
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA family of Cd(II) coordination polymers constructed from 6-aminopicolinate and bipyridyl co-linkers: study of their growth in paper and photoluminescence sensing of Fe3+ and Zn2+ ions†
Oier
Pajuelo-Corral
 *a,
Inmaculada
Ortiz-Gómez
bc,
Jose Angel
García
d,
Antonio
Rodríguez-Diéguez
e,
Iñigo J.
Vitórica-Yrezábal
e,
Alfonso
Salinas-Castillo
*bc,
Jose M.
Seco
f and
Javier
Cepeda
*f
*a,
Inmaculada
Ortiz-Gómez
bc,
Jose Angel
García
d,
Antonio
Rodríguez-Diéguez
e,
Iñigo J.
Vitórica-Yrezábal
e,
Alfonso
Salinas-Castillo
*bc,
Jose M.
Seco
f and
Javier
Cepeda
*f
aPOLYMAT, University of the Basque Country UPV/EHU, Joxe Mari Korta Center, 20018 Donostia-San Sebastián, Spain. E-mail: oier.pajuelo@ehu.eus
bECsens, Department of Analytical Chemistry, Faculty of Sciences, University of Granada, 18071 Granada, Spain. E-mail: alfonsos@ugr.es
cUnit of Excellence in Chemistry Applied to Biomedicine and the Environment, Faculty of Sciences, University of Granada, 18071 Granada, Spain
dDepartamento de Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), 48940, Leioa, Spain
eDepartment of Inorganic Chemistry, Faculty of Sciences, University of Granada, 18071 Granada, Spain
fDepartment of Applied Chemistry, Faculty of Chemistry, University of the Basque Country UPV/EHU, 20018 Donostia-San Sebastián, Spain. E-mail: javier.cepeda@ehu.eus
First published on 29th June 2024
Abstract
In this work, we report on five novel coordination polymers (CPs) based on the linkage of the [Cd(6apic)2] building block [where 6apic = 6-aminopicolinate] by different bipyridine-type organic spacers, forming different coordination compounds with the following formulae: [Cd(μ-6apic)2]n (1), {[Cd(6apic)2(μ-bipy)]·H2O}n (2), {[Cd(6apic)2(μ-bpe)]·2H2O}n (3), [Cd(6apic)(μ-6apic)(μ-bpa)0.5]n (4) and {[Cd2(6apic)4(μ-tmbp)]·7H2O}n (5) [where bipy = 4,4′-bipyridine, bpe = 1,2-di(4-pyridyl)ethylene, bpa = 1,2-di(4-pyridyl)ethane (bpa) and tmbp = 1,3-di(4-pyridyl)propane]. Most of the synthesized compounds form infinite metal–organic rods through the linkage of the building block by the bipyridine-type linker, except in the case of compound 4 whose assembly forms a densely packed 3D architecture. All compounds were fully characterized and their photoluminescence properties were studied experimentally and computationally through density functional theory (DFT) calculations. All compounds display, upon UV excitation, a similar blue emission of variable intensity depending on the linker employed for the connection of the building units, among which compound 2 deserves to be highlighted for its room temperature phosphorescence (RTP) with an emission lifetime of 32 ms that extends to 79 ms at low temperature. These good photoluminescence properties, in addition to its stability in water over a wide pH range (between 2 and 10), motivated us to study compound 2 as a sensor for the detection of metal ions in water, and it showed high sensitivity to Fe3+ through a fluorescence turn-off mechanism and an unspecific turn-on response to Zn2+. Furthermore, the compound is processed as a paper-based analytical device (PAD) in which the phosphorescence emission is preserved, improving the sensing capacity toward Fe3+ ions.
Introduction
Coordination polymers (CPs) are a fascinating class of crystalline solid materials built from the assembly of metal ions or clusters with organic molecules to construct architectures spreading along one, two or three dimensions.1 The attractive aspect of these materials is the versatility and flexibility of their tailorable metal–organic frameworks which can be rationally designed through the appropriate selection of building blocks, which guide the material's structural growth toward the desired applications.2–4 In this sense, the storage of gas or small molecules represents the most exploited application in porous coordination polymers, commonly referred to as metal–organic frameworks (MOFs), thanks to their large surface areas and highly crystalline networks that lead to a well-defined pore size.5,6 Nonetheless, the versatility of CPs makes them relevant for other applications such as catalysis,7–9 magnetism,10,11 electronics12 or photoluminescence.13,14 In particular, luminescent CPs have gained significant attention in the field of materials science in recent years because of their remarkable ability to emit bright and intense light upon excitation, making them valuable for use as solid-state photodevices.15 Besides, some luminescent CPs may exhibit afterglow emissions that are visible to the naked eye after removal of the excitation source, endowing the materials with a long-lasting phosphorescence (LLP) behavior. To achieve persistent luminescence emissions in CPs, it is crucial to not only promote the formation of stable triplet excited states but also minimize non-radiative relaxation pathways responsible for emission quenching. Taking into account that triplet excitation must proceed through intersystem crossing (ISC) from excited single states, which in turn depends on different phenomena such as the El-sayed rule,16 hyperfine coupling17 and the heavy atom effect,18 the selection of suitable building blocks is key to enhance ISC. On the one hand, the organic part should be rigid to prevent vibrational quenching, while also being decorated with functional groups for the establishment of supramolecular interactions within the framework to trap, stabilize and protect the triplet excitons.19 On the other hand, the selection of the metal ion is not of less importance. In this sense, rare-earth ions and Ir(III), Pt(II) and Ru(III) have been traditionally employed for achieving compounds with long-lasting emissions.20–22 Nevertheless, their high costs and limited resources hinder their further development as solid-state lightning devices. In the quest for alternative candidates, closed-shell ions such as Zn(II) or Cd(II) have become popular in recent years because they lack intraionic d–d transitions that detrimentally affect luminescence emission and also enhance the ligand-based emission owing to their coordination in crystalline architectures.23 Nonetheless, the use of these ions in phosphorescent CPs is still in its infancy given that most of the compounds reported so far only show afterglows at low temperatures, and those that exhibit room temperature phosphorescence (RTP) are very limited.24Furthermore, the change of the emissive properties triggered by an external stimulus, caused by either a physical factor or interaction with other species, represents a desirable property in the design of sensors. For instance, the variation of the material's emission properties with temperature has been exploited for the development of luminescent thermometers.25,26 As mentioned, the luminescence variability in CPs toward external changes is not limited to temperature and can be extended to other environmental factors such as the presence of certain molecules, giving the opportunity to develop chemical sensors for the detection of volatile organic compounds (VOCs),27,28 biomolecules,29,30 biomarkers and analytes associated with diseases,31,32 antibiotics,33,34 explosives,35,36 and metal ions37,38 among many other toxic species.39–43 In particular, the detection of toxic metal ions in aqueous solutions by photoluminescent (PL) MOFs and CPs has been largely documented, among which sensing of Fe3+ holds significant importance given that its deficiency or excess in most of the biological systems causes serious functional disorders.44 Additionally, there has been a growing concern over the detection of other less toxic ions in drinking water because the imbalance of some metal ions such as Zn2+, traditionally considered an antioxidant in living systems, has been shown to cause oxidative stress or become toxic in cellular environments.45 As far as we are aware, the vast majority of Zn2+ sensors reported so far are based on organic molecules,46–48 whereas only a few examples of CPs can be found in the literature for the detection of this ion,49,50 thus making the study of novel PL sensors of Zn2+ a priority in this field. Despite the proven high sensitivity and low limits of detection (LOD) shown by luminescent CPs, their application in real analysis is still challenging given their poor mechanical properties that make these materials difficult to handle.51 A potential solution for addressing this problem is to deposit the crystalline solids onto versatile and easy-to-handle substrates such as silica,52 glass53 or polymeric matrices.54 These substrates serve as a support for the CP, generating advanced materials that combine the outstanding sensing capabilities of CPs with the mechanical properties of the substrate. In this sense, cellulose paper can suitably act as the supporting material due to its high availability, ease of manipulation and chemical modification, cheapness, biocompatibility and biodegradability, and versatile surface sustainability, making it an ideal support for fabricating easy-to-use devices.55 Additionally, cellulose substrates can be shaped at will to be integrated into devices, yielding so-called paper-based analytical devices (PADs).
As part of our ongoing research line focused on novel luminescent CPs, we report on a new family of compounds based on an octahedral building unit formed by the assembly of Cd(II) and 6-aminopicolinic acid (H6apic) in which the axial positions can be occupied by a variety of bipyridine-like spacers that modulate the dimensionality and metrics of the metal–organic network (see Fig. S1 in the ESI†). In most cases, the linkage of the building block forms metal–organic rods except for the more flexible bpe ligand which allows for the formation of an efficiently packed 3D framework. The PL properties of all compounds were experimentally and computationally studied, among which compound 2, containing 4,4′-bipyridine as a spacer, displays bright blue fluorescence emission and room temperature green phosphorescence. This compound was selected for a study of its sensing capabilities of different analytes using two different methods: powder suspension in water and growth in cellulose paper to be employed as PADs.
