
Recent progress on electrocatalysts in ammonia electrooxidation reaction for clean hydrogen production
N. S.
Hassan
ab,
A. A.
Jalil
 *ab,
R.
Saravanan
c,
N. M.
Izzuddin
b,
M. B.
Bahari
d,
D.
Prasetyoko
e and
R. E.
Nugraha
f
*ab,
R.
Saravanan
c,
N. M.
Izzuddin
b,
M. B.
Bahari
d,
D.
Prasetyoko
e and
R. E.
Nugraha
f
aCentre of Hydrogen Energy, Institute of Future Energy, 81310 UTM Johor Bahru, Johor, Malaysia. E-mail: aishahaj@utm.my
bFaculty of Chemical and Energy Engineering, Universiti Teknologi Malaysia, 81310 UTM Johor Bahru, Johor, Malaysia
cInstituto de Alta Investigación, Universidad de Tarapacá, Arica-1000000, Chile
dFaculty of Science, Universiti Teknologi Malaysia, 81310 UTM Johor Bahru, Johor, Malaysia
eDepartment of Chemistry, Faculty of Science and Data Analytics, Institut Teknologi Sepuluh Nopember, Keputih, Sukolilo, Surabaya 60111, Indonesia
fDepartment of Chemical Engineering, Faculty of Engineering, Universitas Pembangunan Nasional “Veteran” Jawa Timur, Surabaya, East Java 60294, Indonesia
First published on 7th August 2024
Abstract
Hydrogen (H2) is considered one of the most promising alternative energy resources, serving as a clean energy carrier, replacing fossil fuels. Ammonia (NH3) is recognized as a high-potential H2 carrier and is utilized as a primary material for H2 production. The NH3 electro-oxidation reaction (AOR) is crucial for both direct NH3 fuel cells and NH3 electrolysis. Nevertheless, AOR frequently encounters difficulties including a high overpotential, sluggish reaction rates, and catalyst deactivation as a result of its slow kinetics, even though it is thermodynamically advantageous. In order to overcome these obstacles, numerous approaches have been suggested for the creation of electrocatalysts that can sustain a strong reaction rate even when exposed to low overpotential. The kinetics, mechanisms, and experimental methods of AOR are the primary focus of this overview. It also examines recent advancements in electrocatalyst modifications, with a particular emphasis on single-atom electrocatalysts and transition metal-based catalysts. Additionally, the paper discusses the techno-economic assessment of the AOR reaction for H2 production. Furthermore, future perspectives and recommendations for electrocatalyst development in AOR are provided. Proposing and designing new, resilient electrocatalysts will be significantly aided by the fundamentals and efforts detailed in this review; this, in turn, will propel advancements in AOR for practical applications.
Introduction
Fossil fuels, notably coal, oil, and natural gas, have historically served as primary energy sources across various sectors, such as electricity generation, transportation, and industrial processes. However, their finite nature, coupled with significant environmental and social externalities, underscores the imperative for transitioning towards renewable and alternative energy sources. Amidst this transition, hydrogen (H2) has emerged as a versatile energy carrier with the potential to decarbonize multiple sectors of the economy, including industry, transportation, and power generation.1,2 While H2 can be produced through diverse pathways, including electrolysis of water using renewable electricity,3 biomass gasification,4 and steam reforming of natural gas coupled with carbon capture and storage,5 conventional methods often entail carbon-intensive processes or rely on non-renewable resources thus undermining the environmental benefits of H2 as a clean energy vector.In this context, the electrooxidation of ammonia (NH3) presents a promising pathway toward sustainable H2 production, addressing both environmental and technical challenges associated with conventional methods.6,7 By harnessing the electrochemical reactivity of NH3, researchers endeavour to develop efficient and scalable technologies for on-demand H2 generation, thereby contributing to the transition towards a low-carbon, H2-based economy. Notably, NH3 electrooxidation can be conducted under mild conditions, obviating the need for high temperatures and pressures inherent in conventional steam reforming processes.8 This condition reduces energy consumption and enhances process safety and operational flexibility.
The progression of publications focusing on the electrooxidation of NH3 from 2008 to 2024, depicted in Fig. 1, underscores the increasing research interest in this area. Notably, 449 documents, including articles, review articles, proceeding papers, and book chapters, have comprehensively explored various aspects of NH3 electrooxidation over the past 16 years. The surge in publications since 2010 is indicative of significant advancements in research techniques and the perceived potential applications of NH3 electrooxidation in energy conversion, environmental remediation, and chemical synthesis. This surge is also reflected in the increase in citations by 2023. Furthermore, the overlay visualization in Fig. 2 highlights key terms such as “electrooxidation” and “electrochemical oxidation,” which stand out with the most significant node sizes, indicating their high frequency of occurrence. Additionally, terms represented by yellow nodes, such as catalysts, electrocatalysts, oxidation, mechanism, and fuel cell, underscore the primary focus of current research.
 | ||
| Fig. 1 Publication trends in the area of electrooxidation of NH3 within 2008–2024. | ||
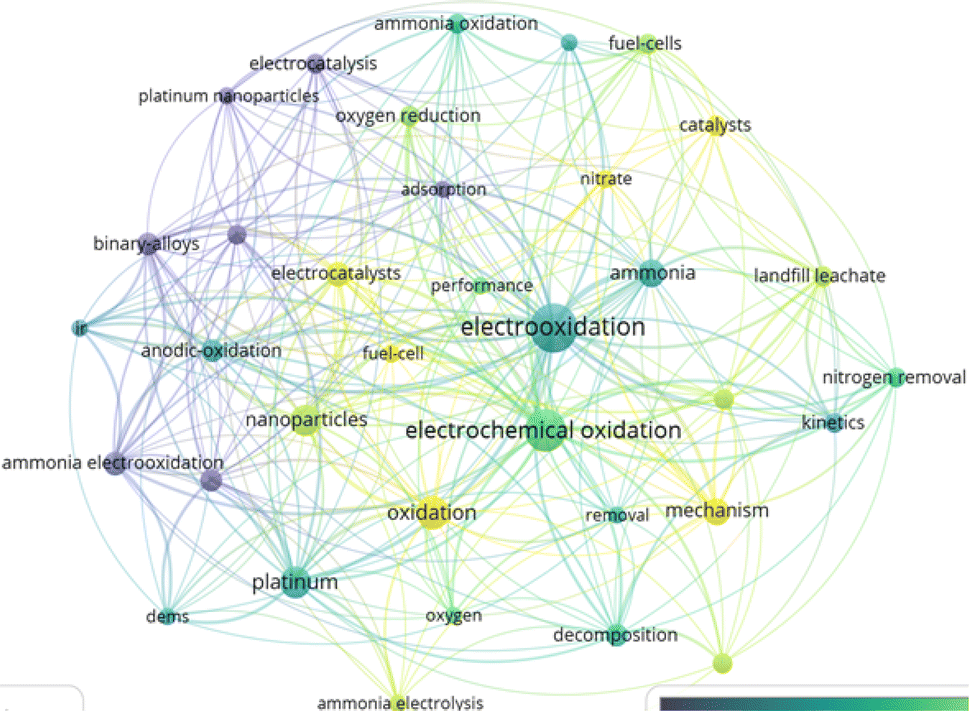 | ||
| Fig. 2 Overlay visualization of most frequent terms attained via VOSViewer v.1.6.19. | ||
Indeed, designing electrocatalysts is pivotal in enhancing the efficiency and selectivity of NH3 electrooxidation. Various materials and strategies have been developed to produce efficient catalysts to replace platinum (Pt),9 which is costly and scarce. Researchers have explored transition metal-based catalysts, such as nickel (Ni),10,11 cobalt (Co),12 and iron (Fe),13 as promising alternatives due to their abundance and lower cost compared to Pt. Additionally, alloying different metals or incorporating dopants into these materials has been investigated to enhance their catalytic activity and stability. Furthermore, non-metallic materials, such as carbon-based14 and metal nitrides15 catalysts have also shown potential for AOR. These materials offer advantages such as high surface area, tunable electronic properties, and chemical stability, making them attractive candidates for electrocatalysis.16,17 Moreover, nanostructuring and surface engineering techniques have been employed to optimize the morphology and active sites of catalyst materials, further improving their performance in catalysing NH3 electrooxidation.
Although many reviews cover catalyst development, this review will focus specifically on recent progress in electrocatalyst development to enhance AOR performance. The review will explore the various materials and strategies explored for electrocatalyst design, including single-atom electrocatalysts and transition metal-based catalysts. Additionally, the review will include a techno-economic analysis, assessing the feasibility and cost-effectiveness of different production methods of H2 for large-scale implementation. Furthermore, the review will discuss the future perspectives and recommendations in electrocatalyst development for AOR. Overall, this review aims to provide insights into the state-of-the-art electrocatalyst design for improving the efficiency and selectivity of NH3 electrooxidation, contributing to the advancement of sustainable H2 production technologies.
Ammonia (NH3) electrolysis for green hydrogen (H2) production
The utilization of NH3 as a H2 carrier undoubtedly is one of the exceptional approaches for the in situ production of H2 fuel. This approach is also noted to exhibit minimal greenhouse gaseous (GHGs) or any hazardous compounds. Besides, NH3 also displays a promising candidate material for large-scale H2 production attributed to its high stability and significant capacity for H2 storage.18 Concerning these intrinsic features, the production of H2 from NH3 has attracted considerable attention and has been deeply studied for its practicability, especially in the industrial process. Interestingly, the transformation of NH3 to H2 can be realized using two distinct pathways which are thermal decomposition and electrolysis approaches. Even though the thermal decomposition method is well-known for being more economical compared to electrolysis, the efficiency of this method yet is still inferior to that of the electrolysis method.19 Moreover, using the high temperature for the decomposition of NH3 to H2 and nitrogen constituents will be vulnerable to the formation of GHGs such as nitrous compounds. On the other hand, the electrolysis of NH3 provides a promising interest in the field of H2 production. This method utilizes a green technology with no GHGs emissions and it also can achieve high efficiency when powered by renewable energy. The efficiency of this approach also can be further enhanced depending on the advancement of the technology used. Furthermore, thermodynamic calculations demonstrated that the conversion of NH3 to nitrogen and H2 requires a negligible quantity of external energy (0.06 V), which is significantly less than the energy demanded by water electrolysis (1.23 V).20 Theoretically, this indicates that the NH3 to H2 conversion is more energy-efficient than water-splitting.21Generally, the electrolysis of water using renewable electricity and the electrooxidation of ammonia are two distinct processes with different objectives, methodologies, and challenges. The primary purpose of water electrolysis is to produce H2 and O2 gases.22 This process involves splitting water into its constituent elements using an electrical current, with the reaction: 2H2O → 2H2 + O2. The H2 produced can be used as a clean fuel for various applications, including fuel cells, chemical synthesis, and energy storage. Typically, noble metal catalysts such as platinum (Pt), iridium (Ir), and ruthenium (Ru) are used in this process due to their high activity and stability. The process can utilize either acidic or alkaline electrolytes, and it usually operates at elevated temperatures and pressures to enhance efficiency and gas production rates.23 The efficiency of water electrolysis systems ranges from 60% to 80%, depending on the specific technology and operating conditions. However, the process requires a high energy input, and challenges include catalyst degradation and overpotential losses. Despite these challenges, water electrolysis is highly suitable for integration with renewable energy sources like solar and wind power, allowing for the storage of intermittent renewable energy in the form of H2 gas. The environmental impact is minimal, as water is the only by-product, making it a sustainable and environmentally friendly process.
