
Square-planar imido complexes of cobalt: synthesis, reactivity and computational study†
Jackson A.
Reyna
a,
V. Mahesh
Krishnan‡
 a,
Roberto
Silva Villatoro
a,
Hadi D.
Arman
a,
Sebastian A.
Stoian
*b and
Zachary J.
Tonzetich
*a
a,
Roberto
Silva Villatoro
a,
Hadi D.
Arman
a,
Sebastian A.
Stoian
*b and
Zachary J.
Tonzetich
*a
aDepartment of Chemistry, University of Texas at San Antonio (UTSA), San Antonio, TX 78249, USA. E-mail: zachary.tonzetich@utsa.edu
bDepartment of Chemistry, University of Idaho, Moscow, ID 83844, USA. E-mail: sstoian@uidaho.edu
First published on 9th July 2024
Abstract
Treatment of [Co(N2)(tBuPNP)] (tBuPNP = anion of 2,5-bis(di-tert-butylphosphinomethyl)pyrrole) with one equivalent of an aryl azide generates the four-coordinate imido complexes [Co(NAr)(tBuPNP)] (Ar = mesityl, phenyl, or 4-tBu-phenyl). X-ray crystallographic analysis of the compounds shows an unusual square-planar geometry about cobalt with nearly linear imido units. In the presence of the hydrogen atom donor, TEMPOH, [Co(NPh)(tBuPNP)] undergoes addition of the H atom to the imido nitrogen to generate the corresponding amido complex, [Co(NHPh)(tBuPNP)], whose structure and composition were verified by independent synthesis. Despite the observation of H atom transfer reactivity with TEMPOH, the imido complexes do not show catalytic activity for C–H amination or aziridination for several substrates examined. In the case of [Co(NPh)(tBuPNP)], addition of excess azide produced the tetrazido complex, [Co(N4Ph2)(tBuPNP)], whose bond metrics were most consistent with an anionic Ph2N4 ligand. Density Functional Theory (DFT) investigations of the imido and tetrazido species suggest that they adopt a ground state best described as possessing a low-spin cobalt(II) ion ferromagnetically coupled to an iminyl radical.
Introduction
Transition metal-catalyzed nitrene addition/insertion is among the most desirable methods for construction of carbon–nitrogen bonds due to its atom economy and compatibility with late stage functionalization.1 Several catalyst systems have proven successful in this regard demonstrating the ability to aminate C–H bonds with high efficiency and selectivity.2–4 Azides are among the molecules most frequently employed as the source of nitrene units in these reactions as their conversion to C–N bonds results in molecular nitrogen as the only by-product. The reaction of organic azides with transition metal centres proceeds via oxidative group transfer of the “NR” fragment to generate metal-imido species.5 The electronic structures of these compounds are often key to their ability to activate C–H bonds. Accordingly, early-metal imido complexes, which feature metals in high formal oxidation states and closed-shell NR2− ligands, are usually eschewed in favour of later metal systems that may feature more electronic flexibility in the M-NR unit.6–12In the area of first-row transition metal chemistry, several compounds of the mid-to-late transition elements have been reported to support well-defined metal imido species while also demonstrating catalytic efficacy in amination protocols.13 Cobalt, in particular, has proven promising as a focal point for further catalyst design with several systems displaying both efficiency and selectivity for C–H amination.14–19 In this vein, recent work with a cobalt PNP pincer system by Chirik and coworkers disclosed one of the first examples of an isolable imido complex possessing a square-planar geometry (Scheme 1).20 The compound was prepared by treatment of a cobalt(I) dinitrogen complex with mesityl azide. As noted by the authors, such geometries are rare for species containing metal–ligand multiple bonds. Moreover, the ground state for the imido was found to be best described as a low-spin Co(II) centre ferromagnetically coupled to an iminyl radical giving an overall ground state of S = 1. Despite this unusual electronic structure, the imido proved unreactive in typical nitrene transfer protocols and instead was found to decompose by migration of the nitrene unit to one of the phosphine ligands. The resulting cobalt(I) species was computed to be stabilized by ca. 9 kcal mol−1 by resonance contributions from the non-innocent PNP ligand (Scheme 1).
 | ||
| Scheme 1 Pincer-supported Co imido complexes. | ||
Encouraged by the precedent of cobalt compounds in nitrene chemistry21–32 and the demonstrated ability of pincer ligands to support novel metal imido species,33–38 we turned our focus to the chemistry of the Co(I) compound featuring the pyrrole-based pincer ligand (Scheme 1).39,40 We have already established the propensity for this species to carry out two-electron oxidative addition reactions with concomitant bond activation. Accordingly, we hypothesized that the corresponding reactions with azides would generate similar imido species to those reported by Chirik, but with a ligand that would not be as prone to formation of the P–N coupling product due to the lack of possible resonance contributors with the pyrrole-based pincer ligand and its greater structural rigidity. We describe our findings herein.
Results and discussion
Synthesis
We have demonstrated previously that the Co(I) species, [Co(N2)(RPNP)] (R = Cy, tBu), is capable of H–H, C–H, C–X, and Si–H bond activation under mild conditions.41–43 These reactions occur via formal oxidative addition to generate Co(III) species. We therefore reasoned that the reducing potential of the Co(I) complex would be similarly effective for activation of organic azides. Treatment of the tert-butyl analogue, 1, with mesityl azide was found to proceed rapidly at room temperature with the evolution of nitrogen to produce a new paramagnetic species formulated as the formally cobalt(III) imido species, 2a (Scheme 2), as determined by NMR spectroscopy. The solution magnetic susceptibility measurements of the compound in benzene provided a magnetic moment of 2.6(2)μB. This value is low but most consistent with a triplet ground state (S = 1). Identical reactions of phenyl azide and 4-tert-butylphenyl azide generated comparable spectra although alkyl azides such as benzyl azide led to intractable mixtures. The bulkier alkyl azide 1-AdN3 failed to react with 1 under the conditions employed. Compound 2a was found to precipitate readily from cold pentane to afford dark olive-green crystals. Slow degradation was observed in solution at room temperature with more rapid decomposition evident at 80 °C in benzene-d6. | ||
| Scheme 2 Azide reactivity of [Co(N2)(tBuPNP)]. | ||
The solid-state structure of 2a is displayed in Fig. 1. The compound displays a square-planar coordination geometry about cobalt much like that found for the related PNP pincer system studied by Chirik and coworkers.20 The cobalt imido distance of 1.728 Å is nearly 0.2 Å shorter than the Co–Npyrrole contact indicating a degree of cobalt–nitrogen multiple bonding consistent with the imido formulation. In addition, the Co(1)–N(2)–C(23) angle is very close to linear at 178.2°. Within the mesityl ring, the C–C distances are not equivalent, with the Cipso–Cortho distances measuring ca. 0.05 Å longer than the remaining bonds. Similar elongation has been observed previously for transition metal imido species containing radical delocalisation into the pi system.44–47 The structure of the phenyl analogue, 2b, is similar to that of 2a with a slightly more bent Co–N–C angle of 155.2° (see the ESI†). The deviation from linearity is likely made possible by the reduced steric congestion encountered by the smaller phenylimido ligand. Cobalt arylimido complexes are known to display a range of Co–N–C angles, with that in 2b falling at the lower end of structurally characterized examples.21 Nonetheless, this angle is substantially more linear than that found for the amido analogue (vide infra).
 | ||
| Fig. 1 Thermal ellipsoid drawing of the solid-state structure of 2a. Hydrogen atoms omitted for clarity. Selected bond distances (Å) and angles (°): Co(1)–N(2) = 1.7278(14); Co(1)–N(1) = 1.8922(14); Co(1)–P(1) = 2.2549(4); Co(1)–P(2) = 2.2521(4) N(2)–C(23) = 1.334(2); Co(1)–N(2)–C(23) = 178.25(13); P(1)–Co(1)–P(2) = 161.654(19); N(1)–Co(1)–N(2) = 169.90(7). | ||
Cyclic voltammetry of 2a in THF demonstrated a quasi-reversible oxidation centred at −0.60 V (vs. Fc+/0) and an irreversible reduction event at −2.38 V (see the ESI†). Attempted chemical oxidation of 2a with ferrocenium salts generated a deep red material that appeared silent in NMR spectroscopy; however, attempts to isolate the material for further characterization were unsuccessful.
During the synthesis of 2b, prolonged reaction with excess phenylazide was found to produce a new compound as studied by NMR spectroscopy. Crystallization of the species and analysis by X-ray diffraction demonstrated the formation of a tetrazido complex (3, Scheme 2), congruent with observations from other metal imido systems.15,44,48 Compound 3 features a distorted square-pyramidal geometry in the solid state with one of the tetrazido nitrogen atoms occupying the axial position (Fig. 2). The 1H NMR spectrum of 3 contains a single broad resonance for the tert-butyl groups indicating a fluxional geometry in solution with the N4 moiety likely rocking back and forth about the PNPCo plane. The bond distances within the N4 metallacycle vary between 1.35 Å for N(2)–N(3) and N(4)–N(5), and 1.29 Å for N(3)–N(4). This shortening of the central N–N contact is most consistent with a ‘closed-shell dianionic’ description of the tetrazido moiety(N42−),49 although calculations of 3 suggest a multi-determinantal ground state (vide infra). Similar tetrazido formation was not observed for 2a, probably because of the enhanced steric bulk of the mesityl substituents. In addition, a species analogous to 3 was not observed by Chirik for the pyridine-based PNP system containing a mesitylimido moiety.20
 | ||
| Fig. 2 Thermal ellipsoid drawing of the solid-state structure of 3. Hydrogen atoms omitted for clarity. Selected bond distances (Å) and angles (°): Co(1)–N(2) = 1.8772(18); Co(1)–N(5) = 1.9650(19); Co(1)–N(1) = 1.9090(18); Co(1)–Pavg = 2.3904(6); N(2)–N(3) = 1.354(3); N(3)–N(4) = 1.288(3); N(4)–N(5) = 1.359(2); P(1)–Co(1)–P(2) = 146.23(2); N(1)–Co(1)–N(2) = 175.74(8); N(1)–Co(1)–N(5) = 97.45(7); N(2)–Co(1)–N(5) = 78.30(8). | ||
Hydrogen atom abstraction by metal imido species is commonly invoked as an initial step in the mechanism for catalytic C–H amination.50 To provide a benchmark for spectroscopic features of such species we next pursued the synthesis of the Co(II) amido species, 4 (Scheme 3). Addition of LiNAr salts to [CoCl(tBuPNP)] afforded the desired amido compounds in moderate isolated yields.
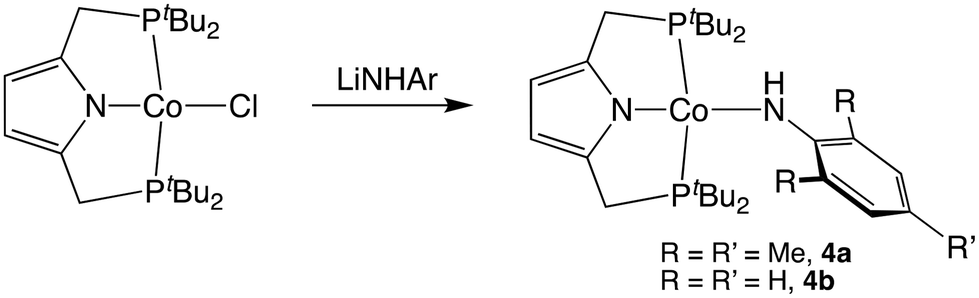 | ||
| Scheme 3 Synthesis of Co(II) amido species. | ||
Compounds 4a and 4b precipitate readily from pentane to afford deep purple crystals. The EPR spectrum of 4b displays a pseudo-axial signal with hyperfine coupling to 57Co. These features are broadly similar to those of other low-spin S = ½ cobalt(II) compounds of PNP pincers containing alkyl and halogen ligands (Fig. 3).51 Simulation of the spectrum produced anisotropic g values of 3.288, 1.994, and 1.853, further consistent with a low-spin square-planar Co(II) system displaying substantial spin–orbit coupling.51–53 In line with this spectroscopic data, the effective magnetic moment of 4a in solution was determined to be 2.2(2)μB, greater than the predicted spin-only value for an S = ½ system.
 | ||
Fig. 3 X-band EPR spectrum recorded at 77 K for a 2-MeTHF solution of 4b (red) recorded at a microwave frequency of ν = 9.44869 GHz and a microwave power of 2 mW. The theoretical spectrum (blue) was obtained using a model consisting of an S = ½ coupled to a I =  59Co nucleus (100% abundance). The parameters used to derive this simulation are gx = 3.288, gy = 1.994, gz = 1.853, σ(gx) = 0.077, σ(gy) = 0.046, σ(gz) = 0.071, Ax = 489 MHz, Ay = 5 MHz, Az = 29 MHz, σ(Ax) = 120 MHz, σ(Ay) = 51 MHz, σ(Az) = 2 MHz and a linewidth of 57 G. Although the simulation was not able to reproduce the small ridges centered around g ∼ 2.2, their peak-to-peak splitting suggests that they originate from a hyperfine splitting of ca. 210 MHz. 59Co nucleus (100% abundance). The parameters used to derive this simulation are gx = 3.288, gy = 1.994, gz = 1.853, σ(gx) = 0.077, σ(gy) = 0.046, σ(gz) = 0.071, Ax = 489 MHz, Ay = 5 MHz, Az = 29 MHz, σ(Ax) = 120 MHz, σ(Ay) = 51 MHz, σ(Az) = 2 MHz and a linewidth of 57 G. Although the simulation was not able to reproduce the small ridges centered around g ∼ 2.2, their peak-to-peak splitting suggests that they originate from a hyperfine splitting of ca. 210 MHz. | ||
The solid-state structure of 4a is depicted in Fig. 4. Most notably, the bond metrics about the cobalt-amido unit show a substantial change from those observed for the cobalt-imido unit of compound 2a. The Co(1)–N(2) bond distance of 4a is over 0.1 Å longer than that in 2a at 1.875 Å. Similarly, the Co(1)–N(2)–C(23) bond angle of 4a is significantly more bent at 135.45°. In addition to the metrics about Co, the C–C intra-ring distances of the mesityl unit and the N(2)–C(23) distance demonstrate less deviation from normality than seen in 2a consistent with less radical delocalization into the arene pi system. The remaining metrics about the CoPNP unit are comparable to those of 2a with the exception of the Co(1)–P(2) distance, which is slightly elongated with respect to the Co(1)–P(1) distance. This distortion most likely takes place to accommodate the bulk of the amido substituent, as it is also observed in the structure of 4b (see the ESI†).
 | ||
| Fig. 4 Thermal ellipsoid drawing of the solid-state structure of 4a. Hydrogen atoms except that attached to N(2) omitted for clarity. Selected bond distances (Å) and angles (°): Co(1)–N(2) = 1.8750(15); Co(1)–N(1) = 1.8877(14); Co(1)–P(1) = 2.2626(5); Co(1)–P(2) = 2.3198(5); N(2)–C(23) = 1.395(2); Co(1)–N(2)–C(23) = 135.44(13); P(1)–Co(1)–P(2) = 158.022(19); N(1)–Co(1)–N(2) = 172.54(6). | ||
Reactivity
With the imido compounds in hand, we turned to an investigation of their reactivity relevant to C–H amination processes. We began by examining the propensity for hydrogen atom abstraction by 2a. Reaction of 2a with H atom donors, 1,4-cyclohexadiene or TEMPOH (2,2,6,6-tetramethylpiperidin-1-ol) in benzene-d6 resulted in no apparent reactivity as judged by 1H NMR spectroscopy. Resonances for 2a were still apparent after prolonged exposure to the H atom donors and extended reaction time and/or elevated temperatures led to decomposition. Reasoning that the steric bulk of 2a was shutting down potential HAT reactivity, we next examined compound 2b. Reaction of 2b with 1,4-cyclohexadiene resulted in the detection of small quantities of aniline and 1 by NMR spectroscopy, although it remains uncertain if direct hydrogen atom transfer (HAT) to the imido nitrogen atom of 2b occurred as no evidence for the formation of 4b was observed.54 In contrast, treatment of 2b with TEMPOH led to the immediate formation of 4b as judged by NMR spectroscopy (Scheme 4). | ||
| Scheme 4 HAT reactivity of 2b with TEMPOH. | ||
In addition to stoichiometric experiments with 2a and 2b, we also examined the potential catalytic activity of 1 with several typical amination substrates. Reactions of 1 with a 5–10 fold excess of styrene or toluene in the presence of phenyl and benzyl azides failed to demonstrate any reactivity consistent with nitrene transfer. In all cases complete consumption of 1 was observed, but no products indicative of C–N bond formation were apparent. We also considered intramolecular nitrene transfer employing the substrate (3-azidobutyl)benzene. As with the intermolecular experiments, treatment of 1 with an excess of the azide led to the immediate consumption of reagents but failed to produce the expected phenylpyrrolidine product as judged by NMR spectroscopy. These results demonstrate that imido formation is facile with the azide substrate, but that subsequent C–H abstraction and amination are not occurring, likely due to the decomposition or orthogonal reactivity of the intermediate alkylimido such as tetrazido formation.
Computational studies
The electronic structures of the imido, amido and tetrazido complexes were investigated by performing a series of DFT and ab initio Complete Active Space Self Consistent Field (CASSCF) calculations on structural models derived from the crystal structures. DFT calculations employed pure BP86 and hybrid B3LYP and TPSSH functionals in conjunction with the Karlsruhe triple-zeta def2-TZVPP basis set.55 | ||
| Fig. 5 Spin density plots of 2a obtained for the F, S = 1 states (left) and the BS, S = 0 (right) obtained at the BP86/def2-TZVPP (left) level of theory. These plots were obtained using an isosurface value of ±0.008. | ||
The energy spacing of the F and BS states suggests a spin coupling constant of J ∼ −2400 cm−1, obtained using the Hamiltonian, Ĥ = J·ŜCo·Ŝimido and the J = (εF − εBS)/2SCoSimido relationship. Just as expected, the large J value is indicative of a direct exchange interaction as opposed to superexchange interactions encountered when the spin coupling is mediated by a diamagnetic bridge.
In the ferromagnetically coupled ground state of 2a the unpaired spin density of the imido ligand is, in most cases, nearly equally distributed between the nitrogen atom and the aromatic core (Fig. 5). The delocalization of the unpaired electron over the latter moiety is consistent with the elongated C–C bonds of the aromatic ring, and likely accounts for the added stability of the arylimido compounds vis-a-vis alkylimidos. The unpaired spin density localized specifically on the nitrogen atom is accommodated by the 2pz and 2py orbitals, which are orthogonal to the Co–N bond. The pz fraction is typically twice as large as py (see the ESI†). This difference may be traced to the fact that only py interacts with the π system of the benzene core. Thus, the two unpaired electrons of both the F and BS states occupy two π*-type MO orbitals: πy* ∼ 3dxy(Co)–[py(Nimido) + p(C6)]; πz* ∼ 3dxz(Co)–pz(Nimido) (see ESI Fig. S12†). Importantly, this difference leads to unequal contributions of the 3d cobalt orbitals to the (πy*, πz*) MOs and, in turn, is responsible for the stabilization of a ferromagnetic-type ground state.
Orbital occupation in the ground state of 2a is best described as having single occupation of the 3dxz orbital with double occupancy of 3dz2, 3dxy and 3dyz (see the ESI†). The metal-based 3dx2−y2 orbital also appears to accommodate one electron; however the population of this orbital is spin unpolarized and is traced to the σ-bonding interaction between the empty metal orbital and doubly occupied ligand orbitals. Therefore, the metal site is best described as a low-spin Co(II) ion. The increased π-bonding interaction is also reflected in a strong Co–Nimido bond. Thus, the bond order calculated for this bond is nearly 1.5 times larger than the other metal–ligand bonds.
The CAS(8,12) calculations of 2a also predict that there is a strong π-bonding interaction between the cobalt ion and the imido ligand (see ESI, Fig. S13†). Unlike DFT, however, the CAS(8,12) calculations forecast that the two unpaired electrons are localized primarily on the metal ion such that, according to the Löwdin population analysis, the cobalt site has a spin density of +1.68 and the imido ligand of +0.22. In addition, these calculations suggest that the S = 1 ground state exhibits an exceptionally large zero field splitting (ZFS), D ∼ 92 cm−1 with a large contribution from the singlet states of ca. 51 cm−1. This D value is similar to that observed experimentally for a S = 1, square-planar Co(III) complex supported by perfluoropinacolates,56 and could potentially explain our inability to detect an EPR signal for this species even when using a microwave frequency greater than 600 GHz (hν > 20 cm−1).
![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) ) has an energy of ca. 10
) has an energy of ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 above the ground state. Moreover, inspection of the orbital populations, indicates that the unpaired electron is accommodated in a 3dxz orbital with double occupation of 3dz2, 3dxy and 3dyz akin to that calculated for the low-spin Co(II) centre in 2a. Therefore, the electronic configuration of the low-spin cobalt(II) ion of the amido complex is quite similar to that of the imido except that the π-bonding interactions between the metal site and the amido ligand are not as important.
000 cm−1 above the ground state. Moreover, inspection of the orbital populations, indicates that the unpaired electron is accommodated in a 3dxz orbital with double occupation of 3dz2, 3dxy and 3dyz akin to that calculated for the low-spin Co(II) centre in 2a. Therefore, the electronic configuration of the low-spin cobalt(II) ion of the amido complex is quite similar to that of the imido except that the π-bonding interactions between the metal site and the amido ligand are not as important.
