
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZeolite catalysts for non-oxidative ethane dehydrogenation to ethylene
Lu
Liu
 ,
Liang
Wang
* and
Feng-Shou
Xiao
*
,
Liang
Wang
* and
Feng-Shou
Xiao
*
Key Lab of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China
First published on 19th March 2024
Abstract
The conversion of ethane to ethylene is crucial for deriving platform chemicals from non-petroleum feedstock. However, it currently relies on steam cracking technology, which involves high temperatures and large reactors. The catalytic dehydrogenation of ethane (EDH) could resolve these issues, but its efficiency is often limited due to thermodynamics, leading to low conversion and coke formation. These challenges make it difficult for catalytic EDH to compete economically with steam cracking. Recent studies show that rational design of catalysts, such as fixing metal nanoclusters within zeolite micropores or isolated metal sites on the zeolite framework, can enhance catalytic performances. These designs lower energy barriers for carbon–hydrogen bond activation, hinder deep dehydrogenation to coke, and provide sinter-resistant metal sites for durability. This review discusses the pivotal role of zeolite structures in catalysis and sums up the principles of catalyst design for efficient non-oxidative EDH. It aims to help in the development of more efficient zeolite catalysts and enhance the viability of catalytic EDH for potential industrialization.
Broader contextEthylene is a critical chemical building block in the modern chemical industry, primarily produced from petroleum feedstock. However, with the rise in shale gas exploitation, converting ethane into ethylene has emerged as a viable non-petroleum alternative. The steam cracking of ethane has yielded over 40 million tons of ethylene, accounting for 20% of the current ethylene production. But this process is endothermic, requiring high reaction temperatures (around 800 °C), leading to 1.2 tons of CO2 emissions per ton of ethylene produced. The non-oxidative catalytic dehydrogenation of ethane (EDH) could help reduce energy costs and yield a stoichiometric hydrogen product. However, existing EDH catalysts still fall short in terms of catalytic activity, selectivity, and durability, hindering the potential for industrialization. Recently, zeolite-based catalysts have shown significant performance improvements compared to the typical oxide-supported catalysts. The rigid and stable zeolite framework, with confined metal species in the micropores or coordinated metal sites on the framework, aids in activating the C–H bond and obstructing coke formation. This review summarizes and discusses these findings. |
1. Introduction
Ethylene is one of the most important platform chemicals in the modern chemical industry, which had a production over 200 million tons in 2022 and was still growing by 2.9% per year. The current ethylene production mainly relies on the steam cracking of naphtha, which has covered more than 70% of the production.1,2 With the large-scale exploitation of shale gas, the supply of ethane has increased more than 15-fold over the past decade in the United States. The low-cost ethane has become a suitable raw material for ethylene production, and the steam cracking of ethane has been utilized as a non-petroleum route for obtaining ethylene.3 However, this process is endothermic, and requires a high reaction temperature (e.g. 800 °C), corresponding to 1.2 tons of CO2 emissions with the production of each ton of ethylene.4Compared with the steam cracking of ethane, catalytic dehydrogenation of ethane could proceed at a relatively mild temperature (e.g. 600 °C). The oxidative EDH has been studied for several decades, but the over-oxidation usually occurs to form non-valuable CO2 with low selectivity to ethylene.5–8 Compared with oxidative EDH, the non-oxidative EDH has high selectivity and could produce the valuable by-product hydrogen, but the single-pass ethane conversion was usually low because of the limit by thermodynamic equilibrium. As shown in Fig. 1A, increasing the reaction temperature and decreasing the partial pressure of ethane are beneficial to obtain significant ethane conversion. However, with increasing reaction temperature, the rates of side reactions such as ethane cracking (eqn (2)), hydrogenolysis (eqn (3)), and coking (eqn (4)) also increase (Fig. 1B), which accelerate the catalyst deactivation and decrease the yield of ethene. For example, the ΔG for deep dehydrogenation of ethane to coke (eqn (4)) is −82.1 kJ mol−1 at 600 °C, and −114.2 kJ mol−1 at 750 °C. Although propane dehydrogenation has been commercialized, the catalysts were not fully appropriate for EDH because ethane has more inert C–H bonds and more severe thermodynamic limitations compared with propane.9–11 Therefore, developing efficient catalysts for active, selective, and durable non-oxidative EDH is still a great challenge.
| C2H6 ↔ C2H4 + H2 (ΔHθ298 = 137 kJ mol−1) | (1) |
| C2H6 ↔ 1/2C2H4 + CH4 (ΔHθ298 = 36.3 kJ mol−1) | (2) |
| C2H6 + H2 ↔ 2CH4 (ΔHθ298 = −64.5 kJ mol−1) | (3) |
| C2H6 ↔ 2C + 3H2 (ΔHθ298 = 84.7 kJ mol−1) | (4) |
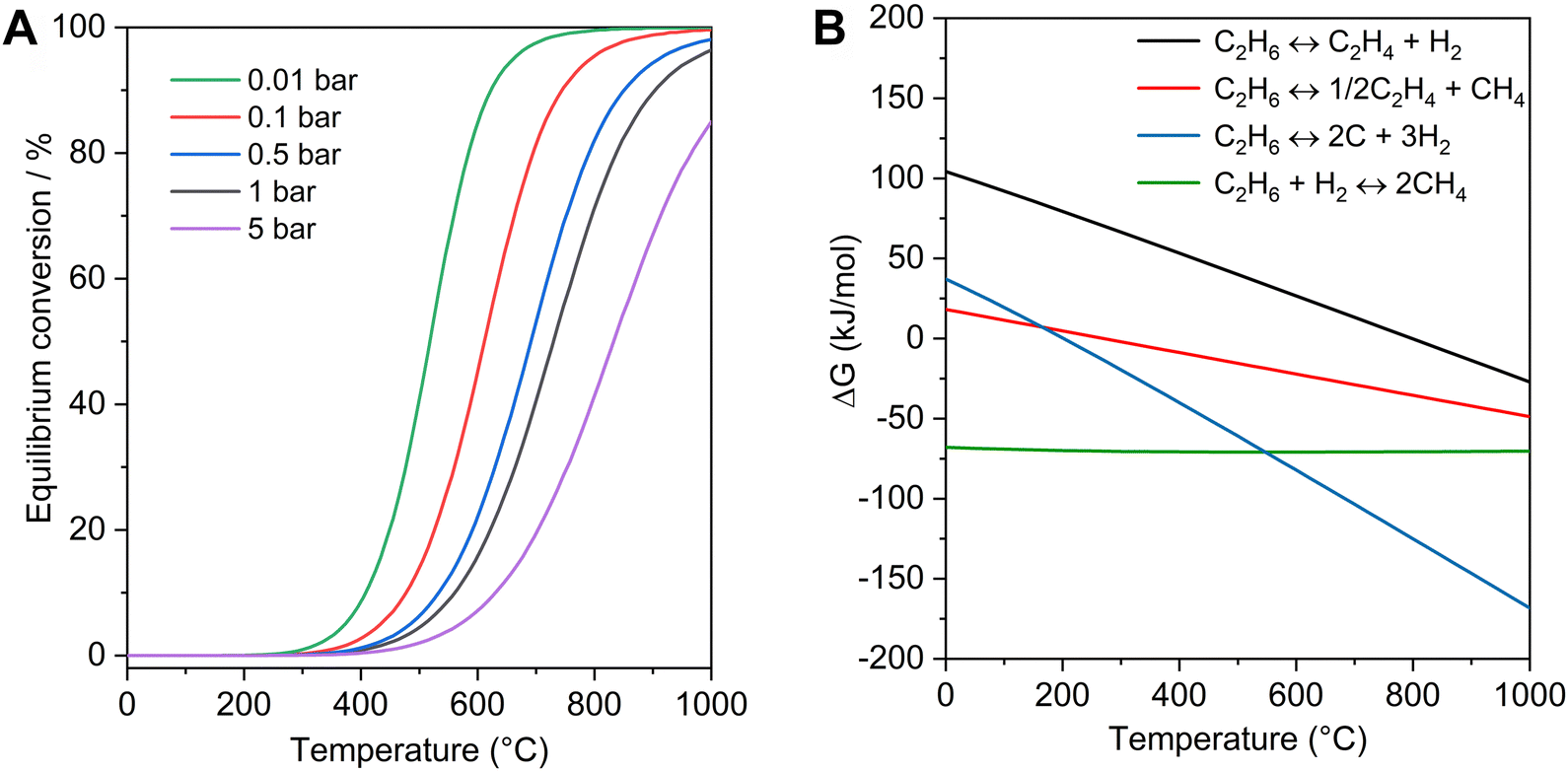 | ||
| Fig. 1 Thermodynamic profiles of non-oxidative EDH. (A) Equilibrium conversion of non-oxidative EDH at different partial pressures as a function of reaction temperatures. (B) ΔG of EDH (eqn (1)) and other side reactions (eqn (2)–(4)) as a function of temperature (1 bar). | ||
Recent findings have proposed several strategies to optimize reaction processes by regulating catalyst structures. Different from the oxide supports, zeolites with rigid, uniform, and stable frameworks provide confinement, coordination, and/or metal–support interaction effects to the metal species, thus improving their catalytic performance.12–18 For example, loading Pt nanoparticles on the surface of titanosilicate zeolites resulted in an ethane conversion of over 43.0% during a 100 hour EDH test due to enhanced metal–support interaction and molecular transport;12 encapsulating Pt nanoparticles within zeolite micropores led to a high electron density of the Pt active phase, weakening the strength of the Pt–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) interaction and promoting rapid olefin desorption. These effects resulted in improved durability compared to supported Pt nanoparticles;13 isolating cobalt in the stable zeolite framework formed H2CoS-1 sites, which exhibited high activity and negligible coke formation in EDH for a long period, outperforming conventionally supported catalysts.14 Based on these strategies, significant advances in improving ethane conversion and catalytic durability have been achieved. Although a number of EDH catalysts have been developed, as summarized in reviews on the dehydrogenation of different light alkanes,3,19 the advantages of zeolite-related catalysts have not been fully realized. To date, there has been no review focusing on zeolite catalysts for non-oxidative EDH yet. Herein, we briefly summarize recent progress and discuss their structure–performance interplay. This minireview not only discusses efficient EDH processes for producing ethylene but also highlights the great potential of zeolites in this field. This work might be important for the development of efficient catalysts in the future.
C) interaction and promoting rapid olefin desorption. These effects resulted in improved durability compared to supported Pt nanoparticles;13 isolating cobalt in the stable zeolite framework formed H2CoS-1 sites, which exhibited high activity and negligible coke formation in EDH for a long period, outperforming conventionally supported catalysts.14 Based on these strategies, significant advances in improving ethane conversion and catalytic durability have been achieved. Although a number of EDH catalysts have been developed, as summarized in reviews on the dehydrogenation of different light alkanes,3,19 the advantages of zeolite-related catalysts have not been fully realized. To date, there has been no review focusing on zeolite catalysts for non-oxidative EDH yet. Herein, we briefly summarize recent progress and discuss their structure–performance interplay. This minireview not only discusses efficient EDH processes for producing ethylene but also highlights the great potential of zeolites in this field. This work might be important for the development of efficient catalysts in the future.
2. Pt–zeolite catalysts
Supported Pt nanoparticle catalysts are widely studied for dehydrogenation of light alkanes due to their high activity in C–H bond activation (Fig. 2A).10,20–23 In the context of Pt-catalyzed EDH, various steps have been experimentally and theoretically identified. These include the dissociative adsorption of ethane, C–H cleavage to generate two hydrogen atoms and olefins, the coupling of two hydrogen atoms to create a hydrogen molecule, and the desorption of both hydrogen and ethylene (Fig. 2B and C).24 Small-sized Pt nanoparticles are known to have more low-coordinated Pt sites with lower energy barriers towards EDH compared to flat Pt surfaces. This characteristic leads to their superior activity in ethane conversion. However, it also triggers the severe deep dehydrogenation of ethylene to form coke, which is considered an inherent disadvantage of Pt catalysts in non-oxidative dehydrogenation.24–26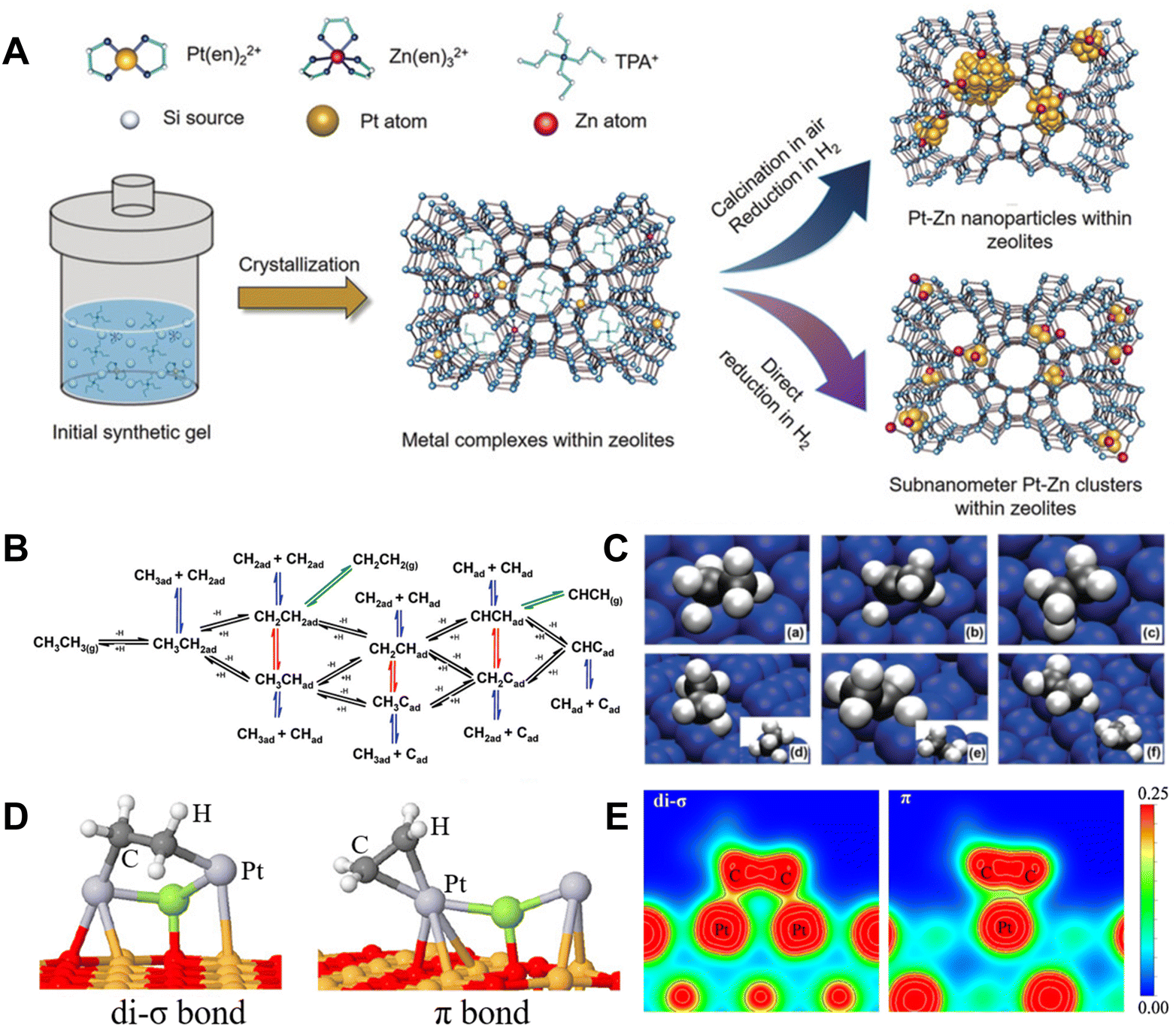 | ||
| Fig. 2 (A) Scheme showing the synthesis of zeolite-encaged subnanometer Pt–Zn cluster catalysts.23 Copyright 2020 John Wiley and Sons. (B) Elementary reactions in the conversion of ethane.24 (C) Geometries of the transition states for the activation of ethane and ethyl C–H bonds on Pt(111): (a) CH3CH2–H, (b) CH3CH–H, and (c) CH2CH2–H; and on Pt(211): (d) CH3CH2–H, (e) CH3CH–H, and (f) CH2CH2–H.24 Copyright 2010 the American Chemical Society. (D) Ethylene adsorbed on Pt2X clusters.27 Copyright 2018 the American Chemical Society. (E) The valence charge density calculated for different propylene adsorption modes.28 Copyright 2010 Elsevier. | ||
The competition between ethylene desorption and deep dehydrogenation is crucial to determine the reaction channels of efficient EDH and coke formation. The unsaturated CC bond could interact with the Pt sites through di-σ and/or π adsorption (Fig. 2D), where the former was stronger.27 This is due to larger valence charge density between the C and Pt surface in the di-σ bond than in the π adsorption, resulting in a stronger C–metal bond (Fig. 2E).28 Similar features were also observed on EDH intermediates. The adsorption energy of ethylene on Pt(111) and Pt(211) is 1.51 and 1.84 eV, respectively. Stronger CH2CH2* adsorption energy on Pt(211) makes the desorption of ethylene more difficult than that on Pt(111). On Pt(111), CH3CH* can be dehydrogenated rapidly to form CH3C* with a reaction barrier of 0.28 eV, which may lead to the loss of selectivity and poor stability.24 To overcome this problem, an efficient strategy is to modify the Pt nanoparticles by promoters such as Pb, Bi, Sb, In, Sn, Ga, and Zn, which could form Pt–M alloyed structures for rapid ethylene desorption.29–31 For example, a Zn-modified Pt nanoparticle catalyst could effectively suppress C–C bond cleavage during ethane dehydrogenation. It could achieve nearly 100% C2H4 selectivity up to the thermodynamically limited equilibrium conversion at 600 °C, while the monometallic Pt catalyst could only obtain a C2H4 selectivity less than 50%.30 Among these various promoters, Sn is the most commonly used one for Pt nanoparticles because it can efficiently interact with Pt to form an alloyed structure, which is stable at high temperatures under redox conditions. The function of Sn species has attracted much interest by studying the Pt–Sn system with different Sn contents, coverages, and locations.13,29,32–35 When the Sn coverages were very low (1/4 mono-layer) on the catalyst, the electronic interaction between supported Sn–Pt species benefits the uniform distribution of Pt nanoparticles. For the catalysts with high Sn coverages (1/2 mono-layer), the geometric effect was dominant and benefited weakening the adsorption of olefins on the Pt surface by reducing the number of Pt–Pt di-σ sites and increasing the activation energy for C–H of ethylene, which contributed to higher ethylene selectivity and suppresses deep dehydrogenation.13,29,33–35 Owing to these features, the Pt–Sn catalysts could catalyze EDH more efficiently relative to the bare Pt catalysts. For example, Pt–Mg(Al)O exhibited an initial ethane conversion of 10.4% and ethylene selectivity of 80%, which were significantly lower than those of PtSn–Mg(Al)O (25.6% ethane conversion and nearly 100% ethylene selectivity) at 550 °C.36 However, these supported Pt–Sn catalysts suffer from metal sintering and Pt–Sn phase separation at high temperatures in air (oxidative conditions that were usually used to remove the coke), which leads to irreversible deactivation.
Encapsulating the Pt nanoparticles within zeolite crystals, even without the Sn promoters, could significantly optimize the performances. For example, the aluminosilicate zeolite (HZSM-5) encapsulated Pt nanoparticles (Pt@HZSM-5) exhibited high ethane conversion (over 15%) and excellent stability (over 50 h) for EDH.37 Under the equivalent reaction conditions, the Pt nanoparticles supported on the zeolite external surface exhibited the initial ethane conversion of 17% but reduced to less than 6% after 20 h. A possible reason is that the encapsulated structure leads to the high electron density of the Pt active phase, which weakens the strength of the Pt–(CC) interaction and promotes rapid olefin desorption. Importantly, Pt@HZSM-5 after regeneration by calcination in air showed an identical structure and catalytic performance relative to the fresh catalyst, because the rigid zeolite structure could stabilize the encapsulated metal nanoparticles under the redox conditions.
In addition to the encapsulated structure, the zeolite containing heteroatoms could also electronically modulate the Pt nanoparticles. Liu et al. reported that the heteroatoms on the TS-1 zeolite could electronically interact with Pt nanoparticles and optimize their performances in EDH.12 As a result, they found that the Pt nanoparticles on two-dimensional multilamellar titanium silicalite-1 zeolite nanosheets exhibited an ethane conversion of 15.7% with ethylene selectivity higher than 99.0%, and such performances were constant in the continuous test up to 50 h. In this case, the Pt–Ti electronic interaction also benefits stabilizing Pt nanoparticles against sintering during EDH at high temperatures. Also, the multilamellar structure with high external surface area benefits the dispersion of Pt nanoparticles and metal–support interaction.
Although the durability of Pt catalysts has been improved through zeolite support, coke formation on these zeolite-related catalysts cannot be fully avoided during long-term EDH tests or under harsh conditions.38–40 Therefore, further development of Pt–zeolite catalysts is necessary to achieve ideal performance with high activity, selectivity, and fully prevented coke formation. To achieve this success, we need to engineer zeolite micropores to optimize mass transfer, and adjust the Pt–zeolite electronic interaction to hinder ethylene cracking and deep dehydrogenation.
3. Non-noble metal–zeolite catalysts
Various non-noble metal-based catalysts, such as Cr-, Ga-, Fe-, Co-, and In–zeolite catalysts, have been studied in EDH, and some showed unusual performances.14,16,18,41–43Table 1 and Fig. 3 briefly summarize the catalytic performances of typical zeolite catalysts for non-oxidative EDH in recent years, with the oxide supported catalysts as references. Non-noble metal zeolite catalysts exhibited excellent activity, some of which even outperform the activity of Pt catalysts. Typically, the Cr/MFI catalyst exhibited an ethane conversion of 36.2% at 650 °C, GHSV = 600 mL gcat−1 h−1, which significantly outperforms the noble metal-based catalysts like Pt@MFI with an ethane conversion of 4.6% under equivalent conditions.41 Besides, the Fe/ZSM-5 catalyst showed an ethane conversion of 15.0%, while Pt/ZSM-5 and Fe/Al2O3 exhibited an ethane conversion of only about 4.2% and 3.2%, respectively.44| Catalyst | Temperature (°C) | Components (%) | Initial C2H6 conversion (%) | Initial C2H4 selectivity (%) | C2H4 productivity (kgC2H4 kgcat−1 hour−1) | Test time (h) | Deactivation rate constant kd (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Pt@HZSM-5 | 550 | C2H6 = 90, N2 = 10 | 17.5 | 69.8 | 0.29 | 50 | 0.003 | 37 |
| Co/S-1 | 590 | C2H6 = 100 | 13.4 | 97.7 | 13.40 | 100 | 0.007 | 14 |
| Pt/Mg(Ga)(Al)O | 600 | C2H6 = 20, H2 = 25, He = 55 | 28.4 | 99.9 | 2.89 | 2 | 0.098 | 45 |
| PtSn/ZrM | 600 | C2H6 = 20, N2 = 80 | 34.2 | 99 | 1.27 | 2 | 0.391 | 46 |
| PtSn/Mg(Al)O | 600 | C2H6 = 19.24, He = 80.76 | 9.8 | 99.8 | 1.02 | 2 | 0.722 | 47 |
| Pt3Ga/SiO2 | 600 | C2H6 = 2.5, H2 = 2.5, N2 = 95 | 47 | 98 | 0.81 | 9 | 0.051 | 31 |
| Fe/ZSM-5 | 600 | C2H6 = 9, Ar = 91 | 28 | 65 | 0.80 | 4.5 | 0.078 | 44 |
| Pt/M-TS-1(EA) | 600 | C2H6 = 75, Ar = 25 | 15.8 | 98 | 0.57 | 50 | ∼0 | 12 |
| Pt/Zn/Al2O3 | 600 | C2H6 = 5, N2 = 90, H2 = 5 | 19 | 98 | 0.22 | 70 | 0.003 | 48 |
| FeS-1-EDTA | 600 | C2H6 = 30, Ar = 70 | 26.3 | 97.5 | 0.19 | 200 | ∼0 | 16 |
| N3GaAl2O3 | 600 | C2H6 = 10, Ar = 90 | 14.6 | 94 | 0.14 | 32 | 0.042 | 49 |
| 2.5P-Mo/ZSM-5 | 600 | C2H6 = 10, N2 = 10, He = 80 | 19.5 | 45 | 0.07 | 4.3 | 0.063 | 50 |
| GaPtHZSM-5 | 615 | C2H6 = 30, N2 = 70 | 67.3 | 11.6 | 1.84 | 2 | 0.512 | 51 |
| GaPt/HZSM-5 | 650 | C2H6 = 30, He = 70 | 92 | 60 | 6.89 | 5 | 0.698 | 52 |
| 5Cr/PE-MCM-41 | 650 | C2H6 = 50, N2 = 50 | 15.7 | 98.8 | 1.78 | 24 | 0.024 | 53 |
| 5Cr@SBA-5 | 650 | C2H6 = 33, Ar = 67 | 45.9 | 99 | 0.87 | 3 | 0.619 | 54 |
| 0.8Cr/MFI | 650 | C2H6 = 20, N2 = 80 | 27.7 | 99 | 0.60 | 110 | 0.003 | 41 |
| Fe/HZ5-HTS | 650 | C2H6 = 5, Ar = 95 | 53.4 | 75.7 | 0.15 | 20 | 0.030 | 55 |
| 3Co/HZ-40 | 650 | C2H6 = 3, N2 = 97 | 45.4 | 91.8 | 0.14 | 6 | 0.052 | 56 |
| Au/TiSi-20 | 650 | C2H6 = 3, N2 = 97 | 16 | 97 | 0.05 | 6 | 0.058 | 57 |
| R-LMN | 700 | C2H6 = 50, N2 = 50 | 41.8 | 97.8 | 7.52 | 50 | — | 58 |
| Pt-ISAS@NaY | 700 | C2H6 = 20, N2 = 80 | 26 | 97 | 0.93 | 2 | 0.036 | 59 |
| CrMgAlOx | 700 | C2H6 = 28.6, N2 = 71.4 | 22 | 80 | 0.86 | — | — | 60 |
| Cr0.92Mg6Al1.88Ox | 700 | C2H6 = 28.6, N2 = 71.4 | 30 | 71 | 0.63 | — | — | 61 |
| 5Cr-10Ce/SBA-15 | 700 | C2H6 = 25, Ar = 75 | 55.5 | 96 | 0.58 | 5 | 0.079 | 62 |
| In-CHA | 700 | C2H6 = 13, He = 87 | 37.4 | 95.1 | 0.26 | 40 | 0.007 | 18 |
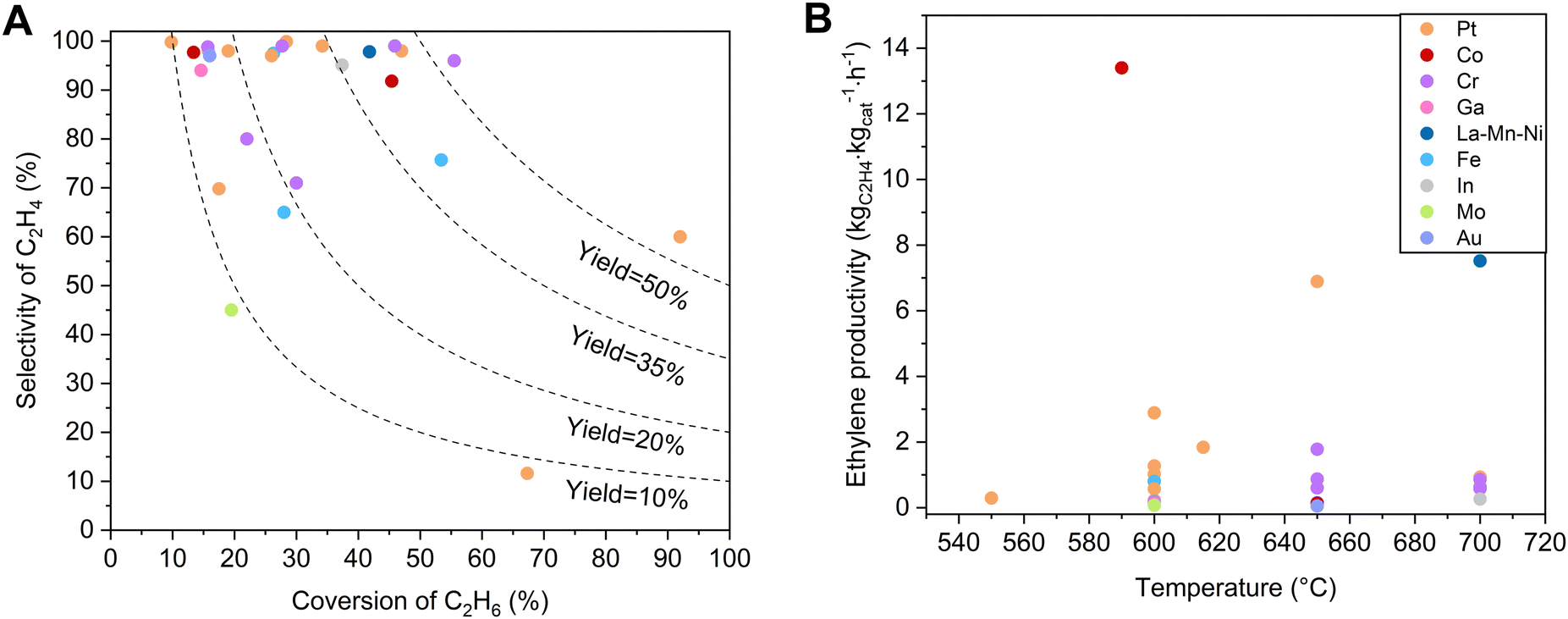 | ||
| Fig. 3 (A) Dependence of ethylene selectivity on ethane conversion over non-oxidative EDH catalysts related to those in Table 1. (B) Comparisons of ethylene productivity as a function of temperature for catalysts from (A). | ||
Indeed, the zeolite morphology, topological structure, micropore environment, and acidity/alkalinity have great impacts on the fine structure of active metals/sites and diffusion of reactants/products, thus affecting the catalytic performances.63–69 From the aspect of the acidity, Brønsted acid sites in zeolites catalyze cracking and dehydrocyclization that form aromatics and coke.70 In order to avoid these side reactions, aluminosilicate zeolites with low contents of alumina species and even siliceous ones that possess low surface acidity are commonly used as supports for the EDH with desired selectivity. In addition, optimization of the fine structure of heteroatom sites in zeolites is also desired. For example, the Fe-containing MFI siliceous zeolite with identical tetracoordinated and isolated iron sites exhibited high activity and selectivity for the EDH, showing a constant ethane conversion of ∼26% and ethene selectivity of ∼97.5% during the continuous EDH reaction for 200 h (Table 1). In contrast, the Fe-containing zeolite with some extra-framework Fe species could form coke species that block the micropores, resulting in relatively poor ethane conversion (<17%) under the equivalent reaction conditions.16
This principle also worked efficiently for the Co–zeolite catalysts. The Co–zeolite with identical tetracoordinated and isolated cobalt sites (Fig. 4A) was highly active, selective, and durable for EDH. As a typical run at 590 °C, WHSV 6.5 h−1, the Co/S-1 zeolite showed an ethane conversion of 13.2%. In contrast, the supported cobalt nanoparticles such as Co/SiO2 and Co/TiO2 exhibited ethane conversions lower than 6%. Considering the different ethane partial pressure and WHSV in the tests, we compared the ethylene productivity of the different catalysts. Co/S-1 gave an ethylene productivity of 13.4 kgC2H4 kgcat−1, much higher than 0.11 and 0.36 kgC2H4 kgcat−1 over Co/SiO2 and Co/TiO2 catalysts. Even the FeS-1-EDTA zeolite catalyst, which was reported as coke-resistant in EDH, gave an ethylene productivity of 0.19 kgC2H4 kgcat−1. These data support the great potential of the Co–zeolite in the EDH.14
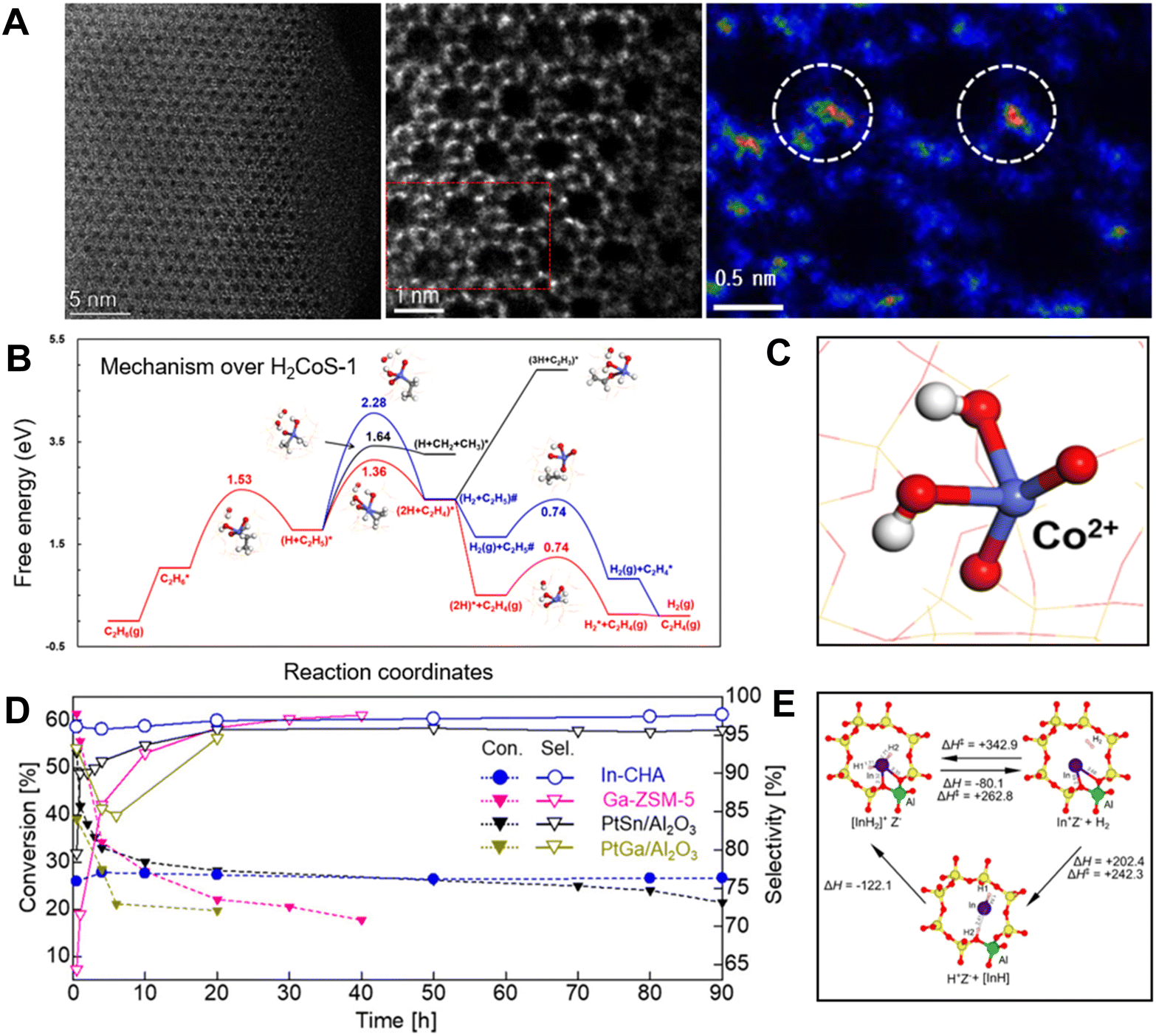 | ||
| Fig. 4 (A) HAADF-STEM images of Co/S-1.14 (B) Calculated free energy diagram of EDH over H2CoS-1.14 (C) The structure of H2CoS-1.14 Copyright 2022 Elsevier. (D) Time course of the nonoxidative dehydrogenation of ethane using In-CHA, PtSn/Al2O3, PtGa/Al2O3, and Ga-ZSM-5.18 (E) Optimized structure of the [InH2]+ ion [AlO4]− site (Z[InH2]) in the CHA zeolite and formation of Z[InH2] from Z[In] via direct and indirect pathways as well as desorption of H2 from Z[InH2]. Copyright 2020 the American Chemical Society.18 | ||
Further insights into the active sites of these Fe and Co zeolite catalysts have been studied by theoretical simulations, where the bridge oxygen of Fe–O–Si or Co–O–Si linkages was regarded as a crucial site for the activation of the first C–H bond. This structure results in high intrinsic activity for ethane dehydrogenation and benefits the desorption of ethylene to avoid coke formation. For example, the dehydrogenation of C2H5* is more favorable than the C–C bond dissociation with the Ga value at 1.64 eV on H2CoS-1 (the isolated Co sites with two protons for charge balance). In addition, the co-adsorption of ethylene and hydrogen atoms (2H + C2H4)* exhibits a repulsive feature, which leads to barrierless desorption rather than the deep dehydrogenation of C2H4* to form coke (Fig. 4B and C). These features could explain the high ethylene selectivity in EDH over these catalysts.
Different from the framework Fe and Co sites in zeolites, the In species could not replace a Si atom in the zeolite framework to form tetracoordinated sites, because of the huge atom size. Maeno et al. found that the isolated In sites could be introduced into the cages of the aluminosilicate CHA zeolite, and formed In hydride species that were active for the dehydrogenation.18 In EDH over the In-CHA zeolite, a durable ethane conversion of 27.2% and ethylene selectivity of 96.9% were achieved in the continuous test for 90 h (Table 1). In contrast, the Ga and Zn zeolites exhibited much lower selectivities (76.1% and 67.3%, respectively) with coke as a by-product. In contrast, the alumina-supported PtSn and PtGa exhibited lower conversions (21.4% for PtSn/Al2O3 at 90 h, and 19.7% for PtGa/Al2O3 at 20 h) than In-CHA (Fig. 4D). These results demonstrate the high selectivity and durability of In-CHA for EDH at high reaction temperatures (700 °C). Based on in situ Fourier transform infrared (FTIR) spectroscopy, kinetic study, and transition state (TS) calculations, the isolated framework [InH2]+ ions served as catalytically active sites for EDH using In-CHA. Theoretical simulations showed that the [InH2]+ ions were thermodynamically unstable but kinetically stable at high temperatures, which supports the formation and stabilization of In hydrides at high temperatures (Fig. 4E). Besides, the [InH2]+ ions reduced the amount of zeolite-coordinated protons and carbenium ions during ethane dehydrogenation compared to Ga–zeolites, resulting in better selectivity and durability.
Different from the precious metals (e.g. Pt) that activated the C–H bond relying on the d orbitals, the heteroatoms in zeolites provided different active sites for the ethane activation, such as the metal d orbital synergising with the adjacent oxygen atoms over Si–O–Feδ+ and Si–O–Coδ+ containing zeolites. This finding paves the way for developing EDH catalysts with non-noble metals and could potentially be extended to the activation of C–H bonds in other hydrocarbon molecules.
4. Summary and outlook
Recent advancements in the non-oxidative ethane dehydrogenation over zeolite catalysts have been briefly reviewed, focusing on the relationship between the structure and catalyst activity. Zeolites with stable and uniform frameworks facilitate the formation of new types of active sites through electronic or geometrical modulation, outperforming traditional metal nanoparticles on oxide supports. In addition, the zeolite-based catalysts have obvious advantages such as fast mass transfer of light alkanes and alkenes, stable active sites in the catalysts, and easy regeneration of metal species, leading to enhanced activity, high selectivity, and excellent durability, compared with current supported metal catalysts. Among various zeolite types, the MFI zeolite shows promise due to its ease of synthesis in its siliceous (alumina-free) form on a large scale.Despite significant progress in catalyst design for non-oxidative EDH, future research still faces important challenges. One is the catalyst's stability under redox conditions. In most studies, the sintering resistance of metal sites is typically tested under dehydrogenation (reductive) conditions. In these cases, the sintering resistance of metal sites under oxidative conditions, which relates to calcination operations to remove coke and influences catalyst regenerability, is often overlooked. For metals used in EDH (e.g., Pt), oxygen-triggered sintering is energy-favorable at high temperatures, often leading to irreversible deactivation during continuous dehydrogenation and calcination operations. This sintering also occurs during calcination, even for zeolite-stabilized metal sites or nanoparticles. The future catalyst design should fully consider these issues. Possibly, a zeolite with a rationally optimized microporous environment to prevent oxygen-triggered Ostwald ripening of metal sites could overcome current limitations. In this case, a promising catalyst could be the MFI zeolite with isolated metal sites, where the siliceous framework might stabilize the metal sites under redox conditions through the Si–O–Mδ+ linkage. Considering the high activity and selectivity of isolated Co in zeolites, the cobaltosilicate zeolite catalyst (CoS-1) could be a promising catalyst for EDH. Furthermore, this catalyst could potentially have widespread applications by avoiding the use of precious metals.
The thermodynamic limitation is another challenge for the EDH and its potential applications. The process economic analysis has shown a significant reduction in operation costs when increasing the one-pass ethane conversions from approximately 15% to 31% in EDH.71 Diluted ethane and a low feed rate are typically used in lab research to increase the apparent ethane conversion. However, such operations result in high separation costs. Therefore, further improvement in one-pass ethane conversion needs to be seriously considered.
Partially removing the hydrogen product could efficiently shift the reaction equilibrium and improve the ethane conversion, which strongly requires catalysts with multiple functions for EDH and hydrogen combustion (Fig. 5).71 Inducing the component for hydrogen combustion to the current EDH catalysts has achieved some success in improving the ethane conversions, but the overoxidation usually occurred to reduce the selectivity of ethylene.
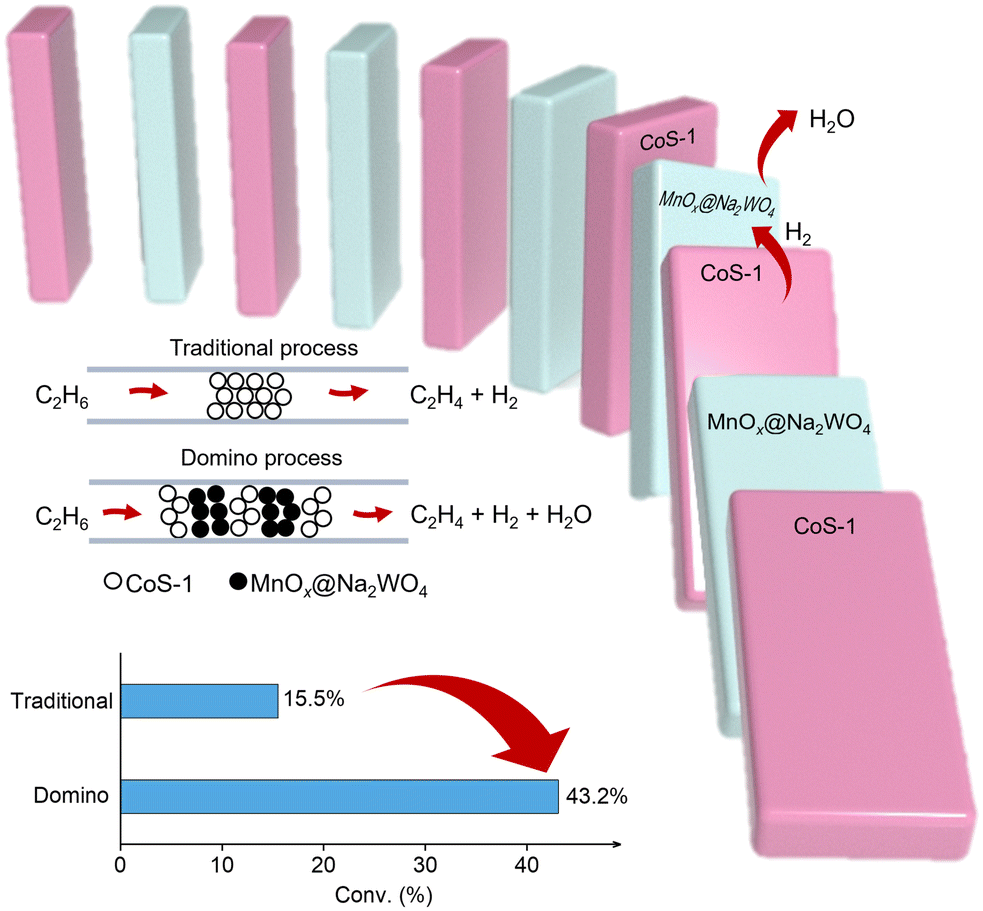 | ||
| Fig. 5 Scheme showing the ethane dehydrogenation and selective hydrogen consumption process proposed by ref. 71. | ||
Multiple functional catalysts with individuals for efficient dehydrogenation, selective hydrogen combustion, and switched-off hydrocarbon combustion are highly desired. For example, Qin et al. developed a reaction process by interleaving CoS-1 for non-oxidative dehydrogenation and sodium tungstate-modified manganese oxide promoter (MnOx@Na2WO4) for selective hydrogen combustion in multiple beds as a domino mode in the reactor, which achieved a per-pass ethane conversion of up to 43.2% and ethylene selectivity of 93.1% at 590 °C with 0.8 bar of ethane feed.71 Such conversion significantly exceeds those in classical non-oxidative EDH, which would guide the development of different catalysts and reaction systems to exceed the conversion limitation.72
In summary, the study on EDH serves as a bridge between the fields of materials, heterogeneous catalysis, and chemical engineering. The zeolite platform paves the way for developing efficient catalysts. This not only enhances the viability of the EDH technique but also broadens the application of zeolite materials.
Author contributions
This manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFA1503502) and the National Natural Science Foundation of China (22288101 and 22241801).Notes and references
- Ethylene demand and production capacity worldwide from 2015 to 2022, Statista, https://www.statista.com/statistics/1246694/ethylene-demand-capacity-forecast-worldwide/ (accessed 2023-09-13).
- China petroleum and chemical bulk products annual report, https://www.cpcifdata.org.cn (accessed 2023-09-13).
- C. Li and G. Wang, Chem. Soc. Rev., 2021, 50, 4359–4381 RSC.
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS.
- S. Yusuf, L. M. Neal and F. Li, ACS Catal., 2017, 7, 5163–5173 CrossRef CAS.
- S. Chen, L. Zeng, R. Mu, C. Xiong, Z.-J. Zhao, C. Zhao, C. Pei, L. Peng, J. Luo, L.-S. Fan and J. Gong, J. Am. Chem. Soc., 2019, 141, 18653–18657 CrossRef CAS PubMed.
- S. Yusuf, V. Haribal, D. Jackson, L. Neal and F. Li, Appl. Catal., B, 2019, 257, 117885 CrossRef CAS.
- J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205–1208 CrossRef CAS PubMed.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- Y. Wang, P. Hu, J. Yang, Y. Zhu and D. Chen, Chem. Soc. Rev., 2021, 50, 4299–4358 RSC.
- X. Tang, X. Jia and Z. Huang, Chem. Sci., 2018, 9, 288–299 RSC.
- Y. Pan, A. Bhowmick, W. Wu, Y. Zhang, Y. Diao, A. Zheng, C. Zhang, R. Xie, Z. Liu, J. Meng and D. Liu, ACS Catal., 2021, 11, 9970–9985 CrossRef CAS.
- M.-L. Yang, Y.-A. Zhu, X.-G. Zhou, Z.-J. Sui and D. Chen, ACS Catal., 2012, 2, 1247 CrossRef CAS.
- L. Liu, H. Li, H. Zhou, S. Chu, L. Liu, Z. Feng, X. Qin, J. Qi, J. Hou, Q. Wu, H. Li, X. Liu, L. Chen, J. Xiao, L. Wang and F.-S. Xiao, Chem, 2023, 9, 637–649 CAS.
- L.-C. Wang, Y. Zhang, J. Xu, W. Diao, S. Karakalos, B. Liu, X. Song, W. Wu, T. He and D. Ding, Appl. Catal., B, 2019, 256, 117816 CrossRef.
- Z. Yang, H. Li, H. Zhou, L. Wang, L. Wang, Q. Zhu, J. Xiao, X. Meng, J. Chen and F.-S. Xiao, J. Am. Chem. Soc., 2020, 142, 16429–16436 CrossRef CAS PubMed.
- S. De, S. Ould-Chikh, A. Aguilar, L.-L. Hazemann, A. Zitolo, A. Ramirez, S. Telalovic and J. Gascon, ACS Catal., 2021, 11, 3988–3995 CrossRef CAS.
- Z. Maeno, S. Yasumura, X. Wu, M. Huang, C. Liu, T. Toyao and K. Shimizu, J. Am. Chem. Soc., 2020, 142, 4820–4832 CrossRef CAS PubMed.
- Y. Dai, X. Gdao, Q. Wang, X. Wan, C. Zhou and Y. Yang, Chem. Soc. Rev., 2021, 50, 5590–5630 RSC.
- L. Shi, G.-M. Deng, W.-C. Li, S. Miao, Q.-N. Wang, W.-P. Zhang and A.-H. Lu, Angew. Chem., Int. Ed., 2015, 54, 13994 CrossRef CAS PubMed.
- M. H. Hansen, J. K. Nørskov and T. Bligaard, J. Catal., 2019, 374, 161–170 CrossRef CAS.
- A. H. Motagamwala, R. Almallahi, J. Wortman, V. O. Igenegbai and S. Linic, Science, 2021, 373, 217–222 CrossRef CAS PubMed.
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou, J. Zhang, T. Zhang, J. Xu, P. Zhang, J. Cheng, D.-C. Yang, R. Jia, L. Li, Q. Zhang, Y. Wang, O. Terasaki and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 19450–19459 CrossRef CAS PubMed.
- Y. Chen and D. G. Vlachos, J. Phys. Chem. C, 2010, 114, 4973–4982 CrossRef CAS.
- G. Peng, D. Gerceker, M. Kumbhalkar, J. A. Dumesic and M. Mavrikakis, Catal. Sci. Technol., 2018, 8, 2159–2174 RSC.
- X. Ding, H. Zhu, H. Ren, D. Liu, Z. Yu, N. Shi and W. Guo, Phys. Chem. Chem. Phys., 2020, 22, 21835–21843 RSC.
- E. Jimenez-Izal, H. Zhai, J.-Y. Liu and A. N. Alexandrova, ACS Catal., 2018, 8, 8346–8356 CrossRef CAS.
- M.-L. Yang, Y.-A. Zhu, C. Fan, Z.-J. Sui, D. Chen and X.-G. Zhou, J. Mol. Catal. A: Chem., 2010, 321, 42–49 CrossRef CAS.
- A. Hook and F. E. Celik, J. Phys. Chem. C, 2017, 121, 17882–17892 CrossRef CAS.
- V. J. Cybulskis, B. C. Bukowski, H.-T. Tseng, J. R. Gallagher, Z. Wu, E. Wegener, A. J. Kropf, B. Ravel, F. H. Ribeiro, J. Greeley and J. T. Miller, ACS Catal., 2017, 7, 4173–4181 CrossRef CAS.
- N. J. Escorcia, N. J. LiBretto, J. T. Miller and C. W. Li, ACS Catal., 2020, 10, 9813–9823 CrossRef CAS.
- J. Wu, Z. Peng and A. T. Bell, J. Catal., 2014, 311, 161–168 CrossRef CAS.
- A. Hook, J. D. Massa and F. E. Celik, J. Phys. Chem. C, 2016, 120, 27307–27318 CrossRef CAS.
- J. Nam and F. E. Celik, Top. Catal., 2020, 63, 700–713 CrossRef CAS.
- T. J. Gorey, B. Zandkarimi, G. Li, E. T. Baxter, A. N. Alexandrova and S. L. Anderson, ACS Catal., 2020, 10, 4543–4558 CrossRef CAS.
- Q. Zhang, K. Zhang, S. Zhang, Q. Liu, L. Chen, X. Li, C. Wang and L. Ma, J. Catal., 2018, 368, 79–88 CrossRef CAS.
- B. Qiu, Y. Zhang, Y. Liu and Y. Zhang, Appl. Surf. Sci., 2021, 554, 149611 CrossRef CAS.
- L. Wang, L. Wang, X. Meng and F. S. Xiao, Adv. Mater., 2019, 31, e1901905 CrossRef PubMed.
- J. Zhu, M.-L. Yang, Y. Yu, Y.-A. Zhu, Z.-J. Sui, X.-G. Zhou, A. Holmen and D. Chen, ACS Catal., 2015, 5, 6310–6319 CrossRef CAS.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- S. De, S. Ould-Chikh, A. Aguilar, L.-L. Hazemann, A. Zitolo, A. Ramirez, S. Telalovic and J. Gascon, ACS Catal., 2021, 11, 3988–3995 CrossRef CAS.
- M. Huang, Z. Maeno, T. Toyao and K.-I. Shimizu, Catal. Today, 2023, 411–412, 113824 CrossRef CAS.
- K. T. Leth, A. K. Rovik, M. S. Holm, M. Brorson, H. J. Jakobsen, J. Skibsted and C. H. Christensen, Appl. Catal., A, 2008, 348, 257–265 CrossRef CAS.
- L.-C. Wang, Y. Zhang, J. Xu, W. Diao, S. Karakalos, B. Liu, X. Song, W. Wu, T. He and D. Ding, Appl. Catal., B, 2019, 256, 117816 CrossRef.
- G. Siddiqi, P. Sun, V. Galvita and A. T. Bell, J. Catal., 2010, 274, 200–206 CrossRef CAS.
- J. Xu, C. Shi, S. Zhang, Q. Zheng, L. Pan, X. Zhang and J.-J. Zou, Chin. J. Chem., 2022, 40, 918–924 CrossRef CAS.
- V. Galvita, G. Siddiqi, P. Sun and A. T. Bell, J. Catal., 2010, 271, 209–219 CrossRef CAS.
- X. Li, Y. Zhou, B. Qiao, X. Pan, C. Wang, L. Cao, L. Li, J. Lin and X. Wang, J. Energy Chem., 2020, 51, 14–20 CrossRef.
- Y. He, Y. Song and S. Laursen, ACS Catal., 2019, 9, 10464–10468 CrossRef CAS.
- Z. Ji, H. Lv, X. Pan and X. Bao, J. Catal., 2018, 361, 94–104 CrossRef CAS.
- A. Caiola, B. Robinson, X. Bai, D. Shekhawat and J. Hu, Ind. Eng. Chem. Res., 2021, 60, 11421–11431 CrossRef CAS.
- A. Samanta, X. Bai, B. Robinson, H. Chen and J. Hu, Ind. Eng. Chem. Res., 2017, 56, 11006–11012 CrossRef CAS.
- T. V. M. Rao, E. M. Zahidi and A. Sayari, J. Mol. Catal. A: Chem., 2009, 301, 159–165 CrossRef CAS.
- B. Eryildirim, H. Arbag, N. Oktar and G. Dogu, Int. J. Hydrogen Energy, 2021, 46, 5296–5310 CrossRef CAS.
- L. Wu, Z. Fu, Z. Ren, J. Wei, X. Gao, L. Tan and Y. Tang, ChemCatChem, 2021, 13, 4019–4028 CrossRef CAS.
- H. Guo, C. Miao, W. Hua, Y. Yue and Z. Gao, Microporous Mesoporous Mater., 2021, 312, 110791 CrossRef CAS.
- Q. Xie, T. Lei, C. Miao, W. Hua, Y. Yue and Z. Gao, Catal. Lett., 2020, 150, 2013–2020 CrossRef CAS.
- X. Yang, T. Wei, B. Chi, J. Pu and J. Li, J. Catal., 2019, 377, 629–637 CrossRef CAS.
- Y. Liu, Z. Li, Q. Yu, Y. Chen, Z. Chai, G. Zhao, S. Liu, W.-C. Cheong, Y. Pan, Q. Zhang, L. Gu, L. Zheng, Y. Wang, Y. Lu, D. Wang, C. Chen, Q. Peng, Y. Liu, L. Liu, J. Chen and Y. Li, J. Am. Chem. Soc., 2019, 141, 9305–9311 CrossRef CAS PubMed.
- A. Tsyganok, P. K. E. Harlick and A. Sayari, Catal. Commun., 2007, 8, 850–854 CrossRef CAS.
- A. Tsyganok, R. G. Green, J. B. Giorgi and A. Sayari, Catal. Commun., 2007, 8, 2186–2193 CrossRef CAS.
- X. Shi, S. Ji and K. Wang, Catal. Lett., 2008, 125, 331–339 CrossRef CAS.
- J. Shin, D. Jo and S. B. Hong, Acc. Chem. Res., 2019, 52, 1419–1427 CrossRef CAS PubMed.
- Y. Wang, C. Wang, L. Wang, L. Wang and F.-S. Xiao, Acc. Chem. Res., 2021, 54, 2579–2590 CrossRef CAS PubMed.
- Y. Li, Q. Zhang, S. Fu, V. A. Kondratenko, T. Otroshchenko, S. Bartling, T. Zhang, A. Zanina, Y. Wang, G. Cui, M. Zhou, Z. Zhao, C. Xu, G. Jiang and E. V. Kondratenko, Chem. Eng. J., 2023, 460, 141778 CrossRef CAS.
- B. Wu, T. Lin, M. Huang, S. Li, J. Li, X. Yu, R. Yang, F. Sun, Z. Jiang, Y. Sun and L. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202204116 CrossRef CAS PubMed.
- W. Fan and M. Dong, Science, 2022, 375, 29 CrossRef CAS PubMed.
- Y. Ding, D. Miao, J. Feng, B. Bai, X. Pan and X. Bao, Appl. Catal., B, 2022, 316, 121628 CrossRef CAS.
- C. Wang, B. Yang, Q. Gu, Y. Han, M. Tian, Y. Su, X. Pan, Y. Kang, C. Huang, H. Liu, X. Liu, L. Li and X. Wang, Nat. Commun., 2021, 12, 5447 CrossRef CAS PubMed.
- V. R. Choudhary, C. Sivadinarayana, A. K. Kinage, P. Devadas and M. Guisnet, Appl. Catal., A, 1996, 136, 125–142 CrossRef CAS.
- X. Qin, H. Wu, R. Wang, L. Wang, L. Liu, H. Li, B. Yang, H. Zhou, Z. Liao and F.-S. Xiao, Joule, 2023, 7, 753–764 CrossRef CAS.
- W. Wang, S. Chen, C. Pei, R. Luo, J. Sun, H. Song, G. Sun, X. Wang, Z.-J. Zhao and J. Gong, Science, 2023, 381, 886–890 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |