
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Macrocyclic receptors for anion recognition
Farhad Ali
Mohammed
 ab,
Tangxin
Xiao
c,
Leyong
Wang
cd and
Robert B. P.
Elmes
*abe
ab,
Tangxin
Xiao
c,
Leyong
Wang
cd and
Robert B. P.
Elmes
*abe
aDepartment of Chemistry, Maynooth University, National University of Ireland, Maynooth, Co, Kildare, Ireland. E-mail: robert.elmes@mu.ie
bSSPC – the Science Foundation Ireland Research Centre for Pharmaceuticals, University of Limerick, V94 T9PX Limerick, Ireland
cSchool of Petrochemical Engineering, Changzhou University, Changzhou, 213164, China
dKey Laboratory of Mesoscopic Chemistry of MOE, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, China
eKathleen Lonsdale Institute for Human Health Research, Maynooth University, National University of Ireland, Co. Kildare, W23 F2H6 Maynooth, Ireland
First published on 19th September 2024
Abstract
Macrocyclic receptors have emerged as versatile and efficient molecular tools for the recognition and sensing of anions, playing a pivotal role in molecular recognition and supramolecular chemistry. The following review provides an overview of the recent advances in the design, synthesis, and applications of macrocyclic receptors specifically tailored for anion recognition. The unique structural features of macrocycles, such as their well-defined structures and pre-organised binding sites, contribute to their exceptional anion-binding capabilities that have led to their application across a broad range of the chemical and biological sciences.
 Farhad Ali Mohammed | Farhad A. Mohammed earned a Bachelor of Science (BSc) degree in Chemistry from Maynooth University in 2021, graduating at the top of his class and receiving the prestigious Kathleen Lonsdale Prize. He is currently pursuing a PhD at Maynooth University as a recipient of the John & Pat Hume Scholarship, under the supervision of Dr Rob Elmes. His research focuses on supramolecular chemistry, specifically the design of macrocyclic receptors employing a novel biomimetic code for anion recognition. |
 Tangxin Xiao | Tangxin Xiao received his PhD in supramolecular chemistry from Nanjing University in 2014, supervised by Prof. Leyong Wang. Following postdoctoral research on fine chemicals at the Zhejiang University-NHU Company United R&D Center, he joined Changzhou University in 2017. He was promoted to Associate Professor in 2020 and to full Professor in 2023. From 2021 to 2022, he was funded by the China Scholarship Council (CSC) as a Visiting Scholar in Prof. Oren Scherman's group at the University of Cambridge. His current research interests focus on supramolecular chemistry and luminescent materials. He has co-authored more than 80 publications with over 4000 citations. |
 Leyong Wang | Leyong Wang received his PhD from Nanjing Univ. in 2000. After postdoctoral stays at the Institute of Chemistry, CAS with Prof. Youliang Hu, Université de Bourgogne with Dr Jean-Claude Chambron and Prof. Roger Guilard, and Mainz Univ. with Dr Voker Böhmer, he then became a Humboldt Research Fellow in Erlangen-Nürnberg Univ., affiliated with Professor John Gladysz. In 2006, he settled in Nanjing. He has been serving on the (advisory) editorial boards of numerous journals, including RSC Adv, Chem. Soc. Rev., Coord. Chem. Rev., Chin. Chem. Lett. et al. He has authored or coauthored over 220 papers in international peer reviewed journals. |
 Robert B. P. Elmes | Rob graduated with a BA Mod. in Medicinal Chemistry from Trinity College Dublin in 2007 and completed his PhD under Prof. Thorri Gunnlaugsson with an IRCSET Embark Scholarship. After a short postdoc at the Trinity Biomedical Sciences Institute, he joined the group of Prof. Kate Jolliffe at The University of Sydney, working on supramolecular platforms for anion sensing and transport. In 2014, Rob returned to Ireland, where is now Associate Professor and Associate Dean of Research for the Faculty of Science and Engineering at Maynooth University. He has held visiting research positions at Université d’Orléans and The Scripps Research Institute, and is a funded investigator at SFI's Pharmaceutical Research Centre, SSPC and the Kathleen Lonsdale Institute for Human Health Research. His research focuses on using supramolecular platforms in health research in areas such as drug delivery, diagnostics, and antimicrobial treatments. |
Introduction
Anionic atoms and molecules play a prevalent role in daily life as part of numerous environmental, chemical, and biological processes.1–4 From fluoride in tap water to nitrates and phosphates in soil fertilizers,5 sulfates in cosmetic products, and chloride and bicarbonate in our own physiology their impact cannot be underestimated. From a biological perspective, anions are not only crucial for maintaining cellular pH6 and volume,7 but they also contribute to the formation of membrane potentials, which facilitate the propagation of electrical signals through our nervous system. For example, a deficiency in iodide can serve as an indicator for thyroid cancer.8 Additionally, an imbalance in chloride ions can result in numerous diseases known as channelopathies including cystic fibrosis and Bartter syndrome.9 This biological relevance of anions, has led to significant research interest in the design of molecules capable of binding and recognising anions and it has become a fundamental aspect of supramolecular chemistry research.10–14Within this context, macrocyclic receptors in particular have emerged as promising scaffolds in this domain of supramolecular chemistry.15–17 Already recognised for their applications in drug-delivery systems, supramolecular polymers and molecular machines and devices, their role in anion recognition stands out prominently,18 where their ability to bind selectively to specific anions through size and shape complementarity has led to their application in various fields, including environmental monitoring for the detection of environmentally relevant anions, such as nitrate, phosphate, and chloride, to monitor water quality and pollution levels.19–21 The field of biomedical diagnostics has also exploited anion binding macrocyclic scaffolds for the recognition of biologically important anions, such as chloride, bicarbonate, and phosphate, for disease diagnosis and monitoring of physiological conditions.22–24 Even industrial processes have begun to exploit these versatile molecules for monitoring levels of anions in chemical manufacturing and waste treatment, to ensure process efficiency and compliance with regulations.25–27 Considering their demonstrated and potential uses, it is unsurprising that research in this area has been so pronounced in recent years.
Macrocyclic receptors possess several key features to make them highly effective for anion recognition. Most importantly, macrocyclic receptors have a preorganized structure that minimizes the entropic cost of binding. This preorganisation aligns the binding sites in a favourable conformation for interaction with the anion, enhancing binding affinity and selectivity.28 Additionally, macrocycles contain a degree of rigidity within the framework that reduces the flexibility of the binding sites, maintaining an optimal geometry for anion interaction. This rigidity helps maintain high binding constants and reduces non-specific interactions.29 Size matching is another key feature in the synthesis of macrocycles whereby the cavity size of the macrocycle can be tailored to match the size of the target anion. Effective size matching coupled with the three-dimensional shape of the macrocyclic cavity can be designed to complement the shape of the target anion, ensuring that the anion fits snugly within the binding cavity, maximizing interaction strength and selectivity as well as enhancing the specificity of the interaction.30 Furthermore, multivalency is another key feature in macrocyclic receptors. The ability for macrocycles to occupy multiple binding sites within their cavity allows for cooperative binding through exhibiting various binding interactions with anions, including electrostatic interactions, hydrogen bond formation, anion–π interactions, and halogen bonding to name but a few.31–34 Such interactions can be exploited in tandem with unique structural features together with chemical modifiability and tailored functionalisation with a vast array of functional groups to achieve exquisite selectivity.35–37 Understanding the key features mentioned above and the nuanced interplay of binding interactions allows the design and optimisation of macrocyclic receptors for precise anion recognition, making macrocyclic receptors a valuable tool for the supramolecular chemist.
While there are recent reviews published on macrocyclic receptors for selective fluoride recognition,38 their applications in precision medicine through sensing biomarkers,22 and selective binding of modified porphyrins towards different substrates.39 No recent reviews have focused on exploring the variety of macrocyclic compounds reported and their recognition of a diversity of anionic guests.
In this review, we examine a range of representative examples of macrocyclic receptors capable of anion recognition. We provide a summary of their synthesis, binding behaviour and explore design considerations across several receptor subtypes from the flexibility of cyclic peptides to more rigid container types of scaffolds such as calix[n]arenes. The objective is not to provide an exhaustive list of literature examples, but rather to establish the strengths and weaknesses of different cyclic scaffolds that take advantage of different binding interactions and the unique opportunities that they can present from the perspective of both anion binding affinity as well as selectivity. Where possible, we have attempted to compare the advantages to be gained by cyclic vs. acyclic receptors and compare with various other supramolecular receptors. Finally, we propose some potential future directions in the field of macrocyclic receptors and where the unique opportunities exist in the field.
Hydrogen bonding macrocycles
Hydrogen bonding macrocycles are a class of supramolecular compounds that are designed to selectively bind and recognise specific guest molecules through hydrogen bonding interactions. These macrocycles typically consist of a cyclic array of hydrogen bond donor and acceptor groups that are arranged in a specific geometry to create a binding pocket for the guest molecule.40,41 Hydrogen bonds are often conceptualised as a dipole–dipole interaction, whereby a hydrogen atom that is bound to an electronegative atom (referred to as the donor), is attracted to the dipole of an adjacent electronegative atom (referred to as the acceptor). The strength of a hydrogen bond can vary significantly, but generally, the stronger the bond can be attributed to the greater the electronegativity of the atoms on either side of the hydrogen.42 Typically, hydrogen bond donors bearing N–H groups such as amides, sulfonamides, ureas, thioureas, squaramides, pyrroles, and indoles, are the most common H-bond donors found in anion-binding units. Ureas, thioureas, and squaramides are particularly valued as anion receptors due to their dual hydrogen bond donor capabilities, which are advantageous for strong anion complexation.43–46Amide – and urea – based macrocycles
Utilising H-bond donors for the synthesis of successful macrocyclic receptors can be seen in the work of Jolliffe and co-workers, whereby they reported a fluorescent macrocyclic receptor (1) with selective binding towards citrate via hydrogen bonding interactions in aqueous media (Fig. 1).47 The macrocyclic receptor comprises of a carbazole-based unit featuring three hydrogen bonding thiourea units with highly preorganization for citrate binding. 1H NMR titrations carried out in 1% H2O/DMSO-d6 with TBA citrate revealed diverse species formation in solution. Attempts to fit the data to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, or 2:1 binding models proved unsatisfactory, yet the absence of further chemical shift changes after adding 1.6 equivalents of citrate strongly indicated robust binding. Throughout the titration, the signals associated with the receptor displayed broadness, and the resonances linked to the thiourea and carbazole NH groups exhibited downfield shifts, indicative of hydrogen-bonding interactions with citrate. A similar shift was noted for the inward-facing aromatic hydrogens of the xylyl linkers, suggesting a conformational change during citrate binding. A fluorescent screen with 1 along a range of dicarboxylates was carried out. Fluorescence quenching, ranging from −20% to −48%, was noted upon introducing terephthalate and various monovalent anions like benzoate, acetate, bicarbonate, nitrate, and halides. Alkyl dicarboxylates of varying lengths from oxalate (C2) to adipate (C6) and dihydrogen phosphate elicited minimal changes in fluorescence response. However, a distinct and significant fluorescence enhancement of over 90% was observed with the addition of citrate. The fluorescence maximum wavelength remained unchanged at 385 nm, while a new shoulder emerged around ∼405 nm, implying the formation of a second, associated excited state complex. These studies provided insights into the selectivity of the probe for citrate over other biologically relevant dicarboxylate species and common anions.
:1, 1:2, or 2:1 binding models proved unsatisfactory, yet the absence of further chemical shift changes after adding 1.6 equivalents of citrate strongly indicated robust binding. Throughout the titration, the signals associated with the receptor displayed broadness, and the resonances linked to the thiourea and carbazole NH groups exhibited downfield shifts, indicative of hydrogen-bonding interactions with citrate. A similar shift was noted for the inward-facing aromatic hydrogens of the xylyl linkers, suggesting a conformational change during citrate binding. A fluorescent screen with 1 along a range of dicarboxylates was carried out. Fluorescence quenching, ranging from −20% to −48%, was noted upon introducing terephthalate and various monovalent anions like benzoate, acetate, bicarbonate, nitrate, and halides. Alkyl dicarboxylates of varying lengths from oxalate (C2) to adipate (C6) and dihydrogen phosphate elicited minimal changes in fluorescence response. However, a distinct and significant fluorescence enhancement of over 90% was observed with the addition of citrate. The fluorescence maximum wavelength remained unchanged at 385 nm, while a new shoulder emerged around ∼405 nm, implying the formation of a second, associated excited state complex. These studies provided insights into the selectivity of the probe for citrate over other biologically relevant dicarboxylate species and common anions.
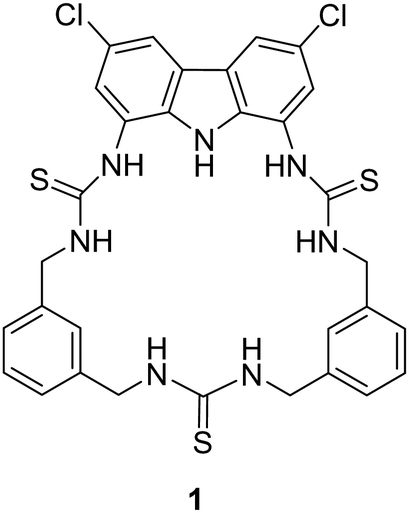 | ||
| Fig. 1 Chemical structure of macrocycle 1 reported by Jolliffe and co-workers. | ||
Another example of utilising H-bond donors into macrocyclic receptors comes from Bao and colleagues whereby they reported the anion binding properties of two macrocycles containing a carbazole and two sulfonamide units that exhibit a strong propensity towards F− binding (Fig. 2).48 Macrocycle 2 contains a 1,3-xylyl linker, while macrocycle 3 features a 2,6-lutidinyl linker. This difference in the linker structure leads to distinct effects on the anion affinity. 1H NMR titrations demonstrated that compound 2, with a 1,3-xylyl bridging unit, exhibits remarkable selectivity and binding strength for F− in CD3CN, outperforming competing anions such as AcO−, Cl−, Br−, NO3−, ClO4−, and H2PO4−. It showed a large binding constant (Ka) of 50878 ± 4721 M−1 in a 1:1 binding model. This could be rationalized by the cooperative binding of fluoride through the strong downfield shifts observed from the sulfonamide NHs, and carbazole NH in 2. Noteworthily, this fluoride complexation was dominated by hydrogen bonding interactions under low F− concentrations (<2.8 equivalents). UV-vis studies were carried out for the macrocycles to confirm the binding affinities with the macrocycle for fluoride. with increasing concentrations of TBA F−, a progressive increase in the absorption peak at 294 nm was observed for 2 Simultaneously, two pseudo-isosbestic points were identified at 305 and 355 nm, confirming the formation of a 1:1 complex between 2 and F−. The UV-vis study provided complementary evidence to the 1H NMR titration study, confirming the strong and selective binding of 2 towards fluoride in acetonitrile solution. A decrease in the affinity was observed for 3 with anions F−, H2PO4, and Br−, with respective decreases of approximately 3.9-fold, 2.0-fold, and more than 2.4-fold. Conversely 3 exhibited a higher affinity compared to 2 towards other anions such as PhCOO−, CH3COO−, Cl−, and HSO4−, with enhancements ranging from approximately 2.0-fold to over 4.2-fold. 2, featuring a 1,3-xylyl linker, benefitted from the presence of an additional aromatic CH group, which served as a potential anion-binding site. Replacing the 1,3-xylyl linker with a 2,6-lutidinyl spacer in compound 3 likely induced some degree of preorganization for its SO2NH protons in solution, enabled by intramolecular hydrogen-bonding interactions with the pyridine nitrogen atom. However, this replacement also led to the loss of an additional anion-binding site. Consequently, distinct changes in affinity towards various anions were observed between the two macrocycles.
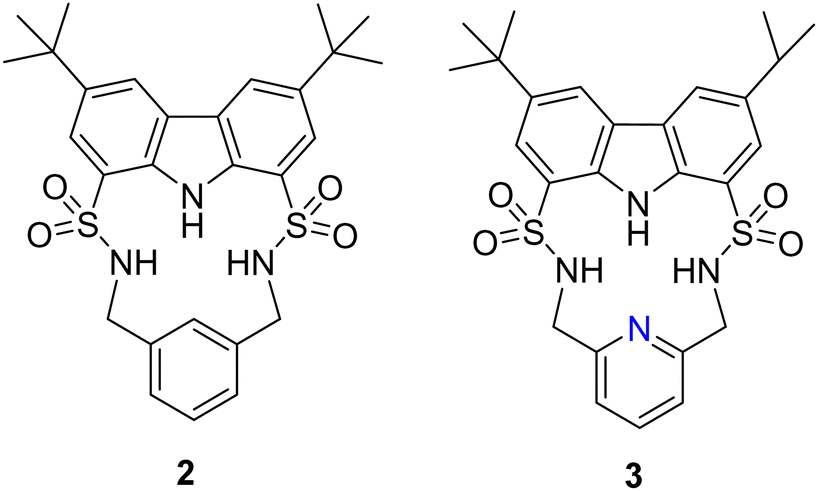 | ||
| Fig. 2 Chemical structures of macrocycles 2 and 3. Reported by Bao and colleagues. | ||
Fluorescent macrocycles can be seen again by work carried out by Kataev and colleagues where they set out to design a macrocyclic receptors (3, 4 and 5) consisting of four hydrogen bond donor amide groups for recognition and a naphthalamide fluorophore as a reporting unit sensitive to F− binding in aqueous solutions (Fig. 3).49 Receptor 3 shows the strongest binding of F− with 100-fold selectivity over other anions in aqueous solution with a remarkable binding constant of 105 M−1 as evidenced by 1H NMR titrations, highlighting its strong interaction with F−. Unlike 4 and 5, receptor 3 exhibited a unique turn-on fluorescence response upon binding with F−, making it a promising visual probe for F− detection.
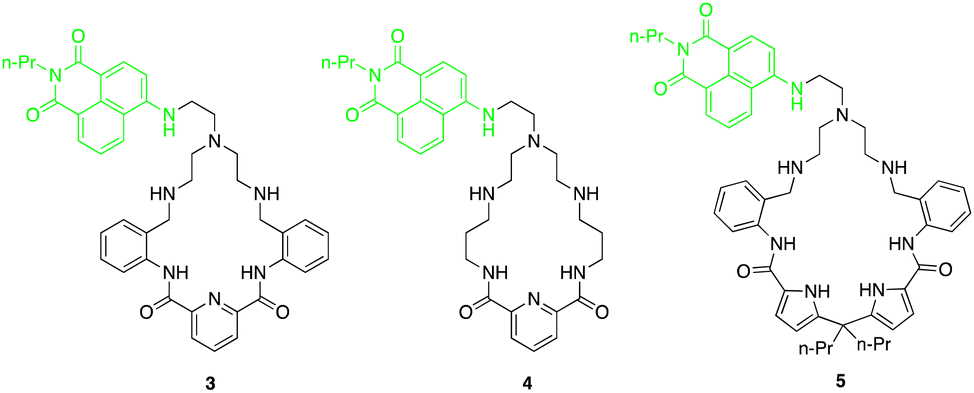 | ||
| Fig. 3 Chemical structures of macrocycles 3–5. Reported by EA. Kataev et al. | ||
One key feature of receptor 3 is its ability to accommodate multiple F− ions, forming stable complexes with 1:1, 1:2, and 1:3 stoichiometries as inferred from the fitting analysis of the fluorescence titration of 3 with F−. This multiple anion coordination leads to the protonation of the tertiary amine group within the receptor, hindering the photoinduced electron transfer process and resulting in a fluorescence enhancement. The size selectivity of receptor 3 allows it to selectively bind F− ions while preventing aggregation, further enhancing its fluorescence response specifically for F− detection. Moreover, the positively charged ammonium groups in receptor 3 play a crucial role in compensating the negative charge of fluoride ions, enhancing the electrostatic interactions and binding affinity. Overall, the binding studies of receptor 3 highlights its exceptional performance as a fluoride-selective receptor, with high affinity, unique fluorescence response, and selective binding mode.
An example of using hydrogen bond donors for dicarboxylate binding can be seen by work carried out by Jollife and co-workers, where they set out to synthesise semi flexible tetrathiourea macrocyclic receptors consisting of bis-carbazole units that are linked by 1,3-xylyl (6) or 1,4-xylyl (7) groups for the purpose of varying the macrocycles cavity shape and size as well as providing some flexibility within the macrocyclic framework (Fig. 4).50 This work gains insight into chelate cooperativity as well as exploring how these semi-flexible receptors interact with various dicarboxylate anions (Mal2−, Suc2−, Glu2−, Adi2−, Pim2−, Sub2−, Aze2−, Ter2−, ttM2− and aKG2−) in a competitive solvent mixture. Initially, fluorescence spectroscopy was carried out in H2O:DMSO (1:9 v/v) in order to explore the spectroscopic responses of 6 and 7 whereby a quenching of fluorescence was observed as a result of PET inhibition for all dicarboxylate anions. Further studies were carried out via Uv-vis titrations. It was found that for saturated dicarboxylates (Mal–Aze), 6 and 7 exhibited minor differences in binding affinity, whereby the highest affinity for both 6 and 7 was observed for Adi (n = 4) with strong 1:1 binding in a competitive aqueous solvent mixture (6: Ka = 7.5 × 104 M−1) (7: Ka = 8.7 × 104 M−1). Despite bearing varying cavity shape, both macrocycles displayed similar selectivity's across the linear dicarboxylate, as a result of the flexibility of the macrocycles. However, binding strength decreased significantly for anions larger than Adi, as they became too large to fit within the macrocyclic cavity. In contrast, the macrocycles differed significantly in their binding affinities for more rigid dicarboxylates. 7 bound to Ter with a high affinity (Ka = 8.7 × 104 M−1), while 6 was significantly weaker (Ka = 3 × 104 M−1), indicating that Ter is a better geometric match for 7. A similar trend was observed with ttM, where 7 (Ka = 5.8 × 104 M−1) bound more strongly than 6 (Ka = 2.8 × 104 M−1). To confirm the binding mechanism, 1H NMR titrations were carried out in DMSO-d6/H2O (0.5%). 6 and 7 showed slow exchange up to 1 equivalent of the Adi after which no further changes were observed. A notable sharpening of the methylene protons indicated a conformational change upon binding, resulting in all 8 methylene protons becoming chemically equivalent in the 1:1 complex. When 6 and 7 was tested with the smallest anion, Mal, intermediate exchange was observed, aligning with UV-vis titration results, with further peak splitting at higher guest concentrations, indicating potential formation of higher-order complexes (oligomers). Finally, a double mutant cycle analysis was carried out to study cooperativity effects of the macrocycles with the dicarboxylate guests. Negative cooperativity (log(Kintra) < 0) was observed for Mal, Sub, and Aze towards both macrocycles, as a result of the dicarboxylates being either too short (mal) to span the macrocycles cavity, or too large (Sub, Aze) to favour a 1:1 binding complex. In contrast, moderate positive cooperativity (log(Kintra) > 0) was observed for Suc, Glu, and Pim, and the strongest binding linear dicarboxylate, Adi, showed the highestlog(Kintra) value, reflecting strong positive cooperativity for both 6 and 7, indicating that Adi can fully fit comfortably within the macrocyclic cavity. Notable differences in cooperativity were observed between 6 and 7 when interacting with rigid anions. For the anion ttM, 7 demonstrated stronger positive cooperativity than that for 6. The same discrepancies were seen with Ter. The strong cooperativity of Ter can be attributed to π–π interactions between the central aromatic rings of the macrocycle and the aromatic guest. This work underscores the intricacies anion recognition and emphasizes the significance of geometric compatibility in improving binding strength and selectivity of macrocycles, offering valuable insights for the development of future macrocyclic receptors.
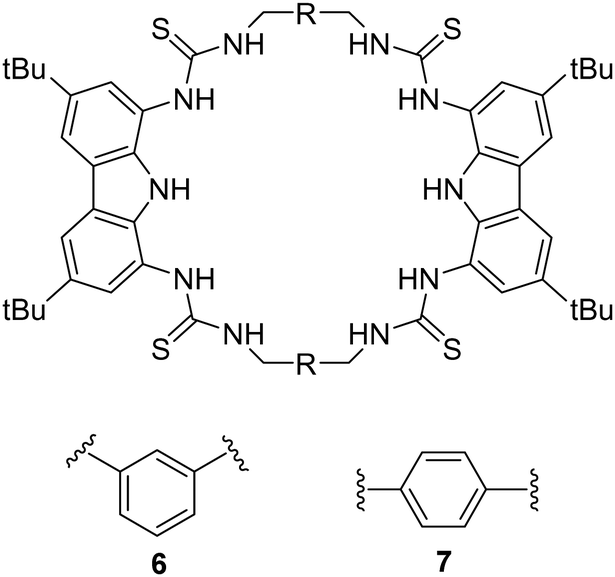 | ||
| Fig. 4 Chemical structures of macrocycles 6 and 7. Reported by Jolliffe and colleagues. | ||
Cyclic peptides
The interaction between peptides and proteins with anionic species constitutes a vital recognition event in biological systems, imparting diverse downstream effects encompassing structural stabilization and catalysis.51–53 Various binding modalities have been discerned in the interaction between peptides/proteins and anions. These interactions include those facilitated by bridging metal ions, electrostatic interactions involving amino acid side chains like lysine and arginine, and hydrogen bonding with side-chain donors such as serine, threonine, and asparagine, as well as the NH groups of the peptide backbone.54–57 This multifaceted spectrum of binding interactions underscores the intricate nature of anion recognition by peptides and proteins in biological contexts. Taking inspiration from natural binding abilities that amino acids possess, cyclic peptides have emerged as suitable macrocyclic receptors.58–60 Mimicking natural subunits, cyclic peptide receptors can provide a rigid and preorganized cavity with multiple directional hydrogen bond donors pointing into a central core. Selectivity can be tuned through backbone modification using different spacers to achieve preference to anions of a variety of shapes and sizes.57 The rest of this section will outline some recent examples of anion recognition achieved through cyclic peptides.Jolliffe and co-workers have made substantial contributions to synthesis of cyclic peptides and peptidomimetics as receptors for molecular recognition.57,61,62 One example is the monocyclic peptide [cyclo(Val-Thr)3] 8 which was shown to bind to Cl−, H2PO4−, SO42− and SeO42− in 1H NMR spectroscopic titration experiments (Fig. 5). SO42− and SeO42− gave the best fit to a 1:1 binding model and under these conditions (H2O:DMSO, 2:8), SO42− (Ka = 500 M−1) is more favoured than SeO42− (Ka = 90 M−1). The authors hypothesized that it could be a result of increased water coordination of the anions which prevented the association of a second peptide, as inferred from density functional theory (DFT) calculations. When sulfate was present, compound 8 assumed a conformation where all six NH protons pointed towards the center of the macrocycle, forming six hydrogen bonds with the sulfate ion. The intramolecular hydrogen-bonding interactions between Thr-OH groups also contributed to the stabilization of the structure, which was consistent with the observed downfield shifts of the signals attributable to these protons in the 1H NMR spectra.63
 | ||
| Fig. 5 (a) Structure of [cyclo(Val-Thr)3] 8 reported by Jolliffe and colleagues. (b) DFT(B3LYP/6-31G*) optimised structure of 1:1 complex of 8 and sulphate indicating hydrogen bonds to the sulphate. Reproduced from ref. 63 with permission from Taylor & Francis, copyright 2020. | ||
Kubik and co-workers have reported several examples of cyclic peptide receptors ranging from monocyclic receptors to sandwich-like ditopic receptors over two decades.64–67 In a more recent study Kubik et al. synthesized a series of palladium(II)-mediated assemblies consisting of one M2L2 (9) macrocycle and one M3L6 cage made up of a cyclic tetrapeptide ligands 8 (Fig. 6).68 During the titrations of 3 with dicarboxylates and disulfonates, upfield shifts of anion protons were observed, indicating that the anions were encapsuled within the macrocycle's cavity thus the protons were shielded by the aromatic rings. The binding constant of 9 to 1,3-benzenedisulfonate was found to belogKa = 4.8 while that to 2,6-naphthalenedisulfonate could not be accurately calculated due to the complexity of equilibrium of the 1:2 binding complex. Such complex equilibria can be a difficulty when trying to assess anion affinity with larger macrocyclic structures.
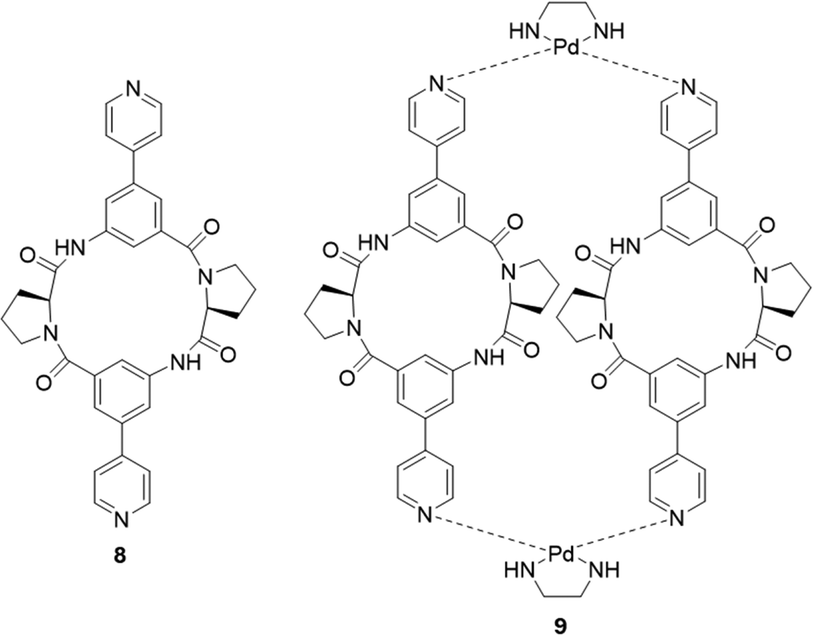 | ||
| Fig. 6 General structure of the ligand L (8), and macrocycle M2L2 (9), reported by Kubik et al. | ||
Tomišić and co-workers have recently reported the synthesis of cyclic pentaphenylalanine 10 as an efficient macrocyclic receptor for a variety of anions in acetonitrile and methanol (Fig. 7). Using a range of experimental techniques and computational methods such as fluorimetry, spectrophotometry, 1H NMR, microcalorimetric titrations and molecular dynamics (MD) simulations, stability constants and anion binding ability of the corresponding complexes were determined.69 The receptor 10 exhibited the strongest affinity towards Cl− with a stability constant of 6.06, followed by that for Br− (4.97), HSO4− (4.18), and H2PO4− (4.31). The stability constants of 10 towards the halogen anions were found to be correlated with anion size, whereby 10 showed a weaker affinity for larger anions. In MeCN, receptor 10 primarily formed 1:1 complexes, except with H2PO4−, where 1H NMR titrations suggested the likely formation of 1:2 species. Coordination of the anions was studied using molecular dynamics simulations, which revealed the binding of nearly all amide protons. This was further supported by circular dichroism titrations in MeCN, showing a significant change in the 220 nm region upon anion complexation. When MeOH was used as a solvent, a moderate to high loss of affinity and selectivity was observed. The main factor contributing to the decreased stability of 10 upon forming anion complexes was predicted to be the strong anion solvation properties of the solvent compared to MeCN. The difference in overall complex stability is contributed to the significantly stronger solvation of free anions in MeOH than in MeCN. The overall results showed that 10 can be a possible candidate as versatile anion binding receptor in protic and aprotic solvents for anion sensing and transport.
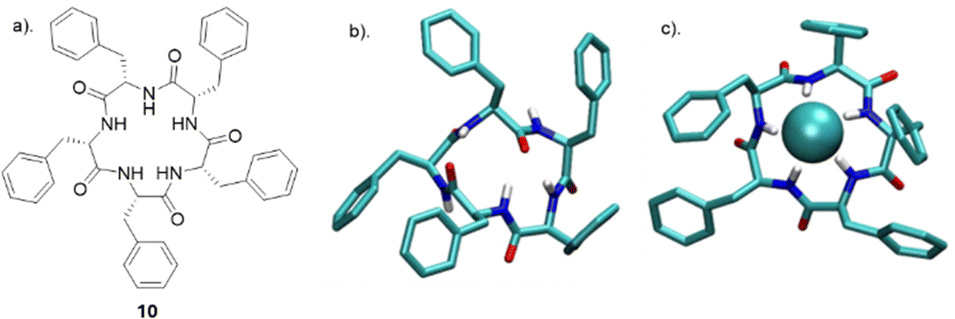 | ||
| Fig. 7 (a) Chemical structure of 10. (b) MD simulation of free 10 (c) MD simulation of anion complexes of 10 with chloride reported by Tomišić and co-workers. Reproduced from ref. 69 under a Creative Commons Attribution (CC BY) license from MDPI 2022. | ||
Squaramide – based macrocycles
Squaramides are a relatively recent addition to supramolecular chemistry and have attracted significant attention as a result of their diverse applications in biological and chemical sciences. Comprising a cyclobutene ring with two carbonyl groups adjacent to two NH groups, squaramides feature hydrogen bond acceptors and donors in close proximity. This unique arrangement makes them particularly useful in molecular recognition and transport.46 Many researchers have focused their attention towards squaramides, such as Catlagirone and co-workers who have worked on squaramide based receptors and ionophores,70–72 Gale and co-workers who have shown squaramides to be successful anion transporters,73–75 and many others in the field of supramolecular chemistry and beyond.An example of the utilisation of squaramides for anion recognition was reported by Jolliffe and co-workers where they synthesised a series of squaramide based macrocycles (11 and 12) comprising of alternating squaramide and benzylic groups tethered with the triethylene glycol monomethyl groups that displays high affinity and selectivity towards SO42− in aqueous media (Fig. 8).76 The binding studies of TEG-MSQs 11 and 12 involved quantitative NMR binding experiments for a range of anions in a 1:2 (v/v) H2O/DMSO-d6 solvent the binding affinities were calculated by fitting to a 1:1 binding model. 11 showed affinity for SO42− in the competitive solvent mixture, however, 12 exhibited a much greater affinity for SO42− that was too high to quantify in these very competitive conditions (Ka = < 104 M−1). The high binding affinity of 12 for SO42− was attributed to having an additional squaramide binding site compared to 11, together with additional hydrogen bonding interactions to the anion. Selectivity studies carried out for 12 exhibited notable selectivity for SO42− over other tetrahedral anions, demonstrating a binding affinity for sulfate that is at least three orders of magnitude higher than that observed for phosphate species presenting itself as an extremely potent and selective binding macrocycle for SO42− ions, outperforming the sulfate binding protein (SBP). In 2019 Jolliffe and colleagues showed MSQ 12 proved to be a highly selective ligand for SO42−, even in the presence of various interferents in aqueous mixtures and across a pH range from 3.2–14.1. This enables potential SO42− separation by the MSQs in a variety of applications including nuclear waste as well as plasma sulfate assays.77 In 2020 Jollife and colleagues showed that by functionalising 12 with aliphatic chains to produce macrocycles 13 and 14, they could efficiently extract SO42− from an aqueous Na2SO4 solution into organic solution, via an anion exchange mechanism with nitrate ions, as well as transport SO42− across a bulk chloroform layer via an anion exchange mechanism with NO3−, allowing the extraction of SO42− from Na2SO4 solutions.78
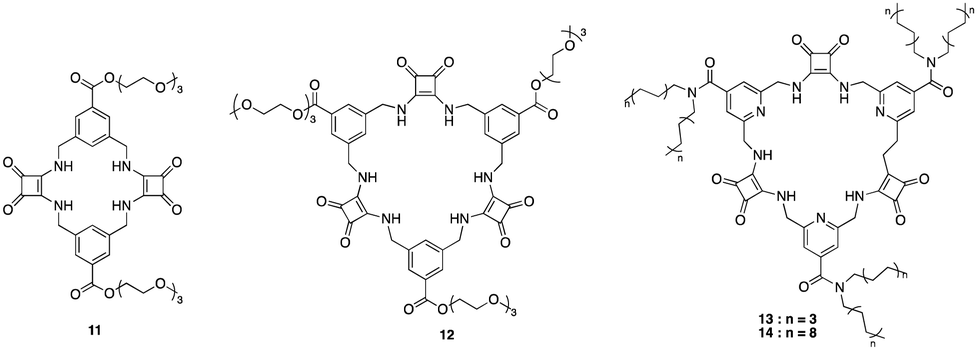 | ||
| Fig. 8 General structures of MSQ's 11–14 reported by Jollife and colleagues. | ||
The utilisation of squaramides as effective ion - pair receptors have been reported by a number of studies carried out by Jan Romanski and colleagues.79–81 An example of a macrocyclic squaramide ion-pair receptor and fluorescent sensor with selectivity towards sulphates was reported by Romanski and colleagues whereby using a combination of specific diamines and methyl squarates under high dilution conditions they synthesised a multi-macrocyclic ion pair receptors 15 and 16 based on a squaramide recognition moiety appended to benzo-18-crown-6. An analogous anion receptor 17, and a fluorescent sensor by incorporating a simple naphthalene unit into the structure of the receptor 18 (Fig. 9).821H NMR titrations were carried out in order to measure binding constants for a variety of anions such as Cl−, Br−, NO2−, NO3−, PhCOO−, CH3COO− and SO42− against each receptor except for 16 due to insolubility issues. 15 displayed an increase in affinity for Cl− (KTBACl = 61 M−1) anions in the presence of one equivalent of Na+ (KNaCl = 78 M−1) and K+ (KKCl = 95 M−1). Interestingly, further strength of Cl− binding was achieved by the addition of 3 equivalents of K+ producing a stability constant of (KKCl = 120 M−1). In order to verify the role of the cation binding domain in the structure of 15, 17 lacking the crown ether unit was tested towards the same range of anions which showed increased anion recognition compared to 15. This is a result of the absence of two electron-donating alkoxy substituents in the structure of anion receptor 17. Compared to 15, this decreases the electron density on the phenyl ring, facilitating a more rigid interaction with the anion. However, 17 could not recognize Cl− in an enhanced manner in the presence of K+ due to the lack of the cation binding domain in the structure (KKCl = 71 M−1vs. KTBACl = 76 M−1). Finally, 18 resulted in an optical ion pair sensor selective towards SO42−, where upon the addition of SO42−, a change in optical properties were observed including a including a bathochromic shift in the maximum absorption, an increase in fluorescence intensity, and the appearance of a new band at 420 nm. All receptors were found to interact with anions by utilizing the squaramide and amide functions of the receptors further supporting the efficiency of squaramides as hydrogen bonding motifs for anion recognition.
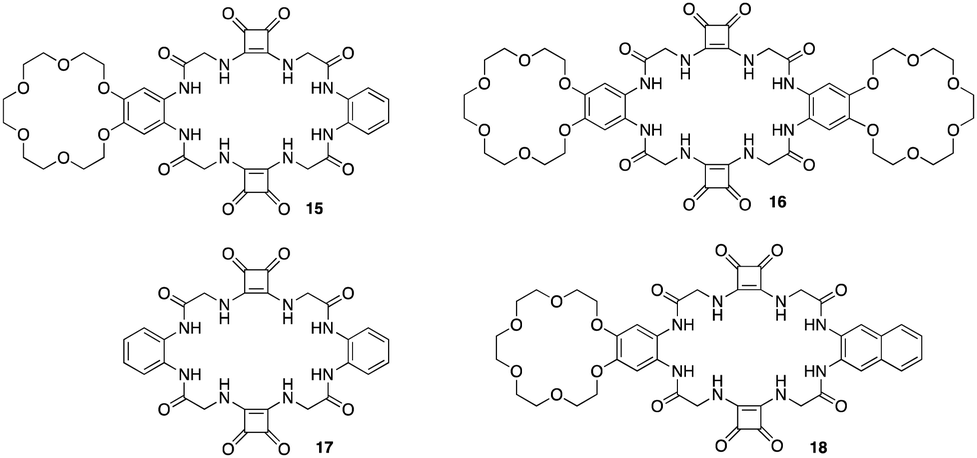 | ||
| Fig. 9 General structures of squaramide based ion – pair receptors 15–18 reported by Jan Romanski and colleagues. | ||
Calix[4]pyrrole – based macrocycles
Pyrrole- and calix[4]pyrrole-based anion receptors differ from the previously discussed macrocyclic receptors. They can only function as hydrogen bond donors as a result of their absence of hydrogen bond acceptor functional groups. However, calix[4]pyrrole-based receptors, adopt a non-planar tetrapyrrolic macrocyclic conformation, enabling them to capably bind to Lewis basic anions such as halides.83 Since Sessler et al. first introduced calix[4]pyrrole into the design of anion receptors in the mid-1990s, calix[4]pyrrole-based receptors have demonstrated high affinity and selectivity for various anions. This has been achieved by adding straps to one side of the calix[4]pyrrole or by incorporating probe units at the periphery of the macrocycle.83,84In 2023, Wezenberg and colleagues reported a dithienylethene-strapped calix[4]pyrrole whereby the dithienylethene (DTE) unit that serves as a photo switch. The DTE unit allows the molecule to isomerize between a ring-open (19) and a ring-closed (20) form upon exposure to 300/630 nm light (Fig. 10).85 This photo switching process brings about a subtle change in the size of the binding pocket, enabling the receptor to selectively bind different halide ions based on their size, thus functioning as an artificial anion receptor with tunable selectivity. 1H NMR titrations were carried out for the receptor in its open-ring isomer 19 in MeCN-d3. Addition of Cl−, Br−, and I− to led to substantial downfield shifts of the pyrrole-NH as a result of strong hydrogen bonding. Isothermal titration calorimetry (ITC) was carried out to quantify the binding affinities which showed a 1:1 binding model with strong affinity for Cl− (Ka = 2.6 × 105 M−1) and Br− (Ka = 8.8 × 103 M−1) however a much lower binding affinity was observed for I− (Ka = 23 M−1). Upon 300 nm irridation where the receptor now presented itself as its closed-ring isomer 20. 1H NMR titrations revealed similar downfield shifts of the pyrrole-NH. When the DTE unit is in the ring-closed form, the binding pocket is smaller, which reduces the binding affinity for larger halide ions, this was seen from the association constants where little change was observed for Cl− (Ka = 1.2 × 105 M−1). As for Br− a significant decrease was observed (Ka = 54 M−1) and the same for I− (Ka = < 1 M−1). This shows that the binding affinity is highly dependent on the photo switching process even though the process brings about a subtle change in the size of the binding pocket, it affects the binding affinity and selectivity of different halide ions.
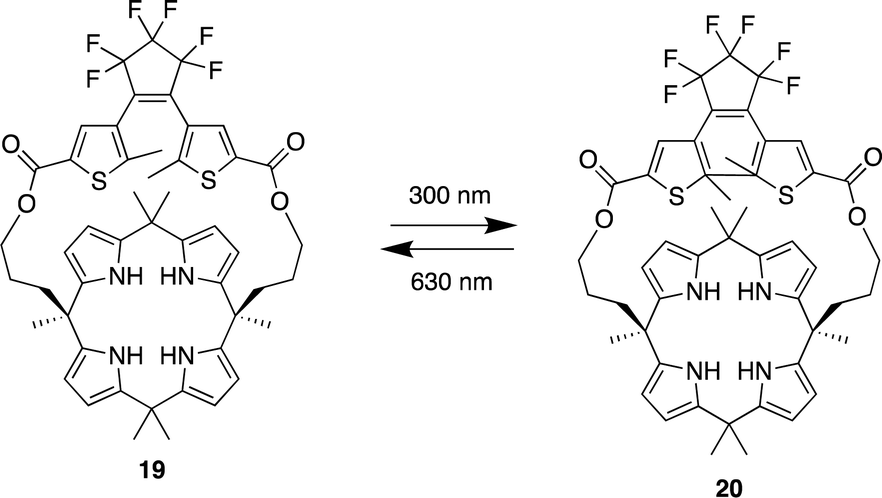 | ||
| Fig. 10 General structure of dithienylethene-strapped calix[4]pyrrole macrocycles in its ring open 19 and ring closed 20 form reported by Wezenberg and colleagues. | ||
Sessler and co-workers set out to synthesise the smallest bis-calix[4]pyrrole (21) comprised of small two-wall bis-calix[4]pyrrole formally linked by two single oxygen atom bridges which proved capable of trapping F− exclusively relative to other anions (Fig. 11).86 The small size of the cavity in bis-calix[4]pyrrole (21) is significant for its selectivity towards F− because it restricts the conformation of the receptor, preventing it from accessing the cone conformation that is most favourable for anion binding. This restriction results in a three-dimensional cavity that is slightly larger than the volume of F−, capable of supporting hydrogen-bonding interactions. As a result, the receptor is able to reject anions larger than F−, resulting in exclusive selectivity for F−. The results of this study were confirmed through various analytical techniques, including X-ray diffraction analysis, DFT calculations, and molecular dynamics simulations. In the solid state, bis-calix[4]pyrrole 21 was found to be capable of binding F−in a 1:2 stoichiometry, with the association constants of K11 = (5.2 ± 0.4) × 103 and K12 = (4.4 ± 1.4) × 102 M−1 where each F− atom trapped itself inside the cavity of the calix[4]pyrrole ring, as evidenced by a single crystal structural analysis. The expected electrostatic repulsion between the two F− atoms was said to be stabilised by multiple cooperative hydrogen bonding interactions. In solution, 21 was found to have favourable binding towards F− but not interact appreciably with other anions, e.g. Cl−, Br−, SCN−, NO3−, HSO4−, H2PO4−, and SO42− as inferred from 1H NMR spectroscopic analyses showing no noticeable changes in the proton signals. DFT calculations and molecular dynamics studies were conducted in the gas phase in order to support the hypothesis that the exclusive selectivity for F− is due to the limited three-dimensional space within receptor 21.
 | ||
| Fig. 11 General structure of bis-calix[4]pyrrole (21) reported by Sessler et al. Image reproduced from ref. 86 with permission from the American Chemical Society, copyright 2020. | ||
Calixarene – based macrocycles
Calix[n]arenes (where n = 4, 5, 6, 8) represent an intriguing class of macrocycles with a chalice-shaped structure, predominantly composed of phenol units. These compounds have been extensively investigated frameworks in supramolecular chemistry.87 The allure arises from their versatile and adjustable characteristics found in both the upper and lower rims, their prearranged nonpolar interior spaces, and prearranged sites for binding ions, all contributing to their well-defined conformations. Calixarenes are known for their ability to selectively bind to various guest molecules within their central cavity through multiple non-covalent interactions, such as hydrogen bonding, van der Waals forces, pi–pi stacking interactions, hydrophobic interactions, electrostatic effects, and others.88 This host–guest interaction is influenced by factors such as the size, shape, and polarity of the guest molecule, as well as the substituents on the calixarene structure. This property makes calixarenes useful in various applications, including molecular recognition, supramolecular chemistry, and drug delivery.87,89,90 Researchers have explored the modification of calixarenes to tailor their properties for specific applications. For example, functionalization of the phenolic hydroxyl groups or the methylene bridges allows for the fine-tuning of their chemical and physical characteristics.22Pulla Rao et al. reported the assembly of a fluorescent based calixarene through the conjugation of 7-chloro 4-nitro benzofurazan (NBD) to calix[4]arene to yield receptor 22 (Fig. 12) in order to study its potential as a sensitive and selective sensor for the detection of F− using a range of techniques such as 1H NMR spectroscopy, UV-vis absorption and fluorescence emission spectroscopy and in biological cells by confocal and fluorescence microscopy.91 To assess the anion-sensing capabilities of 22, various ions, were introduced in titrations conducted in THF. The UV-vis absorption and fluorescence emission spectra were recorded for each titration. Notably, during the titration of 22 with F−, a discernible redshift of the 460 nm absorption band by 6–7 nm was observed, indicating the formation of a new band around 400 nm attributed to the complexation of F− with 22. Similar anion based titrations were carried out by measuring the fluorescence spectra of 22. Although minimal alterations were observed in the emission spectrum of 22 for all studied anions, F− exhibited a substantial ∼95% quenching in fluorescence intensity demonstrating its utility as a selective F− sensor. In order to support the binding of F− to 221H NMR titrations were carried out. Upon addition of F− the peaks corresponding to the NH groups shifted downfield by ∼0.3 ppm, and the peaks corresponding to the protons from the benzofuran ring were upfield-shifted by 0.18 and 0.15 ppm, suggesting the F− binds to the core formed by the two arms of the lower rim region. The association constant for F− was calculated to be 4.2 × 103 and a Job's plot analysis revealed a 1:1 (receptor:anion) complex. Finally confocal and fluorescence imaging of HeLa cells was carried out whereby the cells were treated with 22 showed high intra cellular fluorescence suggesting that 22 enters the cells. Various concentrations of F− (20, 40, 50, 80, and 100 μM), was treated to the cells which showed that the fluorescence intensity decreases with increase in the added F− concentration. This study demonstrated a useful, selective, and sensitive fluorescent sensor 22 for the detection of F− in solution and in the biological cells (Fig. 12).
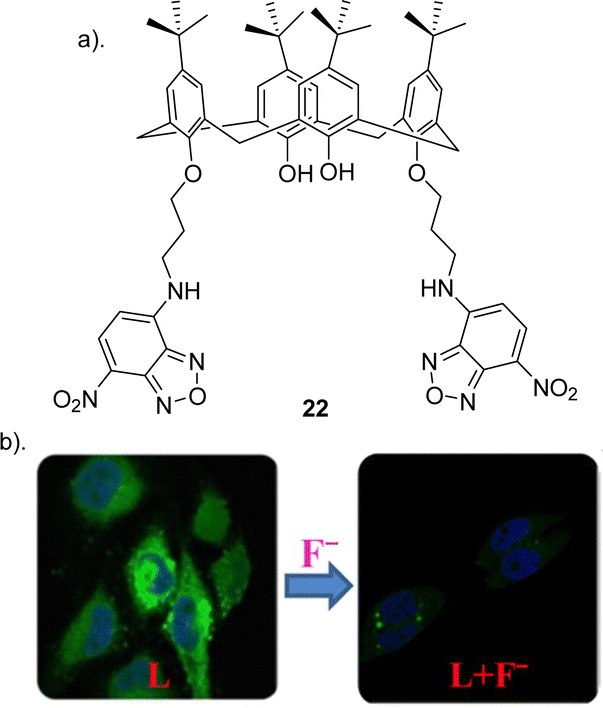 | ||
| Fig. 12 (a) Schematic representation of 22 (b) Change in fluorescence of 22 upon the addition of F− in HeLa cell, reported by Pulla Rao and co-workers. Images reproduced from ref. 91 with permission from the American Chemical Society, copyright 2018. | ||
Another recent study carried out by Wang and co-workers reported the assembly of a novel calix[4]arene based chemosensor 23 (Fig. 13), comprising of a coumarin as a reporter and thiourea groups as the anion-binding site on the calix[4]arene framework for the recognition of fluoride via hydrogen bonding.92 The sensor's selective and sensitive response to fluoride ions was assessed using fluorescence spectroscopy, UV–vis spectroscopy and 1H NMR titrations. Chemosensor 23 exhibited remarkable sensitivity and selectivity towards F− ions, as evidenced by fluorescent and colorimetric studies. Specifically, F− was the only tested anion that significantly diminished fluorescence and induced a visible color change (transparent to orange), discernible to the naked eye (Fig. 13). The UV–vis absorption peak of 23 at 366 nm diminished, and a new red-shifted absorption peak emerged at 475 nm only upon the addition of fluoride ions. Various other anions including I−, Br−, CN−, Cl−, HSO4−, H2PO4−, and CH3COO− did not prompt any notable changes in the absorption spectra, underscoring the sensor's selectivity for fluoride ions. The robust emission band of chemosensor 23 at 481 nm was quenched solely by the addition of F−, while the various other anions tested showed no significant alterations in the emission band. In-depth fluorescence and 1H NMR titrations conducted revealed that 23 forms a 1:1 complex with F−, featuring an association constant of 2.1 × 104 M−1 and a limit of detection (LOD) of 3.5 × 10−6 M. At lower concentrations, fluoride engages with the cavity at the upper rim of calix[4]arene 23 through multiple hydrogen bonds. Conversely, at higher fluoride concentrations, the thiourea units undergo deprotonation induced by F−, thereby enhancing the electron transfer effect during the photoinduced electron transfer (PET) process between the coumarin fluorophore and the thiourea moiety.
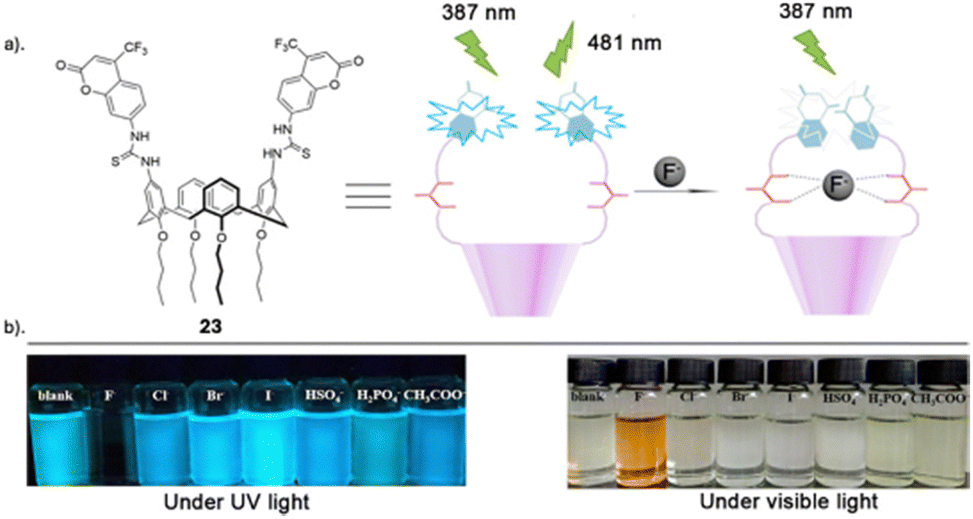 | ||
| Fig. 13 (a) Schematic representation of 23 (b) visible fluorescence emission responses of 23 in the presence of various indicated anions under UV light (365 nm) and visible light, reported by Wang and Colleagues. Images reproduced from ref. 92 with permission from Elsevier, copyright 2018. | ||
Nemati and colleagues reported a novel chemosensor 24 for detecting fluoride, employing a calix[4]arene framework with four sulfonamide-fluorenone subunits attached to the upper rim, exhibiting colorimetric and fluorescent properties (Fig. 14).93 Anion recognition was carried out using a range of techniques such as such as UV–vis, fluorescence and 1H NMR on a range of anions e.g. Cl−, Br−, I−, CN−, Br−, ClO4−, NO3−, F−, H2PO4−, HSO4− and AcO−. Interestingly, 24 exhibited a transition in colour, shifting from yellow to red in the presence of F− anion (Fig. 14b). Conversely, 24 did not display significant colour changes upon the addition of other anions. Through UV–vis absorption spectroscopy, receptor 24 exhibited two prominent absorption bands at 290 nm and a weaker absorption at 520 nm in the absence of anions. Among the tested anions, selective binding was observed with F− ions. The introduction of F− caused a red shift of 25 nm, shifting from 290 nm to 315 nm, along with a reduction in peak intensity in this region. An increase in peak intensity was observed in the 340 to 450 nm range, indicating the formation of a host–guest complex between receptor 24 and F−. The observed red shift in 24 upon the introduction of F− may be attributed to an elevation in the electron density on the nitrogen atom of the sulfonamide group, facilitated by the interaction between F− and the sulfonamide proton (–NH). Furthermore, Fluorescence spectroscopy showed a quenching in fluorescence at the observed emission at 510 nm upon the addition of F− due to complexation. No significant changed were observed for other anions further revealing the selectivity of 24 towards fluoride which was further elucidated through competitive anion titrations where fluorescence emission intensity of [24 + F−] is unaltered in the presence of most of other anions. 1H NMR titrations carried out showed that upon the addition of two equivalents of fluoride to 24, a strong upfield shift was observed for the sulfonamide (N–H) as well as an upfield shift of the Ar–H signal of calix[4]arene. This could be attributed to the formation of a hydrogen bond between N–H groups and F−, resulting in the 24-F− complex. Job's plot shows that 24 forms a 1:2 complex with F−, a result further supported by UV-vis and NMR titrations. The anion F− binds to the chemosensor 24 through a combination of hydrogen bonding and the chemosensor's pre-organized structure, as confirmed by 1H NMR titration. Additionally, the binding constant and detection limit (LOD) of the 24-F− complex were determined to be 1.8 × 105 M−1 and 3.4 × 10−8 M, respectively.
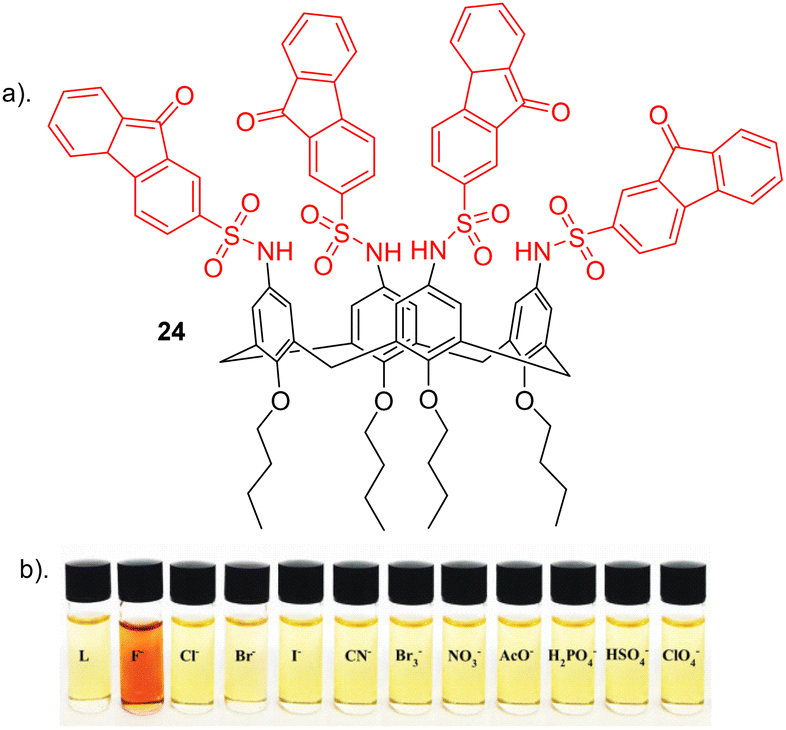 | ||
| Fig. 14 (a) Chemical structure of 24. (b) Visual changes of receptor 24 upon addition of various indicated anions, reported by Nemati and colleagues. Image reproduced from ref. 93 with permission from Elsevier, copyright 2021. | ||
An interesting example of how a simple structural modification within the macrocycle structure can lead to potent anion receptors was demonstrated by Lhoták and colleagues whereby they synthesised a series of intramolecularly bridged calix[4]arene receptors and for comparison, similar receptors without the bridging unit were synthesized to assess the impact of the bridge on anion binding efficiency (Fig. 15).94 Single crystal X-ray analysis of the receptors provided detailed structural insights, confirming the rigidity and preorganization of the bridged receptors, highlighting the importance of the rigidified cavity in enhancing anion binding. This was further evidenced via1H NMR titration experiments to study the complexation of various anions (H2PO4−, AcO−, BzO−, Cl−, Br−, NO3−) with the synthesized receptors. The experiments were performed in DMSO-d6, whereby Upon addition of anions to the receptor solutions, significant downfield shifts in the NH signals were observed, indicating complex formation via hydrogen bonding interactions. Job plot analyses revealed 1:1 stoichiometry for most anions except for 27b which showed a 2:1 stoichiometry. The bridged receptors demonstrated substantially higher binding constants compared to non-bridged analogues. For example, the binding constant for bridged receptor 25a with acetate (K25a(AcO−) = 8270 M−1) was nearly 20 times higher than that for the non-bridged receptor 27a (K27a(AcO−) = 470 M−1). Similar trends were observed for benzoate, K25a(BzO−) = 1600 M−1, while K27a (BzO−) = 330 M−1. and dihydrogen phosphate anions, with bridged receptors showing exceptional binding efficiency for specific anions, particularly carboxylates and dihydrogen phosphate, even in competitive solvents like DMSO-d6, indicating the importance of a rigidified, preorganized structure of the bridged receptors and their role in their superior binding performance.
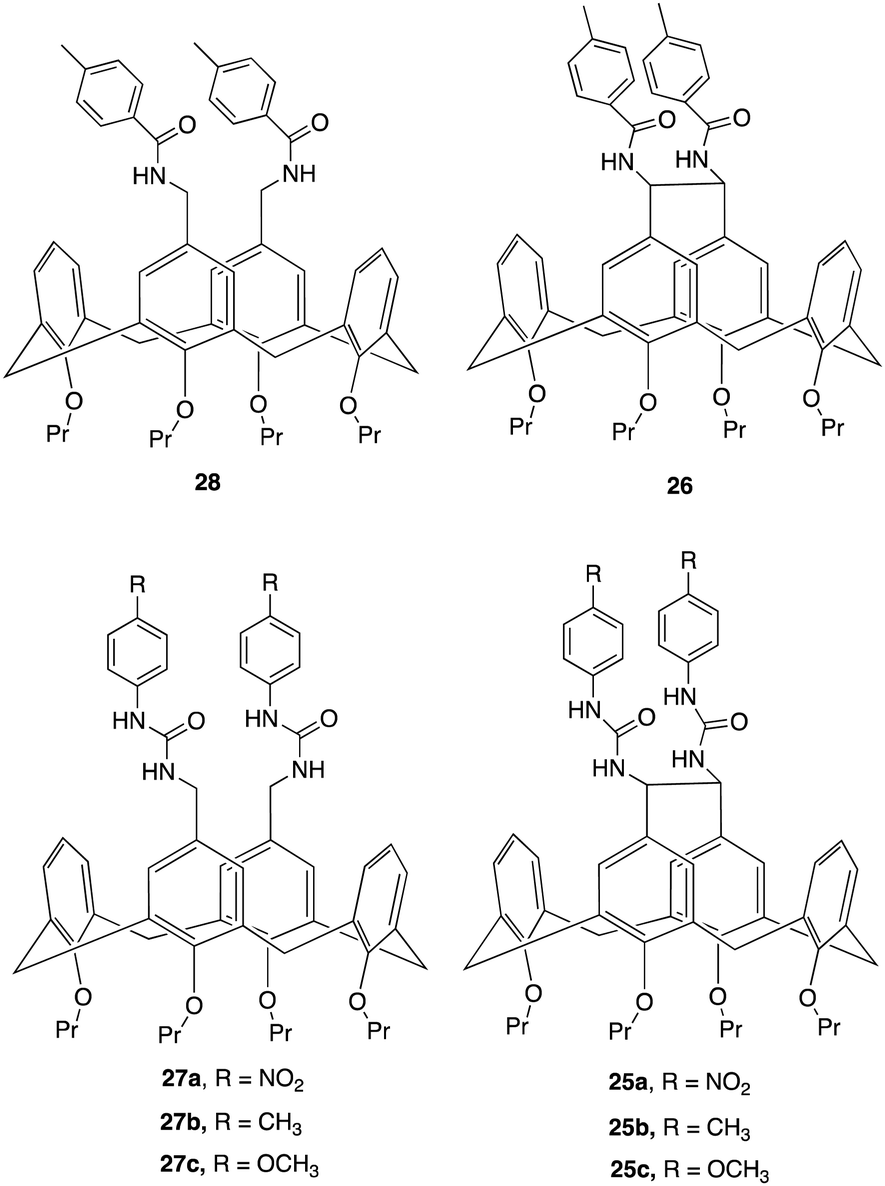 | ||
| Fig. 15 Chemical structures of bridged and non-bridged calix[4]arene receptors reported by Lhoták and colleagues. | ||
Pillararene – based macrocycles
Pillar[n]arenes, are a class of macrocyclic compounds, that have emerged in the realm of supramolecular chemistry. First introduced by Tomoki Ogoshi et al. in 2008,95 these pillar-shaped macrocycles are characterized by their unique columnar structures, connecting hydroquinone or dialkoxybenzene units (ranging from 5 to 10) through methylene bridges in the para position. They share structural similarities with cucurbiturils and calixarenes, both crucial players in host–guest chemistry. The inherent flexibility and tunability of pillarene structures through modifying the hydroxyl groups at all positions or selectively on one or two positions empower pillararenes with a distinct capability to form host–guest complexes with a diverse range of guest molecules, showcasing their potential applications in diverse fields such as drug delivery, molecular recognition, and sensing.26,96 Among the family members, pillar[5]arene stands out as the most conformationally stable. The close arrangement of electron-rich hydroquinones allows the cavity of pillararenes to form robust association complexes with electron-poor species.96 Here we will discuss recent examples in literature of pillararene based macrocycles as receptors for anion recognition.In 2022 Shi, Liu and colleagues reported the synthesis of an electron deficient fluorinated leaning pillar[6]arene 29 consisting of two tetrafluoro-benzene units with a preorganised structure and suitable cavity size capable of forming a 1:1 binding complex with strong selectivity and binding toward iodine ion driven by anion–π interactions (Fig. 16).97 Initially, UV-vis experiments were conducted to confirm the binding interaction between 29 and I−. The UV-vis spectra of 29 in CHCl3 showed a single maximum absorption band at 295 nm. Upon the addition of I−, the emergence of a new absorption band appeared at 246 nm, indicating the formation of a new species in the solution. Importantly, the addition of 5.00 equivalents of F−, Cl−, and Br− did not disrupt the absorption band of 29, demonstrating its specific recognition of iodide anions in chloroform. Furthermore, controlled experiments under competing conditions confirmed the unique selectivity of 29 for I−, unaffected by competing anions such as F−, Cl−, and Br−, thus affirming its selective recognition of iodide anions. 1H NMR and 19F NMR experiments were conducted to further explore the host–guest complexation between 29 and I−. Upon the addition of 5 equivalents of TBA I− to 29, the peaks corresponding to the proton signals on 29 experienced up field shifts, similarly the 19F NMR spectrum of 29 exhibited an upfield shift after the introduction of 5.00 equivalents of TBA I− to 29. These observations are indicative of the heightened electron density of the host molecule 29 resulting from the encapsulation of electron-rich iodide anions within its cavity. The binding data was fitted to a 1:1 binding stoichiometry with an association constant for iodide to be 3.18 × 103 M−1. This data was further verified via DFT calculations as well as high-resolution mass spectrometry (HRMS) whereby the HRMS analysis showed three peaks at m/z for [29 + I]−. This study introduced a novel and selective pillar[n]arene receptor for I−, offering insights into the design of selective pillar[n]arene based anion receptors.
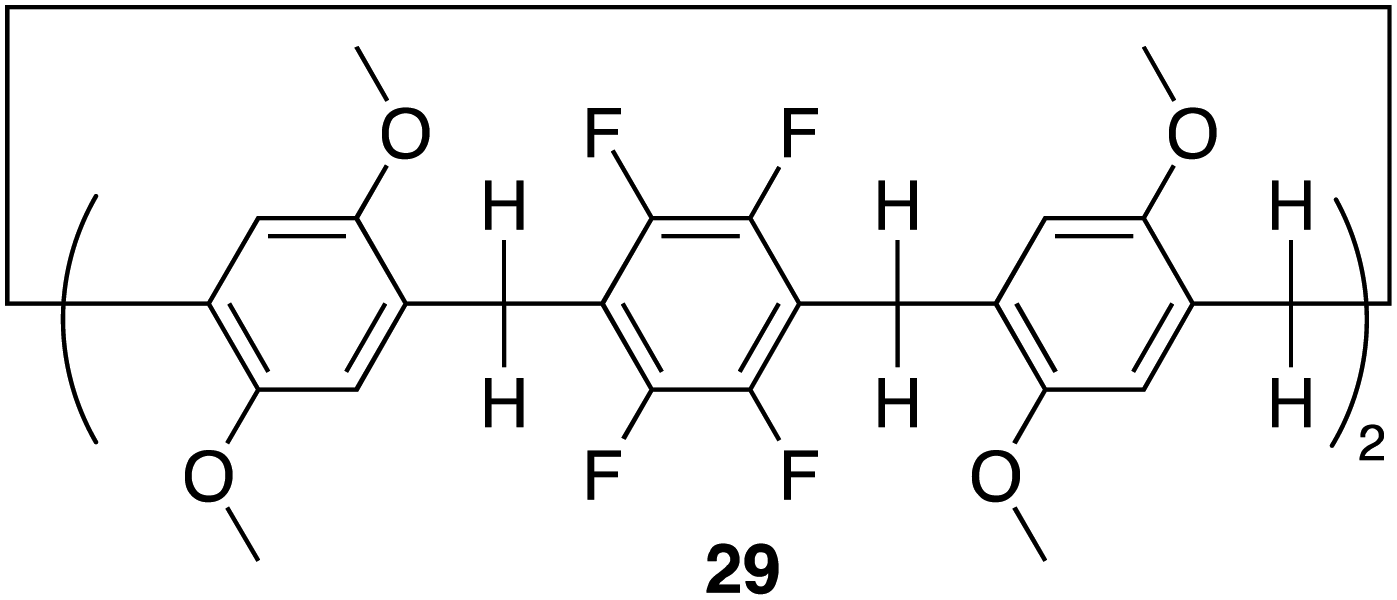 | ||
| Fig. 16 General structure of fluorinated leaning pillar[6]arene 29 reported by Shi, Liu and colleagues. | ||
Another recent study that focuses on functionalising pillararenes for effective anion receptors was reported by Al-Azemi and co-workers whereby they set out to design receptors functionalized with urea, utilizing constitutional isomers of tetra-bromo-functionalized pillar[5]arene (30, 31, 32) as highly selective F− receptors forming a 1:2 host–guest complex (Fig. 17).98 The impact of the pillar[5]arene receptor structure on the binding ability and selectivity towards halide anions was investigated using various experimental techniques, including 1H NMR titration, diffusion-order spectroscopy (DOSY), and isothermal titration calorimetry (ITC). The ITC studies revealed that the binding interactions taking place between the receptors and anions were affected by both the number of urea substituents and their corresponding positions along the pillar[5]arene framework. 32 exhibited a higher affinity towards F− with an association constant of determined to be 4.65 ± 0.16 × 104 M−1, which is one order of magnitude higher than that of 31 (8.21 ± 0.27 × 103 M−1). ITC studies also showed that the receptors resulted in the formation of a 1:2 host-to-guest complex which was further confirmed via Job's plot. Further understanding of host–guest interactions was gained through 1H NMR titration experiments in DMSO-d6 using F− and the pillar[5]arene-based anion receptors (30, 31, 32) Host–guest complexation was confirmed by observable upfield shift of resonances for the urea protons (N–H), as indicated for all the macrocycles. Additionally, the 1H NMR spectra displayed a single set of peaks during the titration experiments, indicating fast-exchange complexation on the 1H NMR time scale. 32 demonstrated the highest binding affinity towards F−. No interactions was observed for Br−, and a significantly lower Ka value towards Cl− (2.27 ± 0.3 × 102 M−1) compared to F− (3.61 ± 0.02 × 104 M−1). The DOSY experiments revealed that the complexation between 32 and F− anions resulted in a distinct set of signals with a specific diffusion coefficient (D), indicating the formation of the host–guest complex. Additionally, DOSY spectra of 32 at different concentrations in DMSO-d6 showed changes in the diffusion coefficient, suggesting the presence of supramolecular assemblies at higher concentrations. The observed change in the diffusion coefficient was attributed to the formation of larger aggregates with larger hydrodynamic radii.
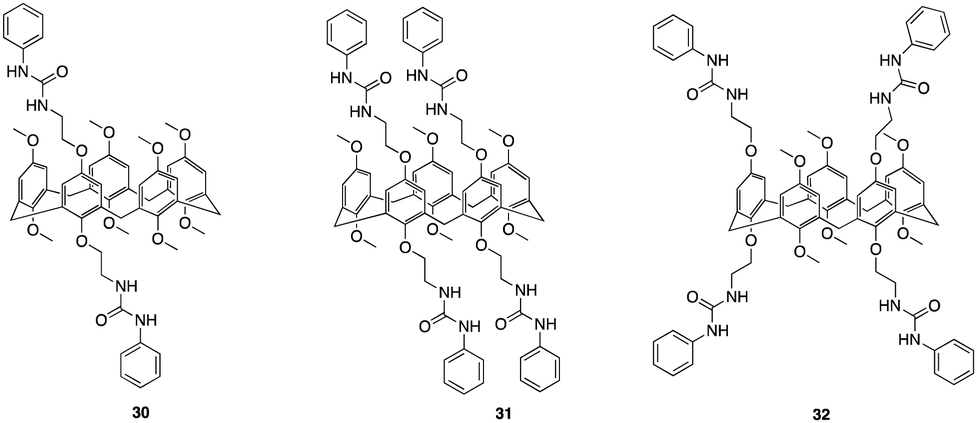 | ||
| Fig. 17 General structures of urea functionalised pillar[5]arenes 30–32 reported by Al-Azemi and co-workers. | ||
C–H Hydrogen bonding macrocycles
C–H hydrogen bonding macrocycles, a class of supramolecular compounds, have captured considerable attention in recent years owing to their unique properties and potential applications in various fields particularly in anion recognition. These macrocycles feature C–H groups that serve as hydrogen bond donors, enabling them to selectively bind to a variety of guest molecules.99 Unlike traditional hydrogen bonding, where hydrogen bridges between a donor and acceptor, C–H hydrogen bonding involves direct interactions between a carbon–hydrogen bond and a suitable acceptor, offering diverse possibilities for designing and synthesizing macrocyclic structures with intriguing properties in the field of supramolecular chemistry.100 Flood and colleagues pioneered the work for C–H Hydrogen bonding macrocycles in 2008, where they introduced a [34]triazolophane macrocycle 33 comprising of four triazoles and four phenylene CH groups orientated directly into the cavity with rigidity and preorganisation (Fig. 18).101 Through multiple triazole C–H-anion and phenyl C–H-anion hydrogen bonding interactions the triazolophane 33 displayed a pronounced affinity for chloride followed by Br− ≫ F− ≫ I−, with an association constant of Ka = (130000 ± 30000) L mol−1 for Cl−.
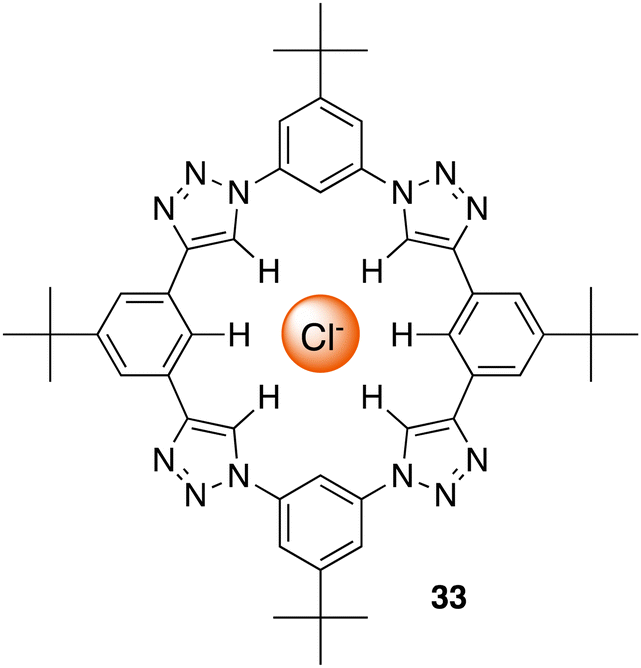 | ||
| Fig. 18 Schematic representation of a [34]triazolophane macrocycle (33) binding to chloride via C–H hydrogen bonding, reported by Flood and colleagues. | ||
Through garnering the information acquired, Flood and colleagues used their understanding of receptor design and CH based hydrogen bonding to explore untapped and uncharted opportunities. From this they set out to create a whole new class of cyano stilbene based macrocyclic receptors, called cyanostars.102 In this section we will discuss examples of C–H Hydrogen bonding macrocycles reported in recent years.
In 2021 G. Bachas, Flood and colleagues set out to design a clamshell-shaped anion-binding ionophore, 35, in order to analyse its performance in ion-selective electrodes (ISE). This ionophore consisted of two cyanostar (34) macrocycles with prearranged cavities connected by a 12-carbon chain (Fig. 19) and plays a crucial role in anion recognition and detection due to its capacity to prearrange cavities and selectively identify anions through CH–hydrogen bonding.103 This distinctive structure and composition facilitate the selective transfer of anions from the solution to the membrane phase, resulting in enhanced selectivity and sensitivity in anion detection. Potentiometric performance of the clamshell ionophore in ISEs was evaluated by measuring its response to different anions (e.g. Cl−, Br−, NO3−, salicylate, I−, SCN−, and ClO4−) and assessing its selectivity and sensitivity. Membrane compositions containing the clamshell ionophore were prepared, and the resulting electrodes were subjected to potentiometric measurements to determine their response characteristics. The electrode based on 35 showed the greatest potentiometric response and excellent selectivity towards I− over various anions, with a selectivity coefficient of 10−2.13 In contrast to the Hofmesiter series, salicylate showed the lowest response. ESI-MS was utilised to evaluate the selectivity of the clamshell ionophore (35) and its parent CNstar (34) towards various anions. Through conducting ESI-MS experiments, observations and analysis the formation of complexes between the ionophores and specific anions were measured. The results revealed the formation of 1:1 complexes between 34 and Cl−, NO3−, Br−, ClO4−, I−, and salicylate, each with intensities less than 4%. Higher stoichiometry complexes, such as [3(34) + 2ClO4]2−, [3(34) + I + ClO4 ]2−, and [3(34) + 2I]2−, were also observed, with relative abundances of 21.7%, 9.7%, and 1.2%, respectively. The most intense peak observed, corresponded to the 2:1 complex [2(34) + ClO4]−. Interestingly, another peak representing the 4:2 heterocomplex [4(34) + I + ClO4]2−, followed by the 2:1 complex [2(34) + I]− were observed with both relative intensities below 10%. Additionally, a peak attributed to a 3:1 complex [3(34) + ClO4]− was also found. Upon increasing the clamshell concentration to 5 μM, ESI-MS analysis revealed predominant peaks at m/z 2147.6 corresponding to 1:1 and 2:2 multimers [(35) + I]− and [2(35) + 2I]2−. However increasing the ionophore concentration to 10 μM caused a shift in the prominent peak of the ESI-MS spectra from iodide to perchlorate with relative contributions of the 1:1 and 2:2 (35) and ClO4− complexes. The findings from ionophore selectivity by ESI-MS provided significant insights into the complexation behaviour of the ionophores, enabling a thorough evaluation of their selectivity towards particular anions and their potential impact on ion detection and quantification in ion-selective electrodes.
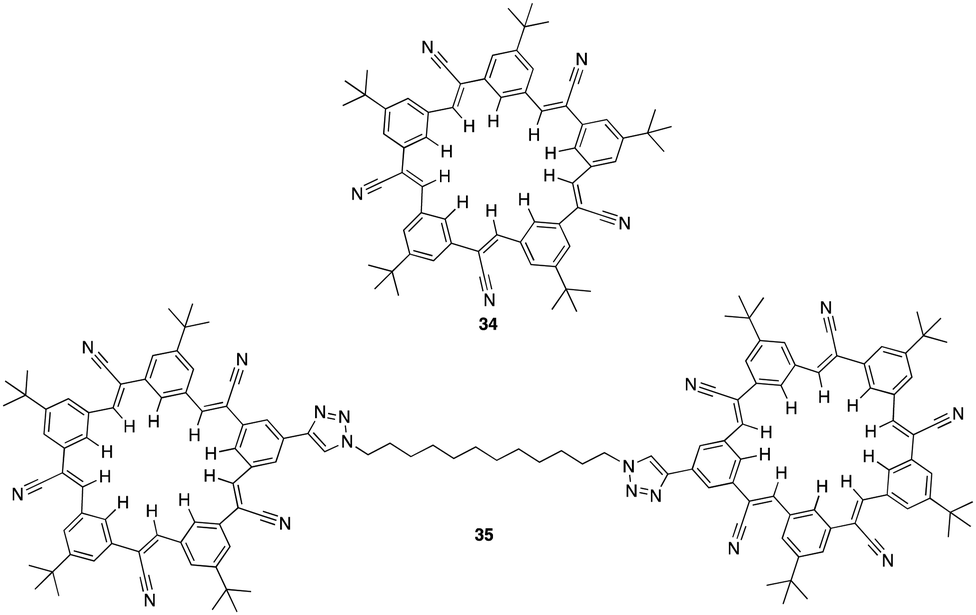 | ||
| Fig. 19 General structure of cyanostar (34) and the clamshell ionophore (35) reported by G. Bachas, Flood and colleagues. | ||
In 2022, Flood and colleagues reported the binding of a series of organotrifluoroborate anions (R-BF3−) e.g. small (methyl, MeBF3−), medium (vinyl, VinBF3−), and large (thiophene, ThBF3− and phenyl, PhBF3−) to cyanostar macrocycles (34) (Fig. 20).104 X-ray crystallography revealed 2:1 complex between 34 and the ThBF3− and PhBF3− anions. As a result of steric interactions taking place between the macrocycle and the ortho hydrogen atoms of the anion, both substituents are prevented from ideally fitting into the binding cavity defined by the center of the two π-stacked macrocycles. Additionally, they are kept further from the bottom macrocycle, which disengages the CH hydrogen bond donors of the second macrocycle, likely reducing stability. From this a combination of techniques, including 1H NMR and UV-vis titrations were carried out to quantify the impact of sterics on the binding affinity of the anions. 1H NMR titrations revealed that the shifts in the peaks of the macrocycle reach a plateau at approximately 0.5 equivalents of the guest. This saturation is accompanied by subtle downfield shifts in the interior protons, which are indicative of the CH⋯anion hydrogen bonding of cyanostars. MeBF3− and VinBF3−, characterized by small steric profiles, exhibited positive cooperativity, favouring the formation of the 2:1 species with association constants of logK2 => 6.5 and > 6.4 respectively. Conversely, the addition of further guest molecules to the larger PhBF3− and ThBF3− anions results in downfield shifts consistent with the conversion to a 1:1 complex and less cooperativity due to greater steric demand. Consequently, the PhBF3− anion forms mixtures that are not suitable for use as designer anions which according to the crystal data, aligns with the hypothesis, that 2:1 anion binding weakens upon disengaging the bottom macrocycle from binding.
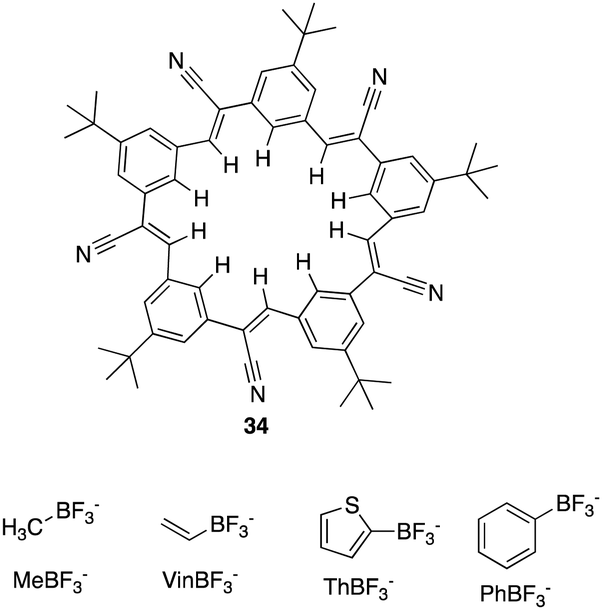 | ||
| Fig. 20 General structure of cyanostar macrocycle (34) and of organotrifluoroborate anions reported by Flood et al. | ||
Bambus[n]urils
Bambus[6]urils are a class of macrocyclic receptors that belong to the broader category of cucurbit[n]urils. They are composed of glycoluril units linked by 2n methylene bridges that constitute the molecular framework. This class of macrocyclic receptors first introduced by Sindelar and co-workers,105 are neutral anion receptors that serve as molecular containers forming stable complexes with a diverse array of guests such as drug molecules, amino acids, peptides, saccharides, dyes, hydrocarbons, perfluorinated hydrocarbons, and even high molecular weight guests like proteins. Notably, they can facilitate cucurbituril-catalysed reactions.106 Bambus[6]urils demonstrate remarkable host–guest binding properties not only in aqueous environments but also in nonpolar solvents.105,107,108 For instance, these macrocycles selectively bind various-sized anions in chloroform,109 showcasing potential applications in real-time NMR analysis of anion mixtures. Moreover, they find utility as carriers in tailor-made liquid membranes for highly selective electromembrane extractions of inorganic anions110 and function as transmembrane Cl−/HCO3− antiporters.111Sindelar and co-workers previously demonstrated that bambusurils are strong macrocyclic receptors with high affinity to anions of up to 3 × 109 L mol−1 in CHCl3. However, difficulties arose during binding studies in more polar solvents, primarily due to the solubility of the macrocycle.107,112,113 In a more recent study carried out by the Sindelar group, they have tweaked the structure of bambusurils by varying the substituetnts on the nitrogen atoms of the macrocyclic portals to synthesis a water soluble macrocyclic receptor (36) with outstanding affinity towards inorganic anions in pure water (Fig. 21).114 Using 1H NMR spectroscopy, the supramolecular properties of 36 were studied in D2O at 30 °C. It was noted that anions such as F−, Cl−, CN−, IO4−, and ReO4− interacted with the macrocycle in a fast exchange regime on the NMR timescale which led to lower affinities, best fitting a 1:1 binding model with association constants all above 2.2 × 106 L mol−1. On the contrary, for strongly bound anions (Br−, NO3−, PF6−, BF4−, I−, ClO4−) slow exchange regime and quantitative complex formation at submilimolar concentrations were recorded with the highest binding and selectivity towards ClO4− having an association constant of 5.5 × 107 L mol−1. The high binding affinities are believed to result from the anion being isolated from water molecules by its encapsulation within the receptor's hydrophobic pocket. The altering glycoluril units from the macrocyclic receptor form a deep electropositive cavity where an anion is included and isolated from water molecules. It's also worth mentioning that in aqueous environments the stability of the complex is reinforced through numerous weak C–H⋯A hydrogen bonding interactions. These interactions involve the methine hydrogen atoms of 36 and the anion. The host cavity's internal volume exhibits adaptability to the anion's size by tilting the glycoluril units. Consequently, the receptor displays versatility in accommodating anions with diverse shapes and radii.
 | ||
| Fig. 21 (A) Chemical structure of 36. (B) Structure of the 36·Cl complex (C) side view of the 36·Cl complex and (D) top view of the 36·Cl complex. Images reproduced from ref. 114 with permission from John Wiley and Sons, copyright 2015. | ||
In a more recent study, Sindelar and co-workers focused on synthesising a series of fluorinated bambusurils to act as highly effective and selective transmembrane Cl−/HCO3− antiporters.111 By introducing electron-withdrawing substituents connected via aryl groups to H-bond donors, the acidity of the H-bond donors generally increases leading to stronger binding properties. It also leads to an increase in lipophilicity which is advantageous for transmembrane transport. In this study, Sindelar set out to investigate the effects on anion binding and transport by designing fluorinated derivatives of bambusuril 37 through various numbers and positions of fluorine atoms or fluourine-containing groups on the benzyl substituents (Fig. 22).
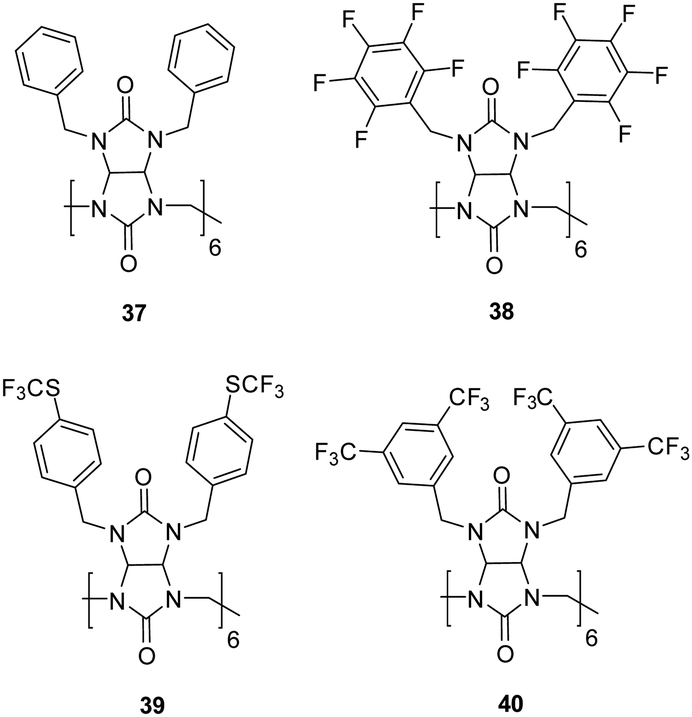 | ||
| Fig. 22 Structures of bambus[6]urils 37–40 reported by Sindelar and co-workers. | ||
1H NMR spectroscopy was carried out in MeCN to study the binding properties and formation of host–guest complexes between bambusurils 38, 39, and 40 and Cl−, NO3−, and HCO3−. The 1H NMR spectra reveal two distinct signal sets for the macrocycle, suggesting that host–guest exchange occurs slowly within the NMR timescale. When 1 equiv. of Cl− or NO3− was added to 40, a significant downfield shift was observed for one set of signals which remained unchanged upon addition of more than 1 equiv indicating the exclusive formation of 1:1 complexes with these anions. Association constants were calculated which showed strongest affinity for NO3− (Ka = 5 × 1011) followed by Cl− (Ka = 6 × 1010), and HCO3− (Ka = 2 × 109). The evaluation of bambusurils as anion carriers involved the use of liposomes in the fluorescence-based lucigenin assay, demonstrated exceptional transport properties. Among these, receptor 40, with the highest affinities, stands out to the authors knowledge, as the most potent Cl−/HCO3− carrier reported to date. It was noted that the Cl−/NO3− antiport exhibited orders of magnitude slower kinetics compared to the Cl−/HCO3− antiport. This disparity can be attributed to the strong affinities towards NO3− as well as its binding selectivity over Cl− which disfavours decomplexation. This work demonstrates the potential of bambusurlis for applications in anion transport and drug delivery through successful incorporation of bambusuril molecules in the bilayers of liposomes followed by membrane fusion.
In 2023, Sinedlar and colleagues showed the effectiveness of bambus[n]urils as anion receptors and the need to develop new well-defined cavitands, which are still extremely under-investigated in the field of host–guest chemistry. This was justified in this study where they reported a series of Bambusurils with varying electron withdrawing groups attached to their benzyl substituents 41 bears –SCF3 moieties, 42 (SOCF3) and 43 (SO2CF3) that exhibited picomolar anion affinity via the inductive effect of distant substituents (Fig. 23).115 Anion binding affinities and associations constants were measured for the bambusuril derivatives in CD3CN via1H and 19F NMR spectroscopy. NMR competition experiments were carried out for 41 with ReO4−vs. ClO4−, then ClO4vs. Cl−, Cl−vs. Br− and I−. 19F NMR spectroscopy revealed a gradual shift of the fluorine signal. The binding data was fitted to a 1:1 host–guest complex for all anions with extremely strong association constants calculated for Cl− (Ka = 4.2 ± 2.1 × 1010 M−1) Br− (Ka = 6.0 ± 3. 1 × 1011 M−1) and I− (Ka = 1.6 ± 0.7 × 1012 M−1). The same approach was carried out in the case for 42 and 43, whereby it was observed that the anion binding strength increases with the increasingly potent electron-withdrawing substituents from 41–43. The enhanced potency is attributed to the distinctive bambusuril structure, setting it apart from other macrocyclic receptors. The well-defined cavity within its structure ensures selectivity, maintaining its efficacy despite alterations to its backbone. Furthermore, even electron-withdrawing groups on the benzyl substituents of macrocycle effectively reduce the electron density within the macrocycle cavity through the inductive effect, exerting a substantial impact on the binding potency of the macrocycle. In the course of NMR titrations, a gradual enhancement in the affinity of bambusurils towards all halides following the sequence 41 < 42 < 43 was noted, aligning with the heightened electron-withdrawing capability of the macrocycle substituent. The strategic modification of receptor structures yielded remarkable binding affinities for 43 with Cl− (logKa = 11.5), Br− (logKa = 12.7), and I− (logKa = 13.1), with the [43 – I−] complex being the most stable 1:1 supramolecular complex for iodide ever reported.
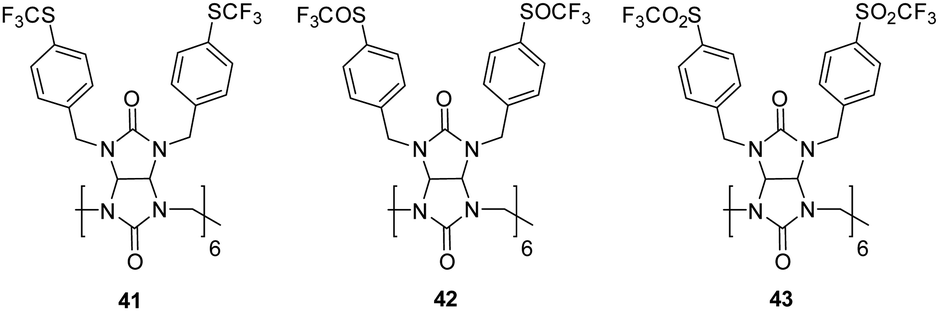 | ||
| Fig. 23 Structures of Bambus[6]urils 41–43 reported by Sindelar and co-workers. | ||
Biotin[6]urils/glycourils
In 2014, Pittelkow and colleagues, through the condensation of d-biotin and formaldehyde in acidic aqueous conditions, synthesized a specialized Q[n] derivative, 6 + 6 macrocycle called biotin[6]uril, (Scheme 1).116 They observed that halide anions act as templates for the formation of biotin[6]uril, and this macrocycle exhibits selective binding to halide anions (I−, Br−, and Cl−) with a preference for iodide in water. Over the subsequent years, Pittelkow and co-workers conducted comprehensive studies on the binding of biotin[6]uril with various anionic guests in water using multiple techniques. They proposed that the binding of anions is influenced by a delicate balance between the size of the anion to fit into the cavity and the hardness/softness of the anion.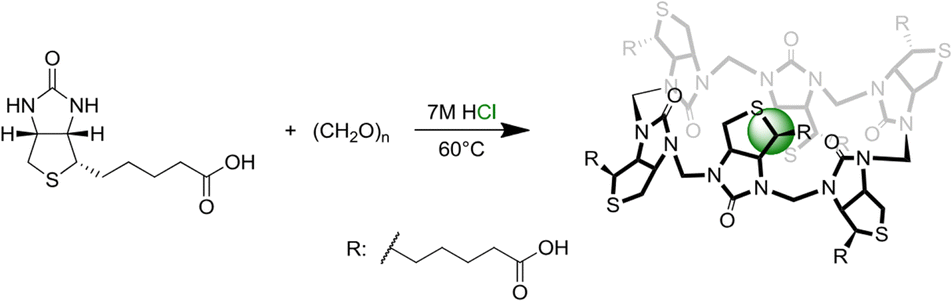 | ||
| Scheme 1 Synthetic scheme of biotin[6]uril reported by Pittelkow and colleagues. Image reproduced from ref. 116 with permission from The Royal Society of Chemistry. | ||
Pittelkow et al., reported the assembly of biotin[6]uril whereby six biotin units of the biotin[6]uril have twelve protons from the convex side of each biotin unit pointing into the cavity of the biotin[6]uril. This makes the cavity distinctly hydrophobic and an binding site for halide anions in water. To this end, the authors set out to show how biotin[6]uril (44) is capable of binding a series of monovalent anions at pH 7.5 in phosphate buffer using 1H NMR spectroscopy (Fig. 24).117 Binding of single charged anions was observed through a change in the chemical shifts of the protons on the convex side of each biotin unit pointing into the cavity with all anions showing a 1:1 binding stoichiometry which was further confirmed via electrospray ionisation mass spectrometry. Binding affinities were measured via1H NMR titrations and isothermal titration calorimetry (ITC) which revealed that 44 binds halide anions in the order of Cl− (log(Ka) = 1.8) > Br− (log(Ka) = 3.0) > I− (log(Ka) = 3.7). The binding of halides showed a trend where the larger halide fits better in the cavity with larger binding affinities for the anions with larger radii as a consequence of the increased softness of the larger ions. 44 prefers larger, softer anions over smaller, harder ones up to a certain size, as the anion must fit within the macrocycles cavity. X-ray crystallography revealed structures of 44 containing two water molecules inside the cavity which must be displaced upon anion binding. The release of the water molecules could contribute favourably to the enthalpically driven binding (non-classical hydrophobic effect) evident from the ITC data. X-ray structures also revealed that the internal volume for the macrocycle is not constant indicating that the binding cavity has some flexibility resulting from the six biotin units tilting slightly to give a narrower binding pocket.
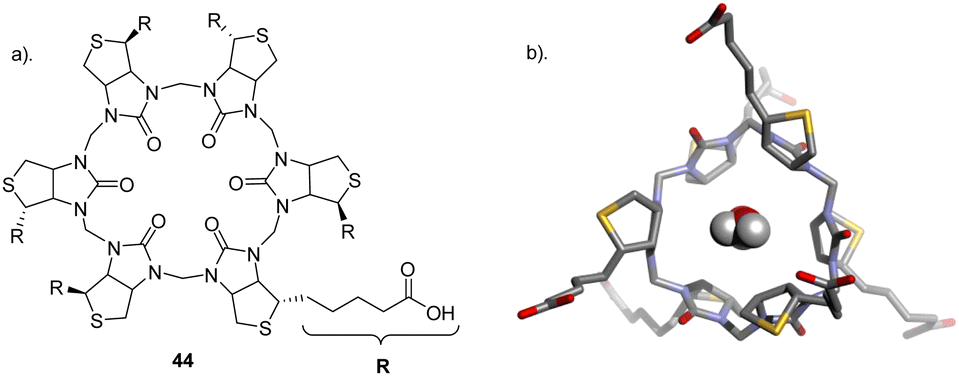 | ||
| Fig. 24 (a) Schematic representation of 44 (b) Crystal structure of 44 H2O containing complexes seen from the top reported by Pittelkow and colleagues. Image reproduced from ref. 117 with permission from The Royal Society of Chemistry. | ||
Another study carried out by Pittelkow and colleagues set out to create hydrophobic analogues of 44 through esterification giving rise to a series of biotin[6]uril hexesters (45, 46, and 47) which are a new class of macrocyclic anionophores that act as chloride-selective transmembrane anion carriers in an organic medium (Fig. 25).118 These macrocycles function exclusively through C–H⋯anion interactions. The exploitation of soft hydrogen bond donors promotes the transport of anions such as Cl− and NO3−, which tend to be less hydrophilic over hard, anions like HCO3− and SO42− which are strongly hydrated. 1H NMR spectroscopy was carried out to study the binding of the hexaesters to Cl−, NO3−, HCO3−, and SO42− in CD3CN. Isothermal titration calorimetry (ITC) indicated 1:1 binding stoichiometries as well as favourable enthalpically and entropically driven binding interactions with Cl− having the highest binding affinity (logKa = 4.5) by roughly two orders of magnitude over −, NO3− (logKa = 2.3), HCO3− (logKa = 2.1) and no detection for SO42−. Lucigenin assay was carried out to study the anion transport by biotin[6]uril hexaesters. The hexaesters demonstrated activity, with the effectiveness influenced by the length of the side chain (47 > 46 > 45). The lipophilicity of the carriers appeared to enhance anionophore effectiveness with leaching tests confirming exclusive membrane location for all carriers, indicating that increased lipophilicity enhances the intrinsic rate of anion transport. The most active transporter (47) promoted Cl− influx with t1/2 = 180 s at transporter:lipid ratio = 1:2500. Bicarbonate transport was tested and showed that the membrane is impermeable to HCO3− implying that 47 shows very high selectivity for Cl−vs. HCO−. This selectivity results from the “soft” nature of CH as a hydrogen bond donor, which should favour the polarizable, more hydrophobic anions (e.g., Cl−, NO3−) over harder, more basic anions (e.g. HCO3−).
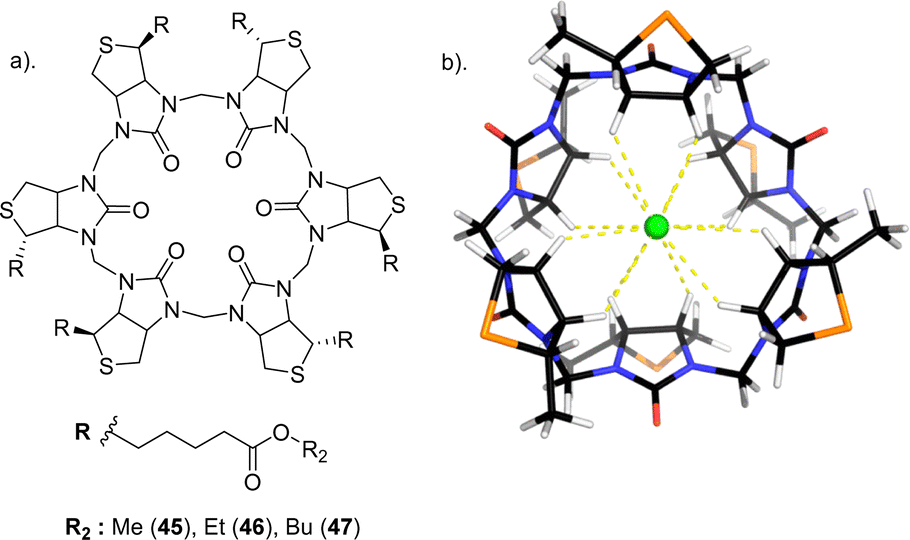 | ||
| Fig. 25 (a) Schematic representation of biotin[6]uril hexaesters 45–47 (b) Macrocyclic core of biotin[6]urils, with side chains replaced by Me groups, modelled binding Cl−, reported by Pittelkow and colleagues. Image reproduced from ref. 118 with permission from The American Chemical Society, Copyright 2105. | ||
Hemicucurbiturils (HcB)
Hemicucurbiturils, also known as HC[n]s, are a class of macrocyclic compounds that have garnered significant attention in supramolecular chemistry due to their unique structural features and versatile host–guest interactions.119–121 These molecules are derived from the parent cucurbituril structure (first synthesized in 1905 by Robert Behrend, by condensing glycoluril with formaldehyde4) by removing one or more glycoluril units, resulting in a partial or incomplete cucurbituril framework. Hemicucurbiturils exhibit intriguing properties that make them promising candidates for various applications in host–guest chemistry, molecular recognition, and material science. Their structural diversity, tuneable binding cavities, and ability to form inclusion complexes with a wide range of guest molecules have positioned them as valuable building blocks for the design of functional supramolecular systems. The host–guest chemistry of hemicucurbiturils generally binds to anions in the cavity via CH⋯anion interactions.122 The methine moieties pointing into the cavity of the hemicucurbit[n]urils are responsible for this behaviour. It's also worth mentioning that depending on the solvent used, a chaotropic effect and/or that the non-classical hydrophobic effect can be involved in achieving high binding affinities.123 Unlike the widely studied cucurbit[n]uril macrocycles, renowned for their proficiency in hosting cations, hemicucbit[n]urils exhibit a distinct capability for hosting anions. In 2017, S. Kaabel et al. reported a comprehensive study on the anion binding properties of a hemicucbit[b]uril derivative, a chiral (all-R)-cyclohexanohemicucurbit[8]uril (48) in protic solvents with exceptional selectivity (Fig. 26).124 The binding selectivity of 48 towards different anions tested in methanol is governed by their size, shape, and charge distribution. The association constants for anion binding vary over five orders of magnitude, with the strongest binding observed for the largest tested octahedral anion SbF6− (Ka = 2.5 × 105 M−1) and weakest binding observed for the smallest octahedral anion BF4− (Ka = 4.8 × 10 M−1). Halides on the other hand showed very weak complexation presumably due to a mismatch in size with the cavity. These results demonstrate the strong size dependency of their anion binding properties. Additionally, anions with symmetric charge distribution that matches the receptor cavity exhibit significantly stronger binding compared to those with asymmetric charge distribution. This is because symmetric charge distribution enables the formation of a greater number of hydrogen bonds simultaneously, resulting in stronger binding. Single crystal X-ray diffraction of the 1:1 host–guest complexes clearly confirms the stoichiometry of the anion inclusion complexes in the solid state. The study also reveals that the host–guest interactions are responsible for the fixed orientation of the encapsulated anions, as evidenced by the Hirshfeld surface analysis. Additionally, the crystallographic study shows that the cavity of 48 can be considered as an octahedrally shaped void, and the anions encapsulate within it in a manner depending on their shape, volume, and complementarity of the interactions. The results presented in this paper provide valuable insights into the unique properties of 48 as an anion receptor, paving the way for potential applications in diverse fields and contributing to the advancement of supramolecular chemistry.
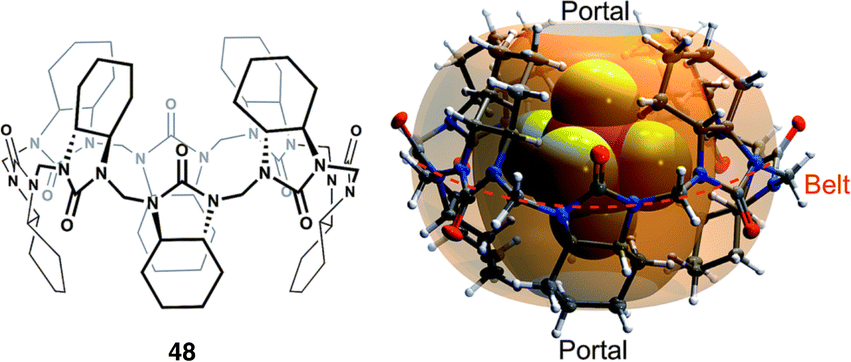 | ||
| Fig. 26 Chemical structure of 48 (left), and the X-ray structure of 48 - SbF6− complex (right) reported by S. Kaabel et al. Image reproduced from ref. 124 with permission from The Royal Society of Chemistry. | ||
Halogen bonding macrocycles
Halogen bonding macrocycles represent a compelling and rapidly evolving area of supramolecular chemistry, characterized by the utilization of halogen bonding interactions within the confines of macrocyclic structures. Halogen bonding, an emerging non-covalent interaction akin to hydrogen bonding, has garnered significant attention due to its unique directional and tuneable nature, offering diverse opportunities for the design of functional macrocyclic systems.125 The interaction is usually represented in literature as R—X⋯Y, where the halogen atom X, as the halogen bonding (XB) donor, is covalently bonded to an electron-withdrawing group R. The entity Y serves as the (XB) acceptor, functioning as an anion.126 The incorporation of halogen bonding motifs into macrocyclic architectures has opened new avenues for the construction of sophisticated host molecules with tailored binding cavities and selective recognition properties. These halogen bonding receptors often exhibit contrasting, and in many cases, superior anion binding affinities and properties compared to their counterparts based on hydrogen bonding.29,127In 2021 Beer and co-workers set out to study lithium halide ion-pair recognition through halogen and chalogen bonding heteroditopic macrocycles. They achieved this by assembling a series of 1,10-phenanthroline-based macrocycles involving XB (49), ChB (50), and HB (51) interactions. These macrocycles act as heteroditopic ion-pair hosts, facilitating the cooperative recognition as well as solid–liquid extraction of lithium halide salts, where the co-bound lithium cation activates the binding of the halide anion. (Fig. 27).128 To investigate the ion-pair binding properties of the macrocycles, initial qualitative 1H NMR binding studies were conducted in a 1:1 CDCl3:CD3CN solvent mixture. Titration experiments with halide anions on 49 revealed no observable halide binding. However, when one equivalent of LiClO4 was added, followed by sequential additions of TBA halide salts, it was observed that the macrocycle's phenanthroline-bound Li+ participated in the binding of halide anions. Bind-fit analysis of the titration binding isotherm data determined 1:1 stoichiometric anion association constant. According to this data, compound 49 is the most effective ion-pair receptor for all halides tested, demonstrating two – seven fold increases in binding affinities for Br− (Ka = 1214 M−1) and I− (Ka = 1236 M−1) compared to the ChB and HB analogues. Notably, the effectiveness of the halogen bonding donor motif is further highlighted by 49's ability to concurrently form a complex with the “hard” LiCl ion-pair, whereas similar experiments with 50 and 51 led to the precipitation of LiCl salt. Overall, the anion binding studies demonstrate the significant impact of the Sigma-hole donor motifs on the binding affinity and selectivity of the heteroditopic macrocycles for lithium halide ion-pairs.
 | ||
| Fig. 27 Chemical structures of phenanthroline-based heteroditopic macrocycles 49, 50, and 51, reported by Beer et al. | ||
In the same year Beer and co-workers reported a series of neutral tetradentate halogen bonding (XB) macrocycles, comprising of two bis-iodotriazole XB donors with varying ring sizes through the integration of meta- and para-substituted xylyl spacer units as well as a naphthyl spacer unit, capable of anion recognition in highly competitive aqueous media (Fig. 28).129 A qualitative 1H-NMR binding study with the meta – xylyl spacer macrocycle (52) with TBA Cl− in 5% D2O/Acetone-d6 revealed that, upon adding 0–1 equivalents of Cl−, the resonances gradually became more resolved, possibly indicating increased macrocycle rigidity due to Cl− complexation. Significant downfield shifts in the macrocycle's internal aromatic protons surrounding the binding site were observed. Importantly, no further proton perturbations occurred after the addition of one equivalent of Cl−, suggesting robust binding in this competitive aqueous-organic solvent mixture. Similar proton signal perturbations were observed in. Binding isotherms were generated by monitoring the changes in shifts from the aromatic proton signals, whereby a 1:1 stoichiometric host–guest association constants for compound 52 were determined via Bindfit, with association constants exceeding 105 M−1 for all halides. The substitution of the m-xylyl spacer unit with p-xylyl and naphthyl groups in XB macrocycles 53 and 54, respectively, was envisioned to yield enlarged cavities with the ability to accommodate larger anionic guest species. Notably, the Bindfit analysis of the 1H-NMR halide titration data with compound 53 showed 1:1 stoichiometric host–guest association constants for Cl− and Br−, both approximately 335 M−1. In contrast, iodide demonstrated a 1:2 host–guest binding stoichiometry, with a slightly higher K1:1 value of 585 M−1. In comparison, the naphthyl macrocycle 54, featuring the most capacious cavity, formed relatively weaker halide complexes, showcasing the Hofmeister selectivity trend. 1H-NMR anion binding studies were extended to dicarboxylate anions, showing that compounds 53 and 54 bind these anions in a 1:1 stoichiometric host–guest manner. The selectivity for dicarboxylate binding was observed as oxalate > malonate > succinate, with compound 52 forming much stronger complexes with higher association constants compared to 54. Fluorescence spectroscopy carried out for 54 in the presence of 10 mM TBA malonate, whereby a notable 38% enhancement in the emission intensity of the naphthyl bands was observed. In contrast, oxalate and succinate induced comparatively smaller enhancements in emission intensity, measuring 6% and 18%, respectively. This trend of heightened emission intensity aligns with the increased length of the dicarboxylate guests, despite the similarity in the magnitude of Ka values determined through NMR titration. This paper illustrates the capability of charge-neutral bis-iodotriazole macrocycles, utilizing halogen bonding, to effectively serve as robust and selective hosts for anion recognition in challenging aqueous environments.
 | ||
| Fig. 28 General structure of the XB macrocycle series (52–54) reported by Beer et al. | ||
In 2022 Sessler and colleagues reported the preparation of tetra iodotriazole halogen bonding macrocycle known as the Ibox (56) through copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC),130 which was inspired from the “Texas-sized” molecular box (55), a tetracationic macrocycle that bound to anions through hydrogen bonding and electrostatic interaction first reported by Sessler et al. in 2010 (Fig. 29).131 The anion binding properties of 56 was measured through 1H NMR spectroscopic studies carried out in CDCl3 and was found to recognise Cl−, Br− and I−. The signals corresponding to the pyridyl proton were seen to shift in an anion and concentration-dependent manner upon the addition of increasing quantities (up to 13 equiv.) of F−, Cl−, Br−, and I− to a 1.25 mM CDCl3 solution of 56. The pyridyl proton signal exhibited an upfield shift, moving from 8.56 ppm to 8.40 ppm for F−, Cl−, Br−, and I−, respectively. Further studies revealed a higher affinity for the heavier halide anions and provided support for a preferred 1:2 binding stoichiometry. The 1H NMR spectral titration data were fitted to a 1:2 binding model and showed a clear binding preference for iodide (I− > Br− > Cl−). The binding mode is thought to involve a pair of iodotriazole subunits within the macrocycle, with each subunit binding one halide anion. This tends to be the opposite of what is typically seen in classic hydrogen bonding anion recognition systems. Additionally, this preference differs significantly from the binding abilities of the “Texas-sized” molecular box 55, which was noted for its capacity to bind both Cl− and NO3− anions.
 | ||
| Fig. 29 Comparison between the structure of the “Texas-sized” molecular box 55 and the Ibox 56. Reported by Sessler and colleagues. | ||
Mechanically interlocked molecules
Mechanically interlocked molecules (MIMs) represent a class of compounds with distinctive structures that involve the threading, interlocking, or linking of molecular components.132 Two prominent examples within this category are [n]rotaxanes and [n]catenanes where n represents the number of mechanically interlocked subcomponents. In rotaxanes, a dumbbell-shaped molecule is threaded through a ring, forming a mechanically bonded structure.133 The interactions between bulky stoppers at the ends of the dumbbell and the ring prevent spontaneous dissociation. Catenanes, on the other hand, consist of two or more rings linked through covalent bonds, resembling interlocked chains. MIMs have gained attention due to their ability to create tailored environments for selective anion binding.134 Rotaxanes, with their threaded structures, and catenanes, with covalently linked rings, offer confined spaces that can selectively encapsulate anions. These mechanically interlocked structures have shown promise in anion sensing applications, molecular switches, and other supramolecular functions.135–137The Beer Research Group have made substantial contributions to the field of supramolecular chemistry through their extensive research over the years on rotaxanes and cantenanes for anion recognition. In 2019 Klein and Beer reported the integration of a novel tetra(iodotriazole)-pyridinium motif into macrocycle and axle components to construct halogen bonding [2]catenane (59) and [2]rotaxanes (mono-cationic (57) and di-cationic (58)) through anion templation (Fig. 30).138 Anion binding properties were studied through 1H NMR titration experiments in competitive aqueous organic solvent mixture 45:45:10 CDCl3/CD3OD/D2O. The anion binding investigations provided valuable insights into the selective binding of I− by the tetra-iodotriazole halogen bonding interlocked hosts. Specifically, the mono-cationic rotaxane 57 displayed a remarkable selectivity for I− (K = 3127 M−1), with an association constant nearly 40 times higher compared to Cl− (K = 89 M−1), as well as an order of magnitude greater than bromide and sulphate. On the other hand, the di-cationic rotaxane 58 displayed greater strength of halide binding but at the expense of a relatively lower degree of I− selectivity over bromide and Cl−. The catenane 59 showed similar results exhibiting preferential binding to iodide however with a lower binding affinity (K = 1176 M−1) than the [2]rotaxanes. These findings highlight the importance of the topological nature and charged state of the interlocked host cavity in dictating the degree of iodide anion discrimination.
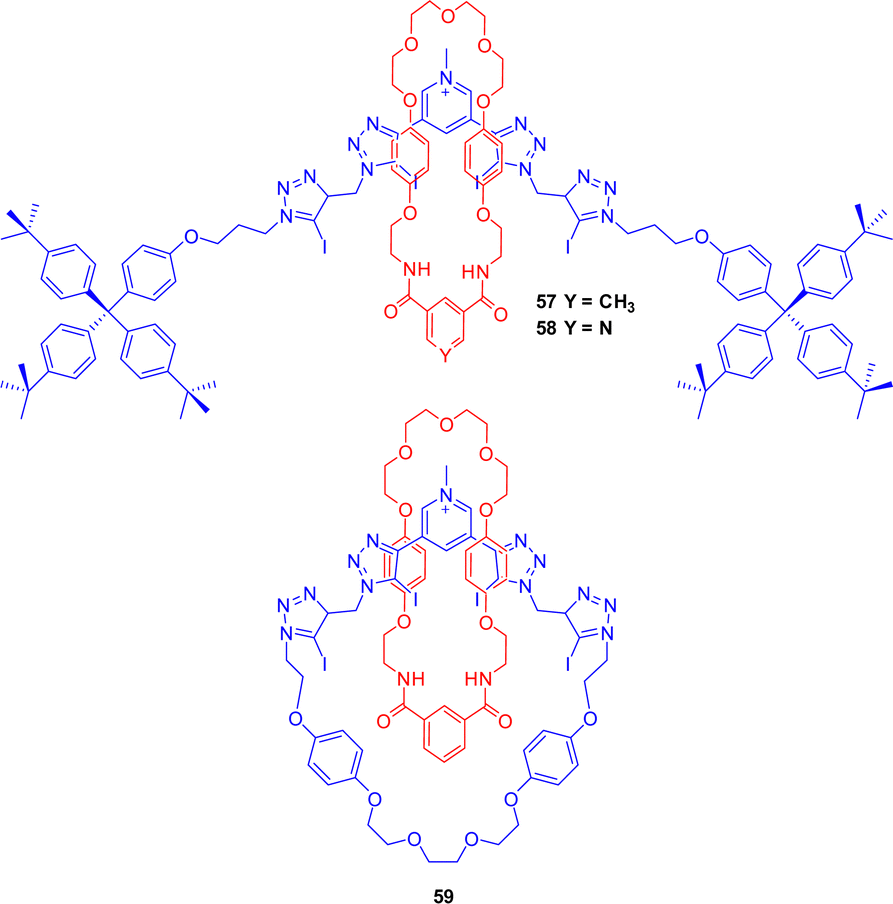 | ||
| Fig. 30 Chemical structures of rotaxanes (57 and 58), and catenane (59) reported by Klein and Beer. | ||
In 2020 Serpell, Beer and co-workers set out to design a novel catenated system 60, with a dense core of cationic hydrogen bonding imidazolium units, providing a high density of cationic hydrogen bonding sites within the molecular structure for enabling effective anion binding through non-covalent interactions (Fig. 31).139 Anion binding studies performed in 9:1 v/v CD3CN:D2O revealed that 60 exhibited an unconventional preference for binding bromide (K = 524 M−1 dm3) over the other halides tested (I− < F− « Cl− < Br−), overcoming halide anion basicity and Hofmeister trends, particularly in aqueous environments. The topology of the cavity in the catenane host 60 is potentially both better defined and more shielded from solvent molecules, resulting in an unusual halide selectivity preference for bromide.
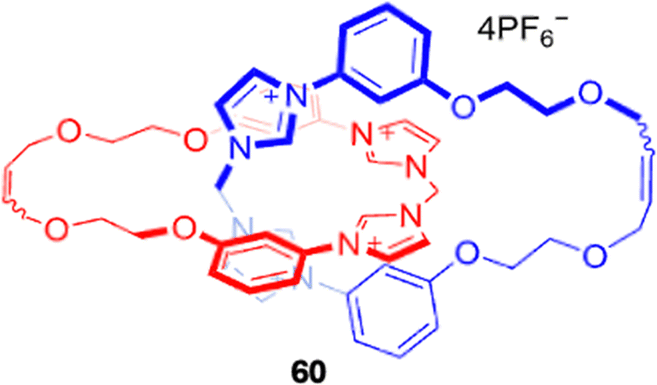 | ||
| Fig. 31 Chemical structure of the catenane 60 reported by Serpell, Beer and co-workers which exhibited anti-Hofmeister bias in halide recognition. Image reproduced from ref. 139 with permission from The Royal Society of Chemistry. | ||
In 2020 Beer and co-workers addressed the paucity of interlocked sensors that incorporate luminophores with long lifetimes and efficient photosensitization. Integrating isophthalamide as the anion-binding group into the dipyridylbenzene ligand structure resulted in the creation of various acyclic and interlocked Pt(II) (61 and 63) and Ru(II) (62 and 64) transition-metal-based hosts. (Fig. 32).140 The anion binding and sensing capabilities of these hosts were thoroughly investigated, shedding light on their potential for anion recognition. The X-ray crystallography analysis revealed the binding of Cl− within the isophthalamide cleft of the receptors, satisfying electrostatic interactions between the anion and the positively charged complex. Additionally, the study demonstrated secondary hydrogen-bonding interactions between the dipyridylbenzene ligand and the isophthalamide carbonyl oxygens, highlighting the role of intra-molecular hydrogen bonding in the preorganization of the metal derivative. Anion recognitions properties were measured through luminescent and 1H NMR binding titrations. Luminescent studies revealed that both acyclic derivatives of the transition-metal dipyridylbenzene complexes were practical sensors for various anionic guests including halides and oxoanions. The near-infrared emitting ruthenium congeners (62) demonstrated increased binding strength compared to platinum (61) as a result of their cationic charge, which provides additional electrostatic interactions. The association constants for all anions studied werelogKa > 5 in CH2Cl2, indicating strong binding affinity. 1H NMR titrations were caried out with the mechanically interlocked host 64 in 30% competitive aqueous mixtures to elucidate the binding mode of Cl−. The observed downfield shifts of the pyridinium axle protons and the perturbation of the internal macrocycle resonance provided insights into the binding of Cl− within the [2]rotaxane cavity. The changes in chemical shifts and peak intensities were indicative of the host–guest interactions and the binding mode of the chloride anion within the interlocked structure.
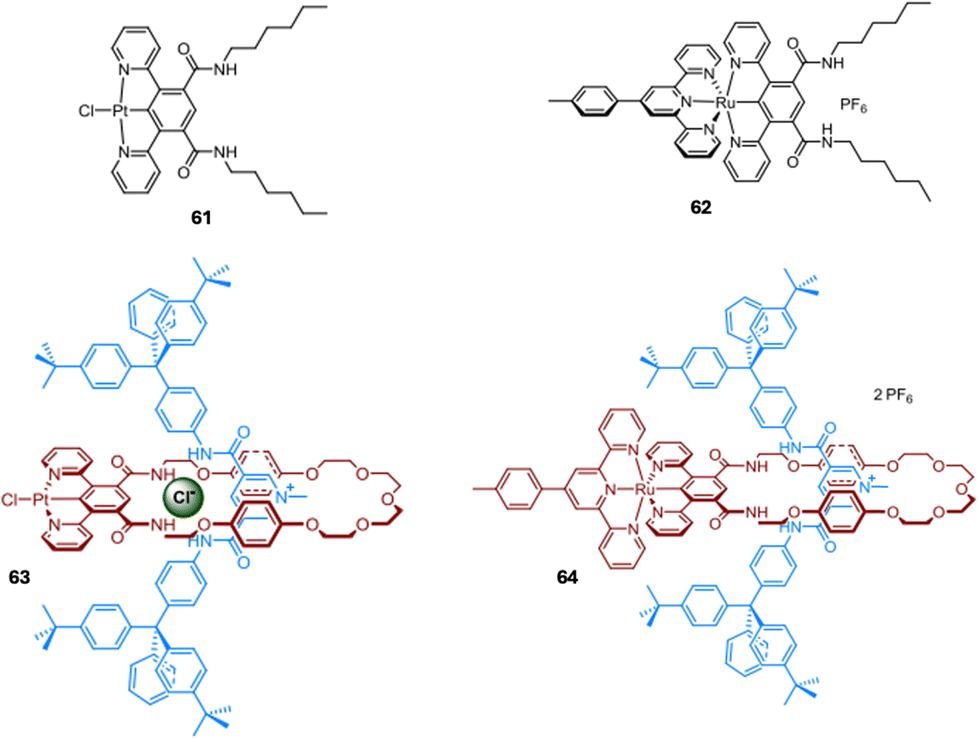 | ||
| Fig. 32 General structures of acyclic Pt(II) (61) and Ru(II) (62) receptors and [2]rotaxane Pt(II) (63) and Ru(II) (64) receptors reported by Beer et al. Images reproduced from ref. 140 with permission from John Wiley and Sons. | ||
Beer and co-workers reported the first examples of halogen-bonding porphyrin BODIPY [2]rotaxanes (65, 66, 67) in 2021, as successful fluorescent and redox electrochemical sensing receptors towards anions (Fig. 33).141 The metalloporphyrin-containing macrocycle component in the rotaxane axle is significant because it serves to pre-organize the rotaxane binding cavity and dramatically enhances anion binding affinities. The triazole group acts as a Lewis base and coordinates to the zinc(II) metal centre of the macrocycle, creating a stable and pre-organized binding site for the anion. This coordination also polarizes the C–H bond of the triazole group, making it a stronger hydrogen bond donor and further enhancing the anion binding affinity. Fluorescence titrations carried out in H2O/acetone (2:98 v/v) with chloride showed that the rotaxanes can optically sense anions through fluorescence quenching of the BODIPY-centered emission of the rotaxane axle. A Larger extent of anion recognition-induced quenching and stronger anion binding strength was observed for the rotaxane containing the electron-withdrawing perfluoroaryl group covalently linked to the BODIPY fluorophore and axle triazole 67. This was attributed to the electron-withdrawing effect of the perfluoroaryl group. Quantitative anion binding studies of 66 were carried out via1H NMR titrations in [D6]acetone. When aliquots of TBA Cl− were added, significant upfield shifts were observed in the aromatic protons on one side of the porphyrin, while the other side exhibited only slight shifts. This observation suggests that the anion is selectively binding to one side of the porphyrin, consistent with the proposed binding mode of the anion perching on one side of the rotaxane cavity. 65 exhibited a 1:1 host–guest binding model with association constants towards all halides (Cl−, Br− and I−) were > 104 M−1. Voltammetric studies showed that rotaxane receptors 66 and 67 detect anions through significant, large-magnitude cathodic shifts in their respective porphyrin P/P+* oxidation couples. This highlights the unique versatility of the MIM synthetic design in integrating complementary optical and electrochemical motifs into the macrocycle and axle components of a rotaxane host, paving the way for potential dual transducer anion sensory systems. Overall, the anion binding studies of [2]rotaxanes provide valuable insights into the anion recognition properties of the rotaxane system, demonstrating the significant impact of the intercomponent interactions between the axle triazole motif and zinc(II) metal centre of the macrocyclic porphyrin on enhancing the anion binding affinity and selectivity of the host system.
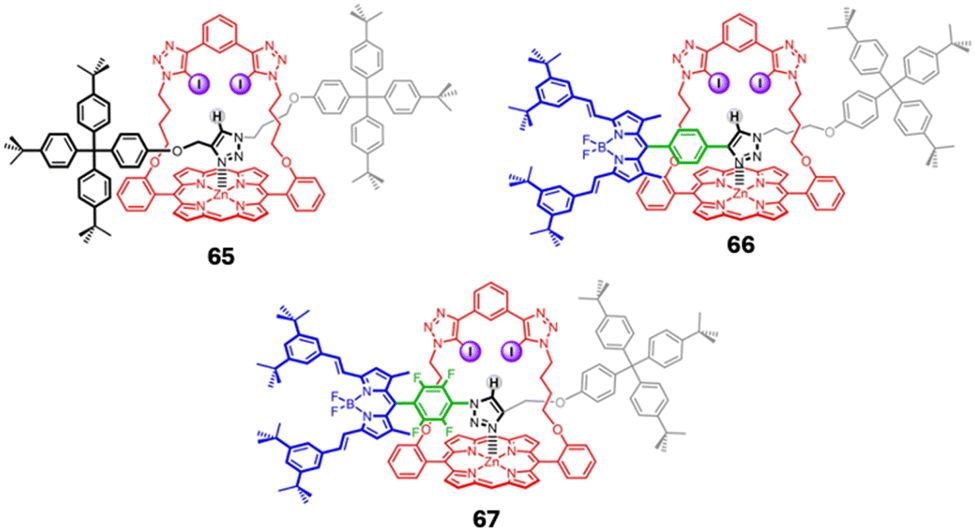 | ||
| Fig. 33 General structures of 65–67 reported by Beer et al. Images reproduced from ref. 141 with permission from John Wiley and Sons. | ||
Cage – based receptors
Cage-based receptors represent a sophisticated class of supramolecular architectures that have garnered significant attention in the field of anion recognition.142,143 Differing from the regular macrocycles mentioned above, cage – based receptors exhibit unique topology and are characterized by their three-dimensional, encapsulating structures. They selectively bind and sense anions through various non-covalent interactions, such as hydrogen bonding, electrostatic interactions, and π–π stacking.144,145 The unique spatial arrangement of these interactions within the confined environment of the cage as well as bearing a well-defined and tuned binding cavity allows for high selectivity and affinity towards specific anions.146 Molecular cages can often be classified into two categories, Metal–Organic cages, which are formed via self-assembly of ligands and metal centres, and organic cages, which are formed through self-assembly of ligands without the presence of a metal centre, or via two distinct bond-forming routes: irreversible bond formation and reversible bond formation.147,148An example of an organic based cage can be seen by Flood and colleagues whereby they reported a design for a cryptand-like triazolo cage (68), which is a type of macrocycle, for their chloride-selective receptor (Fig. 34). The cage was designed to have a size selective cavity for chloride ions and to be stabilized by CH hydrogen bonds originating from 1,2,3-triazoles.149 Introduction of triazole groups increase the conformational rigidity about the cage which in turn would lead to an increase in binding affinity and selectivity. NMR spectroscopy in DMSO-d6 showed that upon addition of TBA Cl− to 68, significant downfield shifts of triazole and phenylene resonances were observed in the 1H NMR spectra, indicating strong CH hydrogen bonding. Liquid–liquid extractions of Cl− from water into nonpolar dichloromethane solvents showed that the chloride-selective receptor had an attomolar affinity (1017 M−1) for CI−. This exceptional affinity exceeded that of other known receptors by several orders of magnitude, demonstrating the high selectivity and binding strength of the CH hydrogen-bonding cage for CI− ions. Moreover, 68 exhibited high selectivity, favouring CI− over larger anions such as I− by a factor of 1 million in certain solvents. The cage's high anion affinity also facilitated the challenging task of extracting alkali metal salts from water into CH2Cl2. The extraction efficiency of 68 was measured which revealed 18% for NaCl, at least 108 times higher than with the solvent alone. This was followed by 8% for Br− while no extraction was observed for NaI. The results from the extraction efficiencies were consistent with the cage's binding selectivity. This indicates not only high affinity but also high selectivity for Cl− over other anions, highlighting the effectiveness of the CH hydrogen-bonding cage in capturing and binding Cl− ions.
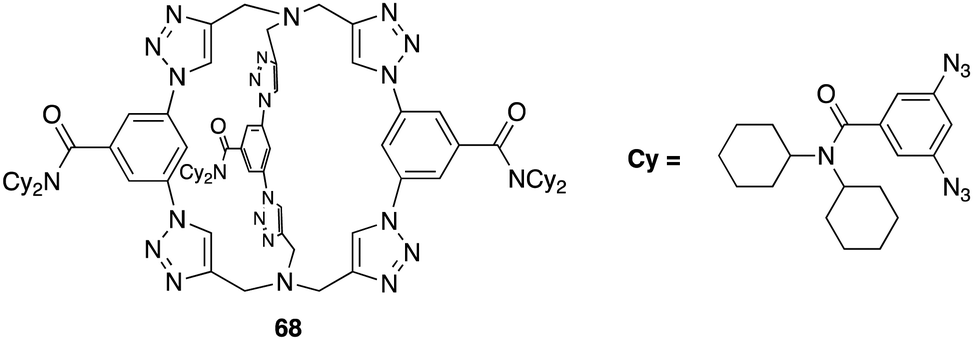 | ||
| Fig. 34 General structure of cryptand-like triazolo cage (68) reported by Flood et al. | ||
Another example of an organic-based cage can be seen by Wang and colleagues whereby they reported the design and synthesis of a triangular prism like cage (69) bearing benzene triimide as its base, and 2,7 naphthalene dimethylene acting as supporting pillars (Fig. 35).150 The articulate structure of 69 possesses a cavity size of 7.6 Å, capable of accommodating and exhibiting strong anion–π binding properties towards a variety of polyhedral anions. X-ray crystallography revealed the three naphthalene planes oriented perpendicularly to the benzene triimide planes, with their lower edges facing inward toward the cavity. The electron-deficient benzene triimide and electron-rich naphthalene moieties engage in intermolecular π–π stacking interactions, leading to an ordered self-assembly in the solid state. The anion binding ability of 69 towards a variety of anions including Cl−, Br−, I−, N3−, SCN−, NO3−, AcO−, BF4−, ClO4−, HSO4−, CH3SO3−, and PF6−, were investigated via UV-vis spectrometry. For anions such as I−, N3−, SCN−, and AcO−, a new charge transfer (CT) band was detected in the range of 400–500 nm, accompanied by a noticeable colour change. In contrast, for Br−, only a weak charge transfer band was observed. For Cl−, NO3−, BF4−, ClO4−, HSO4−, CH3SO3−, and PF6−, the charge transfer band was absent; instead, a hypochromic effect was noted in the range of 360–450 nm. These varying spectral responses likely indicate different positions of the anions on the surface of 69. Binding affinity constants were measured by fitting UV–vis spectrometric titration data to a 1:1 stoichiometry model as revealed via Job's plot. 69 demonstrates a notably high binding affinity for polyhedral anions (BF4−, ClO4−, HSO4−, CH3SO3−, and PF6−), despite their large size and relatively low electron density. The binding constants range from 24651 M−1 for PF6− to 84623 M−1 for HSO4−. The binding strength for SCN− and N3− varies considerably with SCN− demonstrating strong binding at 14878 M−1, whereas N3− shows only moderate binding at 599 M−1. For spherical halides the larger I− is strongly bounded at 2220 M−1, Br− exhibits moderate binding at 58 M−1, and no significant binding was observed for Cl−. As for AcO− and NO3−, weak to moderate binding was observed with binding constants of 73 and 604 M−1, respectively. Wang and colleagues have demonstrated an efficient host (69) for polyhedral anions. According to the authors, the binding constants, ranging from 24651 to 84623 M−1, reflect the strongest anion–π binding recorded for charge-neutral π receptors interacting with individual polyhedral anions.
 | ||
| Fig. 35 General structure of cage-based receptor 69 reported by Wang et al. | ||
Zhang and colleagues set out to develop a new ester-imine cage (68), through efficient imine condensation bearing six functional pyrrole units, as an efficient host for anion binding by utilising the cages three-dimensional cavity (Fig. 36).151 X-ray crystallography revealed 70 to exhibit a slightly twisted conformation, whereby the “top” benzene ring is not aligned parallel to the “bottom” one, and none of the three bipyrrolic fragments are spatially identical. The crystal structure also revealed that two molecules of 70 interact via four intermolecular hydrogen bonds. These bonds are established between an imine-type nitrogen of one molecule and a pyrrolic NH from the other, leading to the formation of a “molecular pair”. The anion binding capability of 70 was measured via Uv-vis specotroscopic titrations across various anions including F−, Br− Cl−, I−, SO42− and AcO−. HSO4− and H2PO4− in chloroform. The optical absorption spectrum of 68, when free of anions, showed a prominent band at 325 nm. The spectrum displayed hyperchromic changes, indicating an increase in absorbance upon the introduction F−, Cl−, I−, SO42−, and AcO−, Conversely, the addition of HSO4− and H2PO4− led to hypochromic changes, reflecting a decrease in absorbance. The presence of Br− resulted in only minor shifts in the absorption band. The binding constants were determined by fitting the UV–vis spectrometric titration data to a 1:1 stoichiometry model as revealed via Job's plot for all the tested anions, except for HSO4−, where both 1:1 and 1:2 binding models were plausible. The binding affinities ranged from 4 × 103 (I−) to 2 × 105 M−1 (F−) for the halides, with H2PO4− having a binding constant of 9 × 103 M−1. Interestingly, the affinity for SO42− was exceptionally high, with a binding constant of 3.46 × 106 M−1, which is roughly 30 times greater than that for HSO4− (9 × 104 M−1), indicating a strong preference of 70 for SO42− compared to other anions. In addition to the aforementioned anions, 70 also exhibits relatively strong interactions with AcO−, having a binding constant of 3.72 × 105 M−1. Overall, the research carried out by Zhang and colleagues underscores the potential of the hexapyrrolic molecular cage (68) as a selective receptor for anions, particularly SO42−, with promising implications for various fields, including environmental and biological applications.
 | ||
| Fig. 36 Synthetic pathway toward hexapyrrolic cage 70, reported by Zhang et al. | ||
Polyhedral oligomeric silesquioxane (POSS) cages are an example of metal–organic cages that have recently emerged as an effective host for anion binding.152,153 These cages are characterized by a silicon–oxygen framework that forms a well-defined polyhedral structure, bearing eight organic groups at each vertex which can allow for extensive functionalization and tuning of properties.154 Their potential as anionic receptors remained unexplored until 2003, when Bassindale, Taylor, and their colleagues unexpectedly discovered the first fluoride-trapped silsesquioxane cage.155 Since then, POSS cages have become a very useful tool for a variety of applications including chemical sensing.
In 2024 Tanaka and co-workers highlighted the innovative use of polyhedral oligomeric silsesquioxane (POSS) as a three-dimensional scaffold for creating effective fluorescence sensors to detect anions, particularly sulfate ions. They carried this out by utilising the hydrogen bonding ability of urea groups, functionalising them onto the POSS cage to yield 71, as well as synthesising a fluorescent derivative using naphthyl urea subunits (72) (Fig. 37).156 Anion binding was determined using 1H NMR spectroscopy in DMSO-d6 by the addition of up to two equivalents for various anions such as Cl−, Br−, I−, HSO4−, SO42−, OH−, AcO−, ClO4−, BPh4−, BF4−, and PF6−. Most of the anions tested lead to insignificant changes of the NH groups of the ureas. In the presence of OH− the peaks disappeared; a slight downfield shift was observed in the presence of AcO−. The most pronounced downfield shift was induced by SO42− indicating hydrogen bonding taking place between the anion and the urea functional groups on 71. The binding mode of 71 was determined via Job's plot which suggested a 1:2 binding stoichiometry whereby four urea groups chelate to SO42−via hydrogen bonding. 71 was further investigated to see if quantifying SO42− ions via fluorescence was possible. This was carried out by modifying 71 by incorporating naphthalene groups to give receptor 72. 1H NMR titrations were carried out with SO42− which showed significant downfield shifts of the urea NH protons indicating that the naphthalene groups on 72 does not have any effect on hindering its binding towards SO42−. The optical properties of 72 were evaluated to further investigate the binding ability and selectivity of SO42−. Notable alterations were detected in the emission spectra of 72. Initially, only monomer emission was observed at approximately 380 nm. However, following the introduction SO42−, the emission intensity at around 450 nm increased, suggesting the formation of excimer states due to the closer arrangement of naphthalene units resulting from SO42− binding. It was also observed that the fluorescence lifetime increased by about 25 ns with the addition of SO42−, indicating that the luminescence detected at this wavelength is linked to excimer formation, which occurs when two naphthalene units are in close proximity. Selectivity studies of 72 were carried out which revealed that in the presence of AcO− and OH− only minor changes in the emission spectra occurred. However, the presence of SO42− resulted in significant alterations, further confirming the sensor's high selectivity for SO42−detection. Overall, Tanaka and colleagues demonstrated the potential of POSS derivatives as effective anion sensors, particularly for quantifying sulfate in various applications.
 | ||
| Fig. 37 Synthetic pathway towards POSS cages 71 and 72, reported by Tanaka et al. | ||
Conclusion and future perspectives
In conclusion, the diverse and rapidly evolving field of macrocyclic receptors for anion recognition presents a compelling panorama of molecular design and functionality. Throughout this review, we have explored the structural diversity, synthetic strategies, and versatile applications of various macrocyclic receptors, such as cyclic peptides, cucurbiturils, pillararenes, and calix[n]arenes, among others. These macrocycles exhibit remarkable anion-binding capabilities attributed to their well-defined cavities, functional groups, and tunable architecture. The extensive research in this area has yielded insights into the intricacies of host–guest interactions and has paved the way for the development of efficient anion sensors and astonishingly selective recognition systems. Furthermore, the adaptability of macrocyclic receptors allows for tailored modifications, enhancing their potential in various fields, including chemical sensing, environmental monitoring, and medicinal chemistry. As we look ahead, the future of macrocyclic compounds in anion recognition looks promising, with several avenues for further research and development. Continued efforts in the synthesis and modification of macrocyclic compounds will enhance their selectivity and binding affinities. Incorporating novel functional groups and exploring hybrid structures may lead to the development of next-generation anion receptors. For example, taking the best of what nature provides and optimising through the incorporation of abiotic elements may provide for optimised designs with further optimisation possible. This is akin to the work conducted in the field of foldmer chemistry, where unnatural helical peptidic foldamers have gained research attention in recent years owing to their interesting folding behaviour, protein binding abilities and potential applications across the chemical sciences; all gains brought about by synthetic mimics of biological systems.157 Developments in this regard may hold significant potential for environmental monitoring and remediation, as well as biological and medical applications. Furthermore, while the vast majority of examples presented are experimental and proof of principle studies, expanding the use of macrocyclic compounds in biological systems for anion sensing and transport is likely to lead to further advances in diagnostic and therapeutic tools. For instance, macrocyclic anion receptors could be employed more widely in drug delivery systems and as biomarkers for various diseases.158 Macrocycles capable of efficient anion transport are also likely to be interesting candidates for exploitation in medicinal chemistry where other transporters have found application.159 The continued exploration of novel macrocyclic structures and innovative design strategies promises to expand the frontiers of anion recognition, providing valuable contributions to the broader landscape of supramolecular chemistry.Author contributions
FAM: conceptualisation, recent progress analysis, writing, editing. TX: reviewing and editing. LW: reviewing and editing. RBPE: writing, reviewing, editing, project supervision.Data availability
No primary data is reported in this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. B. P. E. acknowledges funding from Science Foundation Ireland (SFI) (grant number: 12/RC/2275/P2), which is co-funded under the European Regional Development Fund. SFI are also acknowledged for the funding of Research Infrastructure at Maynooth University through the Research Infrastructure Programme (12/RI/2346/SOF) and the Opportunistic Infrastructure Fund (16/RI/3399). This article is based upon discussions from COST Action CA22131 - Supramolecular LUminescent Chemosensors for Environmental Security, supported by COST (European Cooperation in Science and Technology).References
- P. A. Gale, Coord. Chem. Rev., 2000, 199, 181–233 CrossRef CAS.
- C. R. Bondy and S. J. Loeb, Coord. Chem. Rev., 2003, 240, 77–99 CrossRef CAS.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS PubMed.
- R. Behrend, E. Meyer and F. Rusche, Justus Liebigs Ann. Chem., 1905, 339, 1–37 CrossRef.
- D. L. Correll, J. Environ. Qual., 1998, 27, 261–266 CrossRef CAS.
- R. Dutzler, E. B. Campbell, M. Cadene, B. T. Chait and R. MacKinnon, Nature, 2002, 415, 287–294 CrossRef CAS PubMed.
- A. D. C. Macknight, Eur. J. Appl. Physiol., 1985, 405, S12–S16 CAS.
- Thyroid®, 1994, vol. 4, pp. 107–128.
- M. J. Welsh and A. E. Smith, Cell, 1993, 73, 1251–1254 CrossRef CAS PubMed.
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC.
- P. A. Gale, R. Pérez-Tomás and R. Quesada, Acc. Chem. Res., 2013, 46, 2801–2813 CrossRef CAS PubMed.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- P. A. Gale and C. Caltagirone, Coord. Chem. Rev., 2018, 354, 2–27 CrossRef CAS.
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC.
- A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC.
- X. Wu, A. M. Gilchrist and P. A. Gale, Chem, 2020, 6, 1296–1309 CAS.
- L. Chen, S. N. Berry, X. Wu, E. N. W. Howe and P. A. Gale, Chem, 2020, 6, 61–141 CAS.
- R. Peng, Y. Xu and Q. Cao, Chin. Chem. Lett., 2018, 29, 1465–1474 CrossRef CAS.
- G. Ghale and W. M. Nau, Acc. Chem. Res., 2014, 47, 2150–2159 CrossRef CAS PubMed.
- A. Li, H. Zhai, J. Li and Q. He, Chem. Lett., 2020, 49, 1125–1135 CrossRef CAS.
- Q. Zhang, Y. Zhou, M. Ahmed, N. M. Khashab, W. Han, H. Wang, Z. A. Page and J. L. Sessler, J. Mater. Chem, A, 2022, 10, 15297–15308 RSC.
- Y.-C. Pan, J.-H. Tian and D.-S. Guo, Acc. Chem. Res., 2023, 56, 3626–3639 CrossRef CAS PubMed.
- R. Pinalli, A. Pedrini and E. Dalcanale, Chem. Soc. Rev., 2018, 47, 7006–7026 RSC.
- P. L. Padnya, X. Wu, A. Erxleben and S. S. Braga, Front. Chem., 2021, 9 DOI:10.3389/fchem.2021.730111.
- F. Barbette, F. Rascalou, H. Chollet, J. L. Babouhot, F. Denat and R. Guilard, Anal. Chim. Acta, 2004, 502, 179–187 CrossRef CAS.
- D. Cao, Y. Kou, J. Liang, Z. Chen, L. Wang and H. Meier, Angew. Chem., Int. Ed., 2009, 48, 9721–9723 CrossRef CAS PubMed.
- S.-Q. Cheng, Q. Lin, S.-L. Li, Y.-X. Guo, X.-L. Han, Y. Sun and Y. Liu, Green Chem., 2023, 25, 7026–7040 RSC.
- Z.-Y. Zhang and C. Li, Acc. Chem. Res., 2022, 55, 916–929 CrossRef CAS PubMed.
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS.
- B. Qiao, J. R. Anderson, M. Pink and A. H. Flood, Chem. Commun., 2016, 52, 8683–8686 RSC.
- B. Dietrich, J. Guilhem, J.-M. Lehn, C. Pascard and E. Sonveaux, Helv. Chim. Acta, 1984, 67, 91–104 CrossRef CAS.
- D. H. Sohn, N. Kim, S. Jang and J. Kang, New J. Chem., 2019, 43, 13690–13695 RSC.
- P. Gamez, T. J. Mooibroek, S. J. Teat and J. Reedijk, Acc. Chem. Res., 2007, 40, 435–444 CrossRef CAS PubMed.
- R. S. Carnegie, C. L. D. Gibb and B. C. Gibb, Angew. Chem., Int. Ed., 2014, 53, 11498–11500 CrossRef CAS PubMed.
- A. Sengupta, Y. Liu, A. H. Flood and K. Raghavachari, Chem. – Eur. J., 2018, 24, 14409–14417 CrossRef CAS PubMed.
- T. Zhang, L.-P. Zhou, X.-Q. Guo, L.-X. Cai and Q.-F. Sun, Nat. Commun., 2017, 8, 15898 CrossRef CAS PubMed.
- E. R. Abdurakhmanova, P. Cmoch and A. Szumna, Org. Biomol. Chem., 2022, 20, 5095–5103 RSC.
- S. Xiong, M. V. Nanda Kishore, W. Zhou and Q. He, Coord. Chem. Rev., 2022, 461, 214480 CrossRef CAS.
- G. Mamardashvili, N. Mamardashvili and O. Koifman, Molecules, 2021, 26(17), 5292 CrossRef CAS PubMed.
- Z. Liu, Y. Zhou and L. Yuan, Org. Biomol. Chem., 2022, 20, 9023–9051 RSC.
- K. Choi and A. D. Hamilton, Coord. Chem. Rev., 2003, 240, 101–110 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS PubMed.
- P. J. Smith, M. V. Reddington and C. S. Wilcox, Tetrahedron Lett., 1992, 33, 6085–6088 Search PubMed.
- X. Shang, Z. Yang, J. Fu, P. Zhao and X. Xu, Sensors, 2015, 15, 28166–28176 CrossRef PubMed.
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and M. Milani, Chem. – Eur. J., 2010, 16, 4368–4380 CrossRef CAS PubMed.
- L. A. Marchetti, L. K. Kumawat, N. Mao, J. C. Stephens and R. B. P. Elmes, Chem, 2019, 5, 1398–1485 CAS.
- S. M. Butler, M. Hountondji, S. N. Berry, J. Tan, L. Macia and K. A. Jolliffe, Org. Biomol. Chem., 2023, 21, 8548–8553 RSC.
- N. Luo, J. Li, T. Sun, S. Wan, P. Li, N. Wu, Y. Yan and X. Bao, RSC Adv., 2021, 11, 10203–10211 RSC.
- A. S. Oshchepkov, T. A. Shumilova, S. R. Namashivaya, O. A. Fedorova, P. V. Dorovatovskii, V. N. Khrustalev and E. A. Kataev, J. Org. Chem., 2018, 83, 2145–2153 CrossRef CAS PubMed.
- S. M. Butler and K. A. Jolliffe, Org. Biomol. Chem., 2020, 18, 8236–8254 RSC.
- K. Sikora, M. Jaśkiewicz, D. Neubauer, D. Migoń and W. Kamysz, Pharmaceuticals, 2020, 13, 442 CrossRef CAS PubMed.
- R. Langridge, H. Shinagawa and A. B. Pardee, Science, 1970, 169, 59–61 CrossRef CAS PubMed.
- W. R. Harris, D. Nesset-Tollefson, J. Z. Stenback and N. Mohamed-Hani, J. Inorg. Biochem., 1990, 38, 175–183 CrossRef CAS PubMed.
- S. Kubik, Chem. Soc. Rev., 2009, 38, 585–605 RSC.
- J. F. Riordan, Mol. Cell. Biochem., 1979, 26, 71–92 CrossRef CAS PubMed.
- P. Chakrabarti, J. Mol. Biol., 1993, 234, 463–482 CrossRef CAS PubMed.
- R. B. P. Elmes and K. A. Jolliffe, Chem. Commun., 2015, 51, 4951–4968 RSC.
- S. J. Butler and K. A. Jolliffe, Org. Biomol. Chem., 2011, 9, 3471–3483 RSC.
- S. G. Davey, Nat. Rev. Chem., 2020, 4, 228 CrossRef CAS PubMed.
- S. Kubik, Acc. Chem. Res., 2017, 50, 2870–2878 CrossRef CAS PubMed.
- S. C. Larnaudie, J. C. Brendel, K. A. Jolliffe and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1003–1011 Search PubMed.
- R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe and S. Perrier, Chem. Soc. Rev., 2012, 41, 6023–6041 RSC.
- G. E. Sergeant and K. A. Jolliffe, Supramol. Chem., 2020, 32, 233–237 CrossRef CAS.
- F. Sommer, Y. Marcus and S. Kubik, ACS Omega, 2017, 2, 3669–3680 CrossRef CAS PubMed.
- A. Schaly, R. Belda, E. García-España and S. Kubik, Org. Lett., 2013, 15, 6238–6241 CrossRef CAS PubMed.
- S. Kubik and R. Goddard, Eur. J. Org. Chem., 2001, 311–322 CrossRef CAS.
- T. Fiehn, R. Goddard, R. W. Seidel and S. Kubik, Chem. – Eur. J., 2010, 16, 7241–7255 CrossRef CAS PubMed.
- L. M. Mesquita, J. Anhäuser, D. Bellaire, S. Becker, A. Lützen and S. Kubik, Org. Lett., 2019, 21, 6442–6446 CrossRef CAS PubMed.
- I. Petters, M. Modrušan, N. Vidović, I. Crnolatac, N. Cindro, I. Piantanida, G. Speranza, G. Horvat and V. Tomišić, Molecules, 2022, 27, 3918 CrossRef CAS PubMed.
- G. Picci, I. Carreira-Barral, D. Alonso-Carrillo, C. Busonera, J. Milia, R. Quesada and C. Caltagirone, Org. Biomol. Chem., 2022, 20, 7981–7986 RSC.
- G. Picci, M. C. Aragoni, M. Arca, C. Caltagirone, M. Formica, V. Fusi, L. Giorgi, F. Ingargiola, V. Lippolis, E. Macedi, L. Mancini, L. Mummolo and L. Prodi, Org. Biomol. Chem., 2023, 21, 2968–2975 RSC.
- G. Picci, S. Farotto, J. Milia, C. Caltagirone, V. Lippolis, M. C. Aragoni, C. Di Natale, R. Paolesse and L. Lvova, ACS Sens., 2023, 8, 3225–3239 CrossRef CAS PubMed.
- X. Bao, X. Wu, S. N. Berry, E. N. W. Howe, Y.-T. Chang and P. A. Gale, Chem. Commun., 2018, 54, 1363–1366 RSC.
- I. Marques, P. M. R. Costa, M. Q. Miranda, N. Busschaert, E. N. W. Howe, H. J. Clarke, C. J. E. Haynes, I. L. Kirby, A. M. Rodilla, R. Pérez-Tomás, P. A. Gale and V. Félix, Phys. Chem. Chem. Phys., 2018, 20, 20796–20811 RSC.
- S. Zhang, Y. Wang, W. Xie, E. N. W. Howe, N. Busschaert, A. Sauvat, M. Leduc, L. C. Gomes-da-Silva, G. Chen, I. Martins, X. Deng, L. Maiuri, O. Kepp, T. Soussi, P. A. Gale, N. Zamzami and G. Kroemer, Cell Death Dis., 2019, 10, 242 CrossRef PubMed.
- L. Qin, A. Hartley, P. Turner, R. B. P. Elmes and K. A. Jolliffe, Chem. Sci., 2016, 7, 4563–4572 RSC.
- L. Qin, J. R. Wright, J. D. E. Lane, S. N. Berry, R. B. P. Elmes and K. A. Jolliffe, Chem. Commun., 2019, 55, 12312–12315 RSC.
- L. Qin, S. J. N. Vervuurt, R. B. P. Elmes, S. N. Berry, N. Proschogo and K. A. Jolliffe, Chem. Sci., 2020, 11, 201–207 RSC.
- M. Zaleskaya, M. Karbarz, M. Wilczek, Ł. Dobrzycki and J. Romański, Inorg. Chem., 2020, 59, 13749–13759 CrossRef CAS PubMed.
- M. Zaleskaya-Hernik, E. Megiel and J. Romański, J. Mol. Liq., 2022, 361, 119600 CrossRef CAS.
- M. Zaleskaya-Hernik, Ł. Dobrzycki and J. Romański, Int. J. Mol. Sci., 2023, 24, 8536 CrossRef CAS PubMed.
- M. Zaleskaya, D. Jagleniec and J. Romański, Dalton Trans., 2021, 50, 3904–3915 RSC.
- I. Saha, J. T. Lee and C.-H. Lee, Eur. J. Org. Chem., 2015, 3859–3885 CrossRef CAS.
- P. A. Gale, J. L. Sessler, V. Král and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef CAS.
- D. Villarón, G. E. A. Brugman, M. A. Siegler and S. J. Wezenberg, Chem. Commun., 2023, 59, 8688–8691 RSC.
- S. Xiong, F. Chen, T. Zhao, A. Li, G. Xu, J. L. Sessler and Q. He, Org. Lett., 2020, 22, 4451–4455 CrossRef CAS PubMed.
- R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS PubMed.
- F. Zhang, Y. Sun, D. Tian, W. S. Shin, J. S. Kim and H. Li, Chem. Commun., 2016, 52, 12685–12693 RSC.
- E. S. Español and M. M. Villamil, Biomolecules, 2019, 9, 90 CrossRef PubMed.
- D.-S. Guo and Y. Liu, Acc. Chem. Res., 2014, 47, 1925–1934 CrossRef CAS PubMed.
- B. Uttam, R. Kandi, M. A. Hussain and C. P. Rao, J. Org. Chem., 2018, 83, 11850–11859 CrossRef CAS PubMed.
- Z.-Y. Li, H.-K. Su, H.-X. Tong, Y. Yin, T. Xiao, X.-Q. Sun, J. Jiang and L. Wang, Spectrochim. Acta, Part A, 2018, 200, 307–312 CrossRef CAS PubMed.
- M. Nemati, R. Hosseinzadeh and M. Mohadjerani, Spectrochim. Acta, Part A, 2021, 245, 118950 CrossRef CAS PubMed.
- M. Řezanková, J. Budka, J. Mikšátko, V. Eigner, I. Císařová, P. Cuřínová and P. Lhoták, Tetrahedron, 2017, 73, 742–749 CrossRef.
- T. Ogoshi, S. Kanai, S. Fujinami, T.-A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS PubMed.
- W. Li, P. Qin, X.-X. Zhao, W.-J. Qu, Q. Lin, H. Yao, T.-B. Wei, Y.-M. Zhang, Y. Liu and B. Shi, Org. Biomol. Chem., 2022, 20, 9122–9126 RSC.
- M. Vinodh, F. H. Alipour and T. F. Al-Azemi, ACS Omega, 2023, 8, 1466–1475 CrossRef CAS PubMed.
- S. S. Zhu, H. Staats, K. Brandhorst, J. Grunenberg, F. Gruppi, E. Dalcanale, A. Lützen, K. Rissanen and C. A. Schalley, Angew. Chem., Int. Ed., 2008, 47, 788–792 CrossRef CAS PubMed.
- C. A. Ilioudis, D. A. Tocher and J. W. Steed, J. Am. Chem. Soc., 2004, 126, 12395–12402 CrossRef CAS PubMed.
- Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS PubMed.
- S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef CAS PubMed.
- E. Zeynaloo, E. M. Zahran, E. M. Fatila, A. H. Flood and L. G. Bachas, Anal. Chem., 2021, 93, 5412–5419 CrossRef CAS PubMed.
- E. G. Sheetz, Z. Zhang, A. Marogil, M. Che, M. Pink, V. Carta, K. Raghavachari and A. H. Flood, Chem. – Eur. J., 2022, 28, e202201584 CrossRef CAS PubMed.
- J. Svec, M. Necas and V. Sindelar, Angew. Chem., Int. Ed., 2010, 49, 2378–2381 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- V. Havel, J. Svec, M. Wimmerova, M. Dusek, M. Pojarova and V. Sindelar, Org. Lett., 2011, 13, 4000–4003 CrossRef CAS PubMed.
- V. Havel, V. Sindelar, M. Necas and A. E. Kaifer, Chem. Commun., 2014, 50, 1372–1374 RSC.
- V. Havel and V. Sindelar, ChemPlusChem, 2015, 80, 1601–1606 CrossRef CAS PubMed.
- A. Šlampová, V. Šindelář and P. Kubáň, Anal. Chim. Acta, 2017, 950, 49–56 CrossRef PubMed.
- H. Valkenier, O. Akrawi, P. Jurček, K. Sleziaková, T. Lízal, K. Bartik and V. Šindelář, Chem, 2019, 5, 429–444 Search PubMed.
- Á. Révész, D. Schröder, J. Svec, M. Wimmerová and V. Sindelar, J. Phys. Chem. A, 2011, 115, 11378–11386 CrossRef PubMed.
- J. Svec, M. Dusek, K. Fejfarova, P. Stacko, P. Klán, A. E. Kaifer, W. Li, E. Hudeckova and V. Sindelar, Chem. – Eur. J., 2011, 17, 5605–5612 CrossRef CAS PubMed.
- M. A. Yawer, V. Havel and V. Sindelar, Angew. Chem., Int. Ed., 2015, 54, 276–279 CrossRef CAS PubMed.
- M. Chvojka, D. Madea, H. Valkenier and V. Šindelář, Angew. Chem., Int. Ed., 2024, 63, e202318261 CrossRef CAS PubMed.
- M. Lisbjerg, B. M. Jessen, B. Rasmussen, B. E. Nielsen, A. Ø. Madsen and M. Pittelkow, Chem. Sci., 2014, 5, 2647–2650 RSC.
- M. Lisbjerg, B. E. Nielsen, B. O. Milhøj, S. P. A. Sauer and M. Pittelkow, Org. Biomol. Chem., 2015, 13, 369–373 RSC.
- M. Lisbjerg, H. Valkenier, B. M. Jessen, H. Al-Kerdi, A. P. Davis and M. Pittelkow, J. Am. Chem. Soc., 2015, 137, 4948–4951 CrossRef CAS PubMed.
- X.-Y. Jin, F. Wang, H. Cong and Z. Tao, J. Inclusion Phenom. Macrocyclic Chem., 2016, 86, 241–248 CrossRef CAS.
- S. Kaabel, R. S. Stein, M. Fomitšenko, I. Järving, T. Friščić and R. Aav, Angew. Chem., Int. Ed., 2019, 58, 6230–6234 CrossRef CAS PubMed.
- S. Kaabel and R. Aav, Isr. J. Chem., 2018, 58, 296–313 CrossRef CAS.
- N. N. Andersen, M. Lisbjerg, K. Eriksen and M. Pittelkow, Isr. J. Chem., 2018, 58, 435–448 CrossRef CAS.
- V. Šindelář and T. Lízal, in Cucurbiturils and Related Macrocycles, ed. K. Kim, The Royal Society of Chemistry, 2019 10.1039/9781788015967-00527, p. 0.
- S. Kaabel, J. Adamson, F. Topić, A. Kiesilä, E. Kalenius, M. Öeren, M. Reimund, E. Prigorchenko, A. Lõokene, H. J. Reich, K. Rissanen and R. Aav, Chem. Sci., 2017, 8, 2184–2190 RSC.
- C.-Z. Liu, S. Koppireddi, H. Wang, D.-W. Zhang and Z.-T. Li, Chin. Chem. Lett., 2019, 30, 953–956 CrossRef CAS.
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC.
- N. G. White, A. Caballero and P. D. Beer, CrystEngComm, 2014, 16, 3722–3729 RSC.
- Y. C. Tse, A. Docker, Z. Zhang and P. D. Beer, Chem. Commun., 2021, 57, 4950–4953 RSC.
- T. Bunchuay, K. Boonpalit, A. Docker, A. Ruengsuk, J. Tantirungrotechai, M. Sukwattanasinitt, P. Surawatanawong and P. D. Beer, Chem. Commun., 2021, 57, 11976–11979 RSC.
- T. Zhao, V. M. Lynch and J. L. Sessler, Org. Biomol. Chem., 2022, 20, 980–983 RSC.
- H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406–409 CrossRef CAS PubMed.
- J. Riebe and J. Niemeyer, Eur. J. Org. Chem., 2021, 5106–5116 CrossRef CAS.
- S. J. Nicholson, S. R. Barlow and N. H. Evans, Chemistry, 2023, 5, 106–118 CrossRef CAS.
- A. Caballero, F. Zapata and P. D. Beer, Coord. Chem. Rev., 2013, 257, 2434–2455 CrossRef CAS.
- M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935–1949 CrossRef CAS PubMed.
- T. Takata, ACS Cent. Sci., 2020, 6, 129–143 CrossRef CAS PubMed.
- F. d'Orchymont and J. P. Holland, Angew. Chem., Int. Ed., 2022, 61, e202204072 CrossRef PubMed.
- H. A. Klein and P. D. Beer, Chem. – Eur. J., 2019, 25, 3125–3130 CrossRef CAS PubMed.
- C. J. Serpell, A. Y. Park, C. V. Robinson and P. D. Beer, Chem. Commun., 2021, 57, 101–104 RSC.
- R. C. Knighton, S. Dapin and P. D. Beer, Chem. – Eur. J., 2020, 26, 5288–5296 CrossRef CAS PubMed.
- Y. Cheong Tse, R. Hein, E. J. Mitchell, Z. Zhang and P. D. Beer, Chem. – Eur. J., 2021, 27, 14550–14559 CrossRef CAS PubMed.
- J.-H. Zhang, S.-M. Xie, M. Zi and L.-M. Yuan, J. Sep. Sci., 2020, 43, 134–149 CrossRef CAS PubMed.
- A. Brzechwa-Chodzyńska, W. Drożdż, J. Harrowfield and A. R. Stefankiewicz, Coord. Chem. Rev., 2021, 434, 213820 CrossRef.
- V. Amendola, G. Bergamaschi and A. Miljkovic, Supramol. Chem., 2018, 30, 236–242 CrossRef CAS.
- K. Bowman-James, Science, 2019, 365, 124–125 CrossRef CAS PubMed.
- K. G. Dutton, T. J. Jones, T. J. Emge and M. C. Lipke, Chem. – Eur. J., 2024, 30, e202303013 CrossRef CAS PubMed.
- G. Montà-González, F. Sancenón, R. Martínez-Máñez and V. Martí-Centelles, Chem. Rev., 2022, 122, 13636–13708 CrossRef PubMed.
- G. Montà-González, E. Ortiz-Gómez, R. López-Lima, G. Fiorini, R. Martínez-Máñez and V. Martí-Centelles, Molecules, 2024, 29(7), 1621 CrossRef PubMed.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CrossRef CAS PubMed.
- D.-H. Tuo, Y.-F. Ao, Q.-Q. Wang and D.-X. Wang, CCS Chem., 2022, 4, 2806–2815 CrossRef CAS.
- X. Zhao, S. Xiong, J. Zhang, J. Pu, W. Ding, X. Chen, Q. He and Z. Zhang, J. Mol. Struct., 2023, 1293, 136232 CrossRef CAS.
- Z. Gou, X. Zhang, Y. Zuo, M. Tian, B. Dong, Y. Tang and W. Lin, Sens. Actuators, B, 2021, 338, 129837 CrossRef CAS.
- S. Chanmungkalakul, V. Ervithayasuporn, P. Boonkitti, A. Phuekphong, N. Prigyai, S. Kladsomboon and S. Kiatkamjornwong, Chem. Sci., 2018, 9, 7753–7765 RSC.
- M. Janeta, Ł. John, J. Ejfler, T. Lis and S. Szafert, Dalton Trans., 2016, 45, 12312–12321 RSC.
- A. R. Bassindale, M. Pourny, P. G. Taylor, M. B. Hursthouse and M. E. Light, Angew. Chem., Int. Ed., 2003, 42, 3488–3490 CrossRef CAS PubMed.
- H. Narikiyo, M. Gon, K. Tanaka and Y. Chujo, Polym. J., 2024, 56, 661–666 CrossRef CAS.
- P. Sang and J. Cai, Chem. Soc. Rev., 2023, 52, 4843–4877 RSC.
- M. Yan, S. Wu, Y. Wang, M. Liang, M. Wang, W. Hu, G. Yu, Z. Mao, F. Huang and J. Zhou, Adv. Mater., 2024, 36, 2304249 CrossRef CAS PubMed.
- J. T. Davis, P. A. Gale and R. Quesada, Chem. Soc. Rev., 2020, 49, 6056–6086 RSC.
| This journal is © The Royal Society of Chemistry 2024 |