Results and discussion
Comments on the [Cd(6apic)2] building block
All compounds reported in this work have been synthesized from the linkage of the [Cd(6apic)2] building unit that is formed when Cd(II) and 6apic are combined. The building block is formed by the coordination of two equivalent 6apic ligands through the pyridine nitrogen and the adjacent carboxylic oxygen adopting the κN:κO coordination mode. This configuration seems to be driven by the five-membered chelating ring formed, which is thermodynamically favored over the usually observed four-membered chelating ring formed by the carboxylate moiety. The N,O-chelating mode forces the 6apic ligands to occupy the equatorial plane of an octahedron, thus leaving the axial positions free for the coordination of any other co-ligand (Fig. 1), or the non-chelating oxygen carboxylate atoms of 6apic ligands as observed in compound 1. Taking advantage of the great design capacity of CPs, we rationally introduced additional bipyridine-type co-ligands to act as linkers that join [Cd(6apic)2] building blocks into extended architectures. In particular, 4,4′-bipyridine (bipy) (2), 1,2-di(4-pyridyl)ethylene (bpe) (3), 1,2-di(4-pyridyl)ethane (bpa) (4) and 1,3-di(4-pyridyl)propane (tmbp) (5) were successfully employed to that end, leading to the formation of highly-stable CPs, as shown in Tables S1–S5.† It is worth mentioning that given the rapid assembling capacity of the building block to give rise to the CP of compound 1 and the low solubility of the latter, it was not possible to approach the synthesis of compounds 2–5 by soaking the powder of 1 in a solvent in the presence of the co-linker; instead, these compounds need to be formed following a one-pot reaction process.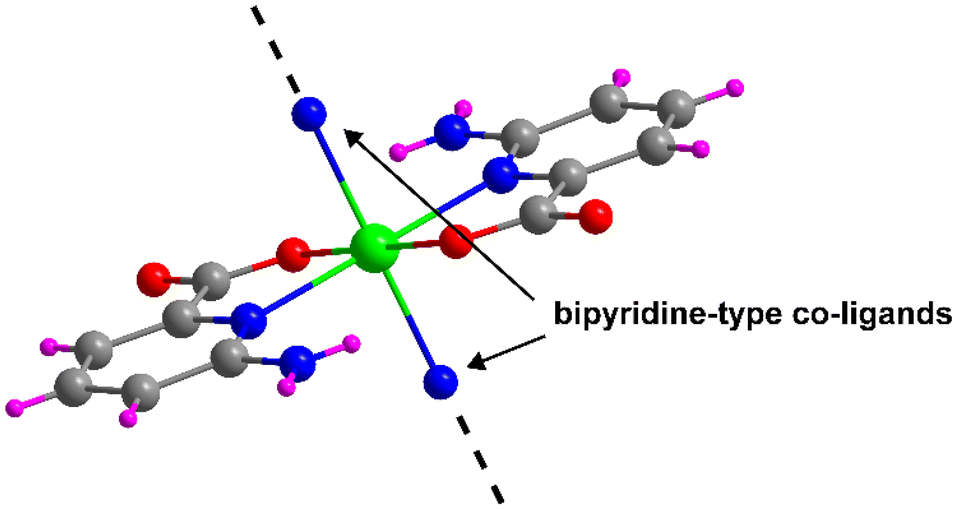 | ||
| Fig. 1 [Cd(6apic)2] building block of the 6apic system and the potential coordination sites for linking the building blocks. Color code: C = grey, H = pink, Cd = green, N = blue, and O = red. | ||
Structural description of 1
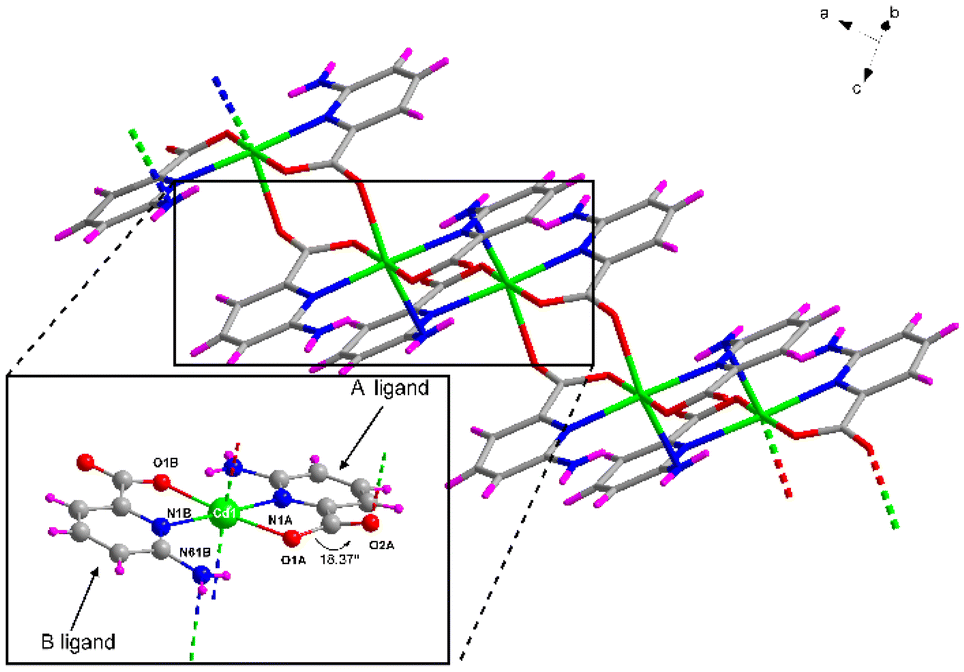
Compound 1 crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with the [Cd(μ-6apic)2]n formula in the form of metal–organic rods linked by 6apic molecules given the absence of additional linkers. Detailed crystallographic information can be found in Tables S6 and 7.† The asymmetric unit contains two spatially independent cadmium atoms (Cd1 and Cd2), each showing the previously described [Cd(6apic)2] building block where two distinct 6apic ligands (showing different coordination modes) can be distinguished, named A and B for the molecules coordinated to the Cd1 atom and C and D in the case of Cd2. In the case of A and C ligands, the 6apic ligand bridges the nodes by the second carboxylic oxygen atom adopting the μ–κ2O,N:κO′ coordination mode, whereas in B and D ligands the connection between the building units is through the amine nitrogen atom with the μ–κ2O,Npyridine:κN′amine coordination mode (Fig. 2). As a result, Cd1 and Cd2 atoms display a distorted octahedral environment formed by a N3O3 chromophore (Table S8†). The additional coordination points, apart from bridging the building blocks successively into infinite chains, promote some distortions in the ligands. In the case of A and C ligands, as a consequence of the coordination of the second carboxylic oxygen, this group is twisted 18.37° with respect to the aromatic ring plane, in contrast with the almost planar disposition observed in the case of B and D ligands. On the other hand, the coordination of the amine group in B and D ligands also places this exocyclic group out of the ring plane (Fig. 2) and establishes larger Cd⋯Namine distances than Cd⋯Npyridine (see Table S9 in the ESI† for more details). This connectivity of 6apic forms infinite metal–organic chains growing along the [100] and [010] directions for the chains containing Cd1 and Cd2, respectively (Fig. S8†). The chains growing in the same direction are further connected through hydrogen bonds between amine and carboxylate groups (Fig. S9 and Table S10†), resulting in supramolecular 2D layers stacked along the crystallographic c axis.
space group with the [Cd(μ-6apic)2]n formula in the form of metal–organic rods linked by 6apic molecules given the absence of additional linkers. Detailed crystallographic information can be found in Tables S6 and 7.† The asymmetric unit contains two spatially independent cadmium atoms (Cd1 and Cd2), each showing the previously described [Cd(6apic)2] building block where two distinct 6apic ligands (showing different coordination modes) can be distinguished, named A and B for the molecules coordinated to the Cd1 atom and C and D in the case of Cd2. In the case of A and C ligands, the 6apic ligand bridges the nodes by the second carboxylic oxygen atom adopting the μ–κ2O,N:κO′ coordination mode, whereas in B and D ligands the connection between the building units is through the amine nitrogen atom with the μ–κ2O,Npyridine:κN′amine coordination mode (Fig. 2). As a result, Cd1 and Cd2 atoms display a distorted octahedral environment formed by a N3O3 chromophore (Table S8†). The additional coordination points, apart from bridging the building blocks successively into infinite chains, promote some distortions in the ligands. In the case of A and C ligands, as a consequence of the coordination of the second carboxylic oxygen, this group is twisted 18.37° with respect to the aromatic ring plane, in contrast with the almost planar disposition observed in the case of B and D ligands. On the other hand, the coordination of the amine group in B and D ligands also places this exocyclic group out of the ring plane (Fig. 2) and establishes larger Cd⋯Namine distances than Cd⋯Npyridine (see Table S9 in the ESI† for more details). This connectivity of 6apic forms infinite metal–organic chains growing along the [100] and [010] directions for the chains containing Cd1 and Cd2, respectively (Fig. S8†). The chains growing in the same direction are further connected through hydrogen bonds between amine and carboxylate groups (Fig. S9 and Table S10†), resulting in supramolecular 2D layers stacked along the crystallographic c axis.
 | ||
| Fig. 2 Excerpt of the structure of compound 1 showing the coordination modes adopted by 6apic ligands around the metal center. Color code as in Fig. 1. | ||
Structural description of 2
Compound 2 crystallizes in the monoclinic P21/c space group through the linkage of the [Cd(6apic)2] building block by the 4,4′-bipyridine linker, resulting in the CP with the general formula {[Cd(6apic)2(μ-bipy)]·H2O}n. Crystallographic details can be found in the ESI.† The asymmetric unit contains two cadmium atoms located on inversion centers (therefore with half occupancy), two 6apic ligands, one 4,4′-bipyridine (bipy) co-linker and one lattice water molecule. Both cadmium(II) ions show an equivalent N4O2 donor set pertaining to two symmetry-related 6apic ligands placed in the equatorial plane (as described above for the [Cd(6apic)2] building block) and to two 4,4′-bipyridines in the axial positions, forming a slightly distorted octahedral geometry (Soc = 1.57) according to the continuous shape measurements (CShMs) carried out with SHAPE software.56 The distances between the metal and donor atoms range between ca. 2.26 and 2.45 Å (see Table S11† for more detailed information). As mentioned, bipy molecules bridge the building units to form metal–organic chains that separate the nodes at a distance of ca. 12.0 Å (Fig. 3). Interestingly, these rods are packed perpendicularly to each other (Fig. S10†), forming π–π stacking interactions between the aromatic rings of the bipy molecules, for which adjacent rods are twisted with a relative rotation angle of ca. 33.40° to maximize the interactions (Fig. S11†). The crystal structure is further stabilized by the intermolecular N61A–H61B⋯O2B hydrogen bond formed between the amine moiety of 6apic ligands and the uncoordinated carboxylic oxygen (Fig. S12 and Table S12†). Finally, the lattice water molecule also establishes hydrogen bonding O–H⋯O interactions with the carboxylate groups of the rods.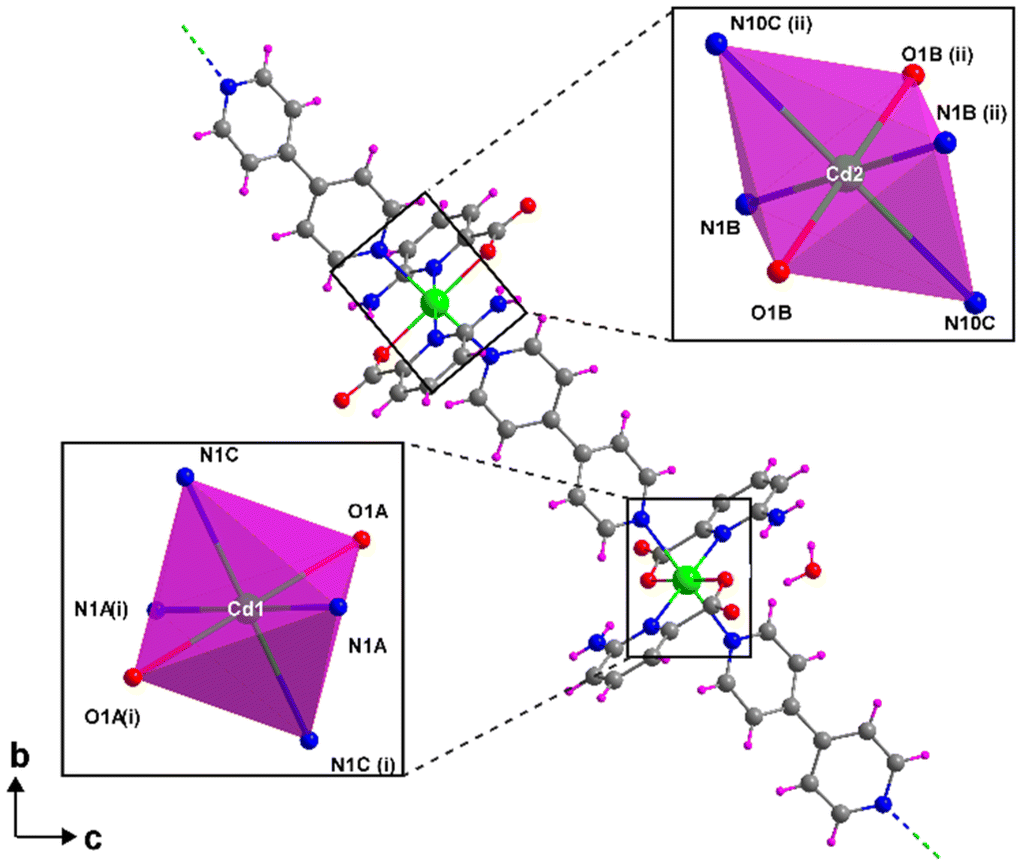 | ||
| Fig. 3 Fragment of the chain of compound 2 showing the coordination environment of cadmium ions. Color code as Fig. 1. | ||
Structural description of 3
Suitable single crystals of compound 3 reveal a similar structure to that of 2 with the formation of metal–organic chains grown by connecting with bpe instead of bipy. The asymmetric unit is composed of a cadmium atom located in an inversion center, one 6apic molecule, half of a bpe molecule and a crystallization water molecule, all of which correspond to the {[Cd(6apic)2(μ-bpe)]·2H2O}n chemical formula. The arrangement adopted around Cd(II) resembles that of 2, with a N4O2 coordination environment displaying a distorted octahedron (Soc = 1.35, see Fig. S13 and Table S14†). The presence of the additional trans C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond increases the Cd⋯Cd distance up to ca. 14.1 Å without breaking the linearity of the chain. In addition, the rigidity of the CC bond prevents the twisting of the bipy molecule observed in 2 preventing the formation of effective π–π interactions. Therefore, the supramolecular building adopts a different packing, in which the chains grow parallel to each other, in such a way that stacking interactions are established among 6apic ligands of adjacent rods, instead (Fig. S14†). The stability of the supramolecular structure is also reinforced in the present compound by the hydrogen bonding interactions between the rods and lattice water molecules intercalated between chains (Fig. S15 and Table S15†).
C bond increases the Cd⋯Cd distance up to ca. 14.1 Å without breaking the linearity of the chain. In addition, the rigidity of the CC bond prevents the twisting of the bipy molecule observed in 2 preventing the formation of effective π–π interactions. Therefore, the supramolecular building adopts a different packing, in which the chains grow parallel to each other, in such a way that stacking interactions are established among 6apic ligands of adjacent rods, instead (Fig. S14†). The stability of the supramolecular structure is also reinforced in the present compound by the hydrogen bonding interactions between the rods and lattice water molecules intercalated between chains (Fig. S15 and Table S15†).
Structural description of 4
Compound 4 shows a different crystalline structure with the formation of a very efficiently packed 3D architecture crystallizing in the monoclinic I2/c space group with the [Cd(μ-6apic)(6apic)(μ-bpa)0.5]n formula. Crystallographic details are shown in Table S7.† Probably, the more flexible sp3 carbon atoms of the ethylene groups of bpa force the 6apic ligand to acquire a different arrangement in the coordination sphere (Fig. 4). The asymmetric unit is composed of two different cadmium atoms, both occupying special positions with half occupancy, two independent 6apic ligands named A and B and half of a bpa co-ligand. Cd1 shows the typical coordination sphere observed for the rest of the described compounds, with two equivalent chelating B ligands occupying the equatorial plane and two bpa linkers in the apical positions of the octahedron (Soc = 1.39). Instead, the Cd2 atom is connected to two pairs of 6apic ligands (two chelating A ligands and two monodentate B ligands), forming an N2O4 coordination sphere in which the geometry is best described as a trigonal prism (STPR = 5.24 vs. Soc = 5.38, see Table S8† for more details). The B ligand, adopting the μ–κ2N,O:κO′ coordination mode, bridges Cd1 and Cd2 atoms, imposing a distance of 5.721 Å. The distances between the central atoms and the donors are summarized in Table S17.† This new arrangement allows the 6apic ligand to bridge multiple metal centers, leading to an intricate and efficiently packed 3D framework (Fig. S16†) induced by the presence of π–π stacking as well as hydrogen bonding interactions between the amine group and the carboxylate group of adjacent ligands (Table S18†). As a consequence of such efficient packing, there are no accessible voids for solvent molecules.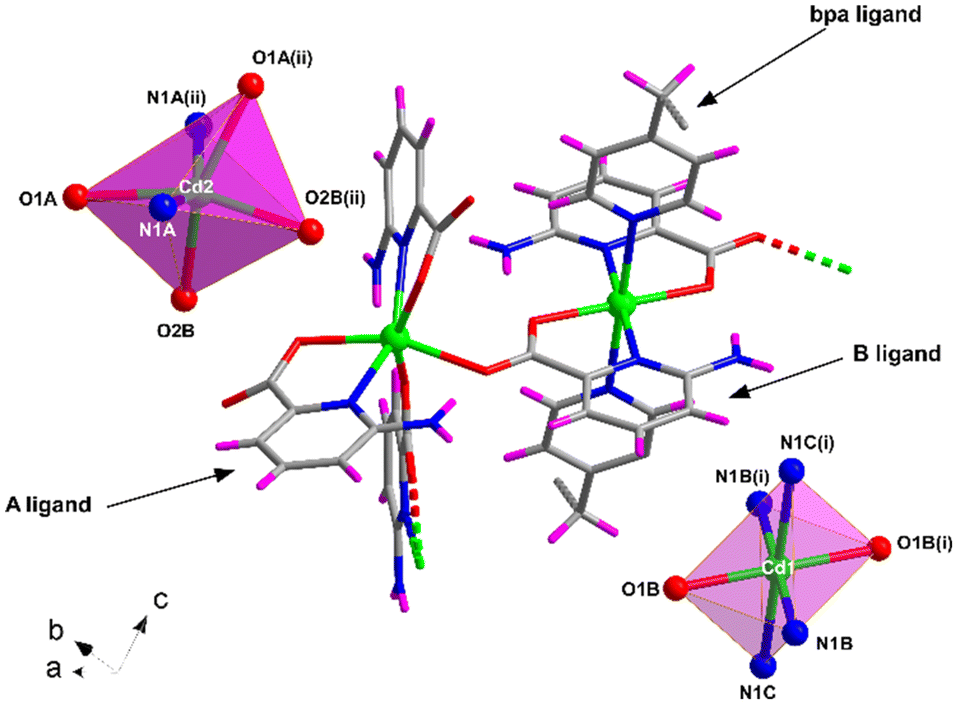 | ||
| Fig. 4 Excerpt of the structure of compound 4 together with the coordination polyhedra for Cd1 and Cd2 atoms. Color code as in Fig. 1. | ||
Structural description of 5
Compound 5, crystallizing in the monoclinic P21/c space group, represents another example of the linkage of the building block by the tmbp ligand with the general formula {[Cd2(6apic)4(μ-tmbp)2]·7H2O}n. The asymmetric unit in 5 is composed of two independent chains, each formed by a cadmium atom, two 6apic ligands, and a bridging tmbp linker, together with 7 lattice water molecules. Both metal centers show a similar coordination sphere to other 6apic-based compounds with a N4O2 donor set formed by two 6apic ligands and two tmbp linkers, acquiring a distorted octahedral geometry (Soc = 1.85 and 2.70 for Cd1 and Cd2, respectively). It is worth noting the mutual disposition of 6apic ligands, which are arranged in cis disposition instead of the previously mentioned trans disposition governing the system (Fig. 5 and S20†). Nonetheless, the different arrangement does not alter the connectivity of the building blocks, still forming one-dimensional chains. The tetrahedral nature of the inner sp3 carbon atoms in the tmbp spacer twists the aliphatic chain, forming a corrugated arrangement (with a “V” shape) that connects cadmium atoms with a distance between them of ca. 12.8 Å. The chains grow simultaneously in two intersecting directions along the crystallographic b and c axes, in such a way that π–π stacking interactions are formed among tmbp molecules as they converge at the same point but with opposite configurations (Fig. S18†). Besides, the lattice water molecules form a hydrogen-bonding network that holds the chains together (Fig. S19 and Table S21†).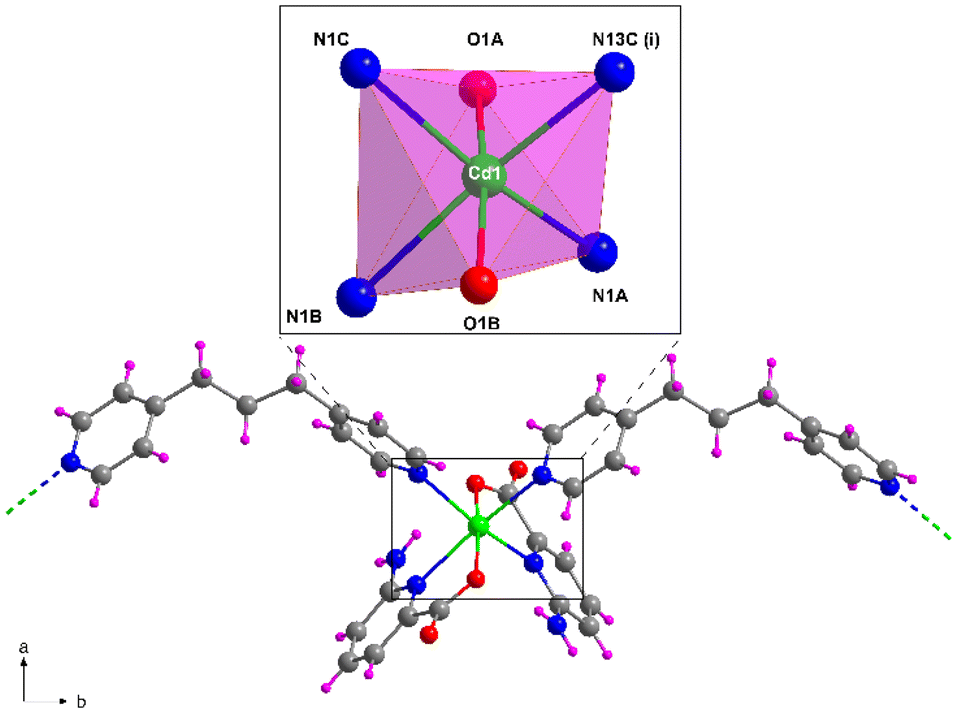 | ||
| Fig. 5 Fragment of the structure of compound 5 together with the coordination polyhedron for Cd1. Color code as Fig. 1. | ||
Photoluminescence properties
The synthesized compounds are excellent candidates to show PL properties since they consist of organic ligands based on aromatic rings decorated and combined with a closed-shell metal ion. In accordance, steady-state emission spectra were recorded at room temperature on polycrystalline samples of compounds 1–5. Upon excitation at 325 nm, all compounds exhibit similar PL emission spectra with a wide band centered at ca. 390 nm that varies only in its intensity (Fig. 6). In order to provide a proper comparison of the emission capacity of the compounds, their absolute quantum yields were measured (Table 1), showing that the emissive efficiency follows the relative order: 4 ≈ 5 ≈ 1 > 2 > 3.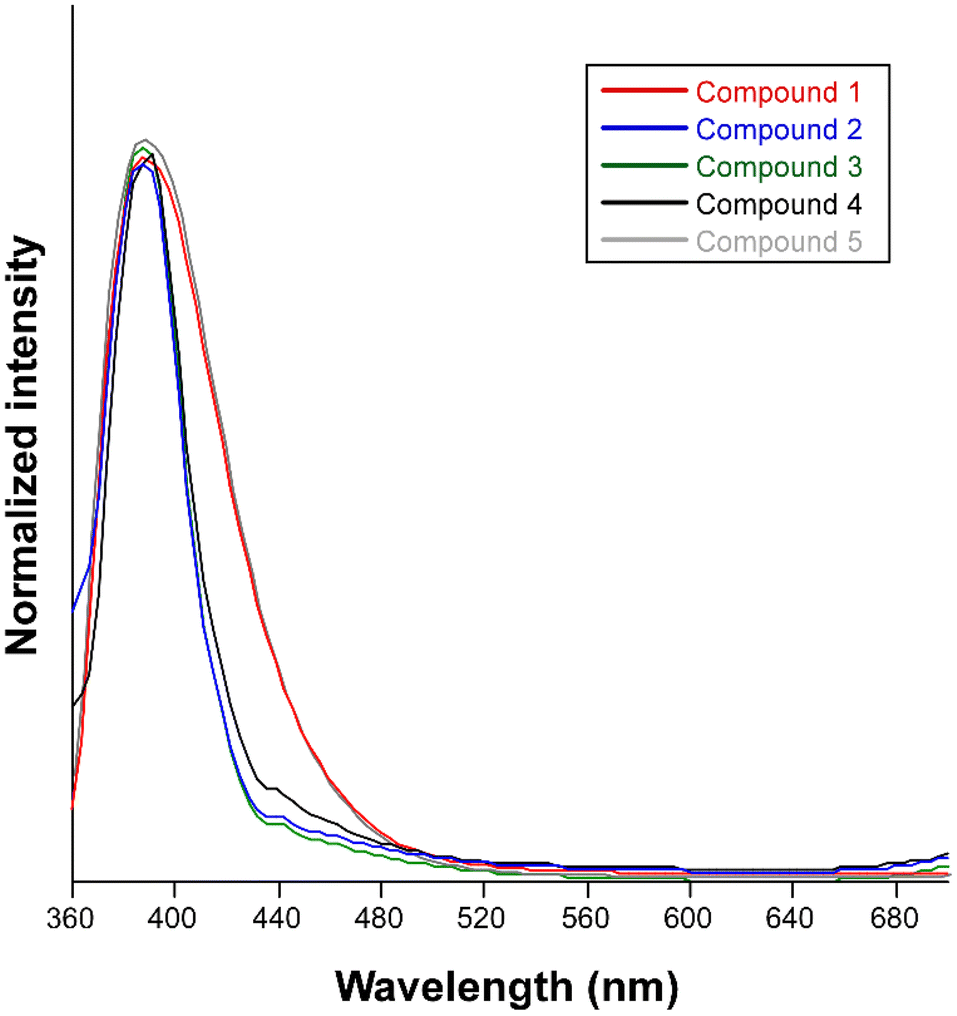 | ||
| Fig. 6 Normalized fluorescence emission spectra of compounds 1–5 at room temperature excited at 325 nm. | ||
The emission spectrum of the free H6apic ligand was also recorded under the same conditions, displaying a similar but slightly red-shifted band (peaking at 410 nm) of comparatively lower intensity (Fig. S20†). Moreover, all bipy-like co-ligands were also characterized for light blue luminescence derived from emission bands falling in the 400–450 nm range. For instance, the emission of bpe shows a main band peaking at 373 nm (Fig. S21†). These similarities between the emission of the free ligand and the compounds suggest that the emission of the compounds could mainly originate from the ligands and that the hypsochromic shift of the compounds could be due to the coordination, as expected for the closed-shell electronic configuration of Cd(II) ions, making this ion unlikely to undergo charge transfer processes.57–59 To provide a deeper insight into the mechanisms that govern the PL properties, computational calculations using time-dependent density functional theory (TD-DFT) were performed on a geometry-optimized fragment of compound 2 as a representative model of the Cd/6apic/bipy-like system in view of its better performance in sensing applications (vide infra) and the almost identical emission pattern shown by all compounds (see the Experimental section for more details about the calculations). The computed excitation spectrum aligns quite well with the absorption spectrum (diffuse reflectance collected on the solid), in which the main contribution of the two experimental overlapped wide bands covering the 220–360 nm range can be theoretically attributed to the vertical excitations peaking at 262 and 312 nm (Fig. 7).
 | ||
| Fig. 7 Photophysical data of compound 2 showing (a) the diffuse reflectance and TD-DFT calculated spectra and (b) a schematic diagram with the most significant electronic transitions and involved molecular orbitals to represent the luminescence mechanism of the compound. | ||
As interpreted from the molecular orbitals involved in these transitions, both of them are assigned to ligand-centered (LC) π–π* excitation, although the first one (from the highest to lowest energy, yielding the excited state 51, referred to as 51ES hereafter) takes place inside the bipy ligand, assigned to the HOMO−13 → LUMO transition, whereas the second (giving rise to the 12 excited state, referred to as 12ES) is attributed to a HOMO → LUMO+2 6apic-centered excitation. Taking into account that two wide bands centered at ca. 255 and 335 nm are also distinguished in the excitation spectra of the compounds (see Fig. S22†), it may be concluded that both sorts of excitations are involved in the PL mechanism of the compounds (see Table S25 and Fig. S28† for further details). It must be noticed that the observed shift of 20–30 nm between the experimental and theoretical spectra falls within the usual mismatch for luminescent MOFs and may be related to the discrete model employed for the calculation (in which supramolecular interactions are not considered) compared to the polymeric nature of the CP. The experimental emission of the compound can be well explained by radiative relaxation taking place from the optimized geometry of 12ES to the ground state (GS). In particular, two main contributions (at 326 and 346 nm) can be highlighted for the emission of the compound. On one hand, the vertical excitation at 326 nm can be described as an interligand electron transfer (ILET) occurring between the LUMO centered on the bipy ligand and the HOMO−2 represented by 6apic ligands. On the other hand, the emission at 346 nm is well described as a HOMO ← LUMO+3 electronic transition involving a ligand-to-ligand charge transfer (LLCT) in the excited state in view of the mixed nature of the LUMO+3 orbital. It is worth highlighting that the relaxation from the high-energy 51ES does not directly result in radiative emissions in the visible range as corroborated from its calculated emission (below 300 nm), so although further calculations of the 51ES-to-12ES transfer rates could not be computed, it may be assumed that such excitation energy could somehow be relaxed to the emitting 12ES. Moreover, no contribution of the metal center was observed in the whole process according to the performed calculations as expected from the closed-shell nature of Cd(II), excluding the possibility of a ligand-to-metal charge transfer. Given the high resemblance of the free bipy-like co-ligands’ emission, we can anticipate that all compounds follow the same luminescence mechanism.
Furthermore, as observed for other positional isomers such as 2-aminonicotinate and 6-aminonicotinate,60–62 the coordination of these pyridine rings decorated with carboxylate groups to closed-shell metal ions may imbue a LLP behavior in the CPs. Therefore, the delayed emission spectra of compounds 1–5 were recorded under the same conditions. These spectra (Fig. S23†), collected with a delay of 200 μs with respect to the reference pulse, show some weak maxima at ca. 390 nm assigned to the residual fluorescence emission and, in the case of 1 and 2, an additional broad band covering the 420–640 nm spectral range, with remarkably high intensity for compound 2. These latter bands correspond, a priori, to phosphorescence emissions originated from lower-lying excited triplet states, which are populated from excited singlet states (higher in energy) by the ISC process.63,64 The remarkable intensity of the phosphorescence emission shown by compound 2 prompted us to further study its longevity, for which decay curves were recorded. At room temperature and at an emission wavelength of 560 nm (λex = 370 nm), the curve fits well to the following multiexponential expression: [It = A0 + A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2)], giving a mean long-lived phosphorescence lifetime of 32 ms, a fact that confirms the presence of RTP in the compound. Such a long lifetime can be compared to other Cd(II)-based CPs that show emission lifetimes of around a hundred milliseconds, for instance Cd-TPA (τ = 158 ms, where TPA = terephthalic acid) and Cd-TMA (τ = 158 ms, where TMA = trimesic acid), which hold the current record of CP-based LLP emitters.65
exp(−t/τ1) + A2exp(−t/τ2)], giving a mean long-lived phosphorescence lifetime of 32 ms, a fact that confirms the presence of RTP in the compound. Such a long lifetime can be compared to other Cd(II)-based CPs that show emission lifetimes of around a hundred milliseconds, for instance Cd-TPA (τ = 158 ms, where TPA = terephthalic acid) and Cd-TMA (τ = 158 ms, where TMA = trimesic acid), which hold the current record of CP-based LLP emitters.65
Seeking for a deeper study of the PL properties of compound 2, additional measurements were performed at low temperature to explore a potential reduction of the vibrational quenching effects. As illustrated in Fig. S24,† under excitation with a monochromatic 325 nm laser beam, the steady-state emission spectrum of 2 is similar to that at room temperature with a slight hypsochromic shift of the maximum of 5 nm (with the main band peaking at 388 nm) and the appearance of a shoulder at 406 nm. A remarkable increase in emission intensity is also noticeable, as expected from the decrease of the molecular motions, which also makes visible a less intense tail of the emission band that extends up to 650 nm, assigned to the phosphorescence. Additional decay curves recorded at 10 K allowed us to further distinguish between the fluorescence and phosphorescence emissions of the compound. On the one hand, the main emission at λm = 406 nm (note that the shoulder was measured instead of the band maximum at 388 nm to properly discriminate the signal from the excitation source, at λex = 370 nm) affords a lifetime of 4.3 ns (Fig. S25†), which confirms the fluorescent nature of the dominant band of the steady-state spectrum. On the other hand, the decay curve collected at 560 nm affords an emission lifetime of 79 ms in good agreement with phosphorescence emission (Fig. S26†). Therefore, the steady-state emission of compound 2 is composed of overlapping bright blue fluorescence (approximately covering the 360–475 nm range, 78.4% of the overall integrated intensity) and green phosphorescence (extended over the 475–640 nm range, with the remaining 21.6% of the total emission) emission bands.
Metal ion sensing performance
Given the excellent PL properties shown by compound 2, the latter was selected to be employed as a luminescent sensor for the detection of cationic species in water. Given the acidic nature of most of the metal ions in water, the stability of the compound dispersed in water was first examined across a variable pH scale, revealing that 2 is stable in a very wide pH range (2–10) when soaked in aqueous solutions for at least 24 h (Fig. S31†). To that end, a screening test was performed by suspending compound 2 in water in the presence of different metal ions at a concentration of 0.1 mM. A first important conclusion of these experiments was that, unlike the solid state, no trace for the RTP could be observed in aqueous dispersion, meaning that the sensing capacity only responds to the fluorescence emission. As observed in Fig. 8, the blue emission band remains almost unchanged in shape and wavelength but strongly varies in its intensity depending on the metal ion present in the dispersion. In particular, at the same metal ion concentration (0.1 mM), the following order of quenching percentage (QP) is observed: Mn2+ > Ni2+ > Co2+ > Cu2+ > Cr3+ > Fe3+. Conversely, the presence of the Zn2+ ion promotes an enhancement of the emission intensity compared to the blank dispersion.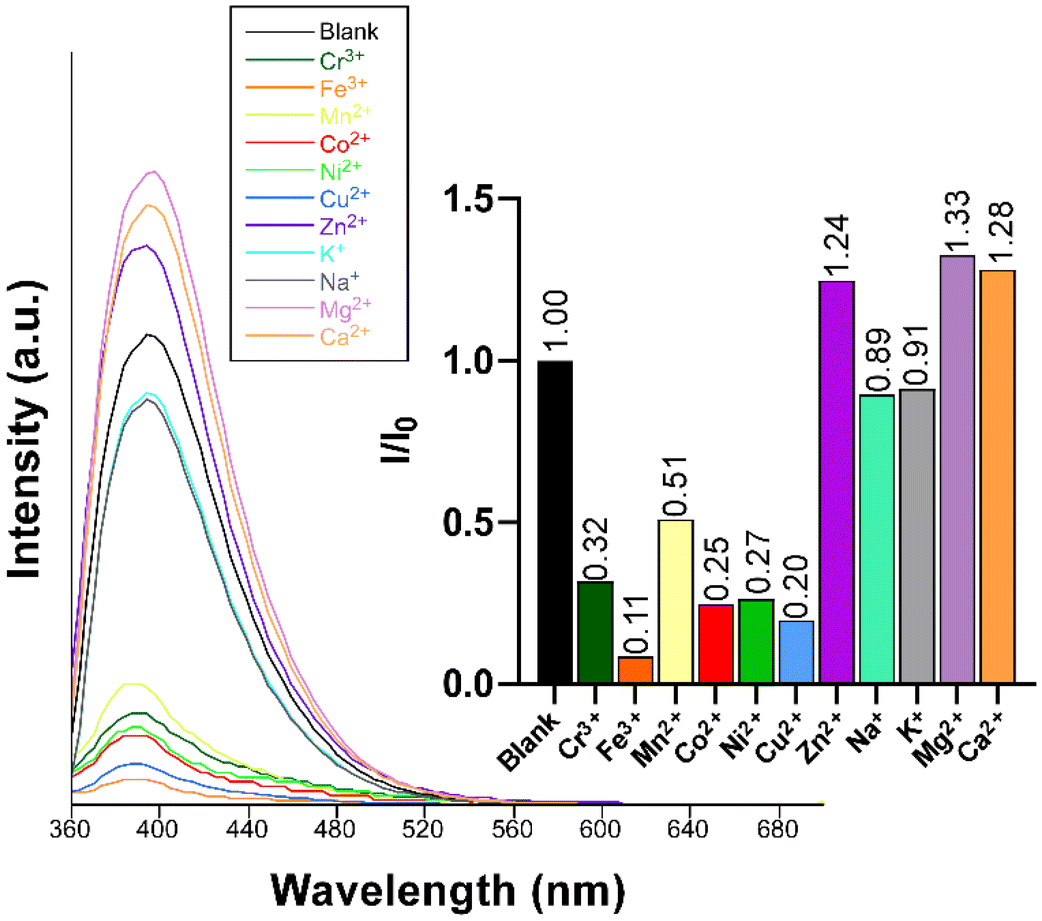 | ||
| Fig. 8 Fluorescence emission spectra of compound 2 (λex = 340 nm) in the presence of various metal ions in aqueous suspensions. The inset shows the quenching/enhancement degree expressed as I/I0. | ||
Clearly, among the ions tested, the Fe3+ ion exerts the highest quenching effect with a QP of approximately 90%. Because of the latter, this ion was selected to monitor the emission intensity of compound 2 as a function of the concentration. Fig. 9 shows a progressive decrease in the intensity of the maximum emission wavelength (λem = 393 nm) upon increasing the concentration of the Fe3+ ion (the quencher, Q) in the solution, which allowed fitting the data with the Sterm–Volmer expression (eqn (1)):
 | (1) |
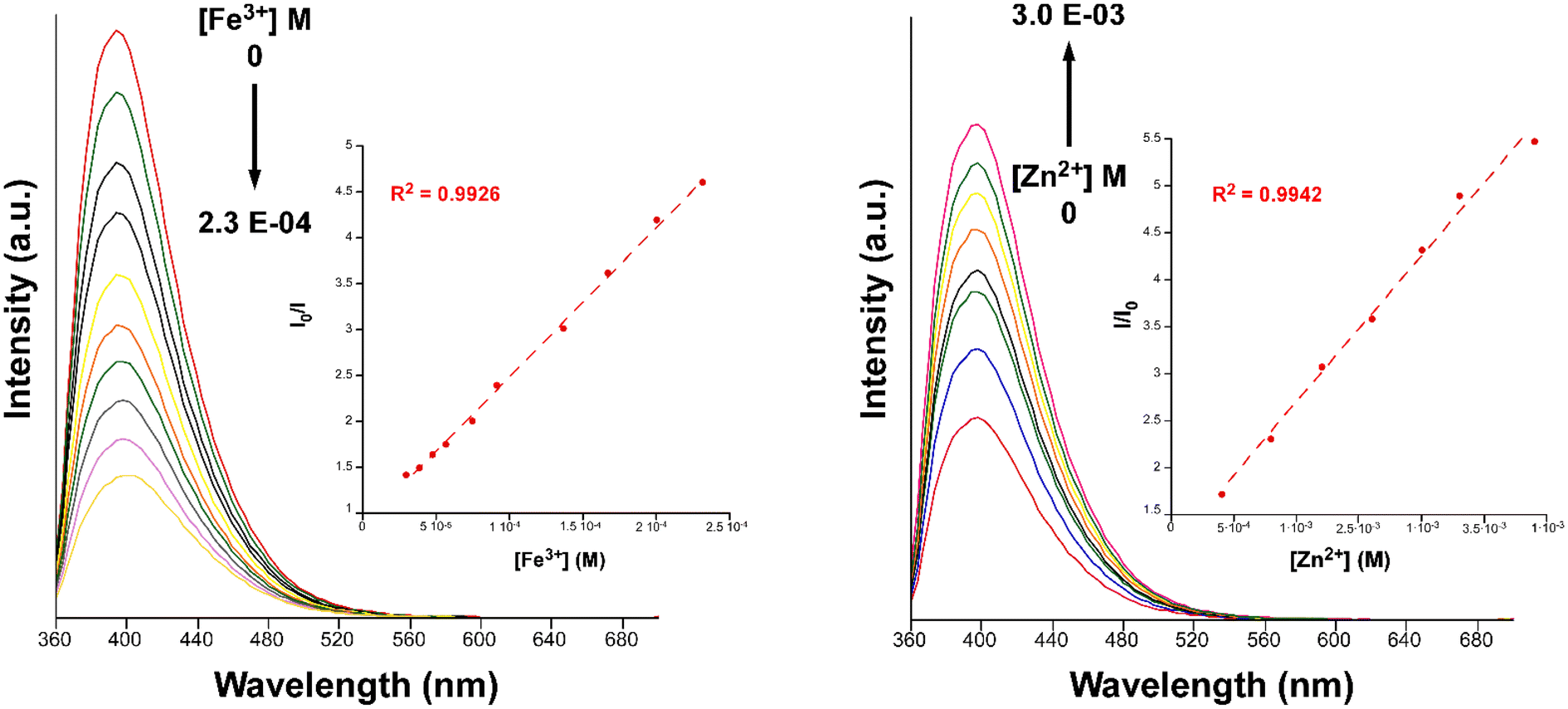 | ||
| Fig. 9 Fluorescence emission spectra of compound 2 (λem = 340 nm) in variable concentrations of Fe3+ (left) and Zn2+ (right). Insets show the Stern–Volmer plots for the titration experiment. | ||
Data were successfully fitted to a linear expression in the 1.0 × 10−5–2.3 × 10−4 M range, obtaining a KSV value of 1.56 × 104 M−1 (R2 = 0.9926) and a limit of detection (LOD) of 7.20 × 10−6 M, calculated following the IUPAC recommendation of the 3σ/slope, where the standard deviation was estimated from the measurement of ten blank samples.66 The obtained values fall in the range of other CPs for the detection of Fe3+ in aqueous media but in the top range of detectors among those performing sensing in water (see Table S28†).40,67–69
Furthermore, we also explored the sensing capacity toward Zn2+ because of its opposite behavior, turning-on response, compared to the rest of transition metal cations (all metal ions except alkaline-earth ions, vide infra). To that end, the emission intensity was studied as a function of the cation concentration. When the concentration of the metal ion was increased, the fluorescence intensity notably enhanced from 4.0 × 10−4 M to 3.0 × 10−3 M, displaying a good linear relationship in the S–V plot, affording a KSV value of 1.6 × 103 M−1 (R2 = 0.9942) and a LOD of 3.5 × 10−4 M (Fig. 9). At higher concentrations, the emission intensity barely changes (Fig. S32†), probably as a consequence of the saturation of the system. As far as we are aware, the vast majority of Zn2+ sensors reported so far are based on organic molecules,46–48 whereas only a few examples of CPs, with similar detection parameters, can be found in the literature for the detection of this ion (see Table S29†).49,50 Given the promising sensing capacity shown by 2 towards the Zn2+ ion, and in an effort to exclude the possible interference by other metal cations usually present in significant concentrations in natural water for a practical application, the PL behavior of the compound was analyzed in the presence of Na+, K+, Ca2+ and Mg2+. As observed in the inset of Fig. 8, alkaline-earth ions revealed a similar trend compared to Zn2+ and with an even higher magnitude, thus discarding the idea of a selective fluorescence detection of Zn2+ ions. These results are not fully unexpected in view of the closed-shell electronic configuration (shared with Zn2+ ions) of alkaline-earth ions, also known to enhance the PL emissions in CPs.70,71
To conclude this section, the time-dependent fluorescence changes for both analytes were evaluated (Fig. S33†). As observed from the time-dependent fluorescence intensity plots, the response time is quite short (around one minute) for both analytes. Moreover, the sensor reusability was also studied by measuring the emission spectra on solid samples collected after three successive cycles involving soaking of the material into aqueous solutions of Fe3+/Zn2+ and subsequent washing into distilled water. As shown in Fig. S34,† the emission intensity hardly varies for both Fe3+ and Zn2+ analytes and the dried sample recovers its initial PL emission after each soaking, while the crystallinity and purity of the samples were also confirmed by PXRD and X-ray fluorescence data (vide infra). Based on these measurements, it can be concluded that the material is reusable for sensing of both ions.
Sensing mechanism
With the aim of understanding the luminescence turning-off and -on responses occurring during the sensing of Fe3+ and Zn2+ ions, respectively, the mechanisms were studied in detail. The opposing trend observed in the signal response suggests that two different sensing mechanisms could be operating. In the first inspection, the collapse of the crystalline structure was initially ruled out given the fact that the diffraction pattern remains unchanged after soaking in both solutions for 24 hours (Fig. S35†).On the one hand, among the most common quenching mechanisms found for Fe3+ sensing by CPs, competitive absorption clearly dominates the scenario.29,72,73 In the present case, it was found that the excitation spectrum of compound 2 overlaps with the absorption spectrum of the analyte (Fig. S36†), corroborating the aforementioned mechanism. On the other hand, the positive emissive response towards Zn2+ is not surprising because the few reported CP-based sensors also display a similar turning-on behavior with this ion, which is presumably derived from a direct analyte–CP interaction.49,50 In order to get deeper insights into this possibility, the PL data were acquired for aqueous dispersions of 2 in the absence/presence of the analytes. As expected from the previous titration results, no significant changes were observed for the excitation and emission bands, meaning that the interaction with these ions does not substantially affect the HOMO–LUMO energy gap of the compound (Fig. S37†). However, the emission lifetime of the fluorescent band is notably affected by the presence of Fe3+ and Zn2+ ions, showing a respective decrease and increase in the average lifetime that is also accompanied by a concordant change in the absolute QY (see Table 2 and Fig. S38†), which suggests that the interaction with the ions could also involve a dynamic mechanism.
| Compound | λ ex,max | λ em,max | τ av | QY |
|---|---|---|---|---|
| a Units of the magnitudes are as follows: wavelength in nm, lifetimes in ns and quantum yields in percentage. | ||||
| 2 @H 2 O | 354 | 379 | 3.6(1) | 18.5 |
| 2 @Fe 3+ (aq) | 358 | 379 | 2.7(1) | 14.6 |
| 2 @Zn 2+ (aq) | 360 | 382 | 4.9(1) | 19.6 |
When considering the crystal structure of compound 2, it must be noted that the hydrogen-bonded framework of packed 1D chains contains Lewis basic uncoordinated carboxylate oxygen atoms exposed to the interstitial voids of the structure (Fig. S39†). Although these sites are not easily accessible in the solid state given that they are involved in strong hydrogen bonding interactions with lattice water molecules, the absence of direct hydrogen bonds with neighboring donor –NH2 moieties could make the dispersion of the solid in water favor the diffusion of ions (possibly aided by the flow of lattice water molecules) along the flexible framework. Under these circumstances, free Zn2+ ions could weakly coordinate to the carboxylate group of the 6apic ligand (already involved in a chelating ring with structural Cd2+ ions) to increase the delocalization in the chromophore, thereby improving the ligand-centered energy transfer efficiency (due to the heavy atom effect)74 and enhancing fluorescence intensity. This hypothesis has been confirmed with a TD-DFT calculation on a model based on the [Cd(6apic)2(bipy)2] complex (previously used for the interpretation of the luminescence mechanism; see Fig. 7b) in which a Zn2+ ion is terminally coordinated to the carboxylate group (see Fig. S43,† somehow reproducing the occupation of the abovementioned site shown in Fig. S39†). As observed in the results, there are no strong shifts for the main calculated excitations, whereas the oscillator strengths are significantly enhanced, especially for the low energy band at ca. 312 nm (Fig. S44†). This fact has been also corroborated by UV-Vis measurements of the dispersed compound 2 in water in the presence/absence of Zn2+ (Fig. S40†), observing an increase of the absorption intensity of the lowest energy band at 314 nm previously assigned to the HOMO → LUMO+2 excitation centered on the 6apic ligand (calculated at 312 nm, see Fig. 7).
In any case, to our surprise, the filtered and gently washed samples of 2 preserve the pristine structure (Fig. S41†) and no trace of Fe or Zn is detected by X-ray fluorescence (Fig. S42†), meaning that these ions are indeed weakly bonded to the framework and do not remain eventually anchored once the material is washed. In order to better understand this interesting coordination equilibrium taking place for the aqueous free Zn2+ ion, we performed an additional calculation to estimate the Zn–O carboxylate(6apic) interaction energy. For this calculation, it was considered that the most plausible ligand substitution reaction (the Zn–OH2 bond replaced by the Zn–Ocarb bond) would take place between a hexaaquozinc(2+) cation and the carboxylate group of 6apic (see Fig. S45†). The detailed analysis of the thermodynamic parameters of this reaction (Table S30†) shows that although the reaction is favorable, the interaction energy (ΔG = −47 kJ mol−1) is not so high compared to other computed interactions reported for Zn2+ detectors (ca. −350 kJ mol−1).75 As observed in the enthalpies of the broken/created bonds, the global enthalpy difference is not very significant (ΔH = −0.1 kJ mol−1) because the reaction is entropy-driven, meaning that factors such as the solvation ability of the species have a dominant effect. Therefore, it may be assumed that the binding energy is not enough to promote permanent coordination to the ligand that breaks the existing double chelating Cd–(O,N) coordination bonds (or even replace the structural Cd2+ ions in the framework), especially considering the difficult access of hexaaquozinc(2+) cations to the interaction sites. Moreover, it must also be remarked that the formation of Zn–O bonds is in a competing equilibrium with the strong hydrogen bonding network, in such a way that the addition of water during washing may revert the equilibrium back, thus allowing the material to show a reversible sensing behavior. In view of the hindered access of hexaaquozinc(2+) cations to the interaction site, it cannot be ruled out that such reactions only occur on the external surface of the solid particles of compound 2 in water. On the basis of these results, and although no direct evidence has been observed in the titration study, we cannot exclude the possibility that the previously mentioned static quenching (competitive absorption) shown by Fe3+ ions is not affected by such interaction mechanisms.
Ion sensing in PADs
In view of the observed capacity of compound 2 to sensing metal ions in water, and with the aim of taking advantage of the phosphorescence emission in solid state, we decided to use paper as the supporting material to create a PAD. This largely manageable system was built by growing compound 2 in cellulose paper (referred to as 2@paper hereafter) after immersing circle-shaped papers in the synthesis vial for 24 hours. More details about the synthetic procedure can be found in the Experimental section. PXRD and SEM images confirm the successful growth of compound 2 in cellulose as shown in Fig. S46 and S47 in the ESI.† Furthermore, the micro-photographs taken under UV excitation and the recorded emission spectra of 2@paper indicate that the PL properties remain unaltered, not only regarding the bright fluorescence emission but also the green afterglow observed in the solid-state measurements (see Fig. S48 and S49 in the ESI†). Then, we investigated the sensing capacity of the PADs for the detection of Fe3+ ions given the high capacity of compound 2 for the detection of this ion. The gradual increase of the analyte concentration led to a quenching of the fluorescence emission as previously observed for the sensing experiment in the suspension approach, giving similar results: KSV = 7.53 × 105 M−1 (R2 = 0.9841) and LOD = 2.38 × 10−7 M in the measured 1.0 × 10−6–1.0 × 10−5 M concentration range (Fig. S50†), suggesting a similar sensing mechanism. Additionally, the delayed emission preserved in the PADs allowed us to study the phosphorescence signal for the detection of Fe3+, which is known to afford some important advantages over fluorescence such as the partial or full elimination of the noise signal from the autofluorescence or light scattering and a larger Stokes shift.76 Accordingly, titration experiments on Fe3+ were repeated using the phosphorescence signal and the same trend, that is, a gradual decrease in intensity upon increasing the quencher concentration, was observed. Fitting the intensity of the maximum emission wavelength (λem = 610 nm) to a linear S–V expression gives a KSV value of 8.29 × 105 M−1 (R2 = 0.9918) and a LOD of 1.47 × 10−6 M in the 1.0 × 10−6–1.0 × 10−5 M range (Fig. S50†), thus improving the results obtained in the suspension method and in PAD-based fluorescence. Despite the advantages of phosphorescence sensing, it is worth noting that only a few reported materials involving quantum dots have proved to be effective for the detection of Fe3+ in water media using delayed emission,77,78 but as far as we are aware, no CP has been reported. Encouraged by the improved results shown by the PAD based on compound 2 and considering its easy manipulation and versatility, the device was explored for its sensing performance in natural water, for which the experiments were repeated on tap water solutions containing low concentrations of the Fe3+ ion (2.5 × 10−5–1.0 × 10−4 M). On the one hand, the fluorescence emission intensity of the irradiated PAD in the solution containing the lowest [Fe3+] is increased (a trend opposite to the expected decrease of the signal in the presence of Fe3+), indicating that the interference of ions present in tap water (possibly alkaline-earth ions, taking into account that the usual concentration of these ions in tap water is ca. 6 × 10−4 M) is more significant than the signal decrease potentially caused by Fe3+ ions (Fig. S51†). On the other hand, the phosphorescence signal is still observed in tap water, and the addition of Fe3+ promotes the expected turn-off response, although the signal's intensity remains constant and does not proportionally evolve when increasing the concentration of the analyte, limiting the performance of the detection process at very low Fe3+ concentrations. Therefore, although the phosphorescence performance of these PADs could be promising in sensing applications, further research is still necessary to improve the hybridization step for achieving PADs with higher sensitivity to properly exploit their practical applications.Conclusions
In this work, we report on five novel coordination polymers (CPs) rationally designed by the linkage of the [Cd(6apic)2] secondary building unit with different bipyridine-like co-linkers. As a result, four 1D and one efficiently packed 3D CPs have been synthesized and characterized and their PL properties have been studied. In all cases, they show similar steady-state emission spectra in the solid state with a bright blue emission, which according to the computed TD-DFT calculation involves two similar charge transfers: an interligand electron transfer involving both 6apic and bipy ligands and a ligand-to-ligand charge transfer with the LUMO extended along both ligands in the excited state. It must be noted that compound 2 exhibits RTP behavior with a naked-eye observable afterglow emission with a lifetime of 32 ms and which can be extended up to 79 ms when the sample is frozen at 10 K. Accordingly, compound 2 has been selected for sensing of metal ions in water following two different approaches given its excellent PL properties. On the one hand, the powdered sample of compound 2 is suspended in an aqueous solution containing different metal ions, in which the presence of Fe3+ and Zn2+ ions promotes a reversible fast quenching and enhancement of the fluorescence emission intensity of 2, respectively, and this material can be reused for several measurements. A detailed study reveals competitive absorption as the sensing mechanism for Fe3+ ions, whereas the presence of Zn2+ ions seems to increase the delocalization in the chromophore, which improves the ligand centered charge transfers. On the other hand, paper-based analytical devices (PADs) have been built by growing 2 in cellulose. These PADs exhibit similar (even slightly improved) binding parameters for fluorescence-based detection of Fe3+ ions in water and, more importantly, they retain phosphorescence emission in such a way that the delayed signal is also employed to achieve more sensitive detection. As far as we are aware, this is the first example of employing fluorescent/phosphorescent PADs for sensing metal ions in water, so they may open a way for the development of easy-to-handle devices, providing easy and fast detection methods for aqueous pollutants.Experimental section
Chemicals
All the chemicals were of reagent grade and were used as obtained. For the PADs, the filter paper (200 μm thick, with a basis weight of 85 g m−2 and pore size of 14–18 μm) was purchased from Filter-Lab (https://www.fanoia.com, ref. 1240).:1, 15 mL) containing H6apic (0.0276 g, 0.2 mmol), Et3N was added dropwise under continuous stirring until the ligand was completely dissolved. Then, 3 mL of an aqueous solution of Cd(NO3)2·4H2O (0.0308 g, 0.1 mmol) was added. Immediately, a white precipitate of compound 1 formed. The solid was filtered off and washed several times with water and methanol. Yield 70% (based on metal). Anal. Calc for C12H10CdN4O4 (%): C, 37.28; H, 2.61; N, 14.49. Found: C, 37.31; H, 2.68; N, 14.42.
Colorless trigonal antiprismatic crystals were obtained via slow diffusion in a test tube. 5 mL of a Cd(NO3)2·4H2O aqueous solution (0.0308 g, 0.1 mmol) was separated by a water interlayer from a methanolic solution containing the H6apic ligand (5 mL, 0.0276 g, 0.2 mmol).
:1, 15 mL) containing the H6apic ligand (0.0276 g, 0.2 mmol). The resulting solution was basified with Et3N until the reagents were dissolved. Finally, an aqueous solution (3 mL) containing Cd(NO3)2·4H2O (0.0308 g, 0.1 mmol) was added dropwise. The solution was left to stand unperturbed and after five days colorless crystals grew, which were filtered off and washed several times with water and methanol. Yield 55% (based on metal). Anal. Calc for C22H20CdN6O5 (%): C, 47.11; H, 3.59; N, 14.98. Found C, 47.42; H, 3.28; N, 14.97.
For the growth of compound 2 in cellulose paper, the same procedure was followed but with the addition of the circle-shaped papers inside the reaction mixture for 24 hours. Then, the papers were removed from the solution and dried in an open atmosphere for an additional 24 hours to finally obtain the PADs.
:1) mixture. The resulting solution was sonicated and kept in an oven for 6 hours at 80 °C. The solution was slowly cooled down inside the oven. Colorless needle-shaped crystals appeared, which were filtered and washed with water and methanol. Yield 40%. Anal. Calc for C24H24CdN6O6 (%): C, 47.65; H, 4.00; N, 13.89. Found; C, 47.63; H, 3.98; N, 13.92.
:1) starting solution of H6apic (0.0276 g, 0.2 mmol), 5 mL of an ethanolic solution containing tmbp (0.0396 g, 0.2 mmol) was added dropwise under continuous stirring. Then, the pH of the solution was basified with Et3N under continuous stirring. Finally, an aqueous solution of Cd(NO3)2·4H2O (0.0308 g, 0.1 mmol) was added and the resulting solution was left to stand unperturbed. After 2 days, high quality colorless needle-shaped crystals were obtained, which were filtered off and washed with water and ethanol. Yield 40% (based on metal). Anal. Calc for C50H62Cd2N12O15 (%): C, 50.19; H, 3.54; N, 21.94. Found C, 50.09; H, 3.38; N, 21.74.
Chemical characterization
Fourier transform-infrared spectra (FT-IR) were recorded on a ThermoNicolet IR 200 spectrometer using KBr pellets in the 4000–400 cm−1 region. Elemental analysis (EA) measurements were conducted using an ELEMENTAR VARIO EL III analyzer, equipped with a thermal conductivity detector. Microscopy images were acquired using a Hitachi TM 3000 scanning electron microscope at 250–320 K. Thermogravimetric analyses (TGA) were carried out using a Mettler-Toledo TGA/SDTA851 thermal analyser from 30 to 800 °C at a heating rate of 5 °C min−1 under a synthetic air atmosphere (79% N2/21% O2) with an outflow of 50 cm3 min−1. X-ray fluorescence measurements on the samples of compound 2 were performed using a wavelength-dispersive X-ray fluorescence sequential spectrometer (PANalytical AXIOS) to rule out the presence of Fe/Zn in the filtered and dried samples. Diffuse reflectance measurements were recorded on a UV-2600 UV/vis Shimadzu spectrophotometer using a polycrystalline sample of 2 at room temperature, with BaSO4 as the reference material.Photoluminescence measurements
Room temperature spectra were recorded using a Varian Cary-Eclipse Fluorescence spectrofluorimeter equipped with a xenon discharge lamp (peak power equivalent to 75 kW), Czerny-Turner monochromators, and an R-928 photomultiplier tube with manual or automatic voltage adjustment. The photomultiplier detector voltage was 600 V and the instrument excitation and emission slits were set at 5 nm. For low-temperature measurements, a closed cycle helium cryostat enclosed in an Edinburgh Instruments FLS920 spectrometer was employed. All samples were first placed under high vacuum (of ca. 10−9 mbar) to avoid the presence of oxygen or water in the sample holder. For steady-state measurements, a Müller-Elektronik-Optik SVX1450 Xe lamp or an IK3552R-G He–Cd continuous laser (325 nm) was used as an excitation source. In contrast, a diode UV pulsed laser (λ = 370 nm, for the fluorescence emission) or a microsecond pulsed lamp coupled to the excitation monochromator (for the phosphorescence emission) was used to measure the decay curves. Photographs of irradiated PADs were taken at room temperature using a micro-PL system integrated into an Olympus optical microscope illuminated with a Hg lamp. Quantum yields were measured on solid samples using a Horiba Quanta-φ integrating sphere coupled by optical fibre to an Oriel Instruments MS257 lamp as the excitation source and a Horiba iHR550 spectrometer for emission analysis. A calibration pattern was applied to correct the emission spectra before signal integration and five measurements were accomplished to properly estimate the mean and standard deviation.X-ray diffraction measurements
Crystals of compounds 1, 2, 4 and 5 were mounted on glass fibre and used for the data collection on an Agilent Technologies Super-Nova diffractometer using micro-focus sealed X-ray tubes emitting Cu or Mo Kα radiation (λ = 1.54184 or 0.71073 Å) and CCD plates as detectors. On the other hand, a well-shaped single crystal of compounds 3 was measured on Bruker D8 Venture with a Photon detector equipped with graphite monochromated Mo-Kα radiation. The data reduction was performed using the APEX2 software and corrected for absorption using SADABS. Crystal structures were solved by direct methods using the SIR97 program and refined by full-matrix least-squares on F2 including all reflections using SHELXL-2014 software as implemented in the WINGX crystallographic package. Anisotropic displacement parameters were correctly assigned to all atoms except for hydrogen atoms, which were geometrically placed according to riding models with respect to their parent atoms with isotropic displacement factors 1.2 or 1.5 times those of their respective parent atoms. In the case of compound 1, it was found to crystallize as a multicomponent twin, so the data were reduced as a 2-component twin, with the (0.0000 −0.9997 −0.0009/−0.9997 −0.0001 −0.0003/−0.0534 0.0640 −0.9993) law. However, the indexation percentage was quite low. In spite of all our efforts, many atoms of the structure required the use of restraints to properly simulate the atomic displacement parameters. All calculations regarding porous structures or supramolecular interactions (hydrogen bonding and π–π interactions) were performed using PLATON software.79 CCDC 2332178–2332182† contain the supplementary crystallographic data for this paper.Powder X-ray diffraction (PXRD) patterns were collected using a Phillips X'PERT powder diffractometer with Cu-Kα radiation (λ = 1.5418 Å) over the range of 5 < 2θ < 50° and using a fixed slit, a step size of 0.026° and an acquisition time of 2.5 s per step at 25 °C. Pattern-matching analysis was carried out using the FULLPROF suite program,80 utilizing the cell parameters and space group obtained from the single-crystal X-ray measurements.
Computational details
TD-DFT calculations were performed using the Gaussian 09 package81 over a geometry-optimized model of compound 2, representative of the 6apic system reported herein. The Becke three-parameter hybrid functional with the non-local correlation functional of Lee–Yang–Parr (B3LYP)82–84 and the 6-31G+(d) basis set85 were employed for non-metallic atoms and the LANL2DZ basis set was used for Cd(II) ions.86,87 Dispersion correction was included systematically using the DFT-D3 method developed by Grimme.88 Results were analysed using the GaussSum package89 and molecular orbitals plotted using the GaussView program.90 Binding energies between the Zn2+ ion and a suitable model of the sensor complex were calculated at the B3LYP/def2-QZVP level of theory to minimize the basis set superimposition effect. Additionally, the polarizable continuum model (PCM) was employed to account for solvent effects in an aqueous environment.Luminescence sensing experiments
For the screening, 5 mg of compound 2 were immersed in 5 mL of 0.1 mM aqueous solutions of different metal ions prepared from M(NO3)n (Mn+ = Cr3+, Fe3+, Mn2+, Co2+, Ni2+, Cu2+ and Zn2+) for transition metals, NaCl and KCl for alkali metals and MgSO4 and CaCO3 for alkaline-earth metals and then the solutions were sonicated for 15 minutes. Then, suspensions were transferred to a quartz Suprasil cuvette and emission spectra were recorded with an excitation wavelength set at 340 nm, with a voltage of 600 V and the excitation and emission slits at 5 nm. For the titration experiments, 1 mM and 10 mM solutions of Fe3+ and Zn2+, respectively, were gradually added (100 μL in each step) to the initial suspension containing 5 mg of 2 in 5 mL of distilled water. Once the solution was homogenized, an emission spectrum was recorded with the same configuration setup.For performing sensing experiments in PADs, 20 μL of aqueous Fe3+ solutions of different concentrations, in the 1.0 × 10−6–1.0 × 10−5 M range, were added. Each PAD was dried in an open atmosphere after adding the aliquot and then emission spectra were collected. For fluorescence titration experiments, the excitation beam was set at 340 nm with the voltage fixed at 500 V and the excitation and emission slits opened at 5 nm. For phosphorescence, PADs were irradiated at 340 nm with the voltage set at 700 V and the excitation and emission slits set at 10 nm, with a delay of 0.2 ms after the pulse light and a total decay time of 20 ms.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Gobierno Vasco/Eusko Jaurlaritza (IT1500-22 and IT1755-22), Ministerio de Ciencia, Innovación y Universidades (MCIN/AEI/https://doi.org/10.13039/501100011033, Grant PID2020-117344RB-I00), Red Guipuzcoana de Ciencia, Tecnología e Innovación (FA385/2023, DG23/16) and Junta de Andalucía (FQM-394 and P21_00386). The authors acknowledge the technical and human support provided by SGIker of UPV/EHU and European funding (ERDF and ESF).References
- S. R. Batten, N. R. Champness, X. M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- L. Jiao, J. Y. R. Seow, W. S. Skinner, Z. U. Wang and H.-L. Jiang, Mater. Today, 2019, 27, 43–68 CrossRef CAS.
- G. Maurin, C. Serre, A. Cooper and G. Férey, Chem. Soc. Rev., 2017, 46, 3104–3107 RSC.
- X. Liu, L. Zhang and J. Wang, J. Materiomics, 2021, 7, 440–459 CrossRef.
- X. Yin, A. Alsuwaidi and X. Zhang, Microporous Mesoporous Mater., 2022, 330, 111633 CrossRef CAS.
- F. Gao, R. Yan, Y. Shu, Q. Cao and L. Zhang, RSC Adv., 2022, 12, 10114–10125 RSC.
- D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- G. Mínguez Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533–557 RSC.
- J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC.
- H. Liu, Y. Wang, Z. Qin, D. Liu, H. Xu, H. Dong and W. Hu, J. Phys. Chem. Lett., 2021, 12, 1612–1630 CrossRef CAS PubMed.
- L. D. Rosales-Vázquez, A. Dorazco-González and V. Sánchez-Mendieta, Dalton Trans., 2021, 50, 4470–4485 RSC.
- L. Jia, Y. C. Hui, Z. Li, H. L. Sun and Z. Wang, CrystEngComm, 2014, 16, 6483–6490 RSC.
- C. Y. Zhu, Z. Wang, J. T. Mo, Y. N. Fan and M. Pan, J. Mater. Chem. C, 2020, 8, 9916–9922 RSC.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS PubMed.
- S. Kuno, T. Kanamori, Z. Yijing, H. Ohtani and H. Yuasa, ChemPhotoChem, 2017, 1, 102–106 CrossRef CAS.
- T. Nyokong, Coord. Chem. Rev., 2007, 251, 1707–1722 CrossRef CAS.
- S. Wu, Z. Pan, R. Chen and X. Liu, Long Afterglow Phosphorescent Materials, Springer Nature, Cham, 2017 Search PubMed.
- X. Zhang, Y. Hou, X. Xiao, X. Chen, M. Hu, X. Geng, Z. Wang and J. Zhao, Coord. Chem. Rev., 2020, 417, 213371 CrossRef CAS.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- B. Pashaei, S. Karimi, H. Shahroosvand, P. Abbasi, M. Pilkington, A. Bartolotta, E. Fresta, J. Fernandez-Cestau, R. D. Costa and F. Bonaccorso, Chem. Soc. Rev., 2019, 48, 5033–5139 RSC.
- E. San Sebastian, A. Rodríguez-Diéguez, J. M. Seco and J. Cepeda, Eur. J. Inorg. Chem., 2018, 2018, 2155–2174 CrossRef CAS.
- H.-R. Wang, X.-G. Yang, J.-H. Qin and L.-F. Ma, Inorg. Chem. Front., 2021, 8, 1942–1950 RSC.
- J. Rocha, C. D. S. Brites and L. D. Carlos, Chem. – Eur. J., 2016, 22, 14782–14795 CrossRef CAS PubMed.
- E. Echenique-Errandonea, R. F. Mendes, F. Figueira, D. Choquesillo-Lazarte, G. Beobide, J. Cepeda, D. Ananias, A. Rodríguez-Diéguez, F. A. Almeida Paz and J. M. Seco, Inorg. Chem., 2022, 61, 12977–12990 CrossRef CAS PubMed.
- Y. Shen, A. Tissot and C. Serre, Chem. Sci., 2022, 13, 13978–14007 RSC.
- H. Y. Li, S. N. Zhao, S. Q. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364–6401 RSC.
- Y. Zhao, H. Zeng, X. W. Zhu, W. Lu and D. Li, Chem. Soc. Rev., 2021, 50, 4484–4513 RSC.
- W. Li, D. Zhao, W. Li, R. Wen, X. Liu, L. Liu, T. Li and L. Fan, Spectrochim. Acta, Part A, 2023, 296, 122637 CrossRef CAS PubMed.
- S. Bej, R. Das, D. Kundu, T. K. Pal and P. Banerjee, CrystEngComm, 2023, 25, 1626–1636 RSC.
- A. Das, S. Bej, N. R. Pandit, P. Banerjee and B. Biswas, J. Mater. Chem. A, 2023, 11, 6090–6128 RSC.
- X. Yu, A. A. Ryadun, D. I. Pavlov, T. Y. Guselnikova, A. S. Potapov and V. P. Fedin, Adv. Mater., 2024, 2311939 CrossRef CAS PubMed.
- X. Yu, A. A. Ryadun, D. I. Pavlov, T. Y. Guselnikova, A. S. Potapov and V. P. Fedin, Angew. Chem., Int. Ed., 2023, 62, e202306680 CrossRef CAS PubMed.
- O. Pajuelo-Corral, L. Razquin-Bobillo, S. Rojas, J. A. García, D. Choquesillo-Lazarte, A. Salinas-Castillo, R. Hernández, A. Rodríguez-Diéguez and J. Cepeda, Nanomaterials, 2022, 12, 3977 CrossRef CAS PubMed.
- A. Dutta, A. Singh, X. Wang, A. Kumar and J. Liu, CrystEngComm, 2020, 22, 7736–7781 RSC.
- C. H. Chen, X. S. Wang, L. Li, Y. B. Huang and R. Cao, Dalton Trans., 2018, 47, 3452–3458 RSC.
- N. R. Pandit, S. Bej, R. Das, N. Ghosal, A. Mondal, R. Pal, M. Ghosh, P. Banerjee and B. Biswas, Dalton Trans., 2023, 52, 11130–11142 RSC.
- S. Wang, B. Sun, Z. Su, G. Hong, X. Li, Y. Liu, Q. Pan and J. Sun, Inorg. Chem. Front., 2022, 9, 3259–3266 RSC.
- L.-L. Qian, Z.-X. Wang, J.-G. Ding, H.-X. Tian, K. Li, B.-L. Li and H.-Y. Li, Dyes Pigm., 2020, 175, 108159 CrossRef CAS.
- H. Xu, H. C. Hu, C. S. Cao and B. Zhao, Inorg. Chem., 2015, 54, 4585–4587 CrossRef CAS PubMed.
- D. I. Pavlov, X. Yu, A. A. Ryadun, D. G. Samsonenko, P. V. Dorovatovskii, V. A. Lazarenko, N. Sun, Y. Sun, V. P. Fedin and A. S. Potapov, Food Chem., 2024, 445, 138747 CrossRef CAS PubMed.
- D. I. Pavlov, X. Yu, A. A. Ryadun, V. P. Fedin and A. S. Potapov, Chemosensors, 2023, 11 Search PubMed.
- J. D. Haas and T. Brownlie IV, J. Nutr., 2001, 131, 676S–690S CrossRef CAS PubMed.
- L. M. Hyman and K. J. Franz, Coord. Chem. Rev., 2012, 256, 2333–2356 CrossRef CAS PubMed.
- K. Boonkitpatarakul, J. Wang, N. Niamnont, B. Liu, L. Mcdonald, Y. Pang and M. Sukwattanasinitt, ACS Sens., 2016, 1, 144–150 CrossRef CAS.
- L.-Z. Liu, L. Wang, M. Yu, Q. Zhao, Y. Zhang, Y.-X. Sun and W.-K. Dong, Spectrochim. Acta, Part A, 2019, 222, 117209 CrossRef CAS PubMed.
- Y. Li, X. Hu, X. Zhang, H. Cao and Y. Huang, Anal. Chim. Acta, 2018, 1024, 145–152 CrossRef CAS PubMed.
- L. Liu, Y. Ran, J. Du, Z. Wang, M. Liu and Y. Mu, RSC Adv., 2021, 11, 11266–11272 RSC.
- N. Wei, R.-X. Zuo, S. Zhang and Z. Han, Inorg. Chim. Acta, 2016, 453, 305–309 CrossRef CAS.
- J. Casaban, Y. Zhang, R. Pacheco, C. Coney, C. Holmes, E. Sutherland, C. Hamill, J. Breen, S. L. James, D. Tufano, D. Wong, E. Stavrakakis, H. Annath and A. Moore, Faraday Discuss., 2021, 231, 312–325 RSC.
- N. Yuan, X. Zhang and L. Wang, Coord. Chem. Rev., 2020, 421, 213442 CrossRef CAS.
- Y. Wang, H. Jin, Q. Ma, K. Mo, H. Mao, A. Feldhoff, X. Cao, Y. Li, F. Pan and Z. Jiang, Angew. Chem., Int. Ed., 2020, 59, 4365–4369 CrossRef CAS PubMed.
- T. Yang and T.-S. Chung, J. Mater. Chem. A, 2013, 1, 6081–6090 RSC.
- A. M. López-Marzo and A. Merkoçi, Lab Chip, 2016, 16, 3150–3176 RSC.
- S. Alvarez, D. Avnir, M. Llunell and M. Pinsky, New J. Chem., 2002, 26, 996–1009 RSC.
- A. K. Gupta, K. Tomar and P. K. Bharadwaj, New J. Chem., 2017, 41, 14505–14515 RSC.
- S. Majumder, L. Mandal and S. Mohanta, Inorg. Chem., 2012, 51, 8739–8749 CrossRef CAS PubMed.
- X. L. Qu, D. Gui, X. L. Zheng, R. Li, H. L. Han, X. Li and P. Z. Li, Dalton Trans., 2016, 45, 6983–6989 RSC.
- A. Rodríguez-Diéguez, S. Pérez-Yáñez, L. Ruiz-Rubio, J. M. Seco and J. Cepeda, CrystEngComm, 2017, 19, 2229–2242 RSC.
- J. M. Seco, A. Rodríguez-Diéguez, D. Padro, J. A. García, J. M. Ugalde, E. San Sebastian and J. Cepeda, Inorg. Chem., 2017, 56, 3149–3152 CrossRef CAS PubMed.
- O. Pajuelo-Corral, A. Rodríguez-Diéguez, J. A. García, E. San Sebastián, J. M. Seco and J. Cepeda, Dalton Trans., 2018, 47, 8746–8754 RSC.
- S. V. Eliseeva, D. N. Pleshkov, K. A. Lyssenko, L. S. Lepnev, J. C. G. Bünzli and N. P. Kuzmina, Inorg. Chem., 2010, 49, 9300–9311 CrossRef CAS PubMed.
- A. De Bettencourt-Dias, P. S. Barber, S. Viswanathan, D. T. De Lill, A. Rollett, G. Ling and S. Altun, Inorg. Chem., 2010, 49, 8848–8861 CrossRef CAS PubMed.
- X. Yang and D. Yan, Adv. Opt. Mater., 2016, 4, 897–905 CrossRef CAS.
- L. Li, S. Shen, R. Lin, Y. Bai and H. Liu, Chem. Commun., 2017, 53, 9986–9989 RSC.
- D. Wang, D. Zhang, S. De Han, J. Pan, Z. Z. Xue, J. H. Li and G. M. Wang, Dalton Trans., 2019, 48, 602–608 RSC.
- T. R. Zheng, V. A. Blatov, Y. Q. Zhang, C. H. Yang, L. L. Qian, K. Li, B. L. Li and B. Wu, J. Lumin., 2018, 199, 126–132 CrossRef CAS.
- K. Talha, Alamgir, T. He, L. H. Xie, B. Wang, M. J. Zhao, Y. Z. Zhang, Q. Chen and J. R. Li, New J. Chem., 2020, 44, 11829–11834 RSC.
- D. Briones, P. Leo, J. Cepeda, G. Orcajo, G. Calleja, R. Sanz, A. Rodríguez-Diéguez and F. Martínez, CrystEngComm, 2018, 20, 4793–4803 RSC.
- O. Pajuelo-Corral, A. Rodríguez-Diéguez, G. Beobide, S. Pérez-Yáñez, J. A. García, E. San Sebastian, J. M. Seco and J. Cepeda, J. Mater. Chem. C, 2019, 7, 6997–7012 RSC.
- H. Xu, J. Gao, X. Qian, J. Wang, H. He, Y. Cui, Y. Yang, Z. Wang and G. Qian, J. Mater. Chem. A, 2016, 4, 10900–10905 RSC.
- J. J. Ma and W. S. Liu, Dalton Trans., 2019, 48, 12287–12295 RSC.
- Z. Xiong, W. Gong, P. Xu, M. Jiang, X. Cai, Y. Zhu, X. Ping, H. Feng, H. Ma and Z. Qian, Chem. Eng. J., 2023, 451, 139030 CrossRef CAS.
- L. Liu, B. Sun, R. Ding and Y. Mao, J. Phys. Chem. A, 2022, 126, 6124–6134 CrossRef CAS PubMed.
- I. Sánchez-Barragán, J. M. Costa-Fernández, A. Sanz-Medel, M. Valledor and J. C. Campo, TrAC, Trends Anal. Chem., 2006, 25, 958–967 CrossRef.
- C. Hao, Y. Bai, L. Zhao, Y. Bao, J. Bian, H. Xu, T. Zhou and F. Feng, Dyes Pigm., 2022, 198, 109955 CrossRef CAS.
- X. Wu, C. Ma, J. Liu, Y. Liu, S. Luo, M. Xu, P. Wu, W. Li and S. Liu, ACS Sustainable Chem. Eng., 2019, 7, 18801–18809 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- J. Rodríguez-Carvajal, Physica B: Condens. Matter, 1993, 192, 55–69 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, 2016.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- N. M. O′boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- R. Dennington, T. Keith, J. Millam, et al., GaussView; Version 5, Semichem Inc., Shawnee Mission, KS, USA, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical characterization of compounds, FTIR spectroscopy, SCXRD refinement details, PXRD data, structural details, PL measurements, TD-DFT calculations, and PL sensing activity. CCDC 2332178–2332182 for 1–5. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00410h |
| This journal is © The Royal Society of Chemistry 2024 |