In contrast, the primary purpose of NH3 electrooxidation is to convert NH3 to N2 gas and water helping to treat ammonia-containing waste and reduce environmental pollution. This process typically uses transition metal catalysts such as Pt, Ni, and Cu, which facilitate the oxidation of NH3. It generally operates at ambient temperature and pressure, making it less energy-intensive than water electrolysis. The efficiency of NH3 electrooxidation depends on the catalyst used and the reaction conditions. Significant challenges include NH3 slip (unreacted ammonia), the formation of unwanted by-products, and electrode poisoning. However, this process converts harmful NH3 into N2 and water, thereby reducing environmental pollution and preventing ammonia emissions into the atmosphere. NH3 electrooxidation can also be integrated with renewable energy sources, particularly where NH3 is produced from renewable H2, creating a closed-loop sustainable process. Its applications include wastewater treatment and nitrogen removal from industrial effluents, with potential uses in H2 production.
Briefly, NH3 electrolysis is a process of dissociating the liquid NH3 or ammonium in electrochemical cells. Fig. 3 illustrates the basic concept of NH3 electrolysis. Unlike the photoelectrochemical water-splitting approach, NH3 electrolysis is a process that is been conducted in the absence of light. This process begins with the direct supply of electricity from the power supply to the system. Typically, in an aqueous NH3 electrolysis system, as the charges flow through the circuit, the anode electrode undergoes the oxidation of NH3 to the nitrates compound which then further oxidizes to N2 molecules. Concurrently, the produced electrons at the anode will be transferred to the cathode electrode for subsequent reduction of water molecules to the H2 products.24 On this side, the H2 evolution reaction (HER) will take place by reducing the water molecules with a consumption of six electron species. Moreover, the mechanism of NH3 electrolysis can be described by the equations below:25
| 2NH3 + 6OH− → N2 + 6H2O + 6e− | (1) |
| 6H2O + 6e− → 3H2 + 6OH− | (2) |
 | ||
| Fig. 3 Principle of electrolysis NH3 in aqueous solution for H2 production. | ||
Adsorption of NH3 onto the surface of the catalyst initiates the six-electron in AOR process as well as takes place in an alkaline environment. Significantly, the AOR is traversed via two discrete pathways (Fig. 4).26 Oswin and Salomon introduced the O–S mechanism, a consistent dehydrogenation process, in 1963 as reported by Kim et al.27 In this mechanism, *NH3 on the surface of catalyst undergoes sequential dehydrogenation by OH− to form *N, which later combines to form N2. Thus, the O–S mechanism can be described as shown in eqn (3)–(7):
| NH3(aq.) → *NH3 | (3) |
| *NH3 + OH− → *NH2 + H2O + e− | (4) |
| *NH2 + OH− → *NH + H2O + e− | (5) |
| *NH + OH− → *N + H2O + e− | (6) |
| *2N → N2 | (7) |
 | ||
| Fig. 4 Oxidation of NH3 by O–S (a) and G–M (b) mechanism. Reproduced from ref. 26 with permission from Elsevier, copyright 2024. | ||
Subsequently to this research, the O–S mechanism was later fine-tuned by Mauerer and Gerischer. Contradict, those researchers proposed that the G–M mechanism in the AOR involves the recombination of *NxHy species as well as OH− dehydrogenation.28 The G–M mechanism is represented by eqn (8) and (9) below, where x = 1 or 2, and y = 1 or 2.
| *NHx + *NHy → *N2Hx+y | (8) |
| *N2Hx+y + (x + y)OH− → N2 + (x + y)H2O + (x + y)e− | (9) |
The dehydrogenated *NH and *NH2 were combined to form *N2Hx+y. Subsequent dehydrogenation of *N2Hx+y leads to the formation of N2. In both major mechanisms, the rate-determining step is the dimerization of *NHx and *N (Nads), which significantly limits the production of N2 from NH3 oxidation. However, Nads can be further oxidized by OH− to generate N–O intermediates, which then form nitrite and nitrate.
| Nads + OH− → HONads + e− | (10) |
| HONads + OH− → HONOads + e− | (11) |
| HONOads + OH− → ONOads + H2O + e− | (12) |
| ONOads + OH− → ONO− (aq.) | (13) |
| ONOads + OH− → ONOOHads + e− | (14) |
Driven by the catalyst, the OHads produced in the electrolyte possess high bonding energies, which halts the dimerization process of Nads, leading to their further oxidation to NO2− and NO3−.26 During this process, under the influence of OH− in the electrolyte, the Nads adsorbed on the catalyst surface are gradually hydroxylated to form species such as HONads, ONOads, etc., which are then converted to ONOads before being smoothly oxidized to NO2− and NO3−. For example, the strongly adsorbed Nads hinder the further adsorption of NHx, resulting in a poor AOR reaction rate. A catalyst, such as CuNi, is expected to exhibit favorable AOR activity for N2 production only if it possesses appropriate adsorption bonding energy for each intermediate (Fig. 5a and b).29
 | ||
| Fig. 5 Ammonia oxidation for the preparation of (a) NO2− and (b) NO3− mechanism. Reproduced from ref. 26 with permission from Elsevier, copyright 2024. | ||
Most research supports the idea that intermediates recombine in the AOR. Additionally, analyses like DEMS and SERS have indicated that N2 cannot be generated into fully dehydrogenated *N; instead, it acts merely as a poisoning species. Several investigations have linked AOR intermediates to negative health effects, which has sparked discussion over AOR intermediates like *Nx, *NxHy, and *NOx. Accordingly, the key to getting high AOR activity is minimizing poisoning by Nx, NOx, and NxHy and improving the recombination of NxHy species.
A straightforward and effective defect-engineering strategy offers a robust approach for AOR mechanistic studies, where oxygen vacancies (Vo) are found to effectively modulate the electronic properties of surface CuO and enhance the binding strength of reaction intermediates.30 Conventional Pt-based electrocatalysts are generally believed to follow the G–M mechanism because the key intermediate of N2H4 was detected. However, the O–S mechanism reaction route via dehydrogenation of NH3 to N and N–N coupling is applied to the rich Vo–CuO. Based on the free energy pathway of the AOR process via DFT simulations, the NH3 adsorption process is energetically unfavourable on the Vo-free CuO surface. The free energy changes of the three dehydrogenation steps are all uphill and higher than 1.4 eV due to weak adsorption towards *NHx species, with the largest free energy change being 1.75 eV, indicating inferior AOR activity (Fig. 6A).30 However, after introducing the Vo, the free energy of each step decreases by 0.34, 1.84, 2.84, and 2.64 eV, respectively. Additionally, the electronic structures of Cu atoms on the different surface CuO showed that the local density of states of surface Cu atoms move significantly upward towards the Fermi level after introducing Vo, indicating stronger adsorption strength towards the adsorbates. Therefore, the presence of the Vo is beneficial to efficient NH3 oxidation by stabilizing reaction intermediates. Similarly, Zhang et al. claimed that the AOR pathways follow the N + N mechanism containing the N2H3 and N2H4 intermediate by using TiO–Vo.32 The introduction of Vo made the N2 desorption a spontaneous exoergic process, which was a good solution to the most critical catalyst poisoning problem in the AOR process.
 | ||
| Fig. 6 Free energy diagrams of NH3 oxidation into N2 on the (A) Vo-rich and Vo-free CuO via O–S mechanism. Reproduced from ref. 30 with permission from Springer, copyright 2022 and (B) CeO2 and CeO2–Vo follow the G–M mechanism. Reproduced from ref. 31 with permission from ACS Publications, copyright 2024. | ||
Differently, Yang et al. reported that CeO2 with Vo species are the active species in the CeOx/Y electrode during ammonia electrolysis and follow the G–M mechanism involving the N2H4 intermediates (Fig. 6B).31 Generally, the dehydrogenation of *NH3 to *NH2 is usually the rate-determining step for AOR over metal oxides. In this study, the energy barrier for the conversion of *NH3 to *NH2 on CeO2 is 2.13 eV, whereas on CeO2–Vo it is only −0.26 eV, indicating that NH3 is readily deprotonated. This shows that the NH3 oxidation process is energetically unfavourable on the CeO2 surface, but the presence of Vo in CeO2 significantly reduces the energy barrier for *NH3 to *NH2, aligning with the enhanced AOR activity of CeO2–Vo. When NH3 adsorbs on Vo sites, one elongated N–H bond points toward a neighboring H atom. The elongation of the N–H bond and the attraction of neighboring O atoms facilitate H transfer. Therefore, it can be concluded that the presence of Vo in CeO2 not only enhances NH3 adsorption but also lowers the energy barrier for NH3 dehydrogenation. This contributes to the increased activity of NH3 electrolysis following the G–M mechanism.
Achieving high NH3 electrolysis performance is the main goal for all researchers. Similar to the water-splitting reaction, optimizing the electrochemical cell of NH3 electrolysis should take into consideration the type, pH and concentration of the electrolyte, and the electrode materials. Frankly speaking, ammonium-based electrolytes such as NH4Cl, NH4Br, NH4I, NH4NO3, and NH4PF6 are the most suitable electrolytes for NH3 electrolysis since this process involves the reduction of ammonium species to generate H2 gas.33 In addition, the influence of pH on AOR is something that needs to be taken into consideration. Typically, the current density measurement in the AOR rises steadily as the pH goes up, while the starting potential of the anode decreases in line with pH increments. In the study of AOR, alkaline-based electrolytes remain the most commonly used because NH3 can stay in its molecular form more effectively in these conditions, generally leading to better AOR performance.26 On the other hand, NH3 indirect oxidation in acidic media is a very slow process because NH4+ is pushed away from the electrodes in acidic ions. Moreover, the oxidation of NH3 in low pH media often made the anode material susceptible to undesirable corrosion which greatly hampers the AOR process. However, an area of potential interest for AOR is the use of non-aqueous electrolytes, which can eliminate undesirable water oxidation side reactions, narrow AOR potential apertures, and the formation of by-products like nitrates. Therefore, regulating the pH of an AOR system logically is a crucial factor that merits investigation.
Moreover, the concentration of NH3 in the electrolyte solution also greatly influences the electrolysis performance. Utilising an electrolyte containing a high concentration of NH3 in this instance can enhance the interaction between NH3 and the catalyst, resulting in substantial production of H2. In a recent study, Tran et al. observed an enhancement in faradaic efficiency along with an increase in NH3 concentration.34 Similarly, Jo et al. also found that the AOR performance is directly proportional to the initial concentration of NH3 which concluded that a high concentration of NH3 is favorable for high H2 production.35 Despite these circumstances, the employment of a high concentration of NH3 in the electrolysis process might upsurge the overall process cost and unsuitable for large-scale implementation. Therefore, optimization of economic cost and electrolysis performance are substantially important for the systematic design of NH3 electrolysis systems.
Furthermore, it has been validated that the electrochemical reaction occurring at the anodic electrode constitutes the rate-limiting step in the NH3-to-H2 conversion process. Hence, the selection of an appropriate anodic electrode exhibiting outstanding physicochemical and electrical characteristics is imperative. Notably, there exist specific prerequisites to be fulfilled when opting for the anodic material. Primarily, the material must possess elevated physicochemical stability, ensuring minimal or negligible degradation throughout the electrolysis procedure, thereby preserving its original attributes. Additionally, the stability of the anodic material plays a pivotal role in preventing undesired side reactions that could pose environmental hazards and compromise the efficacy of the anodic electrode in the AOR.36 Moreover, since the anode electrode undergoes an oxidation reaction, the anode must demonstrate exceptional corrosion resistance to ensure an efficient AOR process. Long-term stability of the anodic material during electrolysis is essential for its viability in commercial applications, particularly in industrial processes. Specifically, a high surface area of anode material is desired for the AOR process. The high surface area with exceptional active sites can enhance the surface electrode–electrolyte interface and accelerate the AOR process, ultimately leading to high production of H2.26 Electronic properties are of paramount importance, with the anodic material requiring excellent electron and ion transport capabilities to sustain high current densities and minimize energy losses during the process. Lastly, to ensure heightened and efficient AOR activity, the anodic material should exhibit low overpotential values and high selectivity towards N2 production over NOx compounds.34
Over the years, due to the extraordinary properties of Pt, such as a lower *N affinity than other noble metals and a superior dehydrogenation capacity of NHx in comparison to non-noble metals, Pt-based materials have garnered considerable interest in the AOR study. This has resulted in superior AOR performance. Nevertheless, the high cost of utilizing Pt has restricted its practical applications. On the other hand, even though non-noble metal demonstrated low AOR efficiency compared to noble metal, its overall efficiency can still be improved by further modification. Therefore, single-atom catalysts and transition-metal-based catalysts have been widely employed for NH3 electrolysis studies.
Electrocatalysts for AOR in NH3-to-H2 conversion
The performance of two classes of AOR catalysts is the primary focus of this section: (1) transition metal-based electrocatalysts and (2) single-atom (SACs) electrocatalysts. Because of their high selectivity, stability, and catalytic activity, SACs electrocatalysts have emerged as promising candidates for NH3 oxidation. Dispersed on a support material are isolated metal atoms that constitute these catalysts. SACs are distinguished by their electronic and geometric configurations, which enable them to facilitate charge transfer with high efficiency and furnish active sites for the oxidation of NH3. Recent research has demonstrated the excellent performance of SACs in NH3 electrooxidation, offering high conversion efficiency and superior selectivity for H2 production. Additionally, SACs exhibit remarkable stability, making them attractive candidates for practical applications.Transition metal-based electrocatalysts have also shown promising performance in NH3 electrooxidation. These electrocatalysts typically consist of transition metal nanoparticles or alloys supported on conductive substrates. Transition metals such as nickel (Ni), cobalt (Co), and copper (Cu) have been extensively studied for NH3 oxidation due to their high catalytic activity and stability. Transition metal-based electrocatalysts offer several advantages, including tunable electronic properties and high surface area, which enhance catalytic activity and facilitate NH3 oxidation.6 Moreover, the abundance and low cost of transition metals make them attractive candidates for large-scale H2 production via AOR. Table 1 shows the summary of single atoms catalysts and transition metal-based electrocatalysts for H2 production via AOR.
| Catalyst | Current density (mA cm−2) | Onset potential (VRHE) | Ref. |
|---|---|---|---|
| Ni films | 0.45 | 0.3 | 33 |
| Pt nanoparticle | 1.45 | 0.9 | 24 |
| Pt nanosphere | 2.6 | — | 37 |
| Pt/Rh | 2.5 | 0.53 | 38 |
| Pt/NC | 2.4 | 0.1 | 39 |
| PtIrCu nanodendrites | 122.9 A g−1 | 0.35 | 40 |
| PtIrCu nanoframe | — | 0.33 | 41 |
| PtRu/C | 0.1 | 0.2 | 42 |
| PtIrZn | 0.0043 | 0.3 | 43 |
| Pt/NiO | 0.24 | −0.48 | 44 |
| Pt/TiO2/S-OLCN | 0.28 | −0.501 | 45 |
| Pt/SnO2/C | — | 0.07 | 14 |
| TM2@g-CN | — | −0.48 | 46 |
| RuO2–ZnO/Al2O3 | 0.74 | 0.2 | 47 |
| TiO2 nanofibers | 0.06 | 0.4 | 32 |
| NiO–TiO2 | 2.93 | 1.24 | 10 |
| Co/Ni–C | 2.0 | 0.45 | 48 |
| NiCO2N nanoneedle | 10 | 0.71 | 21 |
| NiCo2N nanosheets | 10 | 0.55 | 15 |
| NiCuBO/NF | 25 | 1.301 | 49 |
| B-NiFe-LDH/NF | 100 | 1.481 | 50 |
| Ni(OH)2–Cu2O@CuO | 10 | 0.47 | 51 |
| Ni1−xCuxOOH | 35 | 0.28 | 52 |
| Cu2O/NF | 4.6 | 0.55 | 53 |
| Ni3S4@NiCo2O4/NF | 50 | 0.63 | 54 |
| LaNi0.5Cu0.5O3−δ | 10 | 0.55 | 55 |
| La1−yNi0.6Cu0.4−xFexO3−δ | 50 | 0.55 | 56 |
| La0.5Sr1.5Ni0.9Cu0.1O4−δ | 13.4 | 0.53 | 57 |
Single-atom electrocatalysts
Single-atom catalysts, which comprise an isolated active site composed of a single metal atom, have made significant strides in recent years and have been recognized as the most promising electrocatalysts.58,59 In contrast to conventional nanoparticle catalysts, the utilization of metal sites in an isolated dispersion maximizes atom utilization. This not only results in a substantial cost reduction but also imparts exceptional catalytic activity and selectivity. The study conducted by Akagi et al. examines the potential of anodic catalysts such as Pt, Ru, Ir, Co, Ni, Ta, Fe, and Ti in the electrolysis of liquid NH3.33 The current densities were as follows: Ru > Ni > Co > Ir > Pt > Fe at 0.3 V versus H2/NH3 (Fig. 7A). The AOR reaction on Ti, Ta, Fe, and Co is constrained by nitrogen desorption, as these elements possess a low metal nitride formation enthalpy (ΔfHMN) by their high affinity for nitrogen. However, since Ir and Pt have a greater ΔfHMN, H2 scission or NH2-adsorption from NHx to form N atoms restricts the rate at which they occur. At the apex of the volcano correlation, Ru exhibiting a moderate ΔfHMN was identified, indicating the greatest catalytic activity33 (Fig. 7B). Additionally, the authors investigated the impact of Pt and Ni electrode surface configurations, such as plates and films. The current density of the sputtered Pt and Ni films, which possessed a columnar structure and an uneven surface, was observed to be seven and thirteen times greater than that of the Pt and Ni plates, respectively. The sputtered Ni film demonstrated an anodic current density of 0.45 mA cm−2 at an anodic potential of 0.3 V versus H2/NH3 (ref. 33) (Fig. 7C). | ||
| Fig. 7 (A) LSV curves at 10 mV s−1, (B) volcano plot of current density at 0.3 V vs. H2/NH3 as a function of metal nitride formation enthalpy (ΔfHMN (kJ mol−1 N−1)) of various metal plates, and (C) current density for Pt and Ni plates and films for anodic reaction in liquid NH3 with 0.5 M KNH2. Reproduced from ref. 33 with permission from Elsevier, copyright 2022. | ||
The Pt catalyst is the most frequently utilized noble metal catalyst in AOR research.24 The selection of an ideal electrolyte is critical in optimizing the functionality of electrocatalysts in AOR. In addition to providing the electrolyte with the ions required for the electrochemical reaction, it also affects the stability of the catalyst, the rate of the reaction, and the selectivity of the product. In general, the majority of researchers employed non-aqueous electrolytes; nevertheless, this approach resulted in a rapid decrease in current due to the electrode becoming contaminated with nitride that formed on the anode surface. The incorporation of methanol into the PF6–DMF (non-aqueous electrolyte), as suggested by Yang et al., significantly improves the electrochemical characteristics of the system, including NH3 electrolysis stability reduced polarization resistance, and high current density.41 The NH3 electrolysis is conducted for 2 hours in the absence of methanol, during which the current density stabilizes at approximately 0.24 mA cm−2. The abrupt increase in current density occurs when 5 wt% methanol is added to the KPF6–DMF non-aqueous electrolyte. The stabilized current density of 1.45 mA cm−2 is exactly six times that of the KPF6–DMF non-aqueous electrolyte when methanol is not present. Methanol diminishes the intensity of the adsorption of N* species onto the Pt electrode surface, thereby impeding the formation of a nitridation layer.60
In recent times, considerable effort has been devoted to modifying the surface orientation and morphologies of Pt particles in an attempt to expose a greater number of active sites. As an illustration, Liu et al. investigated different morphologies of Pt architectures including flower, sheet, cauliflower-like shapes, and prickly spheres, to demonstrate that the AOR activity is significantly influenced by the Pt surface morphology. Due to better diffusion control, prickly nanospheres had a current density of 2.6 mA cm−2 and better AOR performance than smooth nanoparticles37 (Fig. 8A). Both the high SSA and the high electrocatalytic activity per unit SSA of smooth, spherical Pt particles explain why they are so much more active as electrocatalysts than hierarchical Pt particles37 (Fig. 8B). Because of the high electrochemically accessible area of the electrocatalyst and the increase of the interaction with water, nitrogen-doped carbon (2.4 mA cm−2) and Rh surface entities (2.5 mA cm−2) can improve the AOR on Pt nanoparticles.38,39 An analogous result was observed when PtIrCu hyperbranched concave octahedral nanodendrites improved the electronic effect, exhibiting the highest current density (122.9 A g−1) and the onset potential (0.35 V vs. RHE).40 The exceptional performance of PtIrCu can also be ascribed to the presence of profuse suspended bonds on the high-index facets {553}, {331}, and {221}, which readily interact with NH3 and promote NH3 oxidation. In a separate study, Yang et al. documented the characteristics of a rhombic dodecahedron nanoframe composed of PtIrCu featuring high-index faceted hyperbranched nanodendrites (RDNF-HNDs). These nanodendrites exhibited a high mass activity of 26.1 A gPtIr−1 at 0.50 V, which is 7.5 times greater than the mass activity of commercial Pt/C in the AOR 36 and 140 mV lower than RHE.
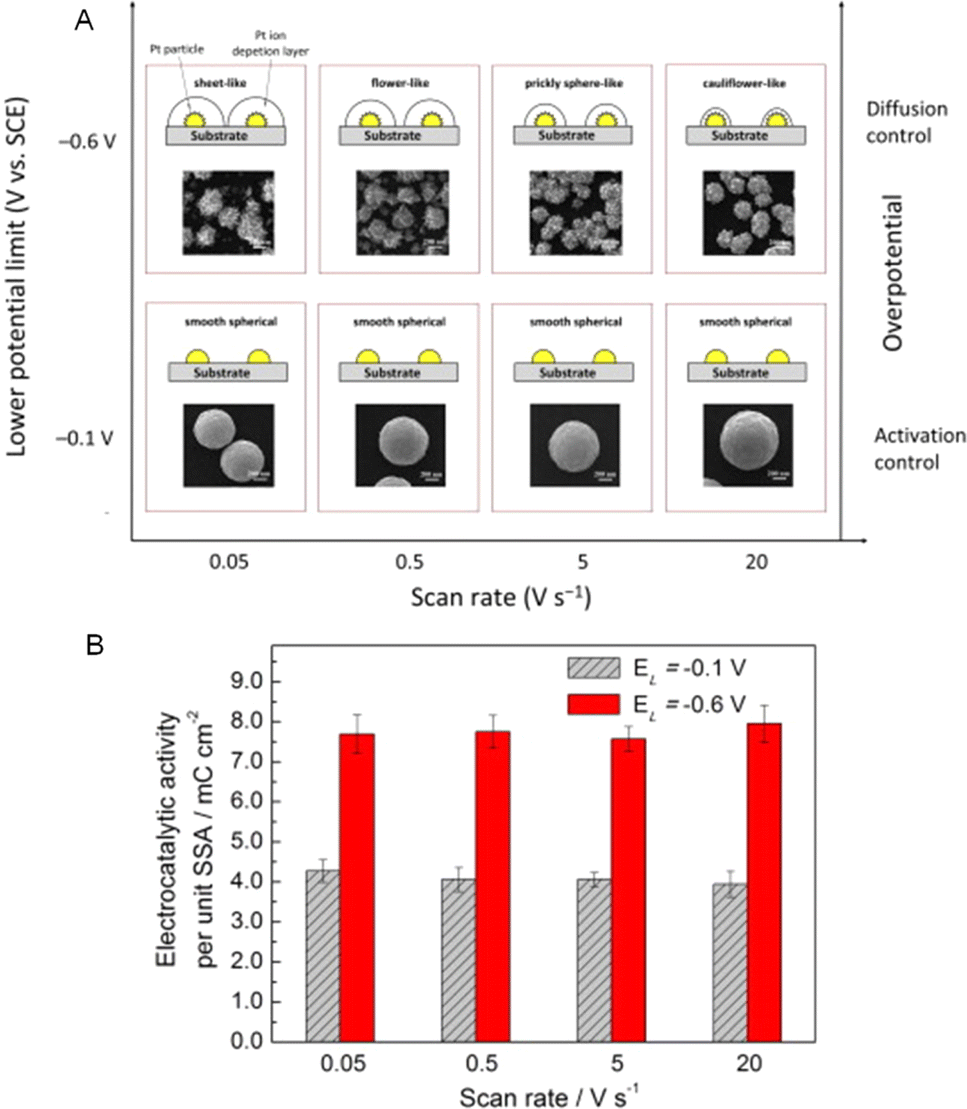 | ||
| Fig. 8 (A) Schematic representation of a two-dimensional map illustrating how the voltage difference and scan rate affect the particle morphologies; and (B) Pt particles with varied morphologies and EL values of −0.1 V and −0.6 V have varying electrocatalytic activity per unit SSA. Reproduced from ref. 37 with permission from Elsevier, copyright 2013. | ||
AOR is influenced by the surface orientation of Pt nanocrystals. AOR occurs mostly on the Pt (100) crystalline plane rather than on the Pt (111) and (110) planes.14,28,61,62 In contrast to low-index surfaces, high-index crystal surfaces are adorned with an abundance of suspended bonds that facilitate reaction with reactants and thereby augment catalytic activity.63,64 For AOR, Vidal-Iglesias et al. utilized PtMe (Me: Pd, Rh, Ir, Ru) with a particular orientation of Pt (1 0 0) nanoparticles. Out of all the bimetallic samples that were examined, only PtIr and PtRh nanoparticles exhibited an increase in the oxidation density current at low potentials, in contrast to the behavior observed in pure Pt nanoparticles synthesized using the identical technique. PtIr and PtRh nanoparticles, which are preferentially oriented with Pt (100), exhibit greater current densities than polycrystalline Pt nanoparticles. This is because AOR is more sensitive to the presence of square-symmetrical surface sites. On roughened surfaces, however, an increased proportion of exposed Pt(311) planes can substantially increase (by up to 550%) the activity of Pt toward AOR.65
Furthermore, one way to manipulate electrical structures is by alloying Pt with other metals.66 Pt is alloyed with noble metals like PtPd, PtRh, PtRu, and PtIr to increase the AOR's catalytic activity and lower the onset potential by lowering the dehydrogenation reaction's dissociation energy.42,67–70 The addition of Ir to Pt, for instance, can substantially decrease the activation energy of NH3 on Pt, thereby decreasing the AOR overpotential.71 On the other hand, according to the free energy calculations, the onset potential for AOR is lower on Ir compared to Pt. Ir-coated Pt nanocubes, according to Siddharth et al., are the surface-engineered model catalysts for AOR.9 A comparison of adsorption energies revealed that reaction intermediates, specifically *NH3, bind more strongly to Ir-decorated Pt (100) with a binding energy of −1.68 eV than to Pt (100) with a binding energy of −1.44 eV. This stronger adsorption on Ir-decorated Pt (100) suggests that the catalyst could enhance AOR kinetics. Fig. 9A shows the Gibbs free energy curves for AOR pathways on Pt(100) and Ir-decorated Pt(100). Among the three pathways (N2H2, N2H3 and N2H4), a N2H3 pathway is the most favourable one, where the *NH2-to-*NH is the rate-determining step with the energy barrier of 0.96 eV for Pt (100).9 Interestingly, a reduced energy barrier of 0.74 eV for *NH2-to-*NH step is observed for Ir-decorated Pt(100). They also investigated the optimized structure of AOR intermediates and discovered that there are five optimized structures for favorable N2H3 pathways on Ir-decorated Pt(100). Comparing the energy levels in Fig. 9B reveals that a greater binding to the chemical intermediates is facilitated by the surface Ir.9 Simulation results show that Ir-decorated Pt NCs/C promote reaction intermediate adsorption and lower the energy barrier during the potential determining phase, increasing their activity.
 | ||
| Fig. 9 (A) Gibbs free energy curves for AOR pathways on Pt(100) and Ir-decorated Pt(100) and (B) optimized structures (top and side views) of reaction intermediates using the N2H3 route during AOR on Ir-decorated Pt(100). Color code: Ir, pink; Pt, dark blue; H, white; N, blue. Reproduced from ref. 9 with permission from Springer, copyright 2020. | ||
In addition, adding 3D transition metals like PtIrZn and PtIrNi to PtIr alloy increased AOR activity.43,72 An instance of this can be seen in the PtIrZn electrode, which exhibits a low onset potential of 0.30 V relative to RHE and a high exchange current density of 0.0043 mA cm−2. This indicates that in contrast to the nanostructuration of PtIr, the most probable source is the introduction of Zn. H2 electrochemistry takes place on Pt-based electrodes between 0 and 0.40 V; therefore, dissociative adsorption of NH3 ought to compete with H2 adsorption. The inhibition of competitive H2 adsorption would be advantageous. This may be accomplished by introducing Zn with a high H2 overpotential into PtIr.
For PtIrNi, the catalyst showed significantly enhanced AOR activity with a reduced onset potential of approximately 0.40 V, compared to the binary PtIr and bare Pt catalysts. This improvement can be explained by the d-band model. The electronic structure analysis revealed that Ir atoms on the surface have a higher d-band center relative to Pt atoms (−1.97 vs. −2.25 eV), resulting in fewer occupied antibonding states and stronger adsorption energies for *NH72 (Fig. 10A). Adding Ni to the surface further shifts the d-orbital density of states up in energy (−1.97 to −1.87 eV), strengthening the adsorption of *NH intermediates and enhancing AOR activity. This group orbital model theoretically supports the promotional role of Ni in PtIr alloys for improved AOR performance. The Gibbs free energy pathways also supported that with the addition of a Ni atom to the Pt3Ir(100) surface, the dehydrogenation of *NH2 to *NH becomes exergonic at 0.3 V vs. RHE which lowering the thermodynamic barrier in the dehydrogenation process72 (Fig. 10B).
 | ||
| Fig. 10 (A) Group d-orbital density of states projected onto the bridge site of model systems and (B) free-energy pathway via constant-potential DFT calculations for initial ammonia dehydrogenation steps at 0.3 V vs. RHE on Pt(100), Pt3Ir(100), Pt3IrNi(100). Reproduced from ref. 72 with permission from ACS Publications, copyright 2020. | ||
In addition to promoting alloy formation, Pt-supported metal oxide at the nanoscale can also accelerate the AOR reaction. Ntais et al. accomplished the synthesis of platinum nanoparticles that were backed by NiO, MnO2, and carbon black.44 As compared to Hg/HgO, Pt/NiO exhibited the greatest current density (0.24 mA cm−2) and the least onset potential for AOR (−0.48 V). Similarly, Pt supported on TiO2 nanoparticles and embellished with sulfur-doped onion-like carbon nanoparticles (S-OLCNs) exhibited superior resistance to charge transfer and enhanced long-term stability.45 Pt/TiO2/S-OLCN demonstrated a current density of 0.28 mA cm−2, which is greater than that of commercial Pt/C. The enhanced catalytic performance of the Pt/TiO2/S-OLCN electrocatalyst can be attributed to the cohesive metal–support interface formed by the S-OLCN-TiO2 composite support material and Pt nanoparticles. Additionally, this may be the result of the “spillover” phenomenon, in which intermediates adsorbed on Pt sites migrate to adjacent Ti–OH species to aid in the acceleration of oxidation. TiO helps keep the Pt catalyst from becoming inactivated by N2 intermediates (Pt–Nads), which are harmful to the Pt that is created during the AOR. In addition, the Pt/TiO2/S-OLCN catalyst will be more able to catalyse the creation of AOR because the Pt NPs and the S groups in the onion structure have robust electron transfer connectivity, which will change the surface's electrical state. Lastly, Barbosa et al. reported that PtSnO2 nanoparticles with a preferential (100) orientation significantly decreased the onset potential for the AOR by approximately 70 mV.14 The high catalytic activity of these materials is attributed to the Pt (100) orientation, which exhibits a higher tolerance for poisoning intermediates like adsorbed nitrogen species (Nads). Additionally, the presence of SnO2 helps prevent the deactivation of the Pt surface caused by intermediates formed during the AOR.
Recently, Zhong et al. conducted a study on a series of transition metal atoms anchored at a g-CN monolayer (TM2@g-CN), including Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, and Pt, to identify high-performance AOR electrocatalysts.46 Compared with bulk Pt, the interaction between metal and support on the TM2@g-CN catalyst leads to a positive charge on the metal atoms, making their corresponding d orbitals easier to polarize and more effective at adsorbing NH3 (ref. 46) (Fig. 11A). This interaction also accelerates the coupling of dinitrogen, reducing the onset potential of NH3 oxidation to 0.48–0.52 eV, compared to the experimentally measured onset potential of 0.62 eV for the Pt (100) surface, without producing surface-poisonous nitrogen. Among all TM2@g-CN electrocatalysts, Fe, Co, Ru, Rh, and Ir anchored in the g-CN monolayer shows the most promise as AOR catalysts due to their low limiting potentials of −0.47, −0.5, −0.48, −0.52, and −0.48 V, respectively. Furthermore, the catalytic performance of these TM2@g-CN candidates strongly correlate with the d band center (εd) of the TM atoms, which is beneficial for designing promising AOR catalysts. When the εd shifts closer to the Fermi energy level, the limiting potential (UL) value becomes less negative, indicating increased activity46 (Fig. 11B). Therefore, the εd can directly predict AOR activity, and a downshift in the metal εd helps maintain long-lasting electrocatalytic activity.
 | ||
| Fig. 11 (A) Adsorption energy of transition metal atoms anchored at a g-CN monolayer (TM2@g-CN) and (B) the scaling relationship between d band center (εd) and the limiting potential (UL). Reproduced from ref. 46 with permission from Elsevier, copyright 2023. | ||
Based on the activation energy barrier, the dimerization of NH2 in Ir2@g-CN is more favorable than in other metals, with an activation energy of 0.93 eV. The activation energies for the other four TM2@g-CN catalysts are 2.17, 1.76, 3.3, and 1.59 eV, all higher than the activation energy for the Pt (111) surface, which is 1.07 eV. Referring to the adsorption and activation energy, the mechanism proposed by G–M is kinetically preferred. This mechanism involves N–N bond formation via hydrogenated NHx species rather than N adatoms because the strong adsorption energy of NH2 on the TM2@g-CN monolayer requires a large potential for NH2 deprotonation into NH. Therefore, the N + N mechanism is not considered in this study.
In summary, recent research focuses on optimizing catalyst materials, morphologies, and surface orientations to enhance AOR performance, with a particular emphasis on single-atom catalysts and alloying strategies.
Transition metal-based electrocatalysts
Active catalysts in AOR have been regarded as Pt and Pt-based noble metals due to their high N2 selectivity and low overpotential; nevertheless, their substantial expenses and vulnerability to deactivation have demonstrated themselves to be formidable obstacles. As an alternative to platinum-based electrocatalysts, non-noble metal-based electrocatalysts have been suggested recently. Non-noble metal catalysts, particularly transition metals (e.g., Ni-based compounds, CuO, Ni–Co bimetallic and Ni–Cu), are of interest in AOR for electrolytic H2 production and effluent cleanup.73 After conducting a large number of theoretical calculations and practical investigations, it was shown that Ni possesses the highest HER exchange current density and the lowest free energy of H2 adsorption (*GH) among a wide variety of nonprecious metals. Based on these findings, it appears that Ni could be a potential candidate for significantly increasing the intrinsic activity of HER. On the other hand, Yao et al. found that pure nickel had a low amount of AOR activity. This was because pure nickel had a high electrode consumption at the voltage that was required for NH3 oxidation.74 It is not possible for NiO to effectively oxidize NH3 because the negative charge on the cation vacancies causes the generation of reverse electrons, which speeds up the deactivation of the anion.In a recent study, Almomani et al. established that the addition of TiO2 to a Ni-based electrode substantially improves its efficacy for AOR.10 The redox potential of Ni(II)/Ni(III) increased from +0.65 to 1.24 V as a result of the pretreatment technique for NiO nanocatalyst. This is in contrast to the redox potential of HgO/Hg, which remained unchanged. The NH3 oxidation potential is the component that is responsible for this increase. As an additional point of interest, it was observed that a potential of 1.24 V is sufficient to fully facilitate the transition of Ni(OH)2 into NiOOH, which is a situation that encourages the oxidation of NH3. In the interim, NiO–TiO2-treated nanopowders demonstrated a notable enhancement in their ability to achieve AOR. As the redox potential for the Ni(II)/Ni(III) reaction increased from +0.72 to 1.35 V, a strongly oxidising environment was formed. This resulted in a rise in the built-on Ni(OH)2 layer, which in turn led to the oxidation of a bigger quantity of NH3. In fact, metallic Ni and NiOOH are not catalytically active unless they are electrochemically converted to NiOOH at approximately 1.3 V vs. RHE. The dehydrogenation of NH3 begins with an electron-coupled proton transfer to NiOOH, resulting in a would-be reversible reduction of NiOOH to Ni(OH)2.75 Huang et al. reported that Cu2O wire-in-Ni(OH)2 plate passivated by a thin CuO surface (Ni(OH)2–Cu2O@CuO) achieves a stable current density as high as ∼80 mA cm−2 for more than 30 h, demonstrating a much superior AOR performance than the previous works.51 This novel wire-in-plate nanostructure, featuring intimate contact among Ni(OH)2, Cu2O, and CuO, develops dual interfaces of Ni(OH)2–Cu2O and Cu2O@CuO. These interfaces effectively protect Cu2O from oxidation and inhibit the dissociation of copper species into the electrolyte. In addition, after adding 1 M NH3 into the electrolyte, a large current density of ∼60 mA cm−2 was detected with an onset potential of 0.47 V vs. Hg/HgO for 10 mA cm−2.
Similarly, Xu et al. found that Cu effectively improve activity of Ni(OH)2 for NH3 electrooxidation which mixed NiCu layered hydroxides achieve a current density of 35 mA cm−2 at 0.55 V vs. Ag/AgCl and 0.28 V of onset potential at higher NaOH concentration, which is much higher than that of bare Ni(OH)2 catalyst (5 mA cm−2).52 This is attributed to the abundant active sites and the synergistic effect between Ni and Cu, likely due to the formation of Ni1−xCuxOOH on the surface of the catalysts through the electrochemical activation of the mixture of Cu(OH)2 and Ni(OH)2. Xu et al. also claimed that NiOOH is the actual active substance.76 NiOOH is formed from β-Ni(OH)2via an electrochemical reaction in an alkaline solution to catalyze NH3 oxidation and then is reduced back to β-Ni(OH)2 (eqn (15) and (16)). This study showed that without Ni element, the Cu catalyst was found to have negligible activity.
| β-Ni(OH)2 + OH− → NiOOH + H2O + e− | (15) |
| NiOOH + NH3 → β-Ni(OH)2 + products | (16) |
In details, Tsai et al. reported that Ni foam (NF) exhibited a redox couple of α-Ni(OH)2(s) ↔ γ-NiOOH(s) at 0.65 V and 0.38 V on the positive and negative scans.53 However, an irreversible anodic peak at approximately 0.95 V (vs. Hg/HgO) was observed when the addition of Cu2O with different three morphologies of flower, particle, and sheet Cu2O. This result indicated that the transformation of β-Ni(OH)2(s) to β-NiOOH(s) via the oxidation process with the addition of Cu2O. At anodic peaks around 0.95 V, the electrodes demonstrated varying peak current densities, highlighting their catalytic activity. Flower Cu2O/NF had the highest peak current density at 4.6 mA cm−2, followed by particle Cu2O/NF at 3.1 mA cm−2, sheet Cu2O/NF at 2.2 mA cm−2, and NF at 1.2 mA cm−2. This order reveals that the presence of Cu2O in combination with NF significantly enhances catalytic activity. In terms of onset potential, it decreased from 0.75 V (for the NF electrode) to 0.55 V for all Cu2O/NF electrodes. This indicates an increase in the NF electrode's activity towards AOR with the introduction of Cu2O electrocatalysts.
Co and its composites, which are equally as extensively utilized as Ni, were also the subject of research due to their exceptional H2 production activity, high electrical conductivity, and durability. After that, the introduction of Co into Ni–C resulted in an improvement in AOR through the reduction of oxidation peaks within the potential range of 0.23–0.56 V, the augmentation of reversible metal-hydroxide production, and the generation of power (Pgen) to maintain the reaction.48 Greeley et al. concluded that, in addition to noble metals such as Pt and Ir, transition metals Ni and Co demonstrated high activities for HER.77
Metal oxides often suffer from insufficient activity and poor electrical conductivity. However, adding electronegative non-metals like phosphorus (P) and sulfur (S) can modify the electronic structure of transition metals and accelerate the surface reconstruction of the catalyst. The high electronegativity of S attracts electrons from the outer layer of the transition metal. This attraction lowers the energy barrier for reaction intermediates, thereby enhancing the catalytic properties. This approach also significantly enhances the electrical conductivity of the catalysts. For example, a heterostructured electrocatalyst, Ni3S4@NiCo2O4/NF, was synthesized using the hydrothermal method for the AOR reaction.54 Electrochemical tests indicated that the heterostructure of Ni3S4 and NiCo2O4 enhanced both the electrocatalytic activity and electrical conductivity compared to single-component catalysts, which demonstrated the highest current response and the lowest onset potential of 0.63 V vs. Hg/HgO at the current density of 50 mA cm−2. In situ Raman spectroscopy revealed that during the AOR process, the surface of the heterostructured catalyst underwent significant reconstruction, forming γ-NiOOH with a more loose and disordered structure. Additionally, the NiCo2O4 phase facilitated reconstruction at lower potentials. DFT calculations demonstrated that electron transfer within the heterostructured catalysts accelerated the reconstruction process and reduced the energy barriers for Ni3S4 and NiCo2O4 in the *NHNH2 generation step. This promoted NH3 adsorption and N2 desorption, resulting in excellent AOR performance and following the G–M mechanism. The production of N2 in the AOR process represents a rapid energy release mechanism that effectively mitigates critical catalyst poisoning issues.
Recently, engineering confined transition metal nitride nanostructures offers an effective approach to directly control the NH3 adsorption/desorption process. This is facilitated by their tunable metal composition, excellent conductivity, high corrosion resistance, and large surface area. For example, after conducting electrochemical measurements, it was determined that the 1D nanoneedle Ni–Co2N electrode exhibited favourable surface area and active sites. This was demonstrated by its near-zero H2 evolution onset potential, 74 mV overpotential at 10 mA cm−2, 138 mV at 100 mA cm−2, and 67 mV dec−1 Tafel slope.21 Furthermore, it exhibited exceptional long-term stability, as evidenced by a marginal decrease in the rate of H2 production following a decade of chronoamperometry measurements. In addition, at 100 mA cm−2, the potential of NH3 electrolysis in a two-electrode system was 0.71 V, which was lower than that of water splitting. This indicates that the H2 produced from NH3 electrolysis may represent a viable alternative to water splitting. Interestingly, the incorporation of transition metal nitrides results in a downshift of the metal d-band center.78 This effectively increases the antibonding electron density in the metal-adsorbate bond, potentially enabling electrocatalysts to achieve targeted moderate metal-adsorbate interactions through d-band contraction. Theoretical simulations indicate that the downshift of the metal d-band on nickel-cobalt nitride (NiCo2N) nanosheets helps maintain long-lasting AOR activity.15 This is due to the moderate metal–NH3 bond strength resulting from the d-band center downshift, optimizing key steps in AOR such as NH3 adsorption and *N2 species desorption. Nitriding metals downshift metal d-bands and create empty eg and t2g states near the Fermi level. These empty states increase the oxidation states of the metal center and act as electron acceptors, reducing the barrier to generate NHx+ cation radicals by facilitating fast electron transfer from the adsorbed NHx to the high oxidation state metal site. NiCo2N nanosheets demonstrate lower charge transfer resistance (4.5 Ω) during AOR and a lower onset overpotential of 0.55 V.
Aside from that, the dendrite-like NiCuBO/NF demonstrates exceptional stability and electrocatalytic activity for AOR. Attainment of 25 mA cm−2 current density and a Tafel slope of 63 mV dec−1 for AOR requires a mere 1.301 V.49 In a similar vein, boron nanoclusters deposited onto NiFe layered double hydroxide nanosheets (B-NiFe-LDH/NF) demonstrated notable efficacy, as evidenced by a current density of 100 mA cm−2 and a voltage of 1.781 V, which is 202 mV lower than the voltage necessary for complete water splitting. Furthermore, after 24 hours of electrolysis, 78.6% of the NH3 was removed50 (Fig. 12A and B). Furthermore, the utilization of density functional theory (DFT) simulation revealed that the formation of boron nanoclusters resulted in a reduction of the energy barrier, thereby promoting the NH3 adsorption–desorption process and the Volmer phase.
 | ||
| Fig. 12 (A) Schematic diagram and (B) LSV curve of B-NiFe-LDH/NF electrode. Reproduced from ref. 50 with permission from Elsevier, copyright 2023. | ||
Fig. 13A illustrates that the effective AOR activity of TiO/Ti foil is demonstrated in an alkaline NH3 solution at a current density of 0.06 mA cm−2 and a voltage greater than 0.4 V.32 The surface Vo in TiO is shown to significantly facilitate AOR activity through the reduction of the energy barrier in the rate-determining –HNNH2 formation phase and the promotion of N2 desorption, as demonstrated by density functional theory (DFT) calculations32 (Fig. 13B). Defective TiO2 is a stable and cost-effective catalyst for AOR, which is a potential electrode for efficient H2 production via NH3 electrolysis and direct NH3 fuel cells, as demonstrated by these results. Similarly, the Vo interfacial site is the main adsorption site for NH3.79 Aside from that, Khan et al. reported that the half-filled d-orbitals of Pd and Ru promoted ZnO/Al2O3 ternary materials as effective and robust electrocatalysts towards AOR.47 The current density was 0.307 mA cm−2 for PdO/ZnO/Al2O3, which was less than that of MnO2/ZnO/Al2O3.
 | ||
| Fig. 13 (A) LSV curves of TiO/Ti foil in 0.5 M NaOH and 0.5 M NaOH with 0.1 M NH3 solution at a scan rate of 10 mV s−1 and (B) schematic diagram of AOR reaction in the presence of an oxygen vacancy on TiO surface. Reproduced from ref. 32 with permission from Elsevier, copyright 2021. | ||
Perovskite oxides, represented by the formula ABO3, are intriguing electrocatalysts due to their significant chemical and structural flexibility. In an ideal cubic system, the larger A-site is typically occupied by an alkaline-earth metal with 12-fold oxygen coordination, while the smaller B-site is occupied by a transition metal with 6-fold BO6 oxygen coordination. Zhang et al. investigated the use of Ni/Cu in the B-site of a stoichiometric LaNi0.5Cu0.5O3−δ perovskite as an AOR catalyst.55 Their findings revealed that annealing the material in an argon atmosphere introduced Vo, which significantly enhanced its catalytic activity (onset potential of 0.55 V vs. Ag/AgCl at 10 mA cm−2). Similarly, Jeerh et al. investigated the optimal amount of Fe doping in La1−yNi0.6Cu0.4−xFexO3−δ and discovered that La0.9Ni0.6Cu0.35Fe0.05O3−δ exhibited superior activity for the AOR.56 This was evidenced by a lower onset potential of 0.55 V vs. Ag/AgCl at 50 mA cm−2, attributed to the presence of Vo introduced by an A-site deficiency, which led to greater exposure of active Ni2+ on the surface. However, compared to the Zhang and co-workers’ study, La0.5Sr1.5Ni0.9Cu0.1O4−δ can act as a robust AOR anode and achieve 13.4 mA cm−2 at a lower onset potential of 0.53 V versus Ag/AgCl.57 The selective substitution of La with Sr enhances electronic conductivity and increases Vo concentration. Additionally, doping Cu into the B-site of the perovskite fosters a synergistic interaction between nickel and copper, leading to excellent AOR activity.
Fig. 14 shows the comparison of onset potential for various transition metal electrocatalysts for AOR. Among all catalysts, RuO2–ZnO/Al2O3 showed the lowest onset potential, followed by the Ni1−xCuxOOH. The half-filled d-orbitals of Ru promoted ZnO/Al2O3 ternary materials as effective and robust electrocatalysts towards AOR, meanwhile the abundant active sites and the synergistic effect between Ni and Cu, likely due to the formation of Ni1−xCuxOOH on the surface of the catalysts through the electrochemical activation of the mixture of Cu(OH)2 and Ni(OH)2. The perovskite oxide also shows a lower onset potential, which is likely due to the presence of Vo, which led to greater exposure of active Ni2+ on the surface. Most Ni-based Cu and Co show comparable onset potential for AOR reaction and the larger onset potential is B-NiFe-LDH/NF.
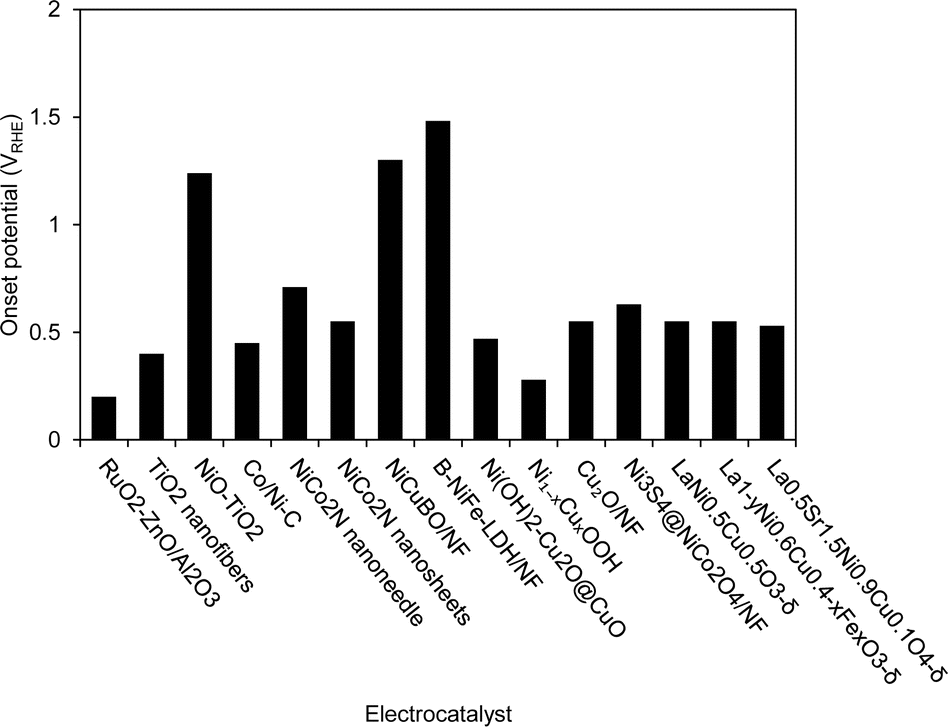 | ||
| Fig. 14 Comparison of onset potential of various transition metal electrocatalysts under different current densities for AOR. | ||
As a conclusion, both single-atom electrocatalysts and transition metal-based electrocatalysts have shown promising performance in NH3 electrooxidation. However, single-atom electrocatalysts offer several advantages over traditional transition metal-based catalysts, including higher catalytic activity, selectivity, and stability. The unique electronic and geometric structures of single-atom catalysts provide highly active and stable catalytic sites, leading to enhanced NH3 conversion efficiency and H2 production selectivity. Additionally, single-atom electrocatalysts can minimize the use of expensive transition metals, reducing production costs and improving the scalability of NH3 electrooxidation technology.
Techno-economic analysis for NH3 electrolysis
It is a promising alternative to use NH3 for the long-distance storage and transportation of H2 that is produced from renewable sources. An evaluation of the technological and economic aspects was carried out to ascertain the levelized cost of H2 (LCOH) that is produced from renewable NH3 feedstocks as tabulated in Table 2. Lee et al. conducted a study in which they evaluated the costs that are connected with the thermal breakdown of NH3 to produce H2 at two distinct rates which are 0.9 m3 h−1 (0.08 kg h−1) for a laboratory scale and 30 m3 h−1 (2.67 kg h−1) for a small-scale H2 refueling station.80 They established a model of the process in their paper and verified its accuracy by comparing it to experimental findings reported by Chung et al.86 The cost of H2 production was assessed by the authors through the utilization of various economic methodologies, including sensitivity analysis, profitability analysis, and itemized cost estimation. By utilising itemised cost estimating and beginning with a green NH3 pricing of 548 USD per tNH3, they were able to calculate that the LCOH on the laboratory scale was 9.97 USD per kgH2, while the LCOH on the refuelling station scale was 6.27 USD per kgH2.80 These figures correspond to reconversion costs of 6.75 USD per kgH2 and 3.14 USD per kgH2, respectively, when the cost of NH3 feedstock is excluded from the LCOH calculation.Nasharuddin et al. conducted a separate study in which they evaluated the technological and economic feasibility of NH3 dissociation into H2 for centralised installations (1000 kgH2 d−1) and replenishment stations (500 kgH2 d−1).81 Due to differences in CAPEX and OPEX for both scales, the authors concluded that breaking NH3 is economically viable for centralised H2 production for 5.5 USD per kgH2.81 On the other hand, the refuelling station is economically viable for 101 USD per kgH2. The price of NH3 was 520 USD per tNH3, which the authors used as the basis for their study. Furthermore, Lin et al. devised a purification system to generate H2 through thermal decomposition at an on-site H2 refueling station supplied with NH3 is 26.7 kg h−1.82 Numerous purification configurations, including PSA, membrane, PSA-membrane, and others, were contrasted in terms of H2 production costs by the authors. Beginning with an NH3 cost of 540 USD per tNH3, the authors came to the conclusion that the PSA followed by the membrane purification system produces the most cost-effective H2 production cost of 5.98 USD per kgH2.82 This is equivalent to a reconversion cost of 0.91 USD per kgH2, which is the most affordable method of producing methane.
Furthermore, for large-scale H2 production, comprehensive modeling was carried out by Makhloufi and colleagues, who determined that the LCOH was 5.2 USD per kg.83 This was determined by assuming that the H2 output rate was 7560 kg h−1 and that the price of NH3 was 488 USD per tNH3.83 A techno-economic modeling study was undertaken by Lullo et al. to examine 32 H2 transport systems, which comprised five fluids (hythane, gaseous H2, NH3, liquid H2 and LOHC) and three modes of transportation (vehicle pipeline, and rail).84 Following the simulation of the process, they concluded that the amount of thermal energy consumed was 4.38 GJ NG per tH2, while the amount of electrical energy consumed was 81 kW h per tH2. In their research, the cost of converting H2 into H2 (reconversion) was calculated to be 2.61 USD per kgH2.84 Cesaro et al. assessed the expenses associated with generating electricity from NH3via various methods, including the conversion of NH3 to H2 for fuel cell applications.87 To determine the energy efficiency and costs associated with the various routes of NH3 thermal breakdown using a large-scale NH3 refinery, a techno-economic assessment was carried out. After everything was said and done, they concluded that the levelized cost of energy would be anywhere between 167 and 197 USD per MW h at a plant capacity factor of 25%, presuming that NH3 would cost 380 USD per tNH3.
Papadias et al. conducted an assessment of the expenses associated with utilizing various liquid H2 carriers, such as toluene, methanol, and NH3, to transport H2.85 At a rate of 50 tpd, the authors found that NH3 had the highest production cost (2.2 USD per kgH2) and the lowest breakdown cost to H2 (0.61 USD per kgH2).85 According to the IEA's “The Future of H2” report, the anticipated cost of converting NH3 to H2 in a centralised facility (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tNH3 per year) would be 1 USD per kgH2. However, the report does not provide any specific assumptions regarding the reconversion process.88
000 tNH3 per year) would be 1 USD per kgH2. However, the report does not provide any specific assumptions regarding the reconversion process.88
In a recent study conducted by Kanaan et al., the most economical ways to produce H2 gas from NH3 were found to be electrolysis while in transit, thermal decomposition using membrane technology at the import terminal, and a fixed bed at the H2 refuelling station.89 Given the significance of the capital cost in conducting the techno-economic assessment, it is imperative to determine the appropriate dimensions for each piece of equipment. They discovered that membrane reactor technology is currently the most cost-effective method for converting NH3 to H2 (7.72 USD per kgH2). The technology exhibits benefit in comparison to traditional fixed bed technologies by its reduced operational expenses. Electrolysis of NH3 on a truck resulted in the lowest LCOH, at 9.59 USD per kgH2. Membrane technology has a somewhat higher LCOH (9.14 USD per kgH2) than fixed bed technology (8.69 USD per kgH2).19 The variations in the process configurations for the various scales account for this. The cost of H2 gas produced at the import terminal using fixed bed and membrane technology is competitive in terms of LCOH, especially when compared to the current green H2 pricing in Europe, which are 4–8 USD per kgH2, according to the IEA.
Summary, future perspectives, and recommendations
Electrocatalysts play a crucial role in the electrooxidation of NH3 for clean H2 production. With the increasing demand for renewable energy sources, electrocatalysis has emerged as a promising method for efficient and sustainable H2 production. NH3, with its high H2 content, has gained attention as a potential carrier for H2. However, the electrooxidation of NH3 faces challenges such as low conversion efficiency, poor selectivity, and catalyst degradation. The commercialization of AOR relies on the development of robust and efficient electrocatalysts. Because of their high NH3 affinity and fast AOR kinetics, Pt electrocatalysts have been investigated in depth using a variety of techniques, including alloying, surface morphology modification, and electrocatalytic reaction optimization. In particular, by successfully lowering the energy barrier, a Pt–Ir greatly enhanced the reaction kinetics of AOR.There has been a lot of talk about using cheaper, non-noble metal catalysts, especially catalysts based on nickel, as a substitute for Pt. When the affinity of NH3 and intermediates on Ni and Cu was changed, the Ni–Cu bimetal catalysts showed the most promising activity in terms of reaction rate and stability. Nevertheless, the development of potent new catalysts and their continued search for are essential for the practical implementation of AOR. Particularly to develop non-noble metal electrocatalysts for AOR that are economical, several obstacles must be surmounted. Initially, selective AOR in regions with minimal overpotential is critical. Alternatively, AOR competes with competing reactions such as water oxidation, NH3 oxidation to NO3− or NO2, and so on, while traditional electrocatalysts based on non-noble metals showed AOR performance at high overpotentials. Conversely, it was discovered that the introduction of heteroatoms into alloys can modify the electronic configuration of catalysts, leading to a decrease in the energy barrier for NH3 adsorption and eventually a reduction in the overpotential.
Improving AOR requires not just creating catalysts, but also a basic understanding of the reaction mechanism. At present, the AOR mechanism is primarily being studied in relation to its applicability to Pt catalysts. Nevertheless, the specific rate-determining steps or reaction mechanisms may vary among distinct electrocatalyst types. The DFT calculations have been extensively utilized in order to delineate the fundamental effect of the catalytic modification and to propose reaction mechanisms. In light of developments in theoretical inquiry, experimental observation of reaction mechanisms through the monitoring of adsorbed intermediates has become crucial. Recent studies have documented endeavors to examine the true mechanisms of action on electrocatalysts through the utilization of in situ or operando characterizations. In light of the advancements made thus far, the AOR possesses considerable practical development potential through the integration of device engineering, catalytic modification, and fundamental research.
The replacement of water with liquid NH3 is another feature that could revolutionize AOR technology for fuel cells. NH3 only becomes corrosive when reacting with water to generate the caustic OH−, however, this gas has a far larger energy density and is resistant to pipeline corrosion. The challenges associated with water oxidation are eliminated when a nonaqueous electrolyte is combined with liquid NH3. Furthermore, it is critical to conduct further mechanistic investigations on nonaqueous liquid NH3, given its considerable potential provided that the challenges impeding its application in AOR can be resolved. Aside from this, the efficient energy generation via direct NH3 fuel cells and the electrochemical synthesis of “green NH3” presents a highly promising pathway towards carbon neutrality by 2050.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Please note that due to confidentiality agreements, certain restrictions apply to the availability of the data.Conflicts of interest
The authors stated that there are no known conflicts of interest in this article.Acknowledgements
The authors are gratefully acknowledging to the Universiti Teknologi Malaysia for the Fundamental Research Grant (No. 22H51) and Professional Development Research University Grant (No. 06E92 and PY/2024/00993).References
- J. A. Okolie, B. R. Patra, A. Mukherjee, S. Nanda, A. K. Dalai and J. A. Kozinski, Int. J. Hydrogen Energy, 2021, 46, 8885–8905 CrossRef CAS.
- A. Kovač, M. Paranos and D. Marciuš, Int. J. Hydrogen Energy, 2021, 46, 10016–10035 CrossRef.
- S. Anwar, F. Khan, Y. Zhang and A. Djire, Int. J. Hydrogen Energy, 2021, 46, 32284–32317 CrossRef CAS.
- L. Cao, I. K. M. Yu, X. Xiong, D. C. W. Tsang, S. Zhang, J. H. Clark, C. Hu, Y. H. Ng, J. Shang and Y. S. Ok, Environ. Res., 2020, 186, 109547 CrossRef CAS PubMed.
- G. Franchi, M. Capocelli, M. De Falco, V. Piemonte and D. Barba, Membranes, 2020, 10, 10 CrossRef CAS PubMed.
- S. A. Lee, M. G. Lee and H. W. Jang, Sci. China Mater., 2022, 65, 3334–3352 CrossRef.
- J. Łuczak and M. Lieder, Adv. Colloid Interface Sci., 2023, 319, 102963 CrossRef PubMed.
- Y. Tian, Z. Mao, L. Wang and J. Liang, Small Struct., 2023, 4, 2200266 CrossRef CAS.
- K. Siddharth, Y. Hong, X. Qin, H. J. Lee, Y. T. Chan, S. Zhu, G. Chen, S.-I. Choi and M. Shao, Appl. Catal., B, 2020, 269, 118821 CrossRef CAS.
- F. Almomani, R. Bhosale, M. Khraisheh, A. Kumar and M. Tawalbeh, Int. J. Hydrogen Energy, 2020, 45, 10398–10408 CrossRef CAS.
- R. Wang, H. Liu, K. Zhang, G. Zhang, H. Lan and J. Qu, Chem. Eng. J., 2021, 404, 126795 CrossRef CAS.
- M.-H. Tsai, T.-C. Chen, Y. Juang, L.-C. Hua and C. Huang, Electrochem. Commun., 2020, 121, 106875 CrossRef CAS.
- F. Quan, G. Zhan, B. Zhou, C. Ling, X. Wang, W. Shen, J. Li, F. Jia and L. Zhang, J. Environ. Sci., 2023, 125, 544–552 CrossRef CAS.
- J. R. Barbosa, M. N. Leon, C. M. Fernandes, R. M. Antoniassi, O. C. Alves, E. A. Ponzio and J. C. M. Silva, Appl. Catal., B, 2020, 264, 118458 CrossRef CAS.
- S. He, Y. Chen, M. Wang, H. Nuomin, P. Novello, X. Li, S. Zhu and J. Liu, Nano Energy, 2021, 80, 105528 CrossRef CAS.
- Z.-H. Lyu, J. Fu, T. Tang, J. Zhang and J.-S. Hu, EnergyChem, 2023, 5, 100093 CrossRef CAS.
- J. H. Jang, S. Y. Park, D. H. Youn and Y. J. Jang, Catalysts, 2023, 13, 803 CrossRef CAS.
- P. Peng, J. Su and H. Breunig, Energy Convers. Manage., 2023, 288, 117166 CrossRef CAS.
- R. Kanaan, P. H. Affonso Nóbrega, P. Achard and C. Beauger, Renewable Sustainable Energy Rev., 2023, 188, 113784 CrossRef CAS.
- H. Mashhadimoslem, M. Safarzadeh Khosrowshahi, M. Delpisheh, C. Convery, M. Rezakazemi, T. M. Aminabhavi, M. Kamkar and A. Elkamel, Chem. Eng. J., 2023, 474, 145661 CrossRef CAS.
- K. Jiang, K. Li, Y.-Q. Liu, S. Lin, Z. Wang, D. Wang and Y. Ye, Electrochim. Acta, 2022, 403, 139700 CrossRef CAS.
- N. Hassan, A. Jalil, N. Khusnun, A. Ahmad, T. Abdullah, R. Kasmani, N. Norazahar, M. Kamaroddin and D. Vo, Environ. Chem. Lett., 2022, 1–23 Search PubMed.
- M. Sawal, A. Jalil, T. Abdullah, N. Khusnun, N. Hassan, F. Aziz, A. Fauzi, M. Kamaroddin, M. Omar and S. Haron, Energy Convers. Manage., 2022, 274, 116456 CrossRef CAS.
- X. Yang, H. Sun, C. Liu, L. Yu and H. Chen, Chem. Eng. J., 2022, 442, 136167 CrossRef CAS.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, J. Electrochem. Soc., 2018, 165, J3130–J3147 CrossRef CAS.
- Y. Tian, H. Tan, X. Li, J. Jia, Z. Mao, J. Liu and J. Liang, Chin. J. Catal., 2024, 56, 25–50 CrossRef CAS.
- H. Kim, S. Hong, H. Kim, Y. Jun, S. Y. Kim and S. H. Ahn, Appl. Mater. Today, 2022, 29, 101640 CrossRef.
- K. Siddharth, Y. Chan, L. Wang and M. Shao, Curr. Opin. Electrochem., 2018, 9, 151–157 CrossRef CAS.
- X. Jiang, D. Ying, X. Liu, M. Liu, S. Zhou, C. Guo, G. Zhao, Y. Wang and J. Jia, Electrochim. Acta, 2020, 345, 136157 CrossRef CAS.
- J. Huang, Z. Chen, J. Cai, Y. Jin, T. Wang and J. Wang, Nano Res., 2022, 15, 5987–5994 CrossRef CAS.
- X. Yang, L. Sun, X. Liu, Z. Yang, H. Sun, W. Liu and H. Chen, ACS Catal., 2024, 14, 6236–6246 CrossRef CAS.
- S. Zhang, Y. Zhao, L. Yan, H. Jiang, X. Yang, Y. Wang, H. Song and X. Zhao, Int. J. Hydrogen Energy, 2021, 46, 39208–39215 CrossRef CAS.
- N. Akagi, K. Hori, H. Sugime, S. Noda and N. Hanada, J. Catal., 2022, 406, 222–230 CrossRef CAS.
- D. T. Tran, T. H. Nguyen, H. Jeong, P. K. L. Tran, D. Malhotra, K. U. Jeong, N. H. Kim and J. H. Lee, Nano Energy, 2022, 94, 106929 CrossRef CAS.
- C. Jo, S. Surendran, M.-C. Kim, T.-Y. An, Y. Lim, H. Choi, G. Janani, S. Cyril Jesudass, D. Jun Moon, J. Kim, J. Young Kim, C. Hyuck Choi, M. Kim, J. Kyu Kim and U. Sim, Chem. Eng. J., 2023, 463, 142314 CrossRef CAS.
- J. H. Jang, S. Y. Park, D. H. Youn and Y. J. Jang, Catalysts, 2023, 13, 803 CrossRef CAS.
- J. Liu, W. Hu, C. Zhong and Y. F. Cheng, J. Power Sources, 2013, 223, 165–174 CrossRef CAS.
- N. N. Fomena, S. Garbarino, E. Bertin, A. Korinek, G. Botton, L. Roué and D. Guay, J. Catal., 2017, 354, 270–277 CrossRef.
- V. A. Ribeiro, I. C. de Freitas, A. O. Neto, E. V. Spinacé and J. C. M. Silva, Mater. Chem. Phys., 2017, 200, 354–360 CrossRef CAS.
- X. Lin, X. Zhang, Z. Wang, X. Zhu, J. Zhu, P. Chen, T. Lyu, C. Li, Z. Q. Tian and P. K. Shen, J. Colloid Interface Sci., 2021, 601, 1–11 CrossRef CAS PubMed.
- H. Yang, X. Huang, Z. Liu, X. Lin, Q. Chen, J. Li, C. Zhang, Z. P. Kan, Z. Q. Tian and P. K. Shen, J. Colloid Interface Sci., 2023, 652, 1764–1774 CrossRef CAS PubMed.
- J. C. M. Silva, S. Ntais, É. Teixeira-Neto, E. V. Spinacé, X. Cui, A. O. Neto and E. A. Baranova, Int. J. Hydrogen Energy, 2017, 42, 193–201 CrossRef CAS.
- J. Jiang, Electrochem. Commun., 2017, 75, 52–55 CrossRef CAS.
- S. Ntais, A. Serov, N. I. Andersen, A. J. Roy, E. Cossar, A. Allagui, Z. Lu, X. Cui, E. A. Baranova and P. Atanassov, Electrochim. Acta, 2016, 222, 1455–1463 CrossRef CAS.
- L. L. Sikeyi, L. R. Bila, T. D. Ntuli, C. T. Selepe, N. W. Maxakato, N. J. Coville and M. S. Maubane-Nkadimeng, Diamond Relat. Mater., 2023, 132, 109612 CrossRef CAS.
- J.-J. Zhong, S.-P. Huang, J.-F. Gu, Y. Li, K.-N. Ding, Y.-F. Zhang, W. Lin and W.-K. Chen, Appl. Surf. Sci., 2023, 609, 155280 CrossRef CAS.
- S. Khan, S. S. Shah, A. B. Yurtcan, A. A. A. Bahajjaj, A. Zafar and N. K. Janjua, Fuel, 2023, 347, 128446 CrossRef CAS.
- F. Almomani and M. A. H. S. Saad, Int. J. Hydrogen Energy, 2021, 46, 4678–4690 CrossRef CAS.
- A. A. Kashale, C.-T. Wu, H.-F. Hsu and I.-W. P. Chen, Chem. Eng. J., 2023, 474, 145907 CrossRef.
- J. Wang, S. Qing, X. Tong, K. Zhang, G. Luo, J. Ding and L. Xu, Appl. Surf. Sci., 2023, 640, 158330 CrossRef CAS.
- J. Huang, J. Cai and J. Wang, ACS Appl. Energy Mater., 2020, 3, 4108–4113 CrossRef CAS.
- W. Xu, R. Lan, D. Du, J. Humphreys, M. Walker, Z. Wu, H. Wang and S. Tao, Appl. Catal., B, 2017, 218, 470–479 CrossRef CAS.
- M.-H. Tsai, Y. Juang, C.-C. Hu, L.-C. Hua and C. Huang, J. Environ. Chem. Eng., 2024, 12, 112339 CrossRef CAS.
- L. Wang, K. Jiang, Z. Wang, T. Li, D. Wang and Y.-Q. Liu, Chem. Eng. J., 2024, 492, 152268 CrossRef CAS.
- M. Zhang, H. Li, X. Duan, P. Zou, G. Jeerh, B. Sun, S. Chen, J. Humphreys, M. Walker and K. Xie, Adv. Sci., 2021, 8, 2101299 CrossRef CAS PubMed.
- G. Jeerh, P. Zou, M. Zhang, S. Chen, J. Humphreys and S. Tao, Sep. Purif. Technol., 2022, 297, 121451 CrossRef CAS.
- M. Zhang, P. Zou, G. Jeerh, B. Sun, M. Walker and S. Tao, Adv. Funct. Mater., 2022, 32, 2204881 CrossRef CAS.
- H. Li, W. Wang, S. Xue, J. He, C. Liu, G. Gao, S. Di, S. Wang, J. Wang and Z. Yu, J. Am. Chem. Soc., 2024, 146, 9124–9133 CrossRef CAS PubMed.
- H. Li, S. Di, P. Niu, S. Wang, J. Wang and L. Li, Energy Environ. Sci., 2022, 15, 1601–1610 RSC.
- C. Liu, F. Yang, A. Schechter and L. Feng, Adv. Sens. Energy Mater., 2023, 100055 CrossRef.
- F. J. Vidal-Iglesias, J. Solla-Gullón, V. Montiel, J. Feliu and A. Aldaz, J. Power Sources, 2007, 171, 448–456 CrossRef CAS.
- H. S. Pillai and H. Xin, Ind. Eng. Chem. Res., 2019, 58, 10819–10828 CrossRef CAS.
- C. Xiao, B.-A. Lu, P. Xue, N. Tian, Z.-Y. Zhou, X. Lin, W.-F. Lin and S.-G. Sun, Joule, 2020, 4, 2562–2598 CrossRef CAS.
- F. J. Vidal-Iglesias, J. Solla-Gullón, P. Rodrıguez, E. Herrero, V. Montiel, J. Feliu and A. Aldaz, Electrochem. Commun., 2004, 6, 1080–1084 CrossRef CAS.
- S. Johnston, B. H. Suryanto and D. R. MacFarlane, Electrochim. Acta, 2019, 297, 778–783 CrossRef CAS.
- J. Zhou, J. S. Chung and S. G. Kang, Int. J. Hydrogen Energy, 2024, 58, 745–752 CrossRef CAS.
- M. H. Assumpção, S. G. da Silva, R. F. de Souza, G. S. Buzzo, E. V. Spinacé, A. O. Neto and J. C. M. Silva, Int. J. Hydrogen Energy, 2014, 39, 5148–5152 CrossRef.
- M. H. Assumpção, R. M. Piasentin, P. Hammer, R. F. De Souza, G. S. Buzzo, M. C. Santos, E. V. Spinacé, A. O. Neto and J. C. M. Silva, Appl. Catal., B, 2015, 174, 136–144 CrossRef.
- T. L. Lomocso and E. A. Baranova, Electrochim. Acta, 2011, 56, 8551–8558 CrossRef CAS.
- Q. Xue, Y. Zhao, J. Zhu, Y. Ding, T. Wang, H. Sun, F. Li, P. Chen, P. Jin and S. Yin, J. Mater. Chem. A, 2021, 9, 8444–8451 RSC.
- A. Estejab and G. G. Botte, Mol. Catal., 2018, 445, 279–292 CrossRef CAS.
- Y. Li, X. Li, H. S. Pillai, J. Lattimer, N. Mohd Adli, S. Karakalos, M. Chen, L. Guo, H. Xu and J. Yang, ACS Catal., 2020, 10, 3945–3957 CrossRef CAS.
- A. Allagui, S. Sarfraz and E. A. Baranova, Electrochim. Acta, 2013, 110, 253–259 CrossRef CAS.
- K. Yao and Y. Cheng, Mater. Chem. Phys., 2008, 108, 247–250 CrossRef CAS.
- J. Łuczak and M. Lieder, Adv. Colloid Interface Sci., 2023, 102963 CrossRef PubMed.
- W. Xu, D. Du, R. Lan, J. Humphreys, D. N. Miller, M. Walker, Z. Wu, J. T. Irvine and S. Tao, Appl. Catal., B, 2018, 237, 1101–1109 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- D. J. Little, D. O. Edwards, M. R. Smith III and T. W. Hamann, ACS Appl. Mater. Interfaces, 2017, 9, 16228–16235 CrossRef CAS PubMed.
- H. Sun, H. Wang and Z. Qu, ACS Catal., 2023, 13, 1077–1088 CrossRef CAS.
- B. Lee, J. Park, H. Lee, M. Byun, C. W. Yoon and H. Lim, Renewable Sustainable Energy Rev., 2019, 113, 109262 CrossRef CAS.
- R. Nasharuddin, M. Zhu, Z. Zhang and D. Zhang, Int. J. Hydrogen Energy, 2019, 44, 14445–14455 CrossRef CAS.
- L. Lin, Y. Tian, W. Su, Y. Luo, C. Chen and L. Jiang, Sustainable Energy Fuels, 2020, 4, 3006–3017 RSC.
- C. Makhloufi and N. Kezibri, Int. J. Hydrogen Energy, 2021, 46, 34777–34787 CrossRef CAS.
- G. Di Lullo, T. Giwa, A. Okunlola, M. Davis, T. Mehedi, A. Oni and A. Kumar, Int. J. Hydrogen Energy, 2022, 47, 35293–35319 CrossRef CAS.
- D. D. Papadias, J.-K. Peng and R. K. Ahluwalia, Int. J. Hydrogen Energy, 2021, 46, 24169–24189 CrossRef CAS.
- D. B. Chung, H. Y. Kim, M. Jeon, D. H. Lee, H. S. Park, S. H. Choi, S. W. Nam, S. C. Jang, J.-H. Park and K.-Y. Lee, Int. J. Hydrogen Energy, 2017, 42, 1639–1647 CrossRef CAS.
- Z. Cesaro, M. Ives, R. Nayak-Luke, M. Mason and R. Bañares-Alcántara, Appl. Energy, 2021, 282, 116009 CrossRef CAS.
- IEA, The Future Hydrogen, 2019, DOI:10.1787/1e0514c4-en.
- R. Kanaan, P. H. A. Nóbrega, P. Achard and C. Beauger, Renewable Sustainable Energy Rev., 2023, 188, 113784 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |