that of 3.20
Chiroptical properties
CD spectra of the enantiomers of dye 13 in DCM (Fig. 5) show multiple peaks, with dissymmetry factors, gabs, lower than 10−2 (see Fig. S10†). The circularly polarized luminescence (CPL) spectra of the enantiomers of 13 in DCM were acquired by exciting the sample at 410 nm. A horizontal polarizer in excitation was employed to prevent artifacts coming from linearly polarized components of emitted lights.54 The CPL spectrum has been smoothed using a Savitzky–Golay filter. The shape of the CPL spectrum roughly corresponds to the shape of the emission spectrum in the same solvent (Fig. 4). The dissymmetry factor of CPL, |glum|, is ca. 1.2 × 10−3, consistent with values observed in helicenes with B←N bonds, and indicative of moderate chiral emission activity. Notably, helicene 13 exhibits minimal structural rearrangement in the excited state, as evidenced by the similarity between the gabs of the lowest-energy CD band (1.7 × 10−3, see Fig. S10†) and |glum|.55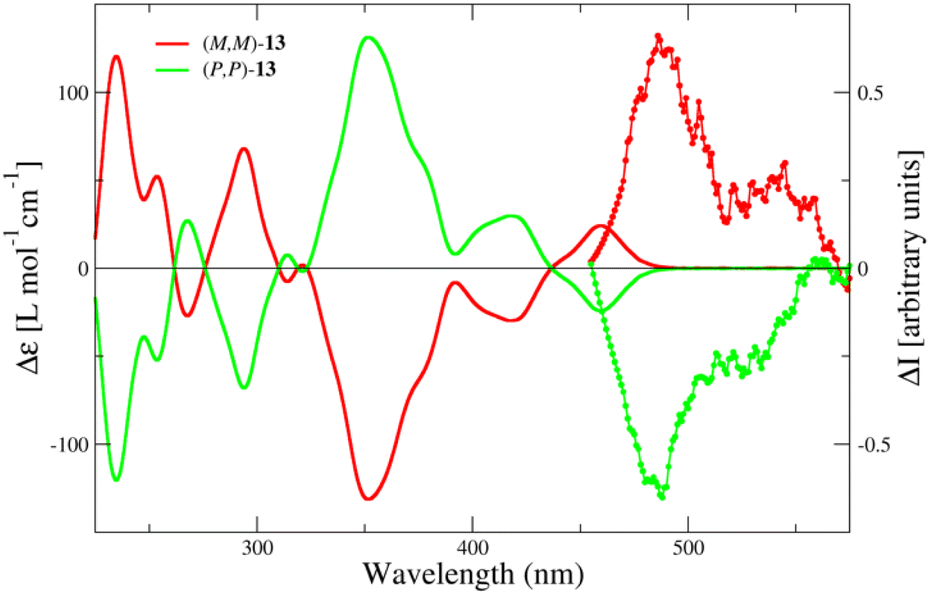 | ||
| Fig. 5 CD (full lines) and CPL (lines with dots) spectra of the enantiomers of 13 in DCM. | ||
To quantify the CPL performance of 13, the CPL brightness (BCPL) was calculated using the formula:56

| g lum (M,M)-13/(P,P)-13 | Φ fl | ε @ 350 nm [M−1 cm−1] | B CPL [M−1 cm−1] | |
|---|---|---|---|---|
| Toluene | +1.32 × 10−3/−1.24 × 10−3 | 36% | 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
15.2 |
| DCM | +1.20 × 10−3/−1.32 × 10−3 | 29% | 66000 |
12.1 |
The chiroptical characterization of 13 was performed in toluene as well (Fig. S10, S11† and Table 2), giving a similar |glum| value but slightly improved BCPL (15.2 M−1 cm−1) thanks to the higher value of Φfl.
In order to assign the right absolute configuration to each eluted fraction of 13, we performed TD-DFT calculations following a procedure detailed in the ESI† and similar to ref. 57 albeit skipping the molecular dynamics simulations sampling because of the high structural rigidity of the chiral chromophoric cores which limits the conformational degrees of freedom. The comparison of the calculated58 and experimental CD spectrum shown in Fig. S12† (mainly the sign of the different peaks) allowed us to assign the absolute configuration, in particular the first eluted fraction of 13 corresponds to the (M,M) enantiomer.
Computational studies
We probed the nature of the lowest excited states of dyes 9, 11, and 13 in toluene using ab initio tools (see the ESI† for computational details). For all three structures, one can envisage both C2 or Ci ground-state structures depending on whether the boron-bearing groups are located on the same side of the DHPP plane or not (see Fig. S13 in the ESI† for representation). For all three compounds, theory foresees that the C2 structures, that are bowl-shaped, are more stable, by 6.9, 6.7, and 7.8 kcal mol−1 (free energy values) than their Ci counterparts, for dyes 9, 11, and 13 respectively. Given that for most “standard” DHPP the two symmetries yield almost the same energies, we attribute this effect to a reduction of the steric stress in the C2 structures. This is consistent with the crystallographic findings. In the excited state, the structures undergo a slight distortion and become C1 though their overall shape remains close to the C2 one. This slight symmetry-breaking is likely at the origin of the solvatofluorochromic effect noticed in the measured emission spectra (vide supra).In the case of dye 9, the four lowest excited states that could be computed are given in Fig. S14† together with electron density difference (EDD) plots. The lowest excited state of B symmetry shows a significant absorption (f = 0.372) and corresponds to the shoulder seen in the experimental absorption at ca. 400 nm (Fig. 4). The second state is an A state with significantly less absorbance. The third transition (B) has also a significant probability (f = 0.300), whereas the fourth transition (B) is very intense (f = 1.442) and corresponds to the main band at ca. 320 nm in the experimental spectra of Fig. 4. In other words, the combination of these closed four excited states explains the observed absorption spectra.
The electron density different plots corresponding to the lowest excited state of all three compounds can be found in Fig. 6. As can be seen, the lowest excited-states are delocalized over the π-conjugated helical structure with quadrupolar-like charge-transfer, the central DHPP acting as the donor (in blue) and the boron-containing rings as the acceptors (in red). While the topology of the transition remains rather preserved in all three compounds, one notices58 a slight delocalization on the additional ring (dye 11) and sulfur atoms (dye 13), consistent with the observed moderate redshifts. This makes the transitions both more intense and more redshifted, explaining the clearer band separation between the small first band and the second intense one in 11 and 13 than in 9 found experimentally.
 | ||
| Fig. 6 EDD plots corresponding to the absorption to the lowest state of 9 (left), 11 (center) and 13 (right). The blue and red lobes represent regions of decreased and increased electron density, respectively (threshold: 0.001 au). | ||
To reach more physically well-grounded comparisons, we have computed the 0–0 energies and performed vibronic calculations. For the former, using a dedicated protocol (see the ESI†), transition energies of 2.64, 2.51, and 2.35 eV are computed for dyes 9, 11, and 13, respectively. These values are in reasonably good agreement (error of ca. 0.2 eV) with the absorption-emission crossing point found experimentally, and reproduce the successive redshifts that are obtained experimentally. For the latter, we computed vibrationally-resolved absorption and emission spectra (see Fig. S15 in the ESI†). These computations successfully reproduce the experimental absorption spectra, with all key peaks, especially the shape of 400–440 nm absorption is correctly foreseen. However, the computed molar absorption coefficient of the most intense peak (at ca. 300 nm) is approximately 200000 cm−1 M−1 for dyes 9 and 11, which significantly exceeds their experimental counterparts of ca. 65000 cm−1 M−1. The same relative error is found for the lowest absorption band. In 13, the computed molar absorption coefficient for the largest peak aligns closely with the experimental value (80000 vs. 67000 cm−1 M−1) though the computed peaks at ca. 400 nm exhibit an excessively large relative intensity in comparison to the experimental value (see Fig. S15†).
Conclusions
Here we disclose that it is possible to build double helicenes possessing both a 1,4-dihydropyrrolo[3,2-b]pyrrole core and nitrogen–boron dative bonds. The efficiency of this convergent approach is underscored by the simplicity of building blocks: two different aromatic aldehydes, a primary aromatic amine, butanedione and a boron source. The method's versatility has been showcased by incorporating such heavily sterically encumbered and demanding moieties as dibenzothiophene-1-yl. Synthesis of N,B-doped nanographenes bearing two [5]helicene scaffolds is very efficient but even in the case of [7]helicene the yields are still acceptable.The helical, ladder-type molecular architecture translates to the fact that even structures possessing 14 conjugated rings have absorption maxima located in the UV-blue part of the electromagnetic spectrum. The double helicenes possessing two B←N dative bonds display strong emission in the green region, are sensitive to solvent polarity, and possess moderate circularly polarized luminescence brightness (BCPL = 15 M−1 cm−1). Collectively, these results highlight the compatibility of B←N dative bonds with the formation of helical molecular architectures. We anticipate that this chemistry will empower the creation of new materials having BN functionality.
Data availability
The data supporting this article have been included as part of the ESI.† Deposition number 2415573 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge via the joint Cambridge Crystallographic Data Centre (CCDC).Author contributions
Conceptualization: W. D. P., D. T. G.; investigation: W. D. P., A. C., F. B., C. N., N. V.; supervision: D. T. G., F. T., M. K. C., D. J.; visualization: W. D. P., A. C., F. B., C. N., N. V.; writing – original draft: W. D. P., F. B., D. J.; F. T., Y. M. P.; D. T. G.; writing – review & editing: W. D. P., F. B., D. J.; C. N., F. T., D. T. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement no. 101007804 and from European Research Council (ARCHIMEDES, 101097337). Views and opinions expressed are, however, those of the authors only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them. The work was also financially supported by the Polish National Science Centre, Poland (OPUS 2020/37/B/ST4/00017) and has benefited from the equipment and framework of the COMP-HUB and COMP-R initiatives, funded by the “Departments of Excellence” program of the Italian Ministry for University and Research (MIUR, 2018-2022 and MUR, 2023-2027). This work was supported by the French Agence Nationale de la Recherche (ANR) under contract ANR-21-CE07-0058-2 (CONDOR). This research used resources of the GLiCID Computing Facility (Ligerien Group for Intensive Distributed Computing, 10.60487/glicid, Pays de la Loire, France). The collaboration between the Jacquemin and Gryko group is supported by the Maria Skłodowska-Curie and Pierre Curie Polish-French Science Award of the Académie des Sciences, the FNP and Zygmunt Zaleski foundation. The X-ray structures was determined in the Advanced Crystal Engineering Laboratory (aceLAB) at the Chemistry Department of the University of Warsaw. MKC and AC acknowledges prof. Ilona Turowska-Tyrk for her support.Notes and references
- A. Abengózar, P. García-García, M. A. Fernández-Rodríguez, D. Sucunza and J. J. Vaquero, in Advances in Heterocyclic Chemistry, Academic Press Inc., 2021, vol. 135, pp. 197–259 Search PubMed.
- J. Huang and Y. Li, Front. Chem., 2018, 6, 341 CrossRef PubMed.
- S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77–89 CrossRef CAS.
- S. Nakatsuka, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 13562–13565 CrossRef CAS PubMed.
- K. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2019, 141, 7453–7462 CrossRef CAS PubMed.
- P. B. Pati, E. Jin, Y. Kim, Y. Kim, J. Mun, S. J. Kim, S. J. Kang, W. Choe, G. Lee, H. J. Shin and Y. S. Park, Angew. Chem., Int. Ed., 2020, 59, 14891–14895 CrossRef CAS PubMed.
- J. Hoffmann, B. Geffroy, E. Jaques, M. Hissler and A. Staubitz, J. Mater. Chem. C, 2021, 9, 14720–14729 RSC.
- M. Franceschini, M. Crosta, R. R. Ferreira, D. Poletto, N. Demitri, J. P. Zobel, L. González and D. Bonifazi, J. Am. Chem. Soc., 2022, 144, 21470–21484 CrossRef CAS PubMed.
- M. Zhao and Q. Miao, Angew. Chem., Int. Ed., 2021, 60, 21289–21294 Search PubMed.
- M. Fingerle, J. Dingerkus, H. Schubert, K. M. Wurst, M. Scheele and H. F. Bettinger, Angew. Chem., Int. Ed., 2021, 60, 15798–15802 CrossRef CAS PubMed.
- A. Wakamiya, T. Taniguchi and S. Yamaguchi, Angew. Chem., Int. Ed., 2006, 45, 3170–3173 CrossRef CAS PubMed.
- S. Luo, J. Wang, N. Li, X. F. Song, X. Wan, K. Li and C. Yang, Angew. Chem., Int. Ed., 2023, 62, e202310943 CrossRef CAS PubMed.
- T. Sakamaki, T. Nakamuro, K. Yamashita, K. Hirata, R. Shang, E. Nakamura and R. Shang, Chem. Mater., 2021, 33, 5337–5344 CrossRef CAS.
- K. Liu, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2017, 139, 18170–18173 CrossRef CAS PubMed.
- M. Vanga, R. A. Lalancette and F. Jäkle, Chem.–Eur. J., 2019, 25, 10133–10140 CrossRef CAS PubMed.
- M. Vanga, A. Sahoo, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2022, 61, e202113075 CrossRef CAS PubMed.
- K. Liu, Z. Jiang, R. A. Lalancette, X. Tang and F. Jäkle, J. Am. Chem. Soc., 2022, 144, 18908–18917 CrossRef CAS PubMed.
- R. Campos-González, P. Vázquez-Domínguez, P. Remón, F. Nájera, D. Collado, E. Pérez-Inestrosa, F. Boscá, A. Ros and U. Pischel, Org. Chem. Front., 2022, 9, 4250–4259 RSC.
- H. Lu, T. Nakamuro, K. Yamashita, H. Yanagisawa, O. Nureki, M. Kikkawa, H. Gao, J. Tian, R. Shang and E. Nakamura, J. Am. Chem. Soc., 2020, 142, 18990–18996 CrossRef CAS PubMed.
- M. Tasior, P. Kowalczyk, M. Przybył, M. Czichy, P. Janasik, M. H. E. Bousquet, M. Łapkowski, M. Rammo, A. Rebane, D. Jacquemin and D. T. Gryko, Chem. Sci., 2021, 12, 15935–15946 RSC.
- Y. Shen and C. F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- Y. Zhang, S. Yu, B. Han, Y. Zhou, X. Zhang, X. Gao and Z. Tang, Matter, 2022, 5, 837–875 CrossRef CAS.
- T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1–32 Search PubMed.
- J. Full, M. J. Wildervanck, C. Dillmann, S. P. Panchal, D. Volland, F. Full, K. Meerholz and A. Nowak-Król, Chem.–Eur. J., 2023, 29, e202302808 CrossRef CAS PubMed.
- P. Stachelek, L. MacKenzie, D. Parker and R. Pal, Nat. Commun., 2022, 13, 553 CrossRef CAS PubMed.
- C. Li, Y. Yang and Q. Miao, Chem.–Asian J., 2018, 13, 884–894 CrossRef CAS PubMed.
- W. W. Yang and J. J. Shen, Chem.–Eur. J., 2022, 28, e202202069 CrossRef CAS PubMed.
- X. Y. Wang, X. C. Wang, A. Narita, M. Wagner, X. Y. Cao, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 12783–12786 CrossRef CAS.
- F. Aribot, A. Merle, P. Dechambenoit, H. Bock, A. Artigas, N. Vanthuyne, Y. Carissan, D. Hagebaum-Reignier, Y. Coquerel and F. Durola, Angew. Chem., Int. Ed., 2023, 62, e202304058 CrossRef CAS PubMed.
- A. Artigas, F. Rigoulet, M. Giorgi, D. Hagebaum-Reignier, Y. Carissan and Y. Coquerel, J. Am. Chem. Soc., 2023, 145, 15084–15087 CrossRef CAS PubMed.
- Y. F. Wu, S. W. Ying, S. Di Liao, L. Zhang, J. J. Du, B. W. Chen, H. R. Tian, F. F. Xie, H. Xu, S. L. Deng, Q. Zhang, S. Y. Xie and L. S. Zheng, Angew. Chem., Int. Ed., 2022, 61, e202204334 CrossRef CAS PubMed.
- M. Akiyama and K. Nozaki, Angew. Chem., Int. Ed., 2017, 56, 2040–2044 CrossRef CAS PubMed.
- M. Schnitzlein, K. Shoyama and F. Würthner, Chem. Sci., 2024, 15, 2984–2989 Search PubMed.
- F. Saal, F. Zhang, M. Holzapfel, M. Stolte, E. Michail, M. Moos, A. Schmiedel, A. M. Krause, C. Lambert, F. Würthner and P. Ravat, J. Am. Chem. Soc., 2020, 142, 21298–21303 CrossRef CAS PubMed.
- C. Dusold, D. I. Sharapa, F. Hampel and A. Hirsch, Chem.–Eur. J., 2021, 27, 2332–2341 CrossRef CAS PubMed.
- S. S. Warthegau, A. E. Hillers-Bendtsen, S. K. Pedersen, C. Rindom, C. Bræstrup, J. S. Jensen, O. Hammerich, M. S. Thomsen, F. S. Kamounah, P. Norman, K. V. Mikkelsen, T. Brock-Nannestad and M. Pittelkow, Chem.–Eur. J., 2023, 29, 1–6 CrossRef PubMed.
- D. Tan, J. Dong, T. Ma, Q. Feng, S. Wang and D. T. Yang, Angew. Chem., Int. Ed., 2023, 62, e202304711 CrossRef CAS PubMed.
- T. Katayama, S. Nakatsuka, H. Hirai, N. Yasuda, J. Kumar, T. Kawai and T. Hatakeyama, J. Am. Chem. Soc., 2016, 138, 5210–5213 CrossRef CAS PubMed.
- A. Nowak-Król, P. T. Geppert and K. R. Naveen, Chem. Sci., 2024, 15, 7408–7440 RSC.
- Y. Appiarius, S. Míguez-Lago, P. Puylaert, N. Wolf, S. Kumar, M. Molkenthin, D. Miguel, T. Neudecker, M. Juríček, A. G. Campaña and A. Staubitz, Chem. Sci., 2023, 15, 466–476 RSC.
- K. Yuan, D. Volland, S. Kirschner, M. Uzelac, G. S. Nichol, A. Nowak-Król and M. J. Ingleson, Chem. Sci., 2022, 13, 1136–1145 Search PubMed.
- J. Full, S. P. Panchal, J. Götz, A. M. Krause and A. Nowak-Król, Angew. Chem., Int. Ed., 2021, 60, 4350–4357 Search PubMed.
- F. Full, Q. Wölflick, K. Radacki, H. Braunschweig and A. Nowak-Król, Chem.–Eur. J., 2022, 28, e202202280 CrossRef CAS PubMed.
- F. Full, M. J. Wildervanck, D. Volland and A. Nowak-Król, Synlett, 2023, 34, 477–482 Search PubMed.
- D. Volland, J. Niedens, P. T. Geppert, M. J. Wildervanck, F. Full and A. Nowak-Król, Angew. Chem., Int. Ed., 2023, 62, e202304291 CrossRef CAS PubMed.
- F. Full, A. Artigas, K. Wiegand, D. Volland, K. Szkodzinska, Y. Coquerel and A. Nowak-Król, J. Am. Chem. Soc., 2024, 146, 29245–29254 CrossRef CAS PubMed.
- A. Janiga, E. Glodkowska-Mrowka, T. Stoklosa and D. T. Gryko, Asian J. Org. Chem., 2013, 2, 411–415 CrossRef CAS.
- M. Krzeszewski, D. Gryko and D. T. Gryko, Acc. Chem. Res., 2017, 50, 2334–2345 CrossRef CAS PubMed.
- G. Sanil, B. Koszarna, Y. M. Poronik, O. Vakuliuk, B. Szymański, D. Kusy and D. T. Gryko, in Advances in Heterocyclic Chemistry, Academic Press Inc., 2022, vol. 138, pp. 335–409 Search PubMed.
- M. Tasior, M. Chotkowski and D. T. Gryko, Org. Lett., 2015, 17, 6106–6109 CrossRef CAS PubMed.
- N. Ishida, T. Moriya, T. Goya and M. Murakami, J. Org. Chem., 2010, 75, 8709–8712 CrossRef CAS PubMed.
- D. L. Crossley, I. A. Cade, E. R. Clark, A. Escande, M. J. Humphries, S. M. King, I. Vitorica-Yrezabal, M. J. Ingleson and M. L. Turner, Chem. Sci., 2015, 6, 5144–5151 RSC.
- G. Longhi, E. Castiglioni, J. Koshoubu, G. Mazzeo and S. Abbate, Chirality, 2016, 28, 696–707 CrossRef CAS PubMed.
- J. L. Greenfield, J. Wade, J. R. Brandt, X. Shi, T. J. Penfold and M. J. Fuchter, Chem. Sci., 2021, 12, 8589–8602 RSC.
- L. Arrico, L. Di Bari and F. Zinna, Chem.–Eur. J., 2021, 27, 2920–2934 CrossRef CAS PubMed.
- G. Pescitelli and T. Bruhn, Chirality, 2016, 28, 466–474 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta Jr, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision A.03), Gaussian Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic protocols, characterization data; 1H, 13C{1H} NMR and HRMS spectra, computational details and analyses, photophysical measurement results, CD and CPL spectra, chiral HPLC separation data, optical rotation values, crystal structures and additional figures as cited in the main text. CCDC 2415573 and 2430266. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00540j |
| This journal is © The Royal Society of Chemistry 2025 |