
Organic–inorganic composite hydrogels: compositions, properties, and applications in regenerative medicine
Xinyu
Wang
a,
Wei
Wei
a,
Ziyi
Guo
a,
Xinru
Liu
a,
Ju
Liu
a,
Tiejun
Bing
b,
Yingjie
Yu
 *a,
Xiaoping
Yang
a and
Qing
Cai
*a
*a,
Xiaoping
Yang
a and
Qing
Cai
*a
aState Key Laboratory of Organic-Inorganic Composites; Beijing Laboratory of Biomedical Materials, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: yuyingjie@mail.buct.edu.cn; caiqing@mail.buct.edu.cn
bImmunology and Oncology center, ICE Bioscience, Beijing 100176, China
First published on 10th January 2024
Abstract
Hydrogels, formed from crosslinked hydrophilic macromolecules, provide a three-dimensional microenvironment that mimics the extracellular matrix. They served as scaffold materials in regenerative medicine with an ever-growing demand. However, hydrogels composed of only organic components may not fully meet the performance and functionalization requirements for various tissue defects. Composite hydrogels, containing inorganic components, have attracted tremendous attention due to their unique compositions and properties. Rigid inorganic particles, rods, fibers, etc., can form organic–inorganic composite hydrogels through physical interaction and chemical bonding with polymer chains, which can not only adjust strength and modulus, but also act as carriers of bioactive components, enhancing the properties and biological functions of the composite hydrogels. Notably, incorporating environmental or stimulus-responsive inorganic particles imparts smartness to hydrogels, hence providing a flexible diagnostic platform for in vitro cell culture and in vivo tissue regeneration. In this review, we discuss and compare a set of materials currently used for developing organic–inorganic composite hydrogels, including the modification strategies for organic and inorganic components and their unique contributions to regenerative medicine. Specific emphasis is placed on the interactions between the organic or inorganic components and the biological functions introduced by the inorganic components. The advantages of these composite hydrogels indicate their potential to offer adaptable and intelligent therapeutic solutions for diverse tissue repair demands within the realm of regenerative medicine.
 Xinyu Wang | Xinyu Wang is currently a Masters student in the College of Materials Science and Engineering at Beijing University of Chemical Technology (BUCT) in China, working under the supervision of Prof. Qing Cai. His research focuses on hydrogels and bone regeneration. |
 Yingjie Yu | Yingjie Yu is an associate professor in the College of Materials Science and Engineering at Beijing University of Chemical Technology (BUCT). He received his Ph.D. from the State University of New York at Stony Brook in 2016, majoring in Materials Science and Engineering. His research interest is in drug delivery systems and tissue engineering. |
 Qing Cai | Qing Cai is a full professor in the College of Materials Science and Engineering at BUCT. She received her Ph.D. in polymer chemistry and physics in 2003 from the Institute of Chemistry, Chinese Academy of Sciences, and has worked at BUCT from then on. Prof. Cai has extensive experience with biodegradable polymers, biocomposites, tissue engineering, and drug delivery, particularly with bone regeneration and dental restoration. |
1. Introduction
Regenerative medicine aims to study the normal characteristics and functions of body organs, mechanisms of tissue regeneration, and guidance for directed stem cell differentiation to develop effective biotechnological treatments for injured tissues and organs, thus maintaining or reconstructing their native functions.1 Regardless of whether it involves constructing new tissues and organs in vitro or direct therapeutic interventions within the body, such as the administration of drugs and active components, suitable material substrates are essential for implementation.2–4 The use of hydrogels has garnered significant attention in the field of regenerative medicine owing to their distinct highly hydrated three-dimensional network, versatile crosslinking techniques, diverse compositions, and exceptional biocompatibility. Common applications for hydrogels include hemostasis,5 wound dressings,6 stem cell encapsulation,7 and more. As scaffolds, hydrogels can mimic the structure and function of the extracellular matrix (ECM) existing in natural tissues, providing friendly microenvironments for cell growth and differentiation, helping to achieve the final tissue regeneration by regulating cell behaviors with different designs.8 As delivery carriers, hydrogels can transport drugs and peptides to target tissues and control their release profiles.9 However, tissue regeneration is a complex dynamic process involving tissues with different mechanical properties and ECM components. Consequently, various demands have been proposed for hydrogel fabrication regarding their compositions, crosslinked networks, mechanical properties, degradation behaviors, and tissue-guiding/inducing activities, etc. To meet these demands, researchers have proposed various material systems, including blends,10 copolymers,11 and graft modifications.12 Simultaneously, several crosslinking strategies such as physical interactions,13 covalent crosslinking,14 and dynamic crosslinking,15 are properly combined to develop composite hydrogels with tailored biological functions. They provide vast feasibilities for regenerating damaged tissues such as skin, cartilage, and bone tissues using the strategy of scaffold-based tissue engineering.In recent years, the incorporation of inorganic fillers into polymeric hydrogels has been a highlighted topic in the field of regenerative medicine.16 Primarily, native bone tissues are composed of organic collagen proteins and inorganic hydroxyapatite (HAp), which necessitates organic–inorganic composites, making them common choices in bone tissue engineering.17 The inorganic fillers can be engineered in the forms of nano/microparticles, rods, whiskers, fibers, and sheets, reinforcing the hydrogels with improved mechanical properties and modulating the hydrogels with desirable tissue compatibility. Apart from bone regeneration, bioceramic filler containing hydrogels are also applied in promoting the regeneration of other tissues (e.g. skin, cartilage). Such filler can be designed to supply therapeutic metallic ions such as magnesium, iron, zinc, and copper, which play crucial roles in processes such as immunomodulation, cell migration, angiogenesis and neurogenesis, and so on.18,19 Inorganic fillers are diverse and rich in compositions (e.g. carbon nanomaterials,20 ceramics,21 metal oxides22) and forms (e.g. dense/porous, particulate/fibrous/sheet-like), and many of them exhibit magnetic, piezoelectric, conductive, photothermal properties that responsive to external stimulus. These diversities associated with inorganic fillers significantly broaden the functionality and smart responsiveness of composite hydrogels, providing a powerful toolbox to address the complexity of tissue/organ-specific repair. The ability to control therapeutic methods is particularly important when treating tissue defects in the presence of symbiotic abnormalities such as infections,23 inflammation,24 diabetes,25 and tumors.26 Multifunctional composite hydrogels offer viable solutions in such scenarios, wherein anti-bacteria, anti-inflammation, and tissue growth promotion effects can be integrated into a single material system by selecting proper combinations of inorganic and organic components, enabling the design for personalized medicine and precision therapeutics.
The formation of hydrogels typically involves physical interactions (e.g. hydrogen bonding,27 chain assembly28) and chemical bonds (e.g. covalent, ionic bonds29,30). Dynamic crosslinking introduces unique reversible connections; among them, host–guest interactions and click reactions are popular choices.31 In some cases, enzyme-responsive hydrogels can be prepared using peptides to crosslink polymeric chains, and the peptides are sensitive to enzymes (e.g. metalloprotease) located on cell membranes.32 All these crosslinking strategies can be applied to fabricate composite hydrogels containing inorganic fillers, while the difference lies in the dispersibility, size, morphology, and surface groups of the fillers, as well as their interactions with the polymer networks. Compared to simple mixing, if inorganic fillers can act as rigid crosslinkers in polymer networks, it will undoubtedly create more possibilities in constructing hydrogel systems with intelligent, programmable, and adaptive properties depending on the nature of the fillers and the polymers. This characteristic presents an excellent opportunity to expand the flexibility and customizability of composite hydrogels suitable for different applications, specifically, meeting the complex situations for diverse regenerative needs of injured tissues. Thus, it is interesting and necessary to know more about organic–inorganic composite hydrogels in the field of tissue regeneration, while the current reviews on hydrogel-based composite materials mainly emphasize the advantages of using hydrogels for cell encapsulation, biomanufacturing, and injectable materials.33,34 There are few systematic reviews summarizing the component selection and network construction strategy in fabricating organic–inorganic composite hydrogels.
In this review, we aim to provide an in-depth and cutting-edge summary of the organic–inorganic composite hydrogels for regenerative medicine. The listed outcomes are closely linked to the inorganic components and the characteristics of the resulting composite hydrogels, with particular emphasis on the dispersion of the inorganic fillers within the hydrogel matrix and their integration with polymer networks. Considering the significant disparity between organic and inorganic components, merely blending these elements without elucidating their interactions may lead to filler precipitation and aggregation, substantially compromising their effectiveness in tissue repair. Accordingly, this review will specifically highlight the functions of inorganic components and the methods applied to build the organic–inorganic composite systems via interfacial construction through physical interactions and chemical bonds (Scheme 1). In sections with corresponding subtitles, we particularly underscore the unique functionalities brought by incorporated inorganic fillers, itemized as mechanical reinforcement, supplier of therapeutic ions, delivery carriers for bioactive substances and responsive modules. These diverse functionalities empower composite hydrogels, endowing them with superior compatibility, enhanced biological activity, and intelligent therapeutic solutions, thereby substantially improving the efficiency in the treatment of wounds and defects. In the last part of this review, comprehensive perspectives are provided for future studies on organic–inorganic composite hydrogels targeting regenerative medicine by looking into the challenges associated with the material preparation and their shortcomings as advanced tissue-inducing biomaterials, providing a roadmap for future research endeavors in this promising field.
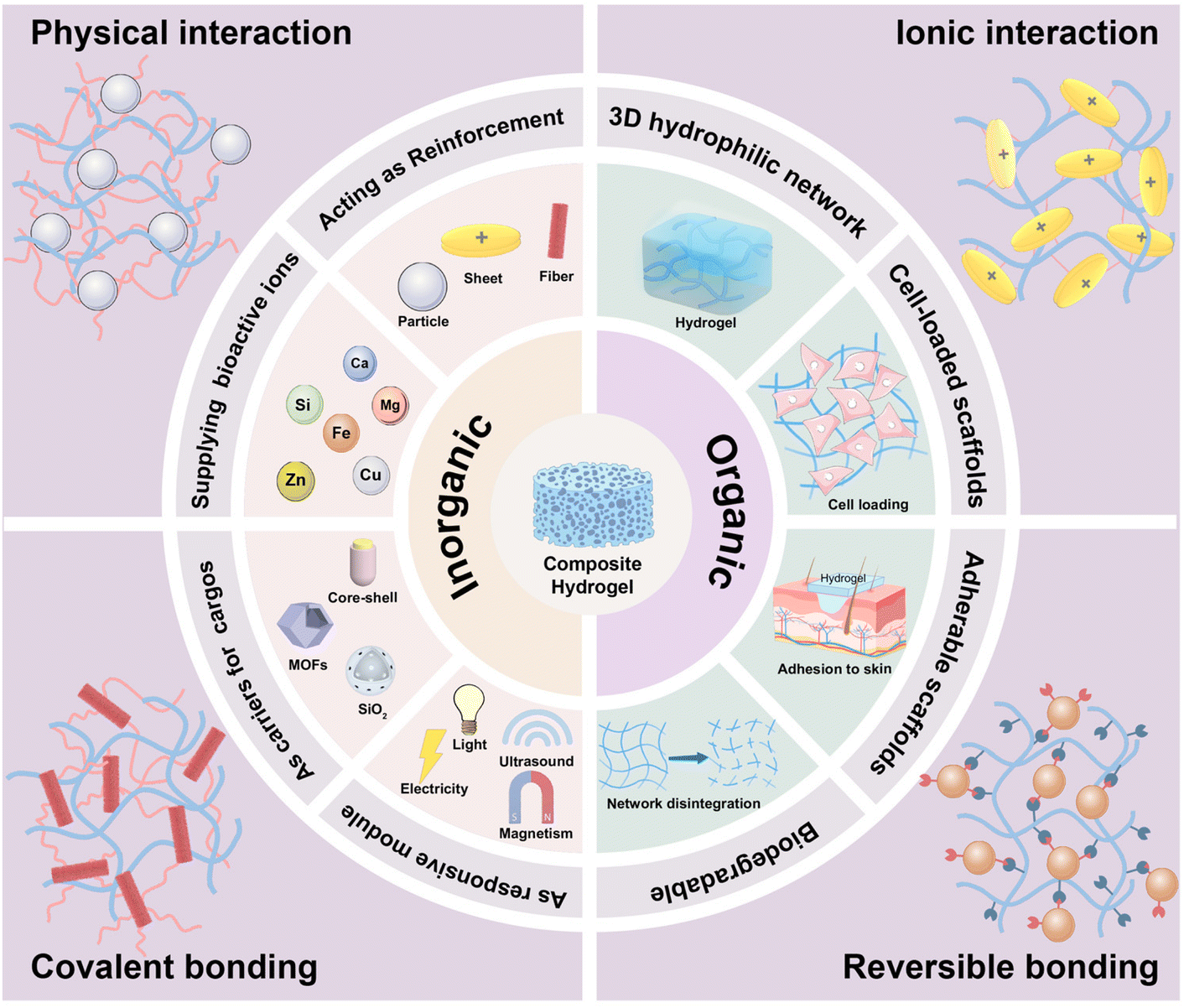 | ||
| Scheme 1 Schematic diagram showing the interaction and functionalization of two constituent components within organic–inorganic composite hydrogels that they exhibit a diverse range of interactions and modulations on hydrogel performances from respective aspects. | ||
2. Materials for constructing composite hydrogels
Hydrogels are polymers that can absorb a significant quantity of water while retaining a three-dimensional network.35 The gelation of precursor solutions can occur through various interactions, including covalent bonds, hydrogen bonds, electrostatic interactions, and ligand binding. These interactions distinguish themselves in strength and stability, allowing hydrogels to exhibit diverse properties and functionalities, even they may contain similar polymeric components.36 Using gelatin as an example, the hydrogel constructed through hydrogen bonding exhibits thermosensitivity, whereas the covalently crosslinked hydrogel created through photo-induced polymerization possesses a stable network. In organic–inorganic composite hydrogels, the inorganic fillers typically serve as a functional core, imparting controllable characteristics like photothermal conversion, magnetothermal conversion, excellent conductivity, and tumor-targeting specificity. One simple example is that cellulose was combined with black phosphorus (BP) nanosheets to prepare injectable nano-composite hydrogels, which exhibit photothermal properties and can efficiently kill tumors.37 In the following subsections, we will briefly introduce the main polymers and inorganic components usually used in fabricating organic–inorganic composite hydrogels.2.1 The organic part – polymers
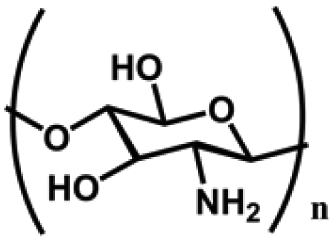
The fundamental properties of organic–inorganic composite hydrogels, such as swelling behavior, degradation, and mechanical performance, are tunable due to the diverse polymer compositions forming the hydrogel networks. These polymers can be broadly categorized into natural polymers and synthetic polymers.38 Natural polymers are derived from abundant natural sources, exhibiting excellent biocompatibility and biodegradability (Table 1). Common examples of polysaccharide-based hydrogels include chitosan, hyaluronic acid, and alginate. These hydrogels offer advantages such as high-water absorption and network tunability. Protein-based hydrogels, like gelatin, collagen, and silk fibroin (SF) hydrogels, possess biochemical properties resembling human tissues, containing specific amino acid sequences conducive to cell adhesion, proliferation, and differentiation. In regenerative medicine, these natural polymer-derived hydrogels are employed as scaffold materials to provide growth support for cells and facilitate tissue regeneration, widely used in wound healing, cartilage and bone repair. The main disadvantages of natural polymeric hydrogels are low mechanical properties, complex molecular chain structure, and risk of immunogenicity depending on sources. Synthetic polymers have well-defined chemical structures and molecular weights, and the corresponding hydrogels have controllable degradation behaviors and adjustable mechanical properties. Common examples include polyethylene glycol (PEG),39 polyacrylamide (PAM),40 and polyvinyl alcohol (PVA) hydrogels.41 However, these synthetic polymers lack biological activity, and some are non-degradable in vivo, which brings obstacles to broadening their applications in regenerative medicine. Both natural and synthetic polymers have unique characteristics in hydrogel preparation, studies to combine them together suggest possible solutions to tackle their individual limitations.42 In Table 1, a brief summary of these polymers is provided in terms of chemical structures, key features, and crosslinking methods to form hydrogels, as well as, their potential biomedical applications.| Polymer | Dominant units | Properties | Crosslinking | Applications | Ref. |
|---|---|---|---|---|---|
| Chitosan (CS) | Glucosamine and acetylamino units | Amino and hydroxyl functional groups; antibacterial, cell adhesion, good water absorption and water retention, degradable, non-toxic, | Chemical crosslinking: amide bonding, double bond crosslinking | Wound healing, cell carrier, drug delivery, bone repair, cartilage repair, etc. | 43–46 |
|
Physical crosslinking: ionic crosslinking, electrostatic interaction, and hydrophobic interaction | ||||
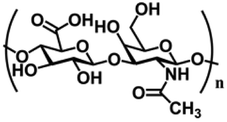
| Hyaluronic acid (HA) | N-Acetylglucosamine and D-gluconic acid units | Hydroxyl and carboxyl functional groups; the main component of ECM, regulate cell adhesion migration, proliferation, and differentiation; good water absorption and water retention, degradable, non-toxic | Chemical crosslinking: Schiff base, borate ester bond, mercaptan, double bond crosslinking | Soft tissue engineering, wound and burn repair, drug delivery, etc. | 47–49 |
|
Physical crosslinking: hydrogen bonding, ionic crosslinking, electrostatic and host–guest interactions | ||||
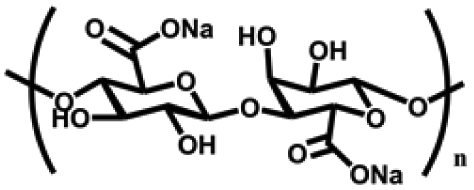
| Sodium alginate (SA) | D-Glucuronic acid and L-glucuronic acid units | Hydroxyl and carboxyl functional groups; rapid gelation with divalent cations; good water absorption and water retention, non-toxic, degradable, reactive handles for functionalization | Chemical crosslinking: Schiff base, double bond crosslinking | Drug delivery, cancer therapy, wound healing, bone repair, cartilage repair, etc. | 50–52 |
|
Physical crosslinking: hydrogen bonding, ionic crosslinking, host–guest interactions | ||||
| Heparin | Monosaccharide units such as glucosamine and gluconic acid | Aldehyde sulfate group, carboxyl group and sulfate group; good water absorption, and water retention, degradable, non-toxic, inhibit platelet adhesion and aggregation both in vivo and in vitro, promote cell proliferation and tissue | Chemical crosslinking: amide bonding, disulfide bond, double bond crosslinking | Anti-inflammation, anti-coagulation, drug delivery, wound healing, cartilage repair, etc. | 53 and 54 |
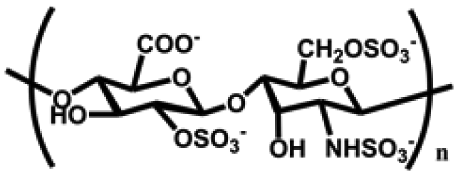
|
Physical crosslinking: hydrogen bonding, electrostatic interaction | ||||
| Collagen/Gelatin | Glycine, proline and hydroxyproline, etc. | Amino and carboxyl functional groups; biocompatible, good water absorption and water retention, degradable, non-toxic | Chemical crosslinking: amide bonding, double bond crosslinking | Cell carrier, bone repair, cartilage repair, drug delivery, wound healing, revascularization, etc. | 55–58 |
|
Physical crosslinking: Hydrogen bonding, electrostatic interaction, and hydrophobic interaction | ||||
| Chondroitin sulfate | N-Acetylgalactosamine and glucuronic acid units | Sulfate group; anti-inflammatory; bioactive and biocompatible, good water absorption and water retention, degradable, non-toxic; binds growth factors and cytokines | Chemical crosslinking: amide bonding, double bond crosslinking | Cartilage repair, anti-inflammation, wound healing, drug delivery, etc. | 59–61 |
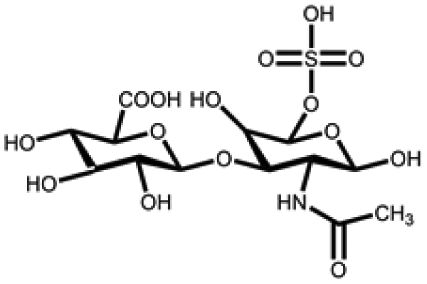
|
|||||
| Silk fibroin (SF) | Glycine, alanine and serine, etc. | Amino and carboxyl functional groups; high mechanical strength and elasticity; cell adhesive; low immunogenicity; good water absorption and water retention, degradable, non-toxic | Chemical crosslinking: amide bonding, double bond crosslinking | Cell carrier, bone repair, cartilage repair, drug delivery, revascularization, wound healing, etc. | 62–64 |
| Gly-Ala-Gly-Ala-Gly-Sern | Physical crosslinking: hydrogen bonding, electrostatic interaction, and hydrophobic interaction | ||||
| Polyethylene glycol (PEG) | –CH2CH2O– | Linear polymer; hydroxyl functional group; polyhydroxy functional groups; anti-protein adsorption and adhesion, good water absorption and water retention, degradable, non-toxic | Chemical crosslinking: ureyl functional groups, double bond crosslinking | Cell carrier, drug delivery, wound healing, cartilage repair, etc. | 39, 65 and 66 |
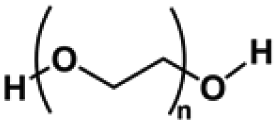
|
Physical crosslinking: hydrogen bonding, electrostatic interaction, and hydrophobic interaction | ||||
| Polyacrylamide (PAM) | –CH2CHCONH2– | Acrylamide functional group; good water absorption and water retention, degradable, non-toxic | Chemical crosslinking: double bond crosslinking | Cell carrier, drug delivery, wound healing, cartilage repair, etc. | 67 and 68 |
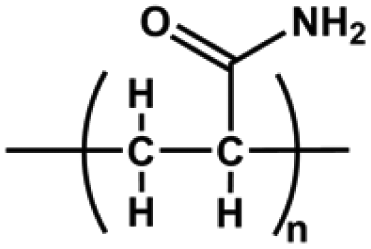
|
Physical crosslinking: hydrogen bonding, electrostatic interaction, and hydrophobic interaction | ||||
| Polyacrylic acid (PAA) | –C3H4O2– | Carboxyl function group; good water absorption, ionic crosslinking, degradable, non-toxic | Chemical crosslinking: double bond crosslinking, Esterification reaction | Drug delivery, wound healing, cell culture, etc. | 69–72 |
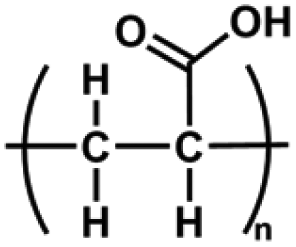
|
Physical crosslinking: Hydrogen bonding, ionic interaction | ||||
| Polyvinyl alcohol (PVA) | –C2H4O– | Hydroxyl functional group; excellent biocompatibility, mechanical properties good water absorption and water retention, degradable, non-toxic | Chemical crosslinking: ether linkage | Cell carrier, drug delivery, wound healing, cartilage repair revascularization, etc. | 41, 73 and 74 |
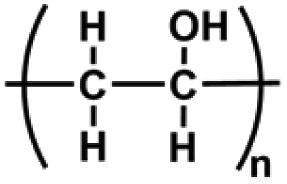
|
Physical crosslinking: hydrogen bonding, electrostatic interaction, and hydrophobic interaction |
Three-dimensional networks are formed through the interactions of functional groups on polymer chains via physical forces and chemical bonds. Different preparation strategies lead to diversity in physicochemical properties, swelling behaviors, and degradation performance of the resulting hydrogels. Selecting appropriate polymer matrices and corresponding crosslinking methods are crucial for the construction of highly cell and tissue-compatible hydrogels. The physical interactions in hydrogel formation are typically polymer chain entanglement, ion interactions, metallic coordination, hydrogen bonding, hydrophobic interactions, and crystallization. Among them, ionic interactions occur under relatively mild conditions for polymers containing cationic/anionic units. Alginate hydrogels are commonly prepared by mixing sodium alginate (SA) solution with CaCl2 solution via the ionic interaction between carboxyl and Ca2+.75 PVA solution can form elastic gel due to crystallization after several circles of freeze–thaw process. Hydrogels formed through physical crosslinking often possess reversibility.76 These methods eliminate the need for toxic small-molecule crosslinking, offering the advantages of good biocompatibility and degradability. Due to the relatively weak forces associated with physical interactions crosslinking, nevertheless, these hydrogels may exhibit insufficient mechanical strength and stability for tissue repair.
Chemical crosslinking refers to the formation of covalent bonds between polymer chains. This process may involve polymer modification to introduce crosslink units, addition of reactive molecules crosslinking, or applying high-energy radiation to initiate addition and condensation reactions, resulting in the formation of a robust polymeric network. Covalently crosslinked hydrogels have higher mechanical strengths and network stability than those physically crosslinked ones, with slower hydrolysis rates if no enzymatic degradation is presented. The introduction of small molecule crosslinking agents, such as glutaraldehyde, genipin, dopamine, and tannic acid, into hydrogels enables the concurrent modulation of the hydrogel's mechanical characteristics. Nevertheless, it is important to acknowledge that chemical crosslinking agents, such as glutaraldehyde, can exhibit cytotoxic effects on cells and tissues, hence restricting their usage within specific concentration ranges.77 To exclude the concern of using toxic small molecular reagents, crosslinking via thermal-, catalytic-, and photo-polymerization are regarded as mild reactions to conduct the crosslinking. Photopolymerization refers to the formation of hydrogels through covalent bond crosslinking between chains under specific light sources with the aid of photo initiators, such as using acrylated gelatin, alginate, and HA, showing less crosslinking biocompatibility problems.
It is not an easy work to directly blend inorganic fillers into these hydrogel systems, because their densely arranged polymeric networks hinder the homogeneous dispersion within the organic matrix. Aggregation and settling may occur, particularly, for nano-scaled inorganic fillers due to their high surface areas, thus compromising the performance of the composite hydrogels for use. This necessitates those studies on proper inorganic filler selection, surface modification, and strengthening the interfacial interactions between the polymers and the inorganic components.
2.2 The inorganic part – rigid fillers
The introduction of inorganic fillers into polymeric hydrogels brings new chances to expand material performance, with a higher potential to meet the requirements of tissue repair in diversely complex situations. The sources of inorganic components are extremely richer than hydrophilic polymers. They can be briefly categorized into metal-based powders, compounds or oxides, and inorganic nonmetal materials, and the latter typically contains ceramics and carbon nanomaterials. Different material types combined with unique characteristics have generated composite hydrogels possessing distinguished natures in microstructure, rheological/mechanical properties, bioactivity, and inherent responses to various factors. These inorganic fillers are also flexible in morphology depending on synthesis techniques; in short, they can be presented in particles, rods, whiskers, fibers, and sheet-like, imparting composite hydrogels with desirable strengths and thermal/electrical/optical properties. Apart from only acting as reinforcement, the functions of inorganic components in composite hydrogels can be extended to drug delivery carriers, therapeutic ion suppliers, and smart modules responsive to environmental or external stimuli. In Table 2, inorganic fillers with different compositions and morphology are summarized from reports concerning composite hydrogels for biomedical applications.| Materials | Physical parameters | Dispersion/Crosslinking | Functions | Applications | Ref. |
|---|---|---|---|---|---|
| Hydroxyapatite (HAp) | Nanoparticles (0D) | Blending, co-precipitation, electro-deposition method | Mechanical reinforcement, osteogenic differentiation | Bone repair, cartilage repair, etc. | 69, 78 and 79 |
| Nanowires, nanorods (1D) | Blending, surface deposition, mineralization, covalent bonding | Mechanical reinforcement, bone regeneration, osteochondral repair | Bone repair, cartilage repair, drug delivery, etc. | 16, 80 and 81 | |
| β-Tricalcium phosphate (β-TCP) | Nanoparticles (0D) | Blending | Osteogenic differentiation, anti-tumor | Bone repair, cartilage repair, cancer therapy, etc. | 82 and 83 |
| Montmorillonite | Nanosheets (2D) | Blending, hydrogen bonding, electrostatic interaction | Mechanical reinforcement, cell recruitment, drug carrier | Drug delivery, bone repair, wound healing, etc. | 27 and 84 |
| LAPONITE® | Nanosheets (2D) | Blending | Mechanical reinforcement, promoting cell adhesion and proliferation, vascular regeneration | Drug delivery, bone repair, revascularization, wound healing, etc. | 85 and 86 |
| Carbon materials | Carbon nanospheres, carbon dots (0D) | Blending | Mechanical reinforcement, lubrication, osteogenic differentiation | Bone repair, cartilage repair, drug delivery, etc. | 87 and 88 |
| Carbon nanotubes (1D) | Blending, covalent bonding | Conductive, mechanical reinforcement, drug carrier, tumor therapy | Drug delivery, bone repair, cancer therapy, etc. | 89 and 90 | |
| Graphene oxide (2D) | Blending | Conductive, antibacterial, mechanical reinforcement, crosslinker, | Bone repair, revascularization, wound healing, etc. | 91–95 | |
| Silicon dioxide (SiO2) | Nanoparticles (0D) | Blending, hydrophobic association, covalent bonding, | Surface functionalization, mechanical reinforcement, drug carrier, anti-tumor, cartilage regeneration | Bone repair, cartilage repair, drug delivery, etc. | 96–99 |
| Nanowires, nanorods (1D) | Blending, silane coupling | Mechanical reinforcement, conductive | Bone repair, drug delivery, neural restoration, etc. | 100 | |
| Silicon (Si) | Nanoparticles (0D) | Blending, covalent bonding | Mechanical reinforcement, crosslinker | Bone repair, drug delivery, etc. | 101 and 102 |
| Silver (Ag) | Nanoparticles (0D) | Blending, hydrogen bonding | Mechanical reinforcement, conductive, antibacterial, pressure-responsive, antibacterial | Wound healing, drug delivery, bone repair, etc. | 70, 103–106 |
| Nanowires, nanorods (1D) | Blending | Conductive, electro-responsive, skin repair, sensors | Wound healing, drug delivery, etc. | 91, 107 and 108 | |
| Gold (Au) | Nanoparticles (0D) | Blending, Michael addition reaction | Photothermal effect, anti-tumor, crosslinker | Cancer therapy, etc. | 109 and 110 |
| Nanowires, nanorods (1D) | Blending | Mechanical reinforcement, photothermal response, anti-tumor | Cancer therapy, etc. | 26, 111 and 112 | |
| Magnesium oxide (MgO) | Nano-particles (0D) | Blending, electrostatic interaction, metal–ligand supramolecular binding | Release of Mg2+, mechanical reinforcement, crosslinker, antibacterial properties, skin repair, osteogenic differentiation, vascular regeneration | Bone repair, revascularization, wound healing, etc. | 22, 113 and 114 |
| Iron oxide (Fe2O3/Fe3O4) | Nanoparticles (0D) | Blending | Cartilage regeneration, magnetic response, photothermal response | Cartilage repair, drug delivery, tissue engineering, etc. | 115 and 116 |
| Nanorods (1D) | Blending, covalent bonding | Mechanical reinforcement, crosslinker, conductivity, magnetic response. | Cartilage repair, drug delivery, magnetothermal therapy, etc. | 117 and 118 | |
| Metal–organic frameworks (MOFs) | Nanoparticles (0D) | Blending, ion coordination binding | Drug loading, photothermal response, nerve regeneration, antibacterial, anti-inflammatory, vascular regeneration, anti-tumor | Drug delivery, anti-infection, revascularization, cancer therapy, etc. | 119–122 |
| Molybdenum disulfide (MoS2) | Nanosheets (2D) | Blending, host–guest connection | Mechanical reinforcement, drug delivery, antibacterial | Biosensor, drug delivery, photothermal therapy, etc. | 123 and 124 |
| Black phosphorus (BP) | Nanosheets (2D) | Blending | Ion and drug delivery, photothermal response, antibacterial, osteogenic differentiation, vascular regeneration, nerve regeneration | Drug delivery, photothermal therapy, bone repair, wound healing, etc. | 120, 125–128 |
In composite fabrication, inorganic components are popularly applied reinforcement to enhance the mechanical properties of materials, which is also suitable for the preparation of composite hydrogels. Compared to polymer networks, inorganic fillers inherently have higher stiffness, enabling them to withstand greater external forces upon compression or stretching. Of note, their reinforcement effect depends significantly on how they interact with polymer chains, that chain entanglement, physical interactions, and chemical bonds are all possible for the two components to make the composite systems tougher.132 When subjected to external forces and deformation, the inorganic phase may help to disperse stress concentration and preserve the structural integrity of the primary polymeric network. Specifically, nano-scaled fillers can inhibit the generation and propagation of cracks to avoid material damage due to their stronger interactions with polymer chains for their high specific surface area.133 It was found that nanoclay can remarkably enhance tensile modulus and elongation at the break of nanocomposite hydrogels at a relatively low addition fraction. The incorporation of nanoclay at 0.03 wt% into polyacrylic acid (PAA) hydrogel could make the composite hydrogel exhibit an elongation exceeding 1000%, and have the ability to endure 90% compressive deformation.71 Lu et al.100 incorporated silica nanofibers and vinyltriethoxysilane into SA/PAM hydrogels, and the vinyltriethoxysilane binds the nanofibers and PAM chains together via its coupling agent nature (Fig. 1a). This composite hydrogel exhibited high elastic modulus (0.11 MPa), high toughness (2.98 MJ m−2), and high elongation at break (close to 2000%). A stronger interfacial bonding between the inorganic fillers and the polymer chains, like covalent bond vs. physical interaction, will lead to more effective reinforcement since the integrity of the organic–inorganic hybrid network was enhanced.
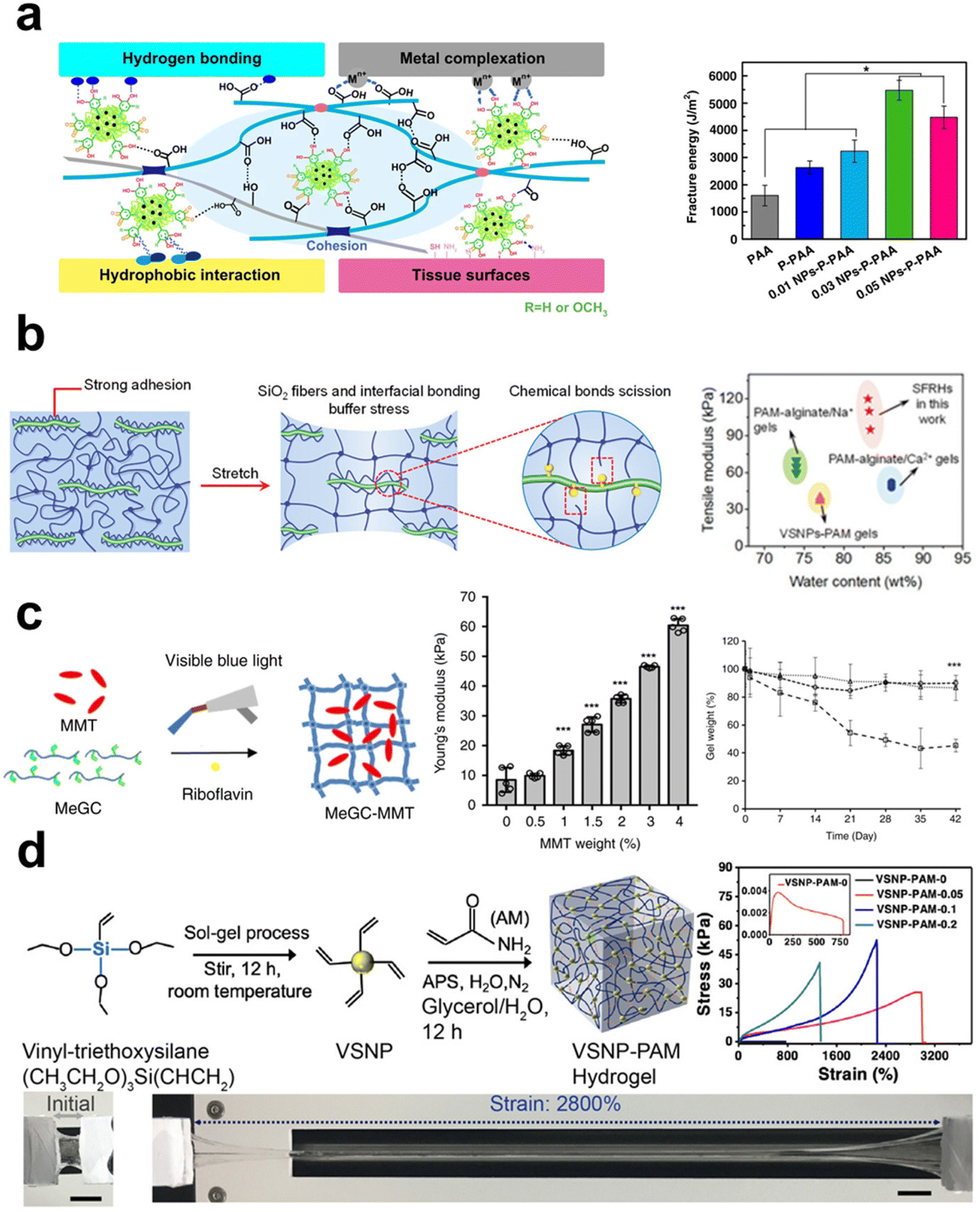 | ||
| Fig. 1 The schematic showing the manner in which inorganic components contribute to the improvement of mechanical characteristics in hydrogels. (a) The introduction of both rigid and flexible silica nanofibers, which are covalently attached to the polymer chains of the PAM hydrogel, leads to the formation of robust interfacial chemical bonds, thereby greatly enhancing the mechanical properties of the hydrogel.100 Copyright 2021, Wiley-VCH. (b) The incorporation of NPs containing catechol groups into PAA hydrogel results in improved tensile properties due to the formation of hydrogen bonding interactions.70 Copyright 2019, Springer Nature. (c) The dispersion of two-dimensional nanosheets within the covalently crosslinked network of the hydrogel leads to an increase in its Young's modulus and a decrease in its degradation rate.84 Copyright 2019, Springer Nature. (d) The simultaneous addition of nanowires and nanosheets to PAM hydrogel significantly enhances its mechanical properties.135 Copyright 2020, Springer Nature. | ||
Inorganic materials used for mechanical enhancement are normally in nano-scale, primarily classified into 0D nanoparticles (NPs), 1D nanomaterials (nanorods, whiskers, nanowires, nanofibers), and 2D nanomaterials (nanosheets). Among them, the types of NPs are the richest, including metal, metal oxide, and ceramic NPs. These NPs can interact with polymers using the cationic ions on their surfaces through ligand bonds, whose dynamic nature imparts the network with high energy dissipation and reversible self-healing properties. Surface modification of inorganic fillers is quite helpful to enhance their interactions with polymer chains, thereby providing stronger reinforcement effect properties. Lu et al.70 incorporated Ag-lignin NPs to create a dynamic pyrogallol redox system, resulting in enhanced mechanical characteristics and long-term adherence of the hydrogel (Fig. 1b). The authors found that the presence of non-covalent interactions among silver-lignin NPs, PAA, and pectin resulted in a substantial dissipation of energy. The fracture energy had a peak value of 5500 J m−2, significantly exceeding the fracture energy of human skin (∼2000 J m−2).
Common 1D inorganic fillers include silicon dioxide (SiO2) nanowires/fibers, boron nitride (BN) nanofibers, carbon nanotubes (CNTs), and metal nanorods/wires, among others. Compared to NPs, 1D nanomaterials have a higher aspect ratio and network-forming ability, effectively preventing gravitational settling. When the aspect ratio of SiO2 nanofibers was increased from 50 to 400, studies have shown that the mechanical strength of composite hydrogels significantly improved from 0.11 MPa to 0.24 MPa.100 Chemical bonding and hydrogen bonding interactions are formed between the SiO2 fibers and PAM chains. This allows for fracture and recombination during stretching and dissipating energy to enhance the hydrogel network's strength. Molecular simulation results revealed that the interaction energy between them increased from 1135 kJ mol−1 to 2241 kJ mol−1 alongside the increasing L/D ratios of the nanofibers, indicating the positive effect of fillers’ aspect ratios on the bonding at organic–inorganic interfaces. Another specific feature of 1D fillers is their ability to form aligned structures, whereas, highly ordered nanostructures are characteristic in natural tissues to provide mechanical supports. Orientated 1D fillers will endow composite hydrogels with even higher mechanical performance.134 Ahadian et al.89 employed dielectrophoresis (DEP) as an effective tool to align conductive CNTs within gelatin methacrylate (GelMA) hydrogels. Compared to hydrogels containing randomly distributed CNTs, those hydrogels with aligned CNTs exhibited increases in Young's modulus from 20.3 ± 1.4 kPa to 50.4 ± 3.8 kPa.
2D materials such as graphene, montmorillonite nanosheets, and silicate nanosheets have recently been highlighted as effective reinforcement for composite hydrogels due to their large specific surface area, rich polar/ionic groups at sheet edge or on the surface, high specific strength by themselves, thus may lead to even stronger interactions with polymeric matrix as compared to 1D nanofillers. Cui et al.84 incorporated layered nanoclay into photo-crosslinked chitosan solution, increasing the Young's modulus of the hydrogel from the original 10 kPa to 30 kPa (Fig. 1c), that the ionic/charge interactions between the two components contributed to this amelioration. Graphene oxide (GO), having reactive functional groups like hydroxyl and carboxyl groups, renders them the capacity to be utilized as crosslinkers for hydrogel formation via both physical and chemical processes. Liu et al.93 modified GO with acrylate groups (mGOa), creating covalently crosslinked hydrogels with poly(2-hydroxyethyl methacrylate-co-acrylamide). Its compressive strength reached 2.3 ± 0.8 MPa, as compared to the 1.4 ± 0.4 MPa of the hydrogel crosslinked with N,N′-methylenebisacrylamide. At a fraction of 2 mg mL−1 mGOa and compressive strain of 90%, the composite hydrogel showed compressive strength as high as 14.1 ± 2.1 MPa. These reports have primarily established the base for using 2D nanomaterials as reinforcements to improve the mechanical behaviors of soft hydrogels.
The diversities in the compositions and morphology of inorganic materials show both advantages and downsides for hydrogel reinforcement. NPs are characterized by a lack of anisotropy and no pulling-out effect, which makes them have a ceiling effect in achieving high mechanical performance. Nanofibers and nanosheets possess anisotropic characteristics, interacting with polymer chains at improved bonding strength, resulting in robust networks and higher strength. The main challenge in preparing composite hydrogels using these inorganic fillers is the difficulty in dispersing them uniformly within the organic matrix, which obviously will compromise their reinforcements. Cai et al.135 noticed this issue and attempted to solve it with an innovative strategy. They developed a multifunctional conductive hydrogel with multidimensional components by simultaneously adding polypyrrole nanowires (PpyNWs, 1D), MXene (2D), and vinyl-hybrid-silica NPs (VSNP, 0D) into PAM hydrogel to form heterostructures. The co-incorporation of multidimensionality of the three inorganic components in different morphology facilitates the integration of intermolecular forces and multilayer structures, thereby mitigating the interfacial repulsion between each other. As a result, these multidimensional configurations made the composite hydrogel exhibit an impressive tensile strain up to 2800% (Fig. 1d). This study provides an alternative approach to tackle the challenges in filler dispersion and maximizing their contributions to improve the mechanical performance of composite hydrogels.
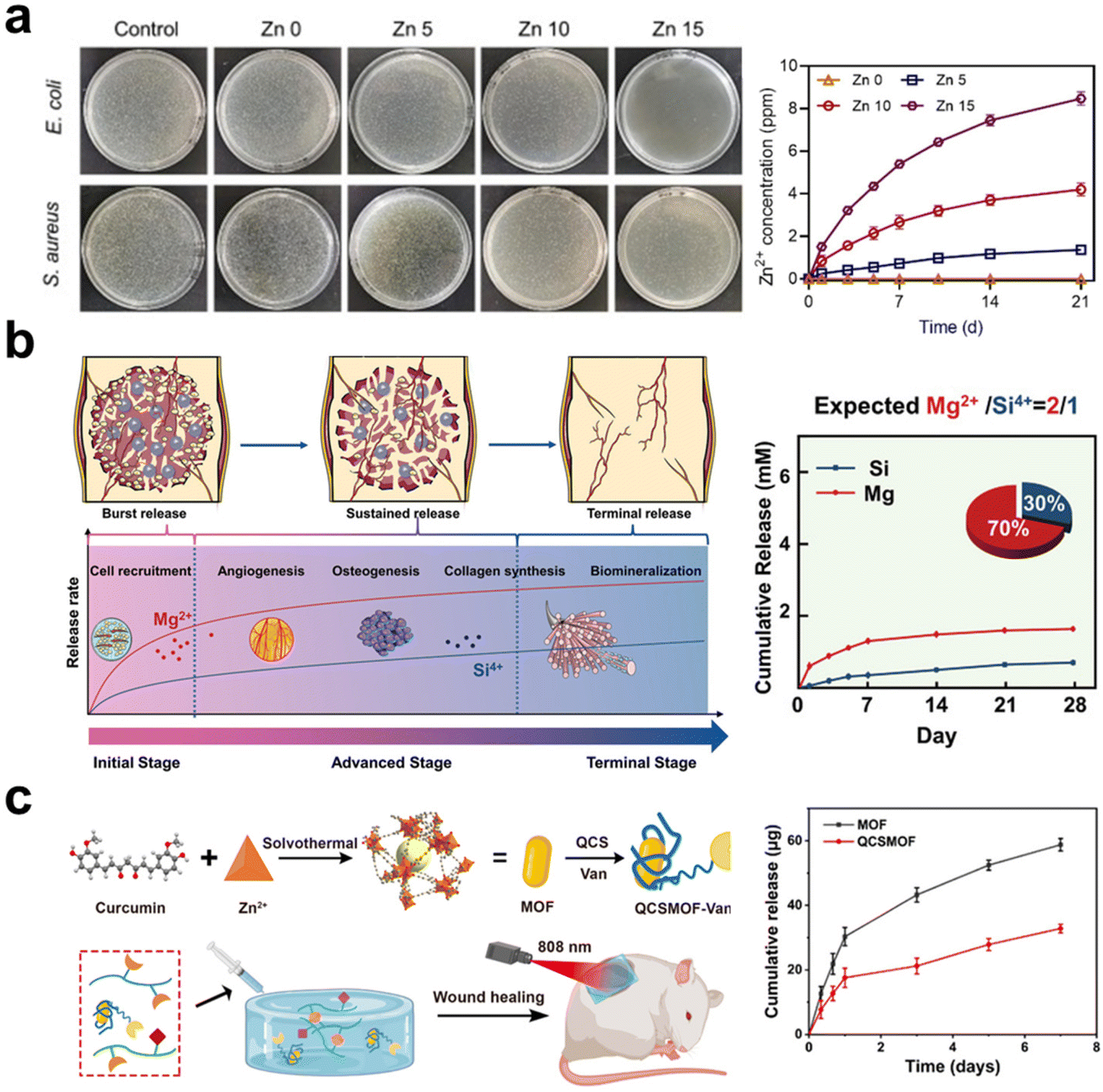 | ||
Fig. 2 The schematic illustrates the inorganic components in the hydrogel releasing and providing therapeutic ions. (a) Hydrogels containing Zn-doped HAp nanorods can continuously release Zn2+ ions, exhibiting excellent antibacterial properties.69 Copyright 2022, Elsevier Ltd. (b) Stably releasing Mg2+ and Si4+ ions in a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, it demonstrates the most significant synergistic effect on vascularization and osteogenesis.18 Copyright 2022, Wiley-VCH. (c) The hydrogel coating on MOF can reduce the release rate of Zn2+ ions from MOF and leverage the photothermal effect of MOF for the gradual healing of chronic wounds.119 Copyright 2022, American Chemical Society. :1, it demonstrates the most significant synergistic effect on vascularization and osteogenesis.18 Copyright 2022, Wiley-VCH. (c) The hydrogel coating on MOF can reduce the release rate of Zn2+ ions from MOF and leverage the photothermal effect of MOF for the gradual healing of chronic wounds.119 Copyright 2022, American Chemical Society. | ||
The organic–inorganic hybrid structure of MOFs provides an ideal platform by coordinating metallic ions (or metallic ion clusters) and organic ligands together.139 In contrast to the straightforward mixing of metallic ions or metal oxide NPs into hydrogels, MOFs offer the potential to simultaneously form physical or chemical connections between organic and inorganic components. This unique characteristic increases the flexibility of controlling ion release. In addition, MOFs possess highly ordered and porous structures, being likely as delivery carriers for drugs and siRNA to bring extra biofunctions.121,140 Huang et al.119 synthesized a vancomycin (Van)-loaded MOF with Zn2+, which was coated with quaternary ammonium chitosan (QCS) and dispersed into hydrogel made of oxidized SA methacrylate (OSAMA) and GelMA (Fig. 2c). This composite hydrogel is targeting chronic would dressing, because the QCS provides positive charges to capture bacteria, while the released vancomycin and Zn2+ rapidly kill the captured bacteria.
Bioceramic components are normally capable of releasing multiple ions, for instance, HAp and β-TCP releasing Ca2+ and phosphate, bioactive glass (BG) releasing Ca2+ and silicate, montmorillonite (MMT) and nanoclay releasing Mg2+, Li+, Ca2+ and phosphate or silicate based on different compositions. These inorganic fillers provide wide choices for designing composite hydrogels in modulating biological responses and promoting tissue regeneration. BG finds wide applications in the regeneration of both soft and hard tissues due to its immunomodulatory, angiogenesis-promoting properties and biomineralization inductivity. Xu et al.141 mixed BG into SA solution to prepare an injectable hydrogel, whose immunomodulatory and angiogenic properties help enhance tendon healing via accelerating blood vessel formation and upregulating the M2/M1 phenotype ratio. MMT is also a kind of silicate-type mineral with a high surface area and aspect ratio; elements such as Ca, Mg, Al, and Na are located within the layered silicate structure. Cui et al.84 incorporated MMT into photo-crosslinkable methacrylated glycol chitosan (MeGC), developing an in situ forming composite hydrogel conducive to osteogenic differentiation of encapsulated mesenchymal stem cells (MSCs) in vitro and calvarial healing in vivo via recruiting native cells. In summary, inorganic materials as therapeutic ion donors, have a high chance to fulfilling the needs of regenerative medicine, and a combination of multiple ions is expected to augment tissue regeneration without the delivery of additional therapeutic agents or stem cells.
In the fabrication of organic–inorganic composite hydrogels, those inorganic fillers can serve as potential drug carriers. Jen et al.142 utilized thiolated oligonucleotides to modify gold NPs, and successfully used the NPs as non-viral DNA carriers, enhancing DNA stability and increasing cellular transfection efficiency. Nakamura et al.143 loaded plasmid DNA into functionalized fullerenes to deliver DNA into cells. Lin et al.144 employed biomimetic mesoporous silica to load vancomycin, and Kim et al.145 applied mesoporous carbon NPs as transmembrane delivery vehicles for cancer treatment. And all these approaches can be combined with composite hydrogels to regulate tissue repair by delivering proper drugs and bioactive agents.146 Compared to directly using inorganic NPs as drug carriers, incorporating drug-loaded NPs into hydrogels offers significant advantages. This method can effectively regulate the drug release rate and prolong the drug release time, thereby improving the drug's bioavailability and therapeutic effects.
In addition, inorganic–organic composite hydrogels are extensively employed in transdermal controlled drug release systems for medications that pose challenges for oral delivery. Chengnan et al.147 successfully mixed GO with metformin hydrochloride, forming a photothermal-activated drug-release hydrogel. Under light stimulation, the graphene substrate in the hydrogel exhibited excellent photothermal properties, leading to drug release. In in vivo experiments, researchers were able to detect the presence of metformin in mouse plasma after 1 h of light activation, demonstrating the effectiveness of this hydrogel as a transdermal controlled drug release system.
To better preserve drugs, An et al.148 employed gold-coated drug NPs, preparing gold-clustered NP (gNP). The gNP exhibited the ability to establish a covalent network structure with dopamine-functionalized HA molecules, hence providing efficient protection for the encapsulated medicines. Drug release was initiated via the photo-thermal action in gold clusters on gNP. In brief, the incorporation of inorganic substances into hydrogel matrices has been found to significantly augment the control of drug release and boost the biocompatibility of the material. This study offers novel perspectives on the development of drug delivery systems and introduces potential possibilities for future research and therapeutic applications within the realm of tissue engineering.
For biomedical applications, light-responsive inorganic materials, including metal and metal oxides, carbon-based nanomaterials, and BP, are common fillers for nanocomposite hydrogels. They are the base for conducting photothermal therapy (PTT), photodynamic therapy (PDT), and targeted drug delivery. Gold NPs have excellent tunability and strong near-infrared (NIR) absorption, Su et al.149 dispersed Ag3AuS2 NPs into CS hydrogel to obtain a composite hydrogel with an effective tumor-killing effect through a one-time PTT treatment. Graphene and GO are also light-responsive materials, possessing excellent photothermal properties, which can be combined with hydrogel preparation to overcome tumor hypoxia by promoting the production of singlet oxygen.92,150 Chen et al.78 prepared a hydrogel with both PPT and PDT functionalities by incorporating amido-modified carbon dots (NCDs). This hydrogel exhibited high photothermal conversion efficiency (77.6%) and singlet oxygen quantum yield (0.37) under 660 nm LED irradiation, benefiting from those amino groups located on the NCDs surface.
Magnetic-responsive inorganic agents are a material that can respond to external magnetic fields, generating local magnetic fields, magnetic heating, and force effects, which have been widely used in disease diagnoses and treatments. Common magnetic-responsive materials include iron oxide, transition metal ferrites, and transition metal alloys. Antman-Passig et al.151 incorporated magnetic NPs into collagen hydrogel. Utilizing an external magnetic field, these magnetic particles aggregated into chains, aligning collagen fibers orderly and directing neuronal regeneration.
Ultrasound is another non-invasive stimulus often used for drug release, tissue regeneration, and imaging technologies.152 An et al.148 designed ultrasound-responsive composite hydrogels containing ultrasound-thermal active gold clusters and HA. Triggered by ultrasound stimulation at 30 W, these gold clusters emitted heat to cause a temperature increase (∼54 °C), inducing apoptosis in adjacent cancer cells. Subsequently, the gold clusters dissociated under heating-up to achieve on-demand drug release.
Chemical stimuli can be conducted by changing medium pH and compositions to modulate the functions of composite hydrogels. Zeng et al.120 reported a MOF-based hydrogel with pH- and ATP-responsive drug release characteristics for cancer therapy. The drug doxorubicin (DOX) was encapsulated in MOF made of Zn2+ and imidazole-2-carboxaldehyde, then this drug-loaded MOF was mixed with bisphosphonate-modified HA (HA-BP). Hydrogel was formed via the dynamic coordination bonds between bisphosphonate groups and Zn2+, and the system presented self-healing, shear-thinning, and localized drug delivery.
All these external stimuli provide extra modulation tools in widening the designs and applications of composite hydrogels. As the core to endow the systems with stimulation-responsive potentials, on the one hand, the choices of inorganic materials are essential to bringing biofunctions that match the diverse needs of different disease treatments and tissue regeneration; on the other hand, the interactions between the organic and inorganic components are important to guarantee therapy effectivity and efficiency of the composite hydrogels.
2.3 Interactions between polymers and inorganic fillers
As illustrated in Scheme 1, the four major interactions that exist between polymers and inorganic fillers in fabricating composite hydrogels are physical interaction, ionic interaction, covalent bonding, and reversible crosslinking. These interactions are among the bases to determine the microstructures, mechanical/biological performance, and targeted application fields of the composite hydrogels. Herein, we will look into these four major interactions in separated subitems.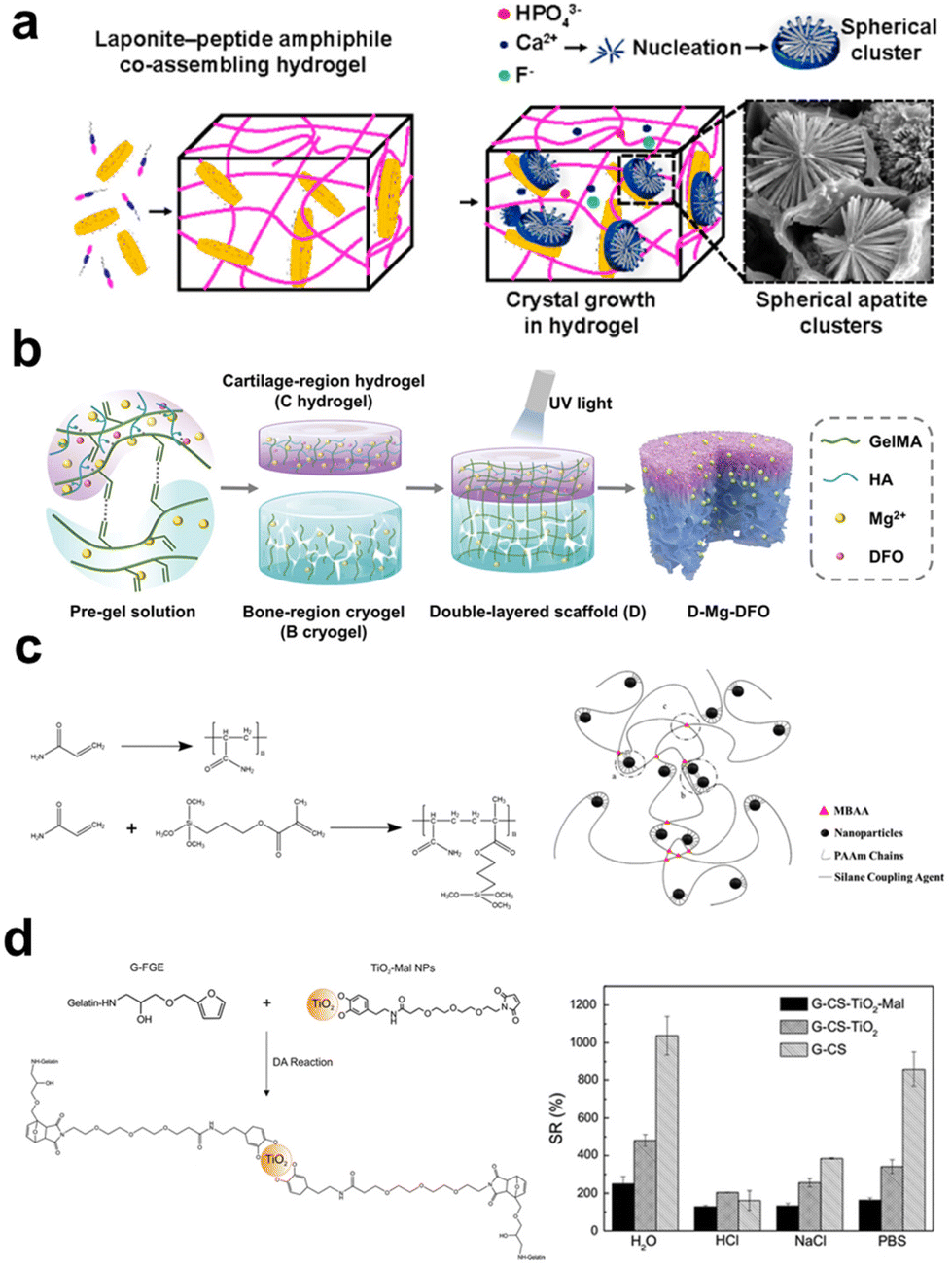 | ||
| Fig. 3 The schematic illustrates the interaction between organic and inorganic components in hydrogel. (a) Supramolecular coassembly of exfoliated Lap nanodisks and PA to create 3D hydrogels able to guide nucleation and hierarchical growth of HAp crystals.154 Copyright 2021, American Chemical Society. (b) The fabrication of a double-layer scaffold with a Mg2+ ion gradient involves the interaction between the carboxyl groups in the hydrogel matrix and Mg2+ ions, enhancing the stability of the hydrogel.157 Copyright 2023, Wiley-VCH. (c) The introduction of magnetically modified NPs, modified with silane coupling agents, into the hydrogel precursor results in the formation of a polymer network through the copolymerization of the silane coupling agent with the hydrogel matrix.160 Copyright 2019, American Chemical Society. (d) Surface-functionalized TiO2 can serve as a crosslinking agent for hydrogels through Diels–Alder cycloaddition reactions, enhancing the swelling performance of the hydrogel.164 Copyright 2016, Springer Nature. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), disulfide bonds (–S–S–), borate bonds (–O–B–O–), and reversible Diels–Alder reactions (–CC–). These bonds undergo repeatedly reverse reactions when countered with specific conditions like hydrolysis, external force, or shearing. The reaction between aldehyde and amino groups leads to the formation of Schiff base bonds, which can undergo hydrolysis to restore their original groups and react again to form the bonds. Yang et al.161 mixed CMC with oxidized dextran NPs (ODex NPs) to prepare hydrogels crosslinked with the dynamic Schiff base bonds. Under cyclic loading/shearing conditions, the CMC-ODex hydrogels exhibit gel–sol transitions repeatedly. Disulfide bonds are formed between two thiol groups (–SH). Yao et al.162 prepared thiolated chitosan (TCH) and thiol-functionalized mesoporous BG NPs (TBG). These two components formed a nanoparticle-crosslinked hydrogel via the thioether bridge network through the reaction between thiol groups, simultaneously showing enhanced strength, elasticity, and improved degradation tolerance for cell-recruiting bone repair and regeneration. Zhu et al.163 synthesized copper-loaded dopamine NPs (CuPDA NPs) and phenylboronic acid-modified HA (HA-PBA), creating injectable and self-healing hydrogels through the reaction between dopamine's hydroxyl groups and phenylboronic acid. This condensation reaction between boronic acid and hydroxyl (or amino) groups can occur under neutral or alkaline conditions to form dynamic borate bonds. Diels–Alder (DA) cycloaddition based on a diene and a dienophile can also be used to crosslink hydrogels via forming a six-membered ring compound, which is liable to be broken down under external stimuli like ultrasound. In a study, TiO2 NPs were modified with dopamine–maleimide-modified and reacted with furan-modified gelatin to obtain composite hydrogels based on the inverse electron-demand DA reaction (Fig. 3d).164 Compared to other dynamic crosslinking bonds, hydrogels prepared based on this strategy exhibit higher mechanical properties.165 In summary, clickable bonds offer high controllability and flexibility in making hydrogels with functional properties beyond self-healing, finding expanded applications for drug delivery, cell culture, and tissue repair.
N–), disulfide bonds (–S–S–), borate bonds (–O–B–O–), and reversible Diels–Alder reactions (–CC–). These bonds undergo repeatedly reverse reactions when countered with specific conditions like hydrolysis, external force, or shearing. The reaction between aldehyde and amino groups leads to the formation of Schiff base bonds, which can undergo hydrolysis to restore their original groups and react again to form the bonds. Yang et al.161 mixed CMC with oxidized dextran NPs (ODex NPs) to prepare hydrogels crosslinked with the dynamic Schiff base bonds. Under cyclic loading/shearing conditions, the CMC-ODex hydrogels exhibit gel–sol transitions repeatedly. Disulfide bonds are formed between two thiol groups (–SH). Yao et al.162 prepared thiolated chitosan (TCH) and thiol-functionalized mesoporous BG NPs (TBG). These two components formed a nanoparticle-crosslinked hydrogel via the thioether bridge network through the reaction between thiol groups, simultaneously showing enhanced strength, elasticity, and improved degradation tolerance for cell-recruiting bone repair and regeneration. Zhu et al.163 synthesized copper-loaded dopamine NPs (CuPDA NPs) and phenylboronic acid-modified HA (HA-PBA), creating injectable and self-healing hydrogels through the reaction between dopamine's hydroxyl groups and phenylboronic acid. This condensation reaction between boronic acid and hydroxyl (or amino) groups can occur under neutral or alkaline conditions to form dynamic borate bonds. Diels–Alder (DA) cycloaddition based on a diene and a dienophile can also be used to crosslink hydrogels via forming a six-membered ring compound, which is liable to be broken down under external stimuli like ultrasound. In a study, TiO2 NPs were modified with dopamine–maleimide-modified and reacted with furan-modified gelatin to obtain composite hydrogels based on the inverse electron-demand DA reaction (Fig. 3d).164 Compared to other dynamic crosslinking bonds, hydrogels prepared based on this strategy exhibit higher mechanical properties.165 In summary, clickable bonds offer high controllability and flexibility in making hydrogels with functional properties beyond self-healing, finding expanded applications for drug delivery, cell culture, and tissue repair.
3. Fabrication techniques for composite hydrogels
Constructing organic–inorganic composite hydrogels is a challenging task. The incorporation of inorganic constituents into polymer systems induces modifications in the interfacial milieu of the material, hence initiating interactions between the two components. Organic and inorganic materials generally exhibit distinct physical and chemical characteristics, such as density, rigidity, polarity, and surface energy. To prevent the self-aggregation and settlement of nano-scaled inorganic fillers, the way of these fillers dispersed in the polymeric matrix and the bonding strength between them with polymer chains are essential in determining the hybrid network formation in composite hydrogels. In view of the contributions of inorganic components in gelation, herein, we will explore methods for constructing organic–inorganic composite hydrogels with examples divided into fillers-crosslinked, physically-crosslinked, and chemically-crosslinked systems.3.1 Inorganic fillers crosslinked hydrogels
In the case of polymers that lack inherent gelation ability, the introduction of inorganic elements can function as crosslinking sites, hence facilitating the network formation of precursor solutions. The utilization of this technique obviates the necessity of employing exogenous chemical agents as crosslinkers in the process of hydrogel production, hence diminishing the likelihood of cytotoxicity arising from chemical reagents.166 During the sol–gel transition, the inorganic constituents occupy a certain volume, and the established hybrid network plays a key role in maintaining the distribution and spatial arrangement of fillers, stabilizing them from undesired settlement. Depending on the types of inorganic materials and their surface modifications, ionic crosslinking, chain segment entanglement, and surface functional group reaction are the main interactions involved in obtaining inorganic filler crosslinked hydrogels (Fig. 4).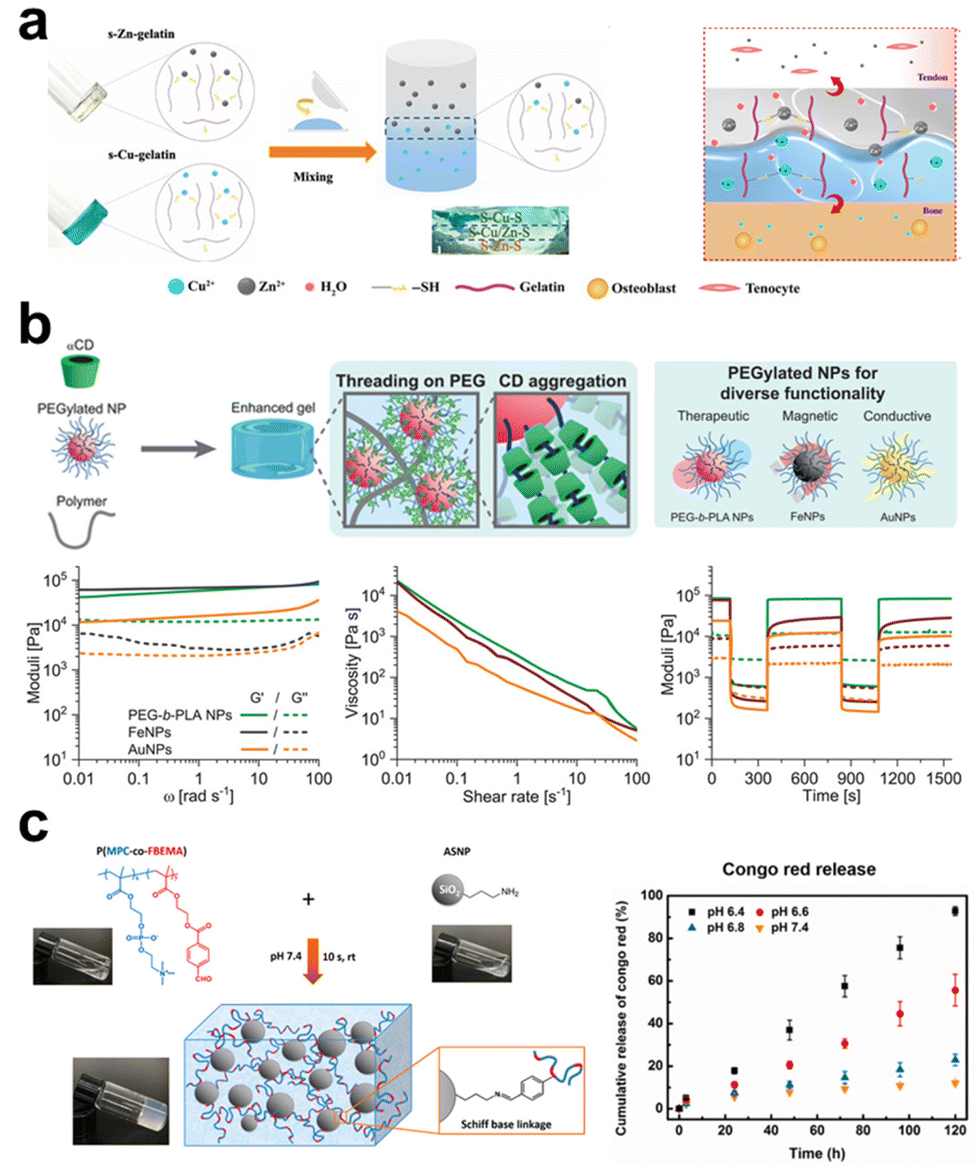 | ||
| Fig. 4 The schematic diagram illustrates three strategies for preparing inorganic filler crosslinked hydrogels: ionic crosslinking, segment entanglement, and surface functional group reaction. (a) Ionic crosslinking involves utilizing ionic interactions between inorganic and organic components. In the presence of copper ions and zinc ions, a one-step coordination crosslinking occurs between thiol groups of thiolated gelatin and copper/zinc ions, leading to the formation of a gradient bimetallic (Cu2+, Zn2+) ion-based hydrogel used for microstructure reconstruction in tendon-bone interfaces.168 Copyright 2021, AAAS. (b) Segment entanglement forms a viscoelastic network due to reversible interactions between polymer chains and NPs. Using α-CD as a supramolecular template enhances the mechanical properties of PNP hydrogels.169 Copyright 2021, Wiley-VCH. (c) Surface modification of inorganic fillers involves attaching specific functional groups, such as amino groups, to the filling materials. Amino functional groups can undergo Schiff base reactions with aldehyde functional groups on polymer chains, forming a pH-sensitive crosslinked network.170 Copyright 2020, American Chemical Society. | ||
Many inorganic compounds or fillers for composite hydrogel preparation contain cationic and/or anionic units, while many polymers that forming hydrogel networks also contain polar or charged groups, these aspects facilitate the electrostatic interactions between them, and ionic interactions can serve as the crosslinking mechanism to stabilize the organic–inorganic hybrid networks. A simple example of this system is adding particles like α-TCP into SA solution, that the carboxyl side groups on the polysaccharide chain form ionic bonds with Ca2+ released from the particles or interact with the particle surface directly.167 Yang et al.168 mixed thiolated gelatin pre-gel solution with CuSO4 (or ZnSO4), and S–Cu/S–Zn bonds formed to constitute one crosslinking mechanism in the hydrogel (Fig. 4a). Lu et al.114 used MgO particles as the fillers to crosslink catechol-mediated polymer instead of using cytotoxic chemical crosslinkers. The presence of an aqueous medium facilitates the formation of Mg2+-catechol complexes. Additionally, the electrostatic interactions between the unbound carboxyl groups inside the polymer and Mg2+ also contribute to strengthening the hydrogel with improved mechanical properties. These procedures exhibit a rather gentle nature, generally, without the need for surface modification on inorganic fillers.
Chain entanglement is a common phenomenon in polymer solutions as the solution concentration exceeds a critical value depending on polymer solubility and molecular weight. It is a kind of physical connection that can also occur between inorganic materials and polymer segments when the added inorganic particles show some affinity to the polymers, such as both of them being hydrophilic. Polar groups (e.g., hydroxyl) located on the surface of inorganic particles will attract polymer chains, leading to increases in entanglement density around the particles to cause gelation. This procedure occurs more easily with macromolecular chains having longer lengths; otherwise, the entanglement interaction force may be relatively weak to obtain a stable crosslinked network. To ameliorate this issue, grafting polymers onto the surface of inorganic materials enhances their self-entanglement interaction with the polymeric chains in forming a hydrogel network. Yang et al.72 covalently grafted PAA chains onto the surface of silica NPs, constructing a hybrid network through the entanglements among the grafted chains with the silica NPs severing as ‘analogous crosslinking points’. The research results indicated that the molecular weight of the grafted chains, the content, and the diameter of the NPs affect the performance of the hydrogel, and it shows heat-induced gel–sol transition due to polymer chain disentanglement, confirming its physically crosslinked nature. Researchers proposed the use of host–guest interactions involving macrocyclic hosts to strengthen this kind of chain entanglements at organic–inorganic interfaces. Bovone et al.169 dissolved hydrogel-forming polymers (e.g. hydroxypropyl methylcellulose, collagen, alginate) at a known mass in PBS, into the solution, cyclodextrins were added, followed by being mixed with PEGylated NPs (e.g. Au, iron oxide) dispersion to form a polymer-NPs (PNP) hydrogel through chain entanglement (Fig. 4b). In the presence of cyclodextrins, they can form poly(pseudo)rotoxanes by threading onto polymer chains, this allows to use poly(pseudo)rotoxanes as supramolecular crosslinks to assemble the formation of PNP hydrogels. In this system, promisingly, the polymers and the NPs can be expanded to a library of choices, even not limiting to inorganic particles and showing high flexibility in composite hydrogel fabrication. In another study,123 the authors also applied the multivalent host–guest interactions between cyclodextrins and adamantane to prepare organic–inorganic composite hydrogels, while they directly conjugated cyclodextrins onto the surface of freshly exfoliated MoS2 nanosheets, constructed an injectable and self-healable hydrogel with adamantane-modified gelatin upon host–guest interactions. The study demonstrated that the incorporation of 1% modified MoS2 increased the storage modulus (G′) and loss modulus (G′′) of the composite hydrogels by 10 and 25 times, respectively, than those for the neat gelatin hydrogel.
Surface modification can bring inorganic fillers with various reactive functional groups, establishing a platform to fabricate composite hydrogels via various chemical bonds. For instance, amine-functionalized inorganic materials can form crosslinks with polymers having aldehyde groups through the Schiff base reaction. Wu et al.170 synthesized an aldehyde-containing copolymer composed of 2-methacryloyloxyethyl phosphorylcholine and 4-formaylbenzoate ethyl methacrylate via free radical polymerization and 3-aminopropyl triethoxysilane-modified silica NPs, injectable and self-healing nanocomposite hydrogels formed as these two gelators being mixed, with tunable mechanical properties by adjusting their ratios (Fig. 4c).
However, a summary made here is to point out the limitation for using inorganic components as crosslinkers for hydrogel fabrication, that the amounts of the inorganic materials introduced are normally not high, as they are the determining factors in influencing crosslinking density, network rigidity, and responses to environmental changes or external stimuli. Thus, in more flexible systems, inorganic fillers with or without surface modification are likely mixed into hydrogels with polymeric networks assembled through physical or chemical crosslinking. Next, we will focus on the modulation effects of the added inorganic components on the crosslinked polymeric networks.
3.2 Modulation on physically crosslinked polymer networks
Physically crosslinked hydrogels, mainly assembled via electrostatic interactions, hydrogen bonding, and hydrophobic interactions, are likely to undergo gel–sol transition with environmental changes. Adding inorganic fillers and their interactions with the polymer chains may strengthen or disintegrate the polymeric networks, depending on the types and surface features of the inorganic components. Ideally, the rigid particles are expected to have strong interactions with the polymers, serving as “analogous crosslinking points” supplementing the first network.Physically crosslinked hydrogels are usually based on polymers with anionic and cationic units; CS, SA, and CMC are common examples. Adding inorganic fillers should not adversely affect their gelation without introducing an extra crosslinking mechanism. Ying et al. intended to prepare HAp/CS composite scaffolds for tissue engineering applications through an environmentally friendly freeze-drying process, which required the homogeneous dispersion of the HAp in CS solution. In the end, the authors synthesized nano-HAp colloidal solution in advance, then mixed it with CS solution for nanocomposite fabrication (Fig. 5a).78 The nano-HAp stabilized with sodium citrate is 40 nm with a narrow size distribution, showing uniform distribution without serious agglomeration, which is crucial for controlling the microstructure, porosity, phase composition, swelling ratio, and mechanical properties of HAp/CS composite scaffolds. Some other studies took an alternative strategy to introduce HAp, circumventing the concern of uneven distribution. Using electrochemical deposition, the surface of the CS scaffold was covered with clustered HAp microspheres of low crystallinity.79 By immersing hydrogels in solutions containing the essential ions (e.g. Ca2+, phosphate) for mineralization, organic–inorganic composite hydrogels were prepared, particularly taking advantage of using peptides and proteins as templates to induce calcium phosphate nucleation and subsequent apatite crystal growth.171 To enhance and control hierarchical mineralization within hydrogels, Okesola et al.85 first established a LAPONITE®-peptide amphiphile co-assembled hydrogel, facilitating apatite nucleation and cluster formation as the LAPONITE® nanodisks provided active sites for mineral crystal growth.
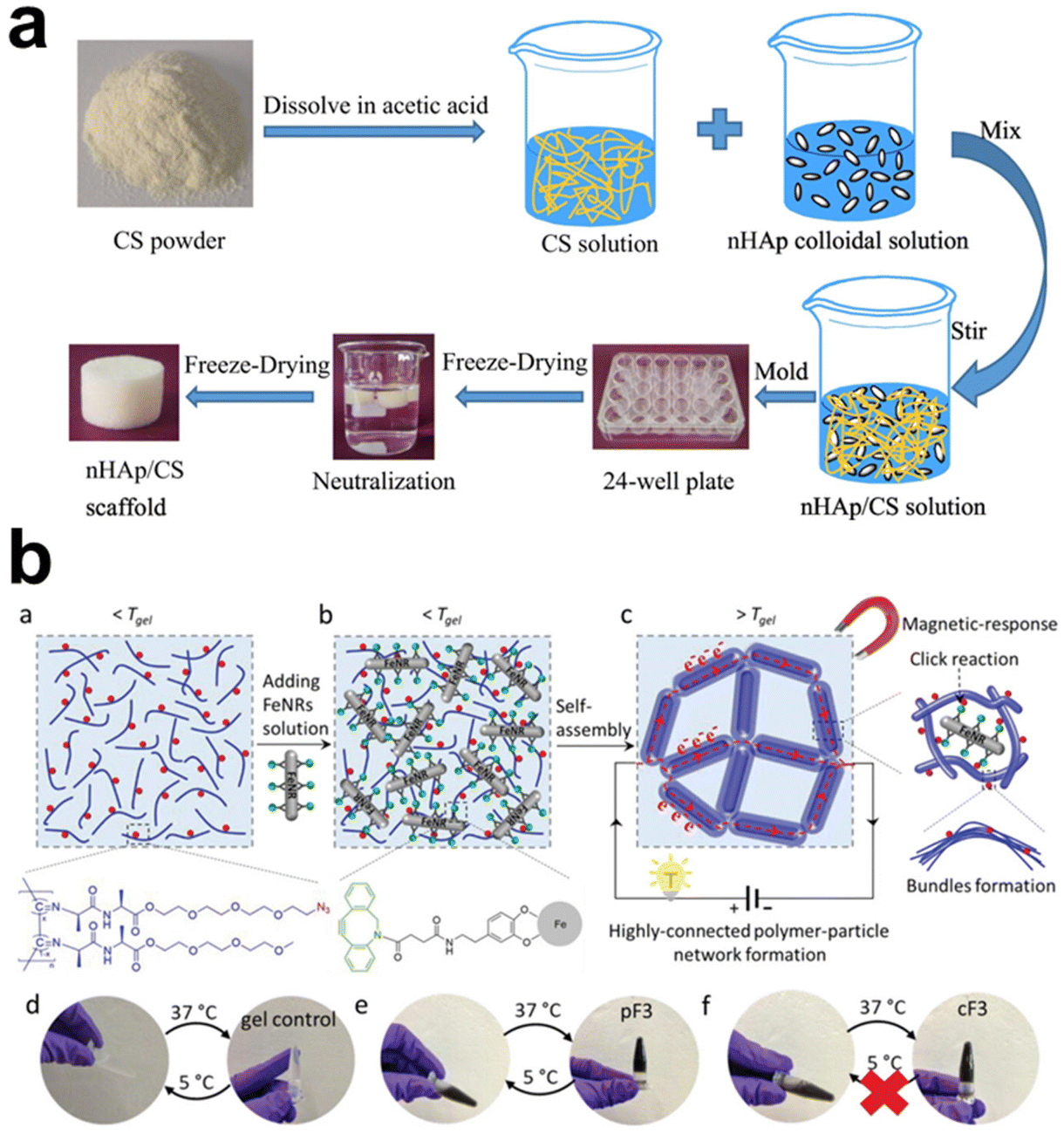 | ||
| Fig. 5 The schematic diagram illustrates the preparation methods of physically crosslinked hydrogels by adding inorganic components. (a) HAp NPs (nHA) incorporated into CS hydrogel: nHA can be incorporated into CS hydrogel through blending, utilizing electrostatic interactions to bind them together.78 Copyright 2022, Elsevier Ltd. (b) Covalent Fe nanorod (FeNR) attachment to polymer chains via surface modification. The modified FeNR can be covalently attached to polymer chains via functional group reaction.117 Copyright 2022, Wiley-VCH. | ||
In a hydrophilic hydrogel system, incorporating a hydrophobic moiety may bring some extra advantages to the hydrogels. Poly(N-isopropyl acrylamide) (PNIPAm) hydrogel is of thermo-responsive nature with a low critical solution temperature (LCST), which means the hydrogel can have transitions between hydrophobic and hydrophilic states upon heating or cooling. Thus, Lee et al.116 incorporated photothermal-responsive Fe2O3 NPs into the hydrogel and adjusted the hydrophobicity of the PNIPAm network by further incorporating N,N-diethylacrylamide moiety, that the hydrogels show rapid light-induced volume change. Xia et al.97 proposed using hydrophobic interaction in preparing hydrogels with high toughness, fatigue resistance, and rapid self-recovery. They grafted SiO2 with poly(n-butyl acrylate) segments to form core–shell inorganic–organic hybrid latex particles at first and used them as hydrophobic centers to crosslink poly(acrylamide-co-lauryl methacrylate) hydrogel via hydrophobic association. The formulated hydrogels are suggested potential applications in biomedical engineering because they have fracture stress of 1.48 MPa, fracture strain of 2511%, and toughness of 12.60 MJ m−3, being high enough to meet anti-fatigue and self-recovery demands for targeted applications.
For most composite hydrogels, inorganic fillers are introduced by simple blending. It usually encounters the difficulty in achieving homogeneous distribution, nevertheless, avoiding the influence of inorganic particles on the self-assembly or crosslinking processes between polymeric chains, particularly at a high fraction of inorganic components. All these issues should be considered in designing and fabricating composite hydrogels. That Chen et al.117 designed a bifunctional linker to enhance the stability of polyisocyanide-based fibrous hydrogel in a non-covalent bundled architecture containing iron oxide nanorods, which may be a good try (Fig. 5b). One end of the linker had a dopamine-mimicking mussel adhesive sequence that could bind to the surface of nanorods, while the other end had a dibenzocyclooctyne (DBCO) group that could react effectively with azide groups on the hydrogel framework. This linker was grafted to the nanorods, which then reacted with polyisocyanide equipped with azide groups to obtain hydrogels with a 40-fold increase in stiffness. It reveals that strong bonding at the organic–inorganic interface is essential in obtaining composite hydrogels with unique properties and advantages for use.
3.3 Modulation on chemically crosslinked polymeric networks
As known, chemically crosslinked hydrogels show high chances of improving material mechanical properties compared to physically crosslinked ones since the former usually provides more reactive sites or functional groups for network formation. When constructing organic–inorganic composite hydrogels, the reactions can simultaneously occur among polymers, between inorganic particles, and at the organic–inorganic interfaces, with proper modification on both components. Formation of covalent C–C bond, C–O–C ether bond, and ester bond, as well as reversible S–S thioether bond and Schiff base bond, etc., is popular in constructing this kind of composite hydrogels, contributing to the vast part in the field.Double-bond modification is extensively employed to graft double-bond functional groups onto the surfaces of inorganic materials through surface modification. This process facilitates the formation of covalently crosslinked networks by enabling the reaction of double-bond functional groups between two components. For instance, Zhou et al.115 synthesized vinyl-coated Fe2O3 NPs, which took part in the free radical crosslinking reaction of GelMA initiated by ammonium persulfate. The crosslinking density is not only related to the methacrylate degree in GelMA, but also depends on the dose of vinyl-coated Fe2O3 NPs; both would decide the stiffness of the formulated hydrogels. Yang et al.101 synthesized vinyl-functionalized silica NPs bearing PEG methyl ether (mPEG) branches, the radical copolymerization of acrylamide and N-isopropyl acrylamide were then conducted in the presence of these modified NPs. The resulting composite hydrogels overcome the shortcomings of traditional neat polymer hydrogels with weak structural strength. The function of the introduced mPEG branches is to accelerate the response rate of PNIPAm hydrogel under temperature change.
To avoid the complex steps in modifying inorganic components with polymerizable vinyl groups, coupling reactions based on ether bridging, amine coupling, and Michael addition can be considered. Covalent ether bridging can form within many biopolymers, like glycosaminoglycan, CS, and alginate, that have abundant hydroxyl groups with the addition of coupling agents like epichlorohydrin. Similarly, this bridging reaction can also occur between hydroxyl-bearing organic and inorganic components to bind them together. Zhao et al.159 made a wound-healing hydrogel dressing with HNTs and chitin by adding epichlorohydrin into their mixed solution, forming covalent ether bridges between hydroxyls including the hydroxyl groups on chitin backbone and the hydroxyl groups on HNTs surface. The covalent bonds are responsible for the formation and enhanced properties of the composite hydrogels. Polydopamine (PDA) coating is an easy way to coat inorganic fillers with the introduction of reactive groups, and these groups will lead to Michael addition reaction with α,β-unsaturated carbonyl units via nucleophilic addition, forming new C–C bonds. Wu et al.109 conducted the preparation of poly(N-isopropylacrylamide-acrylamide) (PNAm) hydrogel via monomers copolymerization with the introduction of PDA-modified gold NPs, that the Michael addition reaction between PDA and amide moieties in the polymers led to the formation of stable covalent bonds, significantly increasing hydrogel's compressive strength (230 kPa) at 4-folds higher than hydrogels without NPs. The coupling between amine (–NH2) and carboxyl (–COOH) groups with the aid of carbodiimide is also feasible in fabricating composite hydrogels since amine and carboxyl groups exist in many biopolymers. Researchers use NPs with the surface carboxyl group with the amino groups on GelMA chains employing 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) as the coupling agents.172 Min et al.173 introduced amino-functionalized BG NPs, and poly(ethylene glycol) diglycidyl ether (PEGDE) into genipin crosslinked glycol chitosan hydrogel to form a secondary crosslinked hybrid network via the high reactivity of the diepoxy groups in PEGDE toward amino and hydroxyl groups. Liu et al.103 utilized lignin–silver NPs to composite with 2-octenyl succinic anhydride modified galactomannan hydrogel, involving reactions related to vinyl groups on galactomannan and quinone/catechol redox reversible transition.
As seen from the above-listed reports, it is proposed that forming a secondary network through reactions between inorganic components may be a crucial supplement to the polymeric or organic/inorganic hybrid network. On the one hand, the content of inorganic components in the hydrogel can be significantly improved. On the other hand, the interweaving of multiple heterogeneous networks may lead to new properties. For example, Chen et al.94 reported an inorganic/organic interpenetrating network (IPN) hydrogel composed of GO and PVA. The authors modified GO sheets with (3-aminopropyl) triethoxysilane (KH550) by silanization to obtain amino-modified GO (GO-NH2), used β-cyclodextrin aldehyde (β-CD-DA) to crosslink the GO-NH2 sheet to form an inorganic β-GO network. This β-GO network interpenetrated with PVA chains, which formed hydrogels using the freeze–thaw method to create the β-GO/PVA IPN hydrogel. Compared to pure PVA hydrogels, the compression modulus of β-GO/PVA hydrogels was increased by 533%, and the elongation at break was improved by 255%. The construction of inorganic networks is still sparsely reported at present, the challenges may lie in how to design crosslinkable inorganic components and their co-assembly or integration with polymeric networks to achieve high-performance composite hydrogels.
4. Inorganic fillers building the base for smart hydrogels
Inorganic materials play pivotal roles in composite hydrogels, endowing the hydrogels with unique mechanical, biological, and responsive properties. These properties are contingent upon the nature and type of inorganic components. Depending on the specific inorganic constituents, organic–inorganic composite hydrogels can exhibit diverse functionalities using inorganic fillers as reinforcements and bioactive sources, such as releasing therapeutic ions or drugs. Owing to the tunable and stable network structure of hydrogels, polymeric chains can effectively modulate their interactions with inorganic components, resulting in mechanical enhancements and various responsive behaviors. These materials are consequently referred to as “smart hydrogels”, which can respond to specific biological or physical trigger factors. Depending on the nature of the responses, herein, smart hydrogels are roughly categorized into environmental-responsive hydrogels and external stimuli-responsive hydrogels for easy summary. The key attention is paid to the noteworthy contributions of inorganic constituents within hydrogel frameworks.4.1 Environmental responsive hydrogels
In order to adapt to alterations in the surrounding environment and cellular demands, compositions and properties of natural ECM undergo dynamic modifications, hence facilitating cell survival, proliferation, and functionality. Cells have the ability to release distinct signaling molecules or substances, like enzymes,174 and glucose,175 which can impact both the mechanical characteristics and biological functionality of ECM, and even change the microenvironmental pH value.176 Inspired by this process, designing hydrogels that can respond to changes caused by cellular events is of paramount importance in tissue regeneration and regenerative medicine. The composite hydrogels have two possible responsive units, i.e., the polymer and the inorganic filler.Furthermore, regulated drug-release hydrogels have been achieved by leveraging the responsiveness of polymers to enzymes and the method of enzymatic breakdown of hydrogel networks. Elevated expressions of MMP-2 and MMP-9 have been observed in some malignant tumor tissues. Nazli et al.177 utilized MMP-sensitive PEG hydrogels to encapsulate drugs, enabling targeted delivery and controlled release of drugs. In enzyme-responsive materials, inorganic NPs can be surface-modified to achieve enzyme-triggered dispersion or crosslinking. Ai et al.178 designed tumor microenvironment-sensitive NPs to enhance the accumulation of NPs. Upon exposure to the tumor microenvironment, tissue proteases that are specific to the tumor break certain sequences located on the surface of NPs. This cleavage event results in the exposure of cysteine residues. The covalent crosslinking process occurs when the cysteine residues located on the surface of NPs react with 2-cyanobenzothiazole present on neighboring particles. This reaction subsequently initiates the aggregation of NPs at the tumor location. In brief, enzyme-responsive hydrogels possess the ability to dynamically respond to tissue microenvironments using enzymatic cleavage or crosslinking mechanisms, hence facilitating the precise delivery of therapeutic agents to specific target sites.
The acidic microenvironment present in tumor tissues enables inorganic components to function as drug transporters and provide controlled medication release. Hu et al.98 developed and synthesized core–shell particles consisting of PNiPAM/AA@SiO2, which exhibited dual responsiveness to changes in temperature and pH. These particles were specifically engineered to enable the controlled release of therapeutic agents, making them suitable for cancer treatment. In acidic environments, actinomycin within the NPs is rapidly released. Apart from acidic environments, the local pH of chronic wounds can be as high as 8–9. Alkaline-sensitive wound dressings are crucial for chronic wounds. Pan et al.181 established a drug-controlled release platform with a relatively high pH (above 8) by adjusting the physical structure of silica NPs (SiNPs). Additionally, including antibacterial compounds within SiNPs has been shown to suppress bacterial proliferation in alkaline pH environments effectively.
Moreover, glucose oxidase (GOx) is a common glucose-sensitive component that catalyzes glucose oxidation to gluconic acid and hydrogen peroxide. Kim et al.25 harnessed GOx and cerium oxide NPs (CeNPs) loaded onto large-pore mesoporous silica, further incorporated into CS hydrogels. The presence of elevated glucose concentrations triggers GOx enzyme to initiate an oxidation reaction, resulting in the production of hydrogen peroxide and subsequent acidification of the hydrogel. As a result, the fall in pH leads to the expansion of the CS hydrogel, facilitating the eventual release of protein medicines encapsulated inside it. CeNPs have the ability to catalyze the decomposition of hydrogen peroxide, which is produced during the oxidation of glucose by the enzyme GOx. The catalytically active CeNPs facilitate oxygen regeneration, hence inhibiting the degradation of GOx. The self-catalytic properties of CeNPs provide more stability to the glucose-responsive closed-loop delivery system, in contrast to catalase.
In utilizing organic components, Zhao et al.184 developed ROS-responsive hydrogels through the reaction between phenylboronic acid and alcohol hydroxyl groups. The prepared hydrogels serve as effective ROS scavengers, promoting wound healing by reducing ROS levels and upregulating M2 phenotype macrophages around the wound area. The hydrogels demonstrate efficacy in scavenging ROS, hence facilitating the healing of wounds through the reduction of ROS levels and the upregulation of M2 phenotype macrophages in the vicinity of the wound site. Conversely, ROS-responsive hydrogels remove ROS using inorganic components and employ these inorganic components as carriers to improve the regenerative wound microenvironment. Wu et al.185 utilized cerium dioxide NPs as both ROS scavengers and carriers for miRNA, creating a composite hydrogel that clears ROS and promotes wound healing. Cerium dioxide NPs play a role in simultaneously regulating the oxidative levels of endogenous cells in the damaged area and protecting miRNA from adverse environmental influences, thus enhancing the diabetic wound healing process.
Significantly, the elevated concentrations of GSH within cancer cells not only function as stimuli for controlled drug release but also exhibit antioxidant properties that effectively neutralize ROS. Hou et al.189 encapsulated sodium humate (SH), chlorin e6 (Ce6), and manganese dioxide (MnO2) NPs into low-melting agarose, forming an agarose@SH/MnO2/Ce6 delivery system. Within this particular system, the utilization of MnO2 NPs extends beyond their role as reducing agents for GSH in tumor tissues. These NPs also exhibit reactivity towards elevated concentrations of H2O2, resulting in the generation of O2. Upon injection into the tumor site, the agarose@SH/MnO2/Ce6 hydrogel exhibits the ability to maintain the production of oxygen, thereby successfully addressing the hypoxic condition of the tumor. This, in turn, leads to an enhancement of Ce6-induced photodynamic treatment. This GSH-responsive hydrogel provides a novel approach for the design of intelligent hydrogels with dual capture capabilities.
Despite the potential of molecular design to confer materials with the ability to adapt to environmental changes, there are still several challenges and limitations in practical clinical applications. The cellular environment is highly complex and diverse, involving various signaling molecules, mechanical forces, and chemical factors. Once smart-responsive materials are introduced into a biological organism or cellular environment, effective monitoring and control methods are needed to ensure their performance and safety. Compared to environmentally responsive hydrogels, the advantages of smart hydrogels responsive to external stimuli in inducing structure or characteristic alterations lie in the wide choices of stimuli such as temperature, light, magnetic field, and ultrasonic, which possess high flexibility in action intensity and time. Of note, ‘on–off’ control is also possible. These provide powerful tools to construct hydrogels with versatile designs for various applications, including but not limited to medication delivery, biosensing, and smart materials.
4.2 External stimuli-responsive hydrogels
Different from the conditional factors (e.g., pH, glucose content) generated in body fluid, which are likely to display fluctuations, the external physical-type stimuli are easier to control by setting different parameters in high or low values. Though polymers with specific chemical structures can be sensitive to changes in temperature, light, magnetic field, and ultrasonic, chain cleavage is also likely to occur, deteriorating other performance of the materials. Incorporating inorganic components with smart responses can provide the composites with ‘switch on’ and ‘switch off’ performance under a selected external stimulus. Temperature variation usually acts on the conformation of thermosensitive polymers, leading to swelling or shrinkage changes in thermosensitive hydrogels. Depending on the nature of introduced inorganic fillers, the external stimuli can also cause a heating effect on polymeric networks; while not limiting to temperature rise, other effects, including photoelectric transition, magnetic/electrical force, and ultrasonic activation are more attractive for composite hydrogels. In the following subtitles, organic–inorganic hydrogels are briefly categorized into photo-responsive hydrogels, magnetic-responsive hydrogels, electric-responsive hydrogels, and ultrasonic-responsive hydrogels, focusing on their compositions and responsive mechanisms.Nanostructures with NIR absorption can be embedded in hydrogels, such as metallic oxide nanosheets, nanorods, and nanotubes.194 When these nanomaterials are exposed to light stimulation, the dominant output is normally photothermal or photoelectric effect in heat or ROS generation, which leads to sol–gel transitions of the composite hydrogels, faster or slower release rates of embedded drugs, higher or lower affinity to normal and tumor cells.195,196 Warm and overheat effects can function differently in modulating cellular behaviors. For mild heat therapy, the temperature is usually maintained between 38–42 °C. This range prevents thermal damage to tissues and organs while also accelerating local blood flow and nutrient exchange. It opens ion channels on cell membranes, enhances ion diffusion rates, and triggers the expression of heat shock proteins (HSPs).197,198 Hyperthermia therapy, on the other hand, requires locally generated temperatures between 42–48 °C, which can induce apoptosis in bacteria and tumor cells.199–201 Within this range, proteins begin to denature and aggregate. Prolonged treatment (>60 minutes) may result in cell inactivation and tissue damage.202 Su et al.149 employed NIR as an illumination source to fabricate injectable hydrogels that are responsive to light. These hydrogels were developed to conduct PTT on tongue for in situ cancer therapy.
Nevertheless, the majority of hydrogel photosensitive motifs necessitate the use of UV light to initiate their activation. UV light possesses inherent harmful properties and exhibits restricted tissue penetrating capabilities. To overcome the constraints associated with UV light, Zheng et al.190 put forth a proposed approach that capitalizes on the remarkable tissue penetration capabilities of NIR light (Fig. 6a). The researchers utilized upconversion NPs (UCNPs) to convert NIR light into UV light, resulting in the development of a scaffold that can be regulated and controlled by light. The hydrogel network was subjected to modification through the incorporation of light-activatable cell adhesion ligands. By exposing the modified hydrogel to NIR light, the researchers were able to exert control over the spatial arrangement of the newly developed vascular network. The utilization of UCNPs in conjunction with light-activatable ligands facilitated the controlled activation and precise localization of cell adhesion and blood vessel development while ensuring the absence of observable cellular harm. In another study, UCNPs were used to activate photosensitive proteins within cells, initiating local siRNA delivery to suppress biological functions.203
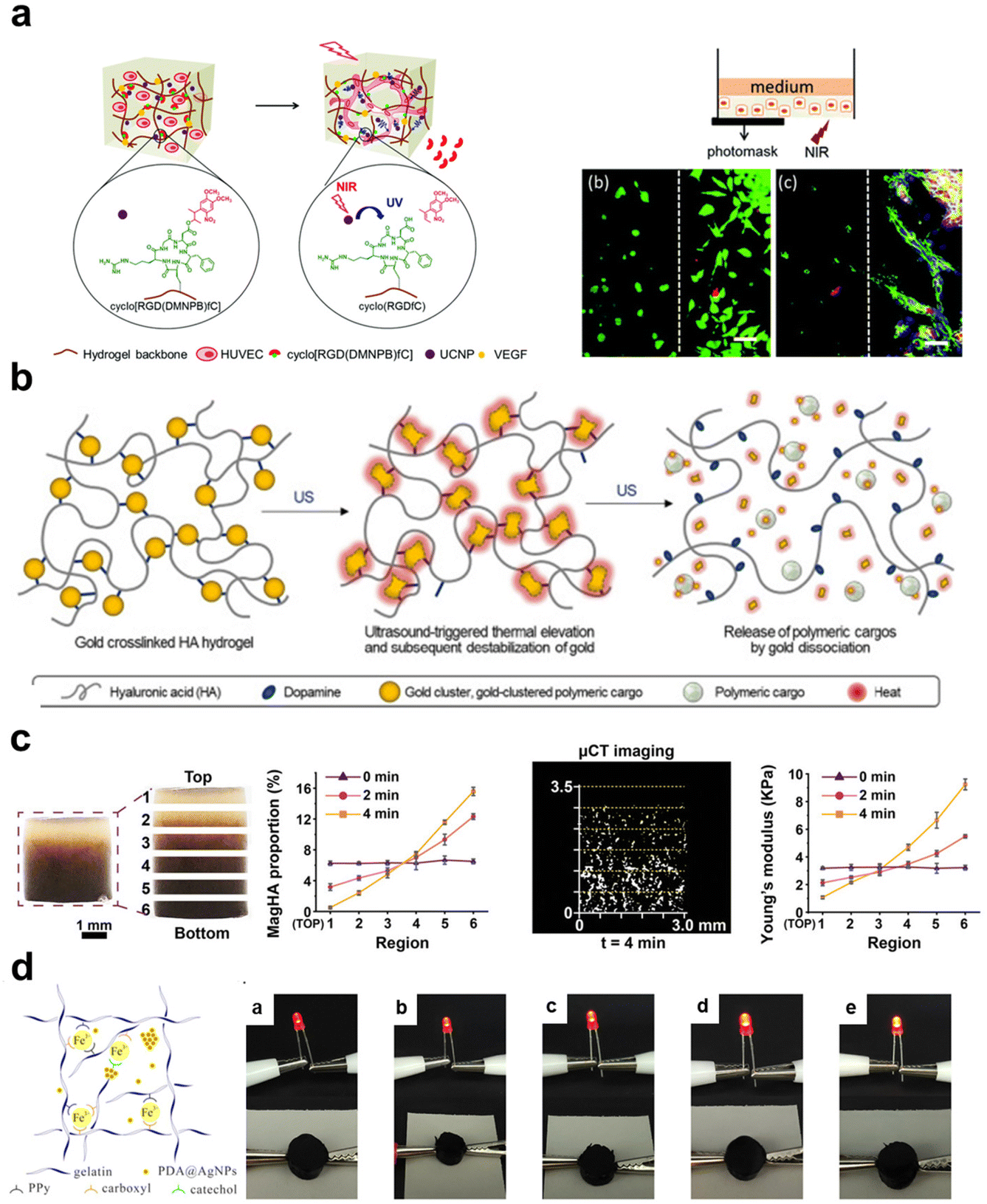 | ||
| Fig. 6 The schematic diagram illustrates four types of stimulus-responsive hydrogels: (a) photo-responsive hydrogels with photoactivatable cell adhesion ligands that can regulate cell spreading and angiogenesis.190 Copyright 2021, Elsevier Ltd. (b) Ultrasound-responsive hydrogels with ultrasound-triggered thermal effects lead to the disintegration of the hydrogel network and drug release.148 Copyright 2021, Elsevier Ltd. (c) Magneto-responsive hydrogels capable of controlling the distribution of magnetic NPs under the influence of a magnetic field, allowing targeted modulation of cellular behavior.16 Copyright 2023, Wiley-VCH. (d) Electro-responsive hydrogels with excellent electrical conductivity can enhance wound healing effects using electrical stimulation.106 Copyright 2020, American Chemical Society. | ||
In brief, the photosensitive inorganic NPs have the capability to selectively absorb particular wavelengths of light and transform the absorbed light energy into thermal energy. This process then induces a thermal reaction inside the hydrogel system, leading to the attainment of photo-responsiveness. Nevertheless, the transmission of light through biological tissues is influenced by the processes of absorption and scattering, which might potentially hinder the effective utilization of light-responsive functions in regions located deep inside the tissues. Despite the superior tissue penetration capabilities of the NIR II area, the utilization of long-wavelength light is constrained by its low energy.204 Nevertheless, using UCNPs to enhance hydrogel photo-response emerges as a viable and effective approach.
Specifically, the use of ultrasonic stimulation has the capability to supply energy that can induce the disruption of the polymer chain or induce alterations in the inorganic crosslinking sites. These alterations subsequently lead to modifications in the swelling and contraction behavior of the hydrogel, ultimately facilitating the release of the encapsulated medication. In a study conducted by An et al.,148 an ultrasonic responsive hydrogel, was developed through the utilization of gold cluster NPs and catechol in conjunction with dopamine-modified HA (Fig. 6b). In this context, gold cluster NPs consist of drug-polymer NPs and gold clusters enriched on the NPs surface. Gold cluster NPs act as an acoustic-thermal crosslinking agent for HA hydrogels. Under conditions when ultrasonic is used as a trigger, gold NPs generate heat, leading to the induction of apoptosis in nearby cancer cells through the thermoacoustic effects. The dissociation of gold clusters leads to drug release and disintegration of the crosslinked network. Furthermore, ultrasonic stimulation can be combined with electrical stimulation and electrical signals. Ultrasonic-stimulated piezoelectric hydrogel is the use of ultrasonic to stimulate the deformation of piezoelectric materials, thereby generating electrical signals, which can be used to promote tissue regeneration and promote drug release in cancer treatment.205 Pucci et al.206 proposed an anti-cancer strategy that combines sonic-responsive drug carriers with piezoelectric NPs. This hydrogel platform enables remote activation of drug release in response to ultrasonic and local delivery of anti-cancer electrical signals. The co-administration of chemotherapy and persistent piezoelectric stimulation has been shown to effectively trigger apoptosis in drug-resistant cells, inhibit tumor migration, and mitigate cell invasiveness.
On the other hand, ultrasonic stimulation can also stimulate inorganic materials to produce ROS for tissue regeneration. Yang et al.207 fabricated a therapeutic hydrogel with acoustic-dynamic capabilities, incorporating antibacterial characteristics. This was achieved by employing mesoporous silica-coated titanium dioxide NPs (TiO2@MS-SH) as a crosslinking agent and ROS generator. The organic–inorganic composite hydrogel system allows for the modification of the inorganic component to serve as a multi-functional module in addition to its existing ultrasonic response capability. Wang et al.208 employed mesoporous silica NPs to load the sonic-sensitive chemical perfluorohexane onto mesoporous silica. They successfully achieved surface self-assembly and enzyme polymerization. The implementation of this novel methodology led to the creation of a composite hydrogel that is well-suited for both sonic imaging and sonic therapy.
In clinical practice, two regularly employed kinds of ultrasound are low intensity pulsed ultrasonic (LIPUS) and high-intensity focused ultrasonic (HIFU). LIPUS is a therapeutic modality characterized by its ability to induce minor temperature fluctuations inside the tissue, hence facilitating the acceleration of fracture healing processes and providing relief from chronic pain. It is worth noting that a single session of LIPUS therapy entails a considerable duration. On the other hand, HIFU is a therapeutic approach of greater intensity, enabling targeted therapy of a localized tumor location while minimizing harm to adjacent tissue. This precision contributes to a reduction in adverse effects and treatment-related problems. Therefore, it is crucial to adopt a prudent methodology while choosing ultrasonic parameters.
The utilization of a static magnetic field has the potential to direct the spatial arrangement of magnetic materials, thereby facilitating the process of cell proliferation and differentiation in localized regions. We utilized a constant static field along with superparamagnetic HAp nanorods to construct a continuous gradient biomimetic scaffold for bone and cartilage (Fig. 6c).16 The multilevel gradient composite hydrogel demonstrates the ability to replicate the progressive transition from cartilage to subchondral bone, resulting in highly favorable outcomes for tissue restoration. Moreover, the presence of a static electric field has the potential to influence the qualitative arrangement of cells, thereby playing a role in controlling the process of tissue regeneration. Shefi et al.151 introduced a technique that entails the direct administration of collagen infused with magnetic NPs into regions of tissue injury. This approach enables the external magnetic field to exert a directed influence on the alignment of fibers and the proliferation of neuronal cells. The experimental findings demonstrated that applying an external magnetic field led to the aggregation of magnetic particles, forming chains of magnetic particles. This phenomenon subsequently resulted in the alignment of collagen fibers in a well-organized manner. The alignment of neurons aided the development of elongated and co-aligned morphologies.
Typically, magnetic-responsive hydrogels often consist of magnetic materials or magnetic NPs that exhibit fast responses to alterations in external magnetic fields. Under the influence of low-frequency alternating magnetic fields (<100 Hz), the magnetic-to-mechanical energy conversion occurs in magnetic NPs as a result of interparticle interactions. The generated mechanical force leads to deformation in the hydrogel, providing the necessary stimulation for cells and tissues, thereby inducing specific biological effects.210 Under the influence of high-frequency alternating magnetic fields ranging from 100 kHz to 1 MHz, magnetic NPs can transform magnetic energy into thermal energy. This phenomenon occurs through mechanisms such as Néel relaxation and Brownian relaxation.211 For instance, Dai et al.212 dispersed iron oxide magnetic NPs into dopamine-modified hyaluronic acid, forming a catechol–iron(III) coordination bond. Drug release occurs via non-covalent interactions between organic and inorganic constituents in the absence of an external alternating magnetic field. The application of an external alternating magnetic field induces a temperature response in magnetic NPs, leading to an accelerated release of drugs.
Nevertheless, it is imperative to acknowledge that the integration of magnetic NPs into the composition of hydrogels is necessary to ensure the efficient utilization of magnetic-responsive hydrogels. Additional research is necessary to reach a consensus between the biocompatibility and functioning of the NPs in question. The task of producing a homogeneous dispersion of these NPs within the hydrogel matrix presents a challenging endeavor.
To address this issue, Zhao et al.214 grafted aniline monomers onto quaternized chitosan and incorporated oxidized dextran as a dynamic Schiff crosslinker, creating a conductive hydrogel with antibacterial properties. The conductivity of the hydrogel synthesized using this method was 0.43 mS cm−1. Moreover, adding inorganic components to the hydrogel can enhance its electrical conductivity and self-healing properties. Wang et al.106 incorporated dopamine-modified silver NPs (PDA@AgNP) into a hydrogel comprising PPy (Fig. 6d). They employed Fe3+ as crosslinkers to fabricate a hydrogel that exhibits self-healing properties, electrical conductivity, as well as antibacterial and antioxidant functionalities. In fact, incorporating conductive inorganic components is the simplest method for constructing electrically responsive hydrogels. Maiz-Fernández et al.215 mixed alginate hydrogel with magnetic zero monovalent iron NPs, producing a time-dependent biomimetic hydrogel responsive to electric field stimulation. The results showed that this hydrogel had high mechanical strength. When exposed to an electric field (15 V), the mixed hydrogel tended to move toward the cathode, providing options for the development of soft robotics and complex biomimetic applications. Therefore, organic–inorganic composite hydrogels with good electrical activity can be prepared using conductive inorganic materials and conductive hydrogels.
In summary, organic–inorganic composite hydrogels possess the ability to adapt to variations in the ECM environment through modifications in network architectures or the controlled release of bioactive chemicals. This characteristic facilitates the precise and timely regulation of cellular responses, medication administration, and therapeutic interventions for various diseases. Hydrogels provide a remarkable capacity for customization in terms of their composition and content, which can meet various application requirements and adapt to multiple scenarios. In contrast to organic components, which passively respond to ECM stimuli, inorganic components offer greater versatility by responding to signals from the external environment. The addition of inorganic substances imparts responsive hydrogels with improved functionality and flexibility, hence broadening their scope of possible applications in areas such as drug delivery, tissue engineering, medical diagnostics, and therapy. Thus, an organic–inorganic composite hydrogel platform can enable multifunctional, multi-responsive hydrogels, significantly broadening their scope of application. In the following sections, we shall explore the various uses of organic–inorganic composite hydrogels within the fields of tissue engineering and regenerative medicine.
5. Applications of composite hydrogels in regenerative medicine
Organic–inorganic composite hydrogels are multifunctional materials widely employed in the fields of regenerative medicine and anti-tumor therapy. The consideration of the interplay between organic and inorganic components is crucial in constructing the hydrogels because of the complex three-dimensional microenvironment produced. In addition, it is imperative to conduct a thorough examination of the impact of these elements on cellular behaviors and tissue regeneration. An ideal hydrogel targeting for disease treatment and regenerative medicine should be produced by precise fabrication methods and functionalization strategies, successfully integrating organic polymers and inorganic components into a tuned whole to meet various needs in diverse biomedical applications. Here, we choose some typical fields in which organic–inorganic composite hydrogels are widely studied and used for summary. The subitems include bone repair, cartilage regeneration, wound healing, and anti-tumor therapy (Fig. 7). | ||
| Fig. 7 The schematic diagram illustrates the applications of organic–inorganic composite hydrogels in regenerative medicine: (a) carboxymethyl agarose (CMA) forms supramolecular hydrogels through hydrogen bonding and can continuously release Ag+, demonstrating excellent antibacterial properties and the ability to promote wound healing.105 Copyright 2020, Wiley-VCH. (b) Hydrogel loaded with BMP-2 functionalized MgFe layered double hydroxide (LDH) nanosheets for efficient bone regeneration.216 2022, Wiley-VCH. (c) Cobalt nanowires (Co NWs) incorporated into poly(ethylene glycol) diacrylate (PEGDA) hydrogel system can effectively simulate a hypoxic microenvironment, guiding umbilical cord-derived MSCs (UMSC) towards chondrogenic differentiation.217 Copyright 2023, American Chemical Society. (d) MOF hydrogel containing amphotericin for sustained drug release, exhibiting pH and ATP-responsive drug release characteristics, effectively killing tumor cells.120 Copyright 2022, Elsevier Ltd. | ||
5.1 Wound healing
Wound healing is a complex and ordered process that can be divided into four sequential and interacting stages: hemostasis, inflammation, proliferation, and remodeling.218 These stages occur sequentially and overlap with each other, directly impacting the time and quality of wound healing. Currently, existing hydrogel dressings can maintain a moist wound environment, aiding in wound healing and alleviating pain around the wound area. However, these hydrogel dressings are often limited in their functionalities and cannot meet the specific requirements of certain wounds, such as wounds in diabetic patients’ skin. To fulfill the dynamic demands of wound healing, introducing inorganic materials presents a promising option. Inorganic components possess functions such as antimicrobial properties, ROS scavenging, ion release promoting angiogenesis, and cell recruitment. These functionalities make organic–inorganic composite hydrogels one of the hotspots in current research on skin dressings.Wounds and incisions are susceptible to infection by bacteria and other microorganisms. Antimicrobial dressings can mitigate the chance of infection and contribute to the preservation of wound or incision cleanliness and sterility. Various metals, metal oxides, and carbon materials possess antimicrobial properties; incorporating them into hydrogels can achieve antimicrobial effects.219 For example, Zhao et al.159 filled gold NPs into the cavities of layered double hydroxides (HNTs) to impart photothermal and antimicrobial effects to the hydrogel. Tang et al.22 employed magnesium oxide (MgO) as a crosslinking agent to fabricate a dual-network hydrogel. The use of MgO as a crosslinking agent led to an improvement in the antibacterial properties of the hydrogel.
In addition, the response to exogenous substances and the onset of acute inflammation result in the development of fibrotic capsules.220 Therefore, anti-inflammatory dressings are crucial for promoting the healing process of wounds or incisions.221,222 Huang et al.,105 proposed CMA hydrogels crosslinked with silver ions (Ag+). The composite hydrogels exhibited remarkable compatibility with cells and blood, alongside the capacity to suppress inflammation (Fig. 7a). Tao et al.23 successfully introduced copper NPs into hydrogels, significantly enhancing their antimicrobial capabilities, reducing inflammation, and promoting angiogenesis. In a chronic wound healing model with Staphylococcus aureus infection, copper NPs notably accelerated the healing process. The achievement was ascribed to the combined impact of laser irradiation on the photothermal characteristics and the swift liberation of copper ions (Cu2+). Additionally, the released Cu2+ stimulated the proliferation of NIH-3T3 fibroblasts without inducing any inflammatory responses. This research offers strong evidence in favor of using hydrogel to attain antibacterial properties, facilitate the healing of wounds, and improve the formation of new blood vessels. It provides innovative perspectives and strategies for addressing wound infections and promoting wound-healing. Currently, metal-based NPs, especially metal oxides, BG, CNTs, GO, and other inorganic components, are being incorporated into hydrogels to stimulate vascular regeneration.223 Nevertheless, an overabundance of angiogenesis result in the development of scar tissue. Consequently, there is a growing interest in the development of smart dressings that possess the ability to be regulated, sensitive, and capable of monitoring the state of wounds. For instance, Jin et al.24 integrated the beneficial characteristics of nanofibers and hydrogels through the incorporation of PLGA nanofibers, VEGF, SiO2, and Mexene into dopamine-modified hyaluronic acid hydrogels. This approach modulates the immune response of wounds, mitigates excessive inflammation, and facilitates the regulated and scarless healing of wounds.
The process of skin healing encompasses several crucial stages, namely hemostasis, antimicrobial activity, antioxidation, angiogenesis, and promotion of epithelialization. These stages have a significant role in determining the overall quality of wound closure. Wound healing typically involves wound contraction, and appropriate tensile forces can accelerate wound closure by creating a cellular sense of stress microenvironment.224 To enhance tissue adhesion and material cohesion, Li et al.95 designed a hydrogel based on dopamine-modified reduced GO (rGO-PDA) and thermos-responsive PNIPAm. Under physiological temperature conditions, this composite hydrogel can undergo self-contraction and generate strong tissue adhesion. The coordination between catechol groups and metal ions can promote high cohesion in hydrogels, enhancing the toughness of adhesives and stabilizing their tissue adhesion. Kim et al.225 utilized quinone-mediated covalent crosslinking and Fe3+-mediated non-covalent crosslinking to create mussel adhesive protein hydrogels with self-healing properties. Moreover, the integration of inorganic constituents facilitates the enhancement of adhesive performance in hydrogels by the augmentation of cohesion energy resulting from molecular interactions between polymer chains and the surfaces of inorganic fillers. Jung et al.226 increased the cohesion of PAM/PDA hydrogel networks by adding 1.5% mesoporous silica NPs with large pore sizes. This enhancement resulted in a 1.4-fold increase in adhesion, reaching a maximum value of 151 J m−2.
In brief, the integration of inorganic and organic constituents augments the efficacy of hydrogel materials in various aspects, including wound hemostasis, adhesion, antibacterial properties, antioxidation, encouragement of vascular regeneration, and intelligent responsiveness. Considering the diversity of wound types that hydrogel dressings need to address, hydrogel dressings with multiple functionalities can achieve an “all-in-one” goal, enabling them to cope with more complex wounds and injuries. Additionally, while efficient adhesion is beneficial for wound closure and repair, excessively strong adhesion hinder the replacement and removal of dressings. Therefore, there is a need to develop hydrogel dressings with reversible adhesion to meet diverse requirements.
5.2 Bone regeneration
Natural bone is a hard organ composed of both organic and inorganic components, serving the function of protecting and supporting other organs. The inorganic constituents of natural bone are commonly sourced from calcium phosphate-based substances, such as HAp and calcium carbonate (CaCO3). Conversely, the organic constituents encompass components like as collagen, proteins, and lipids. Organic–inorganic composite hydrogels possess a compositional structure that closely resembles that of real bone, hence demonstrating favorable attributes such as biocompatibility, biodegradability, and osteoconductive characteristics.227To mimic the composition and structure of natural bone, Kim et al.228 incorporated calcium phosphate NPs into methylcellulose hydrogels to achieve promising bone repair effects. Compared to the method of incorporating NPs, Zhang et al.80 introduced one-dimensional HAp nanorods and HAp nanowires into GelMA hydrogels, significantly improving the compressive and rheological properties of hydrogels for bone regeneration. Aiming to mimic the organic–inorganic characteristics of ECM of bone cells, Wang et al.21 successfully integrated bioceramic components into porous microspheres. The cryogel microspheres demonstrated a sustained release of bioactive elements, including calcium (Ca), phosphorus (P), and silicon (Si), in contrast to the control group consisting solely of bioceramic components. This sustained release can promote the osteogenic differentiation of BMSCs. Natural bone tissues exhibit complex micro-porous and micro-channel structures, which are crucial for the biomechanical properties and biological activity of bones. Hou et al.229 mixed HAp microtubes (HAMT) with GelMA to construct composite hydrogel scaffold simulating the tubular structure of bone tissue. The tubular configuration of HAMT allowed the interconnection of micro-pores within GelMA. The composite scaffold promoted proliferation and differentiation of BMSCs in vitro. However, it is essential to highlight that osteoconductive materials do not possess inherent osteoinductive properties. Once the process of bone callus formation disrupted, the growth factors that promote cell differentiation within the scaffold material decrease.230 This can be unfavorable for the bone tissue repair process.
In clinical practice, bone-inducing factors are commonly used to enhance bone regeneration, including growth factors, drugs, and inorganic ions. Although hydrogels containing growth factors have been extensively utilized in the field of bone defect healing, the barrier to developing intelligent hydrogels with the capability to regulate the release of growth agents remains. Research findings have indicated that incorporating bone morphogenetic protein-2 (BMP-2) into gelatin hydrogel can expedite release of this growth factor.231 Within a time frame of 12 hours, roughly 70% of BMP-2 released in PBS buffer. To address this issue, Mao et al.232 utilized mesoporous silica NPs to load BMP-2, preserving its biological activity within silk methacrylate (SilMA) hydrogel and slowing down its release rate.
To achieve sustained release of BMP-2, researchers utilized two-dimensional layered double hydroxides (LDHs) with a high surface area to load proteins and drugs. Lv et al.216 employed electrostatic interactions to load BMP-2 onto the surface of MgFe-LDH nanosheets, enabling the sustained release of BMP-2 in CS/SF hydrogels (Fig. 7b). The results indicate that the composite hydrogels exhibited a substantial enhancement in bone regeneration when compared to CS hydrogels. Specifically, the composite hydrogels led to a considerable increase in bone volume by a factor of 4.5 and bone density by a factor of 3.6. Furthermore, bone tissue regeneration often involves multiple stages, requiring the sequential release of various active factors to achieve sequential regulation goals. Liu et al.233 encapsulated dexamethasone and recombinant human bone morphogenetic protein (rhBMP) within porous mesoporous BG scaffolds, adjusting their release kinetics to achieve endochondral ossification and ectopic bone formation. Compared to directly mixed growth factors, utilizing inorganic components for topical delivery of bioactive ions is simple and safe.234
Bioactive ions participate in relevant ion channels, stimulate cell differentiation, or engage in secondary signal transduction processes, exerting the therapeutic effects of metal ions.235 In bone metabolism, strontium (Sr) is similar to calcium (Ca), which participates in the differentiation of osteoblasts. Zinc (Zn) involves in the structure, catalysis, and regulation of several important metalloenzymes, such as alkaline phosphatase. Magnesium (Mg) promotes the adhesion, proliferation, and vascular regeneration of BMSCs. Silicon plays a crucial role in collagen synthesis, connecting collagen tissues, and biological mineralization processes. However, directly injecting soluble salts into the gel system is hard to achieve sustained release. This problem can be efficiently addressed using organic–inorganic hydrogels made with inorganic components that release various ions. Chen et al.113 introduced magnesium oxide NPs into a phosphocreatine-functionalized CS solution, using the sustained release of magnesium ions to promote new bone formation. Moreover, bone defects are often accompanied by other diseases, such as infection or osteosarcoma. Therefore, introducing multifunctional inorganic components can achieve a combination therapy effect. Li et al.236 utilized inorganic NPs as crosslinkers, photothermal agents, and Mg2+ reservoirs to construct multifunctional nanocomposite hydrogels. The findings revealed that sustained release of Mg2+ promoted osteogenic differentiation. Furthermore, because of the PDAM coating, the hydrogel exhibits superior photothermal properties for efficient tumor suppression.
Chen et al.237 reported a hydrogel loaded with dexamethasone. The inorganic component in the hydrogel, Mexene nanosheets, is an efficient NIR photo-thermal converter capable of rapidly controlling heat generation. The organic component in the hydrogel is a temperature-sensitive polymer that significantly contracts at temperatures above 42 °C, leading to the ultra-sensitive release of loaded dexamethasone. Upon the NIR light irradiation, the hydrogel exhibited excellent capabilities in suppressing cell apoptosis and promoting osteogenic differentiation. The ultra-sensitive release pattern at 42 °C demonstrates outstanding synergistic osteogenic effects.
In summary, organic–inorganic composite hydrogels exhibit excellent bone conductivity and bone-inducing properties. Composite hydrogels can mimic the nanostructure of primary bone tissue. They are a highly promising platform for bone tissue engineering. Composite hydrogels display numerous advantages, such as increasing vascularization and spatiotemporal control based on the unique requirements at different phases of bone regeneration. When it comes to bone regeneration in the presence of disorders, the flexible design of composite hydrogels suit a wide range of needs. It should be noted that the mechanical qualities of hydrogel systems may not match the demands of weight-bearing areas and are therefore only suited for non-weight-bearing bone restoration.
5.3 Cartilage reconstruction
Unlike many other tissues, cartilage is essentially avascular and exists in a hypoxic environment.238 Due to the lack of sufficient cells and nutrient supply, the self-healing capacity of cartilage tissue is extremely limited. Untreated cartilage defects can lead to irreversible joint damage, potentially causing osteoarthritis and disability.239 To overcome these limitations, numerous hydrogels with embedded cells have been introduced in the field of cartilage tissue engineering. The aim is to recreate the embryonic microenvironment to facilitate the production of cartilage constructions. It is vital to highlight that cartilage tissue does not include any inorganic components. The inorganic components in organic–inorganic composite hydrogels primarily play roles in mechanical enhancement, releasing active substances to promote cartilage regeneration, clearing ROS to inhibit subchondral bone osteoclast activation, and suppressing disease progression synergistically during the cartilage reconstruction process.One of the key design considerations for cartilage regeneration scaffolds is their mechanical performance. Hydrogels made from polymeric materials typically have compression strengths ranging from a few tens to a few hundred kPa. The strength of cartilage ranges from 10 to 20 MPa. Simply enhancing the mechanical qualities of hydrogels by varying the crosslinking density result in an overly dense network, affecting cell spreading. Using active inorganic elements offers a promising solution to this problem. Phosphate glass fibers (PGF) exhibit excellent mechanical properties and biodegradability. Zhu et al.240 introduced PGF into PVA hydrogels, which enhanced the crystallinity and thermal stability of the hydrogel structure. The incorporation of PGF improved the mechanical properties of the composite hydrogel, with a maximum tensile strength of 12.44 MPa and a Young's modulus of 68.35 MPa. This matches the mechanical strength of hyaline cartilage. Additionally, during the degradation process of PGF, magnesium ions are released, promoting cell recruitment and migration. Additionally, inorganic components can regulate the release behavior of bioactive factors to promote cartilage repair. Cui et al.99 successfully demonstrated the achievement of sustained anhydroicaritin (AHI) release to promote cartilage regeneration. The sustained release behavior of AHI can be attributed to the synergistic effect of mesoporous channels in inorganic mSiO2 NPs and the three-dimensional organic hydrogel scaffold. Therefore, multifunctional composite hydrogels greatly induce proliferation and differentiation of articular cartilage stem cells in vitro, promote the production of ECM, and achieve cartilage regeneration in vivo.
Rheumatoid arthritis (RA) and osteoarthritis (OA) are musculoskeletal disorders that affect joints and cartilage, potentially leading to skeletal degeneration. Oxidative stress-induced imbalance in chondrocyte metabolism plays a crucial role in the progression of osteoarthritis. To mitigate damage caused by oxidative stress, Wang et al.241 employed crosslinked chondroitin sulfate hydrogels loaded with manganese dioxide NPs (Mn3O4) and injected the hydrogel into the joint cavity. This study indicate that manganese oxide enzymes can decrease intracellular levels of ROS in chondrocytes by emulating the functions of superoxide dismutase (SOD) and catalase (CAT). As a result, this process aids in alleviating the detrimental effects of chondrocytes’ heightened oxidative stress and impeding the degradation of cartilage during the progression of osteoarthritis. Hypoxic microenvironments play a crucial role in the formation and maintenance of cartilage tissue, where cobalt (Co) can induce hypoxia by stabilizing hypoxia-inducible factor-1α (HIF-1α). Ravi et al.217 incorporated cobalt nanowires (Co NWs) into poly(ethylene glycol) diacrylate (PEGDA) hydrogels to create hydrogels that simulate a hypoxic microenvironment (Fig. 7c). The results demonstrated that these composite hydrogels promoted hypoxia-induced cartilage formation, downregulated hypertrophic/osteogenic marker expression, and facilitated the differentiation of umbilical cord-derived MSCs (UMSC) into chondrocytes. In response to the acidic inflammatory microenvironment, Zhou et al.242 prepared hyaluronic acid (HA) hydrogels through a Schiff base reaction between aldehyde groups on oxidized hyaluronic acid (HA-ALH) and amino groups on hyaluronic acid (HA-ADH). Due to the pH-responsive Schiff base linkages, bovine serum albumin (BSA) encapsulated manganese dioxide NPs (BM NPs) in the hydrogel could be released in the acidic inflammatory microenvironment. In vitro experiments confirmed that BM NPs effectively promoted chondrocyte proliferation and protected chondrocytes from oxidative stress invasion.
Bone-cartilage tissue represents a typical joint interface, and its full-layer defects are challenging to repair after trauma-related injuries or OA. The challenge in bone cartilage repair lies in reconstructing various continuous gradients within the connective tissue. Gao et al.157 addressed this challenge by developing a bilayer scaffold containing gradient magnesium ions, mimicking the layered structure of bone cartilage tissue (Fig. 8a). The upper hydrogel layer had a higher Mg2+ content and smaller pore size, simulating a hypoxic environment. The lower layer of the scaffold had a lower Mg2+ content and an interconnected large pore structure, simulating the bone marrow cavity. Results showed that the upper hydrogel layer could simulate a hypoxic microenvironment, promoting cartilage formation. The lower scaffold provided a pathway for guiding BMSCs to migrate from the basic bone marrow to the implanted scaffold, thereby improving the regeneration of cartilage and subchondral bone tissues. Zhang et al.16 successfully enhanced the capability of hydrogel for cartilage regeneration through external magnetic stimulation and the construction of a continuous nano-hydroxyapatite gradient (Fig. 8b). Moreover, cartilage fulfills the dual role of stress dissipation and joint surface lubrication. The simulation of the lubricating effect on the surface of articular cartilage is of utmost importance in the pursuit of cartilage regeneration. To reduce the friction coefficient, Lu et al.87 incorporated lubricating carbon dots into PEG hydrogels, creating injectable carbon dot/CS/PEG composite hydrogels. The results showed that this composite hydrogel effectively protected the surface of ultra-high molecular weight polyethylene (UHMWPE), thereby extending the lifespan of artificial joints.
 | ||
| Fig. 8 The schematic diagram illustrates the applications of organic–inorganic composite hydrogels in cartilage regeneration: (a) MRI and Micro-CT tests in animal experiments showed that Mg2+ ion gradient double-layer scaffolds promoted osteochondral regeneration.157 Copyright 2023, Wiley-VCH. (b) Nanoindentation tests from animal experiments indicate that the newly formed cartilage in the organic–inorganic composite hydrogel group, under external magnetic stimulation, exhibits a relatively smooth surface, closest to the native cartilage tissue.16 Copyright 2023, Wiley-VCH. | ||
In summary, the regeneration of cartilage in different anatomical sites poses varying demands on materials, aiming to enhance the mechanical properties of hydrogels and promote effective cartilage regeneration. Additionally, it is crucial to consider the impact of the arthritic environment on cartilage repair, necessitating hydrogels to possess therapeutic properties for arthritis. To better mimic the gradient environment of bone-cartilage tissue, the design of scaffold materials with gradients is necessary. Moreover, integrating hydrogel composite systems into the human body may raise concerns about their long-term stability. This potential issue could lead to material degradation or functional decline, thus posing obstacles to the efficcient regeneration of cartilage regeneration.
5.4 Antitumor therapy
Tumor removal frequently leads to tissue injury, hence requiring a dual-pronged strategy in tumor treatment: addressing remaining tumor cells and facilitating tissue regeneration. BG, an example of an inorganic material, possesses the dual capability of effectively eradicating cancerous cells while concurrently promoting the regeneration of tissue. As a result, they have been widely utilized in the treatment of tumors. Based on functional inorganic nanomaterials, in recent years, the dominantly applied mechanism in inducing tumor cell death is phototherapy using NIR activation, including PTT and photodynamic therapy, due to their minimally invasive and rapid efficacy.243,244PTT uses the light absorption properties of nanomaterials to transform light radiation into thermal energy. This conversion process induces a photothermal effect, which ultimately results in the eradication of tumor cells. As mentioned earlier, due to their thermos-responsive properties, light-responsive hydrogels are widely utilized in tumor eradication and tissue regeneration. Using the PTT strategy, dispersing these photothermal-responsive inorganic components within hydrogel networks can facilitate cancer treatment. For instance, Liu et al.110 prepared upconversion lanthanide hybrid NPs (UCNP Au NPs) with a unique layered hybrid structured hydrogel. Through electrostatic interactions between DNA and positively charged UCNP Au NPs, NIR light responsiveness and injectability were achieved. Due to the interactions between DNA chains and UCNP, Au NPs exhibited more pronounced photothermal effects and better cellular compatibility compared to the original inorganic nanomaterials. On the other hand, PTT therapy can be combined with temperature-sensitive hydrogels to develop a photo-responsive hydrogel system to enable triggered release. Wang et al.26 introduced gold nanorods to CS-@puerarine (CP) hydrogel to accomplish PTT and heat-sensitive gel–sol conversion. Upon the irradiation of low-density NIR light, the hydrogel release the gene-targeting medication DC-AC50. The results revealed that CP@Au @DC-AC50 hydrogel, along with moderate PTT and gene therapy, exhibited promising tumor inhibition effect.
PDT utilize light to initiate ROS production, for cancer treatment. This process efficiently elicits immunogenic cell death. Zhang et al.245 developed an anti-tumor hydrogel system using titanium dioxide (TiO2) as a photosensitizer and crosslinker, capable of generating high concentrations of singlet oxygen under NIR light exposure. To address the hypoxic microenvironment of the tumor, Li et al.122 created a synergistic PDT and oxygen-supplying hydrogel system using MOF NPs and CS hydrogel. Zirconium MOF NPs (MnP NPs) with surface-modified MnO2 efficiently convert H2O2 into oxygen, changing the hypoxic microenvironment of the tumor. Due to the biodegradability of CS hydrogel, MnP NPs gradually released at the tumor site, promoting the formation of cytotoxic singlet oxygen, which kills cancer cells under light exposure. Furthermore, tumor treatment also involves wound healing issues. To enhance phototherapy efficiency, PTT can be combined with PDT, leveraging their synergistic effects to combat tumors. Yin et al.246 synthesized palladium NPs (Pd NPs) with PTT and PDT capabilities. They loaded the chemotherapeutic drug DOX onto Pd NPs, creating a dual-functional hydrogel platform for both tumor treatment and wound repair. The photothermal effect of Pd/DOX hydrogel allows light-triggered drug release, effectively inhibiting tumor growth. Additionally, Pd/DOX hydrogel served as a wound dressing, effectively preventing the entry of harmful chemicals and promoting vascular regeneration, thus accelerating the wound-healing process. Hence, the incorporation of NPs into the hydrogel matrix for the purpose of creating organic–inorganic composite hydrogels exhibits significant potential.
In addition to PTT and PDT strategies, organic–inorganic composite hydrogels can modulate the tumor microenvironment, such as pH, oxygen content, and inflammation levels, to achieve tumor treatment and wound healing. For instance, He et al.247 designed a pH-responsive hydrogel. This hydrogel can release manganese sulfide in acidic tumor microenvironments, generating hydrogen sulfide. The phenomenon generates oxidative stress within tumor cells, resulting in an elevation of ROS levels, ultimately leading to apoptosis and necrosis. Additionally, Zeng et al.120 developed a pH-responsive drug release hydrogel utilizing the interaction between bisphosphonate and zinc (Fig. 7d). To achieve pH responsiveness and drug loading, functionalized bisphosphonate hyaluronic acid (HA-BP) molecules were mixed with a suspension of DOX-encapsulated MOF (MOF@DOX). Under acidic conditions, the coordination bonds between Zn2+ ions in MOF and the imidazole groups undergo dissociation, resulting in the structural collapse of the MOF framework and subsequent drug release. Animal experiments showed that compared to the MOF@DOX group, the HA-BP·MOF@DOX hydrogel exhibited enhanced tumor growth inhibition. This was attributed to the hydrogel's immobilization effect on MOF and the sustained drug release. Hypoxia is a prevalent attribute observed within the interior of tumors. To address this issue, hydrogels consisting of microalgae-gold nanorods have been developed to release oxygen specifically within hypoxic conditions.111 Wang et al.248 developed an injectable redox and light-responsive bioinspired manganese dioxide hybrid (BMH) hydrogel. The use of BMH hydrogel has been found to be highly efficient in eradicating bacterial invasion, enhancing wound oxygenation, and mitigating inflammation within the wound microenvironment. Consequently, this treatment has demonstrated a substantial capacity to facilitate wound healing in cases involving multidrug-resistant infections and the suppression of tumors.
In summary, organic–inorganic hydrogels have demonstrated significant anti-tumor properties in animal experiments. These hydrogels have shown promising therapeutic effects in experimental animal models through mechanisms such as modulating the tumor microenvironment, achieving precise drug release, and enhancing immune responses. These studies provide new strategies and directions for tumor treatment. However, there are still challenges to be addressed before practical clinical application. Firstly, despite promising results in animal experiments, further research is needed to address issues related to the biocompatibility, toxicity, and long-term efficacy of hydrogels. Additionally, the applicability and individual variability of hydrogels in different types of tumors require more in-depth research.
6. Conclusion and prospects
Organic–inorganic composite hydrogels hold great promise in the fields of tissue engineering and regenerative medicine. Composite hydrogels offer the potential for innovative solutions to improve the treatment outcomes of various tissue and organ injuries. Nevertheless, the achievement of effective integration of inorganic constituents into hydrogel necessitates the meticulous identification and pairing of appropriate material combinations. As the field of study progresses, it becomes imperative to consider several critical factors. Firstly, hydrogel development necessitates a primary focus on improving the precision in managing its microstructure. Secondly, the forming methods and injectability properties should be carefully considered in the application and design of hydrogels. Thirdly, understanding the interactions between organic and inorganic components is crucial, as they significantly impact the performance and functionalities of hydrogel. Fourthly, the biocompatibility of the composite hydrogel itself, as well as the biological safety of material degradation and leachable are important for in vivo applications. Lastly, hydrogels should be designed to adapt to complex disease models and the processes involved in defect repair treatments. These considerations are important for the future development and application of hydrogels in various fields.The differences in density between organic and inorganic components pose significant challenges in achieving uniformity in preparing organic–inorganic composite hydrogels. This is because achieving the dispersion of inorganic components or preventing their settling before the formation of hydrogels poses significant challenges. Despite the utilization of several techniques such as ultrasound, surface modification, change of solution viscosity, and increased interactions between inorganic particles and polymer chains, a holistic resolution to this problem has not yet been attained. Surface modification stands out as an effective approach to enhance dispersion. Nevertheless, achieving an optimal equilibrium between enhanced dispersion and maintaining contacts among inorganic constituents and polymer chains, together with the subsequent network density, rigidity, and flexibility, necessitates meticulous deliberation. Striking this balance necessitates in-depth research and meticulous control. In the context of injectable hydrogels, ensuring an appropriate viscosity for the pre-gel solution and achieving uniform dispersion of inorganic particles are pivotal. Any non-uniformity in the microstructure may significantly impact cellular behaviors within the gel. Strategies such as utilizing composite microgels offer a potential solution. By miniaturizing the system, issues related to uneven dispersion of inorganic components can be mitigated. Furthermore, the utilization of injectable microspheres or the implementation of organic–inorganic composite crystalline gels could offer viable strategies to tackle the difficulties linked to accurate formulation, control of microstructure, and operability of implantation.
The particle size of inorganic components has an impact on the pore structure of hydrogels. Smaller particles typically result in finer pores, which facilitate cell adhesion and diffusion but may reduce the overall porosity of the hydrogel. Additionally, the morphology of inorganic fillers, such as the shape of NPs (spherical, fibrous, sheet-like, etc.), can affect the thermal conductivity, electrical conductivity, and optical properties of hydrogel. Surface properties of inorganic fillers, such as surface chemistry and functionalization, can influence the hydrogel's hydrophilicity or hydrophobicity, thereby affecting its interactions with cells or drugs. The larger surface area of nanomaterials can enhance the strength and surface activity of the hydrogel. Uniformity and distribution of both components are crucial for the structure and performance of hydrogels. Ordered structures have broad potential applications, such as in the repair of interface tissues, including bone, cartilage, tendon, periodontal tissues, and full-thickness skin defects. The distribution of inorganic components mainly depends on the mixing methods. Evenly distributed particles can provide consistent performance, while uneven distribution may lead to localized performance differences. Achieving the directed movement of inorganic components within the hydrogel is also challenging. Current methods primarily include using magnetic attraction with magnetic particles. However, this method only applies to magnetic materials and requires ensuring that the magnetic material has no adverse effects on biocompatibility and toxicity. Furthermore, it is possible to integrate many techniques, like microsphere stacking, 3D printing, and temperature gradients, among others, to attain an asymmetric dispersion of inorganic constituents inside targeted locations. The choice of which method to use depends on the desired degree of arrangement, the nature of the inorganic component, and the type of hydrogel. In practical applications, a series of experiments and optimizations are required to obtain the best results for directed arrangement or distribution.
The performance of composite hydrogels is substantially influenced by the quantity of inorganic components and their morphology and distribution. Generally, increasing the content of inorganic components enhances the mechanical strength and hardness of composite hydrogels, making them more suitable for bearing loads or supporting tissue engineering. Moreover, the interaction between inorganic components and the hydrogel can improve the stability of the hydrogel, enhancing its resistance to dissolution or degradation. Additionally, the number of inorganic components added can also influence the release rate of drugs, growth factors, or other active molecules. Certain inorganic constituents have the potential to augment the conductivity of composite hydrogels, rendering them well-suited for utilization in electrochemical sensors and optoelectronic devices. Nevertheless, an overabundance of inorganic constituents could potentially reduce the toughness of the hydrogel, rendering it more susceptible to fracturing or breaking. Excessive presence of inorganic substances can also lead to a decrease in the porosity of hydrogels and potentially result in the introduction of harmful elements to cells, hence compromising their biocompatibility. Therefore, the quantity of added inorganic components should be carefully optimized based on the specific requirements of the application. In addition, the microstructure of the material, including factors such as particle size and contact characteristics, exerts a substantial influence on its performance. Enhanced performance can be achieved by optimizing the preparation process, integrating numerous inorganic components, lowering individual dosages, or modifying the shape of inorganic materials.
Biocompatibility is a critical consideration that directly influences the performance of hydrogels in medical applications. The biocompatibility of composite hydrogels is primarily influenced by two main factors: the composition of the materials and the degradation products. The hydrogel matrix is composed of natural or synthetic polymers with excellent biocompatibility. In addition to polymeric materials, composite hydrogels also incorporate inorganic components such as minerals, or metallic compounds. It is noteworthy that the type and concentration of inorganic components may impact biocompatibility, particularly when potential cytotoxic elements are present. Higher concentrations of metallic ions and NPs inhibit cell proliferation or induce cell death as they exceed a critical value, whereas, optimized metallic ions supply can promote cell growth and differentiation. In fabricating composite hydrogels, apart from their functions, the quantity and even distribution of inorganic components are unignorable key issues. Upon implantation, the degradation products generated from scaffold materials will alter the local microenvironment in pH or ionic strength etc., depending on both the organic and inorganic components in the scaffolds. Normally, the degradation products of polymers are acidic, while the degradation of metals, metal oxides, and ceramics release alkaline products. Both peracid and peralkali are not welcomed to maintain normal tissue homeostasis. A proper organic–inorganic combination brings some neutralization effect to ameliorate the concern. Furthermore, it should avoid the sharp degradation of the composite hydrogels. On the one hand, it leads to scaffold collapse; on the other hand, degradation products are released in a short time to cause fluctuation in local microenvironment, adversely affecting surrounding tissues. Also, the long-term effects of the applied polymers, inorganics, and their degradation products on animals and human bodies must be considered before the clinical use of composite hydrogels.
The integration of diverse inorganic constituents into hydrogels can bring a multitude of functionalities. For example, HAp is a bioactive ceramic. The integration of HA into hydrogels has been shown to augment their capacity for bone tissue regeneration, rendering them a highly suitable option for bone repair and implant materials. Additionally, the addition of ZnO NPs can bring antimicrobial and wound-healing properties to hydrogels. The introduction of nano-iron oxide into hydrogels can confer magnetic properties, so enabling precise delivery of drugs or cells to enhance the efficacy of treatments. Multifunctional hydrogels have emerged as crucial components within the realm of biomedicine, particularly in the domains of tissue engineering and regenerative medicine. Nevertheless, the process of tissue regeneration generally encompasses a sequence of distinct phases, hence necessitating the consideration of diverse designs to address the requirements of each step adequately. For instance, bone repair requires processes such as vascular formation, nerve regeneration, and bone tissue regeneration. Similarly, skin repair involves multiple steps, including hemostasis, antimicrobial action, and anti-inflammatory effects. Hence, there is a requirement for hydrogels that possess spatiotemporal control to offer diverse functionalities at distinct temporal intervals. To achieve spatiotemporal control, a promising approach is the application of stratified hydrogel architectures. The hydrogels are organized into many layers or segments, where each segment contains unique functional components. The utilization of stratified hydrogels enables the controlled and gradual release of bioactive agents, allowing for the precise delivery of necessary active compounds at specific time intervals during the different phases of tissue regeneration. Additionally, stimulus-responsive hydrogels can change their properties based on environmental conditions or external stimuli. For example, pH-sensitive hydrogels can release anti-inflammatory drugs in an alkaline environment, while magnetically sensitive hydrogels can control drug release through an external magnetic field. However, inorganic components can provide hydrogels with multiple functions. NPs may undergo cellular internalization, potentially leading to cytotoxicity or uncertainties.
Currently, research on organic–inorganic composite hydrogels has made significant progress. Nevertheless, a dearth of comprehensive research exists regarding systematically examining the interplay between organic and inorganic constituents. The complete understanding of the leaching mechanism of inorganic constituents within the hydrogel matrix remains to be fully clarified. The determination of release kinetics in composite hydrogels can be achieved through experimental measurements. However, the presence of three-dimensional structures in composite hydrogels may introduce variations between anticipated concentrations and observed outcomes. Limited reports exist on the use of organic–inorganic composite hydrogels for embedding cells in cartilage regeneration. Furthermore, differences in the degradation and swelling properties between the two components may affect the adhesion and migration behavior of cells encapsulated in the hydrogel. Hence, the investigation of organic–inorganic composite hydrogels can yield enhanced comprehension and exploration of the interplay between cells and materials. It has great potential to tackle obstacles in tissue engineering.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Beijing Natural Science Foundation (7232100, L232092), the Central Universities (buctrc202220), the National Natural Science Foundation of China (U22A20159, 32371422).References
- K. L. McKinley, M. T. Longaker and S. Naik, Science, 2023, 380, 796–798 CrossRef CAS PubMed.
- D. K. Patel, E. Jung, S. Priya, S.-Y. Won and S. S. Han, Carbohydr. Polym., 2023, 121408 Search PubMed.
- Z. Wang and W. Cui, Adv. Ther., 2021, 4, 2000096 CrossRef CAS.
- A. C. Daly, L. Riley, T. Segura and J. A. Burdick, Nat. Rev. Mater., 2020, 5, 20–43 CrossRef CAS PubMed.
- B. Guo, R. Dong, Y. Liang and M. Li, Nat. Rev. Chem., 2021, 5, 773–791 CrossRef CAS PubMed.
- Y. Liang, J. He and B. Guo, ACS Nano, 2021, 15, 12687–12722 CrossRef CAS PubMed.
- M. Diba, S. Spaans, S. I. S. Hendrikse, M. M. C. Bastings, M. J. G. Schotman, J. F. van Sprang, D. J. Wu, F. J. M. Hoeben, H. M. Janssen and P. Y. W. Dankers, Adv. Mater., 2021, 33, 2008111 CrossRef CAS PubMed.
- F. Wang, W. Zhang, Y. Qiao, D. Shi, L. Hu, J. Cheng, J. Wu, L. Zhao, D. Li and W. Shi, Adv. Healthcare Mater., 2023, 12, 2300192 CrossRef CAS PubMed.
- R. Zhong, S. Talebian, B. B. Mendes, G. Wallace, R. Langer, J. Conde and J. Shi, Nat. Mater., 2023, 22, 818–831 CrossRef CAS PubMed.
- A. Rasool, S. Ata, A. Islam, M. Rizwan, M. K. Azeem, A. Mehmood, R. U. Khan, A. u. R. Qureshi and H. A. Mahmood, Int. J. Biol. Macromol., 2020, 147, 67–78 CrossRef CAS PubMed.
- W. Guo, D. Cao, W. Rao, T. Sun, Y. Wei, Y. Wang, L. Yu and J. Ding, ACS Appl. Mater. Interfaces, 2023, 15, 42113–42129 CrossRef CAS PubMed.
- Y. Chai, Y. Long, X. Dong, K. Liu, W. Wei, Y. Chen, T. Qiu and H. Dai, Colloids Surf., B, 2022, 210, 112220 CrossRef CAS PubMed.
- M. Chen, Y. Cui, Y. Wang and C. Chang, Chem. Eng. J., 2023, 453, 139893 CrossRef CAS.
- C. C. L. Schuurmans, M. Mihajlovic, C. Hiemstra, K. Ito, W. E. Hennink and T. Vermonden, Biomaterials, 2021, 268, 120602 CrossRef CAS PubMed.
- M. Chen, Y. Wu, B. Chen, A. M. Tucker, A. Jagota and S. Yang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203074119 CrossRef CAS PubMed.
- L. Zhang, W. Dai, C. Gao, W. Wei, R. Huang, X. Zhang, Y. Yu, X. Yang and Q. Cai, Adv. Mater., 2023, 35, 2209565 CrossRef CAS PubMed.
- T. M. Koushik, C. M. Miller and E. Antunes, Adv. Healthcare Mater., 2023, 12, 2202766 CrossRef CAS PubMed.
- Z. Yuan, Z. Wan, C. Gao, Y. Wang, J. Huang and Q. Cai, J. Controlled Release, 2022, 350, 360–376 CrossRef CAS PubMed.
- Z. Wan, Z. Yuan, Y. Li, Y. Zhang, Y. Wang, Y. Yu, J. Mao, Q. Cai and X. Yang, Adv. Funct. Mater., 2022, 32, 2113280 CrossRef CAS.
- M. A. Asl, S. Karbasi, S. Beigi-Boroujeni, S. Z. Benisi and M. Saeed, Int. J. Biol. Macromol., 2022, 223, 524–542 CrossRef CAS PubMed.
- C. Gao, W. Dai, X. Wang, L. Zhang, Y. Wang, Y. Huang, Z. Yuan, X. Zhang, Y. Yu and X. Yang, Adv. Funct. Mater., 2023, 33, 2304829 CrossRef CAS.
- X. Tang, X. Wang, Y. Sun, L. Zhao, D. Li, J. Zhang, H. Sun and B. Yang, Adv. Funct. Mater., 2021, 31, 2105718 CrossRef CAS.
- B. Tao, C. Lin, Y. Deng, Z. Yuan, X. Shen, M. Chen, Y. He, Z. Peng, Y. Hu and K. Cai, J. Mater. Chem. B, 2019, 7, 2534–2548 RSC.
- L. Jin, X. Guo, D. Gao, Y. Liu, J. Ni, Z. Zhang, Y. Huang, G. Xu, Z. Yang and X. Zhang, Bioact. Mater., 2022, 16, 162–172 CAS.
- M. Y. Kim and J. Kim, ACS Biomater. Sci. Eng., 2017, 3, 572–578 CrossRef CAS PubMed.
- S. Wang, B. Chen, L. Ouyang, D. Wang, J. Tan, Y. Qiao, S. Ge, J. Ruan, A. Zhuang and X. Liu, Adv. Sci., 2021, 8, 2004721 CrossRef CAS PubMed.
- W. Wang, Y. Zhao, H. Yi, T. Chen, S. Kang, H. Li and S. Song, Nanotechnology, 2017, 29, 025605 CrossRef PubMed.
- T. Guan, J. Li, C. Chen and Y. Liu, Adv. Sci., 2022, 9, 2104165 CrossRef CAS PubMed.
- C. Gao, W. Dai, X. Wang, L. Zhang, Y. Wang, Y. Huang, Z. Yuan, X. Zhang, Y. Yu and X. Yang, Adv. Funct. Mater., 2023, 2304829 CrossRef CAS.
- S. Liu, O. Oderinde, I. Hussain, F. Yao and G. Fu, Polymer, 2018, 144, 111–120 CrossRef CAS.
- W. Dai, L. Zhang, Y. Yu, W. Yan, F. Zhao, Y. Fan, C. Cao, Q. Cai, X. Hu and Y. Ao, Adv. Funct. Mater., 2022, 32, 2200710 CrossRef CAS.
- Y. Li, M. D. Hoffman and D. S. Benoit, Biomaterials, 2021, 268, 120535 CrossRef CAS PubMed.
- M. Jin, A. Gläser and J. I. Paez, Polym. Chem., 2022, 13, 5116–5126 RSC.
- A. E. Emerson, A. B. McCall, S. R. Brady, E. M. Slaby and J. D. Weaver, ACS Biomater. Sci. Eng., 2022, 8, 4002–4013 CrossRef CAS PubMed.
- N. Chirani, L. Yahia, L. Gritsch, F. L. Motta, S. Chirani and S. Farè, J. Biomed. Sci., 2015, 4, 1–23 Search PubMed.
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, eaaf3627 CrossRef PubMed.
- C. Xing, S. Chen, M. Qiu, X. Liang, Q. Liu, Q. Zou, Z. Li, Z. Xie, D. Wang and B. Dong, Adv. Healthcare Mater., 2018, 7, 1701510 CrossRef PubMed.
- Y. Li, J. Rodrigues and H. Tomás, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- A. Chrisnandy, D. Blondel, S. Rezakhani, N. Broguiere and M. P. Lutolf, Nat. Mater., 2022, 21, 479–487 CrossRef CAS PubMed.
- X. Meng, Y. Qiao, C. Do, W. Bras, C. He, Y. Ke, T. P. Russell and D. Qiu, Adv. Mater., 2022, 34, 2108243 CrossRef CAS PubMed.
- M. Hua, S. Wu, Y. Ma, Y. Zhao, Z. Chen, I. Frenkel, J. Strzalka, H. Zhou, X. Zhu and X. He, Nature, 2021, 590, 594–599 CrossRef CAS PubMed.
- S. Utech and A. R. Boccaccini, J. Mater. Sci., 2016, 51, 271–310 CrossRef CAS.
- Y.-G. Chen, C.-X. Li, Y. Zhang, Y.-D. Qi, J. Feng and X.-Z. Zhang, Chin. J. Polym. Sci., 2022, 40, 1050–1061 CrossRef CAS.
- B. Kaczmarek, K. Nadolna and A. Owczarek, Hydrogels Based Nat. Polym., 2020, 12, 151–172 Search PubMed.
- M. L. Pita-López, G. Fletes-Vargas, H. Espinosa-Andrews and R. Rodríguez-Rodríguez, Eur. Polym. J., 2021, 145, 110176 CrossRef.
- S. Peers, A. Montembault and C. Ladavière, J. Controlled Release, 2020, 326, 150–163 CrossRef CAS PubMed.
- C. B. Highley, G. D. Prestwich and J. A. Burdick, Curr. Opin. Biotechnol., 2016, 40, 35–40 CrossRef CAS PubMed.
- X. Yang, B. Wang, D. Peng, X. Nie, J. Wang, C.-Y. Yu and H. Wei, Adv. NanoBiomed Res., 2022, 2, 2200124 CrossRef CAS.
- Y. Peng, Y. Zhuang, Y. Liu, H. Le, D. Li, M. Zhang, K. Liu, Y. Zhang, J. Zuo and J. Ding, Exploration, 2023, 3, 20210043 CrossRef PubMed.
- S. Ahmed, M. Keniry, V. Padilla, N. Anaya-Barbosa, M. N. Javed, R. Gilkerson, K. Gomez, A. Ashraf, A. S. Narula and K. Lozano, Int. J. Biol. Macromol., 2023, 250, 126187 CrossRef CAS PubMed.
- T. Bai, K. Zhao, Z. Lu, X. Liu, Z. Lin, M. Cheng, Z. Li, D. Zhu and L. Zhang, Chin. Chem. Lett., 2021, 32, 1051–1054 CrossRef CAS.
- M. Zhang and X. Zhao, Int. J. Biol. Macromol., 2020, 162, 1414–1428 CrossRef CAS PubMed.
- F. Yang, G. Guo and Y. Wang, Biomaterials, 2022, 289, 121761 CrossRef CAS PubMed.
- Y. Liang and K. L. Kiick, Acta Biomater., 2014, 10, 1588–1600 CrossRef CAS PubMed.
- X. Zhao, Q. Lang, L. Yildirimer, Z. Y. Lin, W. Cui, N. Annabi, K. W. Ng, M. R. Dokmeci, A. M. Ghaemmaghami and A. Khademhosseini, Adv. Healthcare Mater., 2016, 5, 108–118 CrossRef CAS PubMed.
- C. E. Campiglio, N. Contessi Negrini, S. Farè and L. Draghi, Materials, 2019, 12, 2476 CrossRef CAS PubMed.
- X. Wang, W. Dai, C. Gao, L. Zhang, Z. Wan, T. Zhang, Y. Wang, Y. Tang, Y. Yu and X. Yang, ACS Appl. Mater. Interfaces, 2023, 15, 58873–58887 CrossRef CAS PubMed.
- Y. Bian, T. Hu, Z. Lv, Y. Xu, Y. Wang, H. Wang, W. Zhu, B. Feng, R. Liang, C. Tan and X. Weng, Exploration, 2023, 3, 20210105 CrossRef PubMed.
- M. Mihajlovic, M. Rikkers, M. Mihajlovic, M. Viola, G. Schuiringa, B. C. Ilochonwu, R. Masereeuw, L. Vonk, J. Malda and K. Ito, Biomacromolecules, 2022, 23, 1350–1365 CrossRef CAS PubMed.
- C. C. Schuurmans, M. Mihajlovic, C. Hiemstra, K. Ito, W. E. Hennink and T. Vermonden, Biomaterials, 2021, 268, 120602 CrossRef CAS PubMed.
- B. Gong, X. Zhang, A. A. Zahrani, W. Gao, G. Ma, L. Zhang and J. Xue, Exploration, 2022, 2, 20210035 CrossRef PubMed.
- H. Zheng and B. Zuo, J. Mater. Chem. B, 2021, 9, 1238–1258 RSC.
- Y. Zhao, Z. S. Zhu, J. Guan and S. J. Wu, Acta Biomater., 2021, 125, 57–71 CrossRef CAS PubMed.
- H. Zhang, D. Xu, Y. Zhang, M. Li and R. Chai, Smart Med., 2022, 1, e20220011 CrossRef.
- C.-C. Lin, RSC Adv., 2015, 5, 39844–39853 RSC.
- X. Li, R. Yue, G. Guan, C. Zhang, Y. Zhou and G. Song, Exploration, 2023, 3, 20220002 CrossRef PubMed.
- S. Awasthi, J. K. Gaur, M. Bobji and C. Srivastava, J. Mater. Sci., 2022, 57, 8041–8063 CrossRef CAS.
- B. X. Wang, W. Xu, Z. Yang, Y. Wu and F. Pi, Macromol. Rapid Commun., 2022, 43, 2100785 CrossRef CAS PubMed.
- Z. Yuan, Z. Wan, Z. Tian, Y. Han, X. Huang, Y. Feng, W. Xie, X. Duan, S. Huang and X. Liu, Chem. Eng. J., 2022, 450, 138076 CrossRef CAS.
- D. Gan, W. Xing, L. Jiang, J. Fang, C. Zhao, F. Ren, L. Fang, K. Wang and X. Lu, Nat. Commun., 2019, 10, 1487 CrossRef PubMed.
- K. Haraguchi, Curr. Opin. Solid State Mater. Sci., 2007, 11, 47–54 CrossRef CAS.
- J. Yang, X.-P. Wang and X.-M. Xie, Soft Matter, 2012, 8, 1058–1063 RSC.
- M. Wang, J. Bai, K. Shao, W. Tang, X. Zhao, D. Lin, S. Huang, C. Chen, Z. Ding and J. Ye, Int. J. Polym. Mater. Polym. Sci., 2021, 2021, 1–16 CAS.
- M. Yang, Z. Wang, M. Li, Z. Yin and H. A. Butt, J. Vinyl Addit. Technol., 2022, 1, 1–21 Search PubMed.
- J. Tan, Y. Luo, Y. Guo, Y. Zhou, X. Liao, D. Li, X. Lai and Y. Liu, Int. J. Biol. Macromol., 2023, 124275 CrossRef CAS PubMed.
- H. Adelnia, R. Ensandoost, S. S. Moonshi, J. N. Gavgani, E. I. Vasafi and H. T. Ta, Eur. Polym. J., 2022, 164, 110974 CrossRef CAS.
- A. Oryan, A. Kamali, A. Moshiri, H. Baharvand and H. Daemi, Int. J. Biol. Macromol., 2018, 107, 678–688 CrossRef CAS PubMed.
- R. Ying, H. Wang, R. Sun and K. Chen, Mater. Sci. Eng., C, 2020, 110, 110689 CrossRef CAS PubMed.
- Z.-H. Huang, Y.-S. Dong, C.-L. Chu and P.-H. Lin, Mater. Lett., 2008, 62, 3376–3378 CrossRef CAS.
- L. Gu, Y. Zhang, L. Zhang, Y. Huang, D. Zuo, Q. Cai and X. Yang, Biomed. Mater., 2020, 15, 035012 CrossRef CAS PubMed.
- L. Shang, B. Ma, F. Wang, J. Li, S. Shen, X. Li, H. Liu and S. Ge, Cell Proliferation, 2020, 53, e12917 CrossRef CAS PubMed.
- M. Sukul, Y.-K. Min, S.-Y. Lee and B.-T. Lee, Eur. Polym. J., 2015, 73, 308–323 CrossRef CAS.
- P. Karalkin, N. Sergeyeva, I. Sviridova, V. Kirsanova, S. Akhmedova, Y. D. Shansky, N. Leontyev, D. Zuyev, E. Klimashina and P. Yevdokimov, Inorg. Mater. Appl. Res., 2020, 11, 1144–1152 CrossRef.
- Z.-K. Cui, S. Kim, J. J. Baljon, B. M. Wu, T. Aghaloo and M. Lee, Nat. Commun., 2019, 10, 3523 CrossRef PubMed.
- B. O. Okesola, A. K. Mendoza-Martinez, G. Cidonio, B. Derkus, D. K. Boccorh, D. Osuna de la Peña, S. Elsharkawy, Y. Wu, J. I. Dawson, A. W. Wark, D. Knani, D. J. Adams, R. O. C. Oreffo and A. Mata, ACS Nano, 2021, 15, 11202–11217 CrossRef CAS PubMed.
- C. Qin and C. Wu, View, 2022, 3, 20210018 CrossRef.
- H. Lu, S. Ren, P. Zhang, J. Guo, J. Li and G. Dong, RSC Adv., 2017, 7, 21600–21606 RSC.
- T. S. Wheeler, N. D. Sbravati and A. V. Janorkar, Ann. Biomed. Eng., 2013, 41, 2042–2055 CrossRef PubMed.
- J. Ramón-Azcón, S. Ahadian, M. Estili, X. Liang, S. Ostrovidov, H. Kaji, H. Shiku, M. Ramalingam, K. Nakajima and Y. Sakka, Adv. Mater., 2013, 25, 4028–4034 CrossRef PubMed.
- A. Vashist, A. Kaushik, A. Vashist, V. Sagar, A. Ghosal, Y. Gupta, S. Ahmad and M. Nair, Adv. Healthcare Mater., 2018, 7, 1701213 CrossRef PubMed.
- S. Wang, Q. Li, B. Wang, Y. Hou and T. Zhang, Ind. Eng. Chem. Res., 2019, 58, 21553–21561 CrossRef CAS.
- W. Hanif, A. Hardiansyah, A. Randy and L. A. Asri, RSC Adv., 2021, 11, 29029–29041 RSC.
- S. Liu, L. Xu, Z. Yuan, M. Huang, T. Yang and S. Chen, Langmuir, 2022, 38, 8200–8210 CrossRef CAS PubMed.
- J. Chen, X. Shi, L. Ren and Y. Wang, Carbon, 2017, 111, 18–27 CrossRef CAS.
- M. Li, Y. Liang, J. He, H. Zhang and B. Guo, Chem. Mater., 2020, 32, 9937–9953 CrossRef CAS.
- Y. Li, C. Liu, X. Lv and S. Sun, Soft Matter, 2021, 17, 2142–2150 RSC.
- S. Xia, S. Song, X. Ren and G. Gao, Soft Matter, 2017, 13, 6059–6067 RSC.
- X. Hu, X. Hao, Y. Wu, J. Zhang, X. Zhang, P. C. Wang, G. Zou and X.-J. Liang, J. Mater. Chem. B, 2013, 1, 1109–1118 RSC.
- P. Cui, P. Pan, L. Qin, X. Wang, X. Chen, Y. Deng and X. Zhang, Bioact. Mater., 2023, 19, 487–498 CAS.
- X. Lu, Y. Si, S. Zhang, J. Yu and B. Ding, Adv. Funct. Mater., 2021, 31, 2103117 CrossRef CAS.
- B.-Z. Yang, S.-Y. Zhang, P.-H. Wang, C.-H. Liu and Y.-Y. Zhu, Polymer, 2021, 228, 123863 CrossRef CAS.
- L. Guan, J. Chen, Z. Tian, M. Zhu, Y. Bian and Y. Zhu, View, 2021, 2, 20200117 CrossRef CAS.
- C. Liu, X. Cheng, F. Zhang, F. Lei, P. Li, K. Wang and J. Jiang, Mater. Today Commun., 2023, 34, 105256 CrossRef CAS.
- X. Han, Z. Lv, F. Ran, L. Dai, C. Li and C. Si, Int. J. Biol. Macromol., 2021, 176, 78–86 CrossRef CAS PubMed.
- W. C. Huang, R. Ying, W. Wang, Y. Guo, Y. He, X. Mo, C. Xue and X. Mao, Adv. Funct. Mater., 2020, 30, 2000644 CrossRef CAS.
- S. Wang, L. Yuan, Z. Xu, X. Lin, L. Ge, D. Li and C. Mu, ACS Appl. Bio Mater., 2021, 4, 5797–5808 CrossRef CAS PubMed.
- J. H. Ha, J. H. Lim, J. M. Lee and B. G. Chung, Polymers, 2023, 15, 2608 CrossRef CAS PubMed.
- K. S. Kumar, L. Zhang, M. S. Kalairaj, H. Banerjee, X. Xiao, C. C. Jiayi, H. Huang, C. M. Lim, J. Ouyang and H. Ren, ACS Appl. Mater. Interfaces, 2021, 13, 37816–37829 CrossRef CAS PubMed.
- Y. Wu, H. Wang, F. Gao, Z. Xu, F. Dai and W. Liu, Adv. Funct. Mater., 2018, 28, 1801000 CrossRef.
- B. Liu, J. Sun, J. Zhu, B. Li, C. Ma, X. Gu, K. Liu, H. Zhang, F. Wang and J. Su, Adv. Mater., 2020, 32, 2004460 CrossRef CAS PubMed.
- C. Lee, K. Lim, S. S. Kim, E. S. Lee, K. T. Oh, H.-G. Choi and Y. S. Youn, J. Controlled Release, 2019, 294, 77–90 CrossRef CAS PubMed.
- L. Jiao, W. Xu, H. Yan, Y. Wu, W. Gu, H. Li, D. Du, Y. Lin and C. Zhu, Chem. Commun., 2019, 55, 9865–9868 RSC.
- Y. Chen, W. Sheng, J. Lin, C. Fang, J. Deng, P. Zhang, M. Zhou, P. Liu, J. Weng and F. Yu, ACS Appl. Mater. Interfaces, 2022, 14, 7592–7608 CrossRef CAS PubMed.
- X. Lu, S. Shi, H. Li, E. Gerhard, Z. Lu, X. Tan, W. Li, K. M. Rahn, D. Xie and G. Xu, Biomaterials, 2020, 232, 119719 CrossRef CAS PubMed.
- C. Zhou, C. Wang, K. Xu, Z. Niu, S. Zou, D. Zhang, Z. Qian, J. Liao and J. Xie, Bioact. Mater., 2023, 25, 615–628 CAS.
- E. Lee, H. Lee, S. I. Yoo and J. Yoon, ACS Appl. Mater. Interfaces, 2014, 6, 16949–16955 CrossRef CAS PubMed.
- W. Chen and P. H. Kouwer, Adv. Funct. Mater., 2021, 31, 2105713 CrossRef CAS.
- M. Xu, Y. Song, J. Wang and N. Li, View, 2021, 2, 20200154 CrossRef CAS.
- K. Huang, W. Liu, W. Wei, Y. Zhao, P. Zhuang, X. Wang, Y. Wang, Y. Hu and H. Dai, ACS Nano, 2022, 16, 19491–19508 CrossRef CAS PubMed.
- Y. Zeng, C. Zhang, D. Du, Y. Li, L. Sun, Y. Han, X. He, J. Dai and L. Shi, Acta Biomater., 2022, 145, 43–51 CrossRef CAS PubMed.
- Y. Kang, C. Xu, X. Dong, M. Qi and D. Jiang, Bioact. Mater., 2022, 18, 26–41 CAS.
- Y. Li, J. Wang, H. Li, M. Guo, X. Sun, C. Liu and C. Yu, Macromol. Rapid Commun., 2023, 44, 2300268 CrossRef CAS PubMed.
- Z. Zohreband, M. Adeli and A. Zebardasti, Int. J. Biol. Macromol., 2021, 182, 2048–2055 CrossRef CAS PubMed.
- Y. Zu, H. Yao, Y. Wang, L. Yan, Z. Gu, C. Chen, L. Gao and W. Yin, View, 2021, 2, 20200188 CrossRef CAS.
- J. Zhou, T. Li, M. Zhang, B. Han, T. Xia, S. Ni, Z. Liu, Z. Chen and X. Tian, J. Nanobiotechnol., 2023, 21, 330 CrossRef CAS PubMed.
- Y. Xu, C. Xu, L. He, J. Zhou, T. Chen, L. Ouyang, X. Guo, Y. Qu, Z. Luo and D. Duan, Bioact. Mater., 2022, 16, 271–284 CAS.
- K. Huang, J. Wu and Z. Gu, ACS Appl. Mater. Interfaces, 2018, 11, 2908–2916 CrossRef PubMed.
- Q. Ding, T. Sun, W. Su, X. Jing, B. Ye, Y. Su, L. Zeng, Y. Qu, X. Yang and Y. Wu, Adv. Healthcare Mater., 2022, 11, 2102791 CrossRef CAS PubMed.
- Z. Zhao, R. Fang, Q. Rong and M. Liu, Adv. Mater., 2017, 29, 1703045 CrossRef PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- B. Xu, P. Zheng, F. Gao, W. Wang, H. Zhang, X. Zhang, X. Feng and W. Liu, Adv. Funct. Mater., 2017, 27, 1604327 CrossRef.
- J. Yang, C.-R. Han, X.-M. Zhang, F. Xu and R.-C. Sun, Macromolecules, 2014, 47, 4077–4086 CrossRef CAS.
- P. Schexnailder and G. Schmidt, Colloid Polym. Sci., 2009, 287, 1–11 CrossRef CAS.
- R. Sinko, S. Mishra, L. Ruiz, N. Brandis and S. Keten, ACS Macro Lett., 2014, 3, 64–69 CrossRef CAS PubMed.
- Y. Cai, J. Shen, C.-W. Yang, Y. Wan, H.-L. Tang, A. A. Aljarb, C. Chen, J.-H. Fu, X. Wei and K.-W. Huang, Sci. Adv., 2020, 6, eabb5367 CrossRef CAS PubMed.
- E. Zhang, X. Zhao, J. Hu, R. Wang, S. Fu and G. Qin, Bioact. Mater., 2021, 6, 2569–2612 CAS.
- Y. Xie, S. Chen, X. Peng, X. Wang, Z. Wei, J. J. Richardson, K. Liang, H. Ejima, J. Guo and C. Zhao, Bioact. Mater., 2022, 16, 95–106 CAS.
- P. Zhuang, Y. Yao, X. Su, Y. Zhao, K. Liu, X. Wu and H. Dai, Composites, Part B, 2022, 242, 110030 CrossRef CAS.
- O. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- D. Yu, M. Ma, Z. Liu, Z. Pi, X. Du, J. Ren and X. Qu, Biomaterials, 2020, 255, 120160 CrossRef CAS PubMed.
- H. Xu, Y. Zhu, J. Xu, W. Tong, S. Hu, Y. F. Chen, S. Deng, H. Yao, J. Li and C. W. Lee, Bioeng. Transl. Med., 2023, 8, e10345 CrossRef CAS PubMed.
- C.-P. Jen, Y.-H. Chen, C.-S. Fan, C.-S. Yeh, Y.-C. Lin, D.-B. Shieh, C.-L. Wu, D.-H. Chen and C.-H. Chou, Langmuir, 2004, 20, 1369–1374 CrossRef CAS PubMed.
- E. Nakamura, H. Isobe, N. Tomita, M. Sawamura, S. Jinno and H. Okayama, Angew. Chem., 2000, 112, 4424–4427 CrossRef.
- Q. Lin, Q. Huang, C. Li, C. Bao, Z. Liu, F. Li and L. Zhu, J. Am. Chem. Soc., 2010, 132, 10645–10647 CrossRef CAS PubMed.
- T.-W. Kim, P.-W. Chung, I. I. Slowing, M. Tsunoda, E. S. Yeung and V. S.-Y. Lin, Nano Lett., 2008, 8, 3724–3727 CrossRef CAS PubMed.
- M. Sevilla, P. Valle-Vigón, P. Tartaj and A. B. Fuertes, Carbon, 2009, 47, 2519–2527 CrossRef CAS.
- L. Chengnan, Q. Pagneux, A. Voronova, A. Barras, A. Abderrahmani, V. Plaisance, V. Pawlowski, N. Hennuyer, B. Staels and L. Rosselle, Nanoscale, 2019, 11, 15810–15820 RSC.
- J. Y. An, W. Um, D. G. You, Y. Song, J. Lee, N. Van Quy, H. Joo, J. Jeon and J. H. Park, Int. J. Biol. Macromol., 2021, 193, 553–561 CrossRef CAS PubMed.
- J. Su, S. Lu, S. Jiang, B. Li, B. Liu, Q. Sun, J. Li, F. Wang and Y. Wei, Adv. Mater., 2021, 33, 2100619 CrossRef CAS PubMed.
- T. Chen, T. Yao, H. Peng, A. K. Whittaker, Y. Li, S. Zhu and Z. Wang, Adv. Funct. Mater., 2021, 31, 2106079 CrossRef CAS.
- M. Antman-Passig and O. Shefi, Nano Lett., 2016, 16, 2567–2573 CrossRef CAS PubMed.
- A. Schroeder, R. Honen, K. Turjeman, A. Gabizon, J. Kost and Y. Barenholz, J. Controlled Release, 2009, 137, 63–68 CrossRef CAS PubMed.
- E. Guibal, N. V. O. Sweeney, M. Zikan, T. Vincent and J. Tobin, Int. J. Biol. Macromol., 2001, 28, 401–408 CrossRef CAS PubMed.
- B. O. Okesola, A. K. Mendoza-Martinez, G. Cidonio, B. Derkus, D. K. Boccorh, D. Osuna de la Peña, S. Elsharkawy, Y. Wu, J. I. Dawson and A. W. Wark, ACS Nano, 2021, 15, 11202–11217 CrossRef CAS PubMed.
- S. Wang, L. Yu, S. Wang, L. Zhang, L. Chen, X. Xu, Z. Song, H. Liu and C. Chen, Nat. Commun., 2022, 13, 3408 CrossRef CAS PubMed.
- H. Yue, Z. Shang, P. Xu, D. Feng and X. Li, Sep. Purif. Technol., 2022, 280, 119767 CrossRef CAS.
- C. Gao, W. Dai, X. Wang, L. Zhang, Y. Wang, Y. Huang, Z. Yuan, X. Zhang, Y. Yu, X. Yang and Q. Cai, Adv. Funct. Mater., 2023, 33, 2304829 CrossRef CAS.
- R. Barbucci, D. Pasqui, G. Giani, M. De Cagna, M. Fini, R. Giardino and A. Atrei, Soft Matter, 2011, 7, 5558–5565 RSC.
- P. Zhao, Y. Feng, Y. Zhou, C. Tan and M. Liu, Bioact. Mater., 2023, 20, 355–367 CAS.
- X. Hu, G. Nian, X. Liang, L. Wu, T. Yin, H. Lu, S. Qu and W. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 10292–10300 CrossRef CAS PubMed.
- L. Yang, X. Zhao, Y. Kong, R. Li, T. Li, R. Wang, Z. Ma, Y.-m. Liang, S. Ma and F. Zhou, Int. J. Biol. Macromol., 2023, 229, 814–824 CrossRef CAS PubMed.
- H. Yao, Q. Fu, Y. Zhang, Y. Wan and Q. Min, Int. J. Biol. Macromol., 2023, 253, 126619 CrossRef CAS PubMed.
- S. Zhu, B. Zhao, M. Li, H. Wang, J. Zhu, Q. Li, H. Gao, Q. Feng and X. Cao, Bioact. Mater., 2023, 26, 306–320 CAS.
- C. García-Astrain, M. Miljevic, I. Ahmed, L. Martin, A. Eceiza, L. Fruk, M. Corcuera and N. Gabilondo, J. Mater. Sci., 2016, 51, 5073–5081 CrossRef.
- S. M. Morozova, Gels, 2023, 9, 102 CrossRef CAS PubMed.
- W. Aljohani, M. W. Ullah, X. Zhang and G. Yang, Int. J. Biol. Macromol., 2018, 107, 261–275 CrossRef CAS PubMed.
- J. Czechowska, A. Zima, D. Siek and A. Ślósarczyk, Ceram. Int., 2018, 44, 6533–6540 CrossRef CAS.
- R. Yang, G. Li, C. Zhuang, P. Yu, T. Ye, Y. Zhang, P. Shang, J. Huang, M. Cai and L. Wang, Sci. Adv., 2021, 7, eabg3816 CrossRef CAS PubMed.
- G. Bovone, E. A. Guzzi, S. Bernhard, T. Weber, D. Dranseikiene and M. W. Tibbitt, Adv. Mater., 2022, 34, 2106941 CrossRef CAS PubMed.
- M. Wu, J. Chen, W. Huang, B. Yan, Q. Peng, J. Liu, L. Chen and H. Zeng, Biomacromolecules, 2020, 21, 2409–2420 CrossRef CAS PubMed.
- M. C. Mañas-Torres, G. B. Ramírez-Rodríguez, J. I. García-Peiro, B. Parra-Torrejón, J. M. Cuerva, M. T. Lopez-Lopez, L. Á. de Cienfuegos and J. M. Delgado-López, Inorg. Chem. Front., 2022, 9, 743–752 RSC.
- M. K. Jaiswal, J. R. Xavier, J. K. Carrow, P. Desai, D. Alge and A. K. Gaharwar, ACS Nano, 2016, 10, 246–256 CrossRef CAS PubMed.
- Q. Min, C. Wang, Y. Zhang, D. Tian, Y. Wan and J. Wu, Nanomaterials, 2022, 12, 1874 CrossRef CAS PubMed.
- T. Maki, R. Yoshisaki, S. Akama and M. Yamanaka, Polym. J., 2020, 52, 931–938 CrossRef CAS.
- R. Randriantsilefisoa, C. Nie, B. Parshad, Y. Pan, S. Bhatia and R. Haag, Chem. Commun., 2020, 56, 3547–3550 RSC.
- K. M. Rao, A. Kumar and S. S. Han, Polym. Test., 2017, 64, 175–182 CrossRef CAS.
- C. Nazli, G. S. Demirer, Y. Yar, H. Y. Acar and S. Kizilel, Colloids Surf., B, 2014, 122, 674–683 CrossRef CAS PubMed.
- X. Ai, C. Ho, J. Aw, A. Attia, J. Mu, Y. Wang, X. Wang, Y. Wang, X. Liu and H. Chen, Nat. Commun., 2016, 7, 10432 CrossRef CAS PubMed.
- J. Qu, X. Zhao, P. X. Ma and B. Guo, Acta Biomater., 2017, 58, 168–180 CrossRef CAS PubMed.
- X. Lin, L. Miao, X. Wang and H. Tian, Colloids Surf., B, 2020, 195, 111200 CrossRef CAS PubMed.
- F. Pan, G. Giovannini, S. Zhang, S. Altenried, F. Zuber, Q. Chen, L. F. Boesel and Q. Ren, Acta Biomater., 2022, 145, 172–184 CrossRef CAS PubMed.
- Z. Xu, G. Liu, J. Huang and J. Wu, ACS Appl. Mater. Interfaces, 2022, 14, 7680–7689 CrossRef CAS PubMed.
- Y. E. Kim and J. Kim, ACS Appl. Mater. Interfaces, 2021, 14, 23002–23021 CrossRef PubMed.
- H. Zhao, J. Huang, Y. Li, X. Lv, H. Zhou, H. Wang, Y. Xu, C. Wang, J. Wang and Z. Liu, Biomaterials, 2020, 258, 120286 CrossRef CAS PubMed.
- H. Wu, F. Li, W. Shao, J. Gao and D. Ling, ACS Cent. Sci., 2019, 5, 477–485 CrossRef CAS PubMed.
- Z. Fang, E. Yang, Y. Du, D. Gao, G. Wu, Y. Zhang and Y. Shen, J. Mater. Chem. B, 2022, 10, 966–976 RSC.
- K. F. Chu and D. E. Dupuy, Nat. Rev. Cancer, 2014, 14, 199–208 CrossRef CAS PubMed.
- Q. Yi, J. Ma, K. Kang and Z. Gu, J. Mater. Chem. B, 2016, 4, 4922–4933 RSC.
- M. Hou, W. Liu, L. Zhang, L. Zhang, Z. Xu, Y. Cao, Y. Kang and P. Xue, Biomater. Sci., 2019, 8, 353–369 RSC.
- Y. Zheng, Z. Chen, Q. Jiang, J. Feng, S. Wu and A. Del Campo, Nanoscale, 2020, 12, 13654–13661 RSC.
- T. L. Rapp and C. A. DeForest, Adv. Healthcare Mater., 2020, 9, 1901553 CrossRef CAS PubMed.
- Kenry, Y. Duan and B. Liu, Adv. Mater., 2018, 30, 1802394 CrossRef PubMed.
- Y. Yu, D. Tang, C. Liu, Q. Zhang, L. Tang, Y. Lu and H. Xiao, Adv. Mater., 2022, 34, 2105976 CrossRef CAS PubMed.
- L. Li, C. Wang, Q. Huang, J. Xiao, Q. Zhang and Y. Cheng, J. Mater. Chem. B, 2018, 6, 2474–2480 RSC.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, 1807333 CrossRef PubMed.
- E. Wajs, T. T. Nielsen, K. L. Larsen and A. Fragoso, Nano Res., 2016, 9, 2070–2078 CrossRef CAS.
- S. Miyamoto, N. Miyake, L. Jarskog, W. Fleischhacker and J. Lieberman, Mol. Psychiatry, 2012, 17, 1206–1227 CrossRef CAS PubMed.
- J. S. Lopez and U. Banerji, Nat. Rev. Clin. Oncol., 2017, 14, 57–66 CrossRef CAS PubMed.
- L. Wang, Y. Yu, D. Wei, L. Zhang, X. Zhang, G. Zhang, D. Ding, H. Xiao and D. Zhang, Adv. Mater., 2021, 33, 2100599 CrossRef CAS PubMed.
- Y. Huang, D. Wei, B. Wang, D. Tang, A. Cheng, S. Xiao, Y. Yu and W. Huang, Acta Biomater., 2023, 160, 198–210 CrossRef CAS PubMed.
- G. Xiong, D. Huang, L. Lu, X. Luo, Y. Wang, S. Liu, M. Chen, S. Yu, M. Kappen and C. You, Small Methods, 2022, 6, 2200379 CrossRef CAS PubMed.
- S. H. Yun and S. J. Kwok, Nat. Biomed. Eng., 2017, 1, 0008 CrossRef CAS PubMed.
- Y. Zhang, K. Ren, X. Zhang, Z. Chao, Y. Yang, D. Ye, Z. Dai, Y. Liu and H. Ju, Biomaterials, 2018, 163, 55–66 CrossRef CAS PubMed.
- B. Guo, Z. Feng, D. Hu, S. Xu, E. Middha, Y. Pan, C. Liu, H. Zheng, J. Qian and Z. Sheng, Adv. Mater., 2019, 31, 1902504 CrossRef PubMed.
- P. Zhu, Y. Chen and J. Shi, Adv. Mater., 2020, 32, 2001976 CrossRef CAS PubMed.
- C. Pucci, A. Marino, Ö. Şen, D. De Pasquale, M. Bartolucci, N. Iturrioz-Rodríguez, N. di Leo, G. de Vito, D. Debellis and A. Petretto, Acta Biomater., 2022, 139, 218–236 CrossRef CAS PubMed.
- S.-R. Yang, R. Wang, C.-J. Yan, Y.-Y. Lin, Y.-J. Yeh, Y.-Y. Yeh and Y.-C. Yeh, Biomater. Sci., 2023, 11, 4184–4199 RSC.
- X. Wang, L. Qiao, X. Yu, X. Wang, L. Jiang and Q. Wang, ACS Biomater. Sci. Eng., 2019, 5, 5888–5896 CrossRef CAS PubMed.
- W. Shi, J. Huang, R. Fang and M. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 5177–5194 CrossRef CAS PubMed.
- C. Wu, Y. Shen, M. Chen, K. Wang, Y. Li and Y. Cheng, Adv. Mater., 2018, 30, 1705673 CrossRef PubMed.
- S.-h. Noh, S. H. Moon, T.-H. Shin, Y. Lim and J. Cheon, Nano Today, 2017, 13, 61–76 CrossRef CAS.
- G. Dai, L. Sun, J. Xu, G. Zhao, Z. Tan, C. Wang, X. Sun, K. Xu and W. Zhong, Acta Biomater., 2021, 129, 84–95 CrossRef CAS PubMed.
- M. A. Marsudi, R. T. Ariski, A. Wibowo, G. Cooper, A. Barlian, R. Rachmantyo and P. J. Bartolo, Int. J. Mol. Sci., 2021, 22, 11543 CrossRef CAS PubMed.
- X. Zhao, P. Li, B. Guo and P. X. Ma, Acta Biomater., 2015, 26, 236–248 CrossRef CAS PubMed.
- S. Maiz-Fernández, L. Pérez-Álvarez, I. L. de Munain-Arroniz, A. Zoco, A. C. Lopes, U. Silván, D. Salazar, J. L. Vilas-Vilela and S. Lanceros-Mendez, Int. J. Biol. Macromol., 2022, 219, 374–383 CrossRef PubMed.
- Z. Lv, T. Hu, Y. Bian, G. Wang, Z. Wu, H. Li, X. Liu, S. Yang, C. Tan and R. Liang, Adv. Mater., 2023, 35, 2206545 CrossRef CAS PubMed.
- S. Ravi, L. P. Chokkakula, P. S. Giri, G. Korra, S. R. Dey and S. N. Rath, ACS Appl. Mater. Interfaces, 2023, 15, 19921–19936 CrossRef CAS PubMed.
- B. K. Sun, Z. Siprashvili and P. A. Khavari, Science, 2014, 346, 941–945 CrossRef CAS PubMed.
- X. Wang and M. Tang, Smart Med., 2022, 1, e20220032 CrossRef.
- O. R. Mahon, D. C. Browe, T. Gonzalez-Fernandez, P. Pitacco, I. T. Whelan, S. Von Euw, C. Hobbs, V. Nicolosi, K. T. Cunningham and K. H. Mills, Biomaterials, 2020, 239, 119833 CrossRef CAS PubMed.
- W. Li, Y. Zheng, W. Pang and P. Lai, Biomed. Technol., 2023, 1, 65–72 CrossRef CAS.
- J. Nandhini, E. Karthikeyan and S. Rajeshkumar, Biomed. Technol., 2024, 6, 26–45 CrossRef.
- H. Nosrati, R. Aramideh Khouy, A. Nosrati, M. Khodaei, M. Banitalebi-Dehkordi, K. Ashrafi-Dehkordi, S. Sanami and Z. Alizadeh, J. Nanobiotechnol., 2021, 19, 1–21 CrossRef PubMed.
- G. Theocharidis, H. Yuk, H. Roh, L. Wang, I. Mezghani, J. Wu, A. Kafanas, M. Contreras, B. Sumpio and Z. Li, Nat. Biomed. Eng., 2022, 6, 1118–1133 CrossRef CAS PubMed.
- B. J. Kim, D. X. Oh, S. Kim, J. H. Seo, D. S. Hwang, A. Masic, D. K. Han and H. J. Cha, Biomacromolecules, 2014, 15, 1579–1585 CrossRef CAS PubMed.
- H. Jung, M. K. Kim, J. Y. Lee, S. W. Choi and J. Kim, Adv. Funct. Mater., 2020, 30, 2004407 CrossRef CAS.
- Y. Wang, Z. Yuan, Y. Pang, D. Zhang, G. Li, X. Zhang, Y. Yu, X. Yang and Q. Cai, ACS Appl. Mater. Interfaces, 2023, 15, 20661–20676 CrossRef CAS PubMed.
- M. H. Kim, B. S. Kim, H. Park, J. Lee and W. H. Park, Int. J. Biol. Macromol., 2018, 109, 57–64 CrossRef CAS PubMed.
- X. Hou, Y. Chen, F. Chen, J. Liu, T. Wang, Y. Luo, S. Jia, P. Wang, S. Tan and B. Lu, Composites, Part B, 2022, 228, 109396 CrossRef CAS.
- P. J. Harwood and D. O. Ferguson, Orthop. Traumatol, 2015, 29, 228–242 Search PubMed.
- B. H. Shan and F. G. Wu, Adv. Mater., 2023, 2210707, DOI:10.1002/adma.202210707.
- Y. Mao, Y. Zhang, Y. Wang, T. Zhou, B. Ma and P. Zhou, Regener. Biomater., 2023, rbad046 CrossRef CAS PubMed.
- Y. Liu, Z. Yang, L. Wang, L. Sun, B. Y. S. Kim, W. Jiang, Y. Yuan and C. Liu, Adv. Sci., 2021, 8, 2100143 CrossRef CAS PubMed.
- P. Habibovic and J. Barralet, Acta Biomater., 2011, 7, 3013–3026 CrossRef CAS PubMed.
- K. H. Thompson and C. Orvig, Science, 2003, 300, 936–939 CrossRef CAS PubMed.
- C. Li, W. Zhang, R. Wang, X.-F. Du, D. Jiang, B. Liu, Y. Nie, J. Liao, Y. Chen and X. Liang, Chem. Eng. J., 2022, 435, 134896 CrossRef CAS.
- Y. Chen, W. Liu, S. Wan, H. Wang, Y. Chen, H. Zhao, C. Zhang, K. Liu, T. Zhou, L. Jiang, Q. Cheng and X. Deng, Adv. Funct. Mater., 2024, 34, 2309191 CrossRef CAS.
- D. J. Huey, J. C. Hu and K. A. Athanasiou, Science, 2012, 338, 917–921 CrossRef CAS PubMed.
- H. Chen, J. Sun, C. D. Hoemann, V. Lascau-Coman, W. Ouyang, M. D. McKee, M. S. Shive and M. D. Buschmann, J. Orthop. Res., 2009, 27, 1432–1438 CrossRef PubMed.
- C. Zhu, W. Zhang, Z. Shao, Z. Wang, B. Chang, X. Ding and Y. Yang, J. Mater. Res. Technol., 2023, 23, 154–164 CrossRef CAS.
- W. Wang, J. Duan, W. Ma, B. Xia, F. Liu, Y. Kong, B. Li, H. Zhao, L. Wang, K. Li, Y. Li, X. Lu, Z. Feng, Y. Sang, G. Li, H. Xue, J. Qiu and H. Liu, Adv. Sci., 2023, 10, 2205859 CrossRef CAS PubMed.
- T. Zhou, J. Ran, P. Xu, L. Shen, Y. He, J. Ye, L. Wu and C. Gao, Carbohydr. Polym., 2022, 292, 119667 CrossRef CAS PubMed.
- S. Iqbal, M. Fakhar-e-Alam, K. Alimgeer, M. Atif, A. Hanif, N. Yaqub, W. Farooq, S. Ahmad, Y.-M. Chu and M. S. Rana, Saudi J. Biol. Sci., 2021, 28, 1226–1232 CrossRef CAS PubMed.
- Y. He, C. Cong, S. Zhao, Z. Li, D. Wang, J. Gu, L. Liu and D. Gao, Biomater. Sci., 2021, 9, 2313–2321 RSC.
- H. Zhang, R. Shi, A. Xie, J. Li, L. Chen, P. Chen, S. Li, F. Huang and Y. Shen, ACS Appl. Mater. Interfaces, 2013, 5, 12317–12322 CrossRef CAS PubMed.
- X. Yin, T. Fan, N. Zheng, J. Yang, L. Yan, S. He, F. Ai and J. Hu, Nanoscale Adv., 2023, 5, 1729–1739 RSC.
- T. He, X. Qin, C. Jiang, D. Jiang, S. Lei, J. Lin, W.-G. Zhu, J. Qu and P. Huang, Theranostics, 2020, 10, 2453 CrossRef CAS PubMed.
- S. Wang, H. Zheng, L. Zhou, F. Cheng, Z. Liu, H. Zhang and Q. Zhang, Biomaterials, 2020, 260, 120314 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |