DOI:
10.1039/D4TA02299H
(Paper)
J. Mater. Chem. A, 2024,
12, 20077-20087
Electrochemical reduction of nitrate to ammonia on ultra-stable amorphous Co–P electrocatalyst†
Received
4th April 2024
, Accepted 13th June 2024
First published on 14th June 2024
Abstract
Electrocatalytic reduction of nitrate (NO3−) to ammonia (NH3) is garnering increasing interest due to its potential to reduce CO2 emissions as a substitute for the Haber-Bosch process, while also mitigating NO3− pollution. However, it remains a challenge to achieve a current density exceeding 300 mA cm−2 while maintaining the stability of catalysts. Additionally, the anodic oxygen evolution reaction, characterized by slow kinetics and high energy barriers, severely impedes the widespread adoption of NH3 formation from NO3− reduction. Therefore, in this study, we introduce amorphous phosphorus-doped cobalt catalysts (Co–P@NF) prepared via a facile electrodeposition process for efficient NO3− reduction and hydrazine oxidation. The incorporation of phosphorus in Co–P@NF facilitates electron migration from phosphorus to cobalt, enhancing *H provision for efficient hydrogenation of the intermediate *NO2−. This results in a current density of 2 A cm−2 at −0.3 V, with a faradaic efficiency for NH3 of 91% in an electrolyte containing 1 M NO3−. Moreover, the Co–P@NF catalyst exhibits remarkable long-term stability, maintaining an NH3 faradaic efficiency exceeding 90% and a current density of 799 mA cm−2 after 82 hours of electrolysis. Furthermore, Co–P@NF displays high catalytic activity in promoting the rate-determining step of hydrazine oxidation, from *N2H2 to *N2H. The incorporation of the HzOR (hydrazine oxidation reaction)-assisted NO3−RR (nitrate reduction reaction) unit significantly reduces the cell voltage to 0.34 V at 300 mA cm−2.
 Yao-Yin Lou | Yao-Yin Lou earned his PhD degree in 2019 from Ecole Nationale Supérieure de Chimie de Rennes, France, where he received joint training from Université de Rennes 1. From 2020 to 2022, he served as an associate researcher at Xiamen University and subsequently worked as an associate professor at Soochow University. Presently, he is a professor at the Institute of Urban Environment, Chinese Academy of Sciences. His research focuses on the development of novel environmental electrocatalysts and electroreduction methods for the elimination of environmental contaminations. Dr Lou has authored over 30 papers in renowned international journals such as Nature Communications, ACS Catalysis, Environmental Science & Technology, Water Research, and Chemical Engineering Journal. |
1. Introduction
Ammonia (NH3) is a vital chemical utilized in various industrial applications, such as chemical production, fuel synthesis, and as a fertilizer. Approximately 50% of global food production depends on NH3 fertilizers.1 The industrial-scale production of NH3 predominantly relies on the Haber-Bosch process, which operates at high temperatures (400–600 °C) and high pressures (200–350 atm).2 This production process is responsible for approximately 1.2% of annual global CO2 emissions, surpassing emissions from other industrial chemical processes.3 To address decarbonization challenges in the ammonia sector, an alternative approach known as the dinitrogen reduction reaction has been developed [4]. However, the dinitrogen reduction reaction exhibits a significantly low reaction rate (<10 mmol gcat−1 h−1),2 which is two to three orders of magnitude lower than that of the Haber-Bosch process. In marked contrast, nitrate anions (NO3−) require only 204 kJ mol−1 of energy to break the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, a significantly lower energy requirement compared to the N
O bond, a significantly lower energy requirement compared to the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond in dinitrogen gas (941 kJ mol−1). Additionally, NO3− anions exhibit excellent solubility in aqueous solutions.4 Consequently, the electrochemical reduction reaction of NO3− (NO3−RR) for NH3 production has garnered increasing attention.
N bond in dinitrogen gas (941 kJ mol−1). Additionally, NO3− anions exhibit excellent solubility in aqueous solutions.4 Consequently, the electrochemical reduction reaction of NO3− (NO3−RR) for NH3 production has garnered increasing attention.
NO3− pollution is a pressing environmental issue, resulting from agricultural runoff, wastewater discharge, and industrial processes. It poses risks to human health and ecosystems, making the development of effective NO3− removal strategies critical. The electroreduction of NO3− aims to produce ammonia, serving the dual purpose of recycling waste materials and mitigating the adverse environmental and ecological impacts of excessive NO3− discharge.5 Thanks to these advantages, various metal-based electrocatalysts, including Cu, Ru, Ag, Fe, Pd, and Co, have been developed to facilitate the reduction of NO3− to NH3.2,6,7 However, the reduction of NO3− to NH3 is a complex eight-electron reaction (eqn (1)), inevitably resulting in the formation of undesired byproducts such as nitrogen oxyanions and dinitrogen.8,9 Due to significant progress in electrocatalyst development, achieving high faradaic efficiencies towards NH3 (FENH3 > 90%) is feasible at pseudo-ampere-lever current densities. Nevertheless, the long-term stability of catalysts operating at currents exceeding 300 mA cm−2 with FENH3 > 90% remains challenging, falling short of the standards set by the U.S. Department of Energy.10 Meanwhile, the anodic oxygen evolution reaction (OER, 2OH− → H2O + 1/2O2 + 2e−, 1.23 V vs. RHE), suffers from slow reaction kinetics and high energy barriers, necessitating an applied potential exceeding 1.6 V.11 The high voltage requires substantial energy expenditure thereby significantly restricting the widespread application of NH3 formation from NO3− reduction. Substituting sluggish reactions with thermodynamically favorable ones, such as hydrazine oxidation reaction (HzOR, N2H4 + 4OH− → N2 + 4H2O + 4e−, −0.33 V vs. RHE), to assist NO3−RR could greatly lower the electricity consumption for NH3 formation. Moreover, the employment of HzOR-assisted NO3−RR offers environmental benefits, converting toxic, carcinogenic N2H4 in wastewater into dinitrogen and water. Developing bifunctional electrocatalysts toward anodic HzOR and cathodic NO3−RR with high activity and durability is pivotal to promote the practical applications.
| | | NO3− + 6H2O + 8e− → NH3 + 9OH−, pH = 14, Eo = 0.69 V vs. RHE | (1) |
NO3−RR is a typical hydrodeoxygenation reaction. Electrocatalysts, like the first-row transition metal phosphides, with good performance in hydrogen evolution and hydrogenation reactions have been reported to have good NO3−RR catalytic activities.2,6 Javier Vela et al.12 found that over 96% selectivity of NH3 from NO3− reduction was achieved on Ni2P, which was previously reported as a good electrocatalyst for hydrogen evolution reaction (HER).13 Yang et al.,14 found that the Co nanocrystals that could generate plenty of *H had an exceptional performance of electrochemical NO3−RR to NH3. Li et al.15 also proved that the generated *H with moderate adsorption energy on the catalyst can behave as important reactive species for the hydrogenation of reaction intermediates to NH3 and suppress the competing HER. Notedly, the good modulation of the adsorption energy of *H and *NO3 on the catalyst surface is critical for the improvement of the catalytic activity and product selectivity.16 Meanwhile, a synergistic benefit was also proposed by Simpson and Johnson for the CuNi alloys: the Ni sites acted as the active sites for the adsorption of *H, and the Cu sites facilitated the adsorption of NO3− intermediates on these binary alloy electrodes.17
On the other hand, amorphous materials with long-range disordered structures have exhibited superior electrocatalytic performance and durability compared to their crystalline counterparts.18,19 The coordination of surface atoms on amorphous materials is highly unsaturated. Also, amorphous alloy has a high degree of structure flexibility, both of which benefit catalysis. Due to the advanced properties, the amorphous-structure catalysts were widely used in oxygen reduction,20 carbon dioxide reduction,21 and hydrogen evolution reaction.22 However, to our best knowledge, few literature reports exist on the amorphous catalysts for the NO3−RR process. Inspired by the exceptional performance of the Co-based catalyst for HER and hydrodeoxygenation reaction,23 the amorphous Co–P was prepared via a facile one-step electrodeposition procedure for HzOR-assisted NO3−RR. The as-prepared amorphous Co–P exhibited superior catalytic performances, which is one of the most advanced NO3−RR catalysts so far, as (1) a high partial current density for NH3 formation (jNH3) was observed at 2 A cm−2, along with a faradaic efficiency (FENH3) of 91% at −0.3 V versus RHE. The corresponding yield of NH3 production was 2.9 mol gcat−1 h−1, which is almost 11-fold higher than that achieved by the Haber-Bosch process (200 mmol gcat−1 h−1); (2) a good stability for NH3 formation with FENH3 over 90% at current density around 800 mA cm−2 for more than 82 h electrolysis; (3) the cell voltage of HzOR-assisted NO3−RR at 300 mA cm−2 over Co–P@NF‖Co–P@NF was only 0.34 V. Electrochemical in situ shell-isolated nanoparticle enhanced Raman spectroscopy (SHINERS) was conducted to clarify the pathways and mechanisms of NO3−RR.
2. Materials and methods
2.1. Materials
Nickel foam (NF) used as electrode substrate was supplied from Suzhou JiaShide Foam Metal Co., Ltd. Trisodium citrate dihydrate (C6H5Na3O7·2H2O) and sodium hypophosphite (NaH2PO2·H2O) were obtained from Meryer Chemical Technology Co., Ltd. CoCl2·6H2O, KOH and (NH4)2SO4 were purchased from Sinopharm Chemical Reagent Co., Ltd. NaNO3 and NaNO2 were bought from Aladdin Co., Ltd. All reagents were analytical grade without any further purification and all solution were prepared by Milii-Q water (MQ).
2.2. Synthesis of Co@Ni foam and Co–P@Ni foam
Phosphorus doped cobalt on the Ni foam substrate (Co–P@NF) was prepared by a simple constant-potential electrodeposition method in a three-electrode system, where Ni foam, Pt foil, and saturated calomel electrode (SCE) were applied as the working electrode, counter electrode and reference electrode, respectively. Before the electrodeposition process, Ni foam (0.5 × 1.0 cm2) was ultrasonically cleaned in acetone solution, ethanol solution and deionized (DI) water several times. An aqueous solution (55 mL) containing 0.2 M C6H5Na3O7, 0.5 M (NH4)2SO4, 0.6 M NaH2PO2 and 0.1 M CoCl2·6H2O were used as the electrolyte. Co–P was deposited on the Ni foam at −2.0 V vs. SCE in the above electrolyte saturated with N2 at room temperature for 45 min. The preparation of Co@NF followed the same process but without the addition of NaH2PO2.
2.3. Assembly of Zn–nitrate flow battery
The Zn–nitrate flow battery was assembled in a membrane electrode assembly flow reactor wherein Co–P@NF (2 cm × 2 cm) was used as both the anode and cathode, respectively. The anodic and cathodic chambers were separated by a Nafion N115 proton exchange membrane. Bipolar plates were high-purity titanium plates with a single serpentine flow field and guard plates were Perspex sheets. The catholyte was 1 M NaOH/0.1 M KNO3 while the anolyte was 6 M NaOH, and their flow rates were controlled to be 150 mL min−1 by a peristaltic pump. Chronopotentiometry was recorded to plot the polarization curves. The power density (P) was calculated by the following equation: P = j × E, where j is the current density and E is the cell voltage.
2.4. Electrochemical measurements
The electrocatalytic performances of Co@NF and Co–P@NF were investigated in a three-electrode system connected to a DH7000C electrochemical workstation (Donghua, Jiangsu) in an H-type cell with a Nafion 115 membrane as a separator. The Co@NF and Co–P@NF with the same size (0.5 × 0.5 cm2) were used as working electrodes, while a platinum foil and a saturated calomel electrode were used as the counter electrode and reference electrode. 1 M KOH aqueous solution (30 mL) containing different NaNO3 concentrations (i.e., 10, 20, 50, 100 and 1000 mM) and 1.0 M N2H4 were used as the cathodic and anodic electrolyte, respectively. The electrochemical linear voltammetry (LSV) analysis was carried out in a single undivided electrochemical cell. The Tafel slope was obtained by fitting the liner part of the Tafel plots according to the Tafel equation (η = a + b × log(j)). The electrochemical impedance measurement was performed in a frequency range of 0.01 Hz to 1000 Hz with an amplitude of 5 mV. The double layer capacitance (Cdl) of the obtained samples was calculated from CV curves at different scan rates of 10–100 mV s−1 in the non-Faraday area. The current density was normalized to the geometric electrode area (0.25 cm2) unless otherwise specified. All recorded electrode potentials vs. Hg/HgO were transformed to potentials against the reversible hydrogen electrode (RHE) according to the calibration equation expressed as ERHE = EHg/HgO + 0.098 + 0.0591 × pH.
3. Results and discussions
3.1. Catalyst composition and microstructure
The Co–P@NF exhibits a much rougher surface than Co@NF (Fig. 1a and b), as indicated by the irregular nanograins formed on the surface of Co–P@NF (Fig. 1c), compared to the flat surface of Co@NF (Fig. 1d). As shown in the high-resolution transmission electron microscopy (HRTEM) images, the nanograins on Co–P@NF were amorphous (Fig. 1e), while the lattice fringe of Co@NF was observed (Fig. 1f). There was also no diffraction pattern for Co–P@NF, demonstrating the amorphous feature of this catalyst (Fig. S1†). The lattice distance of 1.91 Å was well ascribed to the (101) plane of metallic Co. The element mapping analysis (Fig. 1g) indicated that Co and P were evenly dispersed in the nanograins of Co–P@NF. The different structure morphologies of Co–P@NF and Co@NF were also confirmed by the XRD pattern (Fig. 1h). There was no crystalline peak observed in the XRD pattern of Co–P@NF, except the two peaks belonging to the Ni substrate. In comparison, two small peaks at 41.6° and 44.5° were observed in the XRD pattern of Co@NF, which were assigned to Co(100) and Co(101). The elemental compositions and electronic structure of Co–P@NF were determined using XPS (Fig. 1i and j). For Co–P@NF, P 2p showed a doublet with a binding energy of 128.3 (P 2p3/2) and 130.2 eV (P 2p1/2), assigned to elemental P (P0) and oxidized P (P5+), while Co 2p depicted two peaks at 781.8 eV (Co 2p3/2) and 797.6 eV (Co 2p1/2) assigned to oxidation of Co resulting from the exposition of the catalyst to air. There was no P 2p peaks found in the Co@NF spectra. It was well-fitted with the results from the HRTEM-EDS analysis. There were about 13 at% P in Co–P nanograins (Fig. S2 and Table S1†). Compared to Co–P@NF, the Co 2p3/2 and 2p1/2 peaks of Co@NF exhibited a small shift towards higher binding energies (Fig. 1i). This shift indicated that the introduction of P promotes electron migration from the P to the Co, which makes Co electron-rich and becomes more favorable for the NO3−RR processes. The electron-rich property of Co–P@NF was also demonstrated by EIS analysis (Fig. S3†). Thanks to the electron transfer from P to Co, the impedance of Co–P@NF (0.32 Ω) was lower than that of the Co@NF (0.52 Ω).
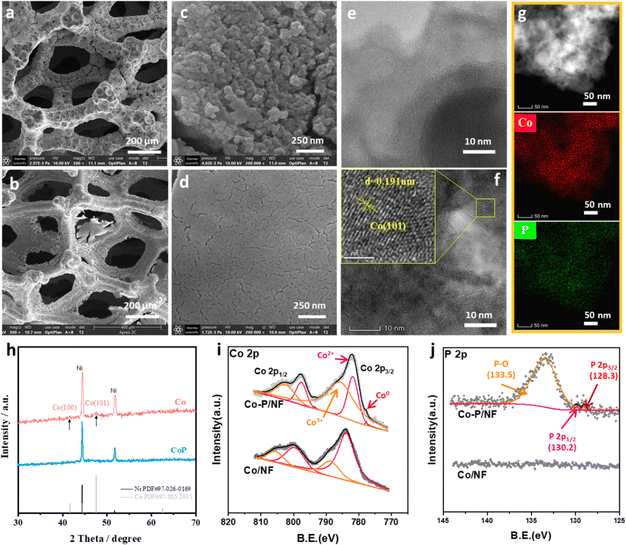 |
| | Fig. 1 SEM imagines of Co–P@NF (a and c) and Co@NF (b and d). HR-TEM imagines of Co–P@NF (e) and Co@NF (f). TEM-EDS mapping of Co–P@NF (g). (h) XRD pattern of Co–P@NF and Co@NF. XPS spectra: Co 2p (i) and P 2p (j) of Co–P@NF and Co@NF. | |
3.2. Electrocatalytic performances of NO3−RR
The linear sweeping voltammetry was firstly applied to investigate the catalytic activity over Co–P@NF and Co@NF during NO3−RR (Fig. 2a). In the absence of NO3−, Co–P@NF has a smaller onset potential (ηHER) and a much higher current density (jHER) of hydrogen evolution reaction than that of Co@NF (ηHER: −0.05 V vs. −0.15 V; jHER at −0.1 V: 69 mA cm−2vs. 12 mA cm−2), indicating the superior catalytic activity of HER on Co–P@NF. The Tafel slop of HER over Co–P@NF (136 mV dec−1) was also smaller than that of Co@NF (164 mV dec−1) (Fig. 2b), demonstrating that Co–P@NF had a faster kinetic for HER and provided more *H for NO3− reduction, according to Volmer step (H2O + e− → *H + OH−). In the presence of NO3−, increased current densities (j) were observed on both Co-based electrodes, since Co was a good candidate for NO3−RR. The larger j on Co–P@NF indicated the enhanced catalytic activity of NO3− reduction after the incorporation of P into Co. Two current reduction peaks were appearing after the addition of NO3−, 0.09 V and −0.03 V on the curve of Co–P@NF, and 0.01 V and −0.15 V on the curve of Co. According to previous studies,24,25 the first peak was assigned to NO3− reduction into *NO2− following a 2-electrons transfer process (eqn (2)), while the second peak was allocated to the *NO2− reduction into NH3 following a 6-electrons transfer process (eqn (3)).| | | NO3− + H2O + 2e− → *NO2− + 2OH− | (2) |
| | | *NO2− + 5H2O + 6e− → NH3 + 7OH− | (3) |
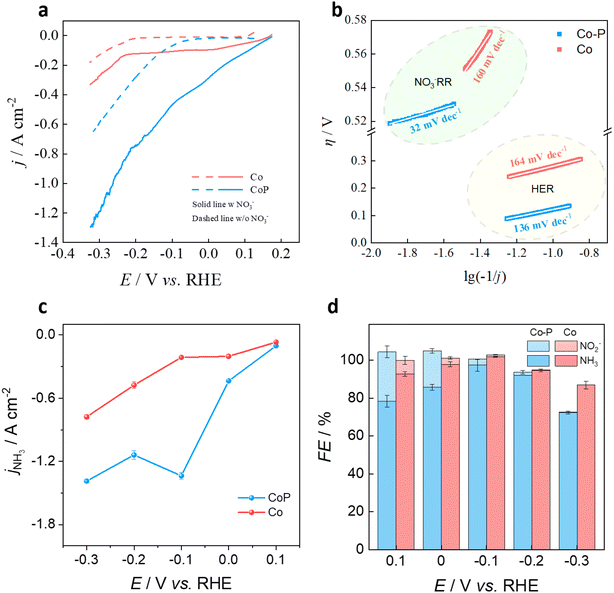 |
| | Fig. 2 (a) Linear sweeping voltammetry over Co@NF and Co–P@NF in the electrolyte of 1 mol L−1 KOH with or without 100 mmol L−1 NO3−. (b) Tafel slop of Co@NF and Co–P@NF in the presence of NO3−. (c) Partial current density of NH3 production over Co@NF and Co-P@NF. (d) FE of NH3 and NO2− over Co–P@NF and Co@NF in the potential range of 0.1–−0.3 V. | |
Co–P@NF displayed positive shifts of 80 mV for the NO3− → NO2− peak and 120 mV for the NO2− → NH3 peak, compared to Co@NF, suggesting a drop of the energy barrier of *NO3− reduction to NH3 on Co–P@NF. According to the Tafel slop in the NO3−-containing electrolyte, Co–P@NF (32 mV dec−1) had a higher kinetic activity for NO3−RR than that of Co (160 mV dec−1). EIS results also revealed the incorporation of P into Co would decrease the electron transfer resistance that was beneficial to facilitate NO3−RR process. The constant-potential electrolysis was performed and the partial current density of NH3 formation (jNH3) at different applied potential was obtained (Fig. 2c). The concentration of NH3 and NO2− formed was detected by colorimetric methods using a UV-vis absorbance spectrophotometer (DR3900) and ion chromatography (ICS-600), respectively (Text S1, Text S2, Fig. S4 and S5†). The substrate of NF was relatively inactive for NO3−RR (Fig. S6†), since the jNH3 on Ni foam was only 7 mA cm−2, compared with the electrodeposited Co-based catalysts (1296 mA cm−2 on Co–P@NF and 212 mA cm−2 on Co@NF). Significantly higher jNH3 was obtained with Co–P@NF, surpassing 1 A cm−2 at potential below −0.1 V vs. RHE. At this potential, the highest faradaic efficiency for NH3 formation (FENH3, 97.5%) was achieved with Co–P@NF (Fig. 2d). Consequently, the Co–P@NF electrode shows superior performance with high FENH3 and jNH3 for NO3− electroreduction to NH3 as compared to other reported electrodes, as exhibited in Table S2.† The low FENH3 over Co–P@NF at the potential of 0.1 V and 0 V, where the HER did not occur and the *H coverage (θH) was too low to further hydrogenation of NO2− to NH3. Until the potential negatively shifted to −0.1 V, the θH increased, based on eqn (4), and therefore the maximum FENH3 was obtained. As potential continued to decreasing, the FENO2– significantly declined but the slight decreased FENH3 was also observed due to the strong HER competition on the Co–P@NF (Fig. 2d). The competitive HER also led to a decrease in jNH3 from 1336 mA cm−2 to 1138 mA cm−2, when potential was negatively shifted from −0.1 V to −0.2 V. The electrochemically active surface area (ECSA) was also measured (Fig. S7a–c†), revealing that Co@NF and Co–P@NF had comparable ECSA. However, a higher ECSA-normalized current density for NH3 production (jNH3(ECSA)) was observed with Co–P@NF (Fig. S7d†), indicating that this catalyst had higher intrinsic activity for NH3 production. To clarify the possible N pollution caused by nitrogen reduction reaction, the electrolysis in the electrolyte free of NO3− ions was performed and little NH3 was produced (Fig. S8†). jNH3 in the solution with NO3− ions was over 133-fold higher than that in the electrolyte free of NO3−, where the FENH3 was only about 1%. In addition, the typical two peaks of 15NH4+ after the electrolysis of 15NO3− also suggested that the NH3 product indeed came from the electrocatalytic reduction of NO3− (Fig. S9†).
| |  | (4) |
where
θH and
θ0H are the *H coverage at
E and
Eo, respectively;
F is the Faraday constant, 96
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
485C mol
−1;
R is the gas constant, 8.314 J mol
−1 K
−1;
T is the kelvin temperature, K.
It should be noted that the sum FE of NO2− and NH3 formation was over 100% (Fig. 2d). The NO3−RR over Co–P@NF and Co@NF were then performed at open circle potential (OCP) and the NO2− and NH3 were both detected in the electrolyte (Fig. 3a and S10†), indicating that there was a spontaneous reduction of NO3− to NO2− and NH3 on Co–P@NF and Co@NF. This spontaneous reduction process may explain the over-100% FE. A similar phenomenon was reported by Han and his colleagues:26 oxidation of Co to Co(OH)2 by NO3− (eqn (5)) and the electroreduction of Co(OH)2 to Co (eqn (6)) proceed simultaneously during the NO3−RR (eqn (2) and (3)), and a dynamic Co valence cycle was achieved. The OCP over Co–P@NF was obviously lower than that of Co@NF (Fig. S11†), indicating that Co–P@NF had a superior ability of the spontaneous reduction of NO3−, thanks to the electron-rich of Co atom in Co–P@NF, giving that the NO2− and NH3 formation on Co–P@NF was approximate 3-times higher than that of Co@NF at OCP.
| | | NO3− + Co + H2O → *NO2− + Co(OH)2 | (5) |
| | | Co(OH)2 + 2e− → Co + 2OH− | (6) |
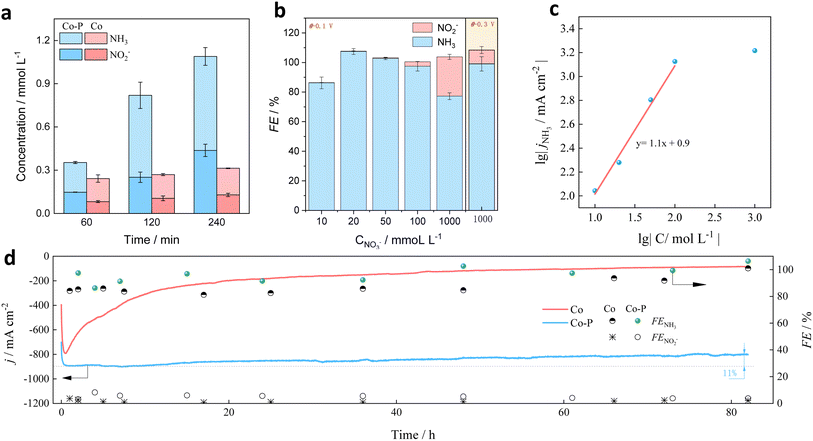 |
| | Fig. 3 (a) Concentration of NO2− and NH3 over Co–P@NF and Co@NF at open circle potential. (b) FE of NO2− and NH3 and (c) lg(−j) vs. lgC plots for Co–P@NF were calculated at −0.1 V vs. RHE. (d) Long-term stability of Co–P@NF in the electrolyte of 1 mol L−1 with 100 mmol L−1 NO3− at −0.1 V. | |
The concentration of NO3− in real wastewater can vary from 1 mmol L−1 to 2000 mmol L−1.27 Therefore, NO3−RR on Co–P@NF was performed in a wide NO3− concentration range. The FENH3 was maintained above 95% in the range of 20 mmol L−1 to 100 mmol L−1 at −0.1 V (Fig. 3b). The low FENH3 in the solution of 10 mmol L−1 and 1000 mmol L−1 was due to the competitive adsorption between *H and *NO3. For example, in the case of 10 mmol L−1, NO3− concentration was so low that catalytic sites were occupied by *H which would be formed into H2via Volmer-Tafel or Volmer-Heyrovsky mechanism and decrease the FENH3. When the NO3− concentration increased, the HER would be gradually hindered so that the FENO2– increased. FENO2– was about 27% in the case of 1000 mmol L−1 NO3−, where *H was insufficient for *NO2− reduction (eqn (3)), leading to the accumulation of *NO2− on the catalytic surface. A quasi-first-order reaction relationship between j and the NO3− concentration was obtained (Fig. 3c), revealing that the rate-determining step of NO3−RR (RDSNO3–RR) was NO3− → NO2− step. Therefore, supplement of sufficient *H was significant for efficient reduction of NO2−. When a potential of −0.3 V was applied in the solution of 1000 mmol L−1, an increase in FENH3 and a decrease in FENO2– were observed, respectively. This is attributed to a higher θH (according to eqn (4)) and a stronger affinity of *H at more negative potentials,28 which promotes further hydrogenation of NO2− to NH3. Meanwhile, jNH3 increased to 2 A cm−2 with the FENH3 as 91% (Fig. S12†). The corresponding NH3 yield rate was 2.9 mol gcat−1 h−1 that was almost 11-fold higher than that of Haber-Bosch route (200 mmol gcat−1 h−1). Long-term stability tests at −0.1 V vs. RHE assessed the durability of Co–P@NF against Co@NF (Fig. 3d). For Co–P@NF, FENH3 (>90%) remained stable after the 82 h electrolysis. The jNH3 experienced a slight decrease from 890 mA cm−2 to 799 mA cm−2 (about 89% of the initial jNH3), which still exceeded U.S. Department of Energy's standard requirement by more than two fold (300 mA cm−2). For Co@NF, the jNH3 quickly decayed from 798 mA cm−2 to 198 mA cm−2 after 20 h electrolysis and its FENH3 was also lower than that of Co–P@NF. To further check the stability of the Co–P@NF catalyst, the cyclic electrolysis was also conducted for NO3− reduction. The results demonstrated that the faradaic efficiency of NH3 formation remained consistently at 100% throughout the 11 electrolysis cycles. Moreover, the yield of NH3 formation of the 11th was still two-times higher than that of Haber-Bosch route (200 mmol gcat−1 h−1) (Fig. S13†). Above stability test results indicated the amorphous-structured Co@P indeed had a satisfactory durability during NO3−RR. According to the TEM analysis, the morphology of Co–P was well kept after the consecutive recycling tests (Fig. S14†). Furthermore, XRD (Fig. S15†) and XPS (Fig. S16†) analysis was also performed on the samples after the consecutive recycling tests, and the results demonstrated negligible changes in the chemical compositions and oxidation states. The excellent stability of the catalysts was possibly ascribed to amorphous feature of the Co–P catalysts.
Tert-butanol (TBA), used as a specific *H radical quenching reagent, was added during the process to evaluate the role of active *H (Fig. 4a). Upon the addition of TBA, the j decreased sharply from 546 mA cm−2 to 373 mA cm−2 on Co@NF and from 886 mA cm−2 to 628 mA cm−2 on Co–P@NF. Meanwhile, the FENH3 also decreased from 96% to 88% on Co@NF and from 106% to 100% on Co–P@NF. The quenching test results demonstrated that the *H radicals truly improved the reaction rate and FENH3. We further used dimethyl-1-pyrroline-N-oxide (DMPO) as the radical trapping reagent to monitor the formation of *H radicals with and without using NO3− (Text S3). As shown in Fig. 4b, the ESR spectra display typical 9 signals with intensity ratios of 1:1:2:1:2:1:2:1:1 without adding NO3− in the catholyte, which confirms the formation of DMPO-H. When NO3− was added, the typical 9 signals nearly disappeared, indicating that the produced *H is consumed by the intermediates in the NO3−RR process. These results strongly prove the key role of the *H radicals in NO3− reduction into NH3. The electrochemical in situ Raman spectra of Co–P and Co catalysts depicted in Fig. 4c and d, were recorded across a potential window of −0.7–−1.13 V versus the RHE with increments of 50 mV. Three distinct peaks, corresponding to A1g, Eg and F2g vibrations models of Co–O band, were visible on Co–P and Co catalysts at around 686.9, 480.5 and 524.9 cm−1.29 The formation of Co–O band was ascribed to the oxygen vacancies over the two catalysts. The coordination of surface atoms on amorphous materials is highly unsaturated.30 According to the electron paramagnetic resonance (EPR) analysis (Fig. S17†), the Co–P@NF had a much higher density of oxygen vacancies. The intensity of Co–O bands on Co–P was much higher than that Co. The Raman spectra were well fitting with the EPR results that the Co–P with amorphous structure had a higher density of oxygen vacancies. As the potential negatively shifted, the Co–O bands gradually decreased. For Co–P catalyst, the Co–O band could be preserved, revealing that the amorphous structure had more stable oxygen vacancies; while no signal could be observed on Co catalyst after the potential lower than −0.9 V. The oxygen vacancies can be used as the adsorption site for the charge transfer, which is conducive to the adsorption/desorption of the reaction intermediates speeding up the NO3−RR.31
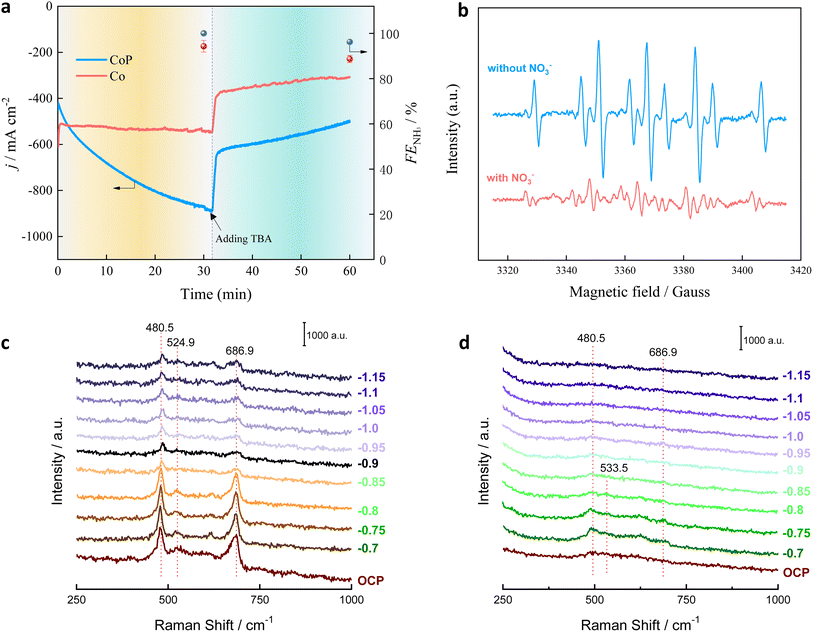 |
| | Fig. 4 (a) j and FENH3 over Co–P@NF and Co@NF at potential of −0.1 V vs. RHE. (b) The DMPO-H* obtained on Co–P@NF after 5 min of electrocatalysis at −0.1 V vs. RHE in 1 M KOH solution with 0.1 M NO3− (bottom) or (b) without NO3− (top). In situ electrochemical–Raman on Co–P (c) and Co (d) in the potential of −0.7 V–−1.15 V vs. Hg/HgO. | |
3.3. Electrocatalytic performances of HzOR
The HzOR electrochemical performance of Co–P@NF was then evaluated in 1.0 M KOH + 1 M N2H4 electrolyte, compared to Co@NF and NF. Co–P@NF showed a higher HzOR catalytic activity with small overpotentials of −67, −16 and 486 mV to reach 10, 100 and 1000 mA cm−2 (Fig. 5a), respectively, compared to Co@NF and NF. The smallest Tafel slope of 24.2 mV dec−1 was obtained in Co–P@NF, indicating the favorable catalytic oxidation kinetics toward HzOR (Fig. 5b). This ultra-low Tafel slope also indicated that the rate-determining step over Co–P@NF was the dehydrogenation from *N2H2 to *N2H. In addition, a comparison between the OER and HzOR performances over Co–P@NF was performed (Fig. 5c and d). Much lower potentials of 16 and 207 mV were needed in HzOR than those in N2H4-free electrolyte (1.66 and 1.95 V) to reach the current densities of 100 and 500 mA cm−2, respectively, indicating the potential application to replace OER for HzOR-assisted NO3−RR. Therefore, thanks to the excellent NO3−RR and HzOR catalytic performances on Co–P@NF, an HzOR-assisted NO3−RR unit with two-electrode system was assembled by employing Co–P@NF as both anode and cathode. As shown in Fig. 5e, the HzOR-assisted NO3−RR unit exhibits significantly enhanced catalytic activity compared to the stand alone NO3−RR unit, only requiring voltages of 93, 308, and 516 mV to achieve current densities of 100, 300, and 500 mA cm−2, respectively. These values are 1.45, 1.53, and 1.67 V lower than those required by the OER-assisted NO3−RR system. The cell voltage of Co–P@NF‖Co–P@NF at 300 mA cm−2 dramatically dropped from ca. 2.0 V to ca. 0.34 V after addition of N2H4 in the electrolyte containing NO3− ions (Fig. 5f). For comparison, NF as electrodes for the HzOR-assisted NO3−RR unit was also assembled (Fig. S18†). For a current density of 300 mA cm−2, and the cell voltage of NF‖NF (2.40 V) are much higher than that of Co–P@NF‖Co–P@NF (0.34 V), indicating the greatly reduced electricity consumption over Co–P catalysts.
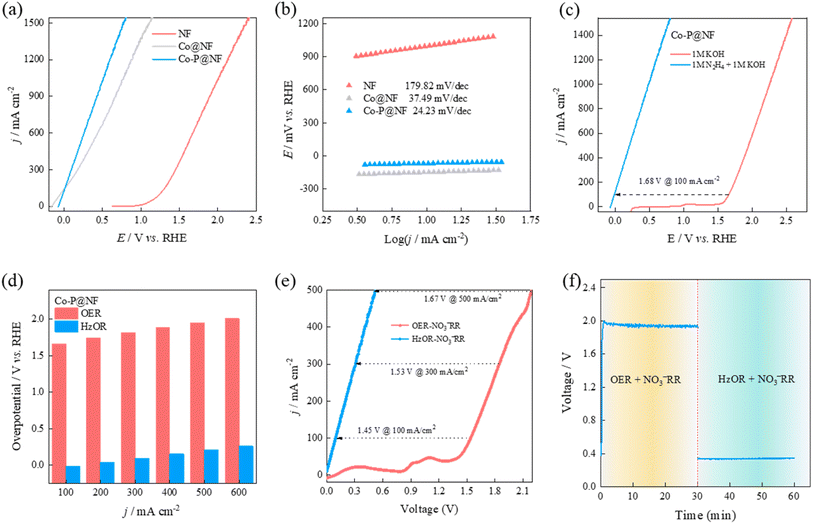 |
| | Fig. 5 HzOR performance in 1.0 M KOH + 1.0 M N2H4 electrolyte. (a) Linear sweep voltammetry analysis on NF, Co@NF and Co–P@NF catalysts and the corresponding (b) Tafel slopes. (c) Compared LSV curves of Co–P@NF between OER (1.0 M KOH) and HzOR (1.0 M KOH and 1.0 M N2H4), and (d) the corresponding histogram catalytic performance comparison. (e) Comparison LSV curves of Co–P@NF between OER-NO3−RR and HzOR-NO3−RR. (f) Cell voltage of Co–P@NF‖Co–P@NF at 300 mA cm−2 in OER-NO3−RR and HzOR-NO3−RR system. | |
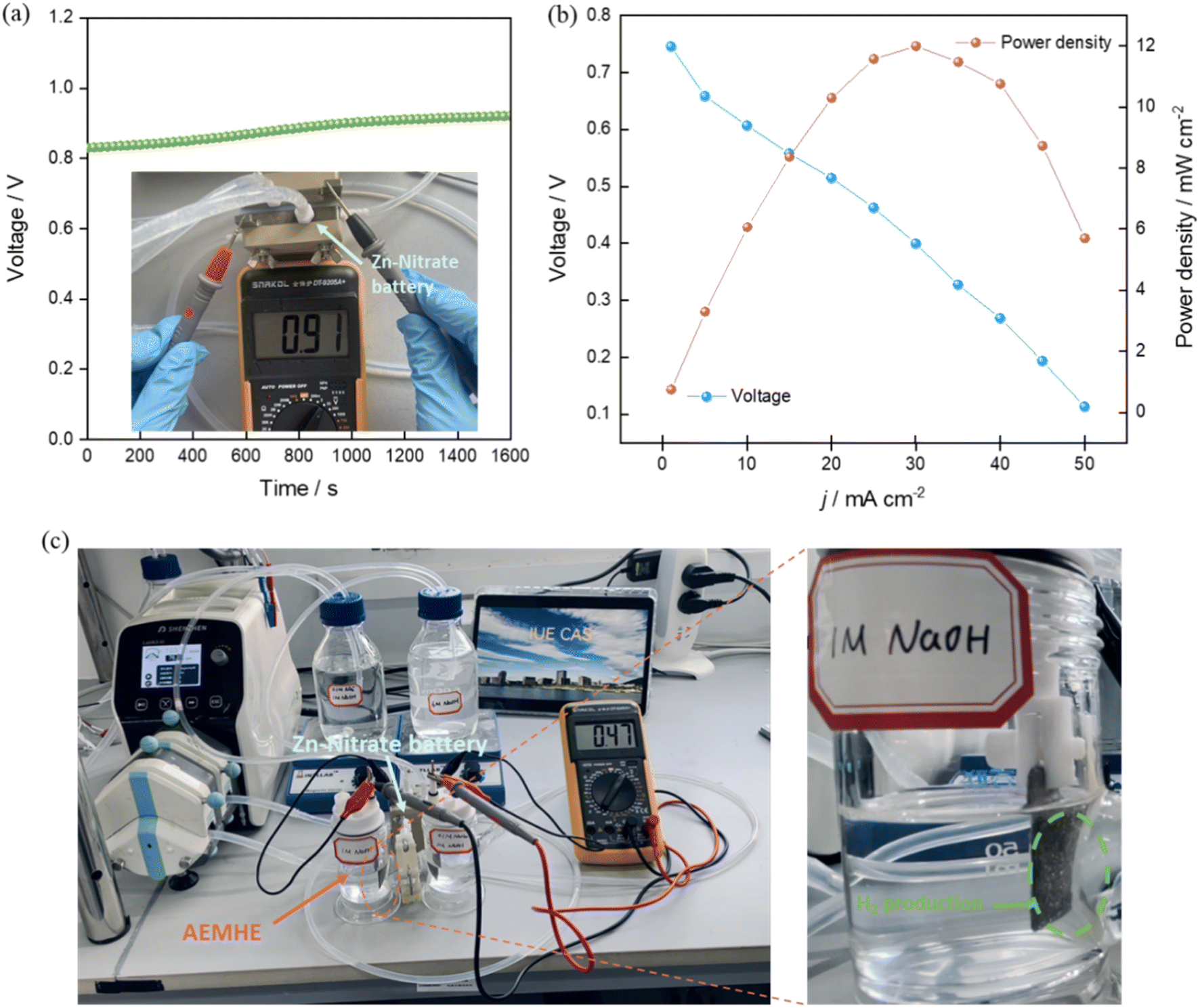
The Zn–nitrate flow battery was assembled with an open circuit voltage over 0.8 V (Fig. 6a). The polarization curves by steady-state chronopotentiometry were obtained. The Zn–nitrate flow battery is practically feasible with the discharging power density up to 12 mW cm−2 (Fig. 6b). To confirm the efficacy of the electricity supply of this battery, the low-voltage-driven anion exchange membrane hydrazine electrolyzer (AEMHE), which used the bimetallic Co–P@NF as both the anode and cathode for the HzOR and HER, respectively (Fig. 6c). The Zn–nitrate flow battery can well drive the AEMHE with only a cell voltage of 0.47 V and give high current density over 35 mA cm−2 for H2 production (Fig. S19†), the corresponding H2 production rate as 0.64 mmol h−1 cm−2. This result shows that the Zn–nitrate flow battery possesses great potential in electricity generation.
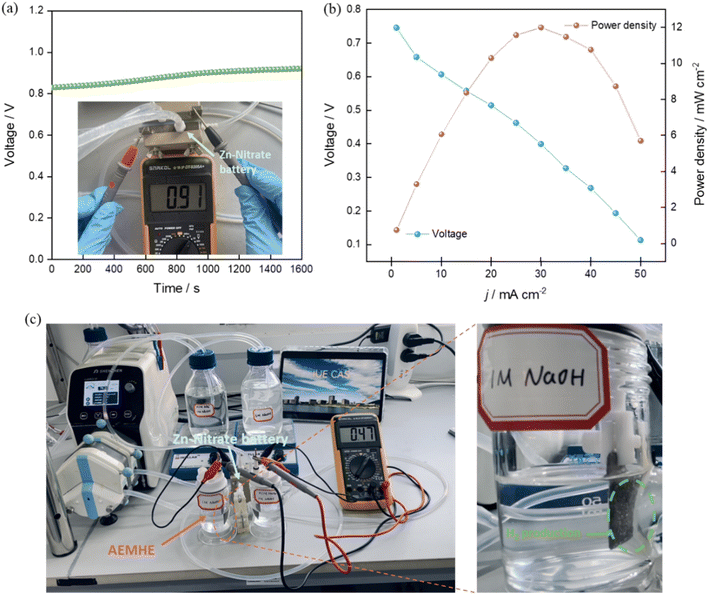 |
| | Fig. 6 Zn–nitrate flow battery application. (a) The open circuit voltage. The cathodic and anodic electrolytes used in Zn–nitrate flow battery were 0.1 M NO3− + 1 M NaOH and 6 M NaOH, respectively. (b) Polarization and power density curves. (c) The digital photograph of the tandem Zn–nitrate flow battery and AEMHE devices for hydrogen production. | |
4. Conclusions
In summary, an amorphous P doped Co (Co–P@NF) was prepared via a facile electrodeposition process. Co–P@NF with a higher density oxygen vacancies sites showed high catalytic activities for both NO3−RR and HzOR. They show a high current density over 2 A cm−2 for NH3 formation and high FE up to 91% at −0.3 V versus RHE in the electrolyte of 1 M NO3−. Thanks to the amorphous structure, the Co–P could preserve the oxygen vacancies sites and give a high stability for NO3− reduction over 82 h electrolysis with the current density over 799 mA cm−2 and the FE over 90%. A low cell voltage of HzOR-assisted NO3−RR at 300 mA cm−2 was also obtained over Co–P@NF‖Co–P@NF and was only 0.34 V. A Zn–nitrate flow battery was assemble using Co–P@NF as electrodes can deliver a peak power density of 12 mW cm2 and well drive an AEMHE, accompanied with an H2 yield rate of ca. 0.64 mmol h−1 cm−2. This work demonstrates the feasibility for boosting the activity and stability toward NO3−RR by constructing the amorphous structure.
Data availability
All relevant data are within the manuscript and its additional files.
Author contributions
Jin-Long Fan: conceptualization, investigation, writing – original draft. Sheng-Bo Liu: investigation. Ming-Liang Chen: writing – review & editing. Zhangxiong Wu: writing – review & editing. Sheng-Peng Sun: investigation, supervision. Yao-Yin Lou: conceptualization, investigation, supervision, writing – review & editing.
Conflicts of interest
No potential conflicts of interest were reported by the authors.
Acknowledgements
This research was financially supported by National Natural Science Foundation of China (NSFC) (No. 22002131) and China Postdoctoral Science Foundation (Grant No. 2020 M671963).
References
- L. K. Boerner, Industrial ammonia production emits more CO2 than any other chemical-making reaction. Chemists want to change that, Chem. Eng. News, 2019, 97, 1–9 Search PubMed
 .
.
- S. Ye,
et al., Elucidating the activity, mechanism and application of selective electrosynthesis of ammonia from nitrate on cobalt phosphide, Energy Environ. Sci., 2022, 15, 760–770, 10.1039/D1EE03097C .
- X. Liu, A. Elgowainy and M. Wang, Life cycle energy use and greenhouse gas emissions of ammonia production from renewable resources and industrial by-products, Green Chem., 2020, 22, 5751–5761, 10.1039/D0GC02301A .
- Q. Hu,
et al., Reaction intermediate-mediated electrocatalyst synthesis favors specified facet and defect exposure for efficient nitrate–ammonia conversion, Energy Environ. Sci., 2021, 14(9), 4989–4997, 10.1039/D1EE01731D .
- J. Gao,
et al., Electrocatalytic Upcycling of Nitrate Wastewater into an Ammonia Fertilizer via an Electrified Membrane, Environ. Sci. Technol., 2022, 56, 11602–11613, DOI:10.1021/acs.est.1c08442 .
- G. Wen,
et al., Ambient ammonia production via electrocatalytic nitrite reduction catalyzed by a CoP nanoarray, Nano Res., 2021, 15(2), 972–977, DOI:10.1007/s12274-021-3583-9 .
- R. Zhang,
et al., Efficient Ammonia Electrosynthesis and Energy Conversion through a Zn-Nitrate Battery by Iron Doping Engineered Nickel Phosphide Catalyst, Adv. Energy Mater., 2022, 2103872, DOI:10.1002/aenm.202103872 .
- J. Zhou,
et al., Linear Adsorption Enables NO Selective Electroreduction to Hydroxylamine on Single Co Sites, Angew. Chem., Int. Ed., 2023, e202305184, DOI:10.1002/anie.202305184 .
- Z. Zhang,
et al., Electrochemical-catalytic reduction of nitrate over Pd-Cu/gamma Al2O3 catalyst in cathode chamber: Enhanced removal efficiency and N-2 selectivity, Chem. Eng. J., 2016, 290, 201–208, DOI:10.1016/j.cej.2016.01.063 .
-
G., S. Renewable, Energy to Fuels through Utilization of Energy Dense Liquids (REFUEL) [EB/OL]
.
- N. K. Oh,
et al., Highly efficient and robust noble-metal free bifunctional water electrolysis catalyst achieved via complementary charge transfer, Nat. Commun., 2021, 12, 4606, DOI:10.1038/s41467-021-24829-8 .
- L. Wei, D.-J. Liu, B. A. Rosales, J. W. Evans and J. Vela, Mild and Selective Hydrogenation of Nitrate to Ammonia in the Absence of Noble Metals, ACS Catal., 2020, 10, 3618–3628, DOI:10.1021/acscatal.9b05338 .
- P. Liu and J. A. Rodriguez, Catalysts for hydrogen evolution from the [NiFe] hydrogenase to the Ni2P (001) surface: the importance of ensemble effect, J. Am. Chem. Soc., 2005, 127, 14871–14878 CrossRef CAS PubMed .
- B. Yang,
et al., Electron-deficient cobalt nanocrystals for promoted nitrate electrocatalytic reduction to synthesize ammonia, Nano Energy, 2023, 117, 108901, DOI:10.1016/j.nanoen.2023.108901 .
- J. Li,
et al., Efficient Ammonia Electrosynthesis from Nitrate on Strained Ruthenium Nanoclusters, J. Am. Chem. Soc., 2020, 142, 7036–7046, DOI:10.1021/jacs.0c00418 .
- J.-Y. Fang,
et al., Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature, Nat. Commun., 2022, 13, 7899 CrossRef CAS PubMed .
- B. K. Simpson and D. C. Johnson, Electrocatalysis of Nitrate Reduction at Copper-Nickel Alloy Electrodes in Acidic Media, Electroanalysis, 2004, 16, 532–538 CrossRef CAS .
- Q. Fu,
et al., Unraveling and leveraging in situ surface amorphization for enhanced hydrogen evolution reaction in alkaline media, Nat. Commun., 2023, 14, 6462, DOI:10.1038/s41467-023-42221-6 .
- X. Li,
et al., Amorphous alloys for electrocatalysis: The significant role of the amorphous alloy structure, Nano Res., 2023, 16, 4277–4288, DOI:10.1007/s12274-021-3682-7 .
- M. Liu, Z. Zhao, X. Duan and Y. Huang, Nanoscale structure design for high-performance Pt-based ORR catalysts, Adv. Mater., 2019, 31, 1802234 CrossRef PubMed .
- Y. X. Duan,
et al., Amorphizing of Cu nanoparticles toward highly efficient and robust electrocatalyst for CO2 reduction to liquid fuels with high faradaic efficiencies, Adv. Mater., 2018, 30, 1706194 CrossRef PubMed .
- Y. C. Hu,
et al., A highly efficient and self-stabilizing metallic-glass catalyst for electrochemical hydrogen generation, Adv. Mater., 2016, 28, 10293–10297 CrossRef CAS PubMed .
- J. Wang,
et al., Recent progress in cobalt-based heterogeneous catalysts for electrochemical water splitting, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed .
- S.-E. Bae, K. L. Stewart and A. A. Gewirth, Nitrate adsorption and reduction on Cu (100) in acidic solution, J. Am. Chem. Soc., 2007, 129, 10171–10180 CrossRef CAS PubMed .
- E. Molodkina,
et al., Electroreduction of nitrate ions on Pt (1 1 1) electrodes modified by copper adatoms, Electrochim. Acta, 2010, 56, 154–165 CrossRef CAS .
- S. Han,
et al., Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism, Nat. Catal., 2023, 6(5), 402–414, DOI:10.1038/s41929-023-00951-2 .
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Electrocatalytic Nitrate Reduction for Sustainable Ammonia Production, Joule, 2021, 5, 290–294, DOI:10.1016/j.joule.2020.12.025 .
- J.-X. Liu, D. Richards, N. Singh and B. R. Goldsmith, Activity and Selectivity Trends in Electrocatalytic Nitrate Reduction on Transition Metals, ACS Catal., 2019, 9, 7052–7064, DOI:10.1021/acscatal.9b02179 .
- T. Begildayeva, J. Theerthagiri, S. J. Lee, Y. Yu and M. Y. Choi, Unraveling the Synergy of Anion Modulation on Co Electrocatalysts by Pulsed Laser for Water Splitting: Intermediate Capturing by In Situ/Operando Raman Studies, Small, 2022, 18, 2204309, DOI:10.1002/smll.202204309 .
- J. Kang,
et al., Recent Progress of Amorphous Nanomaterials, Chem. Rev., 2023, 123, 8859–8941, DOI:10.1021/acs.chemrev.3c00229 .
- J. Wang,
et al., Electrocatalytic Reduction of Nitrate to Ammonia on Low-Cost Ultrathin CoOx Nanosheets, ACS Catal., 2021, 11, 15135–15140, DOI:10.1021/acscatal.1c03918 .
|
| This journal is © The Royal Society of Chemistry 2024 |

 a,
Sheng-Bo
Liu
c,
Ming-Liang
Chen
d,
Zhangxiong
Wu
a,
Sheng-Bo
Liu
c,
Ming-Liang
Chen
d,
Zhangxiong
Wu