
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirection of oxygen evolution reaction electrocatalyst evaluation for an anion exchange membrane CO2 electrolyzer
Seontaek
Kwon
a,
Tae-Hoon
Kong
a,
Namgyoo
Park
a,
Pandiarajan
Thangavel
 a,
Hojeong
Lee
a,
Seokmin
Shin
a,
Jihoo
Cha
a and
Youngkook
Kwon
*ab
a,
Hojeong
Lee
a,
Seokmin
Shin
a,
Jihoo
Cha
a and
Youngkook
Kwon
*ab
aSchool of Energy and Chemical Engineering, Ulsan National University of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: ykwon@unist.ac.kr
bGraduate School of Carbon Neutrality, UNIST, Ulsan 44919, Republic of Korea
First published on 21st February 2024
Abstract
CO2 electrolysis in membrane-electrode assemblies (MEAs) has come up one step closer to commercialization through compact cell design and high-current operation. However, while both cathodic and anodic reactions significantly affect the overall cell efficiency, the anodic oxygen evolution reaction (OER) has received much less attention compared to the cathodic CO2 reduction reaction (CO2RR). More importantly, OER electrocatalysts for CO2 electrolysis are being developed independently of system design, despite their interconnected nature. Since the aqueous testing systems in which OER electrocatalysts have been developed do not reflect the complex local anodic environment inside an anion exchange membrane CO2 electrolyzer (AEMCE), electrocatalysts sensitive to local chemistry may have been optimized for incorrect operating conditions. Based on a comprehensive understanding of the local anodic environment inside the AEMCE, in this perspective, we scrutinize the limitations of conventional OER electrocatalyst development resulting from the discrepancy between aqueous testing systems and the existing MEA-type systems. To bridge these gaps, we suggest three electrocatalyst evaluation platforms that integrate reference electrodes to existing AEMCEs for reliable and genuine OER electrocatalyst assessment.
Broader contextThe electrochemical conversion of carbon dioxide into value-added chemicals is emerging as a promising solution to address the climate crisis. Despite initial concerns about practical feasibility due to the slow rate of the CO2 reduction reaction (CO2RR) resulting from the limited solubility of CO2 in aqueous-based systems, recent advancements in gas diffusion electrodes and membrane electrode assemblies (MEAs) have significantly propelled CO2 electrolysis technology toward commercialization, with anion exchange membrane CO2 electrolyzers (AEMCEs) showing their potential as a future MEA-type. However, while considerable attention has been given to the cathodic CO2RR, the development of oxygen evolution reaction (OER) electrocatalysts, which particularly target their application for CO2 electrolysis, remains limited. Moreover, the in-depth understanding of the local anodic environment within AEMCEs emphasizes the urgent need for OER electrocatalysts that can operate effectively in a near pH-neutral environment created due to the high flux crossover of carbonate species. Herein, we introduce innovative catalyst evaluation platforms that integrate reference electrodes into existing AEMCEs to bridge the gap between material-centrically developed OER electrocatalysts and real-world applications. While faithfully replicating the authentic local environment, these platforms will guide the precise optimization of electrocatalysts concerning the commercialization of CO2 electrolysis. |
1. Introduction
The electrochemical conversion of carbon dioxide into value-added commodities is receiving huge attention as an efficient method to address the global climate crisis.1–5 Although its practical feasibility was questioned due to the sluggish CO2 reduction reaction (CO2RR) rate caused by the limited solubility of CO2 in aqueous-based systems,6 the development of gas diffusion electrodes (GDEs) and gas-phase reactors has significantly improved the mass transport limitation issue in aqueous systems by directly providing gaseous CO2 to the catalyst surface, greatly enhancing the CO2 electrolysis rate.7,8 In particular, recent advancements in membrane electrode assemblies (MEAs) have brought CO2 electrolysis one step closer to industrialization with their distinct advantages, including streamlined product separation and gaseous product pressurization achieved through stack assembly, eliminating the need for supplementary equipment dedicated to pressurization. Moreover, the minimized ohmic cell overpotential achieved by liquid electrolyte-free zero-gap cell configuration renders MEAs even more appealing for scalable operations.9–12 However, while the cathodic CO2 reduction reaction receives all the spotlight, only a few reports focus on its counterpart, the oxygen evolution reaction (OER), although both cathodic and anodic reactions play a crucial role in overall cell performance. Considering the commercialization, this emphasizes the necessity for the simultaneous development of OER electrocatalysts, with a particular focus on non-precious metals, capable of surviving in the industrial CO2 electrolysis environment.For the majority of MEA-type CO2 electrolyzer studies, the anion exchange membrane (AEM) has received significant attention as a future CO2 electrolysis membrane type since an AEM-based CO2 electrolyzer can effectively suppress the competing hydrogen evolution reaction (HER) at the cathode, while concurrently facilitating the OER at the anode.13,14 However, the CO2RR-generated OH− at the cathode (eqn (1)) inevitably reacts with supplied CO2, producing HCO3− and CO32− species (eqn (3) and (4)), which not only decreases the CO2 utilization but also crossover through the AEM to alter the local anodic environment (Fig. 1 and Table 1).12,14–18 Along with a proton released from the prolonged OER, this eventually leads to a highly intricate anodic environment at the membrane-electrode interface with varying CO2, HCO3−, CO32−, H2O, K+ concentrations, and pH depending on the operating current.19 Since the resulting anodic environment significantly affects the performance of the local-chemistry-sensitive electrocatalyst, it is necessary to develop and optimize stable OER electrocatalysts tailored to the environment of the corresponding MEA-type cell.20
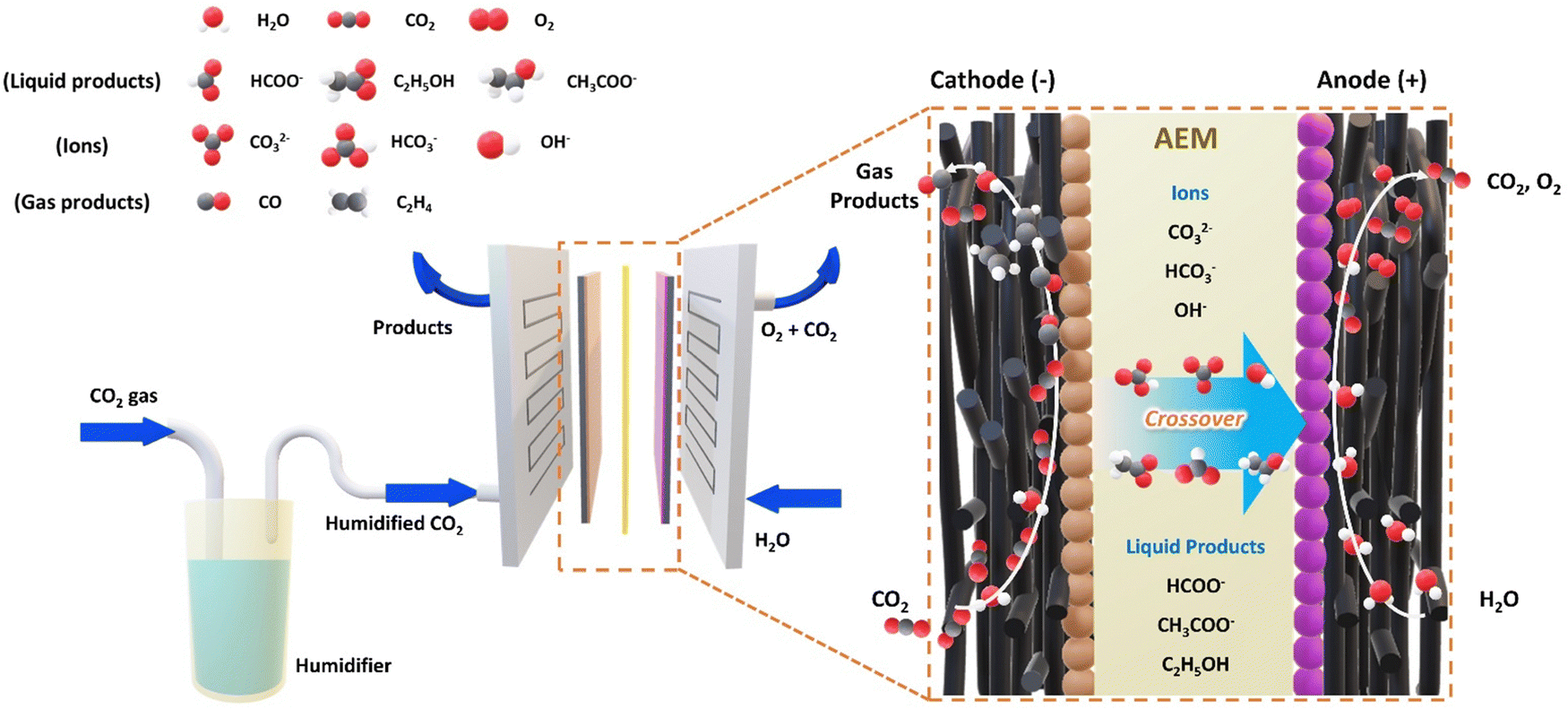 | ||
| Fig. 1 Schematic overview of the crossover of the ions and liquid products in an MEA-type electrolyzer. | ||
| Cathode | Anode | |
|---|---|---|
| Charge transfer reactions | CO2 + H2O + e− → CO + 2OH− (1) | 2H2O → O2 + 4H+ + 4e− (5) |
| 2H2O + 2e− → H2 + 2OH− (2) | ||
| Homogeneous reactions | CO2 + OH− → HCO3− (3) | H+ + CO32− → HCO3− (6) |
| HCO3− + OH− → H2O + CO32− (4) | H+ + HCO3− → H2O + CO2 (7) | |
| Chemical equilibria | CO2 + OH− ↔ HCO3− (8) | |
| HCO3− ↔ CO32− + H+ (9) | ||
Nevertheless, efforts towards OER electrocatalyst developments considering the complex anodic environment within anion exchange membrane CO2 electrolyzers (AEMCEs) are currently lacking. Furthermore, a few OER electrocatalysts recently developed exclusively for CO2 electrolysis have been optimized in aqueous carbonate and bicarbonate electrolytes, resembling traditional H-cells that are not commercially applicable.21–24 This raises the question of ‘how reliable are the conclusions obtained from aqueous systems?’ and urges the additional evaluation of OER electrocatalysts in commercially viable AEMCEs before considering their industrial applications.
In this perspective, our goal is to bridge the gap between the material-centrically developed OER electrocatalysts and the actual behavior of catalysts in MEA-type electrolyzers. As a first step, we scrutinize the local anodic environment inside the AEMCE, which necessitates the development of superior OER electrocatalysts under the high flux of (bi)carbonate crossover. Then, we analyze the discrepancy between aqueous testing and commercially viable systems and the limitations of the conventional evaluation platforms from a systematic point of view. Finally, we propose three catalyst evaluation platforms that integrate reference electrodes to existing AEMCEs to overcome the limitations of the aqueous testing system and facilitate the development of commercially applicable OER electrocatalysts.
2. Complexity of the local anodic environment in the AEMCE
In an AEMCE, which operates without a catholyte, the type of anolyte can significantly impact the anodic and overall cell efficiency. While concentrated alkaline solutions (e.g., 1 M KOH) or near-pH neutral bicarbonate solutions (e.g., 0.5 M KHCO3) are two common types of anolytes, the former can offer an advantage in terms of energy efficiency owing to its lower thermodynamic potential.12,25 Moreover, one might anticipate that well-established Ni-based OER electrocatalysts are directly applicable in alkaline anolyte-operated AEMCEs due to their stability at high anodic potentials in alkaline environments. However, this may not be the case in the actual system. Despite numerous reported cheap and high-performing transition metal-based OER electrocatalysts for alkaline water electrolyzers, a significant proportion of alkaline anolyte-operated AEMCEs still rely on precious metal-based OER electrocatalysts (e.g., IrO2).13 These precious-metal electrocatalysts are known for their gradual dissolution at high anodic potentials under alkaline conditions, yet they are rather recognized for their enhanced stability under near pH-neutral conditions.26 This ironic situation could be explained by understanding the neutralized local anodic environment inside the AEMCE.2.1. Ions and liquid product crossover
The primary factor governing the local anodic environment of the AEMCE is the migration of charge-carrying anions across the AEM. Ideally, the hydroxide ions released from the CO2RR (and the HER) should function as the primary charge carrier, connecting an electrochemical circuit through the AEM (eqn (1) and (2)).26 However, this is not the case. Instead, the fed CO2 undergoes partial neutralization by the liberated OH− ions, generating carbonate or bicarbonate species depending on the pH (eqn (3) and (4)). Consequently, these negatively charged carbonate or bicarbonate species traverse the AEM, altering the local anodic environment while resulting in a substantial reactant loss.14,17,27Through the modeling study, Weber et al. expected that the carbonate ion would become the primary charge carrier in the AEMCE.12 Their expectation was substantiated by Larrazabal et al., who examined the CO2 to O2 ratio at the anode gas outlet,28 allowing the main charge carrier species to be experimentally deduced based on the reaction stoichiometry.7,11 The observed anodic CO2 to O2 ratio clearly indicates that the primary charge carrier is the carbonate ion rather than hydroxide or bicarbonate ions, although this ratio may alter depending on the current density. As a result, the neutralized reactants transported to the anode through the AEM in the form of carbonate ions bring about a significant change in the local anodic chemical environment. This necessitates the development of stable OER electrocatalysts capable of functioning under the lowered local pH caused by the high flux of carbonate ions.
In addition, the complexity of the local anodic environment within the AEMCE is further exacerbated by the crossover of liquid products from the cathode to the anode and their subsequent anodic oxidation.29 Typically, liquid products can partially traverse the AEM through electromigration for negatively charged species (e.g., formate/acetate) and electroosmotic drag for neutral species (e.g., ethanol), and their anodic oxidation is inevitable due to their more cathodic equilibrium potential compared to the competing OER, leading to the production of CO2 or other organic species.5,18 For instance, when ethanol produced at the cathode reaches the anode, its oxidation significantly alters the anolyte composition where its oxidation process involves sequential steps, converting ethanol first into aldehydes and then carboxylates, introducing additional complexity to the anolyte.29 Furthermore, the extra consumption of OH− during the oxidation of ethanol to acetate accelerates the transition towards a near pH-neutral condition, coupled with the high carbonate crossover flux, to result in an increase in the AEMCE's ohmic resistance and changes in the kinetics of anodic reactions.30 Consequently, OER catalysts inside the AEMCE must be robust enough to withstand changes in the anolyte induced by the accumulation and oxidation of liquid products. While strategies to prevent liquid product crossover are essential, such as developing MEA fabrication methods,31 reinforced ionomers,32 and controlling AEM water uptake,33 the impact of anodic oxidation of liquid products should never be overlooked in the development of OER electrocatalysts for CO2 electrolysis.
2.2. Challenges of replacing precious metal-based electrocatalysts
Understanding the local anodic pH drop resulting from the crossover of ions and liquid products enables us to comprehend the paradoxical use of precious metal-based OER electrocatalysts (e.g., IrO2, RuO4) in commercially viable AEMCEs regardless of the anolyte type despite their disadvantages of scarcity and high price. Vass et al. experimentally demonstrated the superior stability of Ir-based materials over Ni as anode catalysts in AEMCEs using online ICP-MS measurements with two types of anolytes.15 When 0.1 M CsHCO3 and 0.1 M CsOH solutions were circulated as the neutral and alkaline anolytes, respectively, there was a notable dissolution of the Ni catalyst in neutral solution, whereas negligible dissolution of the Ir catalyst was observed. Interestingly, the opposite situation occurred in alkaline solution, demonstrating that Ni is more stable under alkaline conditions while Ir is more stable under neutral conditions. Under neutral conditions, the dissolution of Ni catalysts during long-term electrolysis results in a significant increase in the anodic potential over time. In addition, a portion of dissolved Ni2+ ions may pass through the membrane, precipitating the Ni2+ salts at the cathode, which not only increases the overall cell voltage but also alters the CO2RR selectivity. This experiment explains why Ir-based electrocatalysts are more resilient in the real anodic environment within AEMCEs, where local pH decreases due to the crossover of carbonate species and the release of protons from the prolonged OER, highlighting the challenges of replacing precious metal-based catalysts with unstable transition metal-based materials under near pH-neutral conditions in this commercially applicable device.These results motivate us to prioritize a comprehensive understanding of the local anodic environment within the AEMCE, where the anodic catalyst layer encounters under actual operating conditions, rather than solely focusing on the bulk anolyte for future OER electrocatalyst development. For the successful commercialization of CO2 electrolysis, dedicated focus should be directed towards the development of stable transition metal-based OER electrocatalysts that can effectively function under near-pH neutral conditions with exceptional tolerance towards carbonate ions.
3. Incompatibility between aqueous testing systems and AEMCEs
With a special effort to replace precious metals, there have been a few OER electrocatalysts developed in aqueous carbonate electrolytes to simulate the CO2 electrolysis environment. Fig. 2 summarizes the OER electrocatalysts developed in near-pH neutral (bi)carbonate electrolytes, arranged in the ascending order of overpotentials at 10 mA cm−2. Notably, the conventional aqueous OER electrocatalyst testing systems closely resemble the H-cell setup for CO2 electrolysis. Despite being commercially inapplicable, this system offers the advantage of rapid initial screening in the early stage of electrocatalyst development while enabling reliable catalyst assessment through standardized testing procedures under well-defined conditions, ensuring consistency across laboratories, especially in the field of fuel cells and water electrolysis with fixed product and conducting ion species. Furthermore, by utilizing a non-reactive substrate and mitigating mass transport limitations through RDE or mechanical stirring, the aqueous testing system specializes in measuring the inherent activity of the catalyst itself. However, despite their easy accessibility and reliability, drawing conclusions about local chemistry-sensitive electrocatalysts from this aqueous testing system, which does not accurately reflect the complex anodic chemistry within MEAs, inevitably presents several challenges when attempting to translate their catalytic performance into a commercially viable device.34 Particularly, when developing OER electrocatalysts for CO2 electrolysis, where the target product and crossover species vary depending on the operation conditions and cathodic CO2RR electrocatalysts, the complexity and incompatibility between the two systems increase compared to analogous fuel cells or water electrolysis. Taking Fig. 2 as an example, there are no fixed aqueous electrolyte conditions for CO2 electrolysis, and the different pH values resulting from inconsistent electrolytes prevent a fair comparison between different electrocatalysts. Moreover, some of the listed substrates are not applicable as GDEs in MEA-type systems due to inappropriate electrode architecture. Also, when aiming for commercialization, measuring an overpotential at a benchmark current density of 10 mA cm−2 may not provide insight into their performance under industrial-relevant operating conditions, which requires at least 200 mA cm−2 for economic viability of CO2 electrolysis systems.35,36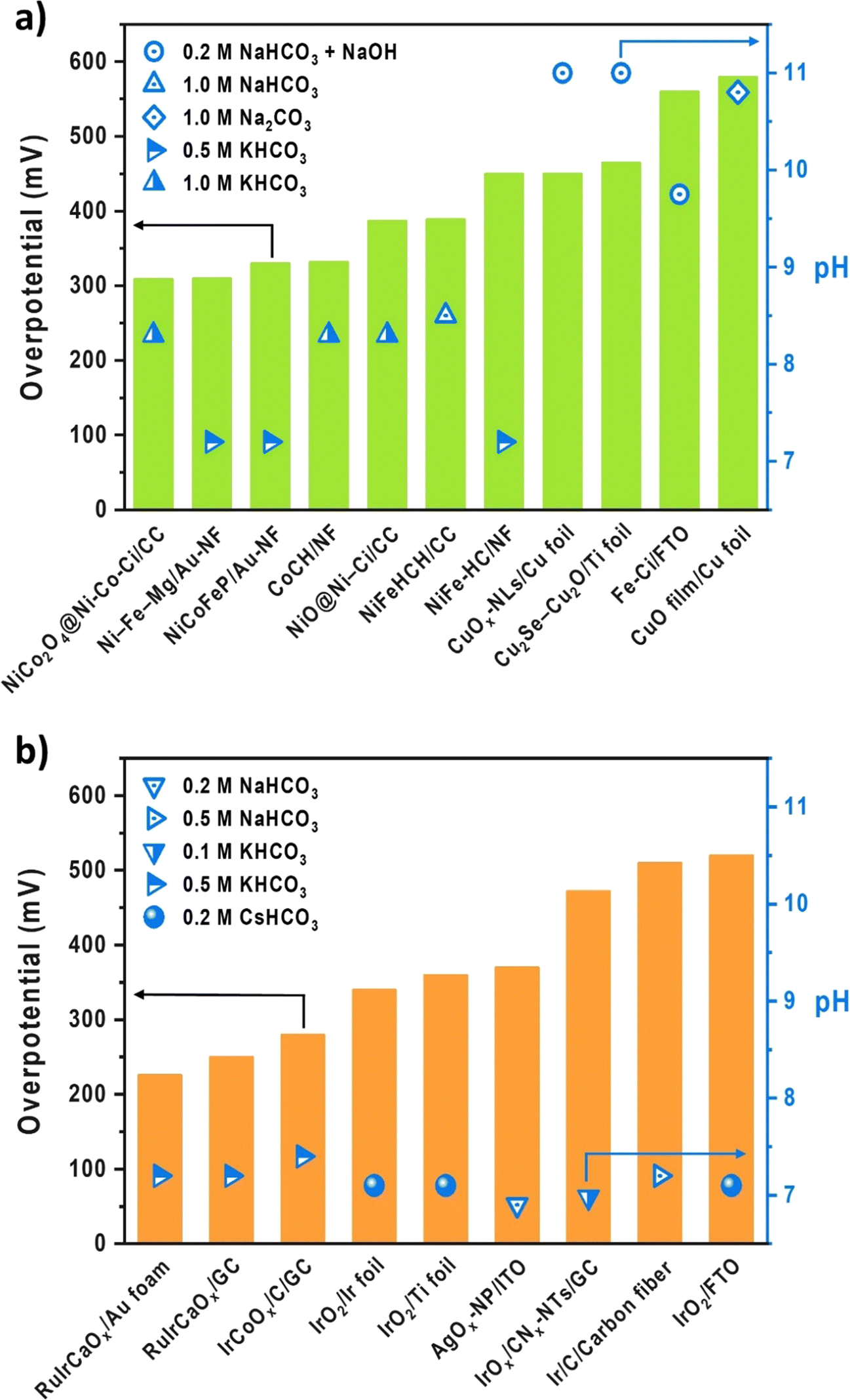 | ||
| Fig. 2 Performance and experimental conditions of (a) non-precious37–47 and (b) precious48–53 OER electrocatalysts in the aqueous carbonate system. | ||
Such incompatibility motivates us to speculate whether our current catalyst evaluation platform results in the independent development of OER electrocatalysts regardless of the system design and under inappropriate conditions for CO2 electrolysis. While the general challenges of near pH-neutral water oxidation and material-centric approaches for catalyst designs have been comprehensively covered in previous reviews,21–24 our focus here is to systematically explore the discrepancies between these two systems beyond mere catalyst performance and address the limitations of the conventional aqueous testing system. The differences between the two systems are summarized in Fig. 3.
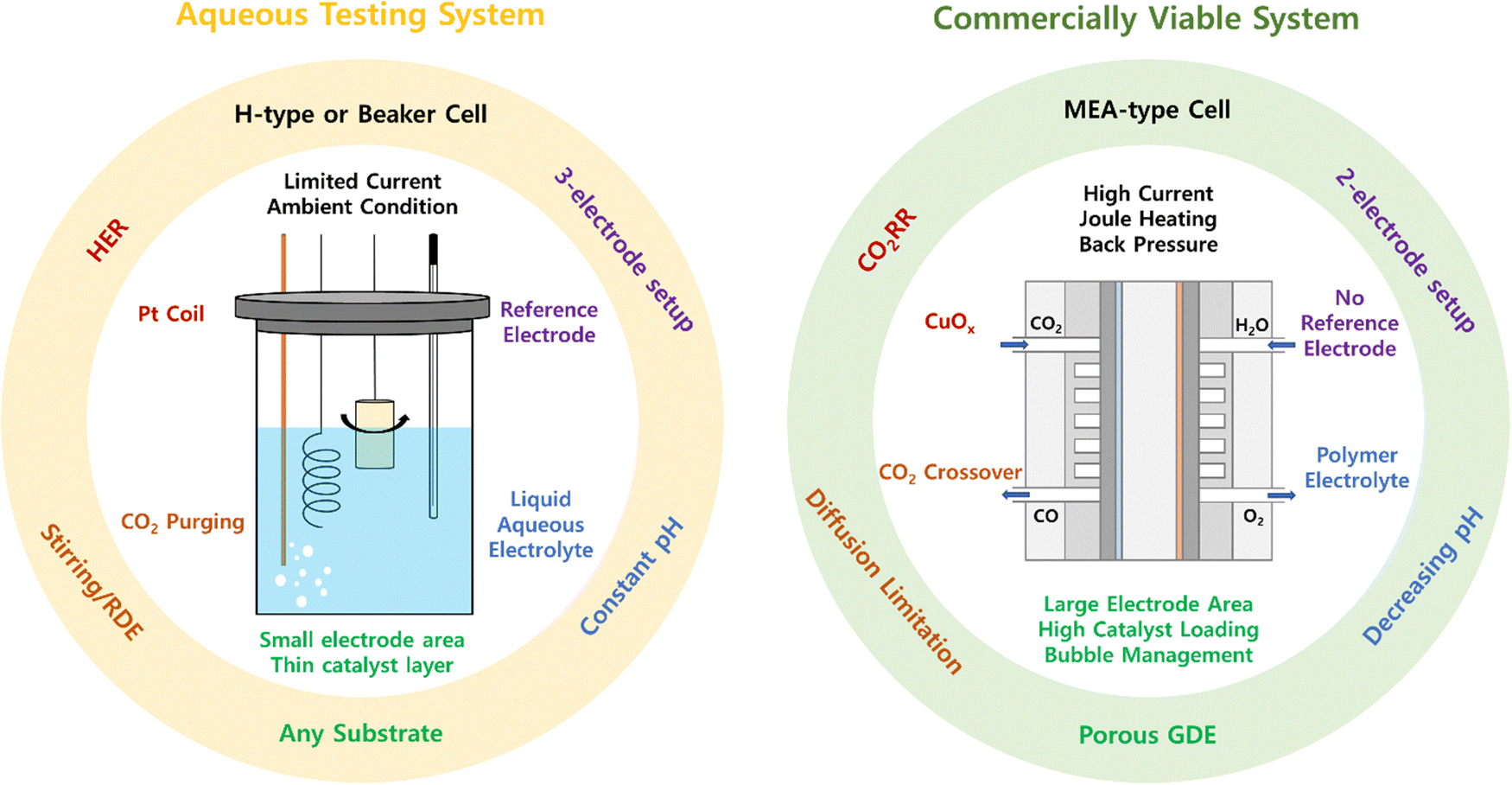 | ||
| Fig. 3 Schematic comparison of the aqueous testing system and commercially viable system. | ||
3.1. Electrolyzer and electrode configuration
In the typical aqueous testing system, OER electrocatalysts have been developed using a standard three-electrode setup in H-cells or beaker cells. Under these well-defined conditions, the employment of a reference electrode with a known potential allows the separation of the OER from the full-cell reaction. However, despite their intended application in CO2 electrolysis, HER facilitating electrodes (e.g., platinum coil) are used as cathodes instead of CO2RR electrodes to prevent the cathodic reaction from becoming the rate-limiting step. In addition, it is not feasible to implement electrode kinetics at the interface of the membrane and electrodes in an aqueous testing system. In contrast, although the AEMCE can replicate a genuine anodic environment comparable to an industrial setting, its structural limitations in the MEA-type cell configuration only permit full-cell experiments with a two-electrode configuration. Unfortunately, the absence of a reference electrode prevents the separate assessment of electrocatalysts for anodic and cathodic reactions within a commercially viable device, limiting the evaluation to identify the overall cell voltage of the entire electrolyzer. Consequently, most of the catalyst development so far have been carried out using conventional aqueous testing systems, in which half-cell studies are available, despite the drawbacks of oversimplification and the inability to reflect the actual anodic environment.The discrepancy between the two systems also arises from differences in the electrode architecture. While there are no restrictions on the electrode architecture in the aqueous system, the porosity of the substrate becomes a primary factor for its utilization as a GDE, thereby limiting the employment of substrates with a flat solid backbone for an AEMCE.8,54,55 Furthermore, carbon-based substrates should be avoided under industrial-relevant operation conditions, as carbon corrosion occurs when a high anodic potential is applied, leading to the dissolution of the catalyst layer on top of the carbon substrate.56,57 While this phenomenon is rarely encountered in an aqueous testing system operating at relatively low current densities, it is inevitable in an industrial environment requiring high current densities with high anodic potentials.
3.2. Electrolyte and operating conditions
Another discrepancy between the two systems arises from the different electrolyte conditions. In the aqueous testing system, mass transport occurs through the liquid (bi)carbonate electrolyte with or without CO2 saturation. Here, the overpotential at a geometric current density of 10 mA cm−2 has been adopted as a typical benchmark descriptor for catalyst activity.58,59 At times, the rotating disk electrode (RDE) or mechanical stirring is utilized to alleviate mass transport limitations arising from a shortage of reactants (i.e., OH−) in a neutral electrolyte.60 Although it is generally understood that the low proton-accepting ability of the carbonate ion cannot effectively neutralize the generated protons during the OER, resulting in an increase in local acidity at the anode,22 the rapid diffusion of the electrolyte facilitated by the RDE setup or mechanical stirring helps in assessing the intrinsic activity of OER electrocatalysts in an aqueous testing system by maintaining a relatively constant pH and reaction environment near the electrode, along with limited current density. Nevertheless, despite its well-defined conditions, the conventional aqueous testing system has at least two limitations. One is the inconsistency in the electrolyte pH caused by disagreements in the type and concentration of electrolytes (Fig. 2), which disturbs a fair comparison of various electrocatalysts. Another is the excessively simplified electrolyte of the aqueous testing system, which cannot accurately represent the complex local anodic environment of the AEMCE.In an AEMCE, mass transport occurs through the porous catalyst layer and polymer electrolyte, where a high current density operation of at least a few hundred mA is necessary for economic feasibility.61,62 This underscores the importance of the kinetics of the entire system over the thermodynamics of the electrocatalyst, which raises questions about the significance of measuring the activity of electrocatalysts at the present benchmark current density, 10 mA cm−2.15 Moreover, the high current density operation under industrial-relevant operating conditions will not only result in an accumulation of released protons at the anode but also alter the cathode reactivity, leading to changes in the quantity and ratio of negatively charged species crossing over the polymer electrolyte. This causes a gradual transition in the local anodic chemistry from the bulk during the long-term operation, ultimately affecting the activity and stability of the OER electrocatalysts.28 In addition, Joule heating can result in a significant increase in the local internal temperature of the AEMCE under industrial-relevant operating conditions. Particularly in CO2 electrolysis devices, the elevated temperature can directly affect the activity and stability of the OER electrocatalysts and indirectly due to the altered crossover species across the polymer membrane by changing the equilibrium constant of electrochemical and homogeneous reactions near the cathode (eqn (1)–(4)).12 While temperature changes due to Joule heating are often overlooked in aqueous systems, our catalyst development should consider these temperature effects within the AEMCE.63,64
Besides, other operational parameters including the electrolyte circulation rate, CO2 flow rate, and backpressure closely interact with each other, further complicating the actual anodic environment within the AEMCE.65,66 Consequently, the oversimplified aqueous testing system, which fails to reflect this complexity, is susceptible to false optimization and raises doubts about the validity of the current direction of catalyst development. In particular, the impact of the membrane, which doesn’t allow its complete separation from the catalyst and ionomer layers in an MEA-type cell configuration, adds to the complexity when attempting to translate catalytic behavior from an aqueous testing system to the AEMCE. Therefore, despite the advantage of simplicity for rapid pre-screening of high-performance OER electrocatalysts, the conventional aqueous evaluation system cannot be directly employed for industrial implementation without additional validation in the AEMCE. To address the incompatibility of the two systems, there is a pressing need to introduce new catalyst evaluation platforms designed to more closely resemble a commercially viable device rather than a simulated aqueous environment while enabling the separation of a single anodic half-reaction.
4. Bridging the gap between the aqueous testing system and AEMCE: reference electrode integrated systems
If OER electrocatalysts could be developed directly within the AEMCE from the initial catalyst screening, it would enable their optimization to suit the local anodic environment, aligning with the actual operation conditions of a context-sensitive device without additional validation. However, unlike the aqueous testing system with a typical three-electrode setup, assessing the electrocatalyst in an MEA-type cell relies on a two-electrode setup, involving the analysis of polarization curves and electrochemical impedance spectrometry (EIS) of the entire device due to the geometrical constraints that prevent the insertion of a reference electrode. This presents a challenge in the optimization of OER electrocatalysts, stemming not only from the mistaken assumption that altering a single parameter will not affect other parameters and the overall cell performance but also from the difficulty of precisely attributing obtained electrochemical data (polarization curve and EIS) to individual components (anode, cathode, and membrane). Therefore, the primary task in addressing this issue is to differentiate between the anodic and cathodic reactions and accurately assess the performance of each respective electrode.In this context, we introduce three distinct methods enabling the integration of reference electrodes into the MEA-type cell configuration. While a few studies on reference electrode integration in MEA-type electrolyzers have been conducted in analogous fuel cells, where the crossover product consists solely of H+ or OH−,67–70 these integration methods are expected to hold higher value and usefulness in the context-sensitive CO2 electrolysis devices. While accurately matching the local anodic environment encountered by OER electrocatalysts, these innovative testing platforms will differentiate anodic (and cathodic) contributions from the overall cell voltage. Furthermore, through the localized EIS, the detailed processes governing the electrocatalyst performance are expected to be unveiled and addressed.
4.1. Three-electrode techniques in AEMCEs
Thangavel and Kim et al. first introduced the extended MEA method in an analogous alkaline AEM water electrolyzer.71 In comparison to existing MEAs, their extended membrane enables electrical contact with an external aqueous electrolyte. This innovative strategy permits the provision of additional OH− to restore the original conductivity of the electrolyzer while maintaining a relatively constant pH during continuous operation. Using a similar approach, Xu and Boettcher et al. achieved the integration of a reference electrode. The geometrical limitation of the MEA-type cell configuration could be overcome by employing a salt bridge as an ionic conductor, which enabled the external reference electrode to be ionically 'wired' to the polymer membrane inside the cell (Fig. 4a).72 In this cell configuration, an additional membrane strip is attached to the edge of the AEM and partially immersed in a 1 M KOH solution containing the external reference electrode.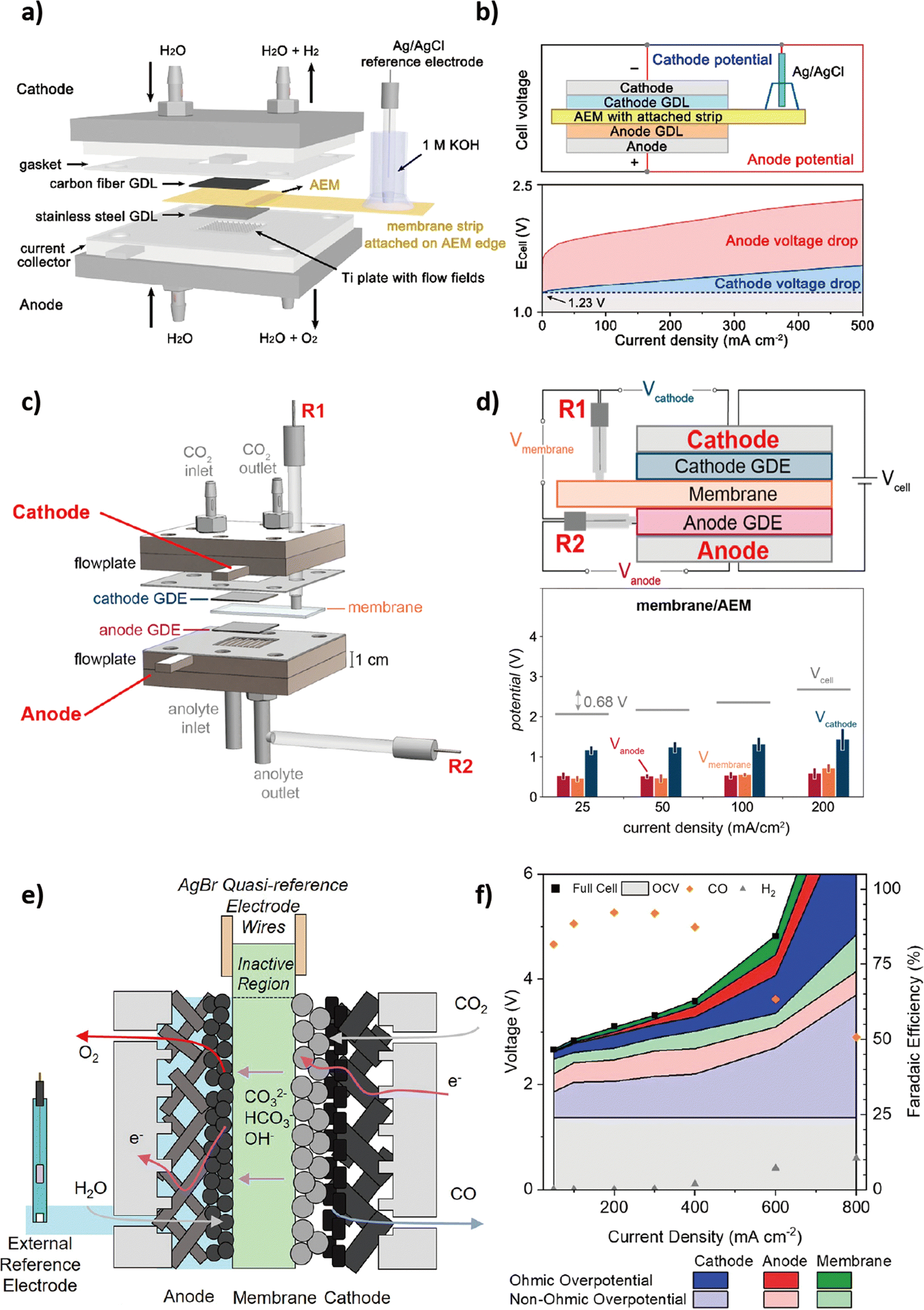 | ||
| Fig. 4 Schematic electrolyzer configuration and corresponding voltage breakdown of reference electrode integration methods. (a) and (b) 3-electrode technique. Reproduced with permission from ref. 67. Copyright 2021 American Chemical Society. (c) and (d) 4-electrode technique. Reproduced with permission from ref. 73. Copyright 2020 American Chemical Society. (e) and (f) 5-electrode technique. Reproduced with permission from ref. 74. Copyright 2022 American Chemical Society. | ||
With the employment of the reference electrode, the potential of each electrode can be assigned (Fig. 4b). Firstly, the overall cell voltage of the electrolyzer, Ecell = Ec − Ea, can be obtained either directly or by adding up the potential of each electrode with respect to an external reference electrode. Then, the anodic and cathodic overpotentials are calculated by taking the absolute value of the difference between the potential of each electrode obtained through an external reference electrode and its corresponding thermodynamic potential. Besides, the charge-transfer resistance (Rct) values obtained for each electrode via an external reference electrode allow a clear assignment of specific resistive or capacitive components within the entire circuit to the variation of a single parameter. While this capability was previously constrained in the typical two-electrode system due to overlapping features in the impedance spectra of both electrodes,67 the localized EIS obtained through this integration method can achieve a substantial reduction in the uncertainty associated with an equivalent circuit model and its interpretation, serving as an efficient tool for optimizing OER electrocatalysts in the complex environment of AEMCEs.72
Nevertheless, there are several concerns regarding this integration method due to the positioning of the reference electrode outside the cell. Firstly, to ensure the stability of the reference electrode, the membrane strip should maintain a hydrated condition throughout the experiment.73 Secondly, the potential measurements with respect to the external reference electrode are susceptible to distortion and potential drop as the reference electrode is positioned outside the main current flow path. While maintaining alignment between the anode and cathode can minimize potential distortion caused by edge effects, positioning the reference electrode as close as possible to the working electrode is expected to reduce potential drop.74–76
The three-electrode technique can be readily implemented in existing MEA-type CO2 electrolyzers without requiring structural modifications, facilitating the straightforward development of OER electrocatalysts suitable for industrial-relevant settings. However, this technique has the limitation of not being able to separate the voltage and resistance associated with the membrane itself. If the separation of the membrane that interfaces with the catalyst layer is possible, it could be more advantageous for achieving advanced catalyst development. This could be realized by incorporating two or more reference electrodes.
4.2. Multi-reference electrode techniques
| Vcell = Vcathode − Vanode − Vmembrane | (10) |
While this analytical flow cell requires slight modifications to existing MEA-type electrolyzers, this distinctive testing platform, which facilitates independent electrochemical measurements of the membrane itself, offers advantages in the catalyst optimization process by providing a deeper comprehension of the membrane and the membrane–catalyst interface.
As depicted in Fig. 4f, the five-electrode technique achieved the deconvolution of the cathode, anode, and membrane contributions during CO2 electrolysis, similar to the four-electrode technique. Moreover, it successfully differentiated various types of overpotential through EIS analysis, categorizing them into three sources: ion/electron transport (ηcond), electrode kinetics (ηkin), and mass transport (ηmt) (eqn (11)). The ion/charge conduction overpotential (ηcond), or ohmic overpotential, can be determined by analyzing impedance at a high frequency (>1000 Hz) during operation at a specific current density, denoted as ‘i’ (eqn (12)). On the other hand, non-ohmic overpotentials related to electrode kinetics and mass transport can be extracted by measuring the impedance at medium and low frequencies, respectively. In this manner, conducting quantitative localized EIS analysis for each component through the five-electrode technique can effectively overcome the constraints of qualitative interpreting the existing MEA-type cell configuration.78 As a result, combining the five-electrode technique with EIS presents a route for systematic modeling of ion/charge conduction, electrode kinetics, and mass transport resistance independently at each electrode with enhanced accuracy via disentangling electrode dynamics from membrane impedance, aiming to expedite the advancement of more efficient components, including optimized OER electrocatalysts for CO2 electrolysis.
| η = ηkin + ηmt + ηcond | (11) |
| ηnon-ohmic = ηkin + ηmt, ηcond = iRcond | (12) |
While much of the research on CO2 electrolysis has predominantly focused on the cathodic CO2RR, the anodic OER is equally crucial for achieving high energy efficiency, Faraday efficiency, and overall durability at the full-device level. Especially in this context-sensitive AEMCE, where charge carriers and products vary based on the targeted cathodic product and corresponding operational conditions, adopting OER electrocatalysts developed in aqueous alkaline and carbonate systems may not be suitable for the local anode environment. In the realm of CO2 electrolysis, we believe that the three evaluation platforms presented above can pave the way for the development of OER electrocatalysts guided by the correct directionality, aligning with industrially relevant cathodic environments and operating conditions, ultimately facilitating the achievement of high performance in a full-device level.
5. Conclusions
In this perspective, we have discussed the direction of OER electrocatalyst evaluation to achieve optimal performance in commercially viable CO2 electrolysis systems. As a first step, we delved into an analysis of the localized anodic environment that a commercially applicable device utilizing OER electrocatalysts would encounter. This exploration revealed that the local anodic pH experiences a continuous decrease due to the crossover of carbonate species generated by homogeneous reactions near the cathode, leading to a near-neutral condition regardless of the initial pH of the bulk anolyte. This comprehensive understanding of the internal anodic environment highlights the pressing need to develop a stable OER electrocatalyst that can operate effectively within this near pH-neutral environment, particularly when subjected to a high flux of carbonate ions, instead of the long-developed alkaline water oxidation electrocatalysts.Until now, there have been a few efforts to develop OER electrocatalysts in aqueous carbonate electrolytes. However, the substantial discrepancies between the aqueous testing system and the AEMCE, encompassing factors such as the cell configuration, electrode architectures, electrolyte properties, and their distinct operational parameters, impede the seamless translation of the exceptional catalytic performance achieved in the conventional aqueous system to actual MEA-type electrolyzers. This challenge delays the swift integration of novel OER electrocatalysts into industrial applications, as developed electrocatalysts must ultimately undergo testing for their activity and stability within an MEA-type electrolyzer to ensure their practical viability, even though the simplicity of the aqueous testing system offers advantages for the rapid preliminary screening of promising electrocatalysts.
To bridge this gap, we introduced three evaluation platforms with integrated reference electrodes to implement three-, four-, and five-electrode setups within the AEMCE, overcoming the geometrical constraints of existing MEA-type cell configurations. While faithfully replicating the genuine operational environment, these platforms provide clear insight into the electrochemical results of individual electrodes to facilitate the accurate optimization of electrocatalysts to align with real-world conditions, guiding the correct developmental trajectory for OER electrocatalysts intended for commercialization. Although the developed OER electrocatalysts may not yet perform as efficiently within the MEA framework as in the aqueous testing system, we believe that the combination of extensive knowledge accumulated over decades in water oxidation catalysts and the precise guidance provided by these innovative evaluation platforms from the early stage of catalyst screening can accelerate industrial implementation of CO2 electrolysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Council of Science & Technology (NST) grant by the Korean government (MSIT) (No. CAP21011-201) and by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korean government (MOTIE) (20213030040520). This work was also supported by the Carbon Neutral Institute (CNI) in UNIST (1.230049.01).References
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- D. U. Nielsen, X. M. Hu, K. Daasbjerg and T. Skrydstrup, Nat. Catal., 2018, 1, 244–254 CrossRef CAS.
- M. K. Kim, H. Lee, J. H. Won, W. Sim, S. J. Kang, H. Choi, M. Sharma, H. S. Oh, S. Ringe, Y. Kwon and H. M. Jeong, Adv. Funct. Mater., 2022, 32, 2107349 CrossRef CAS.
- S. Sultan, H. Lee, S. Park, M. M. Kim, A. Yoon, H. Choi, T. H. Kong, Y. J. Koe, H. S. Oh, Z. Lee, H. Kim, W. Kim and Y. Kwon, Energy Environ. Sci., 2022, 15, 2397–2409 RSC.
- R. K. Miao, Y. Xu, A. Ozden, A. Robb, C. P. O’Brien, C. M. Gabardo, G. Lee, J. P. Edwards, J. E. Huang, M. Fan, X. Wang, S. Liu, Y. Yan, E. H. Sargent and D. Sinton, Joule, 2021, 5, 2742–2753 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- L. Ge, H. Rabiee, M. Li, S. Subramanian, Y. Zheng, J. H. Lee, T. Burdyny and H. Wang, Chem, 2022, 8, 663–692 CAS.
- C. T. Dinh, T. Burdyny, G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. Pelayo García De Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- L. C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2020, 13, 3592–3606 RSC.
- L. C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 RSC.
- Á. Vass, A. Kormányos, Z. Kószo, B. Endrődi and C. Janáky, ACS Catal., 2022, 12, 1037–1051 CrossRef PubMed.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O’Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Nat. Energy, 2021, 6, 339–348 CrossRef CAS.
- Á. Vass, B. Endrödi, G. F. Samu, Á. Balog, A. Kormányos, S. Cherevko and C. Janáky, ACS Energy Lett., 2021, 6, 3801–3808 CrossRef PubMed.
- Y. Xu, R. K. Miao, J. P. Edwards, S. Liu, C. P. O’Brien, C. M. Gabardo, M. Fan, J. E. Huang, A. Robb, E. H. Sargent and D. Sinton, Joule, 2022, 6, 1333–1343 CrossRef CAS.
- J. Zhang, W. Luo and A. Züttel, J. Catal., 2020, 385, 140–145 CrossRef CAS.
- C. M. Gabardo, C. P. O’Brien, J. P. Edwards, C. McCallum, Y. Xu, C. T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- C. McCallum, C. M. Gabardo, C. P. O’Brien, J. P. Edwards, J. Wicks, Y. Xu, E. H. Sargent and D. Sinton, Cell Rep. Phys. Sci., 2021, 2, 100522 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- N. Jiang, Z. Zhu, W. Xue, B. Y. Xia and B. You, Adv. Mater., 2022, 34, 2105852 CrossRef CAS PubMed.
- Y. Dong and S. Komarneni, Small Methods, 2021, 5, 2000719 CrossRef CAS PubMed.
- S. Anantharaj and V. Aravindan, Adv. Energy Mater., 2020, 10, 1902666 CrossRef CAS.
- P. Li, R. Zhao, H. Chen, H. Wang, P. Wei, H. Huang, Q. Liu, T. Li, X. Shi, Y. Zhang, M. Liu and X. Sun, Small, 2019, 15, 1805103 CrossRef PubMed.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C. T. Dinh, T. Burdyny, M. G. Kibria, C. P. O’Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- G. Kastlunger, L. Wang, N. Govindarajan, H. H. Heenen, S. Ringe, T. Jaramillo, C. Hahn and K. Chan, ACS Catal., 2022, 12, 4344–4357 CrossRef CAS.
- G. O. Larrazábal, M. Ma and B. Seger, Acc. Mater. Res., 2021, 2, 220–229 CrossRef.
- G. O. Larrazábal, P. Strøm-Hansen, J. P. Heli, K. Zeiter, K. T. Therkildsen, I. Chorkendorff and B. Seger, ACS Appl. Mater. Interfaces, 2019, 11, 41281–41288 CrossRef PubMed.
- Q. Xu, S. Liu, F. Longhin, G. Kastlunger, I. Chorkendorff and B. Seger, Adv. Mater., 2024, 36, 2306741 CrossRef CAS PubMed.
- Q. Xu, S. Garg, A. B. Moss, M. Mirolo, I. Chorkendorff, J. Drnec and B. Seger, Nat. Catal., 2023, 6, 1042–1051 CrossRef CAS.
- T. Kong, P. Thangavel, S. Shin, S. Kwon, H. Choi, H. Lee, N. Park, J. Woo and Y. Kwon, ACS Energy Lett., 2023, 8, 4666–4673 CrossRef CAS.
- Y. Zhao, X. Zu, R. Chen, X. Li, Y. Jiang, Z. Wang, S. Wang, Y. Wu, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2022, 144, 10446–10454 CrossRef CAS PubMed.
- C. McCallum, C. M. Gabardo, C. P. O’Brien, J. P. Edwards, J. Wicks, Y. Xu, E. H. Sargent and D. Sinton, Cell Rep. Phys. Sci., 2021, 2, 100522 CrossRef CAS.
- J. Fan, M. Chen, Z. Zhao, Z. Zhang, S. Ye, S. Xu, H. Wang and H. Li, Nat. Energy, 2021, 6, 475–486 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustainable, 2021, 4, 911–919 CrossRef.
- J. Du, Z. Chen, S. Ye, B. J. Wiley and T. J. Meyer, Angew. Chem., Int. Ed., 2015, 54, 2073–2080 CrossRef CAS PubMed.
- H. Chen, Y. Gao, L. Ye, Y. Yao, X. Chen, Y. Wei and L. Sun, Chem. Commun., 2018, 54, 4979–4982 RSC.
- Y. Meng, X. Zhang, W. H. Hung, J. He, Y. S. Tsai, Y. Kuang, M. J. Kenney, J. J. Shyue, Y. Liu, K. H. Stone, X. Zheng, S. L. Suib, M. C. Lin, Y. Liang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23915–23922 CrossRef CAS PubMed.
- K. S. Joya and H. J. M. De Groot, ACS Catal., 2016, 6, 1768–1771 CrossRef CAS.
- M. Ma, Y. Liu, X. Ma, R. Ge, F. Qu, Z. Liu, G. Du, A. M. Asiri, Y. Yao and X. Sun, Sustainable Energy Fuels, 2017, 1, 1287–1291 RSC.
- M. Xie, L. Yang, Y. Ji, Z. Wang, X. Ren, Z. Liu, A. M. Asiri, X. Xiong and X. Sun, Nanoscale, 2017, 9, 16612–16615 RSC.
- F. Li, L. Bai, H. Li, Y. Wang, F. Yu and L. Sun, Chem. Commun., 2016, 52, 5753–5756 RSC.
- R. Ge, M. Ma, X. Ren, F. Qu, Z. Liu, G. Du, A. M. Asiri, L. Chen, B. Zheng and X. Sun, Chem. Commun., 2017, 53, 7812–7815 RSC.
- K. Karthick, S. Anantharaj, S. R. Ede and S. Kundu, Inorg. Chem., 2019, 58, 1895–1904 CrossRef CAS PubMed.
- N. Wang, Z. Cao, X. Zheng, B. Zhang, S. M. Kozlov, P. Chen, C. Zou, X. Kong, Y. Wen, M. Liu, Y. Zhou, C. T. Dinh, L. Zheng, H. Peng, Y. Zhao, L. Cavallo, X. Zhang and E. H. Sargent, Adv. Mater., 2020, 32, 1–6 Search PubMed.
- X. Zheng, B. Zhang, P. De Luna, Y. Liang, R. Comin, O. Voznyy, L. Han, F. P. García De Arquer, M. Liu, C. T. Dinh, T. Regier, J. J. Dynes, S. He, H. L. Xin, H. Peng, D. Prendergast, X. Du and E. H. Sargent, Nat. Chem., 2018, 10, 149–154 CrossRef CAS PubMed.
- Gurudayal, J. Bullock, D. F. Srankó, C. M. Towle, Y. Lum, M. Hettick, M. C. Scott, A. Javey and J. Ager, Energy Environ. Sci., 2017, 10, 2222–2230 RSC.
- K. S. Joya, Z. Ahmad, Y. F. Joya, A. T. Garcia-Esparza and H. J. M. De Groot, Nanoscale, 2016, 8, 15033–15040 RSC.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- W. Lv, S. Liu, R. Zhang, W. Wang, Z. Wang, L. Wang and W. Wang, J. Mater. Sci., 2018, 53, 4939–4948 CrossRef CAS.
- W. H. Lee, H. N. Nong, C. H. Choi, K. H. Chae, Y. J. Hwang, B. K. Min, P. Strasser and H. S. Oh, Appl. Catal., B, 2020, 269, 118820 CrossRef CAS.
- L. Zhang, L. Wang, Y. Wen, F. Ni, B. Zhang and H. Peng, Adv. Mater., 2020, 32, 2002297 CrossRef CAS PubMed.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Energy Environ. Sci., 2021, 14, 1959–2008 RSC.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, ChemSusChem, 2020, 13, 400–411 CrossRef CAS PubMed.
- S. G. Ji, H. Kim, W. H. Lee, H. S. Oh and C. H. Choi, J. Mater. Chem. A, 2021, 9, 19834–19839 RSC.
- I. S. Filimonenkov, C. Bouillet, G. Kéranguéven, P. A. Simonov, G. A. Tsirlina and E. R. Savinova, Electrochim. Acta, 2019, 321, 134657 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- C. Wei, S. Sun, D. Mandler, X. Wang, S. Z. Qiao and Z. J. Xu, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- T. Shinagawa, M. T. K. Ng and K. Takanabe, ChemSusChem, 2017, 10, 4155–4162 CrossRef CAS PubMed.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H. S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- S. Verma, B. Kim, H. R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. Chan, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- A. Buttler and H. Spliethoff, Renew. Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- C. P. O’Brien, R. K. Miao, S. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C. T. Dinh, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef.
- M. Hren, M. Božič, D. Fakin, K. S. Kleinschek and S. Gorgieva, Sustainable Energy Fuels, 2021, 5, 604–637 RSC.
- E. Engebretsen, G. Hinds, Q. Meyer, T. Mason, E. Brightman, L. Castanheira, P. R. Shearing and D. J. L. Brett, J. Power Sources, 2018, 382, 38–44 CrossRef CAS.
- G. Li and P. G. Pickup, Electrochim. Acta, 2004, 49, 4119–4126 CrossRef CAS.
- P. Piela, T. E. Springer, J. Davey and P. Zelenay, J. Phys. Chem. C, 2007, 111, 6512–6523 CrossRef CAS.
- T. V. Reshetenko, G. Bender, K. Bethune and R. Rocheleau, Electrochim. Acta, 2013, 88, 571–579 CrossRef CAS.
- P. Thangavel, M. Ha, S. Kumaraguru, A. Meena, A. N. Singh, A. M. Harzandi and K. S. Kim, Energy Environ. Sci., 2020, 13, 3447–3458 RSC.
- Q. Xu, S. Z. Oener, G. Lindquist, H. Jiang, C. Li and S. W. Boettcher, ACS Energy Lett., 2021, 6, 305–312 CrossRef CAS.
- W. He and T. Van Nguyen, J. Electrochem. Soc., 2004, 151, A185 CrossRef CAS.
- A. A. Kulikovsky and P. Berg, J. Electrochem. Soc., 2015, 162, F843–F848 CrossRef CAS.
- T. V. Reshetenko, G. Bender, K. Bethune and R. Rocheleau, Electrochim. Acta, 2011, 56, 8700–8710 CrossRef CAS.
- R. Zeng, R. C. T. Slade and J. R. Varcoe, Electrochim. Acta, 2010, 56, 607–619 CrossRef CAS.
- D. Salvatore and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 215–220 CrossRef CAS.
- K. U. Hansen, L. H. Cherniack and F. Jiao, ACS Energy Lett., 2022, 7, 4504–4511 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |