
Rational design of a phenothiazine-based donor–acceptor covalent organic framework for enhanced photocatalytic oxidative coupling of amines and cyclization of thioamides†
Yang
Liu
a,
Xinyue
Jiang
a,
Likun
Chen
b,
Yan
Cui
b,
Qiu-Yan
Li
 *a,
Xinsheng
Zhao
a,
Xiguang
Han
a,
Yong-Chao
Zheng
*b and
Xiao-Jun
Wang
*a
*a,
Xinsheng
Zhao
a,
Xiguang
Han
a,
Yong-Chao
Zheng
*b and
Xiao-Jun
Wang
*a
aJiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, School of Chemistry and Materials Science, Jiangsu Normal University, Xuzhou 221116, P. R. China. E-mail: xjwang@jsnu.edu.cn; qyli@jsnu.edu.cn
bState Key Laboratory of NBC Protection for Civilian, Beijing, 102205, PR China. E-mail: zhengycfh@163.com
First published on 18th November 2022
Abstract
Owing to the high structural designability and functional tunability, covalent organic frameworks (COFs) have emerged as a promising platform for designing excellent photocatalyst candidates. However, their photocatalytic performances are still hindered by insufficient separation and transfer of photo-generated charge carriers. The ordered alignment of the electron donor (D) and acceptor (A) in two-dimensional COFs can promote charge carrier separation upon photoexcitation and provide a favorable pathway for exciton transport, and thus is advantageous for photocatalysis. In this work, we report a new D–A COF that was constructed from the electron-rich phenothiazine (PTZ) and electron-deficient triazine (TTA) subunits, giving rise to a segregated bicontinuous D–A heterostructure within the framework. In comparison to the triphenylamine (TPA) based analogue COF with smaller D–A contrast, this newly designed PTZ–TTA-COF exhibited lower exciton binding energy and enhanced charge separation/transfer. As a consequence, it revealed remarkably enhanced photocatalytic ability in oxidative amine coupling and cyclization of thioamide to 1,2,4-thiadiazole reactions under visible light and in air. This study will provide a rational guidance for developing high-performance polymeric photocatalysts based on D–A structural COFs through the molecular-level design strategy.
Introduction
Covalent organic frameworks (COFs), as a fascinating class of crystalline porous polymers, are built by covalently linking organic building blocks in two-/three-dimensional extended networks through the reticular chemistry.1 On account of their periodically ordered structures, high stability and porosity, COFs have emerged as a highly designable and tailorable material platform for various potential applications,1,2 such as gas adsorption/separation,3 chemical sensing,4 energy storage5 and heterogeneous catalysis.6 In particular, two-dimensional COFs with π–π stacking between their layers can promote exciton migration and charge carrier transport through the aligned π columnar arrays,7 and thus these materials have become attractive in photocatalytic applications.8 However, their photocatalytic performances, to a certain degree, are still impeded by insufficient charge separation and transfer as well as fast charge carrier recombination.On the other hand, using COFs provides a feasible approach to precisely assemble various molecular building units into their rigid network in a highly controllable and predictable manner. The appropriate combination and ordered alignment of electron-rich donor (D) and electron-deficient acceptor (A) units in the two-dimensional COF lattice can give rise to the formation of an intrinsic heterostructure of spatially separated D–A columns.9 It has been revealed that such D–A dyad structural design allows for lowering exciton binding energy while enhancing the charge separation and migration of photoinduced carriers,9,10 thereby resulting in a notably improved photocatalytic activity.11 Along this research line, some D–A pairs, such as pyrene–thiazole,12 pyrene–benzothiadiazole,13 tetrathiafulvalene–porphyrin,14 anthracene–porphyrin15 and tetraphenylethylene–triazine,16 have been developed and encoded into COF frameworks.17 Although a range of organic building blocks have been selected to construct various D–A COFs as exemplified above, they are still very limited when considering the huge number of functional moieties in organic chemistry. Moreover, the band gap and energy levels of COFs can be finely tuned at the molecular level by tailoring the organic monomers.18 Thus, it is highly essential to seek more novel building units so as to provide abundant possibilities in designing D–A structural COFs with excellent photocatalytic performances.
Among the family of heterocyclic moieties, phenothiazine (PTZ) is a well-known electron-rich building block with distinct features, such as strong electron-donating characteristics, tunable redox properties and versatility of functionalization.19 These merits have made it find extensive application in various optoelectronics.19,20 More recently, this intriguing PTZ subunit has been decorated and conjugated into porous organic polymers (POPs) for pursuing higher photocatalytic activities.11d,21 For example, Yu and co-workers reported the first PTZ based POP for photocatalytic Ugi-type reaction and selenation of indoles.21a Our group introduced the PTZ moiety into covalent triazine frameworks to modulate their band structures and photo-physical properties, leading to enhanced photocatalytic aerobic oxidation of sulfides.21h Just recently, Gu et al. for the first time reported PTZ-based COFs with impressively low exciton binding energies for photocatalytic polymerization.11d These studies revealed that the integration of the PTZ center as a core scaffold into POPs is beneficial to their photocatalytic performances.21 However, most of them are amorphous conjugated polymers with randomly bonded networks, thus lacking long-range order for efficient charge carrier transport. Therefore, much effort should be put into developing more PTZ-based crystalline COF materials and further exploring their structure–property relationships.
In this work, we report the rational design of a PTZ-based D–A COF by a Schiff-base [3 + 3] condensation reaction of N-(4-formylphenyl)-phenothiazine-3,7-dicarbaldehyde (PTZ-CHO) with 1,3,5-tris(4-aminophenyl)triazine (TTA-NH2) as donor and acceptor subunits, respectively (Fig. 1a). The resulting PTZ–TTA-COF exhibited good crystallinity and porosity, a broad light absorption range, and highly effective charge separation and transport, thanks to the ordered D–A heterostructure within the framework. Moreover, it exhibited enhanced photo-catalytic activities for oxidative amine coupling and cyclization of thioamide to 1,2,4-thiadiazole reactions under visible light and in air, in relation to the triphenylamine (TPA) based analogue TPA–TTA-COF with smaller D–A contrast. Furthermore, this activity increase can be rationalized by low exciton binding energy and enhanced charge separation/transfer as observed in the PTZ–TTA-COF.
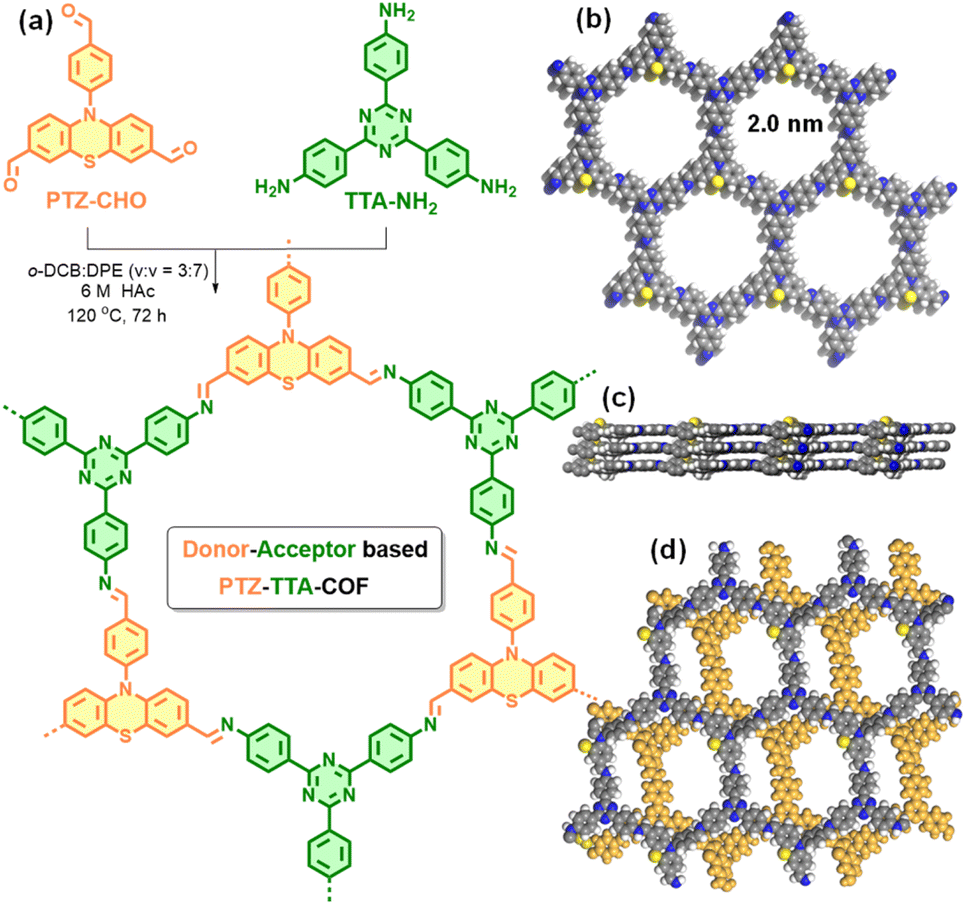 | ||
| Fig. 1 (a) Synthesis of the donor–acceptor PTZ–TTA-COF. Top (b) and side views (c) of the AA stacking mode as well as the top view (d) of the AB stacking mode for the PTZ–TTA-COF (carbon, gray; nitrogen, blue; sulphur, yellow; hydrogen, white). | ||
Results and discussion
Because of the flexible nonplanar butterfly structure and variable conformation of the PTZ unit, it is difficult to crystallize it into the COF lattice.21 Thus, a triazine based trianiline (TTA-NH2) with a more planar structure was used as a complementary acceptor building block to assist the crystallization of the PTZ donor part into the COF skeleton (Fig. S7–S10 in the ESI†). After optimizing synthetic reaction conditions (Table S1 in ESI†), we successfully obtained the target D–A type PTZ–TTA-COF material possessing good crystallinity. Specifically, the PTZ–TTA-COF was prepared by the acid-catalytic Schiff-base condensation of TTA-NH2 and PTZ-CHO in a mixed o-dichlorobenzene (o-DCB)/diphenyl ether (DPE) (3/7, v/v) in the presence of aqueous HAc (6 M) at 120 °C for 72 h. It is worth mentioning that the DPE solvent played an important role in synthesizing crystalline PTZ–TTA-COF samples or even at room temperature, though the related mechanism is still unclear at the present stage. Besides, a control TPA–TTA-COF with a similar hexagonal reticular structure but small D–A contrast was prepared according to the previous reports,10b,22 and used as a comparative sample in the following study (Fig. S14–S22 in the ESI†).The chemical structure of the PTZ–TTA-COF was determined by FT-IR and solid-state 13C CP-MAS NMR analyses. As shown in Fig. 2a, a characteristic signal of the CN stretching peak at ∼1620 cm−1 was found in the PTZ–TTA-COF, confirming the imine-bridged framework structure. A comparative FT-IR analysis of the COF and its monomers revealed the dramatic attenuation of N–H (3200–3400 cm−1) and CO (1680 cm−1) stretching bands after COF formation, indicative of the high polycondensation degree of amine and aldehyde precursors. In the solid-state 13C CP-MAS NMR spectrum of the PTZ–TTA-COF (Fig. 2b), a peak at around 156 ppm can be assigned to the carbon atoms of CN bonds, which further verified the formation of imine linkages in the COF. A signal at 170 ppm is attributed to the carbon atoms of the triazine ring from the TTA part, while peaks located at 144 and 118 ppm should belong to PTZ carbons, which validates the presence of intact PTZ donor and TTA acceptor moieties in the COF skeleton. Additionally, the elemental composition of the PTZ–TTA-COF was further confirmed by X-ray photoelectron spectroscopy (XPS, Fig. S23 in the ESI†). Its high-resolution N1s XPS spectrum exhibited three binding energy peaks at 398.4, 399.7 and 400.2 eV, which can be ascribed to the nitrogen atoms from the triazine core, imine bond, and PTZ moiety, respectively. These above results proved the successful conversion of corresponding monomers (PTZ-CHO and TTA-NH2) into an imine-linked structure.
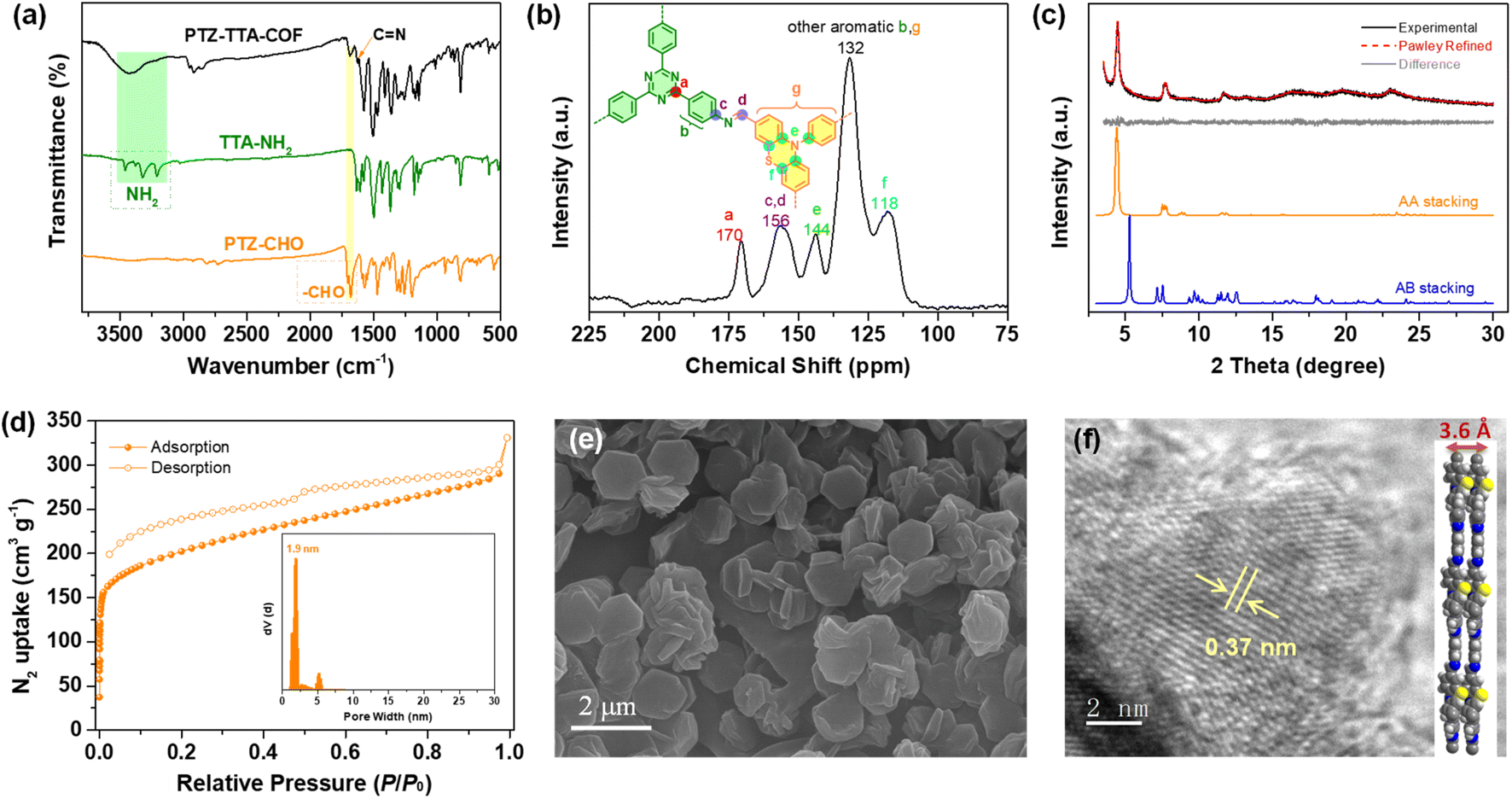 | ||
| Fig. 2 (a) FT-IR spectra of PTZ-CHO, TTA-NH2 and the PTZ–TTA-COF. (b) Solid-state 13C CP-MAS NMR of the PTZ–TTA-COF. (c) Experimental PXRD, Pawley refinement and the difference of the PTZ–TTA-COF, and their simulated PXRD profiles in AA and AB stacking modes. (d) Nitrogen sorption isotherm of the PTZ–TTA-COF at 77 K; inset: pore size distribution derived from the QSDFT model. (e) SEM and (f) HR-TEM images of the PTZ–TTA-COF. | ||
Powder X-ray diffraction (PXRD) analysis of the PTZ–TTA-COF confirmed its ordered structure and good crystallinity. As shown in Fig. 2c, it revealed three main diffraction peaks with a predominant peak at 2θ = 4.5° corresponding to a d spacing of 2.0 nm, which could be assigned to the reflection of the (100) facet. Besides, a weak and broad diffraction peak at around 24° corresponds to the two-dimensional COF interlayer distance of ∼0.37 nm. This distance was enlarged relative to the commonly observed π–π packing distance of 0.33 to 0.35 nm,23 which could be ascribed to the butterfly configuration of the PTZ subunit. In addition, Pawley refinement for the PTZ–TTA-COF was performed to give rise to optimized unit cell parameters with negligible profile differences and acceptable low residual values (Table S2 in the ESI†). Moreover, it can be found that the COF experimental PXRD data were in good agreement with the simulated pattern of the eclipsed AA stacking mode, but exhibited a large offset with the simulated AB stacking mode. The AA stacking arrangement of the PTZ–TTA-COF can ensure the formation of ordered donor–acceptor columnar arrays in the COF framework, which is expected to promote charge separation and transport, and will be beneficial to the subsequent photocatalytic applications.
The porosity of the PTZ–TTA-COF was assessed by nitrogen sorption isotherm measurements at 77 K. As shown in Fig. 2d, it exhibited a type I sorption isotherm with a rapid rise in the low relative pressure range, suggesting the microporous character of the PTZ–TTA-COF. Its Brunauer–Emmett–Teller (BET) surface area was calculated to be 740 m2 g−1. The pore size distribution analysis for the PTZ–TTA-COF based on the quenched solid density functional theory (QSDFT) showed one main distribution centered at 1.9 nm, which is in good agreement with the theoretical value of 2.0 nm in the AA stacking model (Fig. 1b). In addition, scanning electron microscopy (SEM) measurements demonstrated that the COF assumed an interesting hexagonal-like morphology with a fairly uniform size of about 1.5 μm (Fig. 2e). High-resolution transmission electron microscopy (HR-TEM) images of the PTZ–TTA-COF showed clear lattice fringes (Fig. 2f and S24 in the ESI†), confirming its good crystallinity and long-range ordered microstructure. The interplanar crystal spacing of ∼0.37 nm could be attributed to the COF interlayer distance (Fig. 2f), which is consistent with that observed in PXRD.
The electronic structures of the PTZ–TTA-COF was systematically investigated by various methods and also compared with the control TPA–TTA-COF. As shown in Fig. 3a, the ultraviolet-visible diffuse reflectance spectra (UV-vis DRS) of COFs revealed broad absorption bands with maximum peaks at 550 nm for the PTZ–TTA-COF and 450 nm for the TPA–TTA-COF, confirming their strong visible-light harvesting abilities. In comparison to UV-vis absorption spectra of their corresponding monomers (Fig. S25 in the ESI†), a very significant red-shift was observed for COF samples, which can be attributed to the intramolecular charge transfer effect caused by the formation of the D–A heterostructure in COF backbones. Besides, the absorption edge of 630 nm for the PTZ–TTA-COF was red-shifted by 60 nm over that of 570 nm for the TPA–TTA-COF, in line with the color change from yellow to red. The optical band gaps of both COFs were calculated using the Kubelka–Munk function (Fig. 3b), revealing a smaller band gap (Eg) of 2.04 eV for the PTZ–TTA-COF than that of the TPA–TTA-COF (2.31 eV). Photoluminescence (PL) measurements for solid COF samples revealed the emission maxima of the PTZ–TTA-COF at 635 nm, distinctly red-shifted by 87 nm over that of 548 nm for the TPA–TTA-COF (Fig. 3c). Additionally, their PL decay curves were fitted to give the average lifetimes of 11.4 ns for the PTZ–TTA-COF and 9.8 ns for the TPA–TTA-COF (Fig. 3d). The longer lifetime of the PTZ–TTA-COF suggests the suppressed radiative recombination of photogenerated excitons.24 The above observations should be ascribed to the enhanced D–A interaction in the PTZ–TTA-COF, owing to the stronger electron donating ability of PTZ than TPA moieties.
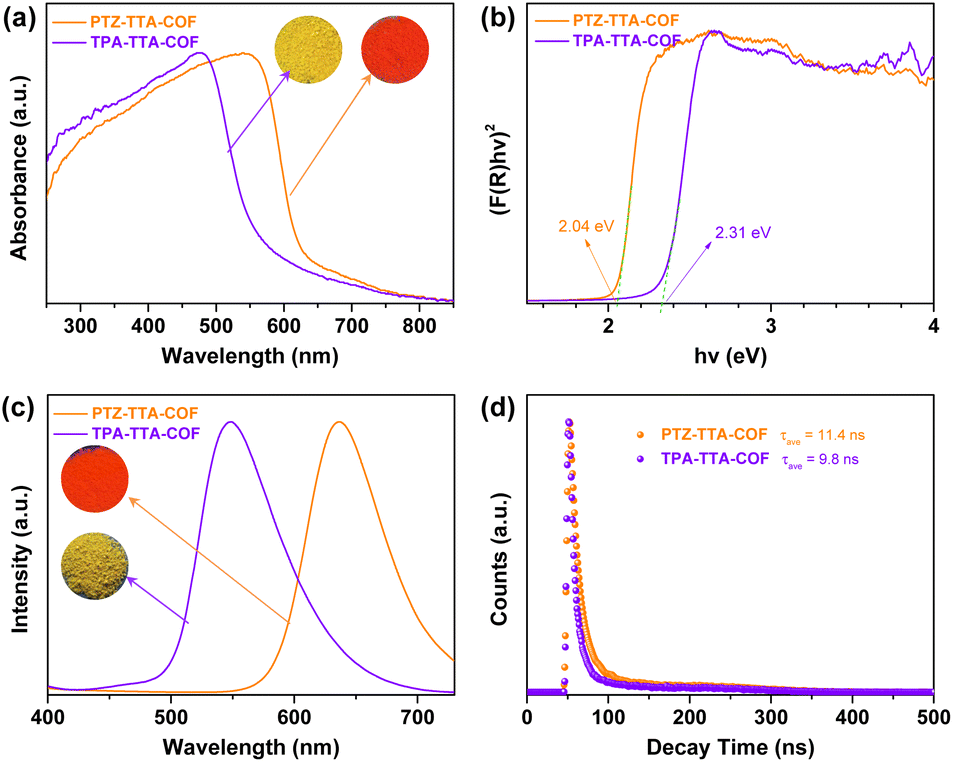 | ||
| Fig. 3 (a) UV-vis DRS of the PTZ–TTA-COF and TPA–TTA-COF; inset: photographs of the COF samples in daylight. (b) Band gaps calculated using Kubelka–Munk transformed reflectance spectra. (c) PL spectra of the PTZ–TTA-COF and TPA–TTA-COF measured in the solid state; inset: photographs of the COF samples under 365 nm UV light. (d) PL decay profiles of the PTZ–TTA-COF and TPA–TTA-COF upon excitation at 400 nm. | ||
In addition, the density functional theory (DFT) calculations were performed to theoretically study the electronic properties of both COFs. Their electronic structures were calculated using the HSE06 hybrid functional25 and theoretical band gaps of 2.18 and 2.32 eV were found for the PTZ–TTA-COF and TPA–TTA-COF, respectively (Fig. S26 in the ESI†), which are consistent with their experimental values. Furthermore, in order to elucidate the difference between the electronic structures of the two COFs, we plotted their total and partial densities of states (DOS) as well as the charge density distribution of the valence band maximum (VBM) and conduction band minimum (CBM) (Fig. S27 in the ESI†). As for the charge distribution of the VBM in the PTZ–TTA-COF, it is predominantly distributed in the PTZ donor part, whereas it is almost delocalized across the whole conjugated framework in the TPA–TTA-COF. The distinct distribution of the VBM and their different band gaps should be mainly attributed to the p orbital of the S atom in the PTZ–TTA-COF as illustrated by its DOS analysis. On the other hand, a similar charge distribution of the CBM was observed for both COFs, which is largely located over the TTA acceptor together with some minor distribution around donor benzene parts. The charge distribution profiles for the two COFs suggested that electrons could be readily excited from PTZ or TPA to the TTA part along the D–A pathway in COF skeletons upon light irradiation. However, through further comparing their charge distribution, a better separation from the VBM to the CBM can be witnessed in the case of the PTZ–TTA-COF on account of its stronger D–A interaction, which implied more effective electron–hole separation and dissociation in the photocatalytic process.
Furthermore, ultraviolet photoelectron spectroscopy (UPS) was utilized to experimentally analyse the band structure alignments of the PTZ–TTA-COF and TPA–TTA-COF (Fig. 4a and S28 in the ESI†). The energy levels of the VBM (EVBvs. the vacuum level) were determined by subtracting the UPS width from its excitation energy (He I, 21.22 eV) to be −5.59 eV and −5.79 eV for the PTZ–TTA-COF and TPA–TTA-COF (Fig. 4b), respectively. The energy values of the CBM (ECB) were calculated using the formula (ECB = EVB − Eg) to be −3.55 eV and −3.48 eV for the PTZ–TTA-COF and TPA–TTA-COF, respectively. The band structures manifest that both COFs possess sufficient reductive and oxidative potentials to reduce O2 into its superoxide O2˙− active species (Ered = −4.16 eV) and oxidize benzylamine (BnNH2) into its cationic radical form (Eox = −4.96 eV) by using the photogenerated electrons and holes, respectively.26 In addition, the electrochemical impedance spectroscopy (EIS) measurements of two COFs were performed in the dark and under visible light. As shown in Fig. 4c, the PTZ–TTA-COF presented a much smaller arc radius than that of the TPA–TTA-COF in the Nyquist plot, indicating the lower resistance of charge transportation in the PTZ–TTA-COF framework. The arc radius further decreased under visible light, suggesting its increased charge transfer rate upon irradiation, in line with the efficient charge separation efficiency. Moreover, the transient photocurrent responses also exhibited a notably higher photocurrent density on the PTZ–TTA-COF (Fig. 4d), further confirming the more efficient separation and mobility of photogenerated charge carriers.
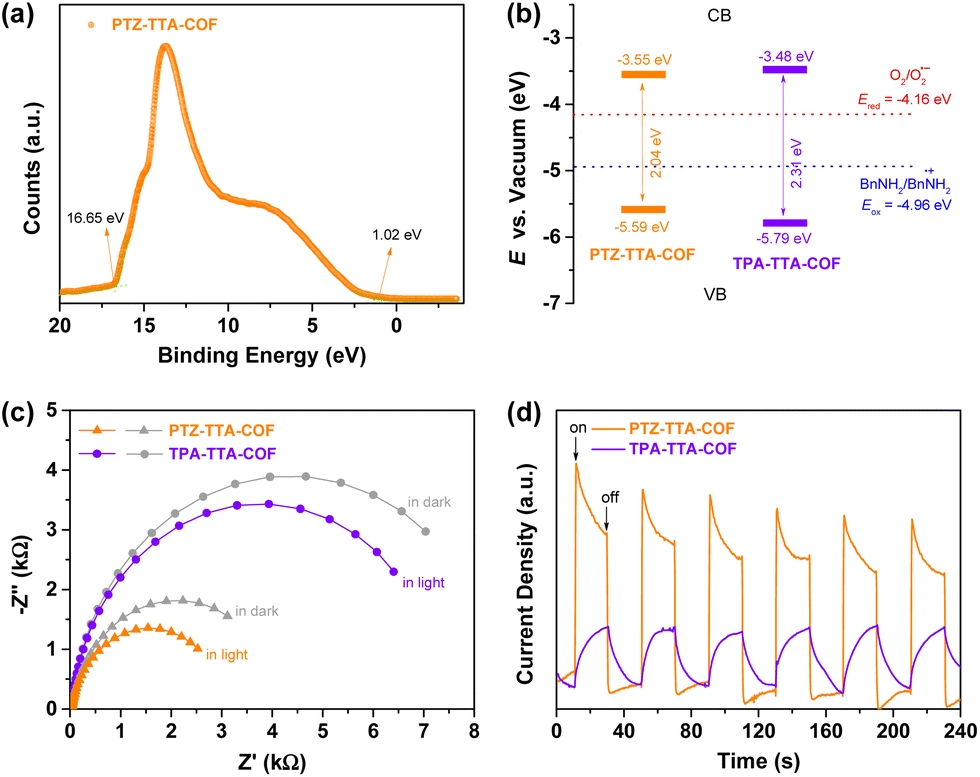 | ||
| Fig. 4 (a) UPS of the PTZ–TTA-COF. Band structures (b), EIS Nyquist plots (c) and transient photocurrent response (d) of the PTZ–TTA-COF and TPA–TTA-COF. | ||
In order to further investigate and compare the kinetics of photo-generated charge carriers in the two COFs, temperature-dependent PL and electron paramagnetic resonance (EPR) measurements were carried out. The exciton binding energy (Eb) that is a key parameter for mediating charge separation in photochemistry, was obtained by fitting integrated temperature-dependent PL intensity to the Arrhenius equation (Fig. S29 in the ESI†).11b–d As the temperature increased from 80 to 250 K, the integrated PL intensity monotonically decreased for both COFs. This suggests that excitons will be likely to dissociate into charge carriers at high temperature and undergo radiative decay at low temperature. Accordingly, the Eb value of the PTZ–TTA-COF was estimated to be 48 meV by fitting the experimental data (Fig. 5a), which is much lower than that of the TPA–TTA-COF with 89 meV. This result manifests that the excitons in the PTZ–TTA-COF are more prone to dissociate into free charge carriers upon photoexcitation. Subsequently, EPR measurements for both COFs were conducted to further examine the production of free electrons under visible light irradiation.11c,27 As shown in Fig. 5b, EPR spectra for COFs have been recorded and compared in the dark and under light conditions to evaluate their charge separation and transfer properties. It was found that both of them showed a distinctly increased EPR signal under visible light, which should be attributed to photoexcited electrons in the CB band, indicative of the photo-induced generation of electron–hole pairs in the COF lattice. In comparison to the TPA–TTA-COF, the PTZ–TTA-COF showed a much higher increase of signal intensity, indicating its remarkable enhancement of charge separation efficiency. Based on these above photoelectrochemical and photophysical measurements along with the theoretical calculation, it can be concluded that the introduction of the PTZ subunit into the COF skeleton with enhancing the D–A interaction is beneficial to reduce the exciton binding energy as well as improve the separation and transfer of charge carriers, thereby ultimately giving rise to the enhancement of photocatalytic activity.
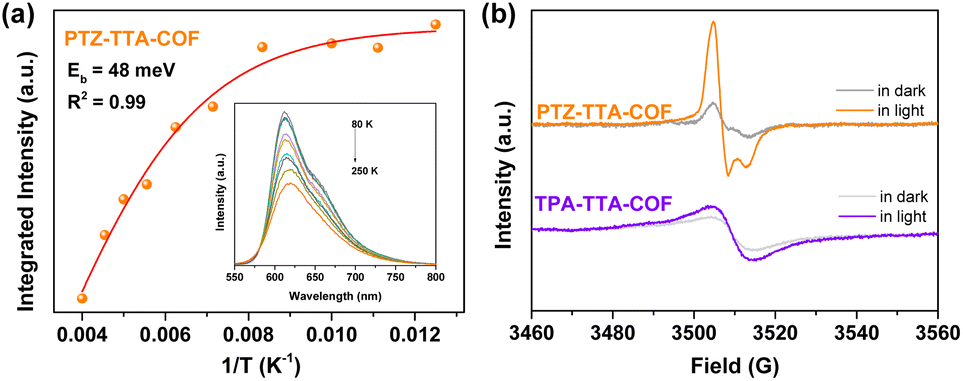 | ||
| Fig. 5 (a) Integrated PL intensity of the PTZ–TTA-COF as a function of temperature (inset: temperature-dependent PL spectra from 80 to 250 K). (b) EPR spectra of the PTZ–TTA-COF and TPA–TTA-COF in the dark and under visible light irradiation. | ||
Imines as valuable intermediates have been widely applied in organic and pharmaceutical synthesis.28 The direct photocatalytic oxidative coupling of amines to imines with oxygen as a green oxidant is emerging as a promising alternative approach for traditional reductive amination and thus has received significant attention in recent years.29 After the experimental and theoretical studies of the photoelectric properties of the PTZ–TTA-COF and TPA–TTA-COF, their photocatalytic activities were subsequently examined and compared by using oxidative amine coupling and cyclization of thioamide to 1,2,4-thiadiazole as two model reactions. First, benzylamine (1a) and three representative 4-substituted ones (1b–1d) with the respective loading of the two COFs as photocatalysts in acetonitrile were illuminated with blue LEDs at room temperature in an air atmosphere. After 1 h irradiation, the product yield of resultant imines (2a–2d) was determined by 1H NMR analysis to be >80% for the PTZ–TTA-COF and <50% for the TPA–TTA-COF (Fig. 6), respectively. Besides, such reaction can even go well under natural sunlight (Fig. S30 in ESI†). Second, the two COFs were further explored and compared as photocatalysts for oxidative cyclization of thioamides to 1,2,4-thiadiazoles (Table S3 in the ESI†). The mixture of thioamide and COFs in acetonitrile was similarly irradiated for 2 h, which was subsequently subjected to 1H NMR analysis. The product yield of 1,2,4-thiadiazoles was in the range of about 60–70% for the PTZ–TTA-COF, whereas that was only around 30% for the TPA–TTA-COF. These above results firmly demonstrated that the PTZ–TTA-COF possessed much higher photo-catalytic activities than its analogue TPA–TTA-COF, which should be attributed to the prominent photoelectric properties of the PTZ–TTA-COF, such as the narrower band gap, lower exciton binding energy, and enhanced charge separation/transfer.
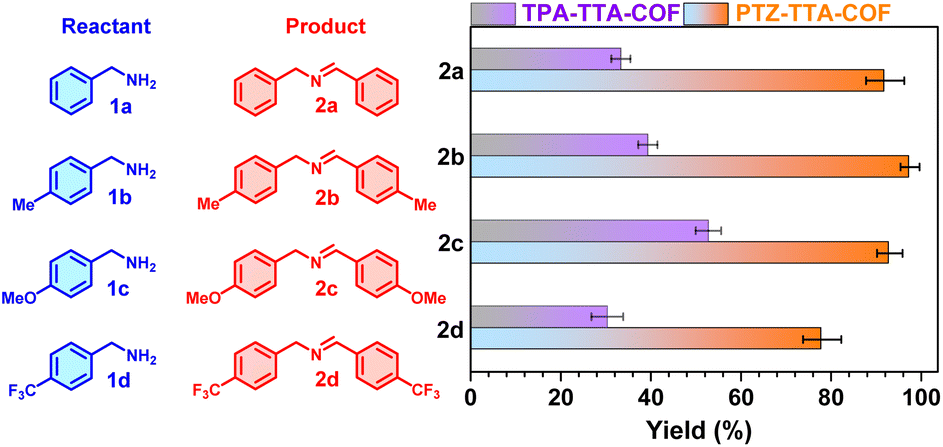 | ||
| Fig. 6 Comparison of the PTZ–TTA-COF and TPA–TTA-COF in photocatalytic oxidative coupling of four benzylamines into imines at room temperature under air. Reaction conditions: amine (0.1 mmol), COF photocatalyst (2 mg), CD3CN (1 mL), blue LEDs (λmax = 450 nm, 3 W × 4), 1 h. The yields were determined by 1H NMR. | ||
To gain insights into the photochemical reaction mechanism and the key active species involved in this process, several control and trapping experiments using different scavengers were carried out as shown in Table 1. Control experiments demonstrated that the COF photocatalyst, light and oxygen are all indispensable for this conversion; otherwise, only little product can be detected (Table 1, entries 2–4). After separately adding KI, NaN3 and p-benzoquinone into the reaction mixture as scavenger agents for holes (h+), singlet oxygen (1O2) and superoxide radical anions (O2˙−), the product yield significantly decreased to be 10%, 40% and 23%, respectively (Table 1, entries 5–7). Thus, these quenching experimental results suggested that all of h+, 1O2, and O2˙− as the key active intermediate species played a crucial role in this photocatalytic transformation. Additionally, EPR measurements were further conducted to detect the reactive oxygen species (ROS) of 1O2 and O2˙− by using 2,2,6,6-tetramethylpiperidine (TEMP) and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as radical capturers. As expected, the characteristic signals for 1O2 and O2˙− adducts can be witnessed in EPR spectra under the irradiation of PTZ–TTA-COF with TEMP and DMPO, respectively (Fig. S31 in the ESI†). These observations evidenced that the phenothiazine based COF can serve as an effective photocatalyst for the facile generation of ROS in the presence of oxygen under light irradiation, which could be widely used in a variety of photooxidation reactions.
| Entry | |||||||
|---|---|---|---|---|---|---|---|
| 1 | 2c | 3d | 4e | 5f | 6g | 7h | |
| a Reaction conditions: benzylamine (0.1 mmol), PTZ–TTA-COF (2 mg), solvent acetonitrile (CD3CN, 1 mL), 1 h, blue LEDs (3 W × 4), room temperature. b Determined by 1H-NMR analysis. c No COF photocatalyst. d No light. e Under a nitrogen atmosphere. f KI as the hole scavenger. g NaN3 as a singlet oxygen scavenger. h p-Benzoquinone as a superoxide scavenger. | |||||||
| PTZ–TTA-COF | + | − | + | + | + | + | + |
| hν | + | + | − | + | + | + | + |
| Air | + | + | + | − | + | + | + |
| Yieldb (%) | 92 | Trace | Trace | Trace | 10 | 40 | 23 |
Based on the above control and trapping experimental results along with related literature reports,13,15 the plausible mechanism over the PTZ–TTA-COF toward photocatalytic oxidative coupling and cyclization reactions was tentatively proposed as illustrated in Fig. 7. Under light irradiation, the COF was excited to generate excitons of bound electron–hole pairs, which could be rapidly and spatially separated owing to the segregated bicontinuous D–A heterostructure in the PTZ–TTA-COF skeleton. The photogenerated electrons with main distribution on TTA acceptor parts can activate O2 molecules by an electron and energy transfer process, leading to the generation of reactive oxidative species, O2˙− and 1O2. On the other hand, the photogenerated holes primarily participated in the oxidization of benzylamine and thioamide into their corresponding cationic radicals. Subsequently, the benzylamine radical cation reacted with O2˙− and 1O2 gave the imine intermediate. It can further react with the additional benzylamine to form the desired product along with elimination of ammonia (Fig. S32 in the ESI†). Similarly, the thioamide radical cation first transformed into two radical isomers after the proton removal, which were further linked to give a dimer through the radical cross-coupling. The final 1,2,4-thiadiazole product was readily obtained by the intramolecular cyclization of the dimer and subsequent aromatization under the assistance of reactive oxidative species.
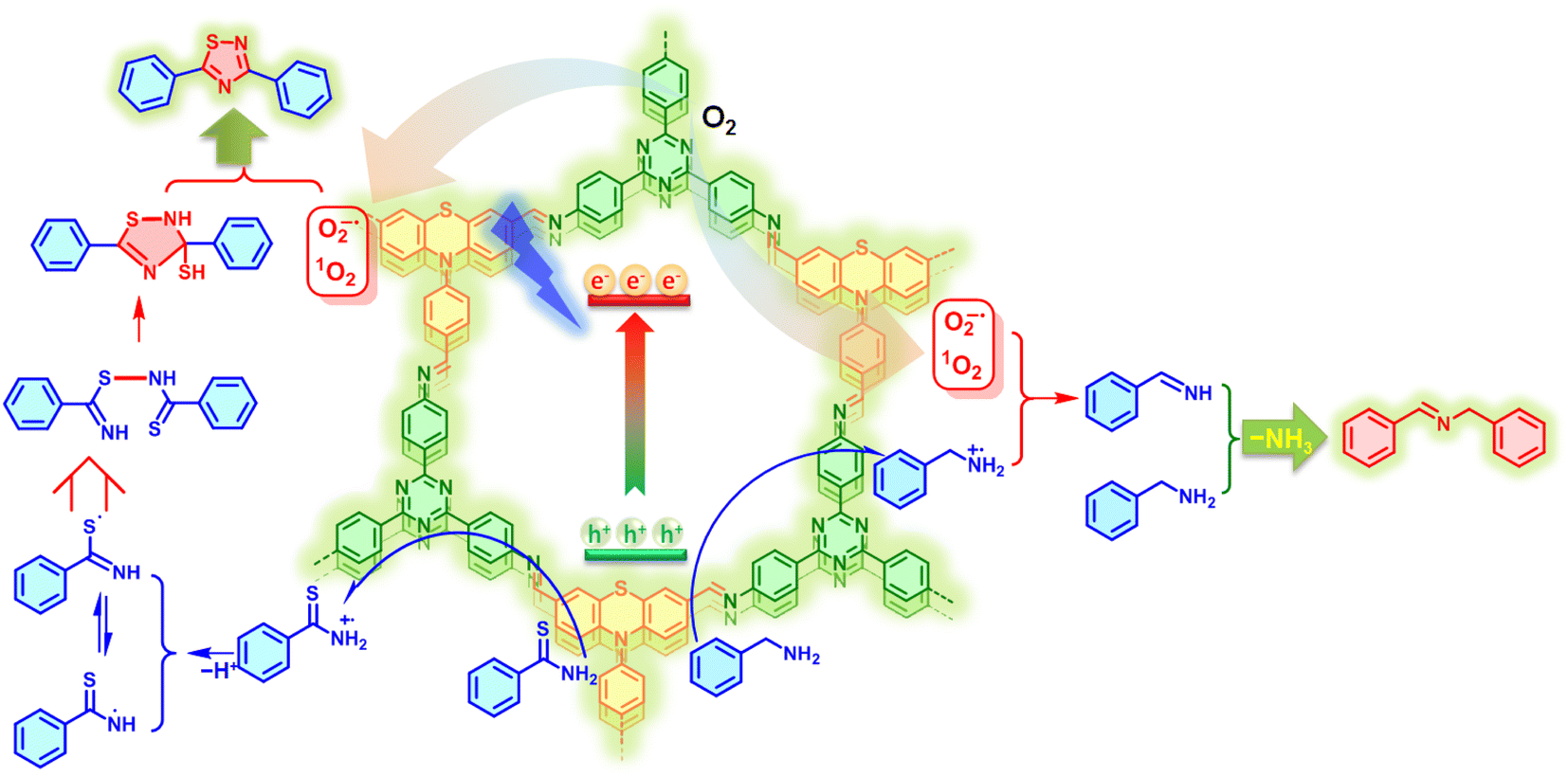 | ||
| Fig. 7 Plausible mechanisms of PTZ–TTA-COF photocatalytic oxidative amine coupling of benzylamine and cyclization of thioamide. | ||
Next, the substrate scope of aerobic oxidative amine coupling reactions catalyzed by the PTZ–TTA-COF was explored for various benzylamines with different substituents (Table S5 in the ESI†). It was found that most of the amine derivatives bearing electron rich or deficient groups can be efficiently transformed into the corresponding imine products in high yields (>90%), although the irradiation time for each amine was some different (1–2 h). Besides, polycyclic aromatic and heterocyclic amines, including 1-naphthalenemethylamine, 2-thenylamine and 3-picolylamine, can also be readily converted into their corresponding imines with high yields (entries 12–14 in Table S5†). Moreover, after recovery from the reaction mixture by centrifugation, the PTZ–TTA-COF can still maintain its high photocatalytic activity for at least three cycles (Fig. S34 in the ESI†). The structure and porosity of the PTZ–TTA-COF were consistent before and after photocatalysis, which has been validated by the PXRD, FT-IR and solid-state 13C NMR spectra along with nitrogen sorption isotherms (Fig. S21, S22, S35 and S36 in the ESI†), confirming its high stability during the catalysis.
Conclusions
In summary, we have constructed a novel phenothiazine based PTZ–TTA-COF that has a well organized D–A heterostructure derived from the columnar stacking of electron rich PTZ and electron deficient TTA building blocks. In comparison to the analogue TPA–TTA-COF with smaller D–A contrast, the PTZ–TTA-COF exhibited lower exciton binding energy and enhanced charge separation and migration, which in turn lead to the remarkable enhancement of photocatalytic performances in oxidative amine coupling and cyclization of thioamide to 1,2,4-thiadiazole reactions. Overall, this comparative study not only revealed the unique advantages of introducing the phenothiazine subunit as a strong electron donor into the COF skeleton, but also could provide a rational guidance for the future development of high-performance COF photocatalysts through exploring more suitable donor–acceptor pairs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22001100 and 21906182) and Natural Science Foundation of Jiangsu Province (BK20211549). The authors are also very grateful to Prof. Xu-Bing Li, Dr Xue-Wang Gao, Dr Yang Wang and Wen-Qiang Zhang for the technical assistance and very helpful discussions.Notes and references
- (a) K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS; (b) C. S. Diercks and O. M. Yaghi, Science, 2017, 355 Search PubMed; (c) C. Gropp, S. Canossa, S. Wuttke, F. Gándara, Q. Li, L. Gagliardi and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1255–1273 CrossRef CAS; (d) B. Gui, G. Lin, H. Ding, C. Gao, A. Mal and C. Wang, Acc. Chem. Res., 2020, 53, 2225–2234 CrossRef CAS.
- (a) X. Li, S. Cai, B. Sun, C. Yang, J. Zhang and Y. Liu, Matter, 2020, 3, 1507–1540 CrossRef; (b) R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC; (c) H. R. Abuzeid, A. F. M. El-Mahdy and S.-W. Kuo, Giant, 2021, 6, 100054 CrossRef CAS.
- (a) X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC; (b) Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- (a) Z. Meng and K. A. Mirica, Chem. Soc. Rev., 2021, 50, 13498–13558 RSC; (b) S. Wang, H. Li, H. Huang, X. Cao, X. Chen and D. Cao, Chem. Soc. Rev., 2022, 51, 2031–2080 RSC.
- (a) Z. Gao, Q. Liu, G. Zhao, Y. Sun and H. Guo, J. Mater. Chem. A, 2022, 10, 7497–7516 RSC; (b) J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC; (c) D.-G. Wang, T. Qiu, W. Guo, Z. Liang, H. Tabassum, D. Xia and R. Zou, Energy Environ. Sci., 2021, 14, 688–728 RSC.
- (a) Y. Yusran, H. Li, X. Guan, Q. Fang and S. Qiu, Energychem, 2020, 2, 100035 CrossRef; (b) X. Han, C. Yuan, B. Hou, L. Liu, H. Li, Y. Liu and Y. Cui, Chem. Soc. Rev., 2020, 49, 6248–6272 RSC; (c) Y. F. Zhi, Z. R. Wang, H. L. Zhang and Q. C. Zhang, Small, 2020, 16, 2001070 CrossRef CAS.
- (a) N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC; (b) C. Wang, Z. Zhang, Y. Zhu, C. Yang, J. Wu and W. Hu, Adv. Mater., 2022, 34, 202102290 Search PubMed; (c) Y. Zhu, S. Jiang, X. Jing and X. Feng, Trends Chem., 2022, 4, 128–141 CrossRef CAS; (d) C. Li, D. Li, W. Zhang, H. Li and G. Yu, Angew. Chem., Int. Ed., 2021, 60, 27135–27143 CrossRef CAS PubMed.
- (a) H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC; (b) C. Xia, K. O. Kirlikovali, N. Thi Hong Chuong, N. Xuan Cuong, T. Quoc Ba, D. Minh Khoa, D. Minh Tuan Nguyen, N. Dang Le Tri, P. Singh, P. Raizada, N. Van-Huy, S. Y. Kim, L. Singh, C. C. Nguyen, M. Shokouhimehr and Q. V. Le, Coord. Chem. Rev., 2021, 446, 214117 CrossRef CAS; (c) Y. Wang, Y. Zhao and Z. Li, Macromol. Rapid Commun., 2022, 202200108 Search PubMed; (d) T.-X. Wang, H.-P. Liang, D. A. Anito, X. Ding and B.-H. Han, J. Mater. Chem. A, 2020, 8, 7003–7034 RSC; (e) G.-B. Wang, K.-H. Xie, H.-P. Xu, Y.-J. Wang, F. Zhao, Y. Geng and Y.-B. Dong, Coord. Chem. Rev., 2022, 472, 214774 CrossRef CAS.
- (a) S. Jin, M. Supur, M. Addicoat, K. Furukawa, L. Chen, T. Nakamura, S. Fukuzumi, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7817–7827 CrossRef CAS PubMed; (b) M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Löbermann, D. Trauner, A. Hartschuh and T. Bein, J. Am. Chem. Soc., 2014, 136, 17802–17807 CrossRef CAS PubMed; (c) S. Jin, K. Furukawa, M. Addicoat, L. Chen, S. Takahashi, S. Irle, T. Nakamura and D. Jiang, Chem. Sci., 2013, 4, 4505–4511 RSC; (d) X. Feng, L. Chen, Y. Honsho, O. Saengsawang, L. Liu, L. Wang, A. Saeki, S. Irle, S. Seki, Y. Dong and D. Jiang, Adv. Mater., 2012, 24, 3026–3031 CrossRef CAS PubMed.
- (a) S. Jin, X. Ding, X. Feng, M. Supur, K. Furukawa, S. Takahashi, M. Addicoat, M. E. El-Khouly, T. Nakamura, S. Irle, S. Fukuzumi, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 2017–2021 CrossRef CAS; (b) R. Luo, H. Lv, Q. Liao, N. Wang, J. Yang, Y. Li, K. Xi, X. Wu, H. Ju and J. Lei, Nat. Commun., 2021, 12, 6808 CrossRef CAS PubMed; (c) L. P. Guo, Y. L. Niu, H. T. Xu, Q. W. Li, S. Razzaque, Q. Huang, S. B. Jin and B. Tan, J. Mater. Chem. A, 2018, 6, 19775–19781 RSC.
- (a) D. D. Medina, T. Sick and T. Bein, Adv. Energy Mater., 2017, 7, 1700387 CrossRef; (b) C. Li, J. Liu, H. Li, K. Wu, J. Wang and Q. Yang, Nat. Commun., 2022, 13, 2357 CrossRef CAS PubMed; (c) F. Liu, Y. He, X. Liu, Z. Wang, H.-L. Liu, X. Zhu, C.-C. Hou, Y. Weng, Q. Zhang and Y. Chen, ACS Catal., 2022, 12, 9494–9502 CrossRef CAS; (d) W. Wang, H. Wang, X. Tang, J. Huo, Y. Su, C. Lu, Y. Zhang, H. Xu and C. Gu, Chem. Sci., 2022, 13, 8679–8685 RSC.
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y.-B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS PubMed.
- S. Li, L. Li, Y. Li, L. Dai, C. Liu, Y. Liu, J. Li, J. Lv, P. Li and B. Wang, ACS Catal., 2020, 10, 8717–8726 CrossRef CAS.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J.-L. Wang, D.-Q. Yuan and Y.-Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS PubMed.
- W. Qiu, Y. He, L. Li, Z. Liu, S. Zhong and Y. Yu, Langmuir, 2021, 37, 11535–11543 CrossRef CAS PubMed.
- Q. Liao, W. Xu, X. Huang, C. Ke, Q. Zhang, K. Xi and J. Xie, Sci. China: Chem., 2020, 63, 707–714 CrossRef CAS.
- (a) J. Zhao, J. Ren, G. Zhang, Z. Zhao, S. Liu, W. Zhang and L. Chen, Chem.–Eur. J., 2021, 27, 10781–10797 CrossRef CAS PubMed; (b) G.-B. Wang, S. Li, C.-X. Yan, F.-C. Zhu, Q.-Q. Lin, K.-H. Xie, Y. Geng and Y.-B. Dong, J. Mater. Chem. A, 2020, 8, 6957–6983 RSC; (c) W. Chen, Z. Yang, Z. Xie, Y. Li, X. Yu, F. Lu and L. Chen, J. Mater. Chem. A, 2019, 7, 998–1004 RSC; (d) H. Liu, X. Yan, W. Chen, Z. Xie, S. Li, W. Chen, T. Zhang, G. Xing and L. Chen, Sci. China: Chem., 2021, 64, 827–833 CrossRef CAS; (e) Z. Li, S. Han, C. Li, P. Shao, H. Xia, H. Li, X. Chen, X. Feng and X. Liu, J. Mater. Chem. A, 2020, 8, 8706–8715 RSC; (f) S. Bi, P. Thiruvengadam, S. Wei, W. Zhang, F. Zhang, L. Gao, J. Xu, D. Wu, J.-S. Chen and F. Zhang, J. Am. Chem. Soc., 2020, 142, 11893–11900 CrossRef CAS; (g) G.-B. Wang, F.-C. Zhu, Q.-Q. Lin, J.-L. Kan, K.-H. Xie, S. Li, Y. Geng and Y.-B. Dong, Chem. Commun., 2021, 57, 4464–4467 RSC; (h) F. Yang, C.-C. Li, C.-C. Xu, J.-L. Kan, B. Tian, H.-Y. Qu, Y. Guo, Y. Geng and Y.-B. Dong, Chem. Commun., 2022, 58, 1530–1533 RSC.
- (a) S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC; (b) T. Zhang, G. Zhang and L. Chen, Acc. Chem. Res., 2022, 55, 795–808 CrossRef CAS PubMed; (c) Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC; (d) H. Liu, C. Li, H. Li, Y. Ren, J. Chen, J. Tang and Q. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 20354–20365 CrossRef CAS PubMed; (e) S. He, B. Yin, H. Niu and Y. Cai, Appl. Catal., B, 2018, 239, 147–153 CrossRef CAS.
- (a) S. Revoju, A. Matuhina, L. Canil, H. Salonen, A. Hiltunen, A. Abate and P. Vivo, J. Mater. Chem. C, 2020, 8, 15486–15506 RSC; (b) I. J. Al-Busaidi, A. Haque, N. K. Al Rasbi and M. S. Khan, Synth. Met., 2019, 257, 116189 CrossRef CAS; (c) Y. Rout, C. Montanari, E. Pasciucco, R. Misra and B. Carlotti, J. Am. Chem. Soc., 2021, 143, 9933–9943 CrossRef CAS PubMed.
- (a) A. F. Buene and D. M. Almenningen, J. Mater. Chem. C, 2021, 9, 11974–11994 RSC; (b) P. S. Gangadhar, G. Reddy, S. Prasanthkumar and L. Giribabu, Phys. Chem. Chem. Phys., 2021, 23, 14969–14996 RSC.
- (a) W. Zhang, S. Li, X. Tang, J. Tang, C. Pan and G. Yu, Appl. Catal., B, 2020, 272, 118982 CrossRef CAS; (b) S. Li, W. Zhang, S. Yang, F. Chen, C. Pan, J. Tang, K. A. I. Zhang and G. Yu, Chem. Eng. J., 2021, 408, 127261 CrossRef CAS; (c) S. Dadashi-Silab, F. Lorandi, M. J. DiTucci, M. Sun, G. Szczepaniak, T. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2021, 143, 9630–9638 CrossRef CAS PubMed; (d) A. Jiménez-Almarza, A. López-Magano, R. Cano, B. Ortín-Rubio, D. Díaz-García, S. Gomez-Ruiz, I. Imaz, D. Maspoch, R. Mas-Ballesté and J. Alemán, Mater. Today Chem., 2021, 22, 100548 CrossRef; (e) C. Zhou, R. Wang, L. Gao, X. Huang and X. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 30962–30968 CrossRef CAS PubMed; (f) A. Jiménez-Almarza, A. López-Magano, R. Mas-Ballesté and J. Alemán, ACS Appl. Mater. Interfaces, 2022, 14, 16258–16268 CrossRef PubMed; (g) R. Wang, C. Zhou, X. Huang, J.-Y. Wu and X. Zhang, ACS Sustainable Chem. Eng., 2022, 10, 4650–4659 CrossRef CAS; (h) B. Wu, Y. Liu, Y. Zhang, L. Fan, Q.-Y. Li, Z. Yu, X. Zhao, Y.-C. Zheng and X.-J. Wang, J. Mater. Chem. A, 2022, 10, 12489–12496 RSC.
- (a) J. Yang, A. Acharjya, M.-Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. van de Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS PubMed; (b) S. Wang, Y. Yang, H. Zhang, Z. Zhang, C. Zhang, X. Huang, D. Kozawa, P. Liu, B.-G. Li and W.-J. Wang, J. Am. Chem. Soc., 2021, 143, 5003–5010 CrossRef CAS PubMed.
- (a) A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed; (b) Y. Yue, H. Li, H. Chen and N. Huang, J. Am. Chem. Soc., 2022, 144, 2873–2878 CrossRef CAS PubMed.
- (a) S. Wei, F. Zhang, W. Zhang, P. Qiang, K. Yu, X. Fu, D. Wu, S. Bi and F. Zhang, J. Am. Chem. Soc., 2019, 141, 14272–14279 CrossRef CAS PubMed; (b) S. Bi, F. Meng, D. Wu and F. Zhang, J. Am. Chem. Soc., 2022, 144, 3653–3659 CrossRef CAS PubMed.
- (a) Y. Wan, L. Wang, H. Xu, X. Wu and J. Yang, J. Am. Chem. Soc., 2020, 142, 4508–4516 CrossRef CAS PubMed; (b) L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 29, 1702428 CrossRef PubMed.
- (a) Y. T. Xiao, G. H. Tian, W. Li, Y. Xie, B. J. Jiang, C. G. Tian, D. Y. Zhao and H. G. Fu, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed; (b) H. Liu, C. Y. Xu, D. D. Li and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 5379–5383 CrossRef CAS PubMed.
- (a) P. Pachfule, A. Acharjya, J. Roeser, R. P. Sivasankaran, M.-Y. Ye, A. Brückner, J. Schmidt and A. Thomas, Chem. Sci., 2019, 10, 8316–8322 RSC; (b) M. Traxler, S. Gisbertz, P. Pachfule, J. Schmidt, J. Roeser, S. Reischauer, J. Rabeah, B. Pieber and A. Thomas, Angew. Chem., Int. Ed., 2022, 61, e202117738 CrossRef CAS PubMed.
- (a) B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS; (b) A. Guðmundsson, S. Manna and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2021, 60, 11819–11823 CrossRef PubMed.
- (a) Q. Niu, Q. Huang, T.-Y. Yu, J. Liu, J.-W. Shi, L.-Z. Dong, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2022, 144, 18586–18594 CrossRef CAS PubMed; (b) Y. Su, B. Li, H. Xu, C. Lu, S. Wang, B. Chen, Z. Wang, W. Wang, K.-i. Otake, S. Kitagawa, L. Huang and C. Gu, J. Am. Chem. Soc., 2022, 144, 18218–18222 CrossRef CAS PubMed; (c) J.-L. Shi, R. Chen, H. Hao, C. Wang and X. Lang, Angew. Chem., Int. Ed., 2020, 59, 9088–9093 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta07177k |
| This journal is © The Royal Society of Chemistry 2023 |