
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mg-rich disordered rocksalt oxide cathodes for Mg-ion batteries†
Yuan Quana,
Dingqiao Jia,
Yi Yuana,
Hang Xua,
Rui Qia,
Sofia De Sousa Coutinhoa,
Sibylle Riedelb,
Zhirong Zhao-Karger b,
Lijiang Songc,
Alexander W. Robertsonc,
Peter G. Brucead and
Robert A. House
*a
b,
Lijiang Songc,
Alexander W. Robertsonc,
Peter G. Brucead and
Robert A. House
*a
aDepartment of Materials, University of Oxford, Parks Road, UK. E-mail: robert.house@materials.ox.ac.uk
bHelmholtz Institute Ulm (HIU), Albert-Einstein-Allee 11, D-89081 Ulm, Germany
cDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK
dDepartment of Chemistry, University of Oxford, South Parks Road, UK
First published on 12th August 2024
Abstract
The discovery of new transition metal (TM) oxide cathodes which can act as intercalation hosts for Mg2+ ions is critical for the development of high energy density Mg-ion batteries. In Li-ion batteries, disordered rocksalts with sufficiently high Li+ charge carrier ion concentration, i.e. Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TM >1.1, can support fast Li+ diffusion and therefore deliver high capacities (∼300 mA h g−1) and rate performance. Here, we investigate a range of simple Mg-rich disordered rocksalt cathodes, Mg2TMO3 (TM = Mn, Ni, Co), which possess similar charge carrier ion concentrations and similar ratios between the ion size and interstitial tetrahedron height to Li-rich disordered rocksalts. However, even with high carbon loadings, elevated cycling temperatures and reduced particle and crystallite size, no significant Mg2+ deintercalation was observed, indicating that conventional design rules established for Li-rich DRS cannot be simply translated to Mg-rich systems. Despite the lack of activity in Mg-rich oxides, we demonstrate that Mg2+ intercalation into close-packed cubic disordered rocksalts, such as Li2−xMnO2F (x = 1), is possible, opening possible routes to activating Mg-rich systems.
:TM >1.1, can support fast Li+ diffusion and therefore deliver high capacities (∼300 mA h g−1) and rate performance. Here, we investigate a range of simple Mg-rich disordered rocksalt cathodes, Mg2TMO3 (TM = Mn, Ni, Co), which possess similar charge carrier ion concentrations and similar ratios between the ion size and interstitial tetrahedron height to Li-rich disordered rocksalts. However, even with high carbon loadings, elevated cycling temperatures and reduced particle and crystallite size, no significant Mg2+ deintercalation was observed, indicating that conventional design rules established for Li-rich DRS cannot be simply translated to Mg-rich systems. Despite the lack of activity in Mg-rich oxides, we demonstrate that Mg2+ intercalation into close-packed cubic disordered rocksalts, such as Li2−xMnO2F (x = 1), is possible, opening possible routes to activating Mg-rich systems.
 Robert A. House | Dr Robert House is a materials chemist at the University of Oxford where he holds a Royal Academy of Engineering Research Fellowship (2021) and leads a group researching next generation battery materials. He holds a master’s degree in Chemistry from University of Cambridge and a DPhil in Materials from University of Oxford, and has worked under the guidance of Prof. Dame Clare Grey and Prof. Sir Peter Bruce. His research focuses on the discovery and understanding of novel materials for Li-ion and beyond Li batteries. He is listed as one of Forbes Magazine’s 30 Under 30 (2023). |
1 Introduction
Lithium-ion batteries dominate the electrochemical energy storage market as they offer high energy densities, fast charge/discharge rates and long cycle life. However, global lithium resources are limited and concentrated in a small number of countries, leading to supply chain challenges and questions over their sustainability. To meet the world's future demand for energy storage, there is a need to explore alternative battery technologies based on more Earth-abundant elements. Batteries based on divalent magnesium ions (Mg2+) instead of monovalent lithium ions (Li+) are a promising contender.1 In addition to being 1000 times more abundant than Li on Earth, Mg also possesses a low reduction potential (−2.4 V vs. SHE) and can transfer twice the charge per ion as Li. Owing to its divalent nature, Mg metal anodes offer high volumetric capacity (3833 mA h cm−3), which is nearly twice that for Li (2062 mA h cm−3) and more than three times that for Na (1128 mA h cm−3).2,3However, one critical challenge that Mg-ion batteries face is the lack of cathode materials that show facile Mg2+ intercalation over wide compositional ranges. While reversible Mg2+ intercalation has been realised using cathode materials featuring soft, polarisable anions such as S2− and Se2−,4,5 their energy density remains poor due to their low working voltages. This underscores the urgent need to develop oxide-based cathodes that can operate at elevated potentials. Some oxide systems have been reported as successful Mg2+ intercalation hosts, such as V2O5, MoO3, and MnO2, however, strong coulombic interactions between Mg2+ and the host structures are recognised to be a challenge limiting the diffusion of Mg2+ in oxides.6,7 Possible strategies to address the issue include creating oxides with disordered structures to promote more varied and less stable Mg2+–O2− coordinations.8,9 This may facilitate the redistribution of charges and consequently improve the diffusion of Mg2+ ions.
Introducing structural disorder into cathodes has proven to be an effective strategy to achieve fast ion diffusion and high Li content materials in Li-ion batteries.10,11 Disordered rocksalt (DRS) cathodes such as Li2MnO2F and Li1.2Mn0.4Ti0.4O2 are capable of delivering large capacities (>300 mA h g−1) at high voltages (>3 V), exceeding the energy density of most commercial cathodes.12,13 Li ions can diffuse rapidly through disordered materials leading to exceptional rate performance.14–16 While there has been significant progress in developing Li-based DRS cathodes, there have been comparably few studies understanding whether the fast ion diffusion properties of these systems translate across to Mg-ion batteries.17–19
In this study, we explore the possibility of facile Mg-ion diffusion in Mg-rich disordered rocksalts. The theoretical capacities of Mg-rich DRS are very high by virtue of the divalent charge on Mg2+ (707 mA h g−1 for Mg2MnO3). Given their high crystallographic density (5.4 g cm−3 for Mg2MnO3), they could also theoretically deliver rechargeable batteries with very high volumetric energy density when paired with Mg metal. Here, we have synthesised a series of novel DRS compounds based on TMO–MgO solid solutions (TM = Mn, Ni, and Co) via solid state methods to obtain micron-sized particles. These transition metals offer high voltage, multi-electron redox couples, i.e. TM2+/3+/4+, to compensate the removal of Mg2+. We examined the feasibility of using Mg-rich DRS compounds Mg2MnO3, Mg2NiO3 and Mg2CoO3 as cathode materials for Mg-ion batteries, employing a Mg anode and an electrolyte that is stable to 3.5 V vs. Mg/Mg2+. Given the limited capacities observed, we further explored the influence of reducing the crystallite size to investigate whether substantially shortening the diffusion lengths of Mg2+ ions within the DRS structure might improve performance. Our results show that Mg2+ ion diffusion in Mg-rich DRS is not facile as in the case of Li+, despite nm-sized diffusion lengths and the similarity in ion size and tetrahedral height. Nevertheless, we demonstrate that Mg intercalation into a cubic-close-packed DRS is possible in delithiated Li2−xMnO2F (x = 1) where we achieve a discharge capacity of 120 mA h g−1 at an average voltage of 1.2 V vs. Mg2+/Mg. This indicates that strategies to introduce cation vacancies or mitigate the coulombic repulsion that Mg2+ ions experience when diffusing through interstitial tetrahedral sites might ultimately enable activation of Mg-rich compositions as cathodes.
2 Experimental section
2.1 Materials preparation
A series of MgxMn1−xO samples, namely MgMn3O4, MgMnO2, Mg2MnO3, and Mg3MnO4, were synthesised from MgO (97% Merck) and MnO (99% Merck) mixed in stoichiometric ratios. Prior to use, MgO was subjected to heat treatment at 900 °C overnight to eliminate traces of MgCO3 and Mg(OH)2. The mixed powders were then pelletised and calcined under argon at 930 °C for 12 h to obtain the MgxMn1−xO products. Similarly, MgO was mixed with CoO (99.99% Merck) and NiO (99.8% Merck) to prepare MgxCo1−xO and MgxNi1−xO samples, respectively. Calcination was performed in air at 1000 °C for 5 h to obtain MgxCo1−xO and for 20 h to obtain MgxNi1−xO products.For the study on the effect of crystallite size, the as-prepared Mg2MnO3 product was subsequently ball-milled at 200 rpm and 300 rpm to obtain the ball-milled samples.
Li2MnO2F was synthesised using a mechanochemical ball-milling method as per the reported work.12 LiF (99.995% Sigma Aldrich), Li2O (99.5% Alfa Aesar) and Mn2O3 (99.9% Sigma-Aldrich) were mixed in stoichiometric ratios and then sealed in air-tight zirconia jars in an argon-filled MBraun glovebox with H2O and O2 less than 1 ppm. The mixture was then ball-milled for 18 hours at 750 rpm in a Fritsch Pulverisette 7 planetary ball-mill to form the Li2MnO2F product.
2.2 Electrochemistry
Electrode preparation and cell assembly were carried out in an argon-filled glovebox where O2 and H2O levels were kept below 1 ppm. Self-supporting electrode films were prepared by grinding the as-synthesised materials with acetylene black and polytetrafluoroethylene in a 7:2:1 mass ratio in a pestle and mortar and subsequently calendared to a thickness of ∼150 μm. 0.3 M Mg[B(hfip)4]2 in DME electrolyte was synthesised following the reported procedure,20,21 with DME as the solvent. The solvent was dried with 3 Å molecular sieves for at least one week prior to use. Magnesium metal was polished with carbide paper and cleaned before use. Electrodes were incorporated into CR2032 coin cells with electrolyte-soaked Whatman glass fibre separators and Mg metal counter electrodes. Galvanostatic charge–discharge was carried out at a rate of 10 mA g−1 using a Maccor Series 4000, with cells held in a thermostatted oven at 60 °C. Li-ion cells were assembled using 1 M LiPF6 in 3:7 EC:EMC electrolyte and Li metal foils as the anode and charged at 30 °C.
2.3 Characterisation
Powder X-ray diffraction (PXRD) patterns were obtained for the as-prepared materials using a Cu source Rigaku SmartLab diffractometer equipped with a Ge(220) double bounce monochromator. Rietveld profile refinements were performed using the GSAS suite of programs.22 Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) were carried out using a Carl Zeiss Merlin analytical electron microscope. X-ray pair distribution function (X-PDF) data were collected on the I15-1 beamline at diamond light source. The samples were loaded into silica glass capillary tubes and sealed under an inert atmosphere. Structural refinement was performed using the PDFgui front end software and PDFfit2 refinement program.233 Results and discussion
3.1 Materials characterisation
A range of compositions were synthesised along the MgO–TMO tie-line (TM = Mn, Ni, Co) via the solid-state methods outlined in the Experimental Section. PXRD patterns for all of the as-prepared materials are given in Fig. S1† and the lattice parameters of the samples were acquired by refining the PXRD peaks, Fig. S2–S4 and Tables S1–S3.† Fig. 1a shows the trend of lattice parameters as the ratio of MgO to TMO changes, which varies continuously between the end member phases in line with Vegard's law,24 confirming solid solution behaviour. Fig. 1b shows the patterns for three representative samples Mg2MnO3, Mg2CoO3, and Mg2NiO3, where all peaks can be assigned to a single-phase rocksalt-type structure with space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m without impurities. The corresponding lattice parameters are 4.31 Å, 4.23 Å and 4.20 Å respectively. All of the mixed metal materials prepared conform to the disordered rocksalt structure where the cations (Mg2+ or TM2+) are randomly mixed on the cation site and are octahedrally coordinated to six anions, Fig. 1c.
m without impurities. The corresponding lattice parameters are 4.31 Å, 4.23 Å and 4.20 Å respectively. All of the mixed metal materials prepared conform to the disordered rocksalt structure where the cations (Mg2+ or TM2+) are randomly mixed on the cation site and are octahedrally coordinated to six anions, Fig. 1c.
 | ||
| Fig. 1 Long and short-range structure of Mg-rich disordered rocksalts. (a) Plot showing the trend in lattice parameters across the three analogues as the ratio of Mg to TM changes. (b) Powder X-ray diffraction data of Mg2TMO3 compounds, where all peaks can be indexed to the rocksalt phase. (c) Schematic of the cubic DRS Mg2TMO3 structure, where Mg and TM atoms are randomly mixed among the cation site. (d) X-ray PDF data fitted between 0.5 and 40 Å to an Fmm cation disordered structural model for each Mg2TMO3 sample (TM = Co, Ni, Mn), showing that the samples exhibit an atomically well-mixed cation disordered rocksalt structure. Qmax = 24 Å. (e) Zoomed-in PDF plot comparing the short-range PDF region between the three analogues. | ||
In order to characterise the local structure of these compounds and confirm atomic-level mixing of the Mg and TM ions, Pair Distribution Function (PDF) analysis was performed on X-ray total scattering data. The atomic PDF was fitted between 0.5 and 40 Å to an Fmm cation disordered structural model giving excellent fits across the full range for each sample (Fig. 1d and Tables S4–S6†). Given both the short and medium range structures for Mg2MnO3, Mg2CoO3, and Mg2NiO3 can be well-described by a single cubic rocksalt phase, we conclude that these materials are all atomically well-mixed solid solutions between MgO and TMO. Some broadening of the medium range peaks for Mg2MnO3 is apparent, indicating a greater degree of positional disorder of the atoms on their crystallographic sites and a loss of consistency in the atom–atom distances over longer ranges. This is in accord with the fact that the lattice parameter mismatch between MnO and MgO is greater than either NiO or CoO leading to greater local variability in bond lengths and atomic positions for Mn. Fig. 1e shows the short-range PDF data for each compound overlaid. The first and second peaks correspond to the nearest neighbour metal–oxygen and metal–metal distances in the cubic structure, respectively. The variation in lattice parameters from the PDF refinements, Mg2MnO3 = 4.31 Å, Mg2CoO3 = 4.23 Å and Mg2NiO3 = 4.20 Å, is consistent with the refined PXRD data in Fig. 1a.
To characterise the particle sizes and morphologies of our materials, SEM imaging was performed. Dense primary particles were observed varying in size from 0.1 to 1 μm, Fig. 2a–c. The primary particles of the Mg2NiO3 material in general appear to be smaller than Mg2CoO3 and Mg2MnO3, likely due to the smaller particle size of the NiO precursor used. Homogeneous mixing of Mg and TM ions within and between the primary particles was verified with EDX analysis (Fig. 2d–f).
 | ||
| Fig. 2 Particle morphology and uniform elemental distribution. SEM and EDX of the Mg2TMO3 samples. SEM images of (a) Mg2MnO3, (b) Mg2CoO3, and (c) Mg2NiO3 showing primary particle sizes from 0.1 to 1 μm. EDX elemental mapping of (d) Mg2MnO3, (e) Mg2CoO3, and (f) Mg2NiO3, which shows homogeneous distribution of elements in the samples. | ||
3.2 Electrochemical performance
Following the structural characterisation, the Mg2TMO3 materials were electrochemically evaluated as cathode materials in Mg-ion batteries with a Mg metal anode and 0.3 M Mg[B(hfip)4]2 in DME electrolyte. This electrolyte was chosen due to its high oxidative stability of 4 V vs. Mg/Mg2+ based on findings by Zhao-Karger et al. and ability to plate and strip Mg with a low overpotential.20,25 To confirm the stability of the electrolyte, cyclic voltammetry (CV) in coin cells using an aluminium working electrode was performed. The CV, Fig. S5b,† indicates that decomposition begins at around 3.5 V. Therefore, a limit of 3.5 V was chosen as the upper cut-off to minimise the contribution of parasitic electrolyte decomposition to the observed capacity. Galvanostatic cycling of a standard Mo6S8 cathode against Mg metal at 60 °C,26,27 using this electrolyte, Fig. S5a,† confirms successful cycling with low overpotential.The electrochemical data for the Mg2TMO3 DRS cathode materials is shown in Fig. 3. Despite employing high carbon loadings (70:20:10 of cathode active material:C:PTFE), low current densities (10 mA g−1) and elevated cycling temperatures (60 °C), no appreciable capacity was observed. These results imply that sluggish diffusion of Mg2+ ions through the oxide structures rather than electrolyte or anode plating/stripping is likely to be the performance-limiting factor in these cells. This is in accord with findings from previous studies that reported the poor diffusivity of Mg2+ ions in similar oxide spinels.7,8,28
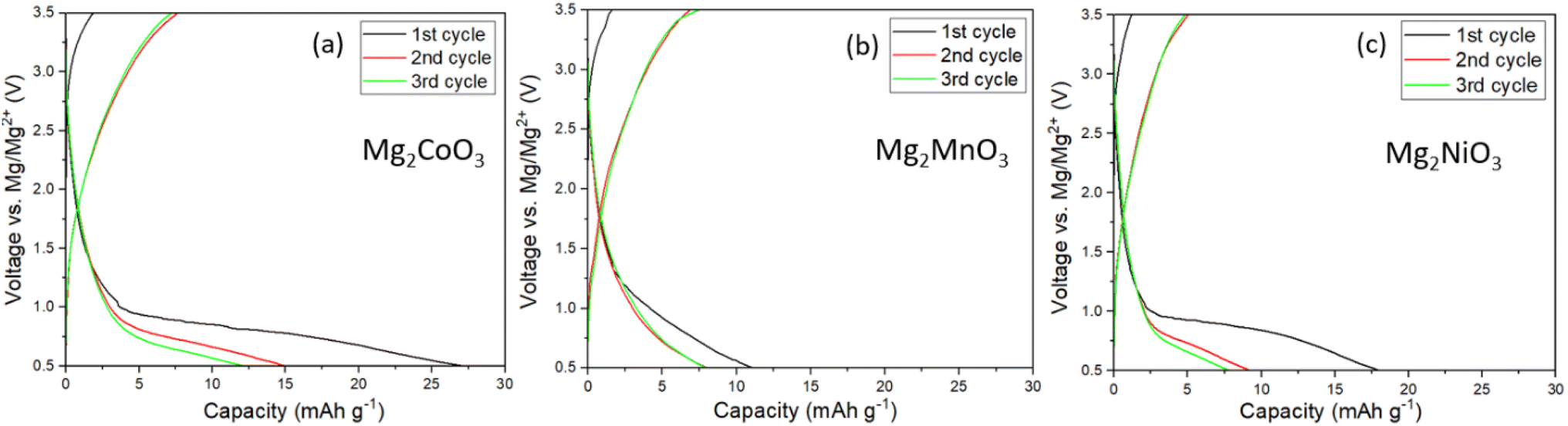 | ||
| Fig. 3 Electrochemistry of Mg-rich disordered rocksalts. Charge/discharge profiles of the first three cycles of (a) Mg2CoO3, (b) Mg2MnO3, and (c) Mg2NiO3 in Mg cells at 60 °C using 0.3 M Mg[B(hfip)4]2 in DME as the electrolyte at a current density of 10 mA g−1. | ||
3.3 Effect of crystallite size on electrochemical performance
In light of the slow rate of Mg2+ diffusion through the as-prepared DRS materials, we explored the impact of reducing the diffusion length by subjecting the Mg2MnO3 sample to an additional ball-milling process to decrease the particle and crystallite sizes. The Mn-based system was chosen for further study given its elemental abundance and reduced toxicity relative to Ni and Co. After ball-milling at 200 rpm and 300 rpm for 3 hours, the average particle sizes decreased systematically, from around 1 to 2 microns to around 100 nm, as depicted in SEM images (Fig. 4a–c). Fig. 4d–f illustrate a broadening of the PXRD peaks after the ball-milling process. Analysis of the peak broadening as a function of scattering angle using refinement (Fig. S6, S7, Tables S7 and S8†) reveals a reduction in average domain sizes after ball-milling from 1 μm before milling to 38 nm (200 rpm) to 16 nm (300 rpm), as well as an increase in strain. A slight shift in peak position to a higher scattering angle induced by the ball-milling process was also noticed, Fig. S8,† which we attribute to strain effects.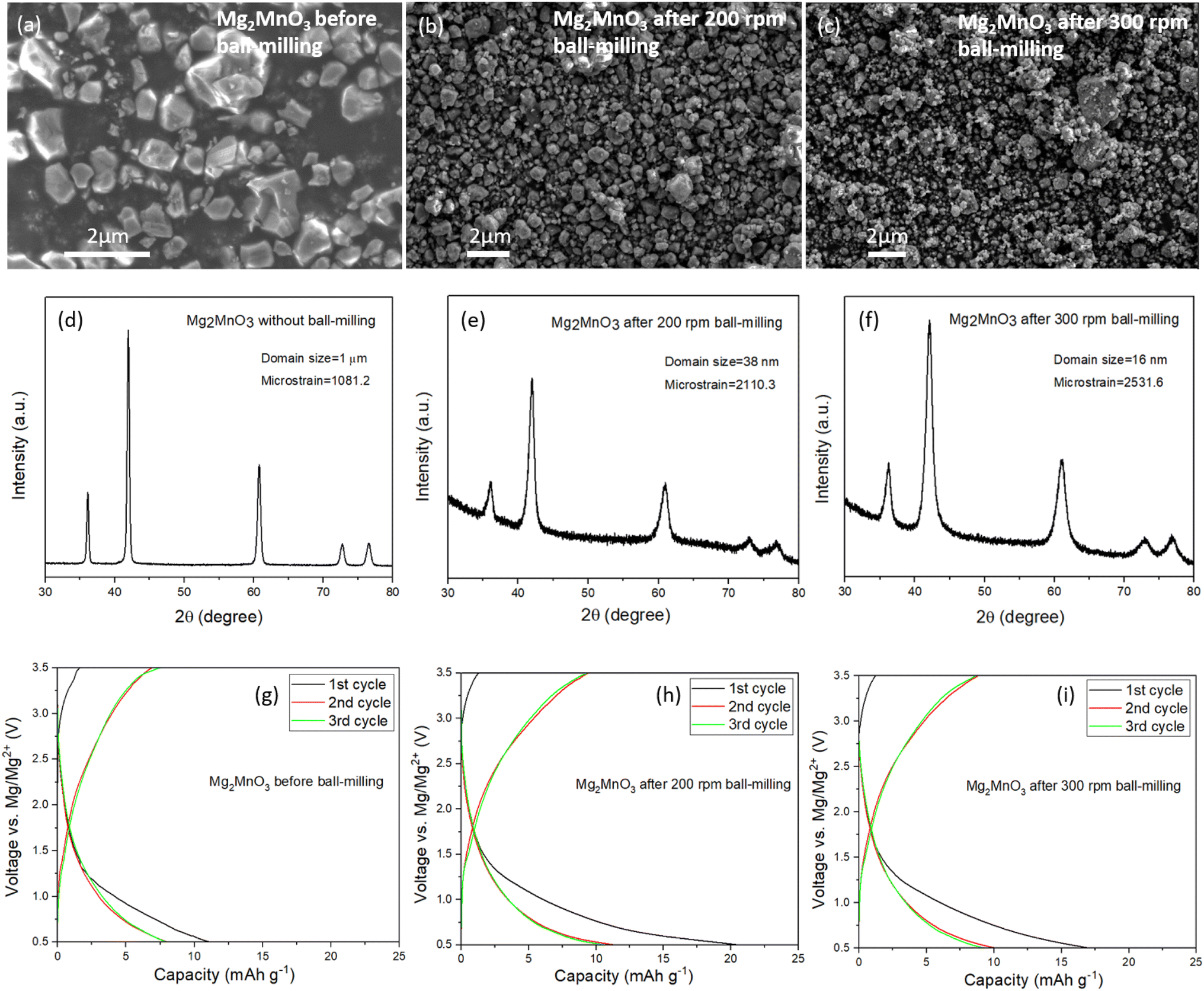 | ||
| Fig. 4 Effect of ball-milling on the structure and electrochemistry of disordered rocksalts. SEM images of Mg2MnO3 (a) before ball-milling, (b) after ball-milling at 200 rpm, and (c) after ball-milling at 300 rpm showing the changes in particle sizes. (d–f) PXRD patterns of Mg2MnO3 before and after ball-milling. The broadening of peaks indicates a decrease in the crystallite size after the ball-milling step. Charge/discharge profiles of Mg2MnO3 (g) before ball-milling, (h) after ball-milling at 200 rpm, and (i) after ball-milling at 300 rpm at 60 °C and at a current density of 10 mA g−1. | ||
The ball-milled materials were processed into electrodes, assembled into coin cells with Mg metal and 0.3 M Mg[B(hfip)4]2 in DME electrolyte and cycled at 60 °C between 0.5 and 3.5 V vs. Mg/Mg2+. The data in Fig. 4g–i show that even despite significantly reduced diffusion lengths through the DRS crystallites and increased interfacial contact area with the electrolyte, there is negligible improvement in the observed capacities. This behaviour appears to be faradaic rather than capacitive, since waiting for 24 hours after charging does not change the discharge capacity. Efforts to over-charge the cathodes to 3.7 V and then holding at this potential for 5 hours yielded only marginal improvements in discharge capacity, Fig. S9.† These results confirm that it is very challenging to remove Mg2+ from a fully magnesiated rocksalt structure.
3.4 Disordered rocksalts as Mg2+ intercalation hosts
So far, our results appear to indicate that the intrinsic fast ion diffusion properties of DRS materials as Li-ion intercalation cathodes do not translate to Mg-ion batteries. Diffusion in Li-rich DRS cathodes is understood to proceed by hopping of Li-ions between octahedral sites via a tetrahedral interstice. The ability of Li+ to diffuse has been linked to: (i) the concentration of carrier ions in the cathode i.e. Li:TM >1.1 for percolating diffusion, which increases the number of tetrahedral interstices surrounded only by Li-ions29 and (ii) the height of the tetrahedral interstitial site, which influences the size of the energy barrier for ion hopping between octahedral sites. The Mg-rich compounds investigated here, e.g. Mg2MnO3, have the same concentration of carrier ions as archetypal Li-rich DRS cathodes such as Li2MnO2F (Li:TM = 2:1), so the availability of an interconnected diffusion network should not be a limiting factor for our cathodes. Furthermore, given Mg2+ and Li+ exhibit similar ionic radii (0.72 Å compared with 0.76 Å), and the tetrahedral heights in our Mg-rich DRS compounds and those of common Li-rich DRS are similar (∼2.43 Å compared with 2.3–2.4 Å respectively),30 the ion size would appear to be compatible with the diffusion channels available in the disordered rocksalt structure.
Another factor which could influence the diffusion of Mg2+ versus Li+ through the DRS structure is coulombic repulsion from other Mg2+ cations which share faces with the interstitial tetrahedral sites. Mg2+ ions diffusing through tetrahedra surrounded by Mg2+ will experience greater coulombic repulsion than Li+ ions would when surrounded by Li+, increasing the energy barrier for ion hopping. To investigate this possibility further, we pre-charged an electrode of Li2MnO2F in a Li-ion cell to a composition of LiMnO2F to create octahedral vacancies for Mg intercalation. The charged electrode was then transferred into a Mg-ion cell and discharged to 0.5 V vs. Mg/Mg2+. The data, Fig. 5a, show that a discharge capacity of 120 mA h g−1 can be achieved at an average voltage of 1.2 V, corresponding to insertion of 0.26 Mg2+. Even at a relatively high current density of 50 mA g−1, Fig. 5b, a discharge capacity of 70 mA h g−1 could be achieved, indicating surprisingly facile Mg2+ intercalation into the cubic close-packed DRS material (cell parameter 4.09 Å).
 | ||
| Fig. 5 Mg2+ intercalation into a disordered rocksalt oxyfluoride. (a) Charge profile for Li2MnO2F (solid line) in a Li-ion battery to 4.2 V vs. Li/Li+. The charged cathode was removed and assembled into a Mg-ion battery, then discharged to 0.5 V vs. Mg/Mg2+ at 10 mA g−1 at 60 °C (dashed line). Over 120 mA h g−1 discharge capacity is observed corresponding to insertion of 0.26 Mg2+ per formula unit, i.e. Mg0.26LiMnO2F. (b) Discharge profile under different current densities for delithiated LiMnO2F in Mg-ion cells. (c) The corresponding PXRD patterns for the pristine Li2MnO2F, delithiated and magnesiated cathodes. There is a clear shift of the DRS diffraction peaks to lower angles confirming intercalation of Mg2+. SEM and EDX of (d) Li charged LiMnO2F and (e) Mg discharged Mg0.26LiMnO2F. | ||
To verify Mg intercalation, ex situ PXRD was performed to examine the lattice parameter changes before and after the discharge process at 10 mA g−1. As shown in Fig. 5c, the discharge process induces a clear shift in the measured peak positions back towards those of pristine Li2MnO2F, confirming the bulk intercalation of Mg2+ into LiMnO2F. EDX and inductively coupled plasma (ICP)-optical emission spectroscopy analyses were also performed before and after discharging in a Mg-ion cell. EDX mapping for LiMnO2F and Mg0.26LiMnO2F, Fig. 5d and e, shows the appearance of Mg uniformly distributed within and between particles after magnesiation. The ICP results (Table 1) also confirm Mg contents of 0.00(1) and 0.27(1) before and after magnesiation, which agrees well with the calculated formula based on the discharge capacity observed and indicates storage of Mg2+.
| Li (atomic) | Mg (atomic) | Mn (normalised atomic) | |
|---|---|---|---|
| LiMnO2F | 0.96(1) | 0.00(1) | 1.00 |
| Mg0.26LiMnO2F | 0.90(1) | 0.27(1) | 1.00 |
These results indicate that the coordination environment around the interstitial tetrahedral sites could play an important role in determining the ability of DRS compounds to act as intercalation hosts for Mg2+. By mitigating the coulombic repulsion experienced by Mg ions diffusing through interstitial tetrahedral sites in Mg-rich DRS compositions, it may be possible to activate these as intercalation cathodes. This would require compositional modifications which substitute some Mg2+ for less highly charged ions or create vacant sites in the structure. It has also been proposed that concerted ion migration of Li+ with the Mg2+ might play a role in increasing the rate of Mg-ion intercalation.31,32 While we cannot easily separate the motion of different ions within the bulk, given the lack of Li-ions in the Mg electrolyte, we can be confident that the Mg2+ intercalates without co-intercalation of Li+. Further work to more fully understand the factors controlling Mg-ion diffusion in the bulk of DRS cathodes is required.
4 Conclusions
A series of novel Mg-rich disordered rocksalt oxide structures based on Co, Mn and Ni were prepared and investigated as potential high-voltage cathodes for Mg-ion batteries. PXRD, X-ray PDF and EDX show that MgO can form solid solutions with CoO, NiO and MnO with well-mixed Mg/TM occupying the cation sites. When incorporated into Mg-ion cells with Mg metal and an electrolyte stable to at least 3.5 V Mg/Mg2+, there is little evidence of successful deintercalation of Mg2+ from Mg2MnO3, Mg2CoO3 or Mg2NiO3, even with high carbon loadings and elevated cycling temperatures (60 °C). Efforts to significantly reduce the Mg-ion diffusion lengths by decreasing particle and crystallite sizes to nanometre length-scales did not yield significant improvements in capacity. Despite this, we demonstrate that other disordered rocksalts, such as Li2−xMnO2F (x = 1), can act as intercalation hosts for Mg2+, raising hope that it might be possible to activate Mg-rich cathodes through compositional modifications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. Q. planned and conducted all aspects of the synthesis and characterisation work with the help of D. J. S. R. and Z. Z. K. synthesised the Mg electrolyte salt. Y. Y. collected the SEM images and R. Q. and S. D. S. C. collected the X-ray total scattering data. Y. Q. and R. A. H. wrote the manuscript with contributions from all authors. R. A. H. supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. A. H. acknowledges the Royal Academy of Engineering for support through the Research Fellowship scheme, the John Fell Fund, the Engineering and Physical Sciences Research Council (EPSRC), and the Henry Royce Institute (EP/R00661X/1, EP/S019367/1, EP/R010145/1). We acknowledge Diamond Light Source for time on I15-1 beamtime under proposal CY35103-1. For the purpose of open access, the authors have applied a CC BY public copyright licence to any author accepted manuscript (AAM) version arising from this submission.References
- J. A. Blázquez, R. Maça, O. Leonet, E. Azaceta, A. Mukherjee, Z. Zhao-Karger, Z. Li, A. Kovalevsky, A. Fernández-Barquín, A. R. Mainar, P. Jankowski, L. Rademacher, S. Dey, S. E. Dutton, C. P. Grey, J. Drews, J. Häcker, A. Latz, D. Sotta, M. R. Palacin, J.-F. Martin, J. M. García Lastra, M. Fichtner, S. Kundu, A. Kraytsberg, Y. Ein-Eli, M. Noked and D. Aurbach, Energy Environ. Sci., 2023, 16, 1964–1981 RSC.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- Z. Zhao-Karger and M. Fichtner, Front. Chem., 2019, 7, 1–12 CrossRef PubMed.
- X. Sun, P. Bonnick and L. F. Nazar, ACS Energy Lett., 2016, 1, 297–301 CrossRef CAS.
- X. Sun, P. Bonnick, V. Duffort, M. Liu, Z. Rong, K. A. Persson, G. Ceder and L. F. Nazar, Energy Environ. Sci., 2016, 9, 2273–2277 RSC.
- Z. Rong, R. Malik, P. Canepa, G. Sai Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 6016–6021 CrossRef CAS.
- M. Liu, Z. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder and K. A. Persson, Energy Environ. Sci., 2015, 8, 964–974 RSC.
- T. Ichitsubo, T. Adachi, S. Yagi and T. Doi, J. Mater. Chem., 2011, 21, 11764–11772 RSC.
- Z. Lun, B. Ouyang, Z. Cai, R. J. Clément, D. H. Kwon, J. Huang, J. K. Papp, M. Balasubramanian, Y. Tian, B. D. McCloskey, H. Ji, H. Kim, D. A. Kitchaev and G. Ceder, Chem, 2020, 6, 153–168 CAS.
- Z. Lun, B. Ouyang, D. H. Kwon, Y. Ha, E. E. Foley, T. Y. Huang, Z. Cai, H. Kim, M. Balasubramanian, Y. Sun, J. Huang, Y. Tian, H. Kim, B. D. McCloskey, W. Yang, R. J. Clément, H. Ji and G. Ceder, Nat. Mater., 2021, 20, 214–221 CrossRef CAS PubMed.
- R. Sharpe, R. A. House, M. J. Clarke, D. Förstermann, J. J. Marie, G. Cibin, K. J. Zhou, H. Y. Playford, P. G. Bruce and M. S. Islam, J. Am. Chem. Soc., 2020, 142, 21799–21809 CrossRef CAS PubMed.
- R. A. House, L. Jin, U. Maitra, K. Tsuruta, J. W. Somerville, D. P. Förstermann, F. Massel, L. Duda, M. R. Roberts and P. G. Bruce, Energy Environ. Sci., 2018, 11, 926–932 RSC.
- N. Yabuuchi, M. Nakayama, M. Takeuchi, S. Komaba, Y. Hashimoto, T. Mukai, H. Shiiba, K. Sato, Y. Kobayashi, A. Nakao, M. Yonemura, K. Yamanaka, K. Mitsuhara and T. Ohta, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- H. Ji, J. Wu, Z. Cai, J. Liu, D. H. Kwon, H. Kim, A. Urban, J. K. Papp, E. Foley, Y. Tian, M. Balasubramanian, H. Kim, R. J. Clément, B. D. McCloskey, W. Yang and G. Ceder, Nat. Energy, 2020, 5, 213–221 CrossRef CAS.
- H. Liu, Z. Zhu, Q. Yan, S. Yu, X. He, Y. Chen, R. Zhang, L. Ma, T. Liu, M. Li, R. Lin, Y. Chen, Y. Li, X. Xing, Y. Choi, L. Gao, H. S. yun Cho, K. An, J. Feng, R. Kostecki, K. Amine, T. Wu, J. Lu, H. L. Xin, S. P. Ong and P. Liu, Nature, 2020, 585, 63–67 CrossRef CAS PubMed.
- Z. Lun, B. Ouyang, D. H. Kwon, Y. Ha, E. E. Foley, T. Y. Huang, Z. Cai, H. Kim, M. Balasubramanian, Y. Sun, J. Huang, Y. Tian, H. Kim, B. D. McCloskey, W. Yang, R. J. Clément, H. Ji and G. Ceder, Nat. Mater., 2021, 20, 214–221 CrossRef CAS PubMed.
- Y. Idemoto, T. Takahashi, N. Ishida, M. Nakayama and N. Kitamura, Inorg. Chem., 2019, 58(9), 5664–5670 CrossRef CAS PubMed.
- N. Kitamura, Y. Konishi, W. Ma, N. Ishida, T. Mandai, C. Ishibashi and Y. Idemoto, Sci. Rep., 2022, 12, 1–8 CrossRef PubMed.
- T. Kawaguchi, M. Yasuda, N. Nemoto, K. Shimokawa, H. Li, N. L. Okamoto and T. Ichitsubo, J. Mater. Chem. A, 2024, 12, 9088–9101 RSC.
- Z. Zhao-Karger, R. Liu, W. Dai, Z. Li, T. Diemant, B. P. Vinayan, C. Bonatto Minella, X. Yu, A. Manthiram, R. J. Behm, M. Ruben and M. Fichtner, ACS Energy Lett., 2018, 3, 2005–2013 CrossRef CAS.
- W. Ren, D. Wu, Y. Nuli, D. Zhang, Y. Yang, Y. Wang, J. Yang and J. Wang, ACS Energy Lett., 2021, 6, 3212–3220 CrossRef CAS.
- L. A. C., Rep. LAUR 86-748, Los Alamos Natonal Lab., 1990, pp. 121–124 Search PubMed.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Boin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS PubMed.
- S. Raj, Int. J. Mater. Res., 2007, 98, 776–779 CrossRef.
- Z. Zhao-Karger, M. E. Gil Bardaji, O. Fuhr and M. Fichtner, J. Mater. Chem. A, 2017, 5, 10815–10820 RSC.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- O. Chusid, Y. Gofer, H. Gizbar, Y. Vestfrid, E. Levi, D. Aurbach and I. Riech, Adv. Mater., 2003, 15, 627–630 CrossRef CAS.
- B. J. Kwon, S. H. Lapidus, J. T. Vaughey, G. Ceder, J. Cabana and B. Key, Acc. Chem. Res., 2024, 57(1), 1–9 CrossRef CAS PubMed.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS PubMed.
- H. Li, R. Fong, M. Woo, H. Ahmed, D. H. Seo, R. Malik and J. Lee, Joule, 2022, 6, 53–91 CrossRef CAS.
- H. Li, N. L. Okamoto, T. Hatakeyama, Y. Kumagai, F. Oba and T. Ichitsubo, Adv. Energy Mater., 2018, 8, 1–8 Search PubMed.
- H. Li, T. Ichitsubo, S. Yagi and E. Matsubara, J. Mater. Chem. A, 2017, 5, 3534–3540 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02348j |
| This journal is © The Royal Society of Chemistry 2024 |