
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nano-in-nano enteric protein delivery system: coaxial Eudragit® L100-55 fibers containing poly(N-vinylcaprolactam) nanogels†
Ana
Sonzogni
 a,
Guadalupe
Rivero
b,
Verónica
Gonzalez
a,
Gustavo
Abraham
b,
Marcelo
Calderón
*cd and
Roque
Minari
*a
a,
Guadalupe
Rivero
b,
Verónica
Gonzalez
a,
Gustavo
Abraham
b,
Marcelo
Calderón
*cd and
Roque
Minari
*a
aINTEC (UNL-CONICET), Güemes 3450, Santa Fe 3000, Argentina. E-mail: rjminari@santafe-conicet.gov.ar
bINTEMA (UNMDP-CONICET), Av. Colón 10850, B7606BWV Mar del Plata, Argentina
cPOLYMAT, Applied Chemistry Department, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo M. de Lardizabal 3, 20018 Donostia-San Sebastián, Spain. E-mail: marcelo.calderonc@ehu.eus
dIKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Spain
First published on 22nd November 2023
Abstract
Oral protein delivery holds significant promise as an effective therapeutic strategy for treating a wide range of diseases. However, effective absorption of proteins faces challenges due to biological barriers such as harsh conditions of the stomach and the low permeability of mucous membranes. To address these challenges, this article presents a novel nano-in-nano platform designed for enteric protein delivery. This platform, obtained by electrospinning, involves a coaxial arrangement comprising poly(N-vinylcaprolactam) nanogels (NGs) enclosed within nanofibers of Eudragit® L100-55 (EU), a pH-responsive polymer. The pH-selective solubility of EU ensures the protection of NGs during their passage through the stomach, where the fibers remain intact at low pH, and releases them in the intestine where EU dissolves. The switchable characteristic of this nano-in-nano platform is confirmed by using NGs loaded with a model protein (ovalbumin), which is selectively released when the intestinal pH is achieved. The versatility of this nano-in-nano delivery platform is demonstrated by the ability to modify the fibers dissolution profile simply by adjusting the concentration of EU used in the electrospinning process. Furthermore, by tuning the properties of NGs, the potential applications of this platform can be further extended, paving the way for diverse therapeutic possibilities.
1. Introduction
Therapeutic proteins have emerged as crucial pharmaceutical agents because of their high success in different disease treatments.1 Proteins exhibit high selectivity through specific interactions with targets, reducing side effects of treatments compared to small drugs.2 While oral administration is the preferred administration route because its safety, patient compliance, ease of application, pain avoidance, and versatility, proteins are typically delivered via parenteral routes. This is primarily due to their poor oral bioavailability, as proteins face multiple challenges such as acid inactivation, enzymatic degradation, and low mucosal permeability.3 Therefore, the development of formulations that overcome gastrointestinal barriers and facilitate oral protein delivery is imperative.The use of nanoparticles for targeted and controlled drug delivery has played a key role in achieving effective treatments and minimizing adverse effects.4 Nanogels (NGs), crosslinked polymer nanoparticles have shown promise in controlled and targeted release due to their high water retention, large loading capacities, stability in biological fluids, and biocompatibility.5–7 However, their application in oral delivery is limited by their large exposed area and porosity, which limit the protection of their cargo from the harsh conditions of the stomach. Previous studies have explored strategies to bypass gastric conditions, such as coating PLGA nanoparticles with Eudragit® FS30D, a pH-responsive polymer soluble at a pH above 7, leading to their degradation and release in the large intestine.8 In the same way, mannosylated chitosan nanoparticles coated with Eudragit® L100, which dissolves at pH 6, have demonstrated resistance to acid and enzyme degradation in gastrointestinal fluid and selective release of an antigenic protein in the intestine.9 Although these nanoparticulate systems allowed to protect the protein from stomach conditions, the release in the intestine was not complete, reaching approximately 30% protein released after 12 h. The use of nanofibers constitutes a promising approach for accelerating the release time of a cargo molecule during oral delivery, due to their large surface-to-volume ratio.10
As there remains unexplored the effective combination both systems (NGs and nanofibers) in a coaxial arrangement, in this work we investigate the synthesis of a nano-in-nano polymer system based on Eudragit® L100-55 (EU) and poly(N-vinylcaprolactam) (PVCL) NGs. The system consists of coaxial nanofibers formed by a continuous EU sheath, a pH-responsive polymer that dissolves at pH above 5.5,11 that contains protein–laden PVCL NGs axially distributed. Fig. 1 shows the two-step process employed to obtain the system. Initially, PVCL NGs were synthesized by emulsion polymerization (Fig. 1a), followed by coaxial electrospinning to fabricate fibers (Fig. 1b). The selective solubility of EU throughout the gastrointestinal tract ensures the protection of NGs during passage through the stomach, where fibers remain unaltered, and it facilitates their release in the duodenum (pH 6) where the EU dissolves (Fig. 1c). Although fibers loaded with NGs were previously synthesized to improve the biological activity of the protein after the electrospinning process,12 this article pursues the coaxial arrangement to ensure the protection of the protein during the stomach passage. Indeed, coaxial electrospinning was specifically chosen to achieve good coverage of NGs by internally injecting their dispersion along with an external EU flow.13 In addition, this processing technique prevents the protein from coming into contact with organic solvents of EU phase, which could potentially damage it. This nano-in-nano platform combines the advantages of using electrospun fibers for oral delivery,10 providing a matrix-protective structure with a large surface area and interconnected porosity,14 along with the versatile properties of NGs as a protein delivery and targeting system.15–17
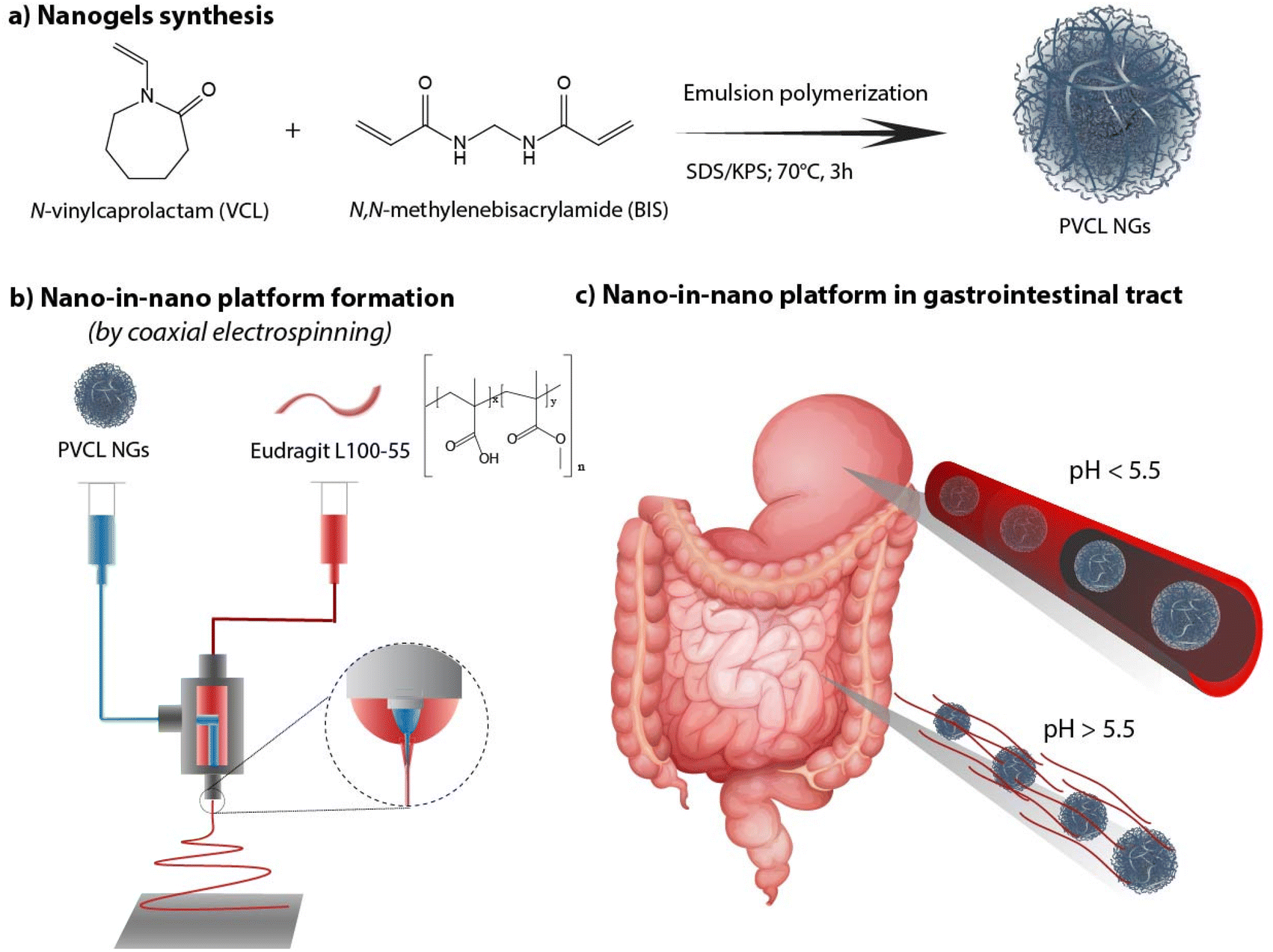 | ||
| Fig. 1 PVCL NGs synthesis (a), fibers formation (b) and nano-in-nano system in the gastrointestinal tract (c). | ||
2. Materials and methods
2.1. Materials
For NGs synthesis the following reagents were used: N-vinylcaprolactam (VCL, 98% purity, Sigma) as monomer, N,N-methylenebis(acrylamide) (BIS, 99.9% purity, Genbiotech) as cross-linker, potassium persulfate (KPS, 99% purity, Mallinckrodt) as initiator, sodium bicarbonate (NaHCO3, 99.7% purity, Cicarelli) as buffer, sodium dodecyl sulfate (SDS, Anedra) as emulsifier, and hydroquinone as polymerization inhibitor (99% purity, Fluka AG). Eudragit® L100-55 was gently provided by Evonik. Ethanol and N,N-dimethylformamide (DMF) were purchased from Cicarelli. Ovalbumin from chicken egg (OVA, 98% purity, Sigma) was used as model protein. All reagents were employed as received without purification.2.2. Methods
2.2.1.1. Nano-in-nano system preparation. For fibers preparation, a coaxial electrospinning process was employed. All electrospinning experiments were conducted under ambient conditions, at a temperature of 23 ± 4 °C and relative humidity of 58 ± 6%. The EU solution was prepared using ethanol
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF (4:1) as a binary solvent and two EU concentrations (150 and 200 mg mL−1) were tested. On the other hand, the NGs concentration was maintained at 10 mg mL−1 in all syntheses. The polymer solutions were electrospun in a YFLOW® 2.2.D-350 equipment with single or coaxial nozzles, and an aluminum flat collector placed 17 cm away. The flow rates were varied from 0.1 to 2 mL h−1, while the voltage was adjusted between 15 and 19 kV to achieve a stable electrospinning process. After processing, all membranes were dried and stored protected from light until further use.
:DMF (4:1) as a binary solvent and two EU concentrations (150 and 200 mg mL−1) were tested. On the other hand, the NGs concentration was maintained at 10 mg mL−1 in all syntheses. The polymer solutions were electrospun in a YFLOW® 2.2.D-350 equipment with single or coaxial nozzles, and an aluminum flat collector placed 17 cm away. The flow rates were varied from 0.1 to 2 mL h−1, while the voltage was adjusted between 15 and 19 kV to achieve a stable electrospinning process. After processing, all membranes were dried and stored protected from light until further use.
2.2.2.1. NGs characterization. Monomer conversion and average particle diameter were measured in the withdrawn samples during the reactions. Conversion of VCL and BIS was determined by 1H NMR spectroscopy, following the method reported by Imaz et al.18 The spectra were recorded in a Bruker 300 MHz spectrometer using a Watergate (83919GP, relaxation time (D1): 1.00 s) technique to eliminate the water signal. VCL conversion calculation was based on the peak areas corresponding to the VCL vinyl bond and the internal standard (AcNa). Average hydrodynamic particle diameter (Dp) was measured by dynamic light scattering (DLS) at a detection angle of 90° in a Brookhaven BI-9000 AT photometer. The thermal response of the final NGs was also determined by DLS by measuring the Dp of dilute samples (1 mg mL−1) at increasing temperatures (from 20 up to 55 °C with 0.5 °C intervals and 2 min of stabilization between measurements). The volume phase transition temperature (VPTT) was defined as the temperature of the inflection point of the Dpvs. temperature curve.
The morphology of the NGs was evaluated using transmission electron microscopy (TEM, JEOL JEM-2100 Plus). Sample preparation involved depositing a diluted NGs dispersion with a concentration of 1 mg mL−1 onto Formvar®-coated copper grids and allowing them to dry at room temperature. NGs size distributions were obtained from TEM images. Image-Pro Plus® software was used for measuring NGs diameters, with at least 300 measurements per sample.
An OVA loaded PVCL NGs dispersion (with a final NGs content of 5 mg mL−1) was prepared by re-dispersing the required mass of lyophilized NGs in a 4 mg mL−1 OVA solution and incubating it for 48 h at 4 °C. After the incubation period, the suspension was filtered using a VIVASPIN centrifugal filter (molecular weight cut-off, MWCO, 300 kDa) at 7227 g and for 15 min to separate the loaded NGs from the unloaded protein solution. The concentration of OVA in the supernatant before the NGs re-dispersion and after filtration was measured using the Bradford assay,20 to determine the loading capacity (LC) and encapsulation efficiency (EE, %) of the NGs as follows:
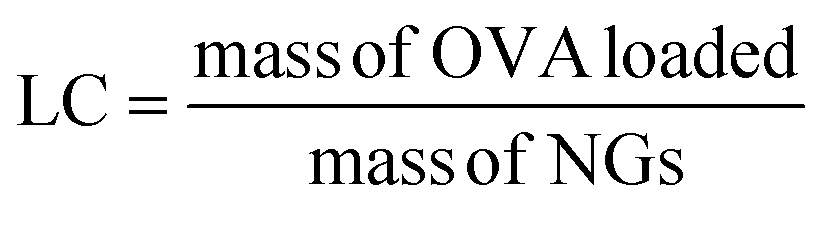
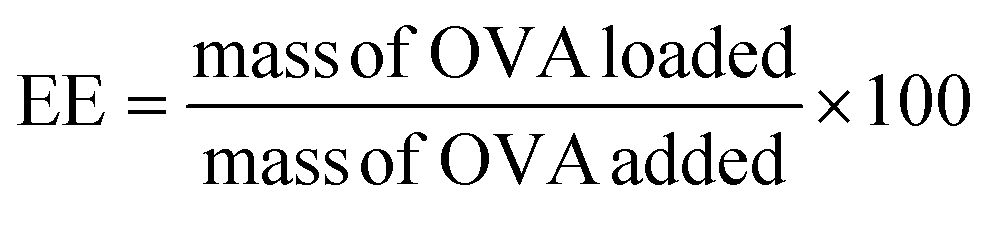
OVA release study was conducted by incubating 1 mL of PVCL NGs dispersion (with a NGs concentration of 5 mg mL−1) loaded with 3.8 mg of OVA in 6 mL of PBS (pH = 7.4) at 25 and 37 °C for 1 week. Assays were carried out per triplicate. The release medium was replaced at regular intervals, and the amount of released protein was measured using the Bradford assay.20
2.2.2.2. Fibers characterization. Fiber morphology was examined by scanning electron microscopy (SEM, Jeol JSM-6460LV), after sputtering with gold/palladium. Fibers size distributions were obtained from SEM images. Image-Pro Plus® software was used for measuring fiber diameters, with at least 300 measurements per sample. The internal morphology was studied using TEM. For this purpose, fibers were directly electrospun and collected on a copper grid for 10 seconds. The sample was stained with a 1% phosphotungstic acid solution and observed a 200 kV.
To determine the degradation profiles of the nano-in-nano systems, nanofibrous membranes were incubated at 37 °C in HCl 0.1 N and PBS pH 6.8 to simulate gastric and intestine pH conditions, respectively. The weight of the membranes was recorded at different times. In the same assay, NGs release was also studied. Media samples were withdrawn to measure the transmittance at 500 nm using a PerkinElmer Lambda 950 UV/Vis spectrophotometer after incubation for 5 min at 40 °C. NGs concentration was determined by correlating a calibration curve under the same conditions. The surface morphology of the membranes was examined by SEM on a representative sample immersed in PBS 6.8 for 5 min to observe fibers morphology evolution during degradation.
The interactions between NGs and EU was investigated by FTIR and TGA. FTIR analysis was performed using a Thermo Scientific Nicolet 6700 Fourier transform infrared spectrometer. The spectra were recorded over a range of 500–4000 cm−1 with a resolution of 2 cm−1 and averaged over 64 scans. TGA was conducted by heating the samples from 40 to 500 °C with a constant heating rate of 10 °C min−1 under nitrogen atmosphere using a Q500 equip from TA instruments.
In vitro OVA release was evaluated by immersing 35 mg of fibers in 10 mL of HCl 0.1 N (pH 1.2) for 2 h, followed by 8 h in 10 mL of PBS (pH 6.8). Experiments were performed in a shaking bath at 37 °C per triplicate. The release medium was replaced at regular time intervals, and the amount of released protein was measured using the Bradford assay.20 For the transition from HCl 0.1 N to PBS environment, the medium was simply replaced in the same vial while maintaining assessed fibers remained during the whole experiment. For fibers containing NGs, samples were centrifugated for 10 min at 10000 rpm and 10 °C to separate loaded NGs from released OVA. Then, the OVA concentration was measured from the supernatant.
3. Results and discussion
3.1. Nanogels synthesis
PVCL NGs were synthesized through batch emulsion polymerization using BIS as a crosslinker. A fast polymerization kinetic was observed, reaching a maximum conversion of VCL and BIS at around 20 min (Fig. S1a†). The growth of NGs diameter correlated with monomer and BIS conversion, indicating that particles nucleation occurred only at the early stages of the reaction (Fig. S1b†). The resulting PVCL NGs exhibited a spherical morphology with irregular edges and an average diameter of 87.3 nm (Fig. 2a), as determined by TEM.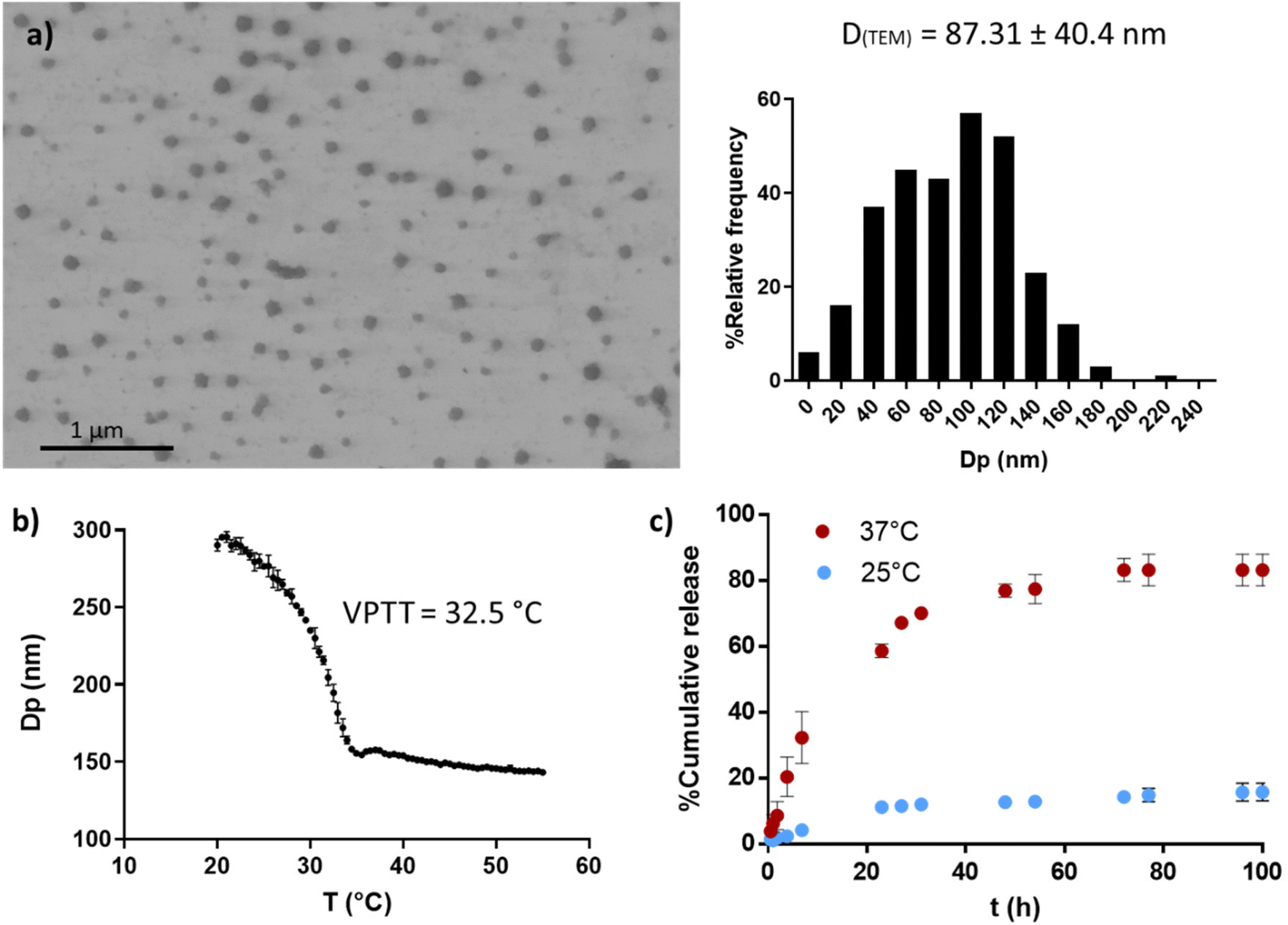 | ||
| Fig. 2 PVCL NGs properties. TEM image and size distribution (a), thermal response determined by DLS (b) and OVA release profile at 25 and 37 °C (c). | ||
The NGs demonstrated thermoresponsive behavior, exhibiting a transition temperature (VPTT) of 32.5 °C (Fig. 2b) in aqueous media. This suggests that the NGs are expected to be collapsed at the human body temperature. Furthermore, PVCL NGs displayed the ability to efficiently load OVA with a LC and EE% of 0.94 and 94%, respectively. As it can be seen in Fig. 2c, the release of OVA from the NGs showed temperature dependence. At 37 °C, OVA was released at a faster rate compared to 25 °C, attributed to the enhanced diffusion of the protein driven by the collapse of the NGs. The minimal release of OVA at 25 °C suggests the presence of strong interactions between the protein and the NGs network. Consequently, a precise temperature-controlled ON–OFF effect was observed. This result is important because NGs should be able to retain the loaded OVA during the nano-in-nano system formation, a process developed at temperatures near 25 °C.
3.2. Fibers preparation
For the preparation of the nano-in-nano system through electrospinning, PVCL NGs and EU L100-55 solutions were simultaneously injected using a coaxial nozzle. Compositional and processing parameters were adjusted to achieve a stable Taylor cone, as illustrated in Fig. S2,† where two nearly conical menisci enable the formation of concentric jets. In the initial stage, different flow rates of both phases (referred to as FNG and FEU, representing the flow rates of NGs dispersion and EU solution, respectively) were tested while maintaining a constant NGs concentration of 10 mg mL−1 and EU concentration of 150 mg mL−1, along with a voltage ranging from 15 to 17 kV. The resulting average fiber diameters obtained with different flow rates are presented in Table 1, and SEM images along with size distribution are shown in Fig. 3.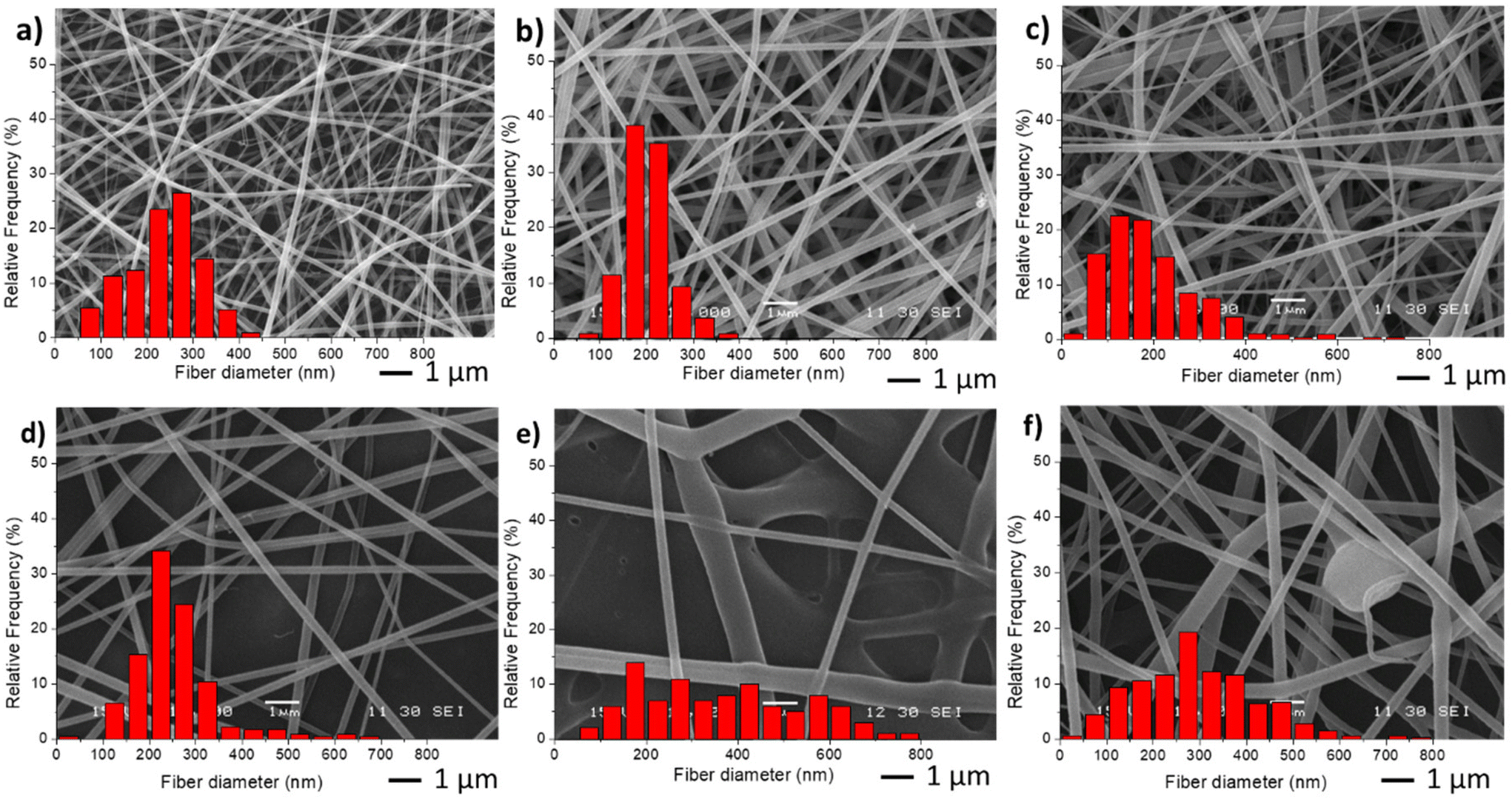 | ||
| Fig. 3 SEM images and diameter distribution from fiber samples M0 (a), M1(b), M2 (c), M3 (d), M4 (e) and M5 (f). | ||
| Fiber code | Flow rate NGs (FNG) | Flow rate EU (FEU) | D (nm) |
|---|---|---|---|
| M0 | — | 0.6 | 233.7 ± 79.6 |
| M1 | 0.1 | 0.6 | 199.8 ± 48.4 |
| M2 | 0.1 | 0.8 | 197.4 ± 100.4 |
| M3 | 0.5 | 1.5 | 248.6 ± 89.9 |
| M4 | 0.5 | 2.0 | 395.4 ± 214.4 |
| M5 | 0.6 | 0.6 | 297.9 ± 141.5 |
The incorporation of NGs as the inner phase, reduces the diameter of fibers, with respect to EU pure fibers (M0) because in the coaxial electrospinning the presence of NGs dispersion, and hence of water, enhances conductivity. As anticipated, an increase in both flow rates (M3 and M4) led to larger fiber diameters and greater size dispersion. Similar results were observed when increasing FEU while keeping FNG constant. Additionally, when the same flow rate of both phases was applied (sample M5), the stability of the Taylor cone was compromised, resulting in fibers with some defects (Fig. 3f).
In the second stage, different NGs dispersion media were used during the electrospinning process, while maintaining the optimal values of FNG = 0.1 and FEU = 0.6, which yielded the thinnest fibers with a narrow distribution (Table 2). In all cases, the average diameter of the fibers remained similar. The obtained results demonstrate that when using both distilled water and PBS, the resulting fibers and Taylor cone exhibited close resemblance (Fig. 4a and b). Conversely, when ultrapure water was used, a higher applied voltage was required, likely due to the lower conductivity of the dispersion (Table 2). Consequently, the Taylor cone changed into a more conical shape (Fig. 4c), and the electrospinning process became less stable. As it is known, in the case of low conductivity solutions, the surface of a charged droplet can not form a Taylor cone which leads to microjet formation. Increasing solution conductivity enables charged droplet to overcome surface tension and to eject microjets of polymer solutions that produce nanofibers with lower diameter.21
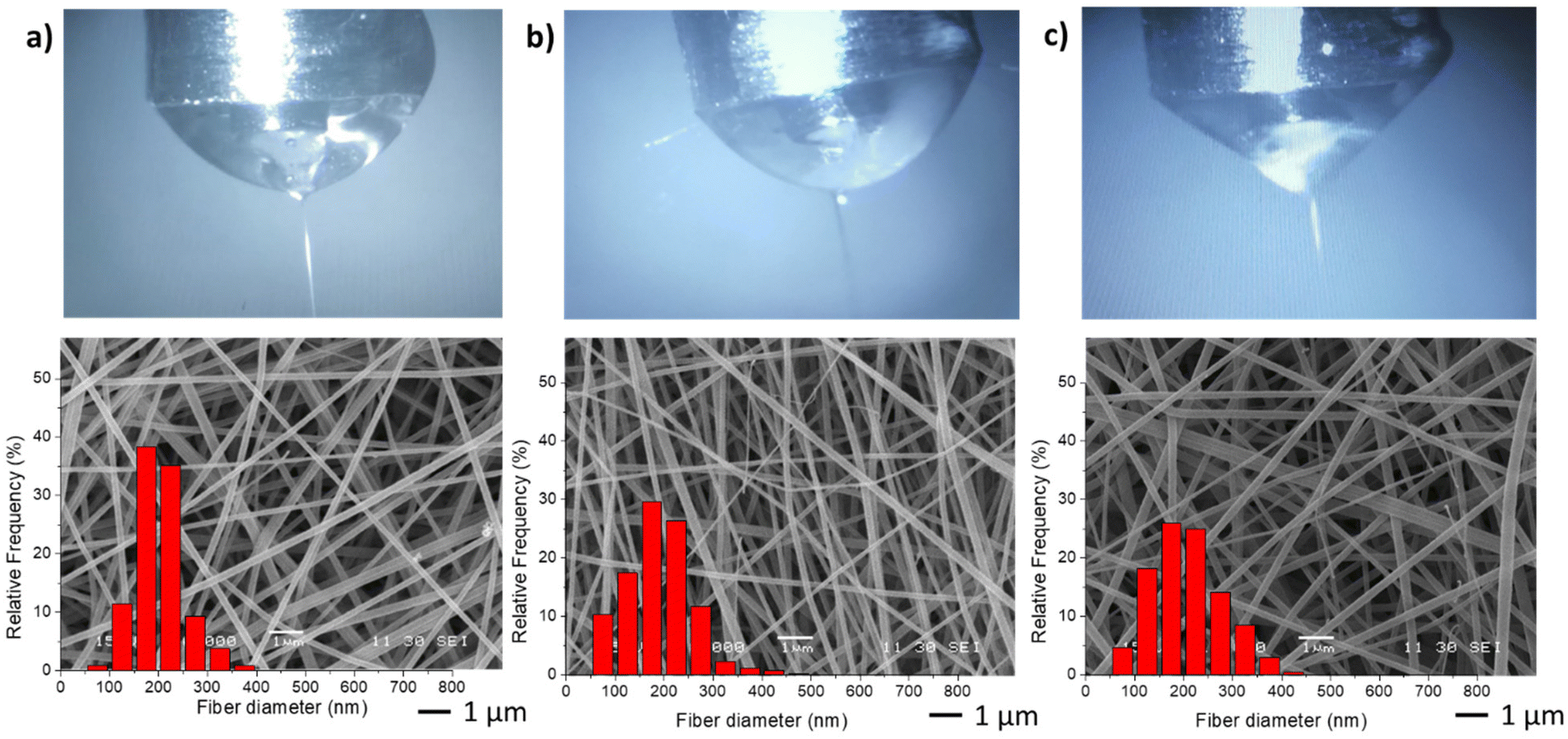 | ||
| Fig. 4 Taylor cones and SEM image and diameter distribution of resulting fibers samples M1 (a), M6 (b) and M7 (c). | ||
| Code | Dispersion media | Conductivity (S m−1) | D (nm) |
|---|---|---|---|
| M1 | Distilled water | 0.119 ± 0.002 | 199.8 ± 48.4 |
| M6 | PBS | 1.280 ± 0.056 | 185.1 ± 54.2 |
| M7 | Ultrapure water | 0.083 ± 0.003 | 199.5 ± 69.2 |
TEM observations were conducted to examine the internal morphology of the fibers. Fig. 5 displays TEM images of membrane M0 and M6. It can be seen in Fig. 5a EU fibers (M0) whose interior is homogeneous, while within the M6 fibers (Fig. 6b and c), elongated domains can be observed, which correspond to the PVCL NGs. Indeed, as it can be seen in Fig. S3,† size of these ellipsoids agree with NGs size observed by TEM (Fig. 2), with an approximate length of the major axis of 118 nm and of the minor axis of 70 nm. The elongation suffered by the NGs is likely due to their softness and flexibility, consistent with the deformation of the polymer matrix during the fiber formation. Similar morphologies were observed in other coaxial fibers (prepared by coaxial or emulsion electrospinning) as a result of droplet stretching from the discrete phase fluid during solvent evaporation.22 It is crucial to note that the NGs are fully encapsulated by an EU layer and not exposed to the surrounding medium. This result confirms the achievement of the desired morphology when employing the coaxial electrospinning method.
 | ||
| Fig. 5 TEM images of membranes M0 (a) and M6 (b, c). | ||
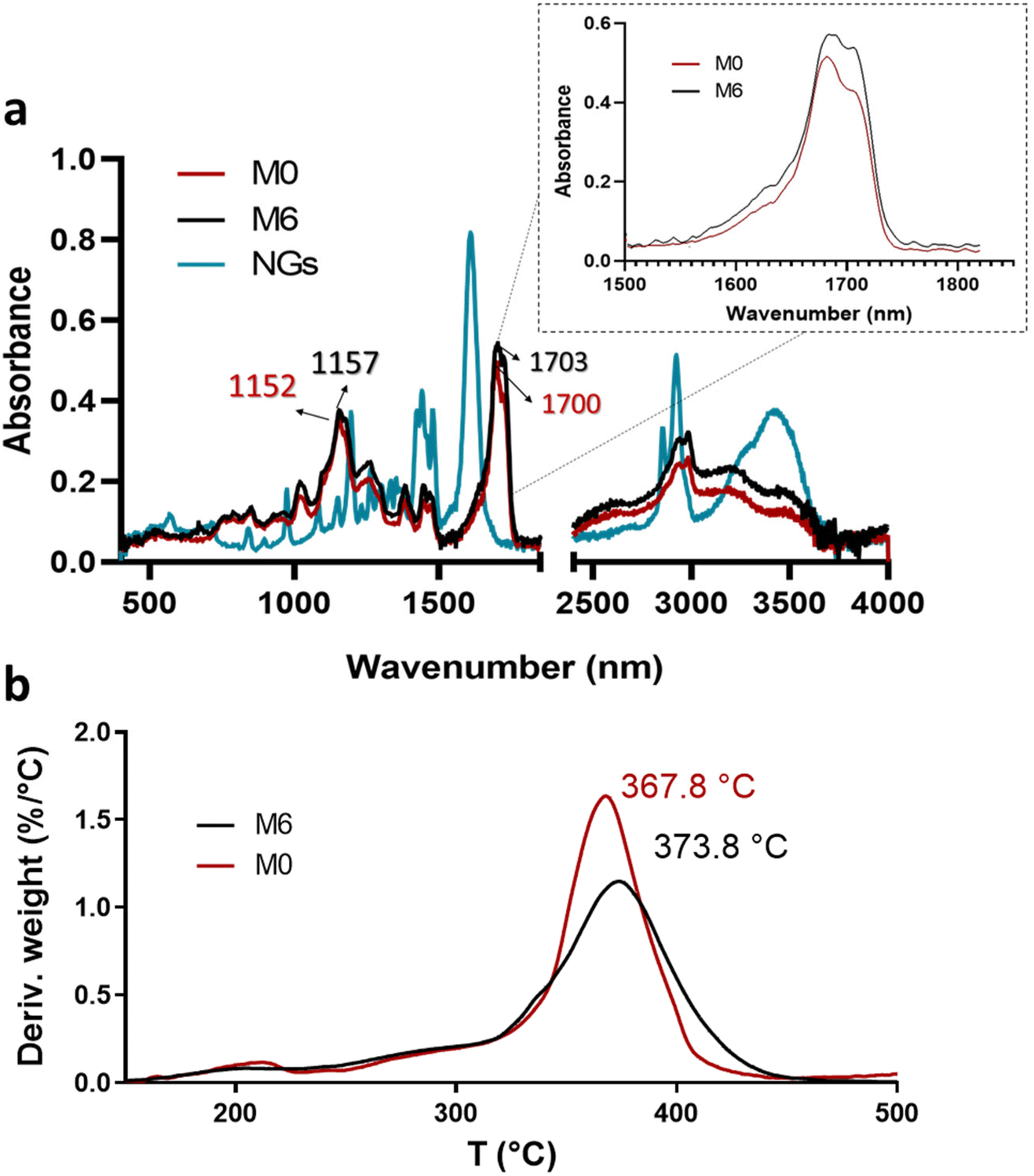 | ||
| Fig. 6 ATR-FTIR spectra of PVCL NGs, M0 and M6 (a) and profiles and DTG thermograms of membranes M0 and M6 (b). | ||
3.3. Interactions between fibers components
Although the mass ratio NGs/EU is very low in the synthesized nano-in-nano systems, FTIR and TGA analyses pursue to find evidence of interactions between these two materials. The ATR-FTIR spectra of the starting materials and electrospun nanofibers are presented in Fig. 6. The spectrum of the NGs exhibits signals attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N groups of PVCL at 1630 and 1479 cm−1 (Fig. 6a).18 In the case of pure EU (M0 fiber), the most prominent peaks are at 1152 and 1700 cm−1, corresponding to the stretching vibrations of C–O–C and CO groups, respectively. While the signals of PVCL NGs are not distinguishable in the spectra of the nano-in-nano systems (as they overlap with those from the major component), a small shift in the signals associated with the ester group of EU can be seen, and a change in the relative intensity of the carbonyl double peak around 1700–1703 cm−1. Even though these signal differences are not substantial, they represent an indication of the possible interactions between EU ester groups and NGs in the system, which is further supported by TGA analysis, where fibers containing PVCL NGs exhibited a higher temperature of maximum degradation rate compared to pure EU fibers (M0) (Fig. 6b).
O and C–N groups of PVCL at 1630 and 1479 cm−1 (Fig. 6a).18 In the case of pure EU (M0 fiber), the most prominent peaks are at 1152 and 1700 cm−1, corresponding to the stretching vibrations of C–O–C and CO groups, respectively. While the signals of PVCL NGs are not distinguishable in the spectra of the nano-in-nano systems (as they overlap with those from the major component), a small shift in the signals associated with the ester group of EU can be seen, and a change in the relative intensity of the carbonyl double peak around 1700–1703 cm−1. Even though these signal differences are not substantial, they represent an indication of the possible interactions between EU ester groups and NGs in the system, which is further supported by TGA analysis, where fibers containing PVCL NGs exhibited a higher temperature of maximum degradation rate compared to pure EU fibers (M0) (Fig. 6b).
3.4. Fibers degradation
The weight evolution of membranes M0 and M6 when incubated in buffers simulating stomach and intestine pH conditions (HCl 0.1 N and PBS 6.8) was registered to determine their degradation profiles (Fig. 7a). It is observed that pure EU fibers (M0) degraded at a faster rate than fibers containing NGs under both pH conditions. This result provides further evidence of the presence of interactions between NGs and EU in the formed fibers. In all cases, fibers experienced more weight loss when incubated in PBS 6.8 compared to HCl 1.2, due to the pH-dependent solubility of EU, which is soluble above pH 5.5. At stomach pH, fibers lost approximately 20% of their weight, while in PBS (pH = 6.8) membranes were completely degraded after 2 h of incubation. The release profile of NGs, shown in Fig. 7b, exhibits a similar trend to the degradation curves. These findings indicate that the proposed nano-in-nano system demonstrates the desired behavior for its application as an enteric delivery platform, where the cargo needs to be protected for 2 h during passage through the stomach and then released during residence in the intestine (10 h in the small intestine).2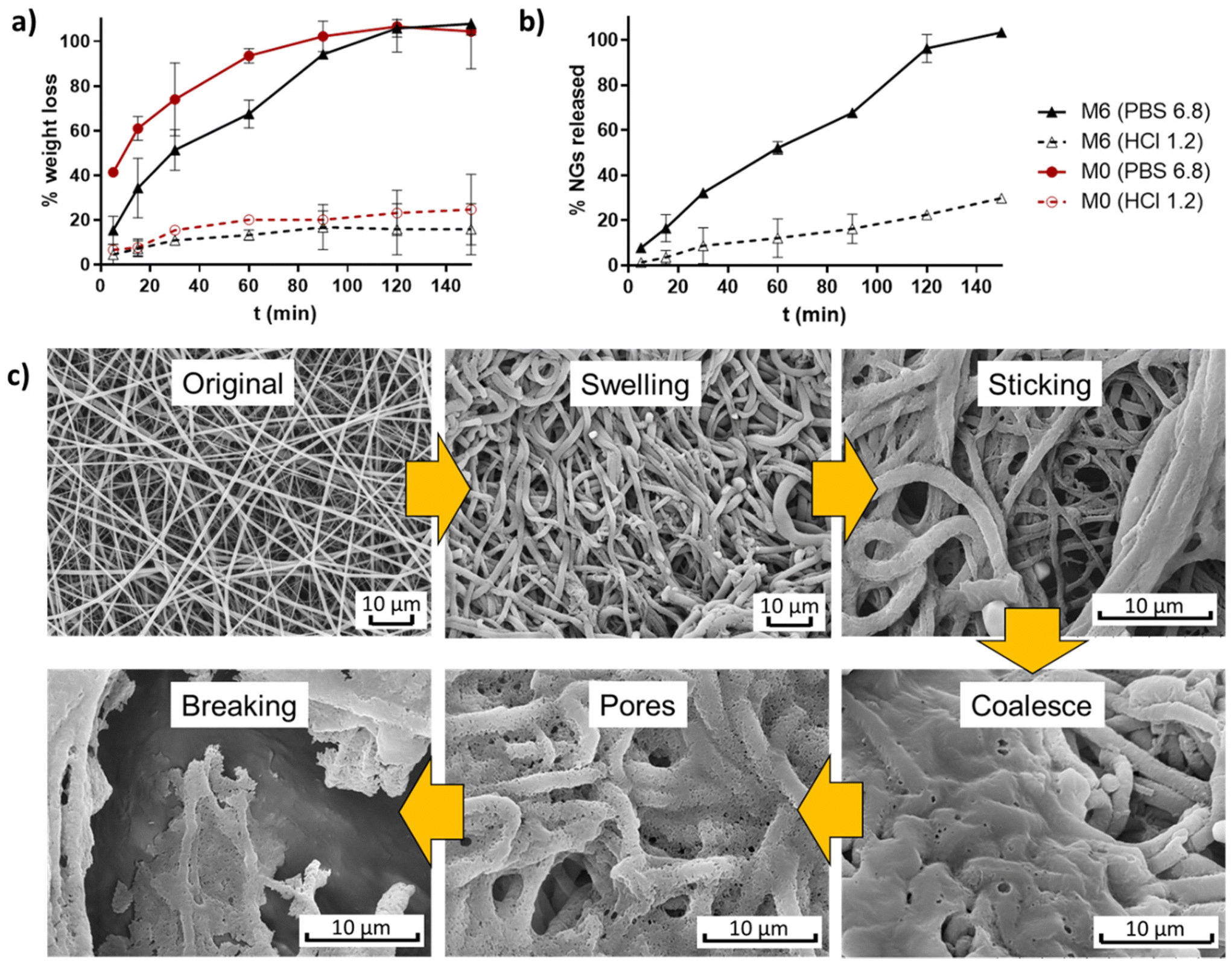 | ||
| Fig. 7 Degradation profiles of membranes M0 and M6 (a), and NGs release of M6 (b); and degradation mechanism proposed from SEM images of incubated M6 (PBS pH 6.8, 5 min) (c). | ||
The possible degradation mechanism of fibers proposed in Fig. 7c stems from the observations of M6 fibers incubated in PBS 6.8 for 5 min (representing the intestine pH, which is higher than the critical pH of EU). In this sample, different stages of degradation can be observed. First, fibers undergo swelling due to water diffusion into their matrix. The absorbed water leads to a significant reduction of the Tg of EU, as a result of the plasticity effect of water,23 thereby promoting membrane modifications such as fibers deformation, adhesion to adjacent fibers, and chain interdifussion that leads to fibers coalescence. Subsequently, the progressive dissolution of EU initiates the formation of pores within the coalesced matrix until the structure eventually breaks down. During this process, the release of NGs from the matrix begins, as evidenced by the appearance of larger pores than the size of NGs.
3.5. Protein loading
For the loading of protein into nano-in-nano system, coaxial fibers were produced by electrospinning using OVA-loaded NGs. In addition, EU fibers without NGs, were also loaded with OVA. To achieve this, an OVA solution in PBS was used as the inner solution in the coaxial nozzle. The electrospinning process employed fixed flow rates (FNG = 0.1 and FEU = 0.6) with varied concentration of EU. As expected, the EU:NGs ratio variation would allow the protein release profile tunning.
Table 3 shows the average fibers diameter obtained from SEM images (shown in Fig. 8) when OVA was encapsulated under different conditions. As expected, fiber diameters were found to be more sensitive to changes in the EU concentration compared to the effect of the encapsulation of OVA or loaded NGs. Increasing the EU concentration from 150 mg mL−1 to 200 mg mL−1 approximately doubled the diameter. The incorporation of loaded NGs into the fibers did not have a significant impact on the homogenous nanofibrous structure and its diameter.
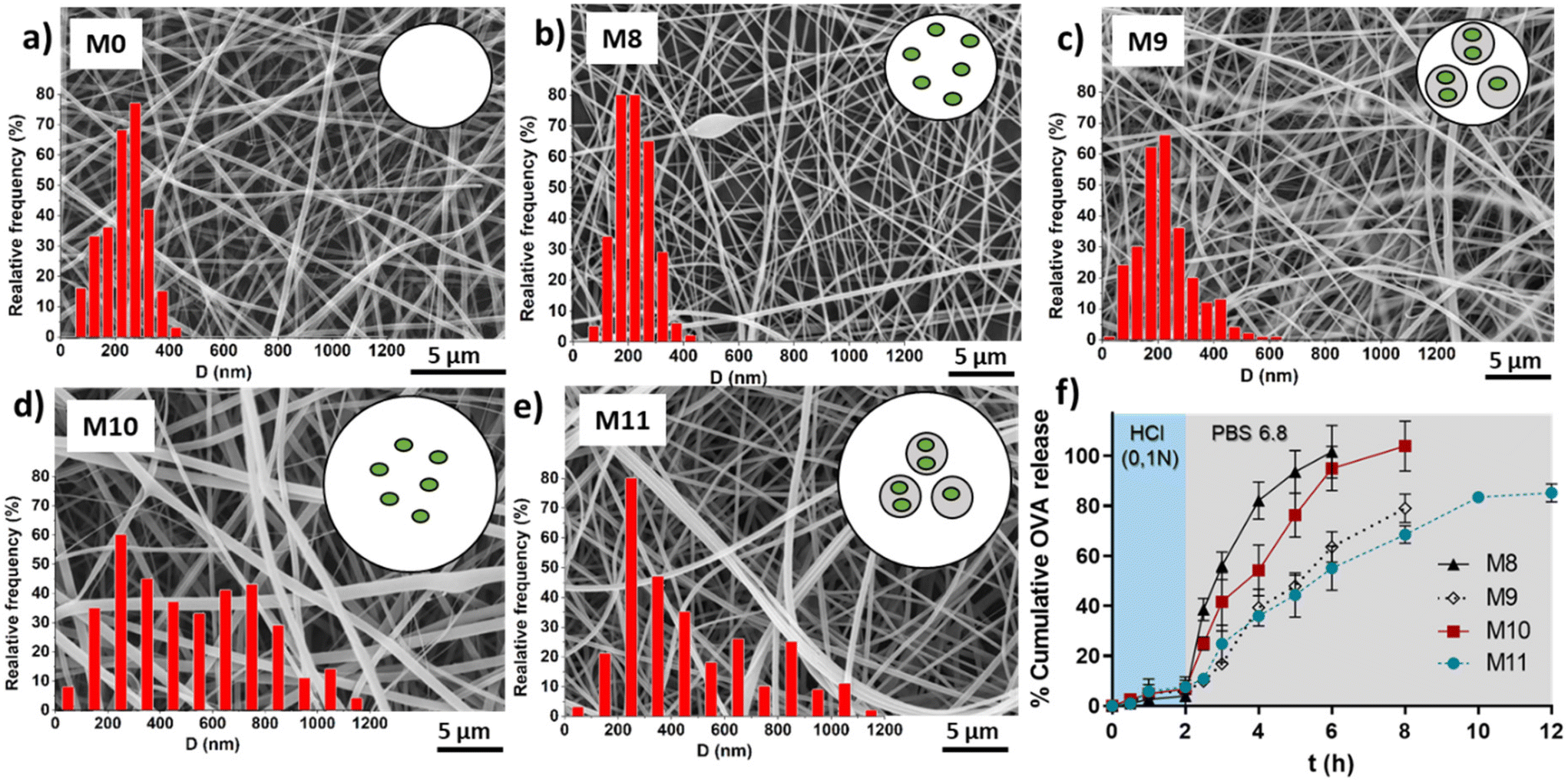 | ||
| Fig. 8 SEM images and scheme of a transversal cut of fibers M0 (a), M8 (b), M9 (c), M10 (d) and M11 (e). OVA release from M8, M9, M10 and M11 incubated for 2 h in HCl 0.1 N and then in PBS pH 6.8 until complete fibers dissolution (f). | ||
| Code | Outer solution | Inner solution | D (nm) |
|---|---|---|---|
| M0 | EU 150 mg mL−1 | — | 233.7 ± 79.6 |
| M8 | EU 150 mg mL−1 | OVA 10 mg mL−1 | 222.4 ± 63.5 |
| M9 | EU 150 mg mL−1 | NGs + OVA 10 mg mL−1 | 229.7 ± 100.6 |
| M10 | EU 200 mg mL−1 | OVA 10 mg mL−1 | 520.2 ± 282.1 |
| M11 | EU 200 mg mL−1 | NGs + OVA 10 mg mL−1 | 462.8 ± 263.9 |
3.6. Ovalbumin release
Finally, in vitro evaluation of OVA release from the loaded fibers was shown in Fig. 8f. OVA release profiles was obtained by incubating fibers M8, M9, M10 and M11 for 2 h in HCl 0.1 N followed by the incubation in PBS (pH 6.8) until complete dissolution of the fibers. It is important to clarify that the experiment extension depended on the tested sample, as it corresponded to the time at which the membrane was completely dissolved. This already provides valuable information, indicating that membranes with lower EU content dissolved earlier, and for the same EU concentration, those with NGs took longer times to dissolve. This observation is directly related to the smaller fiber diameter obtained at lower EU concentrations. Furthermore, as mentioned earlier, the incorporation of NGs makes the fibers more resistant to dissolution, likely due to the presence of physical interactions between NGs and EU.It is worth noting that in all cases, the release of OVA in HCl 0.1 N is lower than 10%, indicating that OVA is retained in the nano-in-nano system during 2 h (i.e., the equivalent residence time in the stomach). Also, once fibers were exposed to pH 6.8, they dissolved earlier with less EU, because of their higher exposed area, and as a consequence, presented a faster OVA release. In fibers containing NGs (M9 and M11), the OVA release is slower due to two factors: (i) fibers with NGs degrade at slower rate than fibers without NGs; and (ii) once NGs released from the fibers, OVA must diffuse from the NGs interior. This significant difference between the release kinetics from fibers with and without NGs evidenced that OVA was effectively loaded inside NGs. It is also notable that the final OVA release from M9 and M11 does not reach 100%. This is because NGs are separated from the released OVA in each sample, interrupting the release of the OVA that remains loaded in the NGs. As demonstrated earlier, free NGs are able to deliver the loaded OVA in a pH 6.8 medium after above 2 h, indicating that the release of the therapeutic protein should occur in the intestinal environment.
It is important to consider that OVA release from nano-in-nano system results are not comparable with OVA release from NGs, since both experiments were conducted in different conditions.
4. Conclusions
A nano-in-nano system for enteric protein delivery was successfully developed by incorporating NGs into electrospun coaxial nanofibers. This designed system demonstrated pH-responsive behavior, enabling the controlled release of NGs with a kinetic profile suitable for therapeutic enteric treatments. It effectively protected the protein cargo in the acidic environment of the stomach and facilitated its release in the duodenum at pH 6. The dissolution mechanism of the pH-responsive nanofibers was hypothesized to trigger the release.The synthesized PVCL NGs exhibited a size suitable for biological applications, allowing for active interaction with cells and the ability to cross biological membranes, among other functions. They were capable of efficiently loading a model protein (ovalbumin, 94%) and retaining it until reaching body temperature, thus ensuring complete release of the loaded cargo. The coaxial electrospinning process was optimized to obtain homogeneous defect-free nanofibrous structures with continuous porosity.
The innovative incorporation of NGs in nanofibers serves as a proof of concept for the potential versatility and applications of this platform. NGs, being highly versatile materials, offer a range of properties that can be tailored to meet specific needs in various fields.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge the financial support provided by ANPCyT (PICT-2016-3876, PICT2018-02334), CONICET (PUE 007, PIP 11220200101353CO), the Universidad Nacional del Litoral (CAI+D 50620190100117LI), IKERBASQUE-Basque Foundation for Science, MINECO project RTI2018-099227-B-I00, and the University of the Basque Country (projects COLLAB22/05 and GIU21/033). Special thanks go to David Esporrin for conducting SEM measurements and Santiago Vaillard for performing NMR measurements.References
- M. Gandhi, R. Srikar, A. L. Yarin, C. M. Megaridis and R. A. Gemeinhart, Mechanistic examination of protein release from polymer nanofibers, Mol. Pharm., 2009, 6, 641–647, DOI:10.1021/mp800160p.
- S. Haddadzadegan, F. Dorkoosh and A. Bernkop-Schnürch, Oral delivery of therapeutic peptides and proteins: Technology landscape of lipid-based nanocarriers, Adv. Drug Delivery Rev., 2022, 182, 114097, DOI:10.1016/J.ADDR.2021.114097.
- D. S. Dimitrov, Therapeutic Proteins, Methods Mol. Biol., 2012, 899, 1, DOI:10.1007/978-1-61779-921-1_1.
- A. L. Martínez-López, C. Pangua, C. Reboredo, R. Campión, J. Morales-Gracia and J. M. Irache, Protein-based nanoparticles for drug delivery purposes, Int. J. Pharm., 2020, 581, 119289, DOI:10.1016/j.ijpharm.2020.119289.
- N. Tiwari, A. S. Sonzogni and M. Calderón, Can dermal delivery of therapeutics be improved using thermoresponsive nanogels?, Nanomedicine, 2019, 14, 2891–2895, DOI:10.2217/nnm-2019-0345.
- A. S. Sonzogni, S. Hamzehlou, V. D. G. Gonzalez, J. R. Leiza and R. J. Minari, Multilobular morphology: the key for biphase multifunctional nanogels, Soft Matter, 2021, 17, 9353–9362, 10.1039/D1SM00968K.
- A. Sonzogni, G. Cabrera, G. Lupi, L. Gugliotta, V. Gonzalez, I. Marcipar and R. Minari, Film forming nanogels for needle-free transdermal vaccination, Macromol. Biosci., 2022, 22, 2100515, DOI:10.1002/mabi.202100515.
- Q. Zhu, J. Talton, G. Zhang, T. Cunningham, Z. Wang, R. C. Waters, J. Kirk, B. Eppler, D. M. Klinman, Y. Sui, S. Gagnon, I. M. Belyakov, R. J. Mumper and J. A. Berzofsky, Large intestine-targeted, nanoparticle-releasing oral vaccine to control genitorectal viral infection, Nat. Med., 2012, 18, 1291–1296, DOI:10.1038/nm.2866.
- B. Xu, W. Zhang, Y. Chen, Y. Xu, B. Wang and L. Zong, Eudragit® L100-coated mannosylated chitosan nanoparticles for oral protein vaccine delivery, Int. J. Biol. Macromol., 2018, 113, 534–542, DOI:10.1016/J.IJBIOMAC.2018.02.016.
- F. Ignatious, L. Sun, C. P. Lee and J. Baldoni, Electrospun nanofibers in oral drug delivery, Pharm. Res., 2010, 27, 576–588, DOI:10.1007/s11095-010-0061-6.
- P. Franco and I. de Marco, Eudragit: A Novel Carrier for Controlled Drug Delivery in Supercritical Antisolvent Coprecipitation, Polymers, 2020, 12, 234, DOI:10.3390/POLYM12010234.
- A. Shimoda, Y. Chen and K. Akiyoshi, Nanogel containing electrospun nanofibers as a platform for stable loading of proteins, RSC Adv., 2016, 6, 40811–40817, 10.1039/c6ra05997j.
- J. Li, Y. Liu and H. E. Abdelhakim, Drug Delivery Applications of Coaxial Electrospun Nanofibres in Cancer Therapy, Molecules, 2022, 27(6), 1803, DOI:10.3390/molecules27061803.
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, A review on polymer nanofibers by electrospinning and their applications in nanocomposites, Compos. Sci. Technol., 2003, 63, 2223–2253, DOI:10.1016/S0266-3538(03)00178-7.
- A. A. Attama, P. O. Nnamani, O. B. Onokala, A. A. Ugwu and A. L. Onugwu, Nanogels as target drug delivery systems in cancer therapy: A review of the last decade, Front. Pharmacol., 2022, 13, 1–23, DOI:10.3389/fphar.2022.874510.
- M. Giulbudagian, G. Yealland, S. Hönzke, A. Edlich, B. Geisendörfer, B. Kleuser, S. Hedtrich and M. Calderón, Breaking the Barrier – Potent Anti-Inflammatory Activity following Efficient Topical Delivery of Etanercept using Thermoresponsive Nanogels, Theranostics, 2018, 8, 450–463 CrossRef CAS https://www.thno.org/v08/p0450/thnov08p0450s1.pdf, (accessed November 23, 2017).
- R. Charbaji, M. Kar, L. E. Theune, J. Bergueiro, A. Eichhorst, L. Navarro, P. Graff, F. Stumpff, M. Calderón and S. Hedtrich, Design and Testing of Efficient Mucus-Penetrating Nanogels—Pitfalls of Preclinical Testing and Lessons Learned, Small, 2021, 17, 2007963, DOI:10.1002/SMLL.202007963.
- A. Imaz and J. Forcada, N -vinylcaprolactam-based microgels: Synthesis and characterization, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2510–2524, DOI:10.1002/pola.22583.
- A. S. Sonzogni, G. Yealland, M. Kar, S. Wedepohl, L. M. Gugliotta, V. D. G. Gonzalez, S. Hedtrich, M. Calderón and R. J. Minari, Effect of Delivery Platforms Structure on the Epidermal Antigen Transport for Topical Vaccination, Biomacromolecules, 2018, 19, 4607–4616, DOI:10.1021/acs.biomac.8b01307.
- M. M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem., 1976, 72, 248–254, DOI:10.1016/0003-2697(76)90527-3.
- C. J. Angammana and S. H. Jayaram, Analysis of the effects of solution conductivity on electrospinning process and fiber morphology, IEEE Trans. Ind. Appl., 2011, 47, 1109–1117, DOI:10.1109/TIA.2011.2127431.
- X. Wang, Y. Yuan, X. Huang and T. Yue, Controlled release of protein from core-shell nanofibers prepared by emulsion electrospinning based on green chemical, J. Appl. Polym. Sci., 2015, 132, 1–9, DOI:10.1002/app.41811.
- S. Rujivipat and R. Bodmeier, Moisture plasticization for enteric Eudragit® L30D-55-coated pellets prior to compression into tablets, Eur. J. Pharm. Biopharm., 2012, 81, 223–229, DOI:10.1016/J.EJPB.2012.01.003.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01422c |
| This journal is © The Royal Society of Chemistry 2024 |