 | ||
| Fig. 6 Spin density plots of 3 obtained for the F, S = 1 states (left) and the BS, S = 0 (right) obtained at the BP86/def2-TZVPP (left) level of theory. The density plots were generated using an isosurface value of ±0.008. | ||
Conclusions
In conclusion, the Co(I) compound [Co(N2)(tBuPNP)] has been found to serve as a precursor to aryl imido species of the type [Co(NAr)(tBuPNP)] through treatment with organic azides. The imido species adopt rare square-planar geometries with bond distances indicative of radical delocalization into the aryl ring. Hydrogen atom transfer was observed in the case of the phenyl imido compound, but only with very active H atom donors. With the smaller phenylamido ligand, addition of two or more equivalents of azide was found to result in the formation of a tetrazido complex. Computational studies on the cobalt imido compounds indicate that the electronic structure is best regarded as arising from low-spin Co(II) centres ferromagnetically coupled to an iminyl radical. The strong ligand field engendered by the PNP ligand likely results in the low-spin configuration for cobalt and may explain the differential reactivity in comparison to other well-defined Co(III) imido complexes.Experimental
General
Manipulations of air- and moisture-sensitive materials were performed under an atmosphere of purified nitrogen gas using a Vacuum Atmospheres glovebox. Tetrahydrofuran, diethyl ether, pentane, and toluene were purified by sparging with argon and passage through two columns packed with 4 Å molecular sieves and/or activated alumina (THF). Benzene-d6 and toluene-d8 were dried over sodium and vacuum-distilled prior to use. NMR spectra were recorded on a Varian INOVA spectrometer operating at 300 or 500 MHz (1H) and referenced to the residual protium resonance of the solvent. UV–Vis spectra were recorded on a Cary-60 spectrophotometer in airtight Teflon-capped quartz cells. Cyclic voltammetry was performed under purified nitrogen at 23 °C on a CH Instruments 620D electrochemical workstation. A three-electrode setup was employed, comprising a glassy-carbon working electrode, platinum-wire auxiliary electrode, and an Ag/AgCl quasi-reference electrode. Triply recrystallized Bu4NPF6 was used as the supporting electrolyte. All electrochemical data were referenced internally to the ferrocene/ferrocenium couple at 0.00 V. Elemental analyses were performed by the CENTC facility at the University of Rochester.Materials
Compound 1 and [CoCl(tBuPNP)] were synthesized according to literature procedures or slight modifications thereof.57 Aryl azides were prepared following published procedures with strict adherence to recommended safety precautions.58 LiNHPh and LiNHMes were prepared by deprotonation of the corresponding anilines with n-butyllithium in pentane. All other materials were purchased from commercial sources and used as received.Crystallography
Crystals suitable for X-ray diffraction were mounted, using Paratone oil, onto a nylon loop. Data were collected at 98(2) K using a Rigaku AFC12/Saturn 724 CCD fitted with Mo Kα radiation (λ = 0.71075 Å) or at 100.0(1) K using a Rigaku XtaLAB Synergy/Dualflex, HyPix fitted with Cu Kα radiation (λ = 1.54184 Å). Data collection and unit cell refinement were performed using the CrysAlisPro software.59 Data processing and absorption correction were ac-complished with CrysAlisPro and SCALE3 ABSPACK,60 respectively. The structure, using Olex2,61 was solved with the ShelXT structure solution program using direct methods and refined (on F2) with the ShelXL refinement package using full-matrix, least-squares techniques.62,63 All nonhydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atom positions were determined by geometry and refined by a riding model. Crystallographic data and refinement parameters for each structure can be found in the ESI.†Computational studies
The electronic structures of 2a, 3 and 4a,b were investigated using Density Functional Theory (DFT) and ab initio Complete Active Space (CAS) Self Consistent Field (SCF) methods as implemented in the ORCA 5 quantum-chemical software package.64,65 All calculations used the def2-TZVPP5,66 basis set and the required auxiliary basis set were included using the autoaux keyword. In addition, resolution of identity (RI) approximation67 and chain of spheres (COSX) numerical integration were employed for the HF exchange to speed up the SCF calculations.68 All SCF calculations used the default tight convergence criteria. In all the cases, the structural models were oriented such that the z axis was set perpendicular to the approximate CoP2 plane, the y axis aligned with the Co–P bonds and the x axis with the co-ligand, as shown in ESI Scheme S1.† Additional computational details appear in the ESI.†Synthesis
General procedure for NMR scale reactions
An NMR tube was charged with 10 mg (0.02 mmol) of the desired cobalt complex and 500 μL of C6D6. The H atom donor was then added (1 eq. 0.02 mmol) as a solution in 200 μL of C6D6. The reaction mixture was transferred to an NMR tube and spectra were recorded via an automated time course every 3 hours over 24 hours.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
Crystallographic data for compounds 2a, 2b, 3, 4a, and 4b have been deposited at the CCDC under 2334970–2334974.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Welch Foundation (AX-1772) for financial support of this work. NMR and X-ray facilities at UTSA were accessed with support from the National Science Foundation (CHE-1625963 and CHE-1920057). Acknowledgement is made to the donors of the American Chemical Society Petroleum Research Fund for support of this research through a DNI grant to SAS (62278-DNI3).References
- Y. Park, Y. Kim and S. Chang, Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- J. L. Roizen, M. E. Harvey and J. Du Bois, Metal-Catalyzed, Nitrogen-Atom Transfer Methods for the Oxidation of Aliphatic C-H Bonds, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed.
- J. M. Alderson, J. R. Corbin and J. M. Schomaker, Tunable, Chemo- and Site-Selective Nitrene Transfer Reactions through the Rational Design of Silver(I) Catalysts, Acc. Chem. Res., 2017, 50, 2147–2158 CrossRef CAS PubMed.
- H. J. Lu and X. P. Zhang, Catalytic C-H functionalization by metalloporphyrins: recent developments and future directions, Chem. Soc. Rev., 2011, 40, 1899–1909 RSC.
- C. T. Saouma and J. C. Peters, ME and M=E complexes of iron and cobalt that emphasize three-fold symmetry (EO, N, NR), Coord. Chem. Rev., 2011, 255, 920–937 CrossRef CAS PubMed.
- V. Lyaskovskyy, A. I. O. Suarez, H. J. Lu, H. L. Jiang, X. P. Zhang and B. de Bruin, Mechanism of Cobalt(II) Porphyrin-Catalyzed C-H Amination with Organic Azides: Radical Nature and H-Atom Abstraction Ability of the Key Cobalt(III)-Nitrene Intermediates, J. Am. Chem. Soc., 2011, 133, 12264–12273 CrossRef CAS PubMed.
- Y. Baek, E. T. Hennessy and T. A. Betley, Direct Manipulation of Metal Imido Geometry: Key Principles to Enhance C-H Amination Efficacy, J. Am. Chem. Soc., 2019, 141, 16944–16953 CrossRef CAS PubMed.
- J. F. Berry, Terminal Nitrido and Imido Complexes of the Late Transition Metals, Comments Inorg. Chem., 2009, 30, 28–66 CrossRef CAS.
- T. Xiong and Q. Zhang, New amination strategies based on nitrogen-centered radical chemistry, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC.
- A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van der Vlugt and B. de Bruin, Complexes with Nitrogen-Centered Radical Ligands: Classification, Spectroscopic Features, Reactivity, and Catalytic Applications, Angew. Chem., Int. Ed., 2013, 52, 12510–12529 CrossRef CAS PubMed.
- A. Kalra, V. Bagchi, P. Paraskevopoulou, P. Das, L. Ai, Y. Sanakis, G. Raptopoulos, S. Mohapatra, A. Choudhury, Z. C. Sun, T. R. Cundari and P. Stavropoulos, Is the Electrophilicity of the Metal Nitrene the Sole Predictor of Metal-Mediated Nitrene Transfer to Olefins? Secondary Contributing Factors as Revealed by a Library of High-Spin Co(II) Reagents, Organometallics, 2021, 40, 1974–1996 CrossRef CAS PubMed.
- R. E. Cowley, N. A. Eckert, S. Vaddadi, T. M. Figg, T. R. Cundari and P. L. Holland, Selectivity and Mechanism of Hydrogen Atom Transfer by an Isolable Imidoiron(III) Complex, J. Am. Chem. Soc., 2011, 133, 9796–9811 CrossRef CAS PubMed.
- A. Grünwald, S. S. Anjana and D. Munz, Terminal Imido Complexes of the Groups 9–11: Electronic Structure and Developments in the Last Decade, Eur. J. Inorg. Chem., 2021, 2021, 4147–4166 CrossRef.
- Y. Baek, A. Das, S. L. Zheng, J. H. Reibenspies, D. C. Powers and T. A. Betley, C-H Amination Mediated by Cobalt Organoazide Adducts and the Corresponding Cobalt Nitrenoid Intermediates, J. Am. Chem. Soc., 2020, 142, 11232–11243 CrossRef CAS PubMed.
- Y. Baek and T. A. Betley, Catalytic C-H Amination Mediated by Dipyrrin Cobalt Imidos, J. Am. Chem. Soc., 2019, 141, 7797–7806 CrossRef CAS PubMed.
- Y. Hu, K. Lang, J. R. Tao, M. K. Marshall, Q. G. Cheng, X. Cui, L. Wojtas and X. P. Zhang, Next-Generation -Symmetric Chiral Porphyrins for Cobalt(II)-Based Metalloradical Catalysis: Catalyst Engineering by Distal Bridging, Angew. Chem., Int. Ed., 2019, 58, 2670–2674 CrossRef CAS PubMed.
- H. J. Lu, V. Subbarayan, J. R. Tao and X. P. Zhang, Cobalt(II)-Catalyzed Intermolecular Benzylic C-H Amination with 2,2,2-Trichloroethoxycarbonyl Azide (TrocN), Organometallics, 2010, 29, 389–393 CrossRef CAS.
- V. Bagchi, A. Kalra, P. Das, P. Paraskevopoulou, S. Gorla, L. Ai, Q. W. Wang, S. Mohapatra, A. Choudhury, Z. C. Sun, T. R. Cundari and P. Stavropoulos, Comparative Nitrene-Transfer Chemistry to Olefinic Substrates Mediated by a Library of Anionic Mn(II) Triphenylamido-Amine Reagents and M(II) Congeners (M = Fe, Co, Ni) Favoring Aromatic over Aliphatic Alkenes, ACS Catal., 2018, 8, 9183–9206 CrossRef CAS.
- L. Capdevila, M. Montilla, O. Planas, A. Brotons, P. Salvador, V. Martin-Diaconescu, T. Parella, J. M. Luis and X. Ribas, C-H Amination Reactions Mediated by Metastable Pseudo-Masked Aryl-Co-nitrene Species, Inorg. Chem., 2022, 61, 14075–14085 CrossRef CAS PubMed.
- Y. Park, S. P. Semproni, H. Y. Zhong and P. J. Chirik, Synthesis, Electronic Structure, and, Reactivity of a Planar Four-Coordinate, Cobalt-Imido Complex, Angew. Chem., Int. Ed., 2021, 60, 14376–14380 CrossRef CAS PubMed.
- W. Q. Mao, Z. H. Zhang, D. Fehn, S. A. V. Jannuzzi, F. W. Heinemann, A. Scheurer, M. van Gastel, S. DeBeer, D. Munz and K. Meyer, Synthesis and Reactivity of a Cobalt-Supported Singlet Nitrene, J. Am. Chem. Soc., 2023, 145, 13650–13662 CrossRef CAS PubMed.
- J. Z. Du, L. B. Wang, M. H. Xie and L. Deng, A Two-Coordinate Cobalt(II) Imido Complex with NHC Ligation: Synthesis, Structure, and Reactivity, Angew. Chem., Int. Ed., 2015, 54, 12640–12644 CrossRef CAS PubMed.
- L. Zhang, Y. S. Liu and L. Deng, Three-Coordinate Cobalt(IV) and Cobalt(V) Imido Complexes with N-Heterocyclic Carbene Ligation: Synthesis, Structure, and Their Distinct Reactivity in C-H Bond Amination, J. Am. Chem. Soc., 2014, 136, 15525–15528 CrossRef CAS PubMed.
- X. L. Hu and K. Meyer, Terminal cobalt(III) imido complexes supported by tris(carbene) ligands: Imido insertion into the cobalt-carbene bond, J. Am. Chem. Soc., 2004, 126, 16322–16323 CrossRef CAS PubMed.
- X. L. Dai, P. Kapoor and T. H. Warren, [MeNN]Co(η-toluene):: O=O, N= N, and O=N bond cleavage provides, β-diketiminato cobalt μ-oxo and imido complexes, J. Am. Chem. Soc., 2004, 126, 4798–4799 CrossRef CAS PubMed.
- E. R. King, G. T. Sazama and T. A. Betley, Co(III) Imidos Exhibiting Spin Crossover and C-H Bond Activation, J. Am. Chem. Soc., 2012, 134, 17858–17861 CrossRef CAS PubMed.
- D. T. Shay, G. P. A. Yap, L. N. Zakharov, A. L. Rheingold and K. H. Theopold, Intramolecular C-H activation by an open-shell cobalt(III) imido complex, Angew. Chem., Int. Ed., 2005, 44, 1508–1510 CrossRef CAS PubMed.
- D. M. Jenkins, T. A. Betley and J. C. Peters, Oxidative group transfer to Co(I) affords a terminal Co(III) imido complex, J. Am. Chem. Soc., 2002, 124, 11238–11239 CrossRef CAS PubMed.
- W. Q. Mao, D. Fehn, F. W. Heinemann, A. Scheurer, M. van Gastel, S. A. V. Jannuzzi, S. DeBeer, D. Munz and K. Meyer, Umpolung in a Pair of Cobalt(III) Terminal Imido/Imidyl Complexes, Angew. Chem., Int. Ed., 2022, 61, e202206848 CrossRef CAS PubMed.
- W. Q. Mao, D. Fehn, F. W. Heinemann, A. Scheurer, D. Munz and K. Meyer, A Pair of Cobalt(III/IV) Terminal Imido Complexes, Angew. Chem., Int. Ed., 2021, 60, 16480–16486 CrossRef CAS PubMed.
- Y. Liu, J. Z. Du and L. Deng, Synthesis, Structure, and Reactivity of Low-Spin Cobalt(II) Imido Complexes [(Me3P)3Co(NAr)], Inorg. Chem., 2017, 56, 8278–8286 CrossRef CAS PubMed.
- B. Wu, R. H. Sánchez, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Formation of Heterobimetallic Zirconium/Cobalt Diimido Complexes via a Four-Electron Transformation, Inorg. Chem., 2014, 53, 10021–10023 CrossRef CAS PubMed.
- T. Schmidt-Räntsch, H. Verplancke, J. N. Lienert, S. Demeshko, M. Otte, G. P. Van Trieste, K. A. Reid, J. H. Reibenspies, D. C. Powers, M. C. Holthausen and S. Schneider, Nitrogen Atom Transfer Catalysis by Metallonitrene C-H Insertion: Photocatalytic Amidation of Aldehydes, Angew. Chem., Int. Ed., 2022, 61, e202115626 CrossRef PubMed.
- L. N. Grant, M. E. Carroll, P. J. Carroll and D. J. Mindiola, An Unusual Cobalt Azide Adduct That Produces a Nitrene Species for Carbon-Hydrogen Insertion Chemistry, Inorg. Chem., 2016, 55, 7997–8002 CrossRef CAS PubMed.
- M. J. Ingleson, M. Pink, H. Fan and K. G. Caulton, Redox chemistry of the triplet complex (PNP)Co, J. Am. Chem. Soc., 2008, 130, 4262–4276 CrossRef CAS PubMed.
- V. Vreeken, L. Baij, B. de Bruin, M. A. Siegler and J. I. van der Vlugt, N-Atom transfer thermal or photolytic activation of a Co-azido complex with a PNP pincer ligand, Dalton Trans., 2017, 7145–7149 RSC.
- W. A. Chomitz and J. Arnold, Reactivity of a Co(I) [N2P2] complex with azides:: evidence for a transient Co(III) imido species, Chem. Commun., 2008, 3648–3650 RSC.
- J. So, S. Kim, K. B. Cho and Y. Lee, Metal-ligand cooperative transformation of alkyl azide to isocyanate occurring at a Co-Si moiety, Chem. Commun., 2021, 57, 3219–3222 RSC.
- C. V. Thompson and Z. J. Tonzetich, Pincer ligands incorporating pyrrolyl units: Versatile platforms for organometallic chemistry and catalysis, Adv. Organomet. Chem., 2020, 74, 153–240 CrossRef CAS.
- L. S. Merz, J. Ballmann and L. H. Gade, Phosphines and -Heterocycles Joining Forces: an Emerging Structural Motif in PNP-Pincer Chemistry, Eur. J. Inorg. Chem., 2020, 2020, 2023–2042 CrossRef CAS.
- H. Alawisi, K. F. Al-Afyouni, H. D. Arman and Z. J. Tonzetich, Aldehyde Decarbonylation by a Cobalt(I) Pincer Complex, Organometallics, 2018, 37, 4128–4135 CrossRef CAS.
- H. Alawisi, H. D. Arman and Z. J. Tonzetich, Catalytic Hydrogenation of Alkenes and Alkynes by a Cobalt Pincer Complex: Evidence of Roles for Both Co(I) and Co(II), Organometallics, 2021, 40, 1062–1070 CrossRef CAS.
- A. L. Amaya, H. Alawisi, H. D. Arman and Z. J. Tonzetich, Well-Defined Cobalt-Silyl Complexes and Their Role in Catalytic Carbonyl Hydrosilylation, Organometallics, 2023, 42, 2902–2909 CrossRef CAS.
- Y. Baek and T. A. Betley, Reversible C-C Bond Cleavage of a Cobalt Diketimide into an Elusive Cobalt Aryl Nitrenoid Complex, Angew. Chem., Int. Ed., 2022, 61, e202115437 CrossRef CAS PubMed.
- G. C. Bai and D. W. Stephan, Formation of C-C and C-N bonds in Ni ketimide complexes via transient Ni aryl imides, Angew. Chem., Int. Ed., 2007, 46, 1856–1859 CrossRef CAS PubMed.
- K. M. Carsch, I. M. DiMucci, D. A. Iovan, A. Li, S. L. Zheng, C. J. Titus, S. J. Lee, K. D. Irwin, D. Nordlund, K. M. Lancaster and T. A. Betley, Synthesis of a copper-supported triplet nitrene complex pertinent to copper-catalyzed amination, Science, 2019, 365, 1138–1143 CrossRef CAS PubMed.
- A. Bakhoda, Q. Jiang, J. A. Bertke, T. R. Cundari and T. H. Warren, Elusive Terminal Copper Arylnitrene Intermediates, Angew. Chem., Int. Ed., 2017, 56, 6426–6430 CrossRef CAS PubMed.
- D. S. Moore and S. D. Robinson, Catenated Nitrogen Ligands .1. Transition-Metal Derivatives of Triazenes, Tetrazenes, Tetrazadienes, and Pentazadienes, Adv. Inorg. Chem. Radiochem., 1986, 30, 1–68 CAS.
- A. C. Bowman, A. M. Tondreau, E. Lobkovsky, G. W. Margulieux and P. J. Chirik, Synthesis and Electronic Structure Diversity of Pyridine(diimine)iron Tetrazene Complexes, Inorg. Chem., 2018, 57, 9634–9643 CrossRef CAS PubMed.
- A. Reckziegel, C. Pietzonka, F. Kraus and C. G. Werncke, C-H Bond Activation by an Imido Cobalt(III) and the Resulting Amido Cobalt(II) Complex, Angew. Chem., Int. Ed., 2020, 59, 8527–8531 CrossRef CAS PubMed.
- S. P. Semproni, C. Milsmann and P. J. Chirik, Four-Coordinate Cobalt Pincer Complexes: Electronic Structure Studies and Ligand Modification by Homolytic and Heterolytic Pathways, J. Am. Chem. Soc., 2014, 136, 9211–9224 CrossRef CAS PubMed.
- A. H. Maki, A. Davison, N. Edelstein and R. H. Holm, Electron Paramagnetic Resonance Studies of Electronic Structures of Bis(Maleonitriledithiolato)Copper)2) -Nickel)3) -Cobalt)2) + -Rhodium)2) Complexes, J. Am. Chem. Soc., 1964, 86, 4580–4587 CrossRef CAS.
- Y. Nishida, A. Sumita and S. Kida, ESR and Optical Spectra of Low-spin Square Planar Cobalt(II) Complexes with Some Quadridentate Schiff Bases of the N2S2 Type, Bull. Chem. Soc. Jpn., 1977, 50, 759–760 CrossRef CAS.
- Y. Park, S. Kim, L. Tian, H. Y. Zhong, G. D. Scholes and P. J. Chirik, Visible light enables catalytic formation of weak chemical bonds with molecular hydrogen, Nat. Chem., 2021, 13, 969–976 CrossRef CAS PubMed.
- J. J. Zheng, X. F. Xu and D. G. Truhlar, Minimally augmented Karlsruhe basis sets, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- J. L. Steele, L. Tahsini, C. Sun, J. K. Elinburg, C. M. Kotyk, J. McNeely, S. A. Stoian, A. Dragulescu-Andrasi, A. Ozarowski, M. Ozerov, J. Krzystek, J. Telser, J. W. Bacon, J. A. Golen, A. L. Rheingold and L. H. Doerrer, Square-planar Co(III) in {O4} coordination: large ZFS and reactivity with ROS, Chem. Commun., 2018, 54, 12045–12048 RSC.
- V. M. Krishnan, H. D. Arman and Z. J. Tonzetich, Preparation and reactivity of a square-planar PNP cobalt(II)-hydrido complex: isolation of the first {Co-NO}-hydride, Dalton Trans., 2018, 1435–1441 RSC.
- S. W. Kwok, J. R. Fotsing, R. J. Fraser, V. O. Rodionov and V. V. Fokin, Transition-Metal-Free Catalytic Synthesis of 1,5-Diaryl-1,2,3-triazoles, Org. Lett., 2010, 12, 4217–4219 CrossRef CAS PubMed.
- CrysAlisPro, Rigaku Oxford Diffraction: Rigaku Corporation, The Woodlands, TX, 2015 Search PubMed.
- SCALE3 ABSPACK -An Oxford Diffraction program, Oxford Diffraction Ltd., 2005 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- F. Neese, Software update: The ORCA program system-Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- G. L. Stoychev, A. A. Auer and F. Neese, Automatic Generation of Auxiliary Basis Sets, J. Chem. Theory Comput., 2017, 13, 554–562 CrossRef CAS PubMed.
- F. Neese, An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix, J. Comput. Chem., 2003, 24, 1740–1747 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra; the cyclic voltammogram of 2a; the EPR spectrum of 4a; additional computational details, figures, and tables. CCDC 2334970–2334974. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01483a |
| ‡ Present address: Eastman Chemical Company, 200 S. Wilcox Dr., Kingsport, TN 37660. |
| This journal is © The Royal Society of Chemistry 2024 |