
Mission immiscible: overcoming the miscibility limit of semiconducting:ferroelectric polymer blends via vitrification†
Aditi
Khirbat‡
 a,
Oded
Nahor‡
b,
Henry
Kantrow
c,
Oladipo
Bakare
a,
Artem
Levitsky
b,
Gitti L.
Frey
*b and
Natalie
Stingelin
*ac
a,
Oded
Nahor‡
b,
Henry
Kantrow
c,
Oladipo
Bakare
a,
Artem
Levitsky
b,
Gitti L.
Frey
*b and
Natalie
Stingelin
*ac
aSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: natalie.stingelin@gatech.edu
bDepartment of Material Science and Engineering, Technion – Israel Institute of Technology, Haifa 3200003, Israel. E-mail: gitti@technion.ac.il
cSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
First published on 18th May 2023
Abstract
Blending offers a versatile processing platform to combine multiple properties in a given material system that may not be realized in one single component, or to induce co-operatively entirely new features. Polymers can, however, be challenging to blend due to their low tendency to mix, especially when processed from the melt. Here, we demonstrate that essentially the entire spectrum of phase morphologies, from basically fully intermixed to strongly phase-separated, can be induced reliably in blends produced from the archetypal polymer semiconductor, poly(3-hexyl thiophene), P3HT, and poly(vinylidene fluoride), PVDF, a polymer that can exhibit ferroelectric polymorphs, despite the intrinsically limited miscibility featured by P3HT and PVDF. We achieve this by manipulating chain entanglements in solution, which in turn dictates the molecular mobility of the two components (i.e., mass transport during solidification), and in extreme cases leads to pronounced vitrification in the solid state. Since partly- to well-intermixed systems can be produced when processed from a good solvent for both components, we conclude that entanglements form between P3HT and PVDF molecules, provided their molecular weight and concentration is sufficiently high. Hence, specific phase morphologies can be targeted towards broad materials discovery via the establishment of reliable interrelationships between structure, phase morphology, and properties.
10th Anniversary StatementWe congratulate the Royal Society of Chemistry for the successful evolution of the Journal of Materials Chemistry into the Journals of Materials Chemistry A, B, C (JMC A, B, C). We have witnessed this transition, and have been delighted to follow the successes of JMC A, B, C – their increased breadth, enhanced reach, and raised visibility. We are frequent authors of the JMC A, B, C family and have always treasured their mission to bring materials chemistry to a broad audience. We also deeply appreciate the assistance JMC A, B, C has provided to the field, via support of conferences, symposia, and workshops, as well as the sponsorship of awards for speakers and poster prizes. We are looking forward to the next ten years of JMC A, B, C and the impact the journals have on the broader materials science field. We will be here to be part of the journals’ success! |
Introduction
Blending polymers is a common industrial strategy for generating new and/or improved properties typically unattainable with the blend's individual components. The blend properties, thereby, strongly depend on the specific characteristics of each component, their interactions, and the degree of intermixing.1Commodity polymer:polymer blends are generally processed from the melt to create bulk structures.1,2 During melt mixing, the number of rearrangement configurations in the Flory–Huggins lattice model is small, resulting in a low entropy of mixing (ΔSmix).3,4 Moreover, the enthalpy of mixing (ΔHmix) is typically positive due to the weak, or lack of, interactions between the segments of the different blend components. The combination of these thermodynamic values governs the Gibbs free energy of mixing (ΔGmix), i.e., the miscibility between blend components. The positive ΔGmix for polymer:polymer blends means that there is limited miscibility between the components leading, in most cases, to phase separation.1,2 Use of polymers with longer chains, i.e., high molecular weights, increases this tendency to phase separate3,4 because it further lowers ΔSmix.
Recently, polymer:polymer blends have found technological use also in the form of thin films, e.g., in the field of organic electronic and optoelectronic devices.5–8 The performance of these devices strictly depends on the microstructure and phase morphology of the active film, often on the nano-scale.9–13 In contrast to commodity polymers, such semiconducting films are produced from solution to realize structures of a thickness of 500 nm and below, which is required for most device platforms. This can complicate reliable processing as the dynamics of structure formation, in addition to the components’ thermodynamic properties, has a very pronounced impact on the resulting phase morphology. Hence, solvent evaporation rate14–18 and solution viscosity (dictated by the number of entanglement and, thus, molecular weight19,20 and solution concentration) play a critical role. As a consequence, small variations during processing can lead to entirely different solid-state structures. Phase transitions such as the glass transition temperature, Tg, need to be taken into account as well. The reason is that mass transport arrests at a temperature below Tg. Hence, the casting temperature, Tcast, plays a paramount role as it dictates how fast Tg is reached upon solvent evaporation and, in turn, can be used to induce vitrification. Vitrification generally limits phase separation because long-range mass transport is frustrated. This effect is enhanced in systems with a high density of chain entanglements, which limits the polymer chains’ molecular mobility/diffusivity.
Here we set out to elucidate whether mass transport can be frustrated in solutions of two functional polymers, i.e., poly(3-hexylthiophene), P3HT, a macromolecular semiconductor,21–23 and the fluoropolymer, poly(vinylidene fluoride), PVDF,24,25 to induce in a controlled manner a specific degree of vitrification and, thus, intermixing. We utilize differential scanning calorimetry (DSC) to obtain information on the phase behaviour and whether blending leads to vitrification. Vapor-phase infiltration (VPI) “staining” and scanning electron microscopy (SEM) are then used to visualize the induced phase morphologies, allowing the establishment of processing guidelines towards desired solid-state structures.
Results and discussion
We selected P3HT:PVDF binaries as model systems as these blends (and blends of their derivatives), when solution-processed, have been gaining interest for applications such as piezoelectric electro-spun fibres for novel water filtering systems,26 nanofibers for triboelectric nanogenerators,27 nanosheets for resistive non-volatile memories,28 and field effect transistors.29 P3HT:PVDF blend systems are expected to have limited or no thermodynamic miscibility due to the limited interactions between the polar PVDF and relatively non-polar P3HT.29 Both polymers also have strong self-interactions and a tendency to crystallize, increasing the likelihood of phase separation when processed from the melt. This may differ when processing from solution, allowing us to test whether we can manipulate the solid-state structure via control of mass transport during solidification, and specifically via solution vitrification.For initial investigations, we started with P3HT:PVDF blends (weight ratio 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25), using a P3HT and PVDF of a high weight-average molecular weight, Mw (130 kg mol−1 and 530 kg mol−1, respectively). High-molecular weight materials were selected to ensure P3HT and PVDF chains entangle so that mass transport during solidification from solution can be limited, which should assist vitrification, especially if entanglements form between the two components.
:25), using a P3HT and PVDF of a high weight-average molecular weight, Mw (130 kg mol−1 and 530 kg mol−1, respectively). High-molecular weight materials were selected to ensure P3HT and PVDF chains entangle so that mass transport during solidification from solution can be limited, which should assist vitrification, especially if entanglements form between the two components.
We scrutinized first whether entanglements occur in the neat polymers via viscometry on solutions of concentrations of 7 mg mL−1 (see ESI† for details). At these concentrations, the critical molecular weight, Mc, above which entanglements start to form is found to be ≈55 kg mol−1 for P3HT. This is deduced from the change of slope in the specific-viscosity-vs.-MwP3HT-plot (see Fig. S1, ESI†). A somewhat higher Mc was measured for PVDF. Hence, both components, P3HT of Mw = 130 kg mol−1 and PVDF of Mw = 530 kg mol−1, are of molecular weight well above Mc at these conditions.
Straight-forward differential scanning calorimetry was used in the second step to obtain information on such high-molecular weight 75:25 P3HT:PVDF blends. We discuss first the melt-processed blend as a reference system. Two well pronounced endotherms were recorded: one around 160 °C and one around 230 °C (end-set of respective endotherm; see Fig. 1a, top thermogram). These features can be assigned to the crystal melting of PVDF and P3HT, respectively.30–32 Tellingly, the DSC thermogram of the blend is essentially a superposition of the ones of the individual components (see Fig. S2 for the thermograms of the neat components of different molecular weight, ESI†), indicating that little or no interactions occur between the blend components. We, thus, conclude that this blend is strongly phase-separated when melt processed.
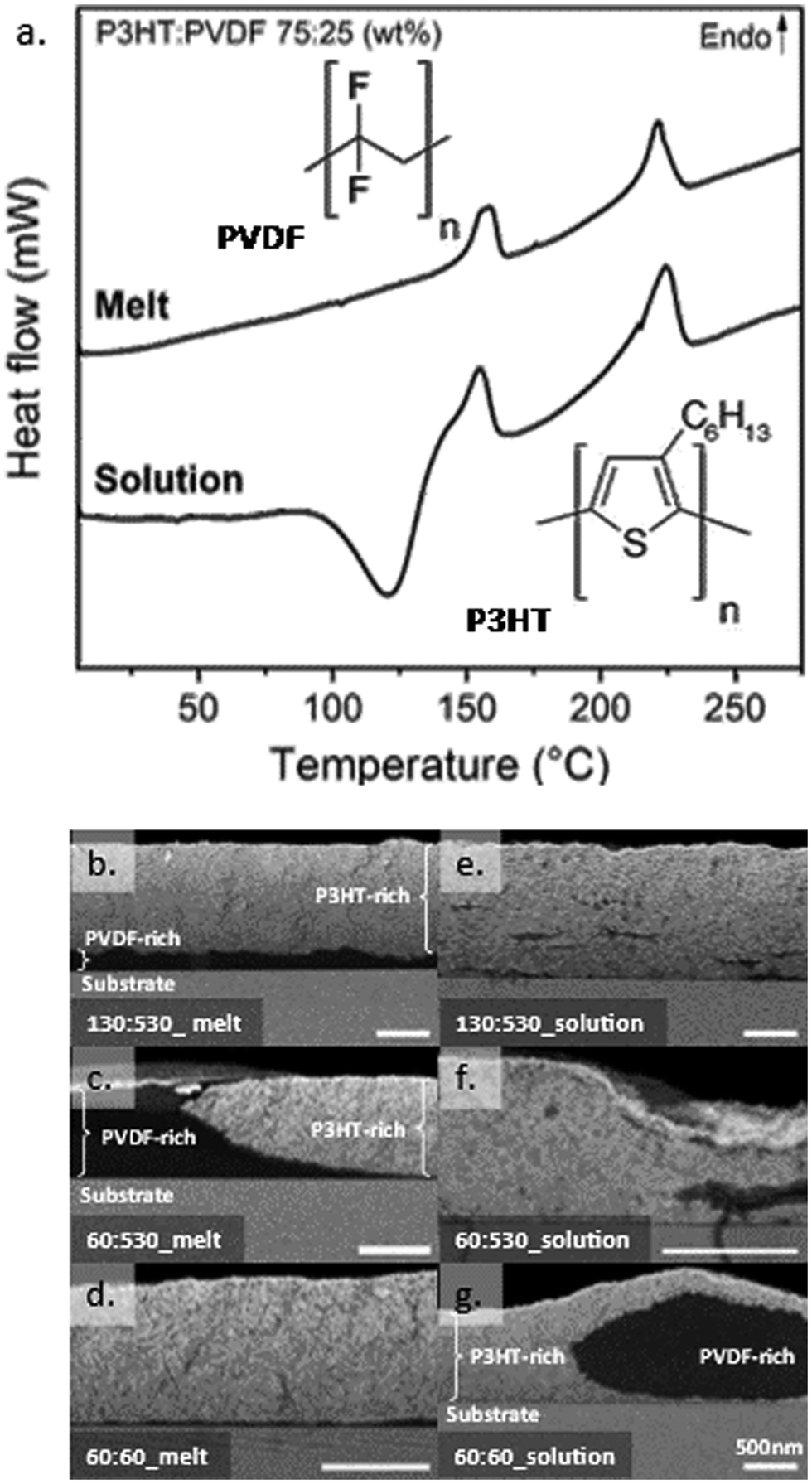 | ||
| Fig. 1 “Compatibilization” of polymer blends when processed from solution compared to melt blending. (a) DSC first and second heating thermograms for, respectively, melt- and solution processed P3HT:PVDF blends (weight ratio 75:25; MwP3HT ≈ 130, MwPVDF ≈ 530 kg mol−1). The chemical structures of P3HT and PVDF are shown in the inset. (b–g) Cross-section high-resolution back-scattering scanning electron micrographs of 75:25 P3HT:PVDF blends after a diethyl zinc/H2O vapor phase infiltration (VPI) process leading to selective deposition of ZnO in the P3HT-rich and intermixed domains (seen as bright regions). In contrast, PVDF-rich domains inhibit ZnO deposition and, thus, are seen as dark regions. Varying the molecular weights of the two blend components leads to different phase morphologies in melt and solution-processed samples: high-molecular-weight blends phase separate, as expected, when melt processing (b and c), but stay intermixed when deposited from solution (e and f); conversely, low-molecular-weight blends phase separate when solution-processed (g) but not when solidified from the melt (d). (The molecular weights of the two components, given in kg mol−1, are indicated in the bottom left of each electron micrograph. The scale bar for all micrographs is 500 nm). | ||
This view is corroborated by VPI where the blend films were exposed to gaseous metal oxide precursors that diffuse into the films and in situ convert to an inorganic product.33 Precursor diffusion is typically permitted in domains with free volume, such as amorphous polymer domains and/or permeable intermixed domains; however, it is restricted in dense regions.34,35 Selective “staining” occurs when the precursors diffuse and are retained only in specific phases. This selectivity offers high contrast when analysing cross-sections of the resulting films by SEM. Specifically, Z-contrast (atomic number) images taken by the back-scattered electron (BSE) detector of an SEM can be used to map the distribution of inorganic-free and “stained” domains36 and complement our DSC data. As described in the ESI,† diethyl zinc (DEZ) and water were used for the present work because these precursors are known to diffuse into the P3HT and react to form zinc oxide (ZnO),34,35 but do not do so in the PVDF. As a consequence, and as is evident from Fig. S3 (ESI†), P3HT films appear bright in electron microscopy due to ZnO deposition, while PVDF stays dark, enabling us to visualize phase separation on the length scales accessible to SEM.
For the melt-processed high-molecular-weight P3HT:PVDF blend, we find a strong and obvious vertical phase separation with a prominent P3HT-rich top-layer (bright part of the film), segregated from a thin, likely highly PVDF-pure layer (dark part of the film; see scanning electron micrograph presented in Fig. 1b). Even when reducing the molecular weight of the P3HT from 130 kg mol−1 to 60 kg mol−1, while keeping the one of PVDF at 530 kg mol−1, the melt-processed blends feature a pronounced phase segregation, see Fig. 1c. Better intermixing seemed to be enabled only when both P3HT and PVDF were of a low weight-average molecular weight of 60 kg mol−1 (i.e. close or below Mc), although a small vertical separation of the PVDF to the bottom is still observed (Fig. 1d).
The picture changes completely when solution coating these high-molecular weight P3HT:PVDF blends from a mixture of cyclohexanone and xylene (volume ratio of 3:1; 7 mg mL−1) and using a casting temperature of 50 °C, conditions that were previously reported for the solution deposition of P3HT:PVDF systems.29 [Note: The choice of a high-boiling point solvent mixture enabled additional degrees of freedom in processing and control over the solidification sequence of the P3HT and PVDF 29.]
We again discuss first the 130 kg mol−1:530 kg mol−1 P3HT:PVDF blend (75:25 weight ratio). In strong contrast to the melt-processed blends, the solution-cast systems display a distinct cold crystallization (CC) exotherm at 140 °C (endset temperature; Fig. 1a, bottom thermogram) in addition to the two crystalline melting endotherms. This suggests that solution blending limits the capability of P3HT, PVDF, or both, to crystallize during solidification from solution, and only upon heating of the produced films do the blend components molecularly order. In other words, solution blending leads to vitrification, resulting in an initially fully or at least partially amorphous structure.37
Importantly, no large-scale phase separation is observed in such solution-processed, vitrified high-molecular-weight binaries after the VPI treatment. Rather, a highly homogenous blend structure is found despite the high-molecular weight of both components (Fig. 1e). Reducing the molecular weight of the P3HT to 60 kg mol−1 has no obvious effect; homogenous films are still obtained, as can be deduced from the electron micrograph taken after VPI staining and presented in Fig. 1f. Only when two low-molecular-weight polymers (Mw = 60 kg mol−1 for both, P3HT and PVDF) are used for blending does large-scale phase separation occur when solution processing (Fig. 1g).
We went on to elucidate whether the homogenous nature of solution-processed P3HT:PVDF blends, and the limited phase separation observed in SEM post-VPI, can be correlated with the extent the blends vitrify. For this purpose, we measured the DSC thermograms of an entire series of solution-cast 75:25 P3HT:PVDF blends, including the above discussed 130 kg mol−1:530 kg mol−1, 60 kg mol−1:530 kg mol−1 and 60 kg mol−1:60 kg mol−1 blends (see Fig. 1e–g), as well as some interim combinations, including 130 kg mol−1:180 kg mol−1 and 60 kg mol−1:180 kg mol−1 P3HT:PVDF systems.
The entire set of DSC thermograms are displayed in Fig. 2a. Clear differences are immediately observed. While no cold-crystallization exotherm is recorded for the low molecular blend (i.e. 60 kg mol−1: 60 kg mol−1), all blends comprising at least one high-molecular-weight component (Mw > 100 kg mol−1) feature a prominent exotherm around 120 °C (Fig. 2a), as already observed for the 130 kg mol−1:530 kg mol−1 P3HT:PVDF binary (Fig. 1a). These findings suggest that blends with high-molecular components tend to vitrify and only crystallize upon heating of the produced, dried films.
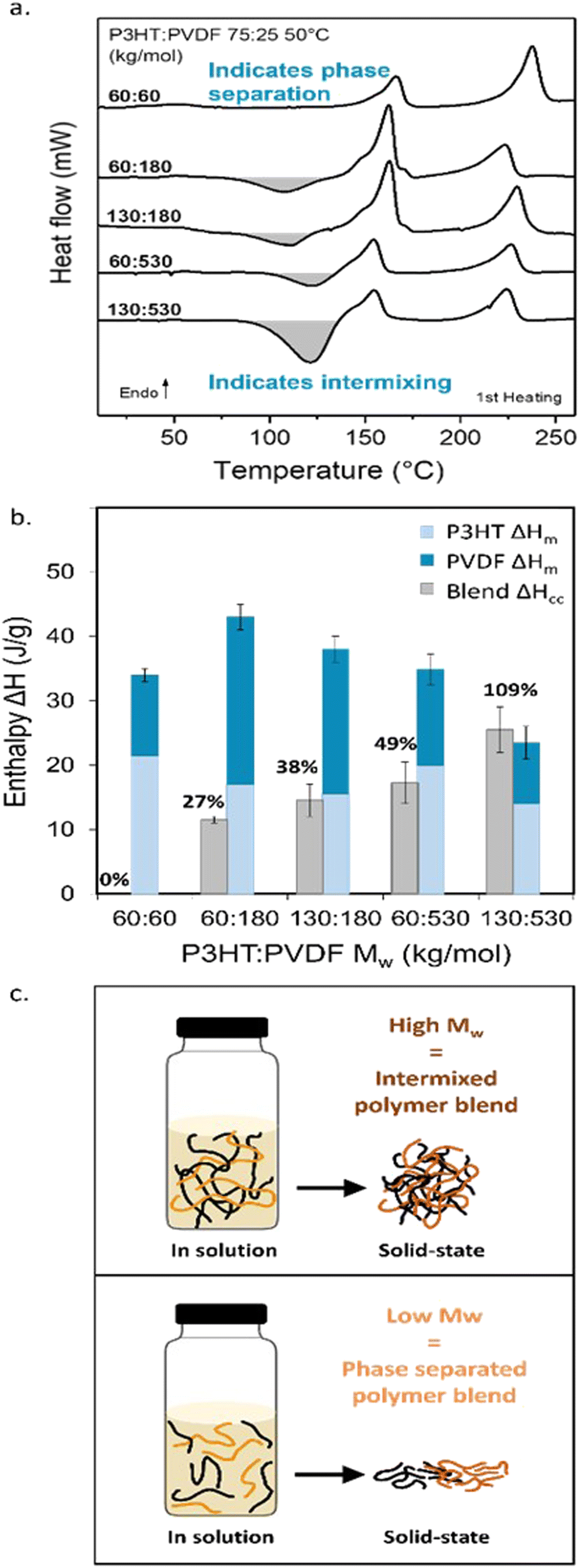 | ||
| Fig. 2 Using molecular weight as a tool to manipulate the extent of vitrification in solution-processed blends. (a) DSC first heating thermograms measured on solution-processed blends of P3HT and PVDF of different molecular weights. More pronounced cold-crystallization exotherms (CC, shaded in grey) are observed for blends comprising higher molecular weight components. (b) Comparison of ΔHcc and the combined melting enthalpies of the individual blend components, (ΔHmP3HT + ΔHmPVDF), allows to estimate the extent of vitrification of the different molecular weight solution-processed blends. Error bars calculated based on baseline fitting. (c) Illustration of the polymer chain arrangement in solution and in the solid state for high- (top) and low- (bottom) molecular weight blends. | ||
Comparison of the cold-crystallization enthalpy (ΔHCC, grey columns in Fig. 2b) with the combined melting enthalpies of the individual blend components, (ΔHmPVDF + ΔHmP3HT) (dark and light blue columns in Fig. 2b), gives us an estimate of the extent of vitrification in a given blend. The reason is that material crystallized during film formation, as well as the material that crystallizes at CC, will melt upon heating. The observation that ΔHCC ≈ (ΔHmPVDF +ΔHmP3HT) implies that the initial film was highly amorphous, i.e., strongly vitrified, directly after film casting/drying. Hence, only the material that crystallized at CC contributes to the melting process. In contrast, if ΔHCC < (ΔHmPVDF + ΔHmP3HT) means that a certain crystalline fraction was produced from solution or the melt with an enthalpy ΔHintial, and thus (ΔHintial + ΔHCC) ≈ (ΔHmPVDF + ΔHmP3HT). In case that Hintial ≈ (ΔHmPVDF + ΔHmP3HT), most of the crystalline content is formed during film formation. [Note: from DSC we can not deduce which blend component crystallizes at CC.]
Tellingly, only for the solution-processed 130 kg mol−1:530 kg mol−1 P3HT:PVDF blend, 75:25 weight ratio, is ΔHCC ≈ (ΔHmPVDF + ΔHmP3HT). Already reducing the molecular weight of one component, leads to ΔHCC < (ΔHmPVDF + ΔHmP3HT), meaning that some crystalline order is induced upon solution casting; though, this does not lead to large-scale phase separation. SEM post-VPI reveals a relatively homogenous, well intermixed film (Fig. 1f; see Fig. S4 (ESI†) for the SEM data on the whole series of P3HT:PVDF blends). ΔHCC for 60 kg mol−1:60 kg mol−1 blends is negligible, in agreement with the large phase separation observed in SEM (Fig. 1g). This observation implies that in as-cast films, minimum vitrification and, thus, maximum possible degree of crystallinity is induced in the low-molecular-weight binaries comprising no high-molecular weight component, while all the other blends at least partly vitrify.
Using the equation ΔHCC/(ΔHmPVDF + ΔHmP3HT), we estimated a normalized degree of vitrification —with respect to the 60 kg mol−1:60 kg mol−1 blend— to be ≈100% for 130 kg mol−1:530 kg mol−1 P3HT:PVDF blends; and, respectively, ≈50%, ≈40% and ≈30% for the 60 kg mol−1:530 kg mol−1, 130 kg mol−1:180 kg mol−1, and 60 kg mol−1:180 kg mol−1 binaries. Since the CC exotherm overlaps with the PVDF melting endotherm, we emphasize that only estimates of ΔHCC and ΔHmPVDF can be deduced from the DSC measurements. Thus, only an estimate of the degree of vitrification can be obtained. Or in other words, our approach leads to a qualitative picture of the extent of vitrification and not a quantitative one.
Nonetheless, the above analysis shows that reduction of the molecular weight of one component notably limits vitrification during solution blending of P3HT and PVDF. In parallel, an increased and more visible phase separation is observed, contrasting starkly with high-molecular-weight blends (Fig. 1e–g; and Fig. S4, ESI†). We deduce from these observations that in blends comprised of high-molecular-weight P3HT and PVDF, chain entanglements form in solution, including between the two components, as schematically illustrated in Fig. 2c, top panel. Indeed, as the polymer molecular weight increases above the critical molecular weight, Mc, the polymer chains start to be sufficiently long to entangle.38 Likely, physical binary “hooks” between PVDF and P3HT chains form as well, all combined hindering chain diffusivity and increasing the solution viscosity. In turn, mass transport is reduced, leading to strong vitrification in some scenarios, the extent of which depending on the number of chain entanglements, including cross-component entanglements, that form. The lower molecular mobility kinetically depresses phase separation and crystallization, resulting in the formation of a vitrified intermixed phase. In contrast, for blends produced with low-molecular weight P3HT and PVDF, with a Mw below Mc, the polymer chains are unentangled and of high molecular mobility (see Fig. 2c, bottom panel). This increased chain mobility in such low-viscosity solutions allows the PVDF and P3HT chains to freely diffuse away from each other during solidification, driven by their low thermodynamic miscibility, resulting in significant molecular ordering and crystallization of the individual components and, in turn, a pronounced phase separation.
Conclusions
Our work illustrates that, during solution processing, the extent of phase separation in P3HT:PVDF blends can be manipulated via the control of chain entanglements in the system, including between PVDF and P3HT macromolecules. This is achieved via the selection of the molecular weights of the blend components. In particular, blends comprising at least one high-molecular-weight material allow to partially trap a kinetically favoured non-equilibrium state because mass transport is reduced due to the presence of chain entanglements. This leads to vitrification, limiting thermodynamically driven phase separation, as SEM of VPI-stained blends of different molecular weight P3HT:PVDF systems demonstrates (Fig. S4, ESI†). Thereby, the vitrification effect is relatively independent of blend composition. Especially, for 130 kg mol−1:530 kg mol−1 P3HT:PVDF blends, the extent of vitrification is unaffected by the blend ratio, i.e.: ΔHCC ≈ (ΔHmPVDF + ΔHmP3HT) except for very high PVDF-content binaries (Fig. S5, ESI†), yet, this can be influenced by the selection of casting temperature (Fig. S6, ESI†). We, hence, conclude that intermixed blends can be obtained when limiting mass transport during solidification so that the material can vitrify.
The close intermixing of the blend components opens various opportunities. For instance, the resulting close molecular intermixing enables local interactions between PVDF and P3HT chain segments. In turn, the chain arrangement of PVDF are affected even in the crystalline regions of the polymers. Indeed, we observe a change in the content of the α- to the β- polymorph of PVDF (see Fig. 3a for schematics) when blended with P3HT, whereby the amount of β- phase that is induced depends on the P3HT:PVDF weight ratio and the extent of vitrification. Most telling, the highest amount of the polar β-polymorph is recorded for the 75:25 P3HT:PVDF blend (see the grazing incidence wide-angle X-ray data presented in Fig. 3b) – i.e., the blend for which the most pronounced vitrification was observed. Clearly, the close intermixing of P3HT and PVDF in solution, and possibly in the semi-dry state, that enables strong(er) interactions between the two polymers, notably affects the overall assembly and, thus, film properties.
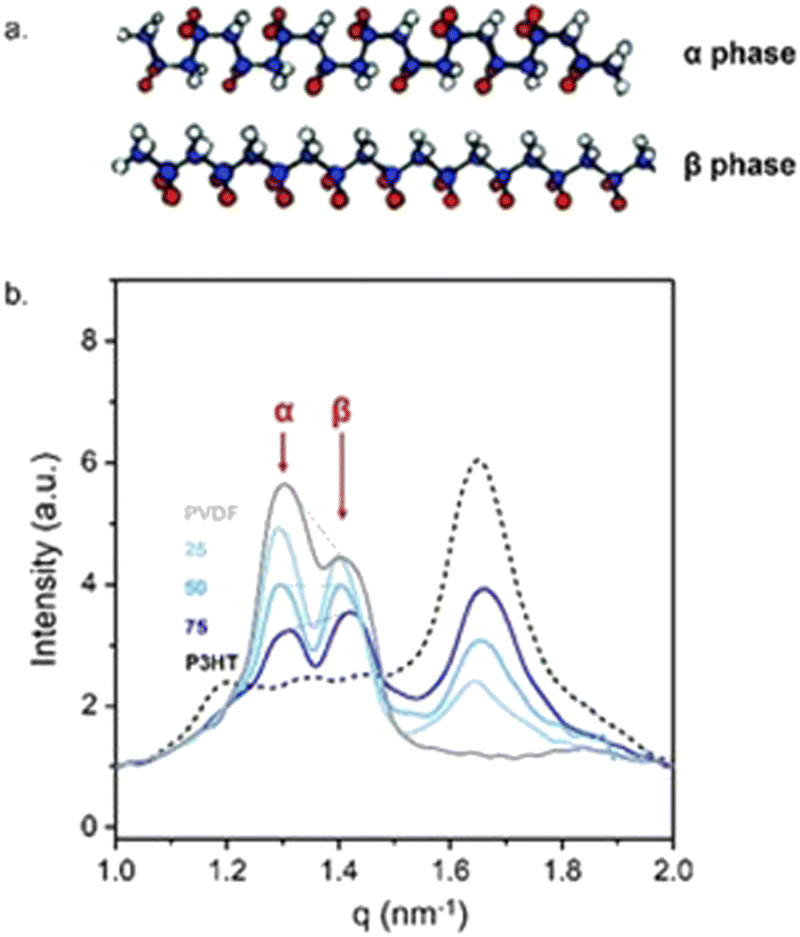 | ||
| Fig. 3 Relating intermixing with macroscopic film properties. (a) Schematic illustrations of the non-polar α and polar β polymorphs of PVDF.39 (b) Grazing-angle wide-angle X-ray scattering in-plane intensity line-cuts of solution-processed P3HT:PVDF thin films (≈60 kg mol−1:350 kg mol−1), showing signatures characteristic of the polar β phase of PVDF becoming most intense for the most vitrified 75:25 P3HT:PVDF blend. | ||
With these obvious benefits of vitrification-induced intermixing, in future, insights need to be gained about the influence of entanglement formation between the two blend components vs. entanglements between macromolecules of the same nature, to obtain full control over the blend assembly. Such understanding will lead to a vitrification control that can open pathways to reliably induce intermixing in polymer blends of even low thermodynamic miscibility, and allow a diverse range of morphologies to be produced. This versatile “knob”, thus, should provide a processing platform to manipulate materials properties and to explore whether novel features can be introduced by full or partial vitrification via solution blending. For example, the fine intermixing of polymer:polymer blends offers a method to control the local dielectric environment of semiconducting polymers via their molecular proximity to the polar polymer, PVDF. Such control over the local dielectric environment may be desirable in applications such as organic photovoltaics to help reduce charge recombination.40 Likewise, manipulating the polar β-polymorph compositions of PVDF via vitrification may be beneficial for ferroelectric applications, such as non-volatile memory storage devices.28 Our work also illustrates that thermal analysis, combined with VPI/SEM, provides a visualization- and interpretation- tool for the characterization of amorphous intermixed phases,41 otherwise difficult to probe with traditional optical, spectroscopic, or X-ray diffraction techniques. We can extract qualitative information such as degree and strength of vitrification, providing a supplementary insight of the solid-state structure of functional polymer blends.
Author contributions
A. K. and O. N. contributed equally to the work to this work, running most of the experiments (A. K. performed all thermal analysis; O. N. executed the VPI staining and conducted SEM on the VPI-stained samples), and they delivered the initial draft of the manuscript. H. K., O. B., and A. L. provided assistance in the experiments. G. F. and N. S. designed and managed the project, and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Marie Skłodowska-Curie Actions Innovative Training Network “H2020-MSCAITN-2014 INFORM – 675867”, and the United States National Science Foundation (NSF)–Israel Binational Science Foundation (BSF) Collaborative Research (DMR Award #1905901). The authors thank Lee Richter at the National Institute of Standards and Technology (NIST) for many invaluable discussions, and Ian Pelse, then at Georgia Tech, with X-ray scattering measurements.Notes and references
- L. A. Utracki and C. A. Wilkie, Polymer Blends Handbook, 2014, pp. 1–2378 Search PubMed.
- M. Rubinstein and R. H. Colby, Polymer physics, Oxford University Press, Oxford, 2003 Search PubMed.
- P. J. Flory, J. Chem. Phys., 2004, 10, 51 CrossRef.
- M. L. Huggins, J. Chem. Phys., 2004, 9, 440 CrossRef.
- A. D. Scaccabarozzi and N. Stingelin, J. Mater. Chem. A, 2014, 2, 10818–10824 RSC.
- H. J. Kim, K. Perera, Z. Liang, B. Bowen, J. Mei and B. W. Boudouris, ACS Macro Lett., 2022, 11, 243–250 CrossRef CAS PubMed.
- H. Fu, Y. Li, J. Yu, Z. Wu, Q. Fan, F. Lin, H. Y. Woo, F. Gao, Z. Zhu and A. K. Y. Jen, J. Am. Chem. Soc., 2021, 143, 2665–2670 CrossRef CAS PubMed.
- L. D. Bozano, K. R. Carter, V. Y. Lee, R. D. Miller, R. DiPietro and J. C. Scott, J. Appl. Phys., 2003, 94, 3061–3068 CrossRef CAS.
- N. Zhou, H. Lin, S. J. Lou, X. Yu, P. Guo, E. F. Manley, S. Loser, P. Hartnett, H. Huang, M. R. Wasielewski, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Adv. Energy Mater., 2014, 4, 1300785 CrossRef.
- C. R. McNeill and N. C. Greenham, Adv. Mater., 2009, 21, 3840–3850 CrossRef CAS.
- D. Kwak, H. H. Choi, B. Kang, D. H. Kim, W. H. Lee and K. Cho, Adv. Funct. Mater., 2016, 26, 3003–3011 CrossRef CAS.
- J. Lee, G. Kim, D. K. Shin, Y. Seo, K. Kim and J. Park, IEEE Trans. Electron Devices, 2018, 65, 3311–3317 CAS.
- L. Huang, Z. Wang, J. Chen, B. Wang, Y. Chen, W. Huang, L. Chi, T. J. Marks, A. Facchetti, L. Z. Huang, L. F. Chi, Z. Wang, J. Chen, B. Wang, Y. Chen, W. Huang, T. J. Marks and A. Facchetti, Adv. Mater., 2021, 33, 2007041 CrossRef CAS PubMed.
- Y. Diao, L. Shaw, Z. Bao and S. C. B. Mannsfeld, Energy Environ. Sci., 2014, 7, 2145–2159 RSC.
- C. Schaefer, P. Van Der Schoot and J. J. Michels, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 91, 022602 CrossRef CAS PubMed.
- T. Inoue, T. Ougizawa, O. Yasuda and K. Miyasaka, Macromolecules, 1985, 18, 57–63 CrossRef CAS.
- S. Kouijzer, J. J. Michels, M. van den Berg, V. S. Gevaerts, M. Turbiez, M. M. Wienk and A. J. Janssen, J. Am. Chem. Soc., 2013, 12057–12067, DOI:10.1021/ja405493j.
- N. D. Treat, P. Westacott and N. Stingelin, Annu. Rev. Mater. Res., 2015, 45, 459–490 CrossRef CAS.
- B. R. Joseph Kline, M. D. McGehee, E. N. Kadnikova, J. Liu, J. M. J. FrØchet, M. D. McGehee, R. J. Kline, E. N. Kadnikova, J. Liu and J. M. J. FrØchet, Adv. Mater., 2003, 15, 1519–1522 CrossRef.
- K. Zhao, H. U. Khan, R. Li, Y. Su and A. Amassian, Adv. Funct. Mater., 2013, 23, 6024–6035 CrossRef CAS.
- M. Valadares, I. Silvestre, H. D. R. Calado, B. R. A. Neves, P. S. S. Guimarães and L. A. Cury, Mater. Sci. Eng., C, 2009, 29, 571–574 CrossRef CAS.
- B. Gburek and V. Wagner, Org. Electron., 2010, 11, 814–819 CrossRef CAS.
- C. Yang, S. Zhang, J. Ren, M. Gao, P. Bi, L. Ye, J. Hou, S. Zhang, L. Ye and J. Hou, Energy Environ. Sci., 2020, 13, 2864–2869 RSC.
- M. Li, I. Katsouras, C. Piliego, G. Glasser, I. Lieberwirth, P. W. M. Blom and D. M. de Leeuw, J. Mater. Chem. C, 2013, 1, 7695–7702 RSC.
- Z. Yin, B. Tian, Q. Zhu and C. Duan, Polymers, 2019, 11, 2033 CrossRef CAS.
- W. Serrano-Garcia, I. Bonadies, S. Thomas and V. Guarino, Mater. Lett., 2020, 266, 127458 CrossRef.
- M.-F. Lin, K.-W. Chang, C.-H. Lee, X.-X. Wu and Y.-C. Huang, Sci. Rep., 2022, 12, 14842 CrossRef CAS PubMed.
- H. Zhu, S. Yamamoto, J. Matsui, T. Miyashita and M. Mitsuishi, RSC Adv., 2018, 8, 7963–7968 RSC.
- D. Sparrowe, M. Baklar and N. Stingelin, Org. Electron., 2010, 11, 1296–1300 CrossRef CAS.
- C. Müller, T. A. M. Ferenczi, M. Campoy-Quiles, J. M. Frost, D. D. C. Bradley, P. Smith, N. Stingelin-Stutzmann and J. Nelson, Adv. Mater., 2008, 20, 3510–3515 CrossRef.
- J. Zhao, A. Swinnen, G. Van Assche, J. Manca, D. Vanderzande and B. Van Mele, J. Phys. Chem. B, 2009, 113, 1587–1591 CrossRef CAS PubMed.
- C. Leonard, J. L. Halary, L. Monnerie and F. Micheron, Polym. Bull., 1984, 11, 195–202 CrossRef CAS.
- C. Z. Leng and M. D. Losego, Mater. Horiz., 2017, 4, 747–771 RSC.
- S. Obuchovsky, H. Frankenstein, J. Vinokur, A. K. Hailey, Y.-L. Loo and G. L. Frey, Chem. Mater., 2016, 28, 2668–2676 CrossRef CAS.
- S. Obuchovsky, M. Levin, A. Levitsky and G. L. Frey, Org. Electron., 2017, 49, 234–241 CrossRef CAS.
- A. Levitsky, G. Maria Matrone, A. Khirbat, I. Bargigia, X. Chu, O. Nahor, T. Segal-Peretz, A. J. Moulé, L. J. Richter, C. Silva, N. Stingelin and G. L. Frey, Adv. Sci., 2020, 7, 2000960 CrossRef CAS PubMed.
- P. Westacott, N. D. Treat, J. Martin, J. H. Bannock, J. C. de Mello, M. Chabinyc, A. B. Sieval, J. J. Michels and N. Stingelin, J. Mater. Chem. A, 2017, 5, 2689–2700 RSC.
- F. P. V. Koch, J. Rivnay, S. Foster, C. Müller, J. M. Downing, E. Buchaca-Domingo, P. Westacott, L. Yu, M. Yuan, M. Baklar, Z. Fei, C. Luscombe, M. A. McLachlan, M. Heeney, G. Rumbles, C. Silva, A. Salleo, J. Nelson, P. Smith and N. Stingelin, Prog. Polym. Sci., 2013, 38, 1978–1989 CrossRef CAS.
- L. Ruan, X. Yao, Y. Chang, L. Zhou, G. Qin and X. Zhang, Polymers, 2018, 10, 1–27 CrossRef PubMed.
- J. Brebels, J. V. Manca, L. Lutsen, D. Vanderzande and W. Maes, J. Mater. Chem. A, 2017, 5, 24037–24050 RSC.
- O. Nahor, A. Khirbat, S. A. Schneider, M. F. Toney, N. Stingelin and G. L. Frey, ACS Mater. Lett., 2022, 4, 2125–2133 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc00071k |
| ‡ Contributed to the same extent to this manuscript. |
| This journal is © The Royal Society of Chemistry 2023 |