
Air-stable ternary organic solar cells achieved by using fullerene additives in non-fullerene acceptor-polymer donor blends†
Elisa
Trippodo
abc,
Vincenzo
Campisciano
 b,
Liang-Wen
Feng
a,
Yao
Chen
*ad,
Wei
Huang
a,
Joaquin M.
Alzola
a,
Ding
Zheng
a,
Vinod K.
Sangwan
*e,
Mark C.
Hersam
*aef,
Michael R.
Wasielewski
*a,
Bruno
Pignataro
*c,
Francesco
Giacalone
*b,
Tobin J.
Marks
*ae and
Antonio
Facchetti
*ag
b,
Liang-Wen
Feng
a,
Yao
Chen
*ad,
Wei
Huang
a,
Joaquin M.
Alzola
a,
Ding
Zheng
a,
Vinod K.
Sangwan
*e,
Mark C.
Hersam
*aef,
Michael R.
Wasielewski
*a,
Bruno
Pignataro
*c,
Francesco
Giacalone
*b,
Tobin J.
Marks
*ae and
Antonio
Facchetti
*ag
aDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA. E-mail: chenyao@cigit.ac.cn; m-wasielewski@northwestern.edu; t-marks@northwestern.edu; a-facchetti@northwestern.edu
bTechnologies (STEBICEF) and INSTM UdR –University of Palermo, Viale delle Scienze, Ed. 17, 90128 Palermo, Italy. E-mail: francesco.giacalone@unipa.it
cDepartment of Physics and Chemistry – Emilio Segrè (DiFC), University of Palermo, bdg. 17, v.le delle Scienze, Palermo, 90128, Italy. E-mail: bruno.pignataro@unipa.it
dChongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences, Chongqing 400714, P. R. China
eDepartment of Material Science and Engineering, Northwestern University, Evanston, Illinois 60208, USA. E-mail: vinod.sangwan@northwestern.edu; m-hersam@northwestern.edu
fDepartment of Electrical and Computer Engineering, Northwestern University, Evanston, Illinois 60208, USA
gLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, Norrköping 60174, Sweden
First published on 15th February 2023
Abstract
Organic solar cells (OSCs) based on donor–acceptor blends have shown a rapid improvement in power conversion efficiency (PCE) now approaching, for small cells, those of the state-of-the art commercial solar modules. However, performance degradation remains one of the most critical impediments for OSC technology commercialization. Ternary solar cells where a third component, for instance an acceptor, is added to a non-fullerene acceptor–polymer donor blend are an effective approach for improving both OSC efficiency and long-term stability. Here, we study the role of two fullerene acceptors, ET18 and PCBM, as the third component in PD:Y6 blends. These fullerene derivatives significantly enhance the cell stability, which retained >90% of their initial PCEs (13–14%) even after storage in air for 6 months, compared to only ∼20% retention for the binary devices. GIWAXS, AFM, in situ impedance spectroscopy and femtosecond transient absorption spectroscopy measurements reveal that the enhanced stability of the ternary devices results from a more robust blend morphology reducing charge recombination in the ternary devices during aging.
10th Anniversary StatementDuring the past 10 years the Journal of Materials Chemistry C has become home of several fundamental and applied research studies, spanning multiple applications, having chemistry and materials science at the core. Considering that applied materials chemistry research requires different expertise, the interdisciplinary readership of this Journal is best suited to appreciate the novelty and impact of the reported studies. Our group has published more than 15 papers in JMCC, mostly addressing the synthesis of organic semiconductors and metal oxides, thin-film processing methodologies and characterization as well as fabrication of unconventional optoelectronic devices such as organic thin-film transistors, circuits, solar cells, and electrochemical transistors. Our work on JMMC has always received considerable attention and has been widely cited. We look forward to continuing publishing in these exciting areas as well as to new bioelectronics and neuromorphic device directions. |
Introduction
Organic solar cells (OSCs) based on non-fullerene acceptors (NFAs) have emerged as a promising alternative for next-generation renewable energy sources due to several advantages such as solution-based low-cost processability, mechanical flexibility, and lightweight.1–4 OSCs are also preferred over conventional silicon photovoltaics for applications involving artificial or diffused light due to large power conversion efficiency (PCE) at low illumination intensities, thus opening new frontiers for indoor and space applications.5–8 Furthermore, there has been considerable enhancement in PCE values during the past five years, now approaching 20%, primarily due to the discovery and optimization of new NFA molecules.6,9–12 There have also been significant advances in understanding degradation mechanisms and improving the ambient stability of OSCs.13–15 However, the stability of binary blends based on NFAs remains insufficient for commercialization. Among the reported stabilization strategies of OSC cells and modules,16 the incorporation of additives is a viable avenue to further enhance the ambient stability of OSCs.16,17Conventional bulk heterojunction (BHJ) OSCs based on a blend of an electron-donor polymer and a fullerene electron-acceptor have failed to reach commercialization mainly due to their limited performance.18–23 These BHJ blends films tend to create isolated domains acting as exciton recombination sites limiting the PCE.24–26 Furthermore, most fullerene-based acceptors also suffer from photo-degradation due to dimerization processes.27,28 Recently, NFAs have risen as a promising alternative due to broader absorption spectral ranges, tunability of the energy levels for alignment with charge transport layers, nanoscale crystallization in the blend, and high carrier mobilities for dramatic PCE enhancement.29–31 Several kinds of NFAs have been developed to address the issues faced by conventional fullerenes,32,33 starting from the pioneering work on perylenediimides34 to the most recent fused-ring electron acceptors consisting of two π-electron-withdrawing moieties linked by a planar π-fused-ring bridge endowed with solubilizing side chains.34 Among them, the most successful are the small molecule acceptors, ITIC and Y6, and their functionalized versions, which have enabled PCEs exceeding 19% in single-junction cells.34–39 While, solubility and electrical properties of Y6-based NFAs can be easily controlled for high-performance OSCs,13,32,33 further reduction of recombination processes and achieving ambient stability, remain outstanding issues.40,41
Several studies have shown that OSCs based on ternary photoactive blends, also known as ternary solar cells (TSCs), where a third donor or acceptor component is added to the binary blend in a minor ratio, have resulted in enhanced PCEs and ambient stability.42–44 The third component can improve PCEs by several mechanisms and enhancing one or more of the main device parameters such as the short circuit current (Jsc), open-circuit voltage (Voc), and fill factor (FF). In addition, the third component can broaden the absorption window, thus enhancing PCE via increasing Jsc without the need for multiple layers as necessary in tandem cells.42,43,45,46 The third component usually exhibits intermediate HOMO–LUMO levels compared to the major donor and the acceptor materials thus affecting energy cascade alignments47,48 and charge transfer mechanisms.49,50 Thus, Voc can also be increased by the third component by promoting energy cascade effects favouring efficient charge transfer process.51,52 Finally, the third component can preferentially dissolve in the donor or in the acceptor rich phase, co-crystallized with one of them or, more frequently, it can be found at the interface between the donor and accept domains. Therefore, enhanced performance can also originate from blend morphology optimization.53
Furthermore, the addition of a third component in binary cells has been explored to address device stability issues,18,54–56 including when processing the blends in air which promotes addition/fragmentation/redox reactions, particularly between superoxide radical species and NFAs,57 as well as BHJ morphological instability.10,20 All these deleterious phenomena drastically enhance OSC burn-in loss rate and aging effects.58,59 Furthermore, it has been shown that a third component can stabilize blend morphology by vitrification of the binary system.37,60 For instance, Tang and co-workers incorporated PC[71]BM as a ternary component in m-INPOIC:PBDB-T blends resulting in an improved PCE value of 14% compared to that of 12% for binary OSCs.61 Moreover, the TSCs retained >95% of the initial PCE value after 20 days in the glove box compared with ≈84% of the binary cells. He et al.62 used PCBM as a ternary additive in PTB7-Th:COi8DFIC cells demonstrating a PCE loss of only 5.7% after about 10 months in glove box versus ≈33% loss in the case of control binary cells. Hao and co-workers63 showed that by adding PDI-2T or DRCN5T as third component in PM6:BTP-4Cl blends, the TSCs retained 70.1% and 78.1%, respectively, of their initial PCE (16.32% and 16.83%, respectively) after 300 h in a nitrogen filled glovebox while the binary control device retained only 61.4% of the initial PCE (15.97%). Recently, Chen et al.64 incorporated PTO2 as the third component in PM6:BTP-eC9 blends resulting in improved long-term stability in an air-filled glove box. After 1352 h, these TSCs retained 82.9% of the initial PCE (18%) outperforming the binary blend. Finally, Anthopoulos and co-workers reported >17% PCE by n-doping PM6,Y6:PC[71]BM TSCs with benzyl viologen and showed that the doped blend also performed better against aging with the binary and ternary devices losing ≈50% and ≈23% of the initial PCE after 1000 hours in nitrogen-filled glove box.65 However, all these important studies were conducted with blend films and devices processed in an inert environment and the OSC stability results could be very different when processing occurs in air.
Here, we study the addition of different amounts of two fullerene acceptor (ET18 or PCBM) to PBTATBT-4F:Y6 blends and investigate device performance and stability of the corresponding binary and ternary blends and devices (Fig. 1). By incorporating the fullerene derivatives in the binary blend, the long-term stability both in a nitrogen-filled glovebox and, more importantly, the ambient stability in the fabrication step and aging outperform the binary control. Most impressively, while the binary blend loss in PCE (initial values ≈13–14%) is up to ∼80%, the ternary blends loss after 6 months in ambient air is negligible. Ultrafast transient absorption spectroscopy confirms that the ultrafast hole transfer process is preserved in the TSC devices. The morphology of the ternary blends was also confirmed to remain unchanged after aging as assessed by Grazing-Incidence Wide-Angle X-ray Scattering (GIWAXS) and atomic force microscopy (AFM), whereas morphology of binary blends undergoes dramatic changes over the same time. Finally, in situ integrated photocurrent device analysis reveals that performance-limiting device parameters such as recombination coefficient and mobility remain unchanged in ET18-based TSCs, while these metrics in binary cells suffer significantly. Overall, a wide range of characterization techniques implicate the same stabilization effects as a fullerene additive in NFA OSCs.
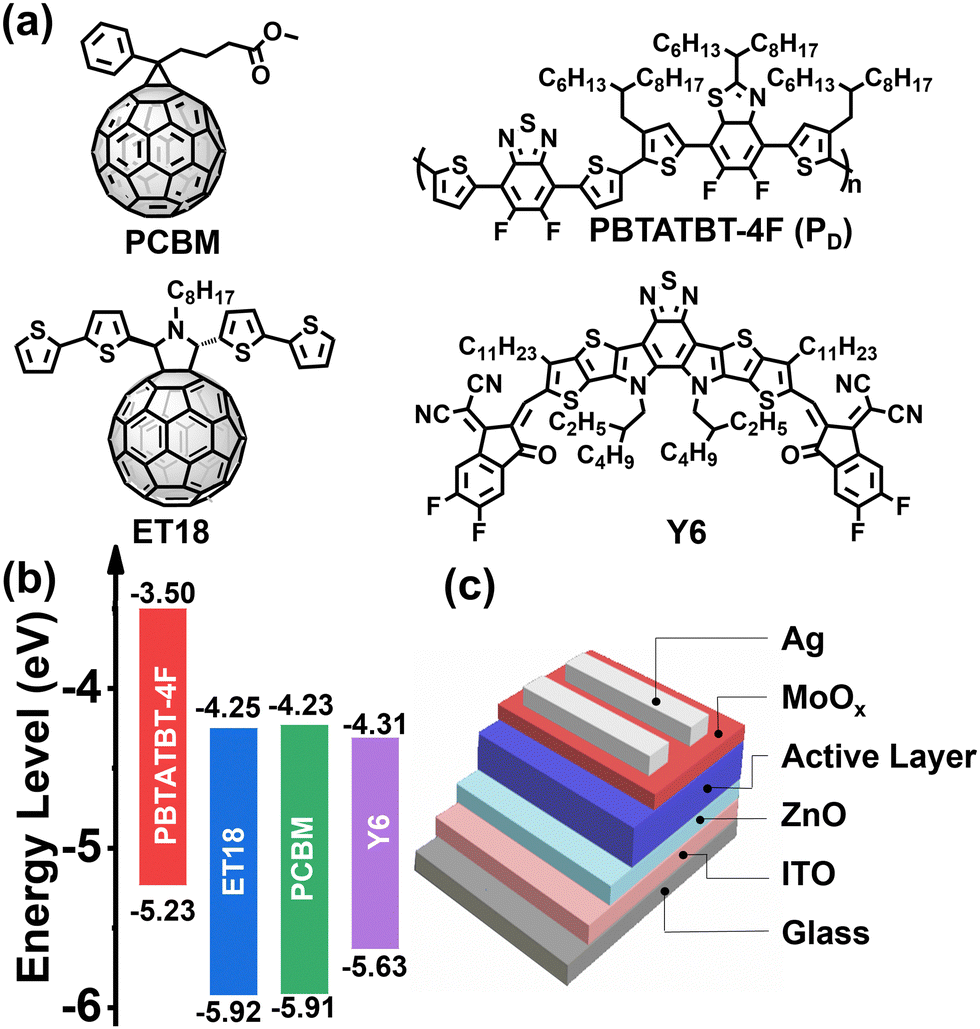 | ||
Fig. 1 Chemical structures (a) and HOMO/LUMO energy levels (b) of PBTATBT-4F (hereafter PD), Y6, ET18, and PCBM. Note, all HOMO energy levels were measured by UPS and the LUMO energy levels calculated by using the optical gap using the equation ELUMO = EHOMO + Eopt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g. (c) Device architecture used in this study. g. (c) Device architecture used in this study. | ||
Results and discussion
In this section we first describe materials selection, binary and ternary OSC fabrication and measurements. Next, we report OSC stability as a function of the device fabrication conditions, bend composition and storage environment. Finally, we investigate the blend film morphology, microstructure, charge transport and trapping mechanisms and how they affect device stability.Device fabrication and photovoltaic characteristics
For device fabrication we selected a high-performing blend consisting of the widely used NFA Y6 and the donor difluorobenzothiadiazole-dithiophene-difluorobenzothiazole based polymer PBTATBT-4F (hereafter named PD for semplicity), which was synthesized according to our previous procedure,66 and employed two fullerene derivatives, ET18 and PC[60]BM, as the third component (see chemical structures in Fig. 1(a)). The fullerene derivative ET18 was previously synthesized by Giacalone et al.26 while PC[60]BM (PCBM hereafter) is one of the most widely studied fullerene acceptors.67Fig. 1(b) reports the schematic of the energy levels of all blend components (for details see ESI,† Fig. S1–S3).68 From a purely energetic standpoint, addition of the fullerene component should not significantly alter light absorption and Voc. Binary OSC and TSC devices, without and with ET18 or PCBM addition, respectively, having an inverted architecture (Fig. 1(c)) were fabricated both in a nitrogen-filled glove box and (except for the vapor deposition steps) in air. Device fabrication and solar cells measurement details can be found in the ESI.† Briefly, the ZnO precursor solution (zinc acetate and 2-ethanolamine in 2-methoxyethanol) was spin-coated on ITO-coated glass substrates. The active layer solution (14 mg mL−1 total concentration in chloroform) was then spin-coated onto the ZnO layer and then annealed for 10 minutes at 130 °C. Previously, we have demonstrated that PD:Y6 device PCEs are almost identical within the D:A weight ratio from 1:1.3 to 1:1.5.66 To exclude the effects on the D:A ratio to the corresponding solar cell efficiency, all binary/ternary devices were prepared with a D:A weight 1:1.5. Thus, for the control binary cells, PD:Y6 = 1:1.5 wt ratio, while for the TSCs, PD:Y6:A2 = 1:1.3:0.2 wt (A2 = ET18, PCBM).66 In all cases the photoactive layer thickness is approximately 120 nm. Finally, the hole transporting layer (MoOx, 10 nm) and the silver electrode (100 nm-thick) were deposited by thermal evaporation. All devices were measured in ambient, without encapsulation, under a simulated AM1.5G irradiation conditions.
Fig. 2(a) reports J–V curves and Table 1 collects all performance parameters for pristine binary and ternary cells fabricated in the glove box (entries 1–3) and in air (entries 4–6). Thus, the binary cells (entry 1) fabricated in a glove box exhibit an average Jsc of 25.4 mA cm−2, a Voc of 0.798 V, a FF of 65.9% resulting in a PCE of 13.4 ± 0.7%. These values are close to our previous study when the devices were fabricated without solvent additives.66 The corresponding ET18/PCBM based TSCs (entries 2 and 3) exhibit slightly larger average Jsc of 25.7/26.0 mA cm−2, minimally lower Voc of 0.785/0.779 V and statistically identical FF of 65.7/66.7% affording comparable PCEs of 13.2 ± 0.5/13.3 ± 0.7%. Thus, the addition of the ET18 or PCBM at the indicated ratio does not affect the performance parameters of the pristine devices. This result is in line with the energy level diagram above and the pristine morphological characteristics of the binary and ternary blend films (vide infra).
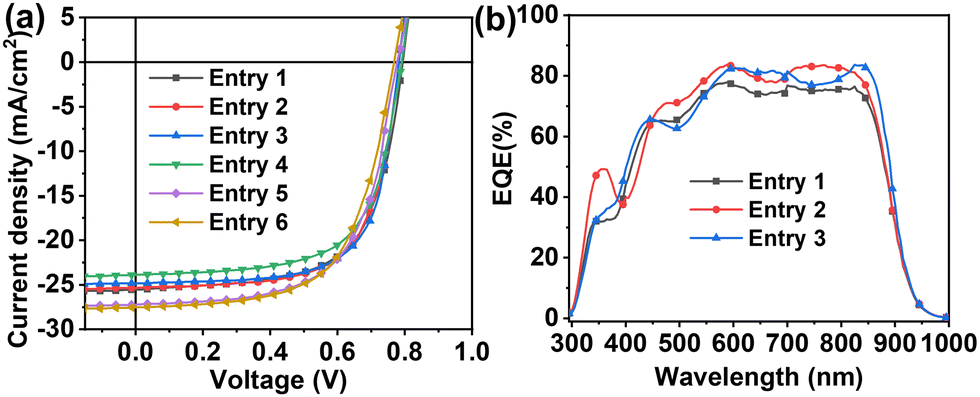 | ||
| Fig. 2 J–V (a) and EQE (b) curves for the indicated devices. D1–D3 devices were fabricated in the glove box, D4–D6 devices were fabricated in air. For D1–D6 blend composition, see Table 1. | ||
| Entry | Blend | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| a Average of 10 devices. b Average of 5 devices. | |||||
| Devices fabricated in glove boxa | |||||
| 1 | PD:Y6 | 25.4 ± 0.9 (26.9) | 0.798 ± 0.006 (0.809) | 65.9 ± 0.7 (66.9) | 13.4 ± 0.7 (14.3) |
| 2 | PD:Y6:ET18 | 25.7 ± 0.9 (26.8) | 0.785 ± 0.002 (0.788) | 65.7 ± 0.9 (67.8) | 13.2 ± 0.5 (14.0) |
| 3 | PD:Y6:PCBM | 26.0 ± 0.9 (27.9) | 0.779 ± 0.006 (0.787) | 66.7 ± 0.9 (68.3) | 13.3 ± 0.7 (14.3) |
| Devices fabricated in airb | |||||
| 4 | PD:Y6 | 23.8 ± 0.2 (24.0) | 0.789 ± 0.002 (0.791) | 65.3 ± 0.7 (66.9) | 12.2 ± 0.2 (12.4) |
| 5 | PD:Y6:ET18 | 26.6 ± 0.4 (27.4) | 0.776 ± 0.003 (0.781) | 63.7 ± 0.7 (64.9) | 13.2 ± 0.3 (13.5) |
| 6 | PD:Y6:PCBM | 27.7 ± 0.3 (28.3) | 0.760 ± 0.009 (0.773) | 62.5 ± 0.8 (63.5) | 13.4 ± 0.2 (13.7) |
Additionally, we also fabricated OSCs in air (entries 4–6, Fig. 2(a) and Table 1), a procedure compatible with R2R of large area OSCs. Interestingly, when these devices were fabricated in air, the binary cells (entry 4) exhibit a slightly lower PCE of 12.2 ± 0.2%, mainly originating by the lower Jsc (23.8 mA cm−2), whereas the PCEs of the ternary cells (Entries 5 and 6) are statistically identical to those of the devices fabricated in the glove box. This result suggests that the benefits of ET18 or PCBM in term of temporal stability originate from the initial stage of the device fabrication.
Photovoltaic stability measurements
Next, temporal stability experiments were carried out for both the devices fabricated in the glove box (entries 7–9) and in air (entries 10–12). All devices (without encapsulation) were stored in ambient air and in the dark and were tested regularly over the period of 6 months. Table 2 collects details of the performance parameters for the glove box-processed devices after 6 months aging while Fig. 3(a) reports selected PCE percentage variations over the same time period. These data clearly demonstrate that the PCE of the binary OSCs progressively decreases from 13.4% to 4.5% (∼70%) as the result of severe degradation of both Jsc (25.4 → 16.0 mA cm−2) and FF (65.9 → 38.1%). In contrast, both TSC devices were found to be fully functional after 6 months with a slight PCE enhancement to 13.6% for ET18 (+∼3%) and 13.5% for PCBM (+∼1.5%).| Entry | Blend | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| a Average of 10 devices. b Average of 5 devices. | |||||
| Devices fabricated in glove boxa | |||||
| 7 | PD:Y6 | 16.0 ± 1.7 (19.0) | 0.725 ± 0.016 (0.750) | 38.1 ± 3.2 (43.9) | 4.5 ± 0.9 (5.7) |
| 8 | PD:Y6:ET18Y6:ET18 | 26.8 ± 0.8 (28.6) | 0.774 ± 0.007 (0.782) | 65.4 ± 3.0 (68.6) | 13.6 ± 0.4 (14.7) |
| 9 | PD:Y6:PCBM | 26.5 ± 1.3 (28.3) | 0.779 ± 0.008 (0.790) | 65.3 ± 2.8 (69.1) | 13.5 ± 0.7 (14.7) |
| Devices fabricated in airb | |||||
| 10 | PD:Y6 | 13.7 ± 1.4 (15.6) | 0.623 ± 0.115 (0.740) | 35.5 ± 6.0 (42.2) | 3.2 ± 1.1 (4.9) |
| 11 | PD:Y6:ET18 | 26.2 ± 0.4 (26.7) | 0.751 ± 0.005 (0.757) | 61.0 ± 1.9 (62.9) | 12.0 ± 0.6 (12.7) |
| 12 | PD:Y6:PCBM | 26.7 ± 0.1 (26.9) | 0.755 ± 0.002 (0.757) | 58.0 ± 1.5 (59.6) | 11.7 ± 0.3 (12.1) |
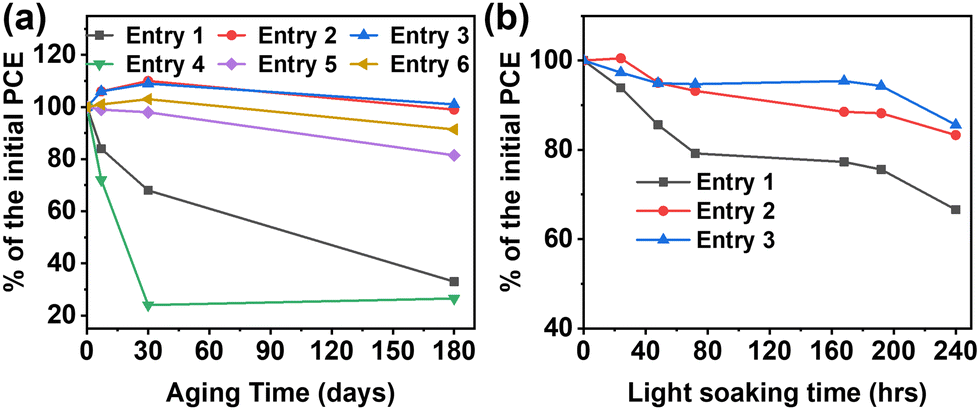 | ||
| Fig. 3 PCE variation in percentage for the indicated cells on the day of fabrication and after storage in the air/dark for the indicated time (a), and the light soaking stability of devices prepared in glove box (b). | ||
Remarkably, similar trends were found for the devices fabricated in air (Fig. 3(b)). Thus, the binary OSCs exhibit dramatic reduction of the PCE from 12.2% to 3.2% (>− 70% of the initial PCE) as the result of strong reduction of all performance parameters, including the Voc. Note, the binary cell PCE falls dramatically (∼80%) even after 1 month of preparation. On the other hand, the ternary OSCs fabricated in air also exhibit remarkably stable performance, with negligible PCE reduction after 1 month test and, even after 6 months of storage in ambient conditions, retaining PCEs of ∼12%.
Finally, we investigated the light stability for the devices fabricated in the glove box by carrying out light soaking experiments (Fig. 3(b)). The unencapsulated samples were exposed to a 5000 K white light source with 100 mW cm−2 intensity (substrate temperature = 58.8 °C; nitrogen-flushed bag with a RH% < 0.5%). The results further corroborate the stabilizing effect of the ternary component, with the binary and ternary devices losing ∼35% and ∼15% of the initial PCE after 240 h of irradiation. Notable is the considerable degradation of the binary cells (>−20%) after only ∼80 h.
Blend morphological and photophysical characterization
To rationalize device performance variation, atomic force microscopy (AFM), GIWAXS and transient absorption spectra (TA) measurements were carried out to investigate the film morphology and microstructure of the blends. First, the surface morphology was assessed by AFM. As shown in Fig. S4 in the ESI,† the PD:Y6 blends display a relatively smooth surface with a small root-mean-square roughness (σRMS) of 1.02 nm. In comparison, the σRMS of the PD:Y6:ET18 and PD:Y6:PCBM ternary blends are 0.72 and 0.73 nm, respectively. After aging in air for 6 months, the PD:Y6 surface roughness increases slightly to 1.13 nm. Similarly the corresponding PD:Y6:ET18 binary blend film becomes rougher with σRMS increasing to 0.79 nm. In contrast, the aged PD:Y6:PCBM film remains smooth (σRMS = 0.59 nm). Overall, all blends exhibit smooth surface morphology with slight difference after aging. Thus, the AFM characterization could not explain the PCE variations after aging.The aging process is further monitored by GIWAXS. The 2D-GIWAXS plots and the corresponding linecuts are shown in Fig. 4 and Tables S1, S2, ESI† summarize details of the diffraction parameters. As shown in the Fig. 4, all fresh samples exhibit well-defined (100) reflections in the in-plane (IP) direction and (010) reflections in the out-of-plane (OoP) direction, indicating their preferential π-face-on packing of the polymer backbone relative to the substrate surface, which is similar to previous findings.66 Thus for the freshly prepared PD:Y6, PD:Y6:ET18, PD:Y6:PCBM blends, the reflections located at qxy = ∼0.30 Å−1 with lamellar d-spacing (dl) of 20.65, 20.72, 20.61 Å, respectively, are attributed to the donor polymer (100) alkyl-chain stacking.69 The corresponding coherence length (CCL) for the binary PD:Y6 blend (54.90 Å) is smaller than that of the PD:Y6:ET18 (69.88 Å) and PD:Y6:PCBM (58.40 Å) blends. Additionally, all fresh blends show a (010) reflection, located at qz = ∼1.75 Å−1 (dπ = ∼3.60 Å) which could be ascribed to the polymer π–π stacking. The ternary blend with ET-18 shows a slightly smaller CCL (28.92 Å) than that of both PD:Y6:ET18 (32.46 Å) and PD:Y6:PCBM (32.76 Å) blends. These results suggest the additions of fullerene derivatives minimally affect the blend microstructure, which rationalize why all fresh blends exhibit similar PCEs. After aging, all blends retain preferential π-face-on packing of the polymer backbone relative to the substrate surface exhibiting well-defined (100) reflections in the IP direction and (010) reflections in the OoP direction. Thus, the aged PD:Y6, PD:Y6:ET18, PD:Y6:PCBM blends exhibit a qxy reflection at ∼0.30 Å−1 corresponding a dl of 20.49, 20.72, 20.61 Å, respectively, also attributed to the donor polymer (100) alkyl-chain stacking. Interestingly, the CCL for the PD:Y6 blend (45.29 Å) is much smaller than that of the PD:Y6:ET18 (64.99 Å) and PD:Y6:PCBM (58.10 Å) blends. Also, all aged blends exhibit a (010) polymer π–π stacking reflection, located at qz = ∼1.75 Å−1 (dπ = ∼3.60 Å). However, those of the ternary PD:Y6:ET18 (30.81 Å) and PD:Y6:PCBM (33.12 Å) blends exhibit larger CCL than that of the PD:Y6 blend (27.48 Å). Interestingly, the CCL of the aged PD:Y6 blend decreases (32.46 → 27.48 Å), while that of the ternary blends slightly increases (28.92 → 30.81 Å for PD:Y6:ET18; 32.76 → 33.12 Å for PD:Y6:PCBM). These results indicate that the addition of fullerene derivatives stabilizes the microstructure, which could promote the corresponding solar cell temporal stability as shown above.
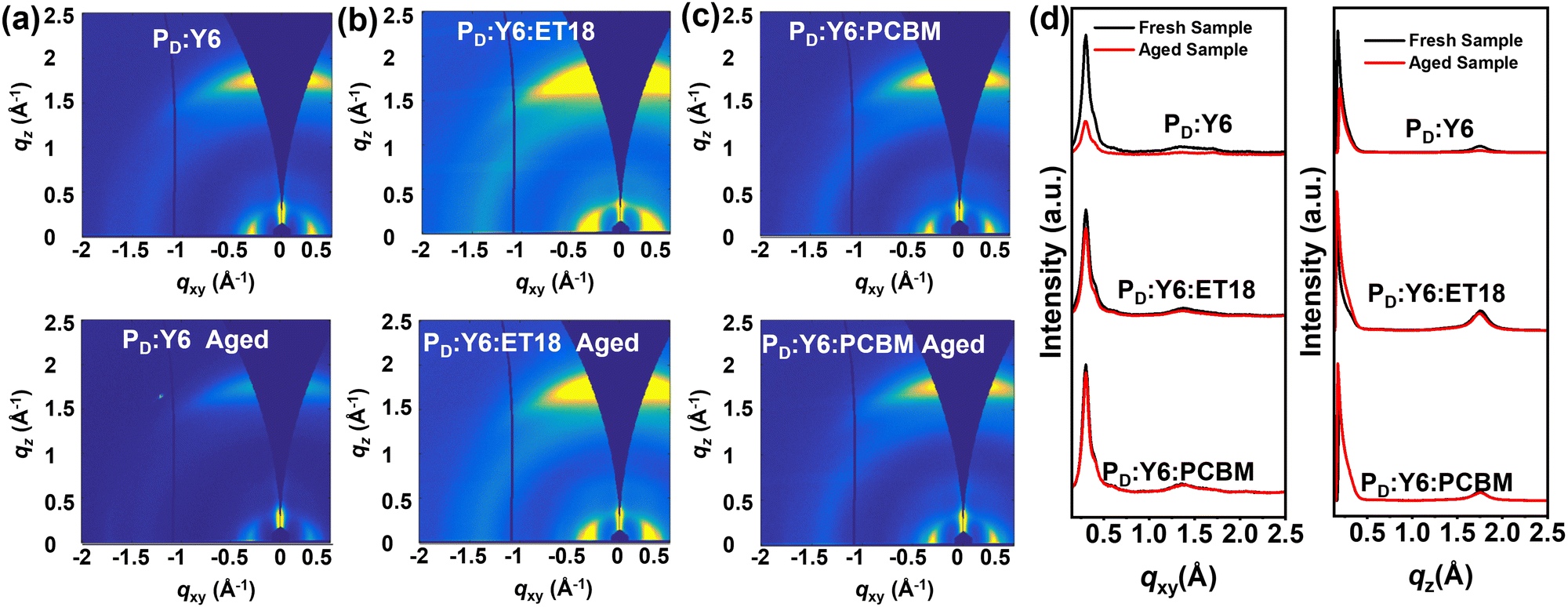 | ||
| Fig. 4 GIWAXS of the investigated fresh and aged blends. 2D GIWAXS images for the indicated blends (a)–(c) and the corresponding in-plane and out-of-plane line cuts (d). | ||
Transient absorption spectroscopy measurements were carried out for selected fresh and aged blend samples. In this experiment, the PD:Y6 and PD:Y6:ET18 blends were coated on a glass substrate and excited at a wavelength of 760 nm, where the donor does not absorb.66 Absorption spectra show that the fresh binary blends produce significantly more holes at long times (6 ns) than the 10 days aged blend (Fig. 5(a)). This is in accordance with the PCE loss due to aging. Therefore, the aged blend is not producing free charge carriers nearly as efficiently as the fresh blend despite the kinetics of the two species are largely similar (Fig. 5(b)). This means that while the fresh blend is producing the charge carriers more efficiently, they do not necessarily live for a longer period of time. On the other hand, the ternary blend absorption spectra change only slightly (∼670 nm) after 6 ns (Fig. 5(c)) suggesting efficient hole generation even after aging. Note that the kinetics of polymer ground state bleaching (660 nm) remain similar between binary and ternary cells (Fig. 5(d)). These results are consistent with the higher recombination, and poor charge transport, in the aged binary blend devices possibly due to increased density of sub-band trap states and morphological changes.69,70 However, these deleterious processes are effectively suppressed in the ternary blends and show remarkable stability in ambient for several months.
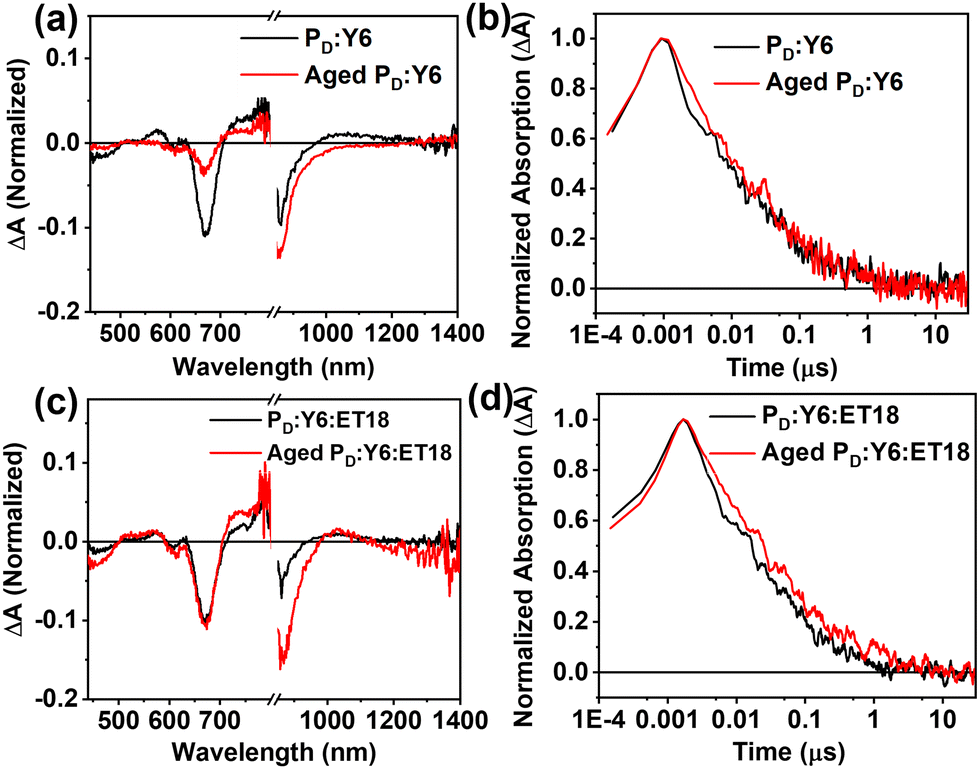 | ||
| Fig. 5 Transient absorption spectra at 6 ns for the binary (a) and ternary (c) blends excited at 760 nm, normalized to the Y6 bleach at 860 nm immediately after excitation; free charge (FC) decay dynamics were monitored at the polymer ground state bleaching (GSB) (660 nm), (b) for binary, (d) for ternary blends. | ||
Impedance and photocurrent measurements
To understand the degradation mechanism, we employed Integrated Photocurrent Device Analysis (IPDA) on binary (PD:Y6) and ternary (PD:Y6:ET18) cells. We conducted in situ impedance and photocurrent measurements immediately after the fabrication of the device and after aging in ambient/dark conditions.71–74 The device parameters were extracted from the measurements of the photocurrent density (Jpc) and chemical capacitance (Cμ) as a function of varying applied voltage (Vapp) and illumination intensity (I).As represented in Fig. 6, the logarithm of Jpc plotted versus the logarithm of the effective voltage (Veff), given from the relation, Veff= Voc − Vapp, shows the qualitative difference in the performance of the two blends. Specifically, the onset of the saturation regime (Vsat) for the binary blend, shifts from 0.2 V (day 0) to ∼0.5–0.8 V (day 10), suggesting a significant increase in the bimolecular recombination (Fig. 6(a) and (b)).
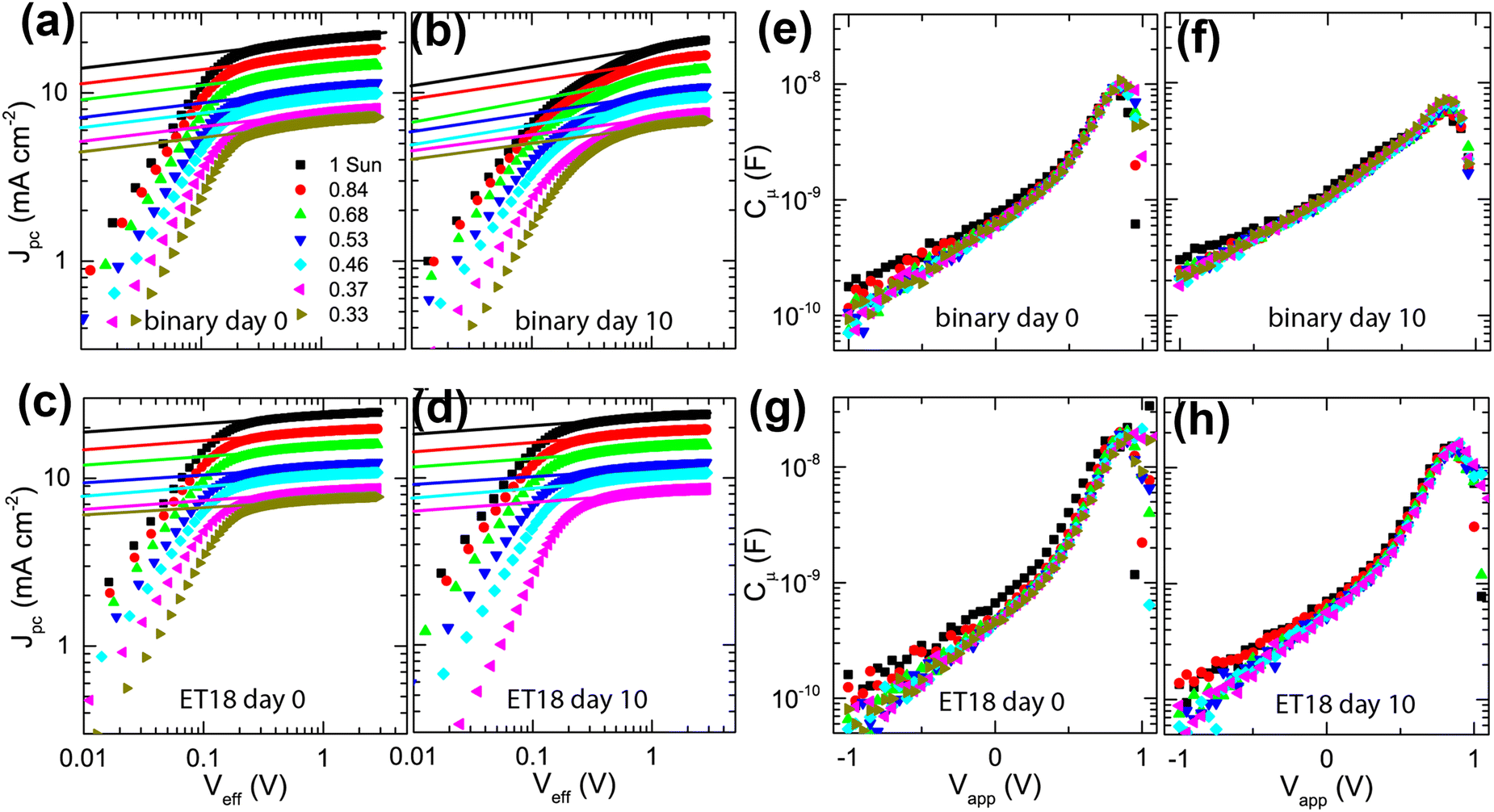 | ||
| Fig. 6 Plots of photocurrent density (Jpc) vs. effective voltage (Veff) for a binary cell, at different illumination intensities, before (a) and after 10 days of ambient exposure (b), respectively. Plots of Jpcvs. Veff for an ET18-based ternary cell before (c) and after 10 days of ambient exposure (d). The legend in (a) shows illumination intensity as a fraction of 1 sun (AM 1.5G light in ambient conditions) and corresponds to all plots (a)–(h). The solid lines are linear fits in the log–log scale to determine the range of saturating regimes. Plots of chemical capacitance (Cμ) versus applied voltage (Vapp) for the same binary cell at the same intensities before (e) and after 10 days of ambient exposure (f), respectively. Plots of Cμversus Vapp for the same ET18-based ternary cell before (g) and after 10 days of ambient exposure (h). | ||
Conversely, for the ternary blend with ET18 (Fig. 6(c) and (d)), the Vsat remains constant even after 10 days, suggesting robust passivation of the blend (Fig. 6(d)). Next, we studied the chemical capacitance (Cμ) of cells given from the relation Cμ = Ctot − Cd, where Ctot is total capacitance and Cd is depletion capacitance measured in the dark at a large negative bias (−2 V) using an AC amplitude of 100 mV at 1 kHz. The binary cells show a clear broadening of the peak in the Cμ–Vapp plot with significantly increased Cμ in the full range of Vapp (from −1 V to Voc) cells after 10 days (Fig. 6(e) and (f)), suggesting the emergence of significant sub-bandgap trap states during degradation. On the other hand, no significant changes were seen in the Cμ–Vapp characteristics for ET18 based ternary cells (Fig. 6(g) and (h)), confirming effective passivation.
Further experiments were performed under varying illumination intensity (0.33–1 sun) to evaluate the relative differences in the aging effects in binary and ET18-based ternary cells. The light intensity dependence of Jsc is given by the relationship Jsc ∝ Iα, where α ∼ 1, and the deviation of this value is attributed to bimolecular recombination.75 First, α for binary cells changes from 0.99 to 0.94 after 10 days (Fig. 7(a)), which suggests increased bimolecular recombination after degradation and correlates well with decreased FF (discussed later). Conversely, ET18-based ternary cells show the same α value (0.99) even after 10 days (Fig. 7(a)). Second, the intensity (I) dependent Voc (∝ β·log(I)) shows a slight decrease in β (from 0.57 to 0.51 mV) for ET18 based ternary cells after 10 days (Fig. 7(b)). Decreasing the value of β closer to 2KBT/e ≈ 50 mV indicates that monomolecular recombination and other non-linear effects seem to ameliorate during the aging of ET18 cells. Similar counter-intuitive effects have been recently reported using impedance spectroscopy of hybrid organic–inorganic perovskite solar cells.76 In contrast, binary cells undergo significant deviation from ideal solar cell characteristics and it was not possible to extract β from the data after 10 days (Fig. 7(b)).
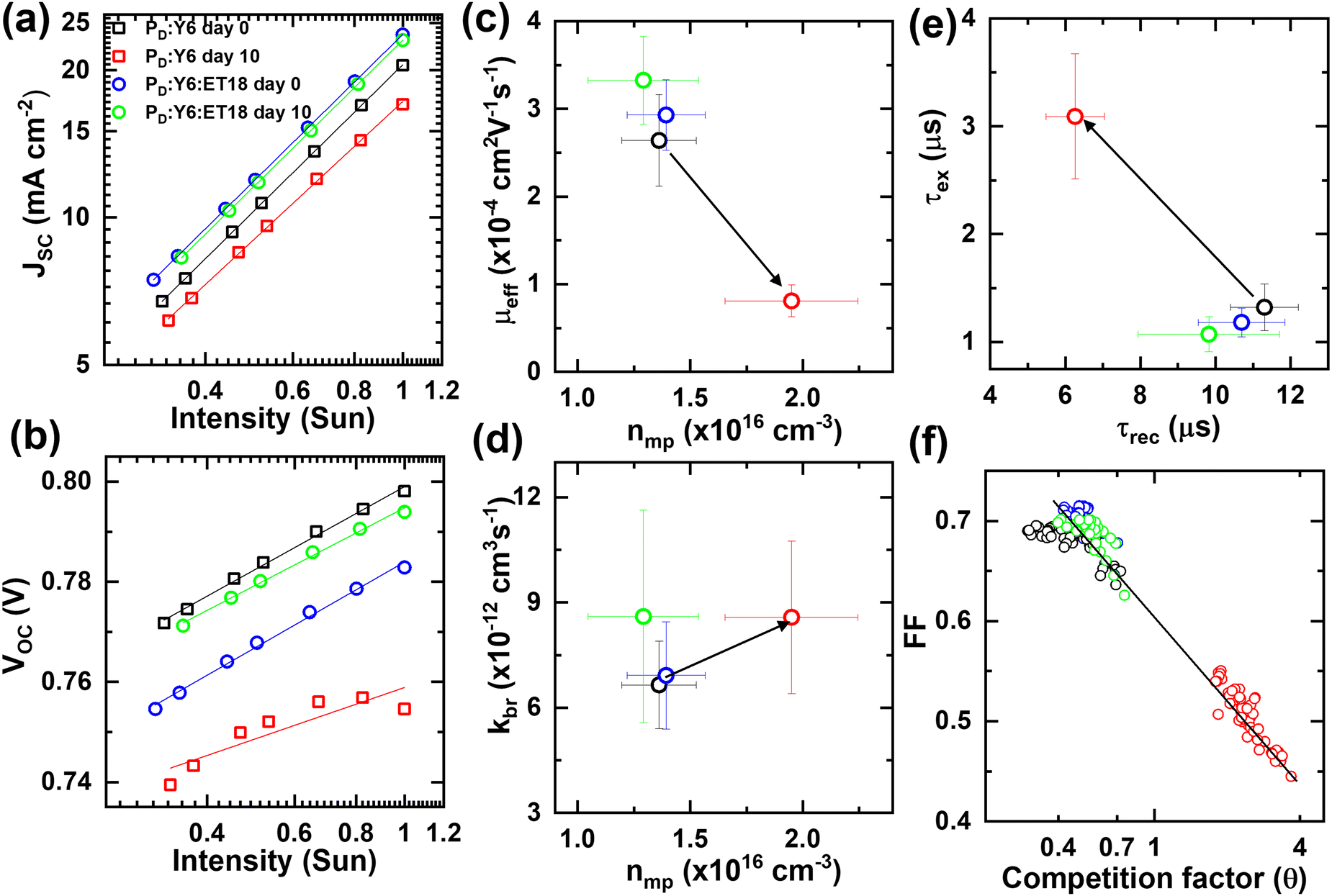 | ||
| Fig. 7 (a) Log–log plot of short-circuit current density (Jsc) vs. illumination intensity (I) in units of 1 sun for binary and ET18 based ternary solar cells before and after 10 days of ambient exposure in dark. The solid lines show fit to log(Jsc) ∼α·log(I) and extracted α values are 1.00 (binary day 0), 0.94 (binary day 10), 0.99 (ET18 day 0), and 0.99 (ET day 10). The legend in (a) corresponds to all plots (a) – (f). (b) Linear-log plot of open-circuit voltage (Voc) versus intensity for the same binary and ET18 cells before and after 10 days. The solid lines show fits to Voc ∼ β·log(I) and the extracted β values are 55 meV (binary day 0), 57 meV (ET18 day 0), and 51 meV (ET18 day 10). (c) A plot of average effective mobility (μeff) vs. carrier density (nmp) extracted from IDPA at the maximum PCE point under 1 sun illumination for binary and ET18 cells before and after 10 days. The error bars show the standard deviation for μeff and nmp from 8 devices of each kind. The black arrow shows a decrease in μeff for binary cells after 10 days. (d) A plot of average recombination coefficient (kbr) versus nmp extracted from the same analysis of binary and ET18 cells before and after 10 days. (e) A plot of average charge extraction time constant (τex) vs. recombination lifetime (τrec) for binary and ET18 cells before and after 10 days. (f) A plot of fill factor (FF) versus competition factor (θ) for all 16 of the measured binary and ET18 based ternary blends before and after 10 days. | ||
Carrier density (n(V)) is calculated by integrating Cμversus Vapp over the range of biases from Vsat to a corrected voltage bias across the active layer that is obtained after subtracting the voltage drop across the interfacial series resistance. Then, we extracted average effective carrier mobilities (μeff = (μe + μh)/2, where μe and μh are electron and hole mobilities, respectively) from eight devices of each kind, binary and ET18 based ternary cells, before and after 10 days (Fig. 7(c)). The calculation of μeff at the maximum PCE point (n = nmp) is a direct measure of the charge extraction capability of the solar cell during operation (1 sun). Binary cells show more than a three-fold decrease in μeff while nmp is increased by more than two-fold after 10 days (Fig. 7(c)). Note, increased nmp in Y6 cells is consistent with increased Cμ at low biases after degradation. On the other hand, ET18-based ternary cells show a slight increase in μeff and concurrent decrease in nmp after 10 days, consistent with a slight improvement in Voc characteristics in Fig. 7(b). However, the bimolecular recombination coefficient (kbr) is increased slightly in both kinds of cells after 10 days (Fig. 7(d)).
The carrier recombination lifetime (τrec) further contrasts the aging process in the two kinds of cells. Note, carrier lifetimes extracted from impedance measurements of operating solar cells (1 sun) reveal significantly longer values as compared to intrinsic decay rates accessed by transient absorption and photoluminescence spectroscopy due to underlying differences in operating conditions.66,71,73,74 The degraded binary cells show 2–3 times decrease in τrec while the extraction time constant (τex) is increased by a similar factor (Fig. 7(e)). In contrast, ET18-based ternary cells again show a similar or slight increase in τrec and τex values after 10 days (Fig. 7(e)). Finally, the combined effects of recombination and extraction processes are more effectively captured in the competition factor (θ ∼ τex/τrec) that scales inversely with FF in a wide range of BHJ solar cells.66,71,73,74 Binary cells show almost an order of magnitude increase in θ after 10 days that correlates well with a roughly 30% decrease in FF (Fig. 7(f)). ET18-based ternary cells, in contrast, do not show any appreciable change in θ and FF after 10 days of ambient exposure (Fig. 7(f)). Thus, the ternary blend can be considered passivated by the presence of ET18 which effectively suppresses the recombination processes.
Conclusions
We investigated the stability in ambient of binary and ternary OSCs comprising a donor polymer, an NFA and, for the latter devices, a fullerene derivative as the ternary component. Our results show that the ternary blends afford far more stable devices than the parent binary blends. Thus, the PCE of the ternary devices is stable up to 6 months storage in ambient while the binary cells exhibit a PCE loss of up to 60% during the same timeframe. Ternary cells can also be fabricated in ambient conditions and they also exhibit similar PCEs and stability when stored in ambient. Ultrafast transient spectroscopy measurements show that the hole transfer process in the aged ternary blends remains unchanged while the binary cells show significant deterioration. GIWAXS measurements confirm that the ternary blends retain the same morphology of the as-prepared films while that of the binary cells undergoes significant variations. From in situ impedance and photocurrent measurements, the degradation in binary cells was found to correlate with increased sub-band traps as well as bimolecular and monomolecular recombination rates. On other hand ET18-based ternary cells did not exhibit any appreciable change in these parameters, in agreement with the stable blend morphology. Overall, the stabilization role of ET18 in ternary solar cells was found to be slightly greater than that of PCBM. Thus, the possibility of fabricating organic solar cells in ambient with subsequent long-term stability in ambient without encapsulation not only encourages further exploration of more stable and higher-performing solar cells but also promotes this technology towards commercialization.Author contributions
ET, YC, VC and L-W. F. synthesized the materials carried out solar cell fabrication and wrote the manuscript. WH, YC and DZ carried out GIWAX and AFM measurements. J. M. A. measured the transient absorption spectra. V. K. S. and M. C. H. conducted the IPDA measurement and the data analysis and wrote the manuscript. BP, FG, TJM, and AF supervised the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the Northwestern University MRSEC (NSF grant DMR-1720139), AFOSR (grant FA9550-18-1-0320), award 70NANB19H005 from U.S. Department of Commerce, National Institute of Standards and Technology as part of the Center for Hierarchical Materials Design, and Flexterra Inc. This work made use of the Keck-II and SPID facilities, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205). This work was supported by the Department of Energy under contract no. DE-AC02-05CH11231 and used resources at beamline 8-ID-E of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. The IPDA measurements were supported by the National Science Foundation Materials Research Science and Engineering Center (MRSEC) at Northwestern University (NSF DMR-1720139). This work (IPDA) made use of the MatCI Facility which receives support from the MRSEC Program (NSF DMR-1720139) of the Materials Research Center at Northwestern University. This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under award DE-FG02-99ER14999 (M. R. W.). University of Palermo acknowledges the Ministry of University and Research (MUR) and its program PON R&I 2014-2020 (project ARS01_00334) for funding. This work was supported by the National Natural Science Foundation of China (22209169).Notes and references
- E. Wang, W. Mammo and M. R. Andersson, Adv. Mater., 2014, 26, 1801–1826 CrossRef CAS PubMed.
- H. Sun, B. Liu, Y. Ma, J. W. Lee, J. Yang, J. Wang, Y. Li, B. Li, K. Feng, Y. Shi, B. Zhang, D. Han, H. Meng, L. Niu, B. J. Kim, Q. Zheng and X. Guo, Adv. Mater., 2021, 33, e2102635 CrossRef PubMed.
- B. Liu, H. L. Sun, J. W. Lee, J. Yang, J. W. Wang, Y. C. Li, B. B. Li, M. Xu, Q. G. Liao, W. Zhang, D. X. Han, L. Niu, H. Meng, B. J. Kim and X. G. Guo, Energy Environ. Sci., 2021, 14, 4499–4507 RSC.
- S. D. Collins, N. A. Ran, M. C. Heiber and T. Q. Nguyen, Adv. Energy Mater., 2017, 7, 1602242 CrossRef.
- H. K. H. Lee, J. Y. Wu, J. Barbe, S. M. Jain, S. Wood, E. M. Speller, Z. Li, F. A. Castro, J. R. Durrant and W. C. Tsoi, J. Mater. Chem. A, 2018, 6, 5618–5626 RSC.
- H. Yin, J. K. W. Ho, S. H. Cheung, R. J. Yan, K. L. Chiu, X. Hao and S. K. So, J. Mater. Chem. A, 2018, 6, 8579–8585 RSC.
- C. L. Cutting, M. Bag and D. Venkataraman, J. Mater. Chem. C, 2016, 4, 10367–10370 RSC.
- L. K. Reb, M. Bohmer, B. Predeschly, S. Grott, C. L. Weindl, G. I. Ivandekic, R. J. Guo, C. Dreissigacker, R. Gernhauser, A. Meyer and P. Muller-Buschbaum, Joule, 2020, 4, 1880–1892 CrossRef CAS.
- X. Zhang, L. Qin, J. Yu, Y. Li, Y. Wei, X. Liu, X. Lu, F. Gao and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 12475–12481 CrossRef CAS PubMed.
- X. Zhang, C. Li, L. Qin, H. Chen, J. Yu, Y. Wei, X. Liu, J. Zhang, Z. Wei, F. Gao, Q. Peng and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 17720–17725 CrossRef CAS PubMed.
- Y. Sun, L. Nian, Y. Kan, Y. Ren, Z. Chen, L. Zhu, M. Zhang, H. Yin, H. Xu, J. Li, X. Hao, F. Liu, K. Gao and Y. Li, Joule, 2022, 6, 2835–2848 CrossRef CAS.
- R. Ma, K. Zhou, Y. Sun, T. Liu, Y. Kan, Y. Xiao, T. A. Dela Peña, Y. Li, X. Zou, Z. Xing, Z. Luo, K. S. Wong, X. Lu, L. Ye, H. Yan and K. Gao, Matter, 2022, 5, 725–734 CrossRef CAS.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- Q. Tai and F. Yan, Adv. Mater., 2017, 29, 1700192 CrossRef PubMed.
- I. Ierides, A. Zampetti and F. Cacialli, Curr. Opin. Green Sustainable Chem., 2019, 17, 15–20 CrossRef.
- Y. Li, X. Huang, K. Ding, H. K. M. Sheriff, Jr., L. Ye, H. Liu, C. Z. Li, H. Ade and S. R. Forrest, Nat. Commun., 2021, 12, 5419 CrossRef CAS PubMed.
- Q. Burlingame, X. Huang, X. Liu, C. Jeong, C. Coburn and S. R. Forrest, Nature, 2019, 573, 394–397 CrossRef CAS PubMed.
- S. O. Oseni and G. T. Mola, Sol. Energy Mater. Sol. Cells, 2017, 160, 241–256 CrossRef CAS.
- E. Pavlopoulou, G. Fleury, D. Deribew, F. Cousin, M. Geoghegan and G. Hadziioannou, Org. Electron., 2013, 14, 1249–1254 CrossRef CAS.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- Y. Chen, L. Yang, J. L. Wu, G. Wang, W. Huang, F. S. Melkonyan, Z. Y. Lu, Y. Huang, T. J. Marks and A. Facchetti, Chem. Mater., 2018, 30, 6810–6820 CrossRef CAS.
- A. Marrocchi, D. Lanari, A. Facchetti and L. Vaccaro, Energy Environ. Sci., 2012, 5, 8457–8474 RSC.
- J. X. He, J. L. Hua, G. X. Hu, X. J. Yin, H. Gong and C. X. Li, Dyes Pigm., 2014, 104, 75–82 CrossRef CAS.
- S. Fabiano and B. Pignataro, Phys. Chem. Chem. Phys., 2010, 12, 14848–14860 RSC.
- C. Sartorio, V. Campisciano, C. Chiappara, S. Cataldo, M. Scopelliti, M. Gruttadauria, F. Giacalone and B. Pignataro, J. Mater. Chem. A, 2018, 6, 3884–3894 RSC.
- A. Distler, T. Sauermann, H.-J. Egelhaaf, S. Rodman, D. Waller, K.-S. Cheon, M. Lee and D. M. Guldi, Adv. Energy Mater., 2014, 4, 1300693 CrossRef.
- F. Piersimoni, G. Degutis, S. Bertho, K. Vandewal, D. Spoltore, T. Vangerven, J. Drijkoningen, M. K. Van Bael, A. Hardy, J. D’Haen, W. Maes, D. Vanderzande, M. Nesladek and J. Manca, J. Polym. Sci., Polym. Phys., 2013, 51, 1209–1214 CrossRef CAS.
- Q. Ma, Z. Jia, L. Meng, J. Zhang, H. Zhang, W. Huang, J. Yuan, F. Gao, Y. Wan, Z. Zhang and Y. Li, Nano Energy, 2020, 78, 105272 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- J. Lee, S.-J. Ko, H. Lee, J. Huang, Z. Zhu, M. Seifrid, J. Vollbrecht, V. V. Brus, A. Karki, H. Wang, K. Cho, T.-Q. Nguyen and G. C. Bazan, ACS Energy Lett., 2019, 4, 1401–1409 CrossRef CAS.
- N. Gasparini, A. Wadsworth, M. Moser, D. Baran, I. McCulloch and C. J. Brabec, Adv. Energy Mater., 2018, 8, 1703298 CrossRef.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed.
- Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Xu, K. Xian, B. Gao, J. Qin, J. Zhang, Z. Wei and J. Hou, Adv. Mater., 2019, 31, e1808356 CrossRef PubMed.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C. C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- L. Zhan, S. Li, Y. Li, R. Sun, J. Min, Z. Bi, W. Ma, Z. Chen, G. Zhou, H. Zhu, M. Shi, L. Zuo and H. Chen, Joule, 2022, 6, 662–675 CrossRef CAS.
