
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating paired Ir–O–Ir via electronic perturbations of correlated Ir single atoms to overcome catalytic selectivity†
Shi-Hua
Chen‡
ab,
Yuan-Fan
Yang‡
ac,
Zong-Yin
Song
ac,
Xiang-Yu
Xiao
ac,
Cong-Cong
Huang
ac,
Xin
Cai
ac,
Pei-Hua
Li
a,
Meng
Yang
a,
Aicheng
Chen
 d,
Wen-Qing
Liu
*e and
Xing-Jiu
Huang
*abc
d,
Wen-Qing
Liu
*e and
Xing-Jiu
Huang
*abc
aKey Laboratory of Environmental Optics and Technology, and Environmental Materials and Pollution Control Laboratory, Institute of Solid State Physics, HFIPS, Chinese Academy of Sciences, Hefei 230031, China. E-mail: xingjiuhuang@iim.ac.cn
bState Key Laboratory of Transducer Technology, Shanghai Institute of Microsystem And Information Technology, Chinese Academy of Sciences, Shanghai 200050, China
cDepartment of Materials Science and Engineering, University of Science and Technology of China, Hefei 230026, China
dDepartment of Chemistry, University of Guelph, Guelph, ON N1G 2W1, Canada
eAnhui Institute of Optics and Fine Mechanics, HFIPS, Chinese Academy of Sciences, Hefei 230031, China. E-mail: wqliu@aiofm.ac.cn
First published on 1st September 2023
Abstract
Single-atom catalysts have been extensively utilized for electrocatalysis, in which electronic metal–support interactions are typically employed to stabilize single atoms. However, this neglects the metal–metal interactions of adjacent atoms, which are essential for the fine-tuning of selective sites. Herein, the high-loading of Ir single atoms (Ir SAs) (8.9 wt%) were adjacently accommodated into oxygen vacancy-rich Co3O4 nanosheets (Ir SAs/Co3O4). Electronic perturbations for both Ir single atoms and Co3O4 supports were observed under electronic metal–support and metal–metal interactions, thus generating Ir–O–Co/Ir units. Electrons were transferred from Co and Ir to O atoms, inducing the depletion of 3d/5d states in Co/Ir and the occupation of 2p states in O atoms to stabilize the Ir SAs. Moreover, the O atoms of Ir–O–Ir functioned as the main active sites for the electrocatalysis of As(III), which reduced the energy barrier for the rate-determining step. This was due to the stronger electronic affinities for intermediates from reduction of As(III), which were completely distinct from other coordinated O atoms of Co3O4 or IrO2. Consequently, the resultant Ir SAs/Co3O4 exhibited far more robust electrocatalytic activities than IrO2/Co3O4 and Co3O4 in the electrocatalysis of As(III). Moreover, there was a strong orbital coupling effect between the coordinated O atoms of Ir SAs and the –OH of H3AsO3, thus exhibiting superior selectivity for As(III) in contrast to other common heavy metal cations. This work offers useful insights into the rational design of intriguing SACs with high selectivity and stability for the electrocatalysis and electrochemical analysis of pollutants on an electronic level.
Introduction
Single-atom catalysts (SACs) have been broadly employed in the synthesis of interfaces for electrochemical catalysis, where various support matrices with unsaturated sites and high surface areas were used to maximize the utilization of atoms to obtain highly active catalysts.1–5 However, SACs still suffer from inadequate long-term stability and selectivity for interfacial catalysis-reactions, which are important for their practical applications.6,7 Numerous attempts have recently been made toward the design and optimization of the electrocatalytic performance of active SACs, whose stability can also be enhanced through the introduction of suitable supports including metal compounds (e.g., oxides,1,8 chalcogenides,9,10 nitrides,11,12 carbides,13,14 and phosphides15,16) and carbon-based supports.17,18 Researchers have focused on the selectivity of SACs for multistep catalytic reactions (e.g., chlor-alkali process,19 the dehydrogenation of ethanol reaction,20 and the preferential oxidation of CO in hydrogen-rich fuel gas streams,21etc.), while the development of practical catalysts for the eradication of complex environmental contaminants (e.g., forever chemicals) remains elusive.22 For instance, many types of heavy metal ions (HMIs) coexist in wastewater generated by industrial or mining activities, including Cu(II), Cd(II), Pb(II), Hg(II), and As(III).23,24 Although SACs with high activity participate well in the catalysis and reduction of HMIs, it remains a challenge to accurately analyze HMIs in ambient waterways due to the poor selectivity of SACs. Thus, the design of selectively stabilized catalysts for the modification of electrodes is extremely significant for the practical application of electrochemical strategies.25The electronic metal–support interactions (EMSIs) provide an efficacious approach to tune the electronic properties of catalysts, thus achieving exceptional catalytic effects.26,27 Inspired by the electronic perturbations of Pt nanoparticles interfaced with CeO2, Campbell advanced research into strong metal–support interactions (SMSIs) to the scale of electrons.28–30 Specifically, there are interfacial charge transfers driven by different chemical potentials between metals and supports, which lead to significantly perturbed electronic structures, thus creating specifically active sites to boost catalytical reactions.31 Furthermore, charge transfers at the interface are dependent on the distribution of metals on the supports, as well as the properties of the supports themselves, which strongly affect the intrinsic activities and selectivity of the catalysts.25 Thus, the selection of metals and supports is critical.31 Single atoms with uniform and explicit active sites provide an ideal platform for the study of EMSIs, which greatly simplifies the exploration of charge transfer within electronic structures.26 In terms of supports, metal oxides are more suitable candidates than other nanomaterials (e.g., carbon-based materials), since the unoccupied d states of single metal atoms tend to interact with O atoms.2,27 As one of the typical transition metal oxides, Co3O4 nanomaterials contain highly oxidized redox couples, which have large specific surface areas and are good for adsorption.24 It has been reported in the literature that the incorporation of metal atoms into a Co3O4 lattice is an effective means by which the catalytic performance of Co3O4 can be significantly improved. Lou et al. incorporated many transition metal atoms (e.g., Mn, Fe, Co, Ni, Cu, and Zn) into a Co3O4 host lattice to obtain a series of metal atom (M) doped Co3O4 nanomaterials (M–Co3O4), which showed excellent catalytic performance for electrocatalytic oxygen evolution.32 Moreover, isolated noble metal atoms (e.g., Au, Ru, Pt, and Ir) were commendable selections for various catalytic reactions due to their advantages of unique electronic structures and irreplaceable activity. For example, a single Au atom on a Co3O4 (Au1/Co3O4) catalyst exhibited excellent catalytic activity and stability for the oxidation of CO, the active center of which was found to be a dynamic domain centered on the single Au atom that also encompassed the surrounding Co and O atoms (and/or O vacancies).33 Wang et al. showed that Ru was incorporated into Co3O4 to occupy the surface octahedral sites of Co3O4 to form Ru–O–Co structures, which significantly increased the surface O and promoted the reducibility of Co3O4 catalysts for the oxidation of chlorinated aromatics.34 Apparently, these studies still focused on the high activity and stability of catalysts. However, these strong metal–oxygen interactions may reduce the activities of single metals due to few binding sites for reactants.1 As for the premise of ensuring the stability and activity of SACs, the regulation of selective active sites still presents immense challenges. Recently, researchers discovered that there were metal–metal interactions between adjacent metal single atoms at a shortened distance, which led to the modified electronic structures of SACs.35,36 Unfortunately, since there have been few studies that focused on metal–metal interactions, due to the difficulty in stably anchoring correlated SACs on metal oxides under high loadings, it has always been difficult to distinguish the electronic perturbations of EMSIs and metal–metal interactions. We believe that the stability, activity, and selectivity of SACs can be improved through synergies between EMSIs and metal–metal interactions. Additional research should also be conducted to elucidate the charge transfer kinetics between metal single-atom sites and their supports. Moreover, for certain complex catalytic reactions, links between the selective adsorption of reaction intermediates and active sites should be established. For example, the catalytic reduction of As(III) requires the removal of three –OH groups, as it exists as a stable H3AsO3 molecule in aqueous media, which requires higher catalytic activities than other HMIs.37,38
The design of SACs based on EMSIs and metal–metal interactions to achieve the selective catalysis of As(III) provides typical modelling that is suitable for the study of the kinetics of selective catalysis. This is expected to establish a connection between the electronic perturbations of selective active sites. Therefore, we elaborately designed highly loaded Ir single atoms (Ir SAs) on oxygen vacancy-rich Co3O4 nanosheets (Ir SAs/Co3O4), where the content of Ir SAs is up to 8.9 wt%. Experimental characterization, involving X-ray absorption fine structure (XAFS) spectroscopy and X-ray photoelectron spectroscopy (XPS), combined with density functional theory (DFT) calculations, revealed that there was an interfacial charge transfer due to the EMSI and metal–metal interactions. This generated electronic perturbations for Ir SAs, Ir–O–Co, and Ir–O–Ir units, where electrons were transferred from Co and Ir sites to O atoms, particularly for Ir–O–Ir structures. This resulted in greatly stabilized Ir SAs with high valence states and active Ir–O–Ir structures. These electron-enriched O atoms were verified to be the essential active sites for electrocatalysis and were completely distinct from the other coordinated O atoms of Co3O4 or IrO2. Consequently, the resultant Ir SAs/Co3O4 nanosheets exhibited dozens of times better electrocatalytic activity than IrO2/Co3O4 and Co3O4 in the electrochemical analysis of As(III). Moreover, there was a strong orbital coupling effect between the coordinated O atoms of Ir SAs and H3AsO3; thus, Ir SAs/Co3O4 exhibited superior selectivity to As(III) compared to other common divalent heavy metal cations.
Results and discussion
Morphological and structural characterization of catalysts
Co3O4, IrO2/Co3O4, and Ir SAs/Co3O4 were prepared and characterized, respectively. The HAADF-STEM image in Fig. 1a excludes the possibility of the aggregation of Ir single atoms with brighter contrast than Co3O4, and the lattice fringes of 0.285 and 0.243 nm belonged to the (220) and (311) planes of Co3O4, respectively.24,39 Moreover, in the high-resolution HADDF-STEM (HADDF-HRSTEM) image (Fig. 1b) bright dots (Ir atoms, marked by red circles) occupied the Co3O4 lattice, where the majority of Ir atoms existed adjacently within the cationic Co3O4 lattice. The line intensity profile along the middle of the green dashed rectangles in the inset further indicated the replacement of Co sites with adjacent Ir SAs. From the scanning electron microscopy (SEM) images and transmission electron microscopy (TEM) images (Fig. S1†), similar Co3O4 and Ir SAs/Co3O4 nanosheets were confirmed, while the surface of IrO2/Co3O4 was rough due to the generation of numerous nanoparticles, which were assigned to IrO2 according to a lattice spacing of 0.201 nm.40 The Ir SAs/Co3O4 nanosheets were devoid of nanoparticles and the corresponding elemental mapping images (Fig. S2a†) revealed the uniform distribution of Ir, O, and Co across the nanosheets. Additionally, the energy-dispersive spectrometry (EDS) results in Fig. S2b† confirmed the coexistence of Ir, O, and Co, which was consistent with the elemental mapping images. The mass ratio of Ir was calculated to be 8.9 wt%, which aligned well with the results obtained by inductively coupled plasma mass spectrometry (ICP-MS). Moreover, in X-ray diffraction (XRD) patterns (Fig. S2c†), the Ir SAs/Co3O4 peaks corresponded well with those of Co3O4 (JCPDS#76-1802),41 which further verified the independent dispersion of Ir atoms. The diffraction peaks were sharper and more substantial than those of Co3O4, which implied that the introduction of Ir atoms improved the crystallinity of Co3O4. Another peak near 40° in IrO2/Co3O4 spectra was assigned to IrO2 (JCPDS#15-0870).42 Electron paramagnetic resonance (EPR) was applied to characterize vacancies in these nanomaterials, and isotropic EPR signals with a g value of 2.003 are presented in Fig. S2d,† owing to the presence of positively charged VO.37 Moreover, for IrO2/Co3O4, the signal intensity of VO slowly decreased with the addition of an Ir source, which meant that the concentration of VO decreased due to the introduction of IrO2 nanoparticles, while the VO content of Co3O4 and Ir SAs/Co3O4 was similar. The change in the Raman spectra (Fig. S2e†) further evidenced the speculation that Ir atoms occupied the Co sites. Notably, the A1g band of the Ir SAs/Co3O4 spectra shifted in contrast to that of the Co3O4 spectra, which indicated that the substitution induced lattice expansion.39 Moreover, as the A1g band depends on octahedral cations, it was demonstrated that Ir dopants occupied the Co(III) sites of octahedral units (Cooct), which was consistent with the tendency that cations with larger radii are preferentially located at octahedral sites.44–46 XPS was employed to examine the surface chemical states of the prepared samples. Fig. S2f–i† present the XPS survey and HR-XPS spectra of the corresponding elements. In the high-resolution XPS spectra of O 1s, the characteristic peaks near 529.8 and 531.6 eV were assigned to Vo and lattice oxygen (OL) for Ir SAs/Co3O4, and the peak at 532.6 eV was due to hydroxyl groups (–OH) or surface-adsorbed water (Oads).43 Compared with Co3O4 and IrO2/Co3O4, the binding energy of OL in Ir SAs/Co3O4 shifted negatively to 0.4 eV, while the VO shifted to a high binding energy of 0.3 eV, which elucidated the strong interaction between Ir and O. In addition, the VO content was reduced in Co3O4 and IrO2/Co3O4, which was consistent with the results revealed by the EPR tests. It could be found that the two fitted peaks of Co 2p3/2 correspond to Co3+ and Co2+, and the Co3+/Co2+ ratio of Ir SAs/Co3O4, Co3O4 and IrO2/Co3O4 was 2.0, 2.0 and 1.8, indicating that the valence of Co in IrO2/Co3O4 was reduced. Moreover, due to the overlap of Co 3p and Ir 4f, we used Ir 4d for comparison, and there was almost no change in the valence state of Ir. BET measurements were used to characterize the surface areas and the porous structures of the prepared samples. The N2 adsorption–desorption isotherms and the suitable aperture distribution of Ir SAs/Co3O4, Co3O4, and IrO2/Co3O4 are presented in Fig. S3.† Their surface areas were calculated to be 122.2, 109.0 and 42.5 m2 g−1, and most of the mesoporous structures were approximately 10.2, 11.6 and 8.8 nm, respectively.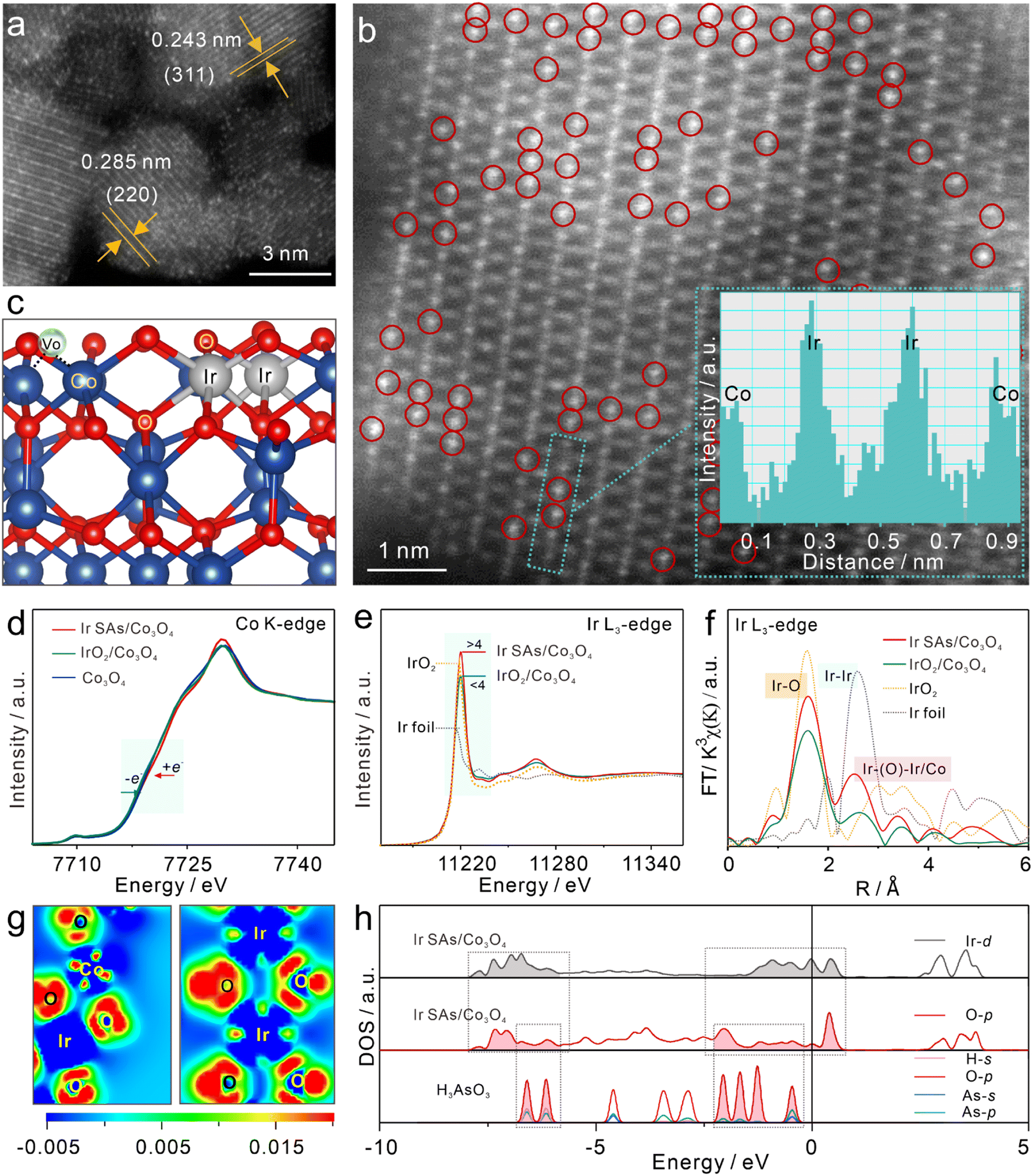 | ||
| Fig. 1 Morphological and structural characterization. (a) HADDF-STEM image of Ir SAs/Co3O4 collected on a randomly selected domain, where bright spots are ascribed to Ir single atoms. (b) HAADF-HRSTEM image with Ir sites marked by red circles (inset: an intensity line profile along the middle of the green dashed rectangles). (c) Side view of Ir SAs/Co3O4. Normalized XANES spectra for (d) Co K-edge and (e) Ir L3-edge. (f) FT-EXAFS k3-weighted χ(k) function spectra (uncorrected for phase shift) for Ir L3-edge of Ir foil, IrO2, Ir SAs/Co3O4, and IrO2/Co3O4. (g) Charge density difference image on the vertical slice through the Ir, O, and Co, where blue and red regions represent the depletion and accumulation of electrons. (h) DOS of Ir and O in Ir SAs/Co3O4 and free H3AsO3. | ||
X-ray absorption fine structure (XAFS) spectroscopy was further employed to characterize the electronic structures and coordination attributes of Co and Ir in the prepared samples. First, the valence states of Co and Ir were determined using X-ray absorption near edge structure (XANES) spectra. As presented in the cyan regions of Fig. 1d, the absorption edge of the Co K-edge in Co3O4 was located in the middle of Ir SAs/Co3O4 and IrO2/Co3O4 spectra, revealing that the order of the valence state of Co was Ir SAs/Co3O4 > Co3O4 > IrO2/Co3O4.37 Therefore, compared with Co3O4, the Co of Ir SAs/Co3O4 lost electrons, while the Co of IrO2/Co3O4 gained electrons. As shown in Fig. 1e, the height of the white line peak of the Ir L3-edge in Ir SAs/Co3O4 was higher than that of IrO2, which indicated that the valence state of Ir SAs exceeded +4. This implied a larger population of unoccupied 5d states of Ir atoms than that of supported single atoms.47,48 However, the height of the white line peak in IrO2/Co3O4 spectra was situated in the middle of the Ir foil and IrO2 peaks, suggesting that the valence state of Ir in IrO2/Co3O4 was between 0 and +4. To be noticed, the higher surface valence state obtained in XPS data compared with the XAFS data may be caused by the surface oxidation. The extended X-ray absorption fine structure (EXAFS) spectra were also fitted for further analysis. Fig. 1f depicts the EXAFS spectra of the Ir L3-edge (uncorrected for phase shift), with the detailed fitting results presented in Fig. S4 and Table S1.† For Ir SAs/Co3O4, the peak near 1.58 Å corresponded to the Ir–O scattering path, while the peak near 2.49 Å was different from the Ir–Ir scattering path of the Ir foil peak located at ∼2.58 Å, which suggested the absence of Ir NPs/clusters, in alignment with the STEM observation.27 In detail, the first shell was fitted as Ir–O scattering (RIr–O = 2.00 Å), surrounded by 6.0 O atoms, indicating that the Ir SAs attained saturated coordination. The scattering of the second shell was fitted as Ir–O–Ir (RIr–O–Ir = 2.98 Å) and Ir–O–Co (RIr–O–Co = 3.00 Å), with coordination numbers of 4.0 and 3.3, which reflected the strong interaction between single atoms and the Co3O4 substrate. For IrO2/Co3O4, the scattering of the first shell at 1.99 Å corresponded to the Ir–O path with unsaturated coordination with a coordination number of 5.1. The second shell as Ir–Co scattering (RIr–Co = 2.57 Å) with a coordination number of 1.0, which implied a strong IrO2 interaction with the Co3O4 substrate. The above analysis was conducive to the construction configurations for analysis of changes in electronic structures. In addition, the Co coordination environment was analyzed. The specific EXAFS spectra of the Co K-edge are shown in Fig. S5 and Table S2,† where peaks at ∼1.53 Å corresponded to the Co–O scattering paths, whereas the peaks at ∼2.46 and 3.02 Å were the Co–Co scattering paths.32 Compared with Co3O4, the coordination number of the Co–Co scattering at RCo–Co = 3.38 Å increased following the incorporation of Ir SAs, with a bond length of 3.35 Å. After compounding IrO2, the Co–Co bond length was 3.36 Å, which indicated that the electronic structure of Co3O4 was adjusted.
Combined with the coordination environment analyzed by XAFS, DFT calculations were used to further explore the differences in the electronic structures of Ir SAs/Co3O4, IrO2/Co3O4, and Co3O4. Fig. S6† shows the front, side, and top views of the optimized configurations. To better distinguish the surface-active sites of IrO2/Co3O4, two configurations were optimized with IrO2 and Co3O4 as the surfaces, respectively, denoted as IrO2/Co3O4(Co) and IrO2/Co3O4(Ir). First, we specifically analyzed the electron transfer process in Ir SAs/Co3O4 (Fig. 1c). As shown in Fig. 1g, the Co and Ir in Ir SAs/Co3O4 were surrounded by blue regions, which suggested that the Co and Ir atoms lost electrons. In contrast, the O atoms were surrounded by red regions, indicating that they gained electrons. The electron transfer process aligned well with the increasing valence states of Ir and Co revealed by the XANES data. It was worth noting that the surrounding area of O atoms in Ir–O–Ir was darker in red than the O atoms in the Co3O4 bulk structure, meaning that the coordinated O atoms of Ir–O–Ir acquired more electrons and may have been more active. Furthermore, Bader charge analysis was utilized to quantify the Co and Ir atoms, with the results listed in Table S3.† In general, the charge of the Co atoms in Ir SAs/Co3O4 shifted from 1.34e in Co3O4 to 1.35e, indicating that the Co atoms acquired 0.01e. Conversely, the charge of Co in IrO2/Co3O4(Co) was 1.30e, which lost 0.04e compared with Co3O4. Furthermore, the charge of Ir atoms in Ir SAs/Co3O4 increased by 0.37e compared with that of IrO2, while the charge of Ir atoms in IrO2/Co3O4(Ir) decreased by 0.02e. These data matched well with the changes in valence states and electron transfer processes revealed by XANES and differential charge density images. In conclusion, with the integration of Ir SAs, electrons were transferred from Co and Ir to the surrounding O atoms, which induced the depletion of the 3d/5d states of Co/Ir and occupation of the 2p states of O atoms, thus stabilizing the Ir SAs, with O atoms being extremely electron-enriched. In contrast, the Ir and Co atoms gained electrons following IrO2 compounding, resulting in reduced valence states. Subsequently, the density of states (DOS) of Ir SAs/Co3O4, IrO2/Co3O4, and Co3O4 are compared in Fig. 1h. There were stable bonding orbitals of Ir and coordinated O atoms in Ir SAs/Co3O4, which ranged from −10 to −2.5 eV. In the dashed box, the coordinated O atoms of the Ir SAs also had an obvious coupling effect with H3AsO3, implying that Ir SAs/Co3O4 had the capacity to interact with H3AsO3. Furthermore, the occupied state of the coordinated O of Ir SAs passed through the Fermi level, which suggested excellent electrical conductivity that is an essential prerequisite for catalytic reactions.49 Similarly, as shown in Fig. S7,† the O of Co3O4 on the surface of IrO2/Co3O4(Co) also passed through the Fermi level, which participated well in the adsorption process. However, the Ir and O bonds in IrO2/Co3O4(Ir) were well coupled between −10 and −2.5 eV, without an occupied state near the Fermi level to accept electrons for adsorption in the O-p orbital, which meant that the Ir–O bond was stable. By integrating the electrons of the Co-d orbitals, it was found that the d-band centers of Co3O4 and IrO2/Co3O4(Co) were located at −3.74 eV and –3.36 eV, respectively, while the d-band center of Ir SAs/Co3O4 obviously shifted to −2.73 eV, which signified that the incorporation of Ir SAs modulated the electronic structure of Co3O4 and the position of its d-band center. Undoubtedly, the distinct electronic structures of these materials can give rise to potential differences in the active sites and interaction modes of catalytic reactions.
Effective electrocatalysis and selective detection of As(III)
Ir SAs/Co3O4/GCE, Co3O4/GCE, and IrO2/Co3O4/GCE were synthesized and used for electrochemical studies. The conductivity of these electrodes was examined via cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS), with the results in Fig. S8† showing that the incoming Ir SAs successfully reduced the resistance of metal oxides, which was of significant benefit for electrochemical detection. Simultaneously, linear sweep voltammetry (LSV) data were used to calculate the turnover frequencies (TOFs) to further reveal the differences in the catalytic activities of these modified electrodes. Their LSV signals toward As(III) of 10 ppb are presented in the inset of Fig. 2a, and the spikes that emerged at about 0 V were the stripping peaks of As(0), which shows that the current on Ir SAs/Co3O4/GCE was far higher than that for Co3O4/GCE and IrO2/Co3O4/GCE. Furthermore, through the shape and area of the LSV peaks, the reaction time (t) and the number of As(III) that contributed to the electrocatalytic reduction reaction (N) were obtained. The TOF was calculated by using the equation TOF = N/(A × t), where A is the active area of electrode interfaces (Fig. S9†).50 Details regarding the TOF calculation are shown in the ESI.† In short, Ir SAs/Co3O4/GCE exhibited the highest activity with an efficiency of 4.47 nm−2 s−1, which was almost 20-fold and 10-fold better than Co3O4/GCE (0.45 nm−2 s−1) and IrO2/Co3O4/GCE (0.27 nm−2 s−1), respectively, which demonstrated the prominent catalytic activity of Ir SAs/Co3O4/GCE for the electroanalysis of As(III). Additionally, the electroanalytical performance of Co3O4/GCE and IrO2/Co3O4/GCE was also confirmed under the same detection conditions using the square wave anodic stripping voltammetry (SWASV) technique with optimized conditions in Fig. S10.† Moreover, the sensitivity and LOD comparisons of different modified electrodes are summarized in Fig. S11,† with detailed SWASV responses and linear equations. Specifically, the sensitivities toward the As(III) of Co3O4/GCE and IrO2/Co3O4/GCE were 0.08 and 0.04 μA ppb−1, respectively, and for the LOD were 4.07 and 4.37 ppb, respectively. Moreover, the electrochemical performance obtained by Ir SAs/Co3O4 for the detection of As(III) was superior to that obtained by other modified cobalt-based nanomaterials (Table S4†). Considering the coexistence of other heavy metal ions, Ir SAs/Co3O4/GCE was implemented for the electroanalysis of Zn(II), Cd(II), Cu(II), Hg(II), and Pb(II). Fig. S12† presents the detailed SWASV responses and corresponding linear equations, and comparisons of the sensitivity and LOD for the HMIs are summarized in Fig. 2b. Explicitly, the sensitivities toward Zn(II), Cd(II), Cu(II), Hg(II), and Pb(II) were 0.001, 0.22, 0.22, 0.07, and 0.23 μA ppb−1, respectively, while the LOD were 360.00, 4.82, 4.32, 3.66, and 4.83 ppb, respectively. The electroanalytical performance of Ir SAs/Co3O4/GCE toward these heavy metal cations was far worse than that toward As(III), which verified that Ir SAs/Co3O4/GCE was highly selective for As(III). Further in-depth analysis was required to uncover the fundamental mechanism of why the Ir SAs/Co3O4/GCE exhibited excellent sensitivity and selectivity for As(III). The stability and reproducibility of the modified electrodes were significant indicators to evaluate their practicability which were further investigated by SWASV toward As(III) at 4 ppb, with the results shown in Fig. S13.† During 10 cycles of SWASV tests of the same electrodes, the relative standard deviation (RSD) was just 1.9% (Fig. S13a†). In addition, the RSD of 10 different Ir SAs/Co3O4 modified electrodes was only 4.0% (Fig. S13b†). Except that these RSDs of the currents were less than 5%, there was almost no change in the shape of the peak currents obtained in the above experiments, indicating the excellent stability and reproducibility of Ir SAs/Co3O4/GCE. Moreover, the morphologies and structures of Ir SAs/Co3O4 after electrochemical tests of 20 times were characterized and analyzed to further certify their durability. First, XRD patterns in Fig. S14† presented unchanged peaks of used Ir SAs/Co3O4 compared with fresh catalysts, and there is no peak match with Ir nanoparticles. HADDF-STEM images presented in Fig. S15a and b† show that the Ir atoms used in Ir SAs/Co3O4 were evenly distributed as single atoms. Besides, k3-weighted and fitting spectra for Ir L3-edge and Co K-edge in Fig. S15c–f† further indicated an unvaried coordination environment of Ir SAs/Co3O4. All in all, Ir SAs/Co3O4 exhibited satisfactory stability, reproducibility and durability. | ||
| Fig. 2 Electrochemical detection of As(III) and selectivity tests toward common heavy metal ions. (a) Comparison of TOF and LSV responses toward 10 ppb As(III) (inset) on Ir SAs/Co3O4, IrO2/Co3O4, and Co3O4 modified electrodes under the same conditions. (b) Sensitivity and LOD of Ir SAs/Co3O4 toward different HMIs. | ||
Charge transfer process in electrocatalysis of As(III)
To reveal the strongly reactive catalytic sites and the analysis of As(III), XPS and XAFS were used to characterize the interactions between Ir SAs/Co3O4 and IrO2/Co3O4 and As(III). After adsorbing As(III) at 10 ppm in 0.1 M HAc–NaAc (pH = 5.0), Ir SAs/Co3O4 and IrO2/Co3O4 were denoted as Ir SAs/Co3O4/As and IrO2/Co3O4/As, respectively. In the XANES spectra of the Co–K edge (Fig. 3a), there was almost no change in the position of the absorption edge of Ir SAs/Co3O4 following the interaction with As(III). Furthermore, the unchanged valence of Co indicated that the Co sites might not be related to the active As(III) site. In contrast, the Co K-edge absorption edge of IrO2/Co3O4 shifted toward higher energy levels after interacting with As(III), which meant that the Co lost electrons. As shown in the Ir-L3 spectra (Fig. 3b), the height of the white line peaks in the Ir SAs/Co3O4/As and IrO2/Co3O4/As spectra were higher and lower, respectively, than those prior to adsorption, which implied that the valence state of the Ir SAs increased. In contrast, the valence state of IrO2 decreased. In the XANES spectra of the As K-edge (Fig. 3c), the position of the absorption edge revealed that the valence state of the adsorbed As was equivalent to that of NaAsO2, indicating that no noticeable oxidation occurred.37 From the analysis of the XANES spectra, it was found that the active sites of Ir SAs/Co3O4 and IrO2/Co3O4 that interacted with As(III) were quite different. In general, the Ir atoms in Ir SAs/Co3O4 lost electrons when interacting with As(III), while the Co and Ir atoms in IrO2/Co3O4 lost and gained electrons, respectively. EXAFS spectra were fitted to analyze the coordination environments of Co and Ir after adsorbing As(III). According to the specifically fitted results of the Co–K edge (Fig. S16 and Table S1†), there was no significant change in the coordination environment of the Co of Ir SAs/Co3O4 and IrO2/Co3O4 prior to and following adsorption. Furthermore, the coordination environment of the Ir was fitted and analyzed, with the fitted data shown in Fig. S17 and Table S2.† There was no significant change in the coordination environment of Ir in IrO2/Co3O4, while the coordination environment of Ir in Ir SAs/Co3O4 was altered. In addition, changes in the O atoms on the surface were further analyzed via using XPS. As displayed in Fig. S18a,† the peak at 531.6 eV of Ir SAs/Co3O4 belonged to that of VO, which shifted to a lower energy level by 0.8 eV after interacting with As(III). Similarly, in Fig. S18b,† the VO peak in the IrO2/Co3O4 spectra also shifted to a lower energy level by 0.5 eV following the interaction with As(III), indicating that the VO in both Ir SAs/Co3O4 and IrO2/Co3O4 interacted with As(III). Although the surface area of IrO2/Co3O4 was reduced by the formation of IrO2 nanoparticles, we found that the physical adsorption toward As(III) was higher than that of Ir SAs/Co3O4 and Co3O4 in the HR-XPS spectra of As 3d (Fig. S19†), which would be further explained by the DFT adsorption configurations. However, the electrochemical sensitivity of IrO2/Co3O4 is lower than that of Ir SAs/Co3O4, due to that the complex catalytic reaction of H3AsO3 not only depended on the adsorption process.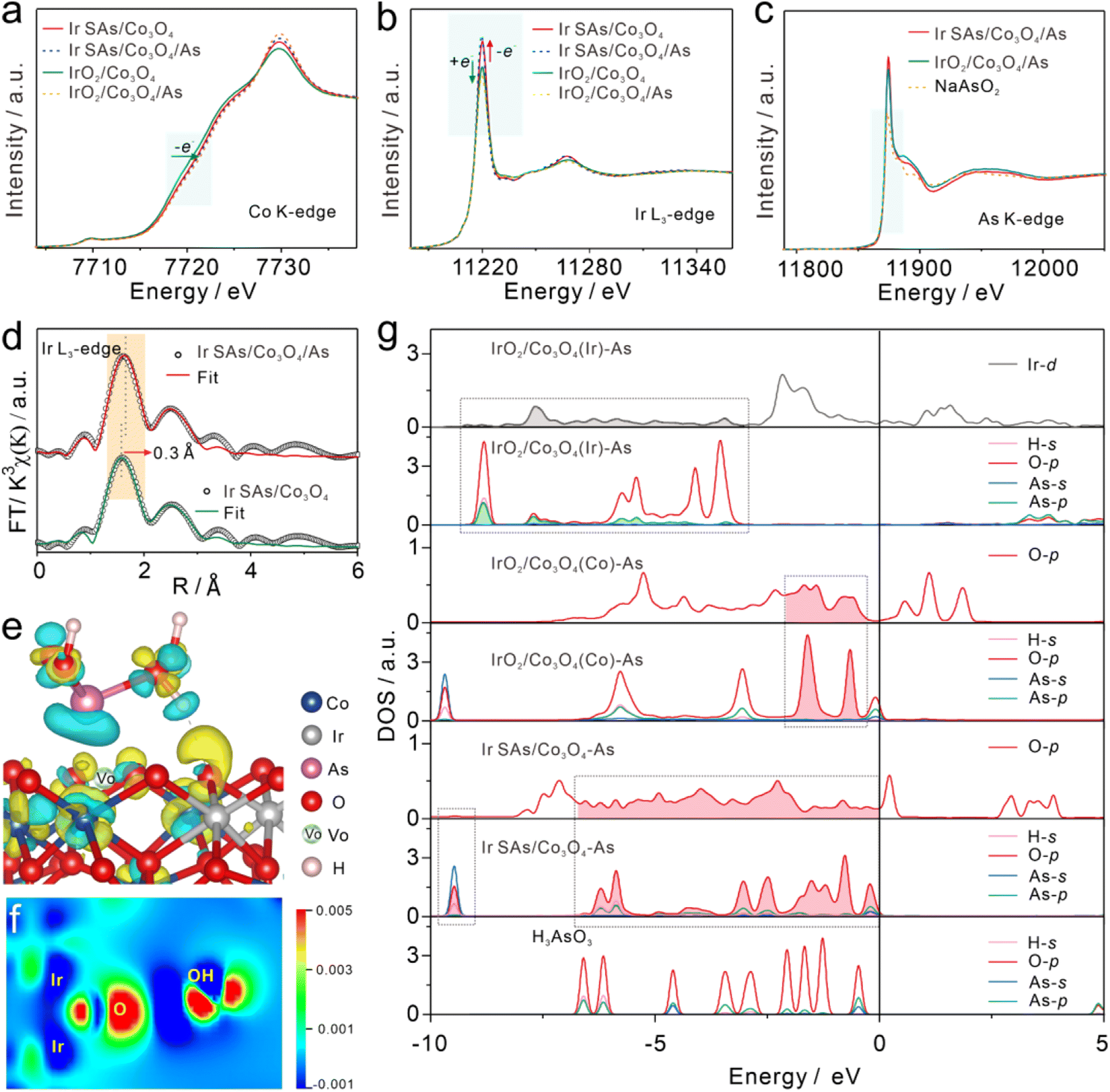 | ||
| Fig. 3 Normalized XANES spectra of Ir SAs/Co3O4 and IrO2/Co3O4 prior to and following interactions with As(III) of (a) Co K-edge, (b) Ir L3-edge and (c) As K-edge. (d) FT-EXAFS k3-weighted χ(k) function spectra (uncorrected for phase shift) for the Ir L3-edge of Ir SAs/Co3O4 and Ir SAs/Co3O4/As. (e) Charge density difference image of Ir SAs/Co3O4/As, the isovalue was 0.005 eV Å−3 (green and yellow regions represent the depletion and accumulation of electrons, respectively). (f) Charge density difference image on the slice through Ir and O in Ir SAs/Co3O4 and O in H3AsO3 (blue and red regions represent the depletion and accumulation of electrons, respectively). (g) PDOS of Ir, O, and H3AsO3 in adsorption configuration. | ||
As shown in Fig. 3d, the Ir–O bond in Ir SAs/Co3O4 was longer by 0.3 Å after the adsorption of As(III), which further suggested that the active sites in Ir SAs/Co3O4 were related to Ir. Combined with the highly active O atoms of Ir–O–Ir and the saturated coordination environment of Ir revealed above, it was speculated that the actual reactive sites were the highly active O atoms. For IrO2/Co3O4, although there was no change in the coordination environment of Ir, considering the change of its valence state, the active sites could be IrO2 or Co3O4. Furthermore, all of the aforementioned materials were involved in catalyzing As(III), and the specific catalytic interactions could be further analyzed with the help of theoretical calculations.
DFT calculations were employed to analyze changes in the electronic structures of catalysts to reveal the actual active sites. First, the adsorption configurations were optimized, including Ir SAs/Co3O4/As, IrO2/Co3O4(Co)/As, and IrO2/Co3O4(Ir)/As, with the specific optimized configurations shown in Fig. S20.† For Ir SAs/Co3O4, the H3AsO3 molecules were spontaneously adsorbed to the concave surface formed by the VO on Ir SAs/Co3O4, and a hydrogen bond was formed between the coordinated O atoms of the Ir–O–Ir structures and the –OH in H3AsO3.51 For IrO2/Co3O4, two hydrogen bonds interacted with H3AsO3; specifically, the coordinating O atoms of unsaturated Co in Co3O4 interacted with –OH in H3AsO3, which was denoted as IrO2/Co3O4(Co)/As. Moreover, the H3AsO3 molecules could interact with unsaturated Ir sites, which was denotted as the IrO2/Co3O4(Ir)/As configuration.
More intuitively, the active sites that interacted with H3AsO3 and the electron transfer process were explored through differential charge density images and corresponding slice differential charge density images. As shown in Fig. 3e and f, the Ir atoms of Ir–O–Ir in Ir SAs/Co3O4 obviously lost electrons, while O gained electrons and further transferred them to the –OH of H3AsO3 to reduce H3AsO3.
As shown in Fig. S21a and c,† in IrO2/Co3O4(Co), the Co lost electrons to its coordinating O atom and further transferred them to the –OH of H3AsO3. As shown in Fig. S21b and d,† in IrO2/Co3O4(Ir), As lost electrons and Ir gained electrons. The electron transfer process revealed by the differential charge density images was consistent with the results revealed by XANES. Bader charge analysis further quantitatively revealed the electron transfer process following the adsorption of H3AsO3 (Table S3†). The Ir atoms of Ir–O–Ir in Ir SAs/Co3O4 lost 0.05e, and the Ir in IrO2/Co3O4(Ir) gained 0.05e, while the Co in IrO2/Co3O4(Co) lost 0.11e.
Variations in the DOS of the Ir and O atoms in these configurations are shown in Fig. 3g. Significantly, the H3AsO3 energy levels adsorbed on the nanomaterials were altered compared with those of free H3AsO3, which also had a strong coupling with the orbital energy levels of specific atoms in the catalysts. Specifically, the O-p orbital of Ir SAs/Co3O4 moved to a lower energy level following the adsorption of H3AsO3. In the dashed box, the O-p orbitals at 0 to −1.67 eV, −5.9 to −6.3 eV, and −9.5 eV matched well with the H3AsO3 energy levels, implying that the O atom of Ir–O–Ir was the active site for interacting with H3AsO3. Similarly, the O atom of the Co3O4 in IrO2/Co3O4(Co)/As also generated an energy level that matched the orbital energy level of H3AsO3. However, in IrO2/Co3O4(Ir)/As it was the orbital energy level of Ir-d that matched with H3AsO3 and not that of O-p, indicating that the interactive H3AsO3 sites in IrO2 were unsaturated Ir sites. In general, the actual reactive sites for catalyzing H3AsO3 in Ir SAs/Co3O4 were the active O atoms in Ir–O–Ir, where the VO on its surface played a role in promoting the adsorption of H3AsO3. For IrO2/Co3O4, both the Ir atoms of IrO2 and the coordinated O atoms of unsaturated Co in Co3O4 were the reactive sites that catalyzed H3AsO3.
Selective reduction reaction of As(III) and the proposed catalytic mechanism
To further explore the electrocatalytic reduction reaction of As(III) on different sensing interfaces, adsorption configurations of the H3AsO3 intermediate during the electrocatalytic reduction were optimized. The adsorption and stepwise reduction configurations of H3AsO3 on Ir SAs/Co3O4, IrO2/Co3O4(Co), and IrO2/Co3O4(Ir) are shown in Fig. 4a (side view) and Fig. S20† (front and top views), and corresponding relative energies were compared in Fig. 4b. The adsorption energies (Eads) of H3AsO3 on Ir SAs/Co3O4, IrO2/Co3O4(Co), and IrO2/Co3O4(Ir) were −1.01, −0.35, and −1.23 eV, respectively. The adsorption capacity of Ir SAs/Co3O4 for H3AsO3 was weaker than that of IrO2/Co3O4(Ir), due to the formation of Ir–As bonds in IrO2/Co3O4(Ir)/As and only hydrogen bonds in Ir SAs/Co3O4. Among the configurations of IrO2/Co3O4(Ir)/As and Ir SAs/Co3O4/As, the largest potential barriers in the stepwise reductive reaction appeared in the *H2AsO2 to *HAsO process, and the energy required by Ir SAs/Co3O4 was 0.23 eV, which was lower than that of IrO2/Co3O4(Ir) (0.42 eV). Furthermore, in IrO2/Co3O4(Co)/As, although the *H3AsO3 was spontaneously converted to *HAsO, the reduction of *HAsO to *As had a potential barrier as high as 1.19 eV, which directly diminished the reduction efficiency of As(III) during electrochemical enrichment. This may have also been the explanation for the high adsorption energy, albeit poor electrochemical performance of IrO2/Co3O4(Co). It was observed that Ir–O–Ir bonded with the *HAsO to spontaneously reduce *HAsO to *As. Overall, the external energy required for the rate-determining step for the catalysis of As(III) by Ir SAs/Co3O4 was the lowest, and the Ir–O–Ir active center had a stronger electronic affinity to intermediates, which was beneficial for the more rapid deposition of As(0) on its surface, thereby boosting the electrochemical signal. Therefore, we proposed the mechanism of the electrochemical detection process in Fig. 4c. When adding a certain amount of As(III), As(III) diffused rapidly onto the surface of Ir SAs/Co3O4 and was confined in the concave surface formed by VO on the surface. Subsequently, the coordinated O atoms of Ir–O–Ir interacted with –OH of H3AsO3, and –OH captured the electrons of the Ir-coordinated O, further accelerating the process by which electrons transferred from the Ir to O atoms in Ir–O–Ir, thereby resulting in the reduced valence state of Ir. Additionally, the Ir–O bonds were elongated by the interaction of H3AsO3 and Ir–O–Ir. –OH was gradually removed from H3AsO3 under the reduction of the active Ir–O–Ir structure. Most of the steps in reducing As(0) were thermodynamically spontaneous, and the external energy required for the rate-determining step was low. In addition, different from other common heavy metal cations, the specific interaction with –OH made Ir SAs/VO-Co3O4 super selective toward As(III). At the applied potential, As(III) was first reduced to As(0) and then oxidized to As(III), and the SWASV signal increased linearly with the increased concentration of As(III), thus realizing the reduction and quantitative detection of As(III) with high-sensitivity.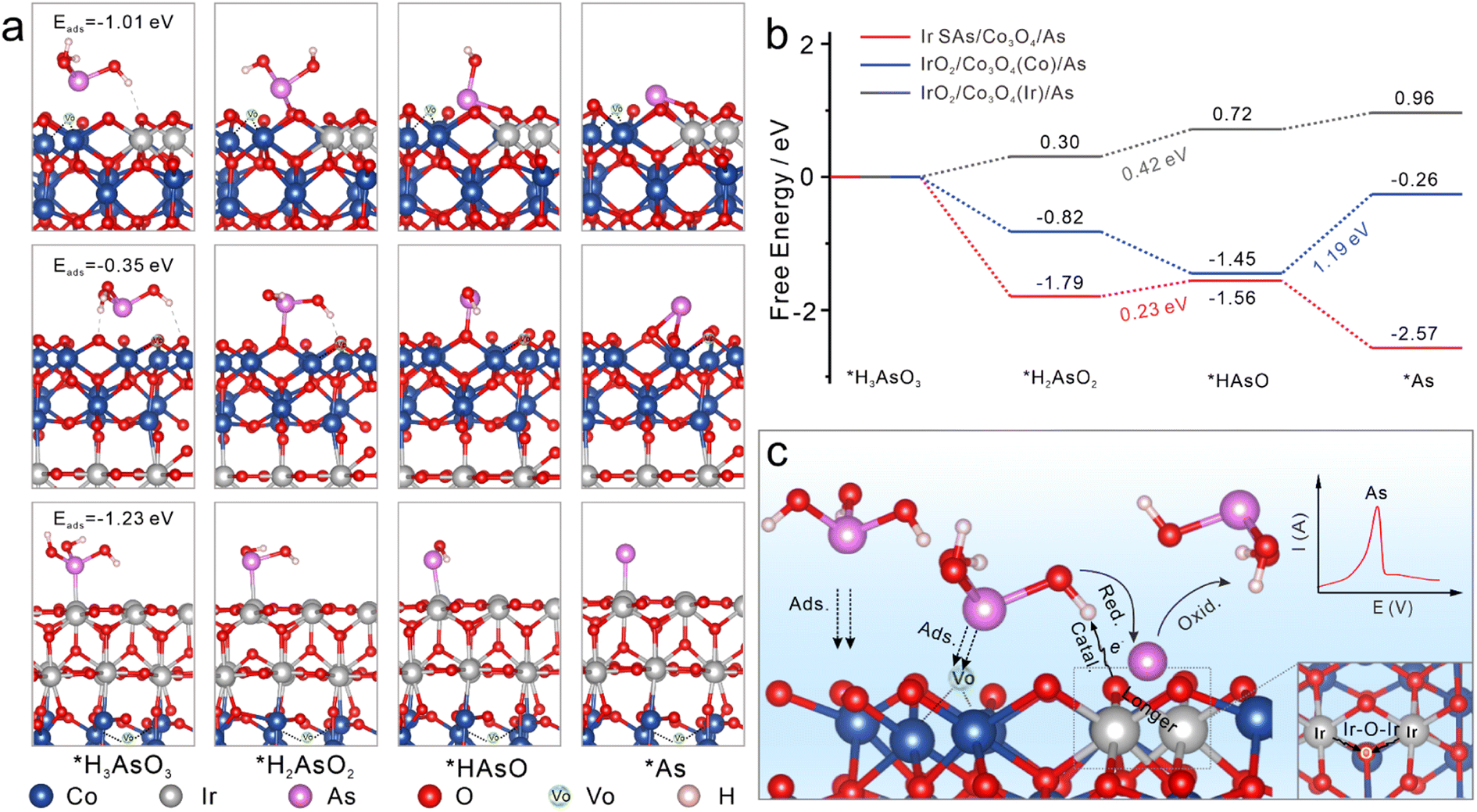 | ||
| Fig. 4 Reduction reaction pathway analyses of As(III) on different catalysts. (a) Adsorption and stepwise reduction configurations of H3AsO3 on Ir SAs/Co3O4, IrO2/Co3O4(Co), and IrO2/Co3O4(Ir). (b) Relative energy of reduction intermediates of H3AsO3 on Ir SAs/Co3O4, IrO2/Co3O4(Co), and IrO2/Co3O4(Ir). (c) Proposed catalytic process of enhanced SWASV signals toward As(III) with Ir SAs/Co3O4. | ||
Conclusions
In summary, high-density Ir SAs were stably anchored on oxygen vacancy-rich Co3O4 that was characterized using HADDF, XAFS, and other techniques to elucidate the uniform and adjacent distribution of Ir SAs and the generation of Ir–O–Ir/Co structures. Combined with DFT calculations, there was an interfacial charge transfer due to electronic metal–support and metal–metal interactions, which resulted in evident electronic perturbations for Ir single atoms and Co3O4. Specifically, electrons transferred from Co and Ir sites to O atoms, particularly in Ir–O–Ir structures. These electron-enriched O atoms were totally different from other coordinated O atoms, which served as essential active sites for electrocatalysis with more robust electronic affinities for the intermediates derived from the reduction of As(III). Consequently, they exhibited dozens of times better electrocatalytic activities than IrO2/Co3O4 and Co3O4 in the electrochemical analysis of As(III). Furthermore, new orbital energy levels matching H3AsO3 appeared in the O atoms of Ir–O–Ir; thus, Ir SAs/Co3O4 exhibited superior selectivity for As(III) compared to other common divalent heavy metal cations. This work revealed the electronic perturbations induced by synergetic interactions between the metal–support and metal–metal from electronic structures and charge transfer processes to provide an innovative strategy for the development of highly active, selective, and stable catalysts.Experimental section
Synthesis of Co3O4, IrO2/Co3O4, and Ir SAs/Co3O4
First, 4.0 mmol Co(NO3)2·6H2O and 5.0 mmol hexadecyl trimethyl ammonium bromide, CTAB were dissolved into 100 mL of water in turn, and 6 mmol sodium borohydride (SB) was quickly added and reacted for 30 min under stirring. Subsequently, 4 mL H2IrCl6·6H2O (40 mg mL−1) was injected into the blue-green solution at an even and slow rate of 12 mL h−1 under continuous stirring. After reacting for another 4 h, 1.0 mmol SB was quickly added and then reacted for another 20 min to obtain a black solution. The resulting products were collected, rinsed with distilled water and acetone, and dried at 60 °C overnight. Finally, Ir SAs/Co3O4 nanosheets were obtained by calcining the precursor at 450 °C for 4 h under air, at a heating rate of 5 °C min−1. For comparison, Co3O4 was synthesized according to the above experimental procedure without the addition of H2IrCl6·6H2O and extra SB. Furthermore, IrO2/Co3O4 was prepared by increasing the H2IrCl6·6H2O content. The specific experimental conditions can be found in the ESI.†Electrochemical adsorption and measurements
All of the adsorption experiments and electrochemical detection were carried out in 0.1 M HAc–NaAc of pH = 5. For SWASV tests, the deposition potential and running time were set at −1.1 V and 150 s and the desorption potential and running time were set at +0.8 V and 150 s.Determination of the turnover frequencies
The TOF value for reduction of As(III) on different electrodes was calculated using the equation where N is the number of As(III) participating in the electrocatalytic reduction process, which could be obtained by LSV tests. A is the active area of the electrode interface, which could be calculated according to the Randles–Sevcik equation. More specific experiments and calculation processes could be found in the ESI.†
where N is the number of As(III) participating in the electrocatalytic reduction process, which could be obtained by LSV tests. A is the active area of the electrode interface, which could be calculated according to the Randles–Sevcik equation. More specific experiments and calculation processes could be found in the ESI.†
Data availability
The data supporting this article and other findings are available from the corresponding authors upon request.Author contributions
X. H., W. L. and S. C. designed this project. S. C. and Y. Y. implemented the experiments. Z. S. implemented the DFT calculations. X. X., P. L. and M. Y. carried out the data analysis. S. C. and A. C. drafted the manuscript. S. C. and A. C. reviewed and edited the manuscript. All authors approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the China Postdoctoral Science Foundation (2022M723188), the Open Research Project of State Key Laboratory of Transducer Technology (SKT2202), the National Natural Science Foundation of China (22306186, 22204166, 22206187), the Youth Innovation Promotion Association of CAS (2023469), the National Key R&D Program of China (2021YFB3201400), the Key R&D Program of Anhui Province (202104i07020006 and 202104i07020011), the Anhui Provincial Natural Science Foundation (2208085QB57), the Postdoctoral Researcher Funding Project of Anhui Province (2021B528), the HFIPS Director's Fund, Grant No., YZJJ202102, YZJJZX202019, YZJJ2022QN26 and YZJJ2023QN33 and 2022YZGH05, the Special Foundation of President of The Chinese Academy of Sciences, and the special project (SXHZ202003) of science and technology cooperation between Fuyang Municipal People's Government and Fuyang Normal University. The authors thank the staff of the BL11B and BL14W beam lines at the Shanghai Synchrotron Radiation Facility (SSRF) for data collection.References
- X. Li, X. I. Pereira-Hernandez, Y. Chen, J. Xu, J. Zhao, C. W. Pao, C. Y. Fang, J. Zeng, Y. Wang, B. C. Gates and J. Liu, Nature, 2022, 611, 284–288 CrossRef CAS PubMed
.
- A. S. Rosen, S. Vijay and K. A. Persson, Chem. Sci., 2023, 14, 1503–1511 RSC
- F. D. Speck, M. T. Y. Paul, F. Ruiz Zepeda, M. Gatalo, H. Kim, H. C. Kwon, K. J. J. Mayrhofer, M. Choi, C. H. Choi, N. Hodnik and S. Cherevko, J. Am. Chem. Soc., 2020, 142, 15496–15504 CrossRef CAS PubMed
- M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS PubMed
- Y. Q. Su, G. J. Xia, Y. Qin, S. Ding and Y. G. Wang, Chem. Sci., 2021, 12, 8260–8267 RSC
- K. Qi, M. Chhowalla and D. Voiry, Mater. Today, 2020, 40, 173–192 CrossRef CAS
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed
- H. Gu, X. Liu, X. Liu, C. Ling, K. Wei, G. Zhan, Y. Guo and L. Zhang, Nat. Commun., 2021, 12, 5422 CrossRef CAS PubMed
- H. B. Zhang, L. Yu, T. Chen, W. Zhou and X. W. Lou, Adv. Funct. Mater., 2018, 28, 1807086 CrossRef
- E. Meza, R. E. Diaz and C. W. Li, ACS Nano, 2020, 14, 2238–2247 CrossRef CAS PubMed
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS PubMed
- S. Yang, Y. J. Tak, J. Kim, A. Soon and H. Lee, ACS Catal., 2017, 7, 1301–1307 CrossRef CAS
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y.-W. Li, C. Shi, X.-D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed
- W. Wang, Y. Wu, Y. Lin, J. Yao, X. Wu, C. Wu, X. Zuo, Q. Yang, B. Ge, L. Yang, G. Li, S. Chou, W. Li and Y. Jiang, Adv. Funct. Mater., 2022, 32, 2108464 CrossRef CAS
- J. Wu, N. Han, S. Ning, T. Chen, C. Zhu, C. Pan, H. Wu, S. J. Pennycook and C. Guan, ACS Sustainable Chem. Eng., 2020, 8, 14825–14832 CrossRef CAS
- H. Song, M. Wu, Z. Tang, J. S. Tse, B. Yang and S. Lu, Angew. Chem., Int. Ed., 2021, 60, 7234–7244 CrossRef CAS PubMed
- Y. Y. Li, Z. Y. Song, X. Y. Xiao, L. K. Zhang, H. Q. Huang, W. Q. Liu and X. J. Huang, J. Hazard. Mater., 2022, 435, 129009 CrossRef CAS PubMed
- K. Elibol, C. Mangler, D. D. O'Regan, K. Mustonen, D. Eder, J. C. Meyer, J. Kotakoski, R. G. Hobbs, T. Susi and B. C. Bayer, ACS Nano, 2021, 15, 14373–14383 CrossRef CAS PubMed
- K. S. Exner, ChemElectroChem, 2020, 7, 1528–1530 CrossRef CAS
- M. Ouyang, K. G. Papanikolaou, A. Boubnov, A. S. Hoffman, G. Giannakakis, S. R. Bare, M. Stamatakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Nat. Commun., 2021, 12, 1549 CrossRef CAS PubMed
- S. Cao, Y. Zhao, S. Lee, S. Yang, J. Liu, G. Giannakakis, M. Li, M. Ouyang, D. Wang, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Sci. Adv., 2020, 6, eaba3809 CrossRef CAS PubMed
- P. Kumar, T. A. Al-Attas, J. Hu and M. G. Kibria, ACS Nano, 2022, 16, 8557–8618 CrossRef CAS PubMed
- S.-H. Chen, J.-J. Zhu, P.-H. Li, Y.-F. Sun, M. Yang and X.-J. Huang, Chem. Eng. J., 2022, 430, 132959 CrossRef CAS
- S.-H. Chen, Z.-Y. Song, P.-H. Li, X.-Y. Xiao, H.-Q. Huang, M. Yang, C.-H. Lin, L.-N. Li and X.-J. Huang, J. Hazard. Mater., 2021, 416, 126157 CrossRef CAS PubMed
- L. X. Wang, L. Wang and F. S. Xiao, Chem. Sci., 2021, 12, 14660–14673 RSC
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS PubMed
- J. Xu, H. Xu, A. Dong, H. Zhang, Y. Zhou, H. Dong, B. Tang, Y. Liu, L. Zhang, X. Liu, J. Luo, L. Bie, S. Dai, Y. Wang, X. Sun and Y. Li, Adv. Mater., 2022, 34, 2206991 CrossRef CAS PubMed
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS PubMed
- A. Bruix, J. A. Rodriguez, P. J. Ramirez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS PubMed
- D. Liu, Q. He, S. Ding and L. Song, Adv. Energy Mater., 2020, 10, 2001482 CrossRef CAS
- S. L. Zhang, B. Y. Guan, X. F. Lu, S. Xi, Y. Du and X. W. D. Lou, Adv. Mater., 2020, 32, 2002235 CrossRef CAS PubMed
- Y. Lou, Y. Cai, W. Hu, L. Wang, Q. Dai, W. Zhan, Y. Guo, P. Hu, X.-M. Cao, J. Liu and Y. Guo, ACS Catal., 2020, 10, 6094–6101 CrossRef CAS
- Y. Lao, N. Zhu, X. Jiang, J. Zhao, Q. Dai and X. Wang, Catal. Sci. Technol., 2018, 8, 4797–4811 RSC
- J. Shan, C. Ye, C. Zhu, J. Dong, W. Xu, L. Chen, Y. Jiao, Y. Jiang, L. Song, Y. Zhang, M. Jaroniec, Y. Zhu, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2022, 144, 23214–23222 CrossRef CAS PubMed
- J. Shan, C. Ye, Y. Jiang, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Sci. Adv., 2022, 8, eabo0762 CrossRef CAS PubMed
- S. H. Chen, Z. Y. Song, X. Y. Xiao, H. Q. Huang, Y. F. Yang, P. H. Li, M. Yang and X. J. Huang, Anal. Chem., 2022, 94, 3211–3218 CrossRef CAS PubMed
- Y. Zhang and R. G. Compton, Talanta, 2022, 247, 123578 CrossRef CAS PubMed
- Y. Lu, W. Zhan, Y. He, Y. Wang, X. Kong, Q. Kuang, Z. Xie and L. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 4186–4195 CrossRef CAS PubMed
- Z. Wang, G. Liu, C. Ding, Z. Chen, F. Zhang, J. Shi and C. Li, J. Phys. Chem. C, 2015, 119, 19607–19612 CrossRef CAS
- B. Varghese, C. H. Teo, Y. Zhu, M. V. Reddy, B. V. R. Chowdari, A. T. S. Wee, V. B. C. Tan, C. T. Lim and C. H. Sow, Adv. Funct. Mater., 2007, 17, 1932–1939 CrossRef CAS
- J. Tang, B. Kong, Y. Wang, M. Xu, Y. Wang, H. Wu and G. Zheng, Nano Lett., 2013, 13, 5350–5354 CrossRef CAS PubMed
- X. Y. Xiao, Z. Y. Song, C. C. Zhang, Y. H. Zhao, Z. W. Gao, S. H. Chen, P. H. Li, Y. F. Sun, M. Yang and X. J. Huang, Chem. Sci., 2023, 14, 2960–2970 RSC
- J. Preudhomme and P. Tarte, Spectrochim. Acta, Part A, 1971, A 27, 1817–1835 CrossRef
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS PubMed
- J. Shan, C. Ye, S. Chen, T. Sun, Y. Jiao, L. Liu, C. Zhu, L. Song, Y. Han, M. Jaroniec, Y. Zhu, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2021, 143, 5201–5211 CrossRef CAS PubMed
- H. Shin, W.-G. Jung, D.-H. Kim, J.-S. Jang, Y. H. Kim, W.-T. Koo, J. Bae, C. Park, S.-H. Cho, B. J. Kim and I.-D. Kim, ACS Nano, 2020, 14, 11394–11405 CrossRef CAS PubMed
- J. Li, Q. Guan, H. Wu, W. Liu, Y. Lin, Z. Sun, X. Ye, X. Zheng, H. Pan, J. Zhu, S. Chen, W. Zhang, S. Wei and J. Lu, J. Am. Chem. Soc., 2019, 141, 14515–14519 CrossRef CAS PubMed
- D. Higgins, M. A. Hoque, M. H. Seo, R. Wang, F. Hassan, J.-Y. Choi, M. Pritzker, A. Yu, J. Zhang and Z. Chen, Adv. Funct. Mater., 2014, 24, 4325–4336 CrossRef CAS
- X. Y. Xiao, Z. Y. Song, H. Xie, Y. H. Zhao, S. H. Chen, Y. Y. Li, M. Yang, P. H. Li, H. Ji and X. J. Huang, Adv. Funct. Mater., 2022, 32, 2209283 CrossRef CAS
- Z. Zhang, C. Feng, D. Wang, S. Zhou, R. Wang, S. Hu, H. Li, M. Zuo, Y. Kong, J. Bao and J. Zeng, Nat. Commun., 2022, 13, 2473 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03285j |
| ‡ S. H. Chen and Y. F. Yang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |