
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefects or no defects? Or how to design 20–25 nm spherical iron oxide nanoparticles to harness both magnetic hyperthermia and photothermia†
Barbara Freisab,
Céline Kiefera,
Maria de los Angeles Ramireza,
Sébastien Harleppcde,
Damien Mertz a,
Benoit Pichona,
Cristian Iacovita*f,
Sophie Laurentb and
Sylvie Begin*a
a,
Benoit Pichona,
Cristian Iacovita*f,
Sophie Laurentb and
Sylvie Begin*a
aUniversité de Strasbourg, CNRS, Institut de Physique et Chimie des Matériaux, UMR CNRS-UdS 7504, 23 Rue du Loess, BP 43, 67034 Strasbourg, France. E-mail: sylvie.begin@unistra.fr
bLaboratoire de NMR et d'imagerie moléculaire, Université de Mons, Avenue Maistriau 19, 7000 Mons, Belgium
cInstitut de Cancérologie Strasbourg Europe, 67000 Strasbourg, France
dEquipe Labellisée Ligue Contre le Cancer, France
eStrasbourg Drug Discovery and Development Institute (IMS), 67000 Strasbourg, France
fDepartment of Pharmaceutical Physics-Biophysics, Faculty of Pharmacy, Iuliu Hatieganu University of Medicine and Pharmacy, 6 Pasteur St., 400349 Cluj-Napoca, Romania. E-mail: cristian.iacovita@umfcluj.ro
First published on 7th October 2024
Abstract
Designing iron oxide nanoparticles (IONPs) to effectively combine magnetic hyperthermia (MH) and photothermia (PTT) in one IONP formulation presents a significant challenge to ensure a multimodal therapy allowing the adaptation of the treatment to each patient. Recent research has highlighted the influence of factors such as the size, shape, and amount of defects on both therapeutic approaches. In this study, 20–25 nm spherical IONPs with a spinel composition were synthesized by adapting the protocol of the thermal decomposition method to control the amount of defects. By tuning different synthesis parameters such as the precursor nature, the introduction of a well-known oxidizing agent, dibenzylether (DBE), in the reaction medium, the heating rate and duration and the introduction of a nucleation step, we thus established two different synthesis protocols, one involving the use of a small amount of DBE leading to IONPs with only a few defects and another that took an optimized route to oxidize the wüstite nuclei during the IONP growth and led to IONPs exhibiting more structural and oxygen defects. IONPs exhibiting fewer defects showed enhanced MH and PTT heating values even when immobilized in a matrix, despite a decrease in MH heating values showing that they release mainly heat through the Brownian mechanism. These MH measurements have also confirmed that defects play a key role in enhancing Néel relaxation. PTT measurements demonstrated higher heating values with IONPs with fewer defects and a correlation between Urbach energy and SAR values suggesting an impact of vacancy defects on PTT performances. Therefore, IONPs exhibiting fewer defects under our synthesis conditions appear as suitable IONPs to combine both MH and PTT treatments with high performances. These findings pave the way for promising applications in combined therapies for cancer treatment.
I. Introduction
Spherical spinel iron oxide nanoparticles (IONPs) with sizes in the range 20–25 nm are promising for magnetic hyperthermia (MH) when the size of IONPs has less impact on photothermia (PTT) performance.1–6 In MH, IONPs, upon exposure to an alternating magnetic field (AMF), generate localized heat effective against cancer cells, which are more sensitive to temperature increases than healthy cells.7–10 However, MH's efficacy is hindered by limited heating power, often requiring high local doses of IONPs.2,7,11–13 Enhancing the performance of IONPs is crucial and is generally achieved through increasing saturation magnetization (Ms) and effective anisotropy.2,8,14–16 Larger-sized IONPs exhibit higher Ms improving MH efficiency.2,11,16,17 Despite their promise, IONPs of 20–25 nm size were shown to primarily release heat through Brownian relaxation rather than Néel relaxation, which diminishes their efficacy when located within cells or tissues.9,18–22 Factors such as NP aggregation and immobilization upon interaction with cells contribute to this decrease, affecting their MH efficiency.7,9,18,19,22,23 Additionally, defects within IONPs have been reported to improve MH performance by favoring Néel relaxation in cellular environments.23–25Recent studies have shown that IONPs are also effective for photothermal therapy (PTT) by converting absorbed light into localized heating when exposed to near-infrared laser irradiation. This process would be facilitated by d–d electron transitions, moving electrons from conduction to valence bands.26 Since Yu et al.'s initial report in 2011,27 IONPs have consistently proved to be efficient in generating heat under laser irradiation.4,5,26,28,29 Factors influencing heat release remain somewhat unclear. While some studies suggest that the IONP shape affects heating,4,5 others argue that the crystallographic structure, rather than size or shape, is the primary determinant of PTT effectiveness.6,26,29,30 Bertuit et al.30 examined iron oxide nanoflowers with varying levels of defects and found that excessive defects (oxygen vacancies) led to diminished PTT performance. They attributed this behavior to the trapping of electrons by oxygen vacancies, hindering electron transfer responsible for heat generation.
The recent use of IONPs as PTT nano-heaters offers a novel dual treatment method by combining MH and PTT. This approach benefits from a reduced IONP dose and enhanced MH through laser irradiation. 20–25 nm iron oxide-based nanospheres hold potential for merging MRI and hyperthermia therapy, including PTT and MH. However, the presence of defects appears to have an antagonistic effect on both MH and PTT. Two main types of defects have been noticed: oxygen vacancy defects as reported by Bertuit et al.30 and structural defects such as dislocation and/or antiphase boundaries. The presence of a wüstite core is often observed using the thermal decomposition method (especially when targeting IONPs with a mean size higher than 15 nm), as the nuclei exhibit a wüstite composition.31,32 Structural defects have been reported to form during the oxidation of Fe1−xO in the magnetite phase.7,11,33 Indeed, our previous studies on core–shell Fe1−xO@Fe3−xO4 nanocubes33,34 and the work of Wetterskog et al.35 have shown that the post-oxidation of Fe1−xO induced the formation of lattice dislocations and/or antiphase boundaries in NPs. Dibenzyl ether has been demonstrated to be an effective oxidizing agent7,36,37 either used as solvent or added to the reaction medium. However, to obtain IONPs with a mean size higher than 15 nm, as the core is wüstite, there is competition between the growth and oxidation kinetics, which may be tuned by adjusting synthesis conditions.31
This work investigates the influence of synthesis parameters and dibenzyl–ether (DBE) quantity on the characteristics of iron oxide nanoparticles (IONPs), focusing on size, composition, and defect levels. The challenge lies in consistently producing, using the thermal decomposition method, spherical IONPs in the range of 20–25 nm to be able to combine both MH and PTT therapies. Our aim is to adapt this synthesis method by tuning synthesis parameters to obtain IONPs with precise control over composition and defect levels. Defects, stemming from oxidation processes, including oxygen vacancies and structural irregularities, are crucial in determining the efficiency of MH and PTT combination. We analyze the impact of various parameters like the precursor nature, reaction temperature, heating rate, and DBE amount on the characteristics of IONPs. Both homemade and commercial iron stearates, along with different DBE levels, influence the size, composition, and defects of IONPs. Additionally, an oxidation treatment on core–shell IONPs yields promising results for magnetic and MH properties. Our goal is to design IONPs capable of effectively combining PTT and MH treatments, assessed separately through MH and PTT performance evaluations.
II. Materials and methods
II.1. Materials
Iron stearate (FeSt3) was either homemade12,38,39 from sodium stearate (purity: 98.8%) purchased from TCI and ferric chloride (purity 99%) purchased from Sigma-Aldrich, or directly purchased from TCI (purity minimum 60% of stearic acid and 5.8 to 7% of iron, and maximum 10% of free acid). Acetone (purity 99.8%), chloroform (purity 99%), hexane (purity 99%) and THF (purity 99.5%) were purchased from Carlo Erba. Dioctyl ether (OE) (purity 99%) was purchased from Sigma-Aldrich. Oleic acid (OA) (purity 99%) was purchased from Alfa Aesar. Dibenzylether (DBE) (purity 98%) and squalane (purity 99%) were purchased from Acros Organics. The dendron D1-2P was provided by SuperBranche.II.2. Precursor synthesis
Iron stearate(III) was synthesized by precipitating sodium stearate (NaSt) and ferric chloride (FeCl3) salts in water.12 Sodium stearate (9.8 g, 32 mmol) was dissolved in distilled water (80 mL) in a round-bottomed flask, heated to reflux, and stirred until fully dissolved. Separately, ferric chloride (2.88 g, 10.6 mmol) was dissolved in water (40 mL) and added to the sodium stearate solution under vigorous stirring, resulting in immediate formation of a light orange precipitate. After 15 minutes of stirring at this temperature, the solution was cooled down to room temperature. The precipitate was washed once by centrifugation (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min), and then filtered with a Büchner funnel and oven-dried at 65 °C for 24 hours, yielding approximately 9 g of the precursor. One batch of the precursor was sufficient for four 20 nm IONP syntheses.
000 rpm, 10 min), and then filtered with a Büchner funnel and oven-dried at 65 °C for 24 hours, yielding approximately 9 g of the precursor. One batch of the precursor was sufficient for four 20 nm IONP syntheses.
II.3. 20 nm spherical IONP synthesis
For 20 nm spherical IONP synthesis, several protocols were tested and are discussed in this work. Only the two main protocols for the synthesis of reproducible 20 nm spherical NPs are described hereafter. All the syntheses were performed in air.II.4. Oxidation process of core–shell IONPs
5 ml of a suspension of core–shell IONPs (Fe1−YO@Fe3−XO4) suspended in THF were mixed with 5 mL of dioctylether and 0.5 g of OA in a two-neck round-bottom-flask of 100 mL. The mixture was heated to 120 °C (heating device temperature: 130 °C) for 30 min and the IONPs were resuspended in 10 mL of THF for their storage or further utilization.II.5. Dendronization step
After synthesis, the IONPs are coated with OA and suspended in THF. Subsequently, a ligand exchange is performed in THF to replace the OA with dendron molecules. The dendron is composed of a bi-phosphonate anchoring group to ensure a robust link to the NP surface, and three PEG chains for biocompatibility and stability in water (Fig. S1†). The central PEG chain is longer and terminated with a carboxylate function, enabling the attachment of additional molecules such as dyes or targeting ligands.44 The ligand exchange between OA and the dendron (D1-2P) (Fig. S1†) was performed in THF. Briefly, 5 mL of IONPs@OA at 1 mg Fe mL−1 was stirred for 48 hours with 10 mg of dendron. To maximize the ligand exchange and remove most of the oleic acid molecules, the suspension was then purified by ultrafiltration. After that, 5 mg of dendron was added to the mixture for another 48 hours of stirring. The suspension was mixed with hexane (volume ratio 1/3:2/3) to precipitate the DNPs. The suspension was centrifuged at 8000 rpm for 5 min and the supernatant was discarded. Finally, the DNPs were collected and dispersed in deionized water. The water suspension was purified by ultrafiltration 3 times.
II.6. Characterization techniques
| ΔT = (Sm/k)(1 − exp(−k(t − t0))) |
| SAR = (c m Sm)/mFe |
where ms and CS are, respectively, the mass (in g) and the heat capacity (in J g−1 K−1) of the sample, mFe3O4 (in g) is the mass of iron oxide present in the sample, and

 is the derivative function of the solvent. Both of these derivatives are determined the same way as that for MH (i.e., a second order polynomial fit of the temperature curve).
is the derivative function of the solvent. Both of these derivatives are determined the same way as that for MH (i.e., a second order polynomial fit of the temperature curve).III. Results
III.1. Effect of the FeSt3 precursor and the reaction temperature range on IONP synthesis
Various synthesis parameters such as the type of precursor, solvent, and ratio were investigated to establish an optimal synthesis protocol for producing 20 nm spherical IONPs.31,38,39 However, there is still a need to understand how to achieve greater reproducibility in size, composition and defects across multiple synthesis attempts. Two iron stearate precursors, namely FeSt2 and FeSt3, with molar ratios of Fe:St of 1:2 and 1:3, respectively, were previously examined. It was demonstrated that only FeSt3 allowed obtaining spherical IONPs with a mean size exceeding 15 nm.31,38 Indeed, FeSt2 is primarily composed of [Fe3-(μ3-O)St6xH2O]Cl, whereas FeSt3 is a mixture composed mainly of [Fe7(μ3-O(H))6(μ2-OH)xSt12−2x]St, [Fe3(μ3-O)St6·xH2O]St and free stearic acid. FeSt3 exhibits a broader distribution of complexes, with larger Fe complexes exhibiting greater thermal stability, which contribute to the growth step of nuclei.38 Consequently, the study dedicated to the synthesis of 20 nm spherical IONPs was carried out using homemade FeSt3, synthesized through the coprecipitation of sodium stearate and ferric chloride salts in an aqueous solution, as described in ref. 12, 38 and 39 and the Materials and methods section. Initially, a previously validated protocol for the synthesis of 20 nm sized IONPs with a spinel composition was replicated.7,31 This protocol (P20_1) involved the use of squalane – which has a boiling point (TB = 470 °C) higher than the targeted temperature36 – and the addition of a small amount of DBE, aiming to slightly lower the reaction temperature and to provide oxidant species to the reaction media.7,36,37 Given that only a limited number of IONP syntheses can be performed with one batch of homemade FeSt3, such a study led us to investigate the reproducibility of homemade FeSt3 synthesis in relation to the obtained IONPs. These syntheses were thus conducted with various batches of homemade FeSt3 (denoted as FeSt3-1, FeSt3-2, FeSt3-3, and FeSt3-4) and with a commercial FeSt3 (denoted as FeSt3–TCI).
Multiple batches of synthesized IONPs underwent characterization via transmission electron microscopy (TEM) and X-ray diffraction (XRD), with results presented in Table S1.† Despite attempts to maintain a setpoint temperature of 360 °C for reaching a reaction temperature of 330 °C, different reaction temperatures were observed, likely due to the solvent mixture's boiling point.31 Nevertheless, the TEM mean size of IONPs remained consistent across different temperatures, ranging from 16 to 27 nm, with no correlation between size and temperature. Despite employing the same protocol, variations in mean sizes were evident among batches showing that they were in fact due to variations between homemade FeSt3 batches.
Concerning the IONP composition, the analysis of XRD patterns allows checking the presence of the wüstite phase and consequently of a Fe1−XO@Fe3−xO4 core–shell structure. The wüstite phase exhibits distinct diffraction peaks at varying Bragg's positions compared to the spinel iron oxide phases, with a different lattice parameter of 4.326 Å (powder diffraction PDF file: 01-089-2468). Thus, this wüstite phase can be identified by XRD. Fig. S2† illustrates a representative XRD pattern of synthesized IONPs using the P20_1 protocol and all diffraction peaks correspond only to the spinel iron oxide phase. Comparison of lattice parameters with bulk magnetite (8.396 Å, PDF file: 00-019-0629) and maghemite (8.351 Å, PDF file: 00-039-1346) indicates that the composition of the NPs is closer to that of magnetite, as expected for NPs with such high mean sizes (Table 1).11,31,40 The average crystallite sizes deduced from XRD patterns are lower than the TEM sizes which strongly suggests the presence of defects within the IONPs synthesized with the P20_1 protocol.7,31 Indeed, nuclei in the thermal decomposition process often exhibit a wüstite composition in which oxidation during the growth step leads to defects.31,32,38 In addition, at the nanoscale, Fe2+ ions at the surface of magnetite are easily oxidized leading to a core–shell structure, typical of IONPs with a mean size higher than 12 nm, consisting of a Fe3−xO4 core surrounded by an oxidized shell.11,13
| Synthesis | NP_P20_2_DBE_A | NP_P20_2_DBE_B | NP_P20_2_DBE_C |
|---|---|---|---|
|
|
|
|
| FeSt3 | FeSt3–TCI | FeSt3–TCI | FeSt3–TCI |
| T1 (°C) | 123 | 123 | 126 |
| T2 (°C) | 327 | 331 | 336 |
| Mean TEM size (nm) | 23.2 ± 2.2 | 26.1 ± 2.2 | 26.0 ± 1.7 |
| XRD crystallite size (nm) | 17.3 ± 1 | 18.5 ± 1 | 26.5 ± 1 |
| Lattice parameter (Å) | 8.395 ± 0.001 | 8.393 ± 0.001 | 8.393 ± 0.001 |



Significant variations in size were observed among different batches of IONPs. A detailed analysis of homemade precursor structures (detailed in the ESI†) aimed to understand these differences. Characterization studies of homemade and commercial batches (FeSt3–TCI) revealed disparities in carboxylate coordination, polycation type, and thermal stability (Fig. S3†). FeSt3 synthesis is particularly delicate due to the presence of larger iron polycations (MADI-TOFF analysis in Table S1†), making consistent polycation composition control challenging. Consequently, the commercial batch was chosen for further experiments. The thermal decomposition synthesis with this commercial FeSt3 and the P20_1 protocol at 326 °C led to IONPs with a similar spinel composition, a TEM size of 23.0 ± 2.4 nm and a crystallite size of 18.3 nm. Such a discrepancy between the mean TEM and crystallite sizes was already observed in earlier studies in NPs with similar sizes7,31 or in nanocubes33,34 and that was shown to be due to the presence of structural defects by HRTEM. Such HRTEM characterization will be performed below on the most suitable NPs.
With the objective to transfer these IONPs in water and to test them for hyperthermia, attempts were made to coat them with the dendron molecule (Fig. S1†). We thus observed that the reaction temperature required for synthesizing larger IONPs can impact the dendronization step. A ligand exchange was performed in THF to replace OA with dendron molecules. However, applying dendronization to 20 nm IONPs synthesized using a specific protocol resulted in large aggregates in water, indicating unsuccessful dendronization. To understand this, we compared batches from this protocol with those from a standard synthesis method for 10 nm IONPs.11 The main difference was the reaction temperature: the standard method heated the mixture to the boiling point of octyl ether (290 °C) for two hours, while the P20_1 protocol heated the reaction mixture to 330 °C for larger IONPs. We hypothesized that prolonged heating at this high temperature may degrade the Fe–OH bonds on the IONPs’ surface (left part of Fig. 1), leading to a phenomenon comparable to passivation and resulting in the conversion of Fe–OH bonds to bridging Fe–O–Fe, ultimately affecting the ligand exchange process.
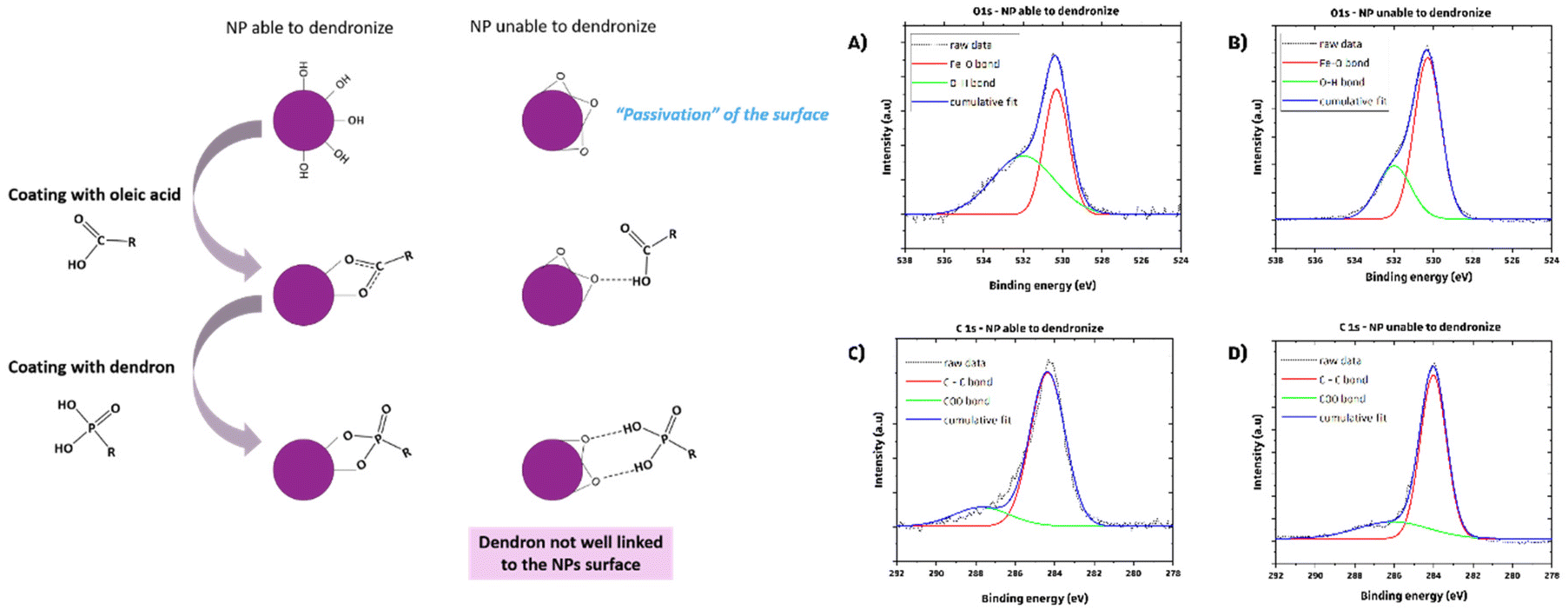 | ||
| Fig. 1 Left: theoretical scheme depicting the comparison of the dendronization process between two types of NPs: one with the Fe–OH surface group and another NP that was synthesized at an excessively high temperature, resulting in the formation of a Fe–O–Fe bridging bond on its surface. Right: deconvoluted O 1s (A and B) and C 1s (C and D) spectra of two sets of NPs: capable of undergoing successful dendronization (A and C) and unable to do so (B and D). | ||
XPS analyses were conducted to verify our hypothesis regarding the surface chemical bonds of two types of IONPs: those produced using the P20-1 protocol (at ≈330 °C) and those synthesized via the 10 nm protocol (at 290 °C). Results indicate a higher intensity of the Fe–OH XPS band (peak at 532 eV) on NPs suitable for dendronization compared to those synthesized at 360 °C for 2 hours (Fig. 1B–D), suggesting a reduced presence of oleic acid molecules and supporting the hypothesis of a bridging Fe–O–Fe bond on their surface. Modifications to the synthesis protocol were necessary to address this dendronization challenge, involving adjustments to heating rate, reaction duration, and temperature. Further studies are needed to optimize the synthesis of 20 nm spherical IONPs for dendronization, including evaluating the role of DBE in the synthesis process.
III.2. Effect of the heating rate and reaction temperature and duration on the size and composition of IONPs
A 2 hour reaction at 330 °C was shown to affect the dendronization step. To address this, two options are proposed: lowering the synthesis temperature to around 310 °C or reducing the reaction time to 330 °C. Lowering the temperature significantly deviates from squalane's boiling point, complicating temperature control and compromising repeatability. It also risks preserving the wüstite core, requiring high temperatures for oxidation and nanoparticle growth. Alternatively, reducing reaction time, though intensifying competition between oxidation and growth kinetics, is more suitable. A new protocol (P20_2) decreases reaction time to one hour and adjusts the FeSt3:OA ratio from 1:2 to 1:3. OA is recognized for its ability to stabilize the iron precursor, leading to a low yield in nuclei and thus resulting in a high concentration of monomers/precursors available for the growth step10,39 but also it was expected to allow better control of the reaction temperature. In addition, these specific reactant ratios and amounts have proven successful in previous studies.1,2,6
Different batches of monodisperse spherical IONPs, named NP_P20_2_DBE_A, NP_P20_2_DBE_B and NP_P20_2_DBE_C given in Table 1, were thus synthesized. However, the reaction temperature was again difficult to precisely control leading to syntheses with slightly different reaction temperatures and thus different mean IONP sizes: 23 nm at 330 °C and 26 nm at 331 °C and 336 °C.
Syntheses at various temperatures were also performed to assess the impact on reaction outcomes. It was found that the mean diameter of IONPs increased with reaction temperature (Fig. 2A), highlighting the stability and consistency of syntheses using commercial FeSt3. Conversely, inconsistent results were observed when using homemade FeSt3 at different temperatures. This reaffirms the choice of commercial FeSt3 as the iron precursor. Nonetheless, variations in the composition of IONPs were noted depending on the reaction temperature.
 | ||
| Fig. 2 (A) Mean diameter of IONPs synthesized using protocol P20_2 as a function of the reaction temperature, (B) HRTEM images of core–shell Fe1−XO@Fe3−xO4 IONPs and (C) its corresponding FFT pattern with plane indication (W = wüstite plane, M = magnetite plane) (A). | ||
When IONPs are synthesized below 325 °C (Table 1), both wüstite and spinel phases are observed in most batches (Fig. S4†), with both phases present in all batches below 322 °C. High-resolution TEM confirmed a core–shell structure (Fig. 2B and C), with surface layers exhibiting different crystallographic structures compared to the core. FFT filtering of HRTEM images identified crystallographic planes for both phases (Fig. 2B and C). These results confirm the formation of a wüstite nucleus under these conditions.31,32,38 A reaction temperature above 325 °C is necessary for obtaining NPs with a mean size of 23–26 nm and an iron oxide spinel composition. Dendronization is feasible with these IONPs synthesized with a shorter heating duration. Protocol P20_2 is suitable for synthesizing ∼20–25 nm IONPs with a monodisperse size distribution and a spinel structure if the reaction temperature is strictly above 325 °C.
III.3. Introduction of defects within IONPs by tuning the synthesis conditions
The impact of dibenzyl ether (DBE) on NP synthesis, particularly regarding NP composition, oxidation process, and defect formation, was further investigated. During the synthesis of IONPs, it has been assessed that the nuclei are constituted of Fe1−XO31,32 and the formation of such Fe1−XO nuclei is again confirmed by these experiments with 0.5 mL of DBE and 19.5 mL of squalene. To achieve spinel iron oxide composition, synthesis conditions need to favor the oxidation kinetics of the wüstite phase. Strategies such as adjusting heating rates31 or employing a “nonaqueous redox phase tuning”36,37 method involving DBE as an oxidizing agent facilitate defect-free NP synthesis. However, DBE's low boiling point limits NP size. Mixing DBE with hydrocarbon solvents enables the production of nearly defect-free NPs in the 10–30 nm range. Additionally, it was found that dense oleic acid (OA) coatings on NP surfaces inhibit oxidation kinetics, while byproducts from DBE thermolysis36,37,41 can disrupt these coatings, thereby promoting oxidation kinetics.31To avoid potentially harmful byproducts in biomedical applications, a synthesis method without DBE has been developed consisting in employing a slower heating rate and introducing a nucleation step around 280 °C to favor oxidation kinetics over growth kinetics and/or enhance the oxidation of nuclei before their growth. In addition, as DBE influences the oxidation process limiting the defect formation in resulting IONPs, by omitting DBE, we can produce IONPs with varying defect levels, offering insights into their MH and PTT properties. DBE also influences the shape and composition of IONPs, favoring a uniform spinel structure. We have investigated the impact of DBE quantity, heating rate, and nucleation step on the size, composition, and defects of IONPs.
The influence of varying volumes of DBE on the shape and oxidation kinetics of IONPs was assessed using the P20_2 protocol (Table S2†). Increasing the DBE volume reduced the reaction temperature due to its lower boiling point compared to squalane (298 °C versus 450 °C). Pure DBE hindered the synthesis of significant-sized IONPs, while creating an oxidizing environment. Adding DBE in volumes up to 1 mL resulted in NPs with an average size of 27–32 nm and a spinel composition, promoting size uniformity and oxidation. Further increase in the DBE volume produced more faceted, smaller NPs, resembling cubic shapes, consistent with previous studies.8,37 The lower reaction temperature and by-products from DBE thermolysis contributed to smaller NPs and a maghemite-like composition. Mixing equal volumes of squalane and DBE yielded well-defined, homogeneous NPs but was deemed less ideal due to safety concerns with the mixture's instability at high temperatures. The ratio of crystallite size to TEM size indicated a positive impact of DBE, although some synthesis conditions introduced an oxidized layer with defects.
To maintain size homogeneity and enhance oxidation kinetics during the synthesis of IONPs with a spinel composition akin to magnetite, incorporating a minimal amount of DBE (0.5 to 1 mL) is necessary due to its crucial role in the oxidation process. Magnetite composition is preferred for its higher Ms and protection against further oxidation is provided by the dendronization thanks to the presence of the two anchoring phosphonate groups.33,42 Efforts have been then made to adjust the protocol to improve oxidation kinetics without relying on DBE while ensuring size uniformity.
The syntheses have been thus conducted by implementing a slower heating rate and incorporating a nucleation step in order to establish a better equilibrium between the growth and oxidation kinetics without relying on an oxidizing component like DBE. The nucleation step was set at 280 °C for 30 min, based on FeSt3 TGA/DTA curves (Fig. 2) and prior studies.31,32,39,43,44 The heating rate was reduced to 1 °C min−1, down from 5 °C min−1, until the 60 minute growth stage at 330 °C. This revised method, labeled P20_3, was evaluated with and without DBE, with results summarized in Table 2. It enables the synthesis of spherical IONPs with a spinel composition and significant average size, eliminating the need for DBE. However, particle size uniformity is slightly reduced compared to the P20_2 protocol, with sizes exceeding 20 nm, adjustable via the reaction temperature.
| Synthesis | With DBE | Without DBE |
|---|---|---|
|
|
|
| FeSt3 | FeSt3–TCI | FeSt3–TCI |
| T1 (°C) | 121 | 121 |
| Tnucleation (°C) | 285 | 282 |
| T2 (°C) | 329 | 326 |
| Mean TEM size (nm) | 33.3 ± 3.2 | 25.4 ± 3.0 |
| XRD crystallite size (nm) | 26.2 ± 1 | 23.3 ± 1 |
| Lattice parameter (Å) | 8.391 ± 0.001 | 8.393 ± 0.001 |
| Ratio of crystallite size to TEM size | 0.79 | 0.929 |


The composition's homogeneity is crucial, being evident from comparable XRD patterns of both batches (Fig. S5†), showing similar characteristics, predominantly iron oxide spinel peaks with lattice parameters akin to magnetite. However, a faint XRD peak in NP_P20_3_no_DBE suggests a minor presence of the wüstite phase. Introduction of DBE leads to larger IONPs (>30 nm), possibly altering nucleation and growth. Reproducibility studies reported in Table S3† confirm promising results with lower reaction temperature, albeit slightly more heterogeneous size distribution compared to the previous protocol (P20_2). Additionally, the ratio of crystallite size to TEM size indicates the presence of defects in these IONPs.
The P20_3_no_DBE protocol allowed for the synthesis of uniform 20–25 nm IONPs without using DBE. However, careful temperature control is still essential to maintain composition uniformity, as low temperatures can hinder wüstite core oxidation. Our study confirms the feasibility of synthesizing these nanoparticles with or without DBE. When DBE is omitted, there is a slight reduction in size uniformity, which is not expected to significantly affect magnetic properties. Nonetheless, its absence may affect oxidation kinetics, potentially altering nanoparticle defect levels.
| Synthesis | NP_CS | NP_CS_ox |
|---|---|---|
|
|
|
| FeSt3 | FeSt3–TCI | FeSt3–TCI |
| Mean TEM size (nm) | 22.6 ± 2.3 | 23.2 ± 1.7 |
| XRD crystallite size (nm) | — | 18.3 ± 1 |
| Lattice parameter (Å) | — | 8.393 ± 0.001 |
| Ratio of crystallite size to TEM size | — | 0.79 |


III.4. Evaluation of the amount of defects in IONPs
The demand to quantify defects in IONPs has grown due to their impact on MH and PTT treatments. Various methods exist for characterizing IONPs to evaluate defects. X-ray pattern refinement determines the crystallite size, reflecting the size of the diffracting domain. A NP's crystallite size typically matches its TEM size if it is monocrystalline. However, structural defects can create smaller crystalline domains. A NP with few defects shows a close match between crystallite and TEM sizes, while numerous defects result in a smaller crystallite size. Comparing these sizes allows for an initial assessment of defects, but other techniques should be used for accuracy, considering variations in measurement methods. Table 4 compares this ratio for batches from different synthesis methods, providing initial insights into potential structural defects.| TEM size (±2 nm) | Crystallite size (±1 nm) | Ratio | Mean ratio | |
|---|---|---|---|---|
| NP_P20_2 (with DBE) | 26.6 | 25.2 | 0.95 | 0.96 ± 0.06 |
| 22.2 | 19.3 | 0.87 | ||
| 26.7 | 26.5 | 0.99 | ||
| 26.0 | 26.5 | 1.01 | ||
| NP_P20_3_no_DBE (without DBE) | 23.9 | 23.4 | 0.98 | 0.85 ± 0.09 |
| 22.9 | 19.2 | 0.84 | ||
| 23.7 | 17.8 | 0.75 | ||
| 18.0 | 15.2 | 0.84 | ||
| NP_P20_CS_ox | 23.2 | 18.3 | 0.79 | — |
Comparing crystallite sizes to determine defect amounts is challenging due to sample variability in size from the P20_3_no_DBE protocol. Among eight samples from two synthesis protocols, NPs from the P20_2 protocol (with DBE) show closely aligned average crystallite sizes with TEM measurements. Additionally, their crystallite size ratio consistently indicates fewer defects, confirming DBE's role in creating an oxidizing environment during synthesis, favorable for fewer defects in IONPs. The oxidation treatment of core–shell IONPs yields a slightly lower ratio than that of P20_3_no_DBE batches, suggesting that the oxidation promoted defects as expected.
The HRTEM images show crystallographic planes in the studied IONPs, with FFT revealing irregularities like stacking faults or dislocations (Fig. 3).7,33 In Fig. 3, inverse FFT results from magnetite lattice planes (111) and (222) in IONPs synthesized with P20_2 and P20_3_no_DBE protocols are compared. Notably, the P20_3_no_DBE protocol showed a higher prevalence of defects, particularly edge dislocations, compared to P20_2. While the analysis is not quantitative, it strongly suggests that IONPs from the P20_3_no_DBE protocol have qualitatively more defects than those from the P20_2 protocol.
 | ||
| Fig. 3 Upper row corresponds to IONPs synthesized with protocol P20_2 and the lower row corresponds to IONPs synthesized with protocol P20_3_no_DBE. (A) and (E) High resolution TEM images of IONPs, (B) and (F) the corresponding FFT images, (C) and (G) applied masked on two reflections plans, and (D) and (H) inverse FFT to visualize the crystallographic planes and eventual defects. Blue squares show crystallographic defects. | ||
A comparative analysis was conducted to determine the band gap and Urbach energy (Fig. S7† and Table 5) of various batches of IONPs from their UV-vis spectra (Fig. S8†). Urbach energy (Eu) levels are closely related to the presence of defects,45,46 particularly in semiconductor oxides like magnetite, where localized defect states,30,47 such as oxygen vacancies, can reduce the band gap. Higher Eu values (Fig. S7–S9†) indicate more defects, suggesting an imperfect conduction band.26,45,47,48 The band gap energy remains consistent across samples (3.5 ± 0.2 eV), but significant differences in Eu are observed between batches synthesized with and without DBE. Samples synthesized with DBE show lower Eu values, indicating fewer defects from oxygen vacancies. Attempts to replicate the effects of DBE by altering synthesis parameters (P20_3_no_DBE protocol) result in spinel structure NPs with both point and structural defects. Urbach energy increases notably in samples synthesized after an oxidation treatment, likely due to more oxidation sites from the wüstite core oxidation process.
| Protocol | Batch | Eg (±0.2 eV) | Eu (eV) |
|---|---|---|---|
| P20_2 (with DBE) | NP_P20_DBE_1 | 3.4 | 0.6 |
| NP_P20_DBE_2 | 3.5 | 0.7 | |
| NP_P20_DBE_3 | 3.7 | 0.7 | |
| P20_3_no_DBE (without DBE) | NP_P20_NO_DBE_1 | 3.5 | 1.2 |
| NP_P20_NO_DBE_2 | 3.4 | 1.2 | |
| NP_P20_NO_DBE_3 | 3.4 | 0.9 | |
| Oxidation process | NP_CS_ox | 3.4 | 1.4 |
III.5. Magnetic properties and heating capabilities of IONP suspensions
The impact of the wüstite core, spinel composition, and defect amount on magnetic properties and heating abilities of 20–25 nm spherical IONPs was investigated. This included IONPs from protocols P20_2 and P20_3_no_BDE (NP_20_DBE and NP_20_no_BDE), core–shell IONPs (NP_20_CS), and their oxidized counterparts (NP_20_CS_ox). Magnetic properties were initially measured using SQUID (Fig. 4). Heating capabilities were then studied via MH after dendronization of IONPs, whose colloidal stability was assessed by DLS measurements (Fig. S10†).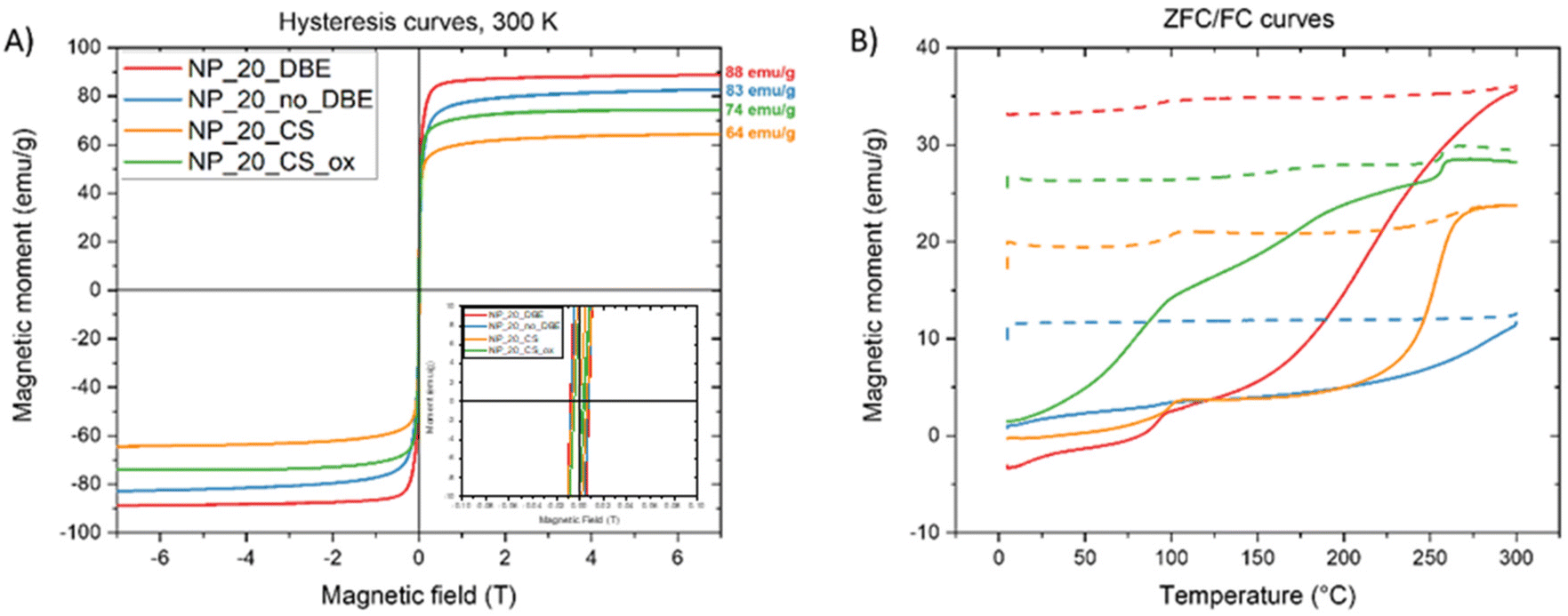 | ||
| Fig. 4 (A) Magnetization curves (insert: zoom between −0.1 and 0.1 T) and (B) ZFC/FC curve of four types of IONPs. | ||
All samples show quite a superparamagnetic behavior at 300 K (Fig. 4A) with a very small hysteresis with a coercive field lower than 0.01T. NP_20_DBE and NP_20_no_DBE exhibit high Ms values of 88 emu g−1 and 83 emu g−1, respectively, close to bulk magnetite (94 emu g−1). The core–shell sample also demonstrates a high Ms of 64 emu g−1, increasing to 74 emu g−1 upon post-oxidation treatment. ZFC and FC curves (Fig. S4B†) reveal high blocking temperatures typical of IONPs of this size range (20–50 nm) due to significant dipolar interactions.49 For the NP_20_DBE sample, there is a noticeable increase in magnetization around 90 K in both the ZFC and FC curves. This increase is attributed to the Verwey transition, which is a characteristic feature of magnetite. The shift of the Verwey transition temperature below 110 K (the usual value for MNPs) has been linked to non-stoichiometric magnetite caused by the presence of Fe2+ vacancies.50 The onset of the Verwey transition is nearly undetectable in the NP_20_no_DBE sample. In contrast, it is quite evident in the core–shell sample. Additionally, the core–shell sample shows a significant increase in magnetization between 225 and 275 K. This feature cannot be related to the Néel transition of wüstite, which is expected around 200 K, but is instead attributed to a highly “disordered” magnetic phase referred to as the X-phase by other groups.51,52 In the oxidized sample, the Verwey transition becomes more prominent than the X-phase transition, indicating the dominance of the magnetite phase, as reflected in the increased Ms.
In the initial stage of the MH study, the heat generation capabilities of four types of iron oxide nanoparticles (IONPs) suspended in water (1 mgFe mL−1) were examined. Specific absorption rate (SAR) values were plotted against the amplitude (H) of alternating magnetic field (AMF), ranging from 5 kA m−1 to 60 kA m−1, at a fixed frequency of 355 kHz. To ensure accuracy, the contribution from pure water at each H was subtracted. IONPs from the NP_20_CS batch, with a core–shell architecture, showed moderate SAR values, increasing slightly with H from 75 W gFe−1 at 5 kA m−1 to 475 W gFe−1 at 60 kA m−1. In contrast, NP_20_CS_ox exhibited a significant enhancement in heating performance, with SAR values nearly doubling at each H value, ranging from 130 W gFe−1 to 850 W gFe−1 as H increased. This improvement is attributed to the conversion of the magnetically inactive würstite core into magnetite/maghemite. IONPs produced using the P20_3_no_BDE protocol, containing defects within their structure and exhibiting a high Ms value, showed higher SAR values over the entire H range compared to previous IONPs, ranging from 80 W gFe−1 to 1525 W gFe−1. The presence of defects within the spinel structure contributed to this increase. IONPs from the NP_20_DBE batch, with reduced defects in their spinel structure and the highest Ms value, yielded the highest SAR values, reaching 1680 W gFe−1 at 30 kA m−1 and 2150 W gFe−1 at 60 kA m−1. These IONPs demonstrate significantly enhanced heating capabilities, surpassing established or revised safety limits for magnetic hyperthermia treatment, with SAR values of 780 W gFe−1 at 15 kA m−1 and 1470 W gFe−1 at 25 kA m−1.
Based on the above analysis, IONPs produced using both P20_2 and P20_3_no_DBE protocols are promising for further in vitro and in vivo MH studies. However, within cells, IONPs are confined to lysosomes, limiting their Brownian motion and relying solely on Néel relaxation for heating. To investigate this, IONPs were immobilized in a PEG8k matrix, either randomly or prealigned under a static magnetic field (strength of 65 kA m−1). SAR values significantly decrease when IONPs are immobilized in PEG, reducing Brownian motion by 68% for NP_P20_DBE and 78% for NP_P20_3_no_DBE (Fig. 5B and C). When the IONPs are prealigned within the solid matrix with their easy axis of magnetization being parallel to AMF lines, the Néel contribution is increased, restoring SAR values by up to 50% compared to water (Fig. 5B and C). However, one may notice that even if the SAR values of IONPs with DBE decrease when they are immobilized, they stay always higher than those of IONPs without DBE. Therefore, IONPs with DBE are the most performant for MH.
 | ||
| Fig. 5 (A) SAR values for NP_P20_DBE, NP_P20_no_DBE, NP_20_CS and NP_20_CS_ox suspensions in water and of (B) NP_20_DBE and (C) NP_20_no_DBE suspended in water and in a PEG8k matrix, random or prealigned, at a concentration of 1 mgFe mL−1. | ||
For photothermal experiments, suspensions of IONPs at 0.05 mgFe mL−1 in chloroform were exposed to a 1064 nm laser at 1 W cm−2 for 3 minutes. A chloroform-only sample served as a control. Temperature was monitored continuously, and temperature changes over time were plotted. IONPs consistently led to significant temperature increases compared to the control. However, discerning a clear trend between synthesis protocols P20_2 and P20_3_no_DBE was challenging, as their temperature–time curves showed similar patterns. Nonetheless, batches synthesized with DBE (P20_2) generally exhibited slightly higher temperature increases compared to those without DBE (P20_3_no_DBE).
Comparing SAR values (Table 6), batches synthesized with the P20_2 protocol show slightly higher values than those with P20-3_no_DBE and a correlation of SAR values with the Urbach energy is observed.30 The SAR values decrease when the Urbach energy increases, but for an Urbach energy of 0.6, the SAR value is 548 W g−1 when it is 412 W g−1 when the energy is two times higher. Further measurements on a broader range of Urbach energy are recommended to confirm this correlation between vacancy defect levels and PTT efficiency. However, IONPs with DBE exhibit notable heating properties even at low concentrations (0.05 mgFe mL−1).
| Protocol | Batch | SAR (W g−1) | Eu (±0.2 eV) |
|---|---|---|---|
| P20_2 | NP_P20_2_DBE_2 | 548 | 0.6 |
| NP_P20_2_DBE_3 | 540 | 0.7 | |
| P20_3_no_DBE | NP_P20_3_no_DBE_1 | 412 | 1.2 |
| NP_P20_3_no_DBE_3 | 497 | 0.9 |
MH measurements indicated that SAR values of IONPs in suspension increase with Ms values, primarily due to Brownian relaxation. Experiments with immobilized IONPs confirmed that defects favor Néel relaxation.7,23 However, IONPs synthesized with DBE with less defects, showed higher SAR values even when immobilized in a matrix and they are thus preferable for both in vitro and in vivo MH applications. Regarding PTT measurements, an impact of defects and especially a correlation with Urbach energy is noted suggesting thus an impact of defects.30 Consequently, the protocol utilizing DBE in our experimental conditions and leading to fewer defects appears optimal for achieving IONPs with superior performance in both MH and PTT.
IV. Conclusion
The objectives were to synthesize IONPs with a mean size of 20–25 nm and a spinel composition exhibiting different “amounts” of defects as defects were more and more demonstrated to impact MH and PTT performances. The synthesis of IONPs with a mean size higher than 20 nm and a spinel composition by the thermal method is always challenging as nuclei with a wüstite composition are formed and thus there is a big issue in controlling both the grain growth and the oxidation kinetics after the nucleation step. The most used approach to face this problem is the addition of DBE, an oxidizing agent, in the reaction media, which avoids obtaining a core–shell structure with a wüstite core and a spinel iron oxide shell and leads to defect free iron oxide spinel NPs. However, DBE can introduce harmful by-products. Therefore, we have tuned different synthesis parameters using the thermal decomposition method to try to avoid the use of DBE and the most important objective here was to synthesize IONPs with a spinel composition and different defect densities.A commercial iron stearate precursor was chosen for reproducibility over homemade ones due to the difficulty in controlling the complex polycation distributions in homemade precursors. Various protocols were tested, revealing optimal conditions of a 1 hour reaction duration and 330 °C reaction temperature to avoid core–shell structure formation. The significance of DBE as an oxidizing agent in producing defect-free IONPs was emphasized by varying its quantity. It was observed that using approximately 0.5 or 1 mL of DBE was adequate to produce IONPs sized around 20–25 nm with a spinel composition. Higher amounts of DBE led to IONPs with cubic shapes and a maghemite composition. Additionally, experiments involving a slower heating rate and a nucleation step, with or without DBE, showed that this protocol could yield IONPs with a spinel composition even in the absence of DBE. Consequently, two distinct protocols have been established for synthesizing IONPs with an average size between 20 and 25 nm, with or without DBE.
The magnetic measurements indicated that IONPs synthesized with DBE had higher Ms values compared to those synthesized without DBE, suggesting a composition similar to that of magnetite. Structural defects were examined using XRD and HRTEM, while point defects like vacancies were assessed by calculating Urbach energy from UV spectra. IONPs synthesized without DBE exhibited more structural defects and higher Urbach energies, indicating the presence of more point defects, in contrast to those synthesized with DBE.
MH and PTT experiments were conducted to evaluate how defects in the spinel structure affect hyperthermia performance. Both experiments indicated that IONPs without defects would show enhanced PTT and MH SAR values. Specifically, IONPs synthesized with DBE showed higher SAR values due to reduced defect content and higher Ms, but SAR values halved when immobilized in a solid matrix. IONPs synthesized without DBE, with more defects, had lower SAR values, but the decrease was less pronounced when immobilized in the solid matrix, indicating less influence of Brownian relaxation. However, even if the SAR values of IONPs with DBE decrease when they are immobilized in a matrix, the SAR values stay higher than those of IONPs synthesized without DBE. PTT measurements showed slightly larger SAR values for IONPs with DBE compared to those without and a correlation is noticed. Other results on a broader range of Urbach energy would be necessary to unambiguously conclude about this correlation. Therefore, IONPs synthesized with DBE exhibiting fewer defects are the best performing ones to combine both MH and PTT in one formulation.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
The Region Alsace, France, and the University of Mons are gratefully acknowledged for the doctoral fellowship to Barbara Freis. This project received funding from ANR (EURONANOMED2020-121 – THERAGET) under the umbrella of the ERA-NET EuroNanoMed (GA No. 723770) of the EU Horizon 2020 Research and Innovation, from Alsace contre le Cancer and ProtherWal “Walloon Region” via the ProtherWal Society (Agreement 7289). SuperBranche is thanked for providing dendron molecules. UMONS acknowledges the financial support of the Fond National de la Recherche Scientifique (FNRS), the ARC Programs of the French Community of Belgium, COST actions and the Walloon region (ProtherWal and Interreg projects). The authors thank the XRD and TEM platform of IPCMS and Dr Vasiliki Papaefthymiou of ICPEES for XPS measurements.References
- G. Cotin, C. B. Andujar, D. V. Nguyen, C. Affolter-Zbaraszczuk, S. Boutry, A. Boos, P. Ronot, B. Uring, P. Choquet, P. E. Zorn, D. Mertz, S. Laurent, R. N. Muller, F. Meyer, D. Felder-Flesch and S. Begin, Nanotechnology, 2019, 30, 374002 CrossRef CAS PubMed.
- C. Blanco-Andujar, A. Walter, G. Cotin, C. Bordeianu, D. Mertz, D. Felder-Flesch and S. Begin-Colin, Nanomedicine, 2016, 11, 1889–1910 CrossRef CAS PubMed.
- A. E. Deatsch and B. A. Evans, J. Magn. Magn. Mater., 2014, 354, 163–172 CrossRef CAS.
- A. Espinosa, R. Di Corato, J. Kolosnjaj-Tabi, P. Flaud, T. Pellegrino and C. Wilhelm, ACS Nano, 2016, 10, 2436–2446 CrossRef CAS PubMed.
- S. Cabana, A. Curcio, A. Michel, C. Wilhelm and A. Abou-Hassan, Nanomaterials, 2020, 10, 1548 CrossRef CAS PubMed.
- B. Freis, G. Cotin, F. Perton, D. Mertz, S. Boutry, S. Laurent and S. Begin-Colin, in Magnetic Nanoparticles in Human Health and Medicine, 2021, pp. 380–429 Search PubMed.
- G. Cotin, C. Blanco-Andujar, F. Perton, L. Asín, J. M. de la Fuente, W. Reichardt, D. Schaffner, D.-V. Ngyen, D. Mertz, C. Kiefer, F. Meyer, S. Spassov, O. Ersen, M. Chatzidakis, G. A. Botton, C. Hénoumont, S. Laurent, J.-M. Greneche, F. J. Teran, D. Ortega, D. Felder-Flesch and S. Begin-Colin, Nanoscale, 2021, 13, 14552–14571 RSC.
- P. Guardia, R. Di Corato, L. Lartigue, C. Wilhelm, A. Espinosa, M. Garcia-Hernandez, F. Gazeau, L. Manna and T. Pellegrino, ACS Nano, 2012, 6, 3080–3091 CrossRef CAS PubMed.
- R. D. Corato, A. Espinosa, L. Lartigue, M. Tharaud, S. Chat, T. Pellegrino, C. Ménager, F. Gazeau and C. Wilhelm, Biomaterials, 2014, 35, 6400–6411 CrossRef PubMed.
- A. Hervault and N. T. K. Thanh, Nanoscale, 2014, 6, 11553–11573 RSC.
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795–3810 CrossRef CAS.
- G. Cotin, C. Kiefer, F. Perton, D. Ihiawakrim, C. Blanco-Andujar, S. Moldovan, C. Lefevre, O. Ersen, B. Pichon, D. Mertz and S. Bégin-Colin, Nanomaterials, 2018, 8, 881 CrossRef PubMed.
- J. Santoyo Salazar, L. Perez, O. de Abril, L. Truong Phuoc, D. Ihiawakrim, M. Vazquez, J.-M. Greneche, S. Begin-Colin and G. Pourroy, Chem. Mater., 2011, 23, 1379–1386 CrossRef CAS.
- A. Sathya, P. Guardia, R. Brescia, N. Silvestri, G. Pugliese, S. Nitti, L. Manna and T. Pellegrino, Chem. Mater., 2016, 28, 1769–1780 CrossRef CAS.
- M. Levy, A. Quarta, A. Espinosa, A. Figuerola, C. Wilhelm, M. García-Hernández, A. Genovese, A. Falqui, D. Alloyeau, R. Buonsanti, P. D. Cozzoli, M. A. García, F. Gazeau and T. Pellegrino, Chem. Mater., 2011, 23, 4170–4180 CrossRef CAS.
- F. Gazeau, M. Lévy and C. Wilhelm, Nanomedicine, 2008, 3, 831–844 CrossRef CAS PubMed.
- J. Mohapatra, A. Mitra, D. Bahadur and M. Aslam, CrystEngComm, 2013, 15, 524–532 RSC.
- J. G. Ovejero, D. Cabrera, J. Carrey, T. Valdivielso, G. Salas and F. J. Teran, Phys. Chem. Chem. Phys., 2016, 18, 10954–10963 RSC.
- D. Cabrera, A. Coene, J. Leliaert, E. J. Artes, N. D. Telling and F. J. Teran, ACS Nano, 2018, 12, 2741–2752 CrossRef CAS PubMed.
- C. Iacovita, G. F. Stiufiuc, R. Dudric, N. Vedeanu, R. Tetean, R. I. Stiufiuc and C. M. Lucaciu, Magnetochemistry, 2020, 6, 23 CrossRef CAS.
- C. Iacovita, R. Stiufiuc, T. Radu, A. Florea, G. Stiufiuc, A. Dutu, S. Mican, R. Tetean and C. M. Lucaciu, Nanoscale Res. Lett., 2015, 10, 391 CrossRef PubMed.
- D. Soukup, S. Moise, E. Céspedes, J. Dobson and N. D. Telling, ACS Nano, 2015, 9, 231–240 CrossRef CAS PubMed.
- A. Lak, M. Cassani, B. T. Mai, N. Winckelmans, D. Cabrera, E. Sadrollahi, S. Marras, H. Remmer, S. Fiorito, L. Cremades-Jimeno, F. J. Litterst, F. Ludwig, L. Manna, F. J. Teran, S. Bals and T. Pellegrino, Nano Lett., 2018, 18, 6856–6866 CrossRef CAS PubMed.
- A. Lappas, G. Antonaropoulos, K. Brintakis, M. Vasilakaki, K. N. Trohidou, V. Iannotti, G. Ausanio, A. Kostopoulou, M. Abeykoon, I. K. Robinson and E. S. Bozin, Phys. Rev. X, 2019, 9, 041044 CAS.
- A. Lak, S. Disch and P. Bender, Adv. Sci., 2021, 8, 2002682 CrossRef CAS PubMed.
- M. E. Sadat, M. Kaveh Baghbador, A. W. Dunn, H. P. Wagner, R. C. Ewing, J. Zhang, H. Xu, G. M. Pauletti, D. B. Mast and D. Shi, Appl. Phys. Lett., 2014, 105, 091903 CrossRef.
- T.-J. Yu, P.-H. Li, T.-W. Tseng and Y.-C. Chen, Nanomedicine, 2011, 6, 1353–1363 CrossRef CAS PubMed.
- A. Espinosa, J. Kolosnjaj-Tabi, A. Abou-Hassan, A. Plan Sangnier, A. Curcio, A. K. A. Silva, R. Di Corato, S. Neveu, T. Pellegrino, L. M. Liz-Marzán and C. Wilhelm, Adv. Funct. Mater., 2018, 28, 1803660 CrossRef.
- C. Lozano-Pedraza, E. Plaza-Mayoral, A. Espinosa, B. Sot, A. Serrano, G. Salas, C. Blanco-Andujar, G. Cotin, D. Felder-Flesch, S. Begin-Colin and F. J. Teran, Nanoscale Adv., 2021, 3, 6490–6502 RSC.
- E. Bertuit, E. Benassai, G. Mériguet, J.-M. Greneche, B. Baptiste, S. Neveu, C. Wilhelm and A. Abou-Hassan, ACS Nano, 2022, 16, 271–284 CrossRef CAS PubMed.
- G. Cotin, F. Perton, C. Petit, S. Sall, C. Kiefer, V. Begin, B. Pichon, C. Lefevre, D. Mertz, J.-M. Greneche and S. Begin-Colin, Chem. Mater., 2020, 32, 9245–9259 CrossRef CAS.
- G. Cotin, B. Heinrich, F. Perton, C. Kiefer, G. Francius, D. Mertz, B. Freis, B. Pichon, J. Strub, S. Cianférani, N. Ortiz Peña, D. Ihiawakrim, D. Portehault, O. Ersen, A. Khammari, M. Picher, F. Banhart, C. Sanchez and S. Begin-Colin, Small, 2022, 2200414 CrossRef CAS PubMed.
- A. Walter, C. Billotey, A. Garofalo, C. Ulhaq-Bouillet, C. Lefèvre, J. Taleb, S. Laurent, L. Vander Elst, R. N. Muller, L. Lartigue, F. Gazeau, D. Felder-Flesch and S. Begin-Colin, Chem. Mater., 2014, 26, 5252–5264 CrossRef CAS.
- B. P. Pichon, O. Gerber, C. Lefevre, I. Florea, S. Fleutot, W. Baaziz, M. Pauly, M. Ohlmann, C. Ulhaq, O. Ersen, V. Pierron-Bohnes, P. Panissod, M. Drillon and S. Begin-Colin, Chem. Mater., 2011, 23, 2886–2900 CrossRef CAS.
- E. Wetterskog, C.-W. Tai, J. Grins, L. Bergström and G. Salazar-Alvarez, ACS Nano, 2013, 7, 7132–7144 CrossRef CAS PubMed.
- R. Chen, M. G. Christiansen, A. Sourakov, A. Mohr, Y. Matsumoto, S. Okada, A. Jasanoff and P. Anikeeva, Nano Lett., 2016, 16, 1345–1351 CrossRef CAS PubMed.
- P. Guardia, A. Riedinger, S. Nitti, G. Pugliese, S. Marras, A. Genovese, M. E. Materia, C. Lefevre, L. Manna and T. Pellegrino, J. Mater. Chem. B, 2014, 2, 4426 RSC.
- F. Perton, G. Cotin, C. Kiefer, J.-M. Strub, S. Cianferani, J.-M. Greneche, N. Parizel, B. Heinrich, B. Pichon, D. Mertz and S. Begin-Colin, Inorg. Chem., 2021, 60, 12445–12456 CrossRef CAS PubMed.
- G. Cotin, C. Kiefer, F. Perton, M. Boero, B. Özdamar, A. Bouzid, G. Ori, C. Massobrio, D. Begin, B. Pichon, D. Mertz and S. Begin-Colin, ACS Appl. Nano Mater., 2018, 1, 4306–4316 CrossRef CAS.
- A. Demortière, P. Panissod, B. P. Pichon, G. Pourroy, D. Guillon, B. Donnio and S. Bégin-Colin, Nanoscale, 2011, 3, 225–232 RSC.
- K. E. Gilbert and J. J. Gajewski, J. Org. Chem., 1982, 47, 4899–4902 CrossRef CAS.
- T. J. Daou, J. M. Grenèche, G. Pourroy, S. Buathong, A. Derory, C. Ulhaq-Bouillet, B. Donnio, D. Guillon and S. Begin-Colin, Chem. Mater., 2008, 20, 5869–5875 CrossRef CAS.
- S. G. Kwon, Y. Piao, J. Park, S. Angappane, Y. Jo, N.-M. Hwang, J.-G. Park and T. Hyeon, J. Am. Chem. Soc., 2007, 129, 12571–12584 CrossRef CAS PubMed.
- L. M. Bronstein, X. Huang, J. Retrum, A. Schmucker, M. Pink, B. D. Stein and B. Dragnea, Chem. Mater., 2007, 19, 3624–3632 CrossRef CAS.
- B. Choudhury and A. Choudhury, Phys. E, 2014, 56, 364–371 CrossRef CAS.
- H. El Ghandoor, H. M. Zidan, M. M. H. Khalil and M. I. M. Ismail, Int. J. Electrochem. Sci., 2012, 7, 5734–5745 CrossRef CAS.
- V. R. Akshay, B. Arun, G. Mandal and M. Vasundhara, Phys. Chem. Chem. Phys., 2019, 21, 12991–13004 RSC.
- S. M. Wasim, C. Rincón, G. Marín, P. Bocaranda, E. Hernández, I. Bonalde and E. Medina, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 195101 CrossRef.
- M. A. Gonzalez-Fernandez, T. E. Torres, M. Andrés-Vergés, R. Costo, P. de la Presa, C. J. Serna, M. P. Morales, C. Marquina, M. R. Ibarra and G. F. Goya, J. Solid State Chem., 2009, 182, 2779–2784 CrossRef CAS.
- A. R. Muxworthy and E. McClelland, Geophys. J. Int., 2000, 140, 101–114 CrossRef.
- I. Castellanos-Rubio, I. Rodrigo, R. Munshi, O. Arriortua, J. S. Garitaonandia, A. Martinez-Amesti, F. Plazaola, I. Orue, A. Pralle and M. Insausti, Nanoscale, 2019, 11, 16635–16649 RSC.
- C. P. Guntlin, S. T. Ochsenbein, M. Wörle, R. Erni, K. V. Kravchyk and M. V. Kovalenko, Chem. Mater., 2018, 30, 1249–1256 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01397b |
| This journal is © The Royal Society of Chemistry 2024 |