- I. Ramirez, M. Causa, Y. F. Zhong, N. Banerji and M. Riede, Adv. Energy Mater., 2018, 8, 1703551 CrossRef.
- D. Baran, T. Kirchartz, S. Wheeler, S. Dimitrov, M. Abdelsamie, J. Gorman, R. S. Ashraf, S. Holliday, A. Wadsworth and N. Gasparini, Energy Environ. Sci., 2016, 9, 3783–3793 RSC.
- T. Ameri, P. Khoram, J. Min and C. J. Brabec, Adv. Mater., 2013, 25, 4245–4266 CrossRef CAS PubMed.
- H. Huang, L. Yang and B. Sharma, J. Mater. Chem. A, 2017, 5, 11501–11517 RSC.
- T. Gokulnath, K. Feng, H. Y. Park, Y. Do, H. Park, R. D. Gayathri, S. S. Reddy, J. Kim, X. Guo, J. Yoon and S. H. Jin, ACS Appl. Mater. Interfaces, 2022, 14, 11211–11221 CrossRef CAS PubMed.
- N. Gasparini, A. Salleo, I. McCulloch and D. Baran, Nat. Rev. Mater., 2019, 4, 229–242 CrossRef.
- L. Xiao, K. Gao, Y. Zhang, X. Chen, L. Hou, Y. Cao and X. Peng, J. Mater. Chem. A, 2016, 4, 5288–5293 RSC.
- W.-L. Xu, B. Wu, F. Zheng, X.-Y. Yang, H.-D. Jin, F. Zhu and X.-T. Hao, J. Phys. Chem. C, 2015, 119, 21913–21920 CrossRef CAS.
- V. Gupta, V. Bharti, M. Kumar, S. Chand and A. J. Heeger, Adv. Mater., 2015, 27, 4398–4404 CrossRef CAS PubMed.
- L. Y. Lu, T. Xu, W. Chen, E. S. Landry and L. P. Yui, Nat. Photonics, 2014, 8, 716–722 CrossRef CAS.
- M. Koppe, H. J. Egelhaaf, E. Clodic, M. Morana, L. Luer, A. Troeger, V. Sgobba, D. M. Guldi, T. Ameri and C. J. Brabec, Adv. Energy Mater., 2013, 3, 949–958 CrossRef CAS.
- W. Y. Su, Q. P. Fan, X. Guo, B. Guo, W. B. Li, Y. D. Zhang, M. J. Zhang and Y. F. Li, J. Mater. Chem. A, 2016, 4, 14752–14760 RSC.
- Y. He, H.-Y. Chen, J. Hou and Y. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed.
- R. Sun, Y. Wu, X. Yang, Y. Gao, Z. Chen, K. Li, J. Qiao, T. Wang, J. Guo, C. Liu, X. Hao, H. Zhu and J. Min, Adv. Mater., 2022, 34, e2110147 CrossRef PubMed.
- H. Q. Cao, W. D. He, Y. W. Mao, X. Lin, K. Ishikawa, J. H. Dickerson and W. P. Hess, J. Power Sources, 2014, 264, 168–183 CrossRef CAS.
- A. J. Parnell, A. J. Cadby, A. D. F. Dunbar, G. L. Roberts, A. Plumridge, R. M. Dalgliesh, M. W. A. Skoda and R. A. L. Jones, J. Polym. Sci., Polym. Phys., 2016, 54, 141–146 CrossRef CAS.
- S. Rafique, S. M. Abdullah, K. Sulaiman and M. Iwamoto, Renewable Sustainable Energy Rev., 2018, 84, 43–53 CrossRef.
- A. Seemann, T. Sauermann, C. Lungenschmied, O. Armbruster, S. Bauer, H. J. Egelhaaf and J. Hauch, Sol. Energy, 2011, 85, 1238–1249 CrossRef CAS.
- X. R. Tong, N. N. Wang, M. Slootsky, J. S. Yu and S. R. Forrest, Sol. Energy Mater. Sol. Cells, 2013, 118, 116–123 CrossRef CAS.
- C. H. Peters, I. T. Sachs-Quintana, W. R. Mateker, T. Heumueller, J. Rivnay, R. Noriega, Z. M. Beiley, E. T. Hoke, A. Salleo and M. D. McGehee, Adv. Mater., 2012, 24, 663–668 CrossRef CAS PubMed.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Rohr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2017, 16, 363–369 CrossRef CAS PubMed.
- H. Feng, X. Song, Z. Zhang, R. Geng, J. Yu, L. Yang, D. Baran and W. Tang, Adv. Funct. Mater., 2019, 29, 1903269 CrossRef.
- Q. Q. Zhao, Z. Xiao, J. F. Qu, L. Z. Liu, H. Richter, W. Chen, L. Han, M. J. Wang, J. M. Zheng, Z. Q. Xie, L. M. Ding and F. He, ACS Energy Lett., 2019, 4, 1106–1114 CrossRef CAS.
- K.-N. Zhang, J.-J. Guo, L.-J. Zhang, C.-C. Qin, H. Yin, X.-Y. Gao and X.-T. Hao, Adv. Funct. Mater., 2021, 31, 2100316 CrossRef CAS.
- B.-H. Jiang, Y.-J. Peng, Y.-W. Su, J.-F. Chang, C.-C. Chueh, T.-S. Shieh, C.-I. Huang and C.-P. Chen, Chem. Eng. J., 2022, 431, 133950 CrossRef CAS.
- Y. Lin, Y. Firdaus, M. I. Nugraha, F. Liu, S. Karuthedath, A. H. Emwas, W. Zhang, A. Seitkhan, M. Neophytou, H. Faber, E. Yengel, I. McCulloch, L. Tsetseris, F. Laquai and T. D. Anthopoulos, Adv. Sci., 2020, 7, 1903419 CrossRef CAS PubMed.
- L.-W. Feng, J. Chen, S. Mukherjee, V. K. Sangwan, W. Huang, Y. Chen, D. Zheng, J. W. Strzalka, G. Wang, M. C. Hersam, D. DeLongchamp, A. Facchetti and T. J. Marks, ACS Energy Lett., 2020, 5, 1780–1787 CrossRef CAS.
- Y. Liu, X. Wan, F. Wang, J. Zhou, G. Long, J. Tian and Y. Chen, Adv. Mater., 2011, 23, 5387–5391 CrossRef CAS PubMed.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- J. Wu, C. Y. Liao, Y. Chen, R. M. Jacobberger, W. Huang, D. Zheng, K. W. Tsai, W. L. Li, Z. Lu, Y. Huang, M. R. Wasielewski, Y. M. Chang, T. J. Marks and A. Facchetti, Adv. Energy Mater., 2021, 11, e2102648 CrossRef.
- N. D. Eastham, J. L. Logsdon, E. F. Manley, T. J. Aldrich, M. J. Leonardi, G. Wang, N. E. Powers-Riggs, R. M. Young, L. X. Chen, M. R. Wasielewski, F. S. Melkonyan, R. P. H. Chang and T. J. Marks, Adv. Mater., 2018, 30, 1704263 CrossRef PubMed.
- W. Zhu, A. P. Spencer, S. Mukherjee, J. M. Alzola, V. K. Sangwan, S. H. Amsterdam, S. M. Swick, L. O. Jones, M. C. Heiber, A. A. Herzing, G. Li, C. L. Stern, D. M. DeLongchamp, K. L. Kohlstedt, M. C. Hersam, G. C. Schatz, M. R. Wasielewski, L. X. Chen, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2020, 142, 14532–14547 CrossRef CAS PubMed.
- S. M. Swick, J. M. Alzola, V. K. Sangwan, S. H. Amsterdam, W. Zhu, L. O. Jones, N. Powers-Riggs, A. Facchetti, K. L. Kohlstedt, G. C. Schatz, M. C. Hersam, M. R. Wasielewski and T. J. Marks, Adv. Energy Mater., 2020, 10, 2000635 CrossRef CAS.
- M. C. Heiber, T. Okubo, S. J. Ko, B. R. Luginbuhl, N. A. Ran, M. Wang, H. B. Wang, M. A. Uddin, H. Y. Woo, G. C. Bazan and T. Q. Nguyen, Energy Environ. Sci., 2018, 11, 3019–3032 RSC.
- D. Bartesaghi, C. Perez Idel, J. Kniepert, S. Roland, M. Turbiez, D. Neher and L. J. Koster, Nat. Commun., 2015, 6, 7083 CrossRef CAS PubMed.
- L. J. A. Koster, V. D. Mihailetchi, H. Xie and P. W. M. Blom, Appl. Phys. Lett., 2005, 87, 203502 CrossRef.
- C. Aranda, J. Bisquert and A. Guerrero, J. Chem. Phys., 2019, 151, 124201 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc04971f |
| This journal is © The Royal Society of Chemistry 2023 |