
Air-stable ternary organic solar cells achieved by using fullerene additives in non-fullerene acceptor-polymer donor blends†
 b
Yao
Chen,
*ad
Wei
Huang,
a
Ding
Zheng,
a
Vinod K.
Sangwan,
*e
Mark C.
Hersam,
*aef
Michael R.
Wasielewski,
*a
Bruno
Pignataro,
*c
Francesco
Giacalone,
*b
Tobin J.
Marks
*ae
and
Antonio
Facchetti
*ag
b
Yao
Chen,
*ad
Wei
Huang,
a
Ding
Zheng,
a
Vinod K.
Sangwan,
*e
Mark C.
Hersam,
*aef
Michael R.
Wasielewski,
*a
Bruno
Pignataro,
*c
Francesco
Giacalone,
*b
Tobin J.
Marks
*ae
and
Antonio
Facchetti
*ag
Abstract
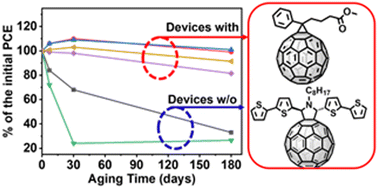
Organic solar cells (OSCs) based on donor–acceptor blends have shown a rapid improvement in power conversion efficiency (PCE) now approaching, for small cells, those of the state-of-the art commercial solar modules. However, performance degradation remains one of the most critical impediments for OSC technology commercialization. Ternary solar cells where a third component, for instance an acceptor, is added to a non-fullerene acceptor–polymer donor blend are an effective approach for improving both OSC efficiency and long-term stability. Here, we study the role of two fullerene acceptors, ET18 and PCBM, as the third component in PD:Y6 blends. These fullerene derivatives significantly enhance the cell stability, which retained >90% of their initial PCEs (13–14%) even after storage in air for 6 months, compared to only ∼20% retention for the binary devices. GIWAXS, AFM, in situ impedance spectroscopy and femtosecond transient absorption spectroscopy measurements reveal that the enhanced stability of the ternary devices results from a more robust blend morphology reducing charge recombination in the ternary devices during aging.
- This article is part of the themed collections: 2023 Journal of Materials Chemistry C HOT Papers and Celebrating ten years of Journal of Materials Chemistry C
 Please wait while we load your content...
Please wait while we load your content...

