DOI:
10.1039/D3RA04809H
(Paper)
RSC Adv., 2023,
13, 33129-33145
The effects of Fe, Mg, and Pt-doping on the improvement of Ni stabilized on Al2O3-CeO3 catalysts for methane dry reforming†
Received
17th July 2023
, Accepted 26th October 2023
First published on 9th November 2023
Abstract
Herein, the promotional effects of Mg, Fe, and Pt on Ni-based catalysts supported on Al2O3-CeO2 (Ni/Al2O3-CeO2) were investigated in the dry reforming of methane (DRM) reaction. The interaction of a suitable amount of MgO and FeO with Ce2O3 stabilized in the catalysts was demonstrated by the temperature-programmed desorption of CO2 (CO2-TPD). Ce2O3 has a high basicity for adsorbing CO2, generating a monoclinic Ce2O2CO3 species in the DRM reaction. Surface oxygen ions were also produced by adding MgO and FeO, as demonstrated by the temperature-programmed reduction of H2 (H2-TPR). Monoclinic Ce2O2CO3 and surface oxygen may both be used to oxidize and remove the carbon that was deposited, maintaining the high activity and stability of the metal Ni and Pt catalysts. The high dispersion and synergistic interactions between the platinum and oxide phases, which are associated with the decrease in reduction temperature and the rise in the number of basic sites, are responsible for the increased activity of Pt with M–Ni/Al2O3-CeO2 catalysts. The co-doped Ni/Al2O3-CeO2 catalysts with Mg and Fe significantly enhanced the activity (more than 80% methane and 84% CO2 conversion), the selectivity toward syngas (∼90%), and maintained the H2/CO ratio at about 0.97 at 700 °C.
1. Introduction
The dry reforming of methane (DRM) with CO2 has attracted attention because of the utilization of two major greenhouse gases (CH4 and CO2) as feedstocks and provides a route for converting them into the low H2/CO ratio syngas, which can be directly used as fuel or to produce chemicals and fuels by the methanol synthesis and Fischer–Tropsch (FT) processes.1–3 DRM is an endothermic reaction and is usually conducted at a very high operation temperature (>800 °C) to ensure high methane conversion and minimize carbon deposition thermodynamically.4,5
The majority of the catalysts investigated for DRM are generally made up of group VIII transition metals, such as Ni due to their high activity.6,7 Promoting nickel-based catalysts with various metals, such as Mg, Fe, Zr, Cr, Ce, V, Mo, Rh, Pt, Pd, and Ru, is the most widely practiced approach for modifying DRM catalysts.7–10 In particular Pt, Rh, and Ru are highly active towards DRM, which enhances the stability against coke deposition as compared to the other non-promoted nickel-based catalysts.11,12 The catalyst performance is dependent on the Ni/metal ratio and the nature of the support.6,13,14 Both the promoters and support play important roles in metal electron transfer, cluster stabilization, and reducibility.15 In particular, it has been shown that Ni catalysts are highly reducible in the presence of noble metals, which enables both methane combustion and reforming to occur simultaneously, thereby resulting in higher energy efficiency and improved catalytic activity.4 Although noble metals are much more resistant to carbon deposition than other metal-based catalysts, they are generally uneconomical. Developing bimetallic catalysts by combining nickel with other metals is an alternative route to obtaining highly coke-resistant Ni-based catalysts for the DRM reaction.16 Several studies have been dedicated to improving the Ni-based CeO2-Al2O3 performance and stability through the addition of second metal promoters, such as Co, Pd, and Pt. It has been confirmed that adding a trace of transition metals can modify Ni surface properties by promoting the reducibility of Ni and thus increasing the number of active sites to achieve better catalytic performance.9,17 For example, several bimetallic catalysts such as Ni–Co, Ni–Pd and Ni–Pt with different supports (e.g., SiO2, Al2O3, CeO2, MgO, TiO2, ZrO2, H-ZSM-5) have exhibited much higher activity and carbon resistance than monometallic Ni catalysts.10,18–20
Developing bi/trimetallic catalysts is a viable and critical method for developing highly coke-resistant Ni-based DRM catalysts, and there has been tremendous research interest in this regard in recent years. Combining nickel with other metals can easily change its surface properties, resulting in improved catalytic activity; this phenomenon is known as the synergistic impact.21 Noble metal catalysts such as Pt, Ru, and Rh exhibit good performance and selectivity for DRM reactions, even though noble metals have a high stability to carbon agglomeration at high temperatures.6 However, due to their high cost and scarcity, they are not economically competitive with other transition metal-based materials. Elsayed et al.22 discovered that supported bimetallic catalysts have good activity and stable DRM reaction capabilities. To assess the stability, platinum (0.2–2 wt%), nickel (8 wt%), and magnesium (8 wt%) were immobilized onto a ceria-zirconia support. Precipitation was used to create ceria–zirconia (0.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.4) solid solutions, and the metals were loaded using the incipient wetness method. The combination of Pt with NiMg/(Ce,Zr)O2 catalysts improved low-temperature dry reforming activity when compared to the control catalysts without Ni, Mg, and Pt. Among the various supports investigated, Al2O3 and HZSM-5 possessed greater surface areas, which enhanced the Ni particle dispersion; nevertheless, their inherent acidity and the greater interactions between the metal and support led to the rapid deactivation of the catalyst due to severe coke deposition. With the excellent thermal stability of CeO2 and the high specific surface area of Al2O3, which promotes the binding of tiny metal nanoparticles while also promoting CO2 binding in the catalytic processes by generating flexible carbonate-like species,23 a combined framework of these two materials was proposed and showed enhanced catalytic performance.7,24,25 In particular, cerium oxide has been widely investigated by many researchers as a promoter and support for nickel-based catalysts due to its unique redox properties (Ce4+/Ce3+) and remarkable oxygen storage capacities for the DRM reaction.26 Fang et al.27 investigated the impact of the promotors (Ce, Zr, and Al) employed to promote Y2O3 as supports for Ni/NiO to obtain more efficient catalysts for the DRM process. The XRD and Raman data showed that all three cations were doped into the Y2O3 lattice to form a solid solution structure, resulting in supports with decreased crystallinity and enhanced surface areas. As a result, all the changed catalysts had increased reaction performance. The Ni-support interaction of the modified catalysts was improved as compared to the unmodified catalyst, resulting in improved Ni dispersion. Furthermore, the modified catalysts had increased alkalinity, which is advantageous for activating CO2 and increasing activity. Most of the published studies reported improvement in catalytic behavior, however, little is known about the nature, structure and performance of Ni–M/CeO2-Al2O3 catalysts, where M = Fe, Fe–Mg, and Fe–Mg–Pt.
:0.4) solid solutions, and the metals were loaded using the incipient wetness method. The combination of Pt with NiMg/(Ce,Zr)O2 catalysts improved low-temperature dry reforming activity when compared to the control catalysts without Ni, Mg, and Pt. Among the various supports investigated, Al2O3 and HZSM-5 possessed greater surface areas, which enhanced the Ni particle dispersion; nevertheless, their inherent acidity and the greater interactions between the metal and support led to the rapid deactivation of the catalyst due to severe coke deposition. With the excellent thermal stability of CeO2 and the high specific surface area of Al2O3, which promotes the binding of tiny metal nanoparticles while also promoting CO2 binding in the catalytic processes by generating flexible carbonate-like species,23 a combined framework of these two materials was proposed and showed enhanced catalytic performance.7,24,25 In particular, cerium oxide has been widely investigated by many researchers as a promoter and support for nickel-based catalysts due to its unique redox properties (Ce4+/Ce3+) and remarkable oxygen storage capacities for the DRM reaction.26 Fang et al.27 investigated the impact of the promotors (Ce, Zr, and Al) employed to promote Y2O3 as supports for Ni/NiO to obtain more efficient catalysts for the DRM process. The XRD and Raman data showed that all three cations were doped into the Y2O3 lattice to form a solid solution structure, resulting in supports with decreased crystallinity and enhanced surface areas. As a result, all the changed catalysts had increased reaction performance. The Ni-support interaction of the modified catalysts was improved as compared to the unmodified catalyst, resulting in improved Ni dispersion. Furthermore, the modified catalysts had increased alkalinity, which is advantageous for activating CO2 and increasing activity. Most of the published studies reported improvement in catalytic behavior, however, little is known about the nature, structure and performance of Ni–M/CeO2-Al2O3 catalysts, where M = Fe, Fe–Mg, and Fe–Mg–Pt.
The aim of this study is to improve the performance of Ni-based CeO2-Al2O3 composite catalysts by synthesizing and structurally characterizing a series of monometallic M–Ni (M = Fe), bimetallic M–Ni (M = Mg–Fe and Pt–Fe) and trimetallic M–Ni (M = Mg–Fe–Pt) supported on CeO2-Al2O3 to suppress coke formation and maintain the H2/CO ratio. The role of the Pt co-promoter in composite catalysts was studied in the DRM reaction to distinguish the catalytic effects of Pt. Among all the synthesized M–Ni/CeO2-Al2O3 catalysts, the Pt/MgFe/Ni/CeO2-Al2O3 catalyst has shown the best performance by enhancing the CH4 and CO2 conversion and selectivity toward syngas, as well as extending the catalyst life.
2. Experimental
2.1. Materials
2.1.1 Reagents. The following compounds were utilized in the manufacture of doped Ni-based catalysts and the construction of composite catalysts. The support for CeO2-Al2O3 with a CeO2 concentration of 50% on Al2O3 (Sigma-Aldrich), and metal precursors such as Ni(NO3)2·6H2O, Fe(NO3)3·9H2O, and Mg(NO3)2·6H2O were bought from Sigma-Aldrich (99.99+). Airgas supplied ultra-high pressure (UHP) H2, CO2, and CH4 gases.
2.1.2 Synthesis of the Al2O3 support. Aluminum hydroxide was precipitated from an aqueous Al(NO3)3 solution using ammonia as the precipitating agent. At a temperature of 80 °C, the precipitation process was carried out by adding ammonia to the nitrate solution until the pH of the solution changed from acidic to basic (pH = 9–10). The Al(OH)3 precipitate was aged for 24 h, then filtered and rinsed with deionized water until the solution's pH was 7. The precipitate was obtained, which was then dried at 100 °C for 1 h and calcined at 550 °C in an oxygen stream for 4 h.Cerium(III) nitrate hexahydrate was used to create the CeO2-Al2O3 support. The aqueous nitrate solution was added dropwise to the previously obtained alumina support, then, the resulting solution was aged for 24 h. After the solvent had evaporated, the solid residue was dried at 100 °C for 1 h before being calcined in air for 5 h at 550 °C. CeO2 was loaded to a 50% level.
2.1.3 Synthesis of monometallic systems. The monometallic Ni catalyst was prepared using the incipient wetness impregnation method. An aqueous solution of nickel(II) nitrate was used to deposit a nickel phase onto CeO2-Al2O3, which was then impregnated for 24 h. The catalyst was produced, dried at 100 °C, and then calcined at 550 °C for 5 h after the solvent had evaporated. The catalyst was obtained with a nominal metal concentration of 4%.
2.1.4 Synthesis of bimetallic systems. The bimetallic catalyst Fe–Ni was obtained by subsequent impregnation using an aqueous Fe(NO3)3 solution. The impregnation process was the same as previously stated. The catalyst was obtained with a nominal Fe concentration of 2%.
2.1.5 Synthesis of trimetallic systems. The trimetallic catalyst Mg–Fe–Ni was obtained by subsequent impregnation using an aqueous Mg(NO3)2 solution. The impregnation process was the same as previously stated. The catalyst was obtained with a nominal Fe concentration of 2%. The aqueous H2PtCl6·6H2O solution was impregnated into (Mg–Fe–Ni and Fe–Ni) catalysts. The impregnation procedure was similar to that reported before. The nominal Pt content in the obtained catalysts was 0.0005%. The metal loadings were set at 4 wt% nickel for Ni/CeO2-Al2O3; 4 wt% nickel, 2 wt% iron for Fe/Ni/CeO2-Al2O3; 4 wt% nickel, 2 wt% iron, and 0.005 wt% platinum for Pt/FeNi/CeO2-Al2O3; 4 wt% nickel, 2 wt% iron, and 0.5 for magnesium for MgFe/Ni/CeO2-Al2O3; 4 wt% nickel, 2 wt% iron, 0.5 for magnesium, and 0.005 wt% platinum for Pt/MgFe/Ni/CeO2-Al2O3.
2.2. Instrumental measurements
X-ray diffraction (XRD) patterns of the catalysts were obtained by a diffractometer using a PANalytical instrument operating at 30 kV and 15 mA. The XRD pattern was evaluated at a step size of 0.026° from 2θ = 5° to 90° and a rate of 2° min−1. N2 physisorption isotherm measurements were carried out in a Micromeritics 3Flex surface characterization analyzer at 77 K. Textural properties such as surface area, total pore volume, micropore volume, and average pore width were determined using Brunauer–Emmett–Teller (BET), Barrett–Joyner–Halenda (BJH), and t-plot methods, respectively. Prior to the measurements, samples were degassed at 250 °C for 6 h using a Smart VacPrep. H2-TPR measurements were carried out in a U-shaped quartz cell using a 5% vol H2/He gas with a flow rate of 50 cm3 min−1 at a heating rate of 10 °C min−1 up to 900 °C by using a Micromeritics 3Flex analyzer. Hydrogen consumption was followed by on-line mass spectroscopy (BELMass) and quantitative analysis was done by comparison of the reduction signal with the hydrogen consumption of a CuO reference. The temperature-programmed desorption of CO2 (CO2-TPD) was performed on the same Micromeritics 3Flex analyzer. Prior to adsorption measurement, all samples were initially reduced at a temperature of 200 °C in a 5% H2 in He gas mixture and held at the reduction temperature for 1 h, then cooled down to 50 °C under He. After the temperature was stabilized, the sample was exposed to 10% CO2 in He for 30 min. To remove physically bound CO2 from the surface, a flow of He (50 cm3 min−1) for 30 min at 50 °C was used. The desorption of CO2 was measured from 50 to 600 °C at a constant heating rate of 10 °C min−1. To determine the nature of surface acid sites, Fourier-transform infrared spectroscopy (FTIR) of pyridine, using a Bruker Tensor spectrophotometer, was employed to determine the types of acid sites present in the samples. All samples were activated at 450 °C for 4 h to release the moisture before the adsorption of pyridine and cooled down to 60 °C for pyridine adsorption until saturation. All the measured spectra were recalculated to a “normalized” wafer of 10 mg. For the quantitative characterization of acid sites, the bands at 1450 and 1550 cm−1 were considered to correspond to Lewis and Brønsted sites, respectively. Furthermore, inductively coupled plasma mass spectrometry (ICP-MS) analyses were used to obtain the chemical composition of the surfaces and bulk before reaction.
2.3. Catalytic tests
Catalyst tests were carried out in a stainless-steel packed-bed reactor with an internal diameter of 10 mm and a length of 300 mm, as depicted in Scheme 1. The feed gas consisted of either pure CH4 or 50% CH4/CO2 and its flow rate was controlled by a digital mass flow controller (MFC, Brocks Instrument) towards the reaction zones. For each run, about 300 mg of the sample (particle size 0.5 μm) was diluted with sand (particle size 0.5 μm) at the ratio of 1:6 and placed in the center of the reactor with quartz wool at both ends. Prior to the reaction, the catalyst was activated in situ at 500 °C in H2 flow for 1 h. Each catalyst was evaluated within a temperature range of 550–700 °C at a constant weight hourly space velocity (WHSV) of 12000 mL g−1cat h−1. The reaction temperature was controlled by embedding a type-K thermocouple inside the catalyst center. The reactions were carried out isothermally for 10 h time-on-stream. The reaction products were analyzed online every 30 min with a gas chromatograph (SRI 8610C) equipped with a flame ionized detector (GC-FID) and thermal conductivity detector (TCD) for H2, CO2, CO, H2O, and hydrocarbons. The effluent line of the reactor to the GC injector was kept at 110 °C to avoid the potential condensation of the hydrocarbons. From Table 1, the Ni, Mg, Fe, and Pt percentages were found to be very close to the theoretical values using ICP elemental analysis, which could be due to the incomplete precipitation of the nickel, magnesium, iron, and platinum metal precursors used during the co-impregnation process.
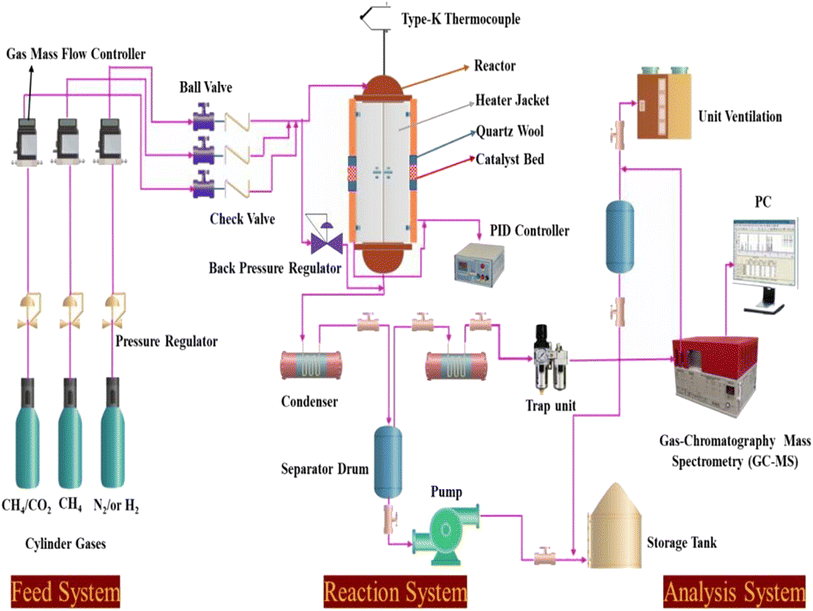 |
| | Scheme 1 Schematic diagram of the experimental setup for the dry reforming of methane. | |
Table 1 XRD and ICP analysis of the Ni-based Al2O3-CeO2 composite catalysts
| Catalysts |
Metal loadinga (wt%) |
Db (nm) |
| Al |
Ce |
Fe |
Ni |
Mg |
Pt |
| Determined by ICP analysis. Estimated by the Debye–Scherrer equation for Ni (200) of XRD. |
| Al2O3-CeO2 |
51.2 |
48.8 |
— |
— |
— |
— |
— |
| Ni/Al2O3-CeO2 |
48.1 |
48.0 |
— |
3.9 |
— |
— |
36 |
| FeNi/Al2O3-CeO2 |
47.1 |
46.8 |
2.1 |
4.0 |
— |
— |
32 |
| Pt/FeNi/Al2O3-CeO2 |
47.5 |
46.5 |
2.0 |
3.9 |
— |
0.005 |
26 |
| MgFe/Ni/Al2O3-CeO2 |
47.0 |
46.8 |
1.9 |
3.8 |
0.5 |
— |
33 |
| Pt/MgFe/Ni/Al2O3-CeO2 |
47.2 |
46.4 |
2.1 |
3.6 |
0.6 |
0.005 |
27 |
3. Results and discussion
3.1. Characterization of the catalyst
The powder X-ray diffraction patterns for the thermally calcined Ni-based Al2O3-CeO2 composite catalysts with metal additives are shown in Fig. 1. The diffraction peaks observed at 2θ = 28, 33, 48 and 57° indicate the presence of the cubic crystal structure of the CeO2 support.28 All samples displayed diffraction peaks at 2θ = 38, 45, 67°, which are attributed to the γ-Al2O3 support. Peaks were barely seen at 2θ = 23.5, 35 and 60.5°, which can be assigned to the NiO (006), NiO (009) and NiO (110) phases, respectively. XRD signatures of MgO, Fe2O3, and Pt were not observed in the Ni-based Al2O3-CeO2 composite catalysts. This could be due to the presence of a small amount of oxide and also the successful incorporation of these metals into the Ni-based Al2O3-CeO2 structure.29 As listed in Table 1, elemental analysis confirmed the presence of these metals. The average crystalline size of nickel was calculated by using the Debye–Scherrer equation (Table 1). As a function of the metal oxide(s) content, the nickel crystal size decreased from 36 to 26 nm. As a result, the crystallite size also produced oxygen vacancies, in addition to the production of oxygen vacancies mediated by dopants.30 This phenomenon could be associated with the effects of the metal oxides and nickel that remained on the surface of the sample and inhibited the growth of Ni crystals. This was confirmed by the ICP elemental analysis results shown in Table 1.
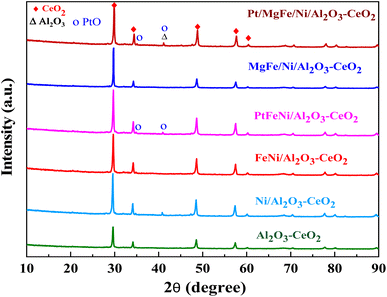 |
| | Fig. 1 XRD patterns of Ni-based Al2O3-CeO2 composite catalysts. | |
The H2-TPR profiles of the reduced Ni-based Al2O3-CeO2 composite catalysts showed four well-defined reduction peaks in the range of 550–850 °C (Fig. 2). These reduction peaks correspond to NiO incorporated within the structure of Al2O3-CeO2.31 The broad peak at 492–793 °C is associated with the bulk reduction of Ce4+ to Ce3+.32 The process of NiO reduction is thought to be a complex solid–gas reaction driven by temperature, reducing gas concentration, and the presence of additives (Mg, Fe, and Pt).33 This is consistent with the findings of Manukyan et al.34 The decrease began with the initial nucleation of Ni in the temperature range from (265 to 545 °C). During this phase, Ni nuclei appeared on the outer surface of NiO particles. This gradual process eventually resulted in the formation of small channels, which began to propagate and initiate the second step of the reduction. A dominant peak was formed between 565 and 900 °C, indicating the second stage of reduction on the majority of the NiO and metal oxide(s) particles. The small channels evolved into bulk NiO. Further Ni nucleation proceeded on the inner surface of these channels, resulting in the development of Ni nano-layers. The nano-layers then begin branching and interconnecting the network until the NiO phase was totally reduced to Ni.32,34,35 The addition of Fe and Mg metals into the Ni-based Al2O3-CeO2 catalyst modified the reduction process and caused the decay of the peak connected to unbound NiO, whereas the observation was made for the Fe2O3-doped systems. It should be noted that the presence of alkaline metal oxide(s) in the catalyst had a substantial impact on the reduction behavior of the NiO catalysts. In addition to the decrease in the temperature for the ceria reduction, promoted by the presence of the Fe2O3, the supplementary reduction of Fe2O3 was observed as previously reported by Wimmers et al.36 who studied the reduction of Fe2O3 and proposed a reduction in two steps, Fe2O3 → Fe3O4 → Fe, with no formation of FeO. For the same oxide, other authors proposed a three-step reduction process, which considered FeO formation dealing with Fe2O3 → Fe3O4 at about 400 °C, Fe3O4 → FeO at about 600 °C and finally, FeO → Fe at higher temperatures.37 Irrespective of the number of reduction steps of the Fe2O3, the separation of its reduction from that of the CeO2 overlapping reduction zones is hard to achieve. However, the Ni-based Al2O3-CeO2 catalyst without any platinum content was reduced at a much higher temperature than catalysts with platinum. The addition of platinum to Ni particles causes easily reducible NiO particles, thereby decreasing the reduction temperature because of the strong contact between Ni and Pt. Therefore, the Pt/FeNi/Al2O3-CeO2 and Pt/MgFe/Ni-based Al2O3-CeO2 catalysts allowed the reduction in the selectivity for carbon and reached values closer to equilibrium at lower reaction temperatures. This implies that the platinum interaction with the support also has a significant effect on increasing the reducibility of the support.38 Platinum helps reduce the oxide phases through its ability to facilitate dissociative hydrogen adsorption. Hydrogen has been observed to adsorb and dissociate on the surface of the platinum, whereby it spills over to the entire surface of the support.7,17 On the metallic surface, hydrogen molecules dissociate into hydrogen atoms that diffuse to the Ni2+ and can react with NiO, resulting in the uptake of the hydrogen. Nevertheless, the rate of reduction of NiO depends not only on its chemical nature but also on the nucleation process by which metallic nuclei are generated.14 Compared with FeNi/Al2O3-CeO2 and MgFe/Ni/Al2O3-CeO2 catalysts, it can be assumed that the highly dispersed and NiO in Pt/FeNi/Al2O3-CeO2 and Pt/MgFe/Ni/Al2O3-CeO2 could be more easily reduced by hydrogen atoms from the spillover effect due to the unique interactions between Ni, and Pt species and served as metallic nuclei to facilitate the reduction of Fe2+ and Ni2+. The Al2O3-CeO2 support plays a key role in the active site dispersion, activity and stability. To improve the reducibility, and enhance the oxygen mobility and metal dispersion, γ-Al2O3 material was modified by adding CeO2 and the formation of the Al2O3-CeO2 support. Further addition of metal oxide promoters improves the reducibility and chemisorption capacity of Ni-based Al2O3-CeO2 catalysts due to a better dispersion of bi/tri-metallic catalysts on the Al2O3-CeO2 support. The better interaction between the nickel particles and MOx-doped ceria supports could be associated with an anchoring effect inside the mesoporous structure of the support, as revealed by the textural characterization results (Fig. 5 and Table 3). It has been demonstrated that metal nanoparticles confined in mesoporous CeO2 enhance the metal support interaction and, therefore, the catalytic activity.39 Specifically, a stronger Ni-based Al2O3-CeO2 interaction inhibits the Ni particle agglomeration and coke deposition, which leads to better stability of the catalysts in the DRM reaction.
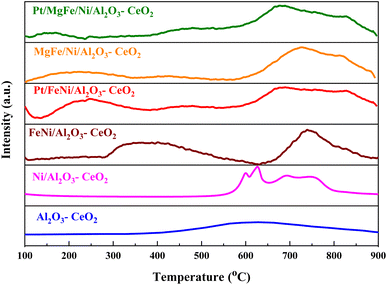 |
| | Fig. 2 H2-TPR profiles of Ni-based Al2O3-CeO2 composite catalysts. | |
The number of surface alkaline centers also has an important influence on the DRM process. According to prior research, surface alkaline sites can adsorb and activate CO2 molecules, resulting in reactive intermediates and surface oxygen species.40 The adsorbed oxygen species are active in removing the initially produced carbon deposition over time, which is beneficial for the DRM process. As a result, CO2-TPD was employed to characterize the surface alkalinity of the catalysts. The CO2-TPD results are shown in Fig. 3 and changes in the weak, moderate and strongly basic sites are presented in Table 2. The CO2-TPD profile of the samples shows weakly basic sites between 60 and 200 °C, moderately basic sites between 200 and 400 °C, and strong sites between 400 and 800 °C, respectively.27 The total amount of CO2 was estimated from the integration of the CO2-TPD peak area desorbed, which obviously increased with the addition of metal oxides as compared to Ni-based Al2O3-CeO2. Typically, it was reported that the peak below 200 °C was attributed to the desorption of CO2 on the weakly alkaline sites. Due to its easy desorption, this part of adsorbed CO2 could have a limited contribution to the reaction. The CO2 adsorbed on strongly basic sites was desorbed at high temperatures.27 As a result, the CO2 desorption peak area improved significantly at higher temperatures, indicating that the amount of adsorbed/desorbed CO2 was increased at 550 °C, which is in the range of the reaction temperature and favorable for improving the reactivity.
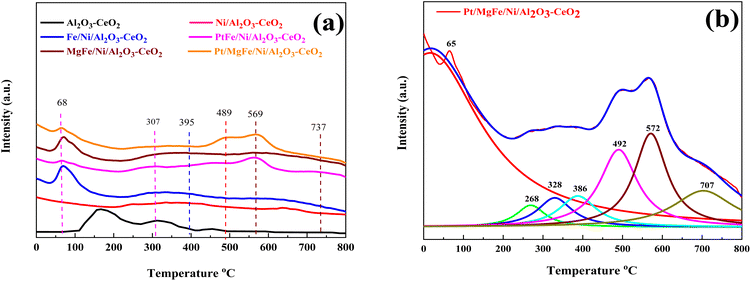 |
| | Fig. 3 CO2-TPD profiles of (a) Ni-based Al2O3-CeO2. (b) The fine structure of the Pt/MgFe/Ni/Al2O3-CeO2 composite catalysts. | |
Table 2 Summary of CO2-TPD of Ni-based Al2O3-CeO2 composite catalysts
| Catalysts |
CO2 desorptiona (mmol g−1) |
| 60–200 °C |
200–400 °C |
400–800 °C |
Total |
| The amount and strength of base sites were estimated from CO2-TPD profiles. |
| Al2O3-CeO2 |
0.11 |
0.15 |
0.09 |
0.35 |
| Ni/Al2O3-CeO2 |
— |
0.34 |
0.11 |
0.45 |
| FeNi/Al2O3-CeO2 |
0.39 |
0.08 |
0.58 |
1.04 |
| Pt/FeNi/Al2O3-CeO2 |
0.03 |
0.26 |
1.25 |
1.54 |
| MgFe/Ni/Al2O3-CeO2 |
0.32 |
0.11 |
0.71 |
1.14 |
| Pt/MgFe/Ni/Al2O3-CeO2 |
0.08 |
0.30 |
2.15 |
2.53 |
Koo et al.41 reported that besides weak basic sites, moderate basic sites, and strong basic sites are favorable for depressing the coke formation in MgO-promoted Ni catalysts. In our study, the catalysts promoted with MOx have weak, moderate and strong basic sites, whereas Ni-based Al2O3-CeO2 catalysts have only weak and moderate basic sites. Therefore, the intensity of TPD peaks became higher, indicating the improved CO2 adsorption capacity. The higher adsorption of acidic CO2 over the surfaces of the MOx-promoted catalysts confirmed that these catalysts are more basic as compared to Ni-based Al2O3-CeO2. It is well-established that the basic catalysts could improve the adsorption of CO2 during the DRM reaction, which supplies the surface oxygen to suppress the coke deposition. This finding is in agreement with previous studies.41,42 It can be concluded that the Mg2+-containing supports have an increased number of basic sites (greater amount of desorbed CO2) with respect to the support. Notably, Mg-containing zirconias showed more basic sites as compared to Ni/Al2O3-CeO2. Similar results over magnesia–zirconia oxides were reported by Moreno et al.43 Moreover, it was observed that Pt/FeNi/Al2O3-CeO2 and Pt/MgFe/Ni/Al2O3-CeO2 catalysts doped with Pt showed high basicity as compared to FeNi/Al2O3-CeO2 and MgFe/Ni/Al2O3-CeO2. This is attributed to the increased basicity of the catalysts, which in turn increased the rate of activation of mildly acidic CO2 and hence assisted in the oxidation of surface carbon and increased the catalyst resistance to deactivation.7 Compared to M–Ni tri/bimetallic catalysts, Pt-modified M–Ni tri/bimetallic catalysts greatly increased the contribution of CO2 deoxidation, and the excellent Pt deposition over M–Ni tri/bimetallic catalysts can be used to explain this, allowing oxygen to diffuse from the metal or support to the Pto in a process known as “reverse oxygen spillover”. This produces PtO and an increase in the concentration of the metal oxide(s) ions, which are the active sites for CO2 reduction.44 The PtO was detected by XRD (Fig. 1), which was proven by previous research.45 Eventually, the catalyst was provided with active oxygen species that suppressed carbon deposition, followed by catalyst deactivation.
Three desorption peaks centered around 395, 489, and 569 °C were also observed for all the catalysts despite the latter two peaks overlapping. The strength of the overlapped peaks was closely related to the addition of the metal oxides. These three peaks might be related to the strongly chemisorbed CO2. To meticulously investigate these TPD profiles, the Lorentz mathematical model was used to resolve the overlapped desorption peaks. For example, as shown in Fig. 3b, four distinct desorption peaks centered at 65, 268, 328, 386, 492, 572, and 707 °C were observed over the Pt/MgFe/Ni/Al2O3-CeO2 catalyst. This implies that more than one type of basic centers with different intensities existed in the mesoporous framework of the Pt/MgFe/Ni/Al2O3-CeO2 composite catalyst. Overall, the categories of the basic centers for Ni-based Al2O3-CeO2 catalysts were abundant due to their own structural features as well as the promotion of the metal oxides and this is in agreement with previously reported data.46
The Brønsted and Lewis sites were found via the ex situ FTIR spectra of pyridine adsorption using a Bruker Tensor spectrophotometer. The FTIR spectra (Fig. 4 and Table 2) showed the impact of the addition of metal oxides (M = Pt, Mg, and Fe) on the Ni-based Al2O3-CeO2 surface acid site. One milliliter of dried pyridine was adsorbed on 30 mg of the catalyst for 14 h. The sample was dried at 120 °C for 1 h to release the loosely adsorbed pyridine from the surface of the catalyst. From that dried sample, 10 mg was taken and, to ensure homogeneity, mixed thoroughly with 200 mg of dry KBr. The mixture was then pelletized using hydraulic pressure and the FTIR spectra of the pellets were then obtained. According to Fig. 4, an absorption band at 1440 cm−1 or 1434 cm−1, which corresponds to the pyridine coordinated on Lewis acid sites on all of these catalysts, was identified in all catalysts, indicating the interaction between the pyridine and metal oxides.47 The bands on these catalysts at 1617 cm−1 and 1596 cm−1 were attributed to pyridinium cations generated after pyridine adsorption on Brønsted acid sites and the interactions between the adsorbed pyridine and metal oxides, respectively.48,49 Fig. 4 and Table 3 demonstrate that the addition of metal oxides resulted in a decrease in the creation of Lewis and Brønsted acid sites, respectively, as compared to the Ni-based Al2O3-CeO2 catalyst. Conversely, it may be said that M–Ni-based Al2O3-CeO2 has greater Lewis basicity than Ni/Al2O3-CeO2, which is the preferred material for CO2 adsorption.50 As a result, the activity performance during the DRM is improved, and carbon deposition during formation is reduced.
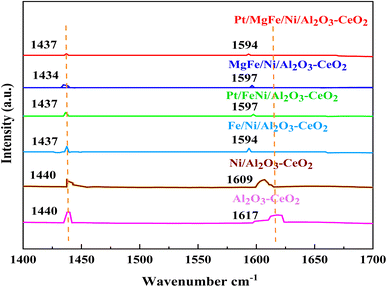 |
| | Fig. 4 Pyridine IR spectra of Ni-based Al2O3-CeO2 composite catalysts. | |
Table 3 Results of FTIR spectra of pyridine adsorption
| Catalysts |
Pyridine desorption amount (mmolPy gcat−1)a |
| 1434–1440 cm−1 |
1596–1617 cm−1 |
Total |
| The amounts were calculated by Emeis.51 |
| Al2O3-CeO2 |
12.991 |
38.069 |
51.060 |
| Ni/Al2O3-CeO2 |
11.241 |
22.215 |
33.456 |
| FeNi/Al2O3-CeO2 |
5.831 |
6.573 |
12.404 |
| Pt/FeNi/Al2O3-CeO2 |
2.302 |
1.209 |
3.511 |
| MgFe/Ni/Al2O3-CeO2 |
2.556 |
2.417 |
4.973 |
| Pt/MgFe/Ni/Al2O3-CeO2 |
1.826 |
0.854 |
2.680 |
N2 physisorption isotherms of the as-prepared samples are shown in Fig. 5 with the corresponding pore size distribution shown as inset figures. All isotherms exhibited a combination of the type IV isotherm with the H4 type hysteresis loop, associated with capillary condensation, and indicated the formation of mesoporous structures in all the Ni-based Al2O3-CeO2 composite catalysts. The BJH method was used to estimate the pore size distributions by using the adsorption branch. Metal oxides doped Ni-based Al2O2-CeO2 catalysts displayed a hierarchically bi-modal porous structure with an apex of 3.8 nm (Fig. 5b) and the other between 34 and 38 nm, both of which fall within the mesoporous range; in contrast, Ni-based Al2O2-CeO2 and Al2O2-CeO2 only presented an apex. It has been established that the presence of the porous architecture in metal oxides-doped Ni-based Al2O2-CeO2 catalysts has a favorable influence on the improvement of catalytic activity because the connected internal voids might potentially function as efficient transport channels.30,52 Table 4 summarizes the total surface area, micropore surface area, external surface area, mesopore volume pore size and diameter of Ni-based Al2O3-CeO2 composite catalysts. For comparison, the Al2O3-CeO2 powder was also measured. The surface areas of Al2O3-CeO2, Ni/Al2O3-CeO2 and Pt/MgFe/Ni/Al2O3-CeO2 were found to be 92, 86, and 76 cm2 g−1, respectively, suggesting that the addition of metal promoters reduced the total surface area. All the investigated metals influenced the textural properties of the Ni-based Al2O3-CeO2 catalyst but the extent was varied from metal to metal. This might result from the added metals physically blocking the support pores.53 The results suggest that the doped metals entered the pores of the Al2O3-CeO2 during the doping process, thereby affecting the mesoporosity and pore volume of the support.
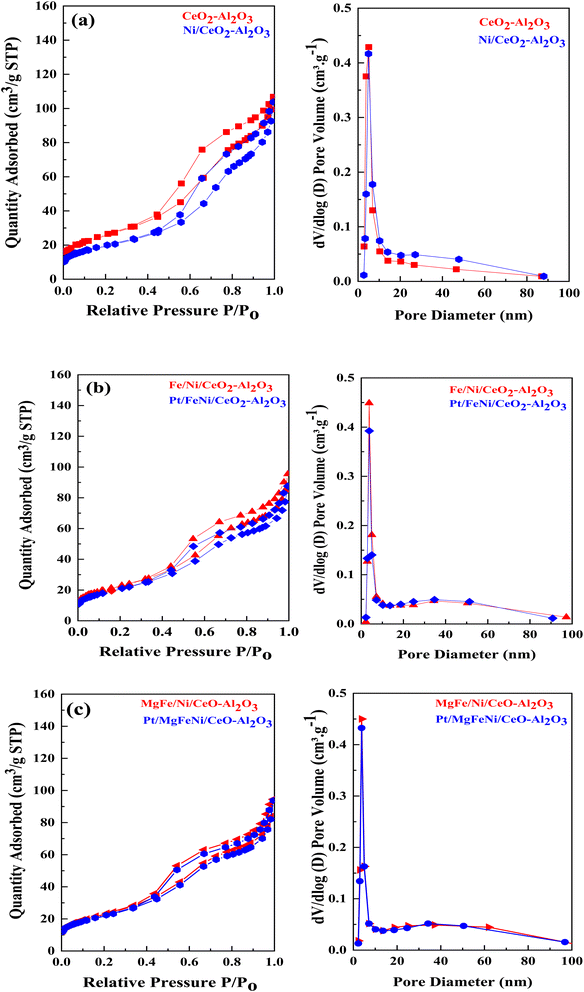 |
| | Fig. 5 ((a–c), left side) N2 physisorption isotherms and ((a–c), right side) BJH pore size distribution of of Ni-based Al2O3-CeO2 composite catalysts. | |
Table 4 Physical properties of the investigated samples obtained from the nitrogen physisorption of Ni-based Al2O3-CeO2 composite catalysts
| Catalysts |
SBETa (m2 g−1) |
Smicob (m2 g−1) |
Smesoc (m2 g−1) |
Vtotd (cm3 g−1) |
Pore sizee (nm) |
| Estimated by the Brunauer–Emmett–Teller (BET) at the p/po in the range of 0.05–0.30. Micropore area and micropore volumes were determined using the t-plot method. Estimated by BJH at the adsorbed amount at the p/po = 0.99 single point. Estimated by a t-plot. Estimated by BJH desorption average pore diameter. |
| Al2O3-CeO2 |
92 |
3 |
89 |
0.04 |
5.59 |
| Ni/Al2O3-CeO2 |
86 |
3 |
66 |
0.03 |
5.56 |
| FeNi/Al2O3-CeO2 |
82 |
1 |
80 |
0.03 |
5.49 |
| Pt/FeNi/Al2O3-CeO2 |
78 |
1 |
76 |
0.02 |
5.55 |
| MgFe/Ni/Al2O3-CeO2 |
80 |
1 |
82 |
0.03 |
5.48 |
| Pt/MgFe/Ni/Al2O3-CeO2 |
76 |
2 |
76 |
0.02 |
5.69 |
3.2. Catalytic performance in DRM
Fig. 6(A)–(C) present catalytic results of the DRM at 550–700 °C in the presence of pure CH4 and a CH4/CO2 mixture (50:50). As shown in Fig. S1 (ESI),† Tables 5 and 6, the activity and stability of the catalysts, as well as the H2/CO product ratio were measured at 550–700 °C for 600 min time-on-stream. For all catalysts, the CO2 conversion was higher than that of CH4 and the conversions of both CO2 and CH4 were enhanced with increasing reaction temperature, indicating the occurrence of side reactions such as the reverse water gas shift reaction (CO2 + H2 → CO + H2O).7,29 In the absence of CO2 feeding (pure CH4), a significant amount of CO was formed over different catalysts due to the decomposition of methane (CH4 → C + H2) and a further oxidative regeneration (C + O2 → COx) process.54 The H2/CO ratios were also increased as the reaction temperature increased because the RWGS reaction would gradually be prevented at elevated temperatures.46 The presence of CO2 promoted CH4 conversion (Fig. 6, Tables 5 and 6), catalyst stability and H2/CO ratio as compared to the catalysts in the absence of CO2 (Fig. S1†) at all reaction temperatures.
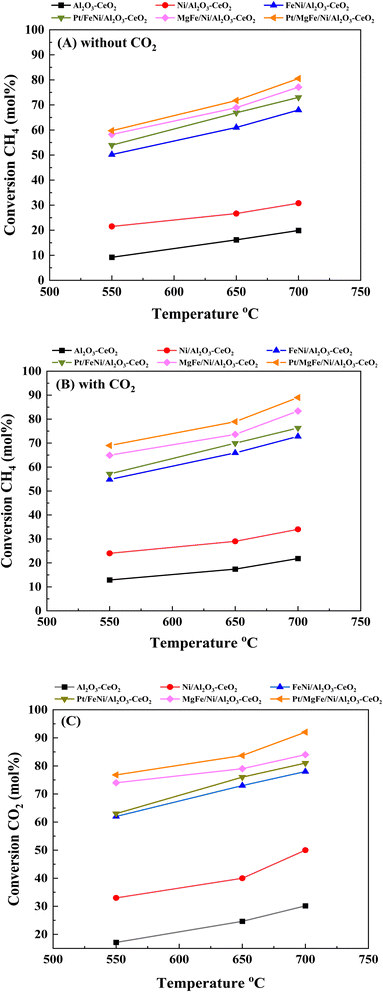 |
| | Fig. 6 Activity of catalysts for DRM: (A) CH4 conversion (without CO2); (B) CH4 conversion (with CO2) and (C) CO2 conversion. Reaction conditions: reaction temperature, 550–700 °C; P = 1 bar; feed gas, pure CH4 (without CO2) and CH4/CO2 = 50/50 (with CO2), flow rate = 60 mL min−1, WHSV = 12000 mL g−1cat h−1. | |
Table 5 Summary of the catalytic testing of Ni-based Al2O3-CeO2 composite catalysts
| Catalysts |
Reaction temp. (°C) |
Conversion (mol%) |
Selectivity (mol%) |
H2/CO |
DFa (%) |
TOFCH4 (S−1) |
TOFCO2 (S−1) |
| CH4 |
CO2 |
H2 |
CO |
| Deactivation Factor (DF) = [(final CH4 conversion − initial conversion CH4)/initial conversion of CH4] × 100. Reaction conditions: CH4/CO2 = 50/50, flow rate = 60 mL min−1, wt. cat. = 0.3 g, P = 1 bar for 10 h. |
| Al2O3-CeO2 |
550 |
13 |
17 |
48 |
77 |
0.58 |
−132 |
1.21 |
1.42 |
| 650 |
17 |
25 |
55 |
86 |
0.63 |
−99 |
1.49 |
1.55 |
| 700 |
22 |
30 |
64 |
88 |
0.73 |
−114 |
1.54 |
1.77 |
| Ni/Al2O3-CeO2 |
550 |
24 |
33 |
48 |
77 |
0.62 |
−120 |
1.93 |
2.64 |
| 650 |
29 |
40 |
55 |
86 |
0.64 |
−103 |
1.98 |
2.77 |
| 700 |
34 |
50 |
64 |
88 |
0.73 |
−111 |
2.13 |
2.92 |
| FeNi/Al2O3-CeO2 |
550 |
54 |
62 |
60 |
86 |
0.70 |
−70 |
2.43 |
2.74 |
| 650 |
65 |
73 |
71 |
90 |
0.79 |
−56 |
2.64 |
2.83 |
| 700 |
71 |
78 |
80 |
91 |
0.87 |
−49 |
2.78 |
2.97 |
| Pt/FeNi/Al2O3-CeO2 |
550 |
57 |
63 |
70 |
88 |
0.78 |
−65 |
2.76 |
2.96 |
| 650 |
69 |
76 |
75 |
90 |
0.83 |
−52 |
2.98 |
3.28 |
| 700 |
75 |
81 |
84 |
92 |
0.92 |
−46 |
3.18 |
3.52 |
| MgFe/Ni/Al2O3-CeO2 |
550 |
65 |
74 |
80 |
96 |
0.82 |
−60 |
2.94 |
3.37 |
| 650 |
74 |
79 |
85 |
94 |
0.90 |
−43 |
3.10 |
3.58 |
| 700 |
83 |
84 |
91 |
95 |
0.96 |
−40 |
3.46 |
3.89 |
| Pt/MgFe/Ni/Al2O3-CeO2 |
550 |
69 |
76 |
85 |
92 |
0.92 |
−56 |
3.33 |
3.64 |
| 650 |
79 |
83 |
87 |
96 |
0.94 |
−42 |
3.63 |
3.96 |
| 700 |
89 |
92 |
93 |
96 |
0.97 |
−36 |
3.99 |
4.46 |
Table 6 Catalytic activity from DRM over various catalystsa
| Catalysts |
DRM reaction conditions |
CH4 conv.% |
CO2 conv.% |
H2/CO |
Synthesis method |
Ref. |
| n.a. (not available). |
| Monometallic system |
| 4%Ni/Al2O3CeO2(Al/Ce = 50/50) |
700 °C, CH4/CO2 = 50/50, 12000 mL g−1 h−1, 1 bar |
≈34 |
≈50 |
≈0.73 |
Impregnation |
This work |
| 13%Ni/Al2O3CeO2(Al/Ce = 50/50) |
700 °C, CH4/CO2 = 50/50, 180000 mL g−1 h−1 |
≈44 |
≈58 |
≈0.82 |
One-pot |
65 |
| 10%Ni/Al2O3CeO2(Al/Ce = 80/20) |
700 °C, CH4/CO2 = 40/40, 90000 mL g−1 h−1, 1 bar |
≈40 |
≈50 |
≈0.78 |
Impregnation |
12 |
|
| Bi-metallic system |
| 4%Ni–Fe/Al2O3CeO2(Al/Ce = 50/50) |
700 °C, CH4/CO2 = 50/50, 12000 mL g−1 h−1, 1 bar |
≈71 |
≈78 |
≈0.87 |
Impregnation |
This work |
| 4%Ni–Mo/Al2O3CeO2(Al/Ce = 50/50) |
700 °C, CH4/CO2 = 50/50, 12000 mL g−1 h−1, 1 bar |
≈71 |
≈75 |
≈0.86 |
Impregnation |
66 |
| 20%Ni–Ru/Al2O3CeO2(Al/Ce = 95/5) |
700 °C, CH4/CO2 = 2/2, 15000 mL g−1 h−1, 1 bar |
≈88 |
≈84 |
n.a. |
Impregnation |
67 |
|
| Tri-metallic system |
| PtFe/4%Ni/Al2O3CeO2(Al/Ce = 50/50) |
700 °C, CH4/CO2 = 50/50, 12000 mL g−1 h−1, 1 bar |
≈75 |
≈81 |
≈0.92 |
Impregnation |
This work |
| MgFe/4%Ni/Al2O3CeO2(Al/Ce = 50/50) |
700 °C, CH4/CO2 = 50/50, 12000 mL g−1 h−1, 1 bar |
≈83 |
≈84 |
≈0.96 |
Impregnation |
This work |
| Pt/MgFe/4%Ni/Al2O3CeO2(Al/Ce = 50/50) |
700 °C, CH4/CO2 = 50/50, 12000 mL g−1 h−1, 1 bar |
≈89 |
≈92 |
≈0.97 |
Impregnation |
This work |
| Pt/FeMo/4%Ni/Al2O3CeO2(Al/Ce = 50/50) |
700 °C, CH4/CO2 = 50/50, 12000 mL g−1 h−1, 1 bar |
≈81 |
≈86 |
≈0.91 |
Impregnation |
66 |
| Ni–Co–Ru/MgO-Al2O3(Mg/Al = 1/4) |
760 °C, CH4/CO2 = 1/1, 111000 mL g−1 h−1, 1 bar |
≈95 |
≈90 |
n.a. |
Impregnation |
68 |
| 4%NiAuPt/AlMg(Al/Mg = 90/10) |
700 °C, CH4/CO2 = 1/1, 60000 mL g−1 h−1, 1 bar |
77.10 |
85.14 |
n.a. |
Impregnation |
69 |
| 4%NiAuPt/AlCe(Al/Ce = 90/10) |
700 °C, CH4/CO2 = 1/1, 60000 mL g−1 h−1, 1 bar |
79.06 |
86.94 |
n.a. |
Impregnation |
69 |
The catalytic stability of the catalysts was investigated over 10 h at varied temperatures (550, 650, and 700 °C) and constant mass of catalyst. Fig. S1† depicts the CH4, and CO2 conversions, as well as the H2/CO molar ratio as a function of time on-stream (TOS). These results confirmed the better performance of the metal oxide(s)-doped Ni/Al2O3-CeO2 catalyst as compared to the Ni/Al2O3-CeO2 catalyst. The results revealed that all catalysts had good activity and stability during 10 h on stream (TOS). It may be concluded that the carbon deposition on the surface of the catalyst was minimal, resulting in the stable conversion and H2/CO ratio. The small size of Ni contributes to its excellent stability. Another reason for these catalysts' high activity is the addition of metal oxides as the second active site. This finding is consistent with previous research.30,55 The DRM reaction was carried out over the Al2O3-CeO2 support (Fig. 6A–C) as a control experiment to evaluate the potential impact of Al2O3-CeO2. The H2/CO ratio was lower than the stoichiometric value for DRM (1 mol mol−1) for Al2O3-CeO2, however, it was increased by the addition of Ni and other metal promoters. As shown in Fig. 6A–C, Ni-based Al2O3-CeO2 catalysts exhibited almost two-fold the CH4 and CO2 conversions as compared the Al2O3-CeO2 support in the temperature range investigated. Previous studies have demonstrated that the high metal dispersion led to a large number of active sites and consequently, high activity.7,29 At all reaction temperatures, the Ni-based Al2O3-CeO2 catalyst performance was further increased significantly and activity follows the trend Pt/MgFe > MgFe > PtFe > FeNi for both CH4 and CO2. The doping of Fe2O3 into the Ni-based Al2O3-CeO2 catalyst resulted in an almost two-fold increase for both the CH4 and CO2 conversions (Fig. 6A–C, Tables 5 and 6). Notably, the formation of CO was higher for all Fe-doped catalysts, thereby precluding the accumulated carbon deposition on nearby Ni atoms and enhancing the catalytic activity for non-oxidative methane dehydrogenation and DRM. The redox mechanism is well-defined as the Mars-van Krevelen (MvK), which consists of two reactions: the first reaction is a reduction of the catalyst via hydrocarbon (methane), and the second is the re-oxidization reaction of the catalyst.56 Doping the FeNi/Al2O3-CeO2 catalyst with 0.005 wt% Pt further improved the catalyst stability and maintained the H2/CO ratio, probably by the initial dissociation of methane (CH4 → CH3 + H) as shown in Fig. 6A–C, Tables 5 and 6. Previous studies11,12 also confirmed that the presence of Pt sites can also initiate the reduction of NiO by the rapid dissociation of H2 and then the migration of atomic H to the NiO surface by the phenomenon of hydrogen spillover, which produces a higher mobility of hydrogen on the support surface, facilitating the access to Ni particles. The Pt/FeNi/Al2O3-CeO2 catalyst showed more resistance to carbon formation than Ni/Al2O3-CeO2 and FeNi/Al2O3-CeO2 catalysts (Fig. 6A–C, Tables 5 and 6). As noted in Table 5, these findings have been confirmed by calculating the deactivation factor and are consistent with literature reports.7 A previous study by Pawelec et al.57 demonstrated that adding 0.005%Pt to the Ni catalyst leads to the generation of nanosized NiO particles, which can be readily reduced. Based on their findings, the authors ascribed the enhancement in the performance and coke resistance over the Pt-Ni catalysts to the increase in the nickel metallic dispersion caused by the intimate contact between nickel and platinum. The effect of Mg doping on the catalytic performance of the FeNi/Al2O3-CeO2 catalyst was also examined, as illustrated in Fig. 6A–C, Tables 5 and 6. The excellent catalytic activity and long catalytic stability were observed over a MgFe/Ni/Al2O3-CeO2 catalyst. As shown in the FTIR characterization, Mg doping enhanced the Lewis basicity, which is in favor of the chemisorption of CO2 that would accelerate the reaction, CO2 + C = 2CO, thus inhibiting the carbon deposition.50,56,58 The Pt-doped MgFe/Ni/Al2O3-CeO2 catalyst showed the best performance for H2 selectivity; it performed better than the other five catalysts in the whole process, and reached 0.97 at 650 °C as shown in Fig. 6A–C, Tables 5 and 6, while both Ni/Al2O3-CeO2 and Al2O3-CeO2 catalysts showed higher CO selectivity at 550 °C. The Pt/Mg-Fe/Ni/Al2O3-CeO2 showed the highest CH4 and CO2 conversions and H2/CO values for all the temperatures, which indicated that the cooperative interaction between metals and the support could suppress the RWGS reaction. Therefore, Pt/MgFe/Ni/Al2O3-CeO2 is considered a promising candidate for the DRM reaction in terms of activity, stability and selectivity. Furthermore, as summarized in Fig. 6A–C, Tables 5 and 6, the highest specific activity was obtained, which was followed by Pt/MgFe/Ni/Al2O3-CeO2 > MgFe/Ni/Al2O3-CeO2 > PtFe/Ni/Al2O3-CeO2 > Fe/Ni/Al2O3-CeO2. Surface basicity, oxygen vacancy and redox properties are crucial for enhancing the CO2 adsorption capacity and carbonate species formation.59 The addition of metal oxide(s) and rare earth elements (Ce2O3) effectively enhance the surface basicity and redox properties of the catalysts, which further affect the CO2 adsorption capacity of the Al2O3-CeO2 catalyst. The characterization analyses and catalytic tests revealed that the introduction of metal oxide(s) into the Ni/Al2O3-CeO2 catalyst generated more coordination unsaturated Ni atoms, oxygen vacancies, defects and active sites for the DRM reaction. The existence of Pt can initiate the NiO reduction process by the rapid dissociation of H2 and migration of atomic H to the NiO surface by the hydrogen spillover phenomenon, which can retrain Ni in the metallic state under DRM conditions. Niu et al.60 demonstrated that the metal with lower electronegativity enhanced CO2 activation, which had a positive impact on the surface oxygen concentration and promoted the oxidation of the surface carbon species, thus reducing the carbon formation and improving the catalyst stability. The addition of Pt to the catalyst stabilized the size of the Ni particles and prevented Ni from becoming encapsulated in carbon. This phenomenon occurs because of nickel particles being anchored by several carbon layers that grow in multiple directions. According to García-Diéguez et al.,61 Pt addition to the Ni/Al2O3 catalyst decreases global carbon synthesis, and the C generated in tri-metallic catalysts (Pt/M–Ni/Al2O3-CeO2) appears to be less tightly attached to the Ni than in the monometallic and bi-metallic Ni/Al2O3-CeO2 catalyst. Furthermore, unlike Ni, carbon is gasified on Pt particles rather than dispersed, resulting in the constant activity of Pt sites. According to Niu et al., the electronic structure of active sites is modified in bimetallic catalysts, which affects the adsorption of specific reagents. When compared to monometallic catalysts, it reduces the activation energy of CH4 dissociation and CO2 activation. Furthermore, it increases the responsiveness of surface oxygen species, which improves carbon species suppression. The results (Table 7) show that Pt/M–Ni has lower carbon deposition than Ni due to (i) a higher energy barrier for the decomposition of the 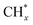 species, which
led to C* (
species, which
led to C* (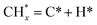 , step (5), plausible catalytic mechanism) and (ii) lower energy barriers for the oxidation of
, step (5), plausible catalytic mechanism) and (ii) lower energy barriers for the oxidation of 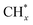 species (
species (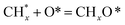 , step (17), plausible catalytic mechanism, where CHxO* is an intermediate for CO*) and C* species (C* + O* = CO* + *). As a result, the carbon concentration on the surface of Pt/M–Ni was lowered. Pt/M–Ni also weakens hydrogen dissociation, lowering the RWGS and increasing H2 generation. To better understand the influence of metal promoters on the activities of the M–Ni/Al2O3-CeO2 catalysts for the DRM, the turnover frequencies (TOFs), which reflect the intrinsic activity of the active sites, were calculated based on the Ni crystallites from XRD and the initial CH4 conversions (XCH4) at 550–700 °C, and the results are listed in Table 5. For all catalysts, the TOF was improved by increasing the reaction temperature, and also by secondary and tertiary metal doping, consistent with the variation in CH4 conversions. These observations are in agreement with the conversion and TPR results, which revealed the best catalytic activity and the highest reducibility for Ni-based catalyst-doped metal oxides. This could be ascribed to the high interaction and dispersion of metal dopants on the catalyst surface in accordance with XRD results (Table 1). The M–Ni tri/bimetallic surface modified with Pt had a lower binding energy for H* than the monometallic Ni-based Al2O3-CeO3 catalyst. This demonstrates that on Pt-modified M–Ni tri/bimetallic catalysts, the surface oxidation step leading to CO production (potentially CH* + O*) became the kinetically significant phase. The electrical modification by generating the Pt-modified M–Ni tri/bimetallic surface can be attributed to the catalyst composition's significant activation energy dependence.62 The trend of shifting the CO stretching frequency as a result of the charge transfer between Pt and Ni fits well with the tendency of variations in the methane activation energy. Additionally, the methane activation energy tends to be similar to the O* adsorption energy, but the CO2 activation energy tends to be similar to the C* adsorption energy with catalyst composition. This emphasizes the relationship between the heat of C* or O* adsorption and the apparent activation energy. The activation energy of CO2 increases as the C* adsorption energy increases, while the activation energy of CH4 increases as the O* adsorption energy increases. This agrees with earlier literature and emphasizes the relationship between the heat of C* or O* adsorption and the apparent activation energy. The activation energy of CO2 increases as the C* adsorption energy increases, while the activation energy of CH4 increases as the O* adsorption energy increases. This agrees with previous literature.63 The results show that a minimal addition of Pt could significantly reduce the energy required to activate CO2. On the surface of catalysts, this will encourage the dissociation of CO2 into CO and O. The oxidation of surface carbon and CHx, which affects the product's H2/CO ratio and the rate of coke generation, is another process that is greatly aided by the presence of O*. Tri/bimetallic Pt-modified Ni-based Al2O3-CeO3 catalyst clusters improve catalytic activity, lower the energy barrier for CO2 activation, and encourage the creation of surface O* via CO2 adsorptive dissociation while lowering the activation energy for CH4 dissociation. Surface oxygen density may be increased by the tri/bimetallic Pt-modified Ni-based Al2O3-CeO3 catalysts. This is consistent with the findings of CO2-TPD (Fig. 3). Enhancing the catalyst's resistance to the generation of coke and extending its useful life during the reaction process is advantageous. According to H2-TPR investigations (Fig. 2), the presence of Pt in the Ni particles causes easily reducible NiO particles, thereby decreasing the reduction temperature because of the strong contact between Ni and Pt. As a result, the Ni–Pt matrix (alloy phase) generated with CO2 dissociation on the metal surface is easier to remove from the surface and subsurface oxygen. Similarly, the Ni-based Al2O3-CeO3 catalyst's reducibility increased and the temperature at which the NiO species were reduced dropped after the addition of metal oxides (M = Mg and Fe). This is consistent with the findings of H2-TPR (Fig. 2). The estimated TOF numbers are consistent with the literature findings that demonstrated higher methane conversion and syngas yield at higher TOF numbers.64 In general, the coke generated by methane decomposition (step (6), plausible catalytic mechanism) and the CO disproportionation reaction (step (19), plausible catalytic mechanism) in the methane dry reforming reaction may cover the metal active sites and cause the catalyst to deactivate quickly. A 10 hours long-term durability test at 700 °C was carried out to further evaluate the potential utilization of M–Ni/Al2O3-CeO2 catalysts. The small decrease in catalytic activity seen in Fig. S1† and Table 7 could be attributed to a change in catalyst surface during the reaction process. The M–Ni/Al2O3-CeO2 catalysts exhibited strong carbon resistance over a 10 hours procedure. Fig. S1† and Table 6 indicate that the M–Ni/Al2O3-CeO2 catalysts maintained their CH4 conversion, CO2 conversion, and H2/CO ratio in the presence of minimal carbon deposition. This is due to the great dispersion of Ni on the support, which prevented Ni active site agglomeration and sintering and the strong metal-support contact, which kept the Ni nanoparticle size stable during high-temperature reactions. This is a well-established H2-TPR profile.
, step (17), plausible catalytic mechanism, where CHxO* is an intermediate for CO*) and C* species (C* + O* = CO* + *). As a result, the carbon concentration on the surface of Pt/M–Ni was lowered. Pt/M–Ni also weakens hydrogen dissociation, lowering the RWGS and increasing H2 generation. To better understand the influence of metal promoters on the activities of the M–Ni/Al2O3-CeO2 catalysts for the DRM, the turnover frequencies (TOFs), which reflect the intrinsic activity of the active sites, were calculated based on the Ni crystallites from XRD and the initial CH4 conversions (XCH4) at 550–700 °C, and the results are listed in Table 5. For all catalysts, the TOF was improved by increasing the reaction temperature, and also by secondary and tertiary metal doping, consistent with the variation in CH4 conversions. These observations are in agreement with the conversion and TPR results, which revealed the best catalytic activity and the highest reducibility for Ni-based catalyst-doped metal oxides. This could be ascribed to the high interaction and dispersion of metal dopants on the catalyst surface in accordance with XRD results (Table 1). The M–Ni tri/bimetallic surface modified with Pt had a lower binding energy for H* than the monometallic Ni-based Al2O3-CeO3 catalyst. This demonstrates that on Pt-modified M–Ni tri/bimetallic catalysts, the surface oxidation step leading to CO production (potentially CH* + O*) became the kinetically significant phase. The electrical modification by generating the Pt-modified M–Ni tri/bimetallic surface can be attributed to the catalyst composition's significant activation energy dependence.62 The trend of shifting the CO stretching frequency as a result of the charge transfer between Pt and Ni fits well with the tendency of variations in the methane activation energy. Additionally, the methane activation energy tends to be similar to the O* adsorption energy, but the CO2 activation energy tends to be similar to the C* adsorption energy with catalyst composition. This emphasizes the relationship between the heat of C* or O* adsorption and the apparent activation energy. The activation energy of CO2 increases as the C* adsorption energy increases, while the activation energy of CH4 increases as the O* adsorption energy increases. This agrees with earlier literature and emphasizes the relationship between the heat of C* or O* adsorption and the apparent activation energy. The activation energy of CO2 increases as the C* adsorption energy increases, while the activation energy of CH4 increases as the O* adsorption energy increases. This agrees with previous literature.63 The results show that a minimal addition of Pt could significantly reduce the energy required to activate CO2. On the surface of catalysts, this will encourage the dissociation of CO2 into CO and O. The oxidation of surface carbon and CHx, which affects the product's H2/CO ratio and the rate of coke generation, is another process that is greatly aided by the presence of O*. Tri/bimetallic Pt-modified Ni-based Al2O3-CeO3 catalyst clusters improve catalytic activity, lower the energy barrier for CO2 activation, and encourage the creation of surface O* via CO2 adsorptive dissociation while lowering the activation energy for CH4 dissociation. Surface oxygen density may be increased by the tri/bimetallic Pt-modified Ni-based Al2O3-CeO3 catalysts. This is consistent with the findings of CO2-TPD (Fig. 3). Enhancing the catalyst's resistance to the generation of coke and extending its useful life during the reaction process is advantageous. According to H2-TPR investigations (Fig. 2), the presence of Pt in the Ni particles causes easily reducible NiO particles, thereby decreasing the reduction temperature because of the strong contact between Ni and Pt. As a result, the Ni–Pt matrix (alloy phase) generated with CO2 dissociation on the metal surface is easier to remove from the surface and subsurface oxygen. Similarly, the Ni-based Al2O3-CeO3 catalyst's reducibility increased and the temperature at which the NiO species were reduced dropped after the addition of metal oxides (M = Mg and Fe). This is consistent with the findings of H2-TPR (Fig. 2). The estimated TOF numbers are consistent with the literature findings that demonstrated higher methane conversion and syngas yield at higher TOF numbers.64 In general, the coke generated by methane decomposition (step (6), plausible catalytic mechanism) and the CO disproportionation reaction (step (19), plausible catalytic mechanism) in the methane dry reforming reaction may cover the metal active sites and cause the catalyst to deactivate quickly. A 10 hours long-term durability test at 700 °C was carried out to further evaluate the potential utilization of M–Ni/Al2O3-CeO2 catalysts. The small decrease in catalytic activity seen in Fig. S1† and Table 7 could be attributed to a change in catalyst surface during the reaction process. The M–Ni/Al2O3-CeO2 catalysts exhibited strong carbon resistance over a 10 hours procedure. Fig. S1† and Table 6 indicate that the M–Ni/Al2O3-CeO2 catalysts maintained their CH4 conversion, CO2 conversion, and H2/CO ratio in the presence of minimal carbon deposition. This is due to the great dispersion of Ni on the support, which prevented Ni active site agglomeration and sintering and the strong metal-support contact, which kept the Ni nanoparticle size stable during high-temperature reactions. This is a well-established H2-TPR profile.
Table 7 Summary of the weight loss of Ni-based Al2O3-CeO2 composite catalysts
| Catalyst |
Weight loss% |
| Ca |
Cb |
| Spent catalyst in the absence of CO2. Spent catalyst in the presence of CO2. |
| Al2O3-CeO2 |
4.81 |
3.38 |
| Ni/Al2O3-CeO2 |
4.01 |
2.82 |
| Fe/Ni/Al2O3-CeO2 |
2.94 |
1.97 |
| PtFe/Ni/Al2O3-CeO2 |
1.6 |
1.112 |
| MgFe/Ni/Al2O3-CeO2 |
1.2 |
0.79 |
| Pt/MgFe/Ni/Al2O3-CeO2 |
0.93 |
0.39 |
3.3. Plausible catalytic mechanism
Metal oxide(s) as promoters are crucial in the development of extremely effective DRM catalysts. All the data show that the additions had a positive effect on the surface basicity, redox characteristics, and dispersion of Ni particles, and knowing these favorable qualities can help to clarify the catalytic mechanism. The activation of CH4 and CO2 is commonly regarded as a critical step.70 As illustrated in Fig. 7, CH4 is activated over the Ni sites, and CO2 can be adsorbed and activated on the support surface, and the metal oxide and support interface. As demonstrated in CO2-TPD profiles, increasing the basicity of catalysts can increase the rate of CO2 activation, which has a major impact on catalytic performance (Fig. 3 and Table 2). According to our findings, the addition of metal oxides can effectively increase the surface basicity of the catalysts, thus improving the CO2 adsorption capacity of metal oxides (M = Pt, Mg, and Fe)-doped Ni/Al2O3-CeO3 catalysts. This is consistent with the in situ DRIFTs analysis via Dengsong et al.71 The adsorbed CO2 can form two kinds of carbonate species on the catalyst surface: bidentate carbonates and monodentate carbonates. Meanwhile, the active intermediate CHx can react with these generated carbonates; bidentate carbonates, in particular, are more suitable for CHx conversion. Furthermore, the produced carbonates can efficiently react with the deposited carbon; therefore the presence of the carbonates may assist in the removal of the deposited carbon, resulting in increased catalytic stability. The redox property and its effect on oxygen vacancy are crucial for improving the catalytic performance. Due to the coexistence of redox pairs, the Pt, Mg, Fe, and Ce-modified catalysts showed enhanced redox properties, thereby resulting in abundant oxygen vacancies among the M–Ni/Al2O3-CeO3 catalysts as shown in the H2-TPR profiles (Fig. 2). The abundant oxygen vacancies can provide additional active oxygen and more active sites for CO2 and CH4 activation. Active oxygen species played an essential role in reducing carbon deposition. Because surface active oxygen species can react with deposited carbon, this helps to prevent catalyst deactivation during the DRM process. Furthermore, the improved redox characteristics can enable electron transport, which can increase the rate of CH4/CO2 conversion as well as the elimination of deposited carbon. Since metal oxides were incorporated into Ni-based catalysts, the homogeneous dispersion of Ni species had a good effect on the anti-coking behavior, as shown in Table 1. The highly dispersed and small Ni nanoparticles could effectively inhibit the carbon nucleation and the subsequent growth, playing an important role in suppressing the coke formation. However, the hypothesized mechanisms for bi- and tri-metallic Ni-based catalysts are shown below for the DRM. The reaction mechanisms postulated are based on relevant compiled literature data, given as the following steps (1)–(23):| |
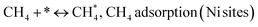 | (1) |
| |
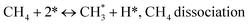 | (2) |
| |
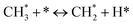 | (3) |
| |
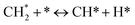 | (4) |
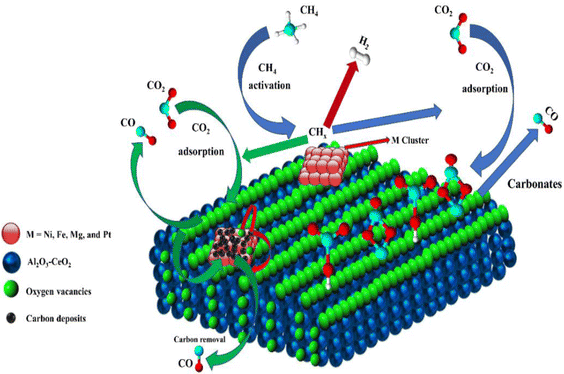 |
| | Fig. 7 Schematic representation of a plausible reaction over the (M = Pt, Mg, and Fe) doped Ni/Al2O3-CeO3 catalysts. | |
Gathering steps (2)–(5) gives step (6):
| |
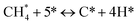 | (6) |
| |
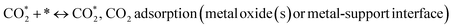 | (7) |
| |
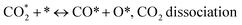 | (8) |
| |
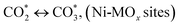 | (9) |
| |
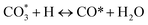 | (10) |
| |
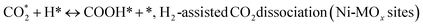 | (11) |
| | |
COOH* + * ↔ HCO* + O* (Ni-MOx sites)
| (12) |
| | |
COOH* + * ↔ CO* + OH*, (Ni-MOx sites)
| (13) |
| | |
HCO* + * ↔ +CO* + H*, (Ni-MOx sites)
| (14) |
| |
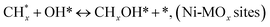 | (15) |
| | |
CHxOH* + (x + 1)* ↔ CO* + (x + 1)H*, (Ni-MOx sites)
| (16) |
| |
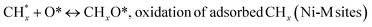 | (17) |
| | |
CHxO* + x* ↔ CO* + xH*, (Ni-MOx sites)
| (18) |
| | |
C* + OLT ↔ CO + OLT + *, (Ni-MOx sites)
| (19) |
| |
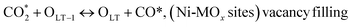 | (20) |
| | |
H2 + Ox ↔ OLT−1 + H2O, (Ni-MOx sites)
| (21) |
| |
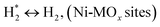 | (22) |
| | |
CO* ↔ CO, (Ni-MOx sites)
| (23) |
(*) and OLT represent unoccupied active sites, and lattice oxygen on the metal oxide surface, respectively. According to the above analysis, CH4 dissociative adsorption occurs on the Ni metal surface (steps (2)–(5)), while CO2 is adsorbed on the surface of the catalysts in the form of the carbonate species (step (7)). As the carbonate species encounter the methane pyrolysis products, they degrade fast into formate species, which subsequently decompose further into CO (step (9) and step (10)). CO2 reacts with H* generated by methane dehydrogenation to yield COOH* or HCOO* (step (11)). COOH* is subsequently decomposed into COH* and O* (step (12)) or CO* and OH* (step (13)) before COH* dehydrogenation to produce CO* and H* (step (14)). Adsorbed CHx is oxidized via OH* groups to create CHxOH* (step (15)), which is then dissociated into CO* and H* (step (16)). The lattice oxygen, produced by the dissociation of CO2, and the facile movement of oxygen could be mobilized to the nearby Ni nanoparticles and then react with 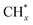 species to give CHxO* (step (17)) and then decompose into CO* and H* (step (18)), eliminating carbon deposition and preventing catalyst deactivation. Both HCO* decomposition and
species to give CHxO* (step (17)) and then decompose into CO* and H* (step (18)), eliminating carbon deposition and preventing catalyst deactivation. Both HCO* decomposition and 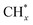 interaction with O* result in the creation of syngas. Reaction step (19) represents solid carbon, C*, primarily from the CH4 molecular dissociation, which may be converted to CO via reacting with the lattice oxygen coming either from the CeAlO3 support or from metal oxide phases (M = Pt, Mg, and Fe)-doped Ni/Al2O3-CeO3 catalysts. Reaction step (20) represents the oxidation of the reduced CeO2-Al2O3 sites (OLT-1) by reaction with CO2 molecules, and step (21) represents the reduction via
interaction with O* result in the creation of syngas. Reaction step (19) represents solid carbon, C*, primarily from the CH4 molecular dissociation, which may be converted to CO via reacting with the lattice oxygen coming either from the CeAlO3 support or from metal oxide phases (M = Pt, Mg, and Fe)-doped Ni/Al2O3-CeO3 catalysts. Reaction step (20) represents the oxidation of the reduced CeO2-Al2O3 sites (OLT-1) by reaction with CO2 molecules, and step (21) represents the reduction via 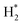 . Finally,
. Finally, 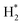 and CO* are desorbed on the catalyst to form H2(g) and CO(g) (step (22) and (23)). Fan et al.72 postulated a different reaction mechanism for catalysts with basic supports, such as CeO2, in which CO2 activation occurs on the surface of the support rather than the metal active site. In this mechanism, CO2 is adsorbed in the vicinity of the metal particles on the catalyst support, CO2 (g) = CO2 (support), resulting in the carbonate species, CO2 (support) + O2− = CO32− (support). The hydrogen is then used to reduce the carbonate (CO32− (support) + 2H = HCO2− (support) + OH−) to produce CO.73 According to the theory, CO2 is adsorbed on promoters like Ce or CeO and dissociates into CO and O (step (5)). Following this, carbon from the decomposition of CH4 is deposited on the catalyst's active sites where it reacts with the adsorbed oxygen on the promoter to generate CO (O* + C* → CO*).
and CO* are desorbed on the catalyst to form H2(g) and CO(g) (step (22) and (23)). Fan et al.72 postulated a different reaction mechanism for catalysts with basic supports, such as CeO2, in which CO2 activation occurs on the surface of the support rather than the metal active site. In this mechanism, CO2 is adsorbed in the vicinity of the metal particles on the catalyst support, CO2 (g) = CO2 (support), resulting in the carbonate species, CO2 (support) + O2− = CO32− (support). The hydrogen is then used to reduce the carbonate (CO32− (support) + 2H = HCO2− (support) + OH−) to produce CO.73 According to the theory, CO2 is adsorbed on promoters like Ce or CeO and dissociates into CO and O (step (5)). Following this, carbon from the decomposition of CH4 is deposited on the catalyst's active sites where it reacts with the adsorbed oxygen on the promoter to generate CO (O* + C* → CO*).
4. Conclusion
Pt, Fe, and Mg were doped into Ni-based Al2O3-CeO2 composite catalysts via an impregnation method and evaluated for DRM reaction. All doped Ni-based Al2O3-CeO2 composite catalysts presented significantly higher activity and H2 selectivity as compared to the Ni-based Al2O3-CeO2 composite catalysts. Characterization results showed that compared to unmodified catalysts, the catalysts treated with metal oxide (Mg, Fe, and Pt) particles had a significant number of basic sites for enhancing CO2 adsorption. The most stable and efficient conversion of CO2 and CH4 was achieved by the modified catalysts because they displayed the highest Ni particle dispersion, which can effectively stop the migration of Ni species. Additionally, as shown by the H2-TPR profile, the co-doped catalysts displayed high levels of redox cycling, which enhanced the generated oxygen vacancies and helped eliminate carbon deposition. Although all metal-doped Ni-based Al2O3-CeO2 catalysts showed slightly lower surface areas than the Ni/Al2O3-CeO2 catalyst and Al2O3-CeO2 support, they presented significantly higher activity and H2 selectivity compared to Ni-based Al2O3-CeO2 composite catalysts. Catalytic activity results revealed the impact of secondary and tertiary metal doping on the Ni-based Al2O3-CeO2 catalyst and the highest methane (>85%) and CO2 (∼90%) conversions and high selectivity towards H2/CO ratio (0.97) were obtained over the Pt/MgFe/Ni/Al2O3-CeO2 composite catalyst. The order of activity of the catalysts, based on the turnover frequencies, was Pt/MgFe/Ni/Al2O3-CeO2 > MgFe/Ni/Al2O3-CeO2 > Pt/FeNi/Al2O3-CeO2 > FeNi/Al2O3-CeO2 > Ni/Al2O3-CeO2. The observed better Pt/MgFe/Ni/Al2O3-CeO2 stability could be due to favorable changes in the distribution of surface basic sites, and better Ni dispersion.
Data availability
All data generated or analyzed during this study are included in this published article in the main manuscript and ESI.†
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors would like to thank the Higher Committee for Educational Development in Iraq (HCED) for its financial support. The authors would also like to acknowledge the Iraqi Ministry of Oil/Midland Refineries Company/AL-Daura Refinery Company for financial their support. The authors thank the Missouri university of Science and Technology for its financial support this work.
References
- P. Schwach, X. Pan and X. Bao, Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects, Chem. Rev., 2017, 117, 8497–8520, DOI:10.1021/acs.chemrev.6b00715.
- M. A. Atanga, F. Rezaei, A. Jawad, M. Fitch and A. A. Rownaghi, Oxidative Dehydrogenation of Propane to Propylene with Carbon Dioxide, Appl. Catal., B, 2018, 220, 429–445, DOI:10.1016/j.apcatb.2017.08.052.
- R. H. Saihod, Z. M. Shakoor and A. A. Jawad, Prediction of Reaction Kinetic of Al-Doura Heavy Naphtha Reforming Process Using Genetic Algorithm, Al-Khwarizmi Eng. J., 2014, 10, 47–61 Search PubMed.
- W. J. Jang, J. O. Shim, H. M. Kim, S. Y. Yoo and H. S. Roh, A Review on Dry Reforming of Methane in Aspect of Catalytic Properties, Catal. Today, 2019, 324, 15–26, DOI:10.1016/j.cattod.2018.07.032.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Homogeneous Functionalization of Methane, Chem. Rev., 2017, 117(13), 8521–8573, DOI:10.1021/acs.chemrev.6b00739.
- Q. L. M. Ha, U. Armbruster, C. Kreyenschulte, H. Atia, H. Lund, H. T. Vuong and S. Wohlrab, Stabilization of Low Nickel Content Catalysts with Lanthanum and by Citric Acid Assisted Preparation to Suppress Deactivation in Dry Reforming of Methane, Catal. Today, 2019, 334, 203–214, DOI:10.1016/j.cattod.2018.11.021.
- D. Pakhare and J. Spivey, A Review of Dry (CO 2 ) Reforming of Methane over Noble Metal Catalysts, Chem. Soc. Rev., 2014, 43, 7813–7837, 10.1039/C3CS60395D.
- S. D. Angeli, F. G. Pilitsis and A. A. Lemonidou, Methane Steam Reforming at Low Temperature: Effect of Light Alkanes' Presence on Coke Formation, Catal. Today, 2015, 242, 119–128, DOI:10.1016/j.cattod.2014.05.043.
- J. Al-Darwish, M. Senter, S. Lawson, F. Rezaei and A. A. Rownaghi, Ceria Nanostructured Catalysts for Conversion of Methanol and Carbon Dioxide to Dimethyl Carbonate, Catal. Today, 2020, 350, 120–126, DOI:10.1016/j.cattod.2019.06.013.
- M. A. Vasiliades, C. M. Damaskinos, K. K. Kyprianou, M. Kollia and A. M. Efstathiou, The Effect of Pt on the Carbon Pathways in the Dry Reforming of Methane over Ni-Pt/Ce 0.8 Pr 0.2 O 2-δ Catalyst, Catal. Today, 2020, 355, 788–803, DOI:10.1016/j.cattod.2019.04.022.
- A. A. Rownaghi and R. L. Huhnke, Producing Hydrogen-Rich Gases by Steam Reforming of Syngas Tar over CaO/MgO/NiO Catalysts, ACS Sustain. Chem. Eng., 2013, 1, 80–86 CrossRef CAS.
- I. Luisetto, S. Tuti, C. Battocchio, S. Lo Mastro and A. Sodo, Ni/CeO2-Al2O3 Catalysts for the Dry Reforming of Methane: The Effect of CeAlO3 Content and Nickel Crystallite Size on Catalytic Activity and Coke Resistance, Appl. Catal., A, 2015, 500, 12–22, DOI:10.1016/j.apcata.2015.05.004.
- Y. H. Taufiq-Yap, S. Sudarno, U. Rashid and Z. Zainal, CeO2-SiO2 Supported Nickel Catalysts for Dry Reforming of Methane toward Syngas Production, Appl. Catal., A, 2013, 468, 359–369, DOI:10.1016/j.apcata.2013.09.020.
- C. C. Chong, S. N. Bukhari, Y. W. Cheng, H. D. Setiabudi, A. A. Jalil and C. Phalakornkule, Robust Ni/Dendritic Fibrous SBA-15 (Ni/DFSBA-15) for Methane Dry Reforming: Effect of Ni Loadings, Appl. Catal., A, 2019, 584, 117174, DOI:10.1016/j.apcata.2019.117174.
- Y. He, A. Jawad, X. Li, M. Atanga, F. Rezaei and A. A. Rownaghi, Direct Aldol and Nitroaldol Condensation in an Aminosilane-Grafted Si/Zr/Ti Composite Hollow Fiber as a Heterogeneous Catalyst and Continuous-Flow Reactor, J. Catal., 2016, 341, 149–159, DOI:10.1016/j.jcat.2016.07.001.
- W. Y. Kim, J. S. Jang, E. C. Ra, K. Y. Kim, E. H. Kim and J. S. Lee, Reduced Perovskite LaNiO3 Catalysts Modified with Co and Mn for Low Coke Formation in Dry Reforming of Methane, Appl. Catal., A, 2019, 575, 198–203, DOI:10.1016/j.apcata.2019.02.029.
- C. Egawa, Methane Dry Reforming Reaction on Ru(0 0 1) Surfaces, J. Catal., 2018, 358, 35–42, DOI:10.1016/j.jcat.2017.11.010.
- T. D. Gould, M. M. Montemore, A. M. Lubers, L. D. Ellis, A. W. Weimer, J. L. Falconer and J. W. Medlin, Applied Catalysis A : General Enhanced Dry Reforming of Methane on Ni and Ni-Pt Catalysts Synthesized by Atomic Layer Deposition, Appl. Catal., A, 2015, 492, 107–116, DOI:10.1016/j.apcata.2014.11.037.
- J. Xin, H. Cui, Z. Cheng and Z. Zhou, Bimetallic Ni-Co/SBA-15 Catalysts Prepared by Urea Co-Precipitation for Dry Reforming of Methane, Appl. Catal., A, 2018, 554, 95–104, DOI:10.1016/j.apcata.2018.01.033.
- A. A. Jawad and N. J. Saleh, Studying the Effect of Both Gas Oil and Diesel Fuel on Polypropylene-Polycarbonate Reinforcement with Carbon Black, Eng. Technol. J., 2013, 31, 2039–2054, DOI:10.30684/etj.31.11A3.
- J. Niu, X. Du, J. Ran and R. Wang, Dry (CO2) Reforming of Methane over Pt Catalysts Studied by DFT and Kinetic Modeling, Appl. Surf. Sci., 2016, 376, 79–90, DOI:10.1016/j.apsusc.2016.01.212.
- N. H. Elsayed, N. R. M. Roberts, B. Joseph and J. N. Kuhn, Low Temperature Dry Reforming of Methane over Pt-Ni-Mg/Ceria-Zirconia Catalysts, Appl. Catal., B, 2015, 179, 213–219, DOI:10.1016/j.apcatb.2015.05.021.
- G. Busca, Structural, Surface, and Catalytic Properties of Aluminas, Elsevier Inc., 1st edn, 2014, vol. 57, DOI:10.1016/B978-0-12-800127-1.00003-5.
- A. Jawad and S. Ahmed, Studying the Influence of the Addition of Nano-Titanium Dioxide on the Rheological, Mechanical, Thermal, and Electrical Properties of Polycarbonate/Wood Flour, J. Thermoplast. Compos. Mater., 2023, 1–31, DOI:10.1177/08927057231162018.
- A. A. Jawad and N. Salah, Studying the Effect of Addition of Carbon Black on Rheological Properties of Polypropylene and Polycarbonate, Eng. Technol. J., 2013, 31, 976–990, DOI:10.30684/etj.31.5A13.
- B. Yan, X. Yang, S. Yao, J. Wan, M. N. Z. Myint, E. Gomez, Z. Xie, S. Kattel, W. Xu and J. G. Chen, Dry Reforming of Ethane and Butane with CO2 over PtNi/CeO2 Bimetallic Catalysts, ACS Catal., 2016, 6(11), 7283–7292, DOI:10.1021/acscatal.6b02176.
- Z. Chen, L. Mao, X. Fang, X. Xu, J. Xu and X. Wang, Methane Dry Reforming over Ni/NiO Supported on Ce-, Zr-, and Al-Modified Y2O3 for Hydrogen Production, Catalysts, 2023, 13, 1–18, DOI:10.3390/catal13020430.
- T. Odedairo, J. Chen and Z. Zhu, Metal-Support Interface of a Novel Ni-CeO2 Catalyst for Dry Reforming of Methane, Catal. Commun., 2013, 31, 25–31, DOI:10.1016/j.catcom.2012.11.008.
- J. Niu, S. E. Liland, J. Yang, K. R. Rout, J. Ran and D. Chen, Effect of Oxide Additives on the Hydrotalcite Derived Ni Catalysts for CO2 Reforming of Methane, Chem. Eng. J., 2019, 119763, DOI:10.1016/j.cej.2018.08.149.
- H. A. Lara-García, D. G. Araiza, M. Méndez-Galván, S. Tehuacanero-Cuapa, A. Gómez-Cortés and G. Díaz, Dry Reforming of Methane over Nickel Supported on Nd-Ceria: Enhancement of the Catalytic Properties and Coke Resistance, RSC Adv., 2020, 10, 33059–33070, 10.1039/d0ra05761d.
- K. Świrk, M. E. Gálvez, M. Motak, T. Grzybek, M. Rønning and P. Da Costa, Dry Reforming of Methane over Zr- and Y-Modified Ni/Mg/Al Double-Layered Hydroxides, Catal. Commun., 2018, 117, 26–32, DOI:10.1016/j.catcom.2018.08.024.
- E. le Saché, L. Pastor-Pérez, D. Watson, A. Sepúlveda-Escribano and T. R. Reina, Ni Stabilised on Inorganic Complex Structures: Superior Catalysts for Chemical CO2recycling via Dry Reforming of Methane, Appl. Catal., B, 2018, 236, 458–465, DOI:10.1016/j.apcatb.2018.05.051.
- M. N. Abu Tahari, F. Salleh, T. S. Tengku Saharuddin, N. Dzakaria, A. Samsuri, M. W. Mohamed Hisham and M. A. Yarmo, Influence of Hydrogen and Various Carbon Monoxide Concentrations on Reduction Behavior of Iron Oxide at Low Temperature, Int. J. Hydrogen Energy, 2019, 44, 20751–20759, DOI:10.1016/j.ijhydene.2018.09.186.
- K. V. Manukyan, A. G. Avetisyan, C. E. Shuck, H. A. Chatilyan, S. Rouvimov, S. L. Kharatyan and A. S. Mukasyan, Nickel Oxide Reduction by Hydrogen: Kinetics and Structural Transformations, J. Phys. Chem. C, 2015, 119, 16131–16138, DOI:10.1021/acs.jpcc.5b04313.
- A. A. Jawad, Studying the Rheological and Diffusion Properties of Polypropylene-Polymethylmethacrylate Reinforced with Bentonite Studying The Rheological and Diffusion Properties of Polypropylene-Polymethylmethacrylate Reinforced with Bentonite, Eng. Technol. J., 2014, 32, 1618–1639, DOI:10.30684/etj.32.7A.2.
- O. J. Wimmers, P. Arnoldy and J. A. Moulijn, Determination of the Reduction Mechanism by Temperature-Programmed Reduction : Application to Small Fe203 Particles, J. Phys. Chem., 1986, 90, 1331–1337, DOI:10.1021/j100398a025.
- S. Paldey, S. Gedevanishvili, W. Zhang and F. Rasouli, Evaluation of a Spinel Based Pigment System as a CO Oxidation Catalyst, Appl. Catal., B, 2005, 56, 241–250, DOI:10.1016/j.apcatb.2004.09.013.
- T. Kobayashi, T. Furuya, H. Fujitsuka and T. Tago, Synthesis of Birdcage-Type Zeolite Encapsulating Ultrafine Pt Nanoparticles and Its Application in Dry Reforming of Methane, Chem. Eng. J., 2018, 377, 120203, DOI:10.1016/j.cej.2018.10.140.
- R. Sokoll, H. Hobert and I. Schmuck, Thermal Desorption and Infrared Studies of Butylamine Adsorbed on SiO2, Al2O3 and CaO, J. Chem. Soc., Faraday Trans. 1, 1986, 82(11), 3391–3399, 10.1039/F19868203391.
- N. A. Abd Ghani, A. Azapour, A. F. Syed Muhammad and B. Abdullah, Dry Reforming of Methane for Hydrogen Production over Ni[Sbnd]Co Catalysts: Effect of Nb[Sbnd]Zr Promoters, Int. J. Hydrogen Energy, 2019, 44, 20881–20888, DOI:10.1016/j.ijhydene.2018.05.153.
- K. Y. Koo, H. S. Roh, U. H. Jung, D. J. Seo, Y. S. Seo and W. L. Yoon, Combined H2O and CO2 Reforming of CH4over Nano-Sized Ni/MgO-Al2O3 Catalysts for Synthesis Gas Production for Gas to Liquid (GTL): Effect of Mg/Al Mixed Ratio on Coke Formation, Catal. Today, 2009, 146(1–2), 166–171, DOI:10.1016/j.cattod.2009.02.002.
- A. A. Jawad, Scholars Mine’ Catalytic Utilization of Carbon Dioxide as Renewable Feedstock for Production of Chemicals and Fuels, PhD Thesis, Missouri University of Science and Technology, 2020, https://scholarsmine.mst.edu/doctoral_dissertations/2958/.
- V. García, J. J. Fernández, W. Ruíz, F. Mondragón and A. Moreno, Effect of MgO Addition on the Basicity of Ni/ZrO2and on Its Catalytic Activity in Carbon Dioxide Reforming of Methane, Catal. Commun., 2009, 11(4), 240–246, DOI:10.1016/j.catcom.2009.10.003.
- P. Jagódka, K. Matus, M. Sobota and A. Łamacz, Dry Reforming of Methane over Carbon Fibre-supported Cezro2, Ni-cezro2, Pt-cezro2 and Pt-ni-cezro2 Catalysts, Catalysts, 2021, 11(5), 1–21, DOI:10.3390/catal11050563.
- Y. Kalvachev, T. Todorova, H. Kolev, D. Merker and C. Popov, Benzene Oxidation over Pt Loaded on Fly Ash Zeolite X, Catalysts, 2023, 13(7), 1–16, DOI:10.3390/catal13071128.
- L. Xu, H. Song and L. Chou, Carbon Dioxide Reforming of Methane over Ordered Mesoporous NiO-MgO-Al2O3 Composite Oxides, Appl. Catal., B, 2011, 108–109, 177–190, DOI:10.1016/j.apcatb.2011.08.028.
- F. Benaliouche, Y. Boucheffa, P. Ayrault, S. Mignard and P. Magnoux, NH3-TPD and FTIR Spectroscopy of Pyridine Adsorption Studies for Characterization of Ag- and Cu-Exchanged X Zeolites, Microporous Mesoporous Mater., 2008, 111, 80–88, DOI:10.1016/j.micromeso.2007.07.006.
- X. Tang, J. Li, L. Sun and J. Hao, Origination of N2O from NO Reduction by NH3 over β-MnO2 and α-Mn2O3, Appl. Catal., B, 2010, 99, 156–162, DOI:10.1016/j.apcatb.2010.06.012.
- R. W. Stevens, S. S. C. Chuang and B. H. Davis, In Situ Infrared Study of Pyridine Adsorption/Desorption Dynamics over Sulfated Zirconia and Pt-Promoted Sulfated Zirconia, Appl. Catal., A, 2003, 252, 57–74, DOI:10.1016/S0926-860X(03)00375-2.
- G. R. Johnson and A. T. Bell, Effects of Lewis Acidity of Metal Oxide Promoters on the Activity and Selectivity of Co-Based Fischer-Tropsch Synthesis Catalysts, J. Catal., 2016, 338, 250–264, DOI:10.1016/j.jcat.2016.03.022.
- C. A. Emeis, Determination of Integrated Molar Extinction Coefficients for Infrared Absorption Bands of Pyridine Adsorbed on Solid Acid Catalysts, J. Catal., 1993, 141, 347–354, DOI:10.1006/jcat.1993.1145.
- J. Wang, Q. Ma, Y. Wang, Z. Li, Z. Li and Q. Yuan, New Insights into the Structure-Performance Relationships of Mesoporous Materials in Analytical Science, Chem. Soc. Rev., 2018, 47, 8766–8803, 10.1039/c8cs00658j.
- X. Li, F. Rezaei and A. Rownaghi, Methanol-to-Olefin Conversion on 3D-Printed ZSM-5 Monolith Catalysts: Effects of Metal Doping, Mesoporosity and Acid Strength, Microporous Mesoporous Mater., 2018, 276, 1–12, DOI:10.1016/j.micromeso.2018.09.016.
- B. Gao, I. Wang, L. Ren, T. Haines and J. Hu, Catalytic Performance and Reproducibility of Ni/Al2O3 and Co/Al2O3 Mesoporous Aerogel Catalysts for Methane Decomposition, Ind. Eng. Chem. Res., 2018, 58, 798–807, DOI:10.1021/acs.iecr.8b04223.
- A. Jawad, F. Rezaei and A. A. Rownaghi, Highly Efficient Pt/Mg-Fe/Ni-Based Al2O3-CeO2 Composite Catalysts for Dry-Reforming of Methane, in 17th International Congress on Catalysis Meeting, 2020, DOI:10.13140/RG.2.2.10830.43845.
- A. Jawad and S. Ahmed, Analysis and Process Evaluation of Metal Dopant (Zr, Cr)-Promoted Ga-Modified ZSM-5 for the Oxidative Dehydrogenation of Propane in the Presence and Absence of CO2, RSC Adv., 2023, 13, 11081–11095, 10.1039/D2RA08235G.
- B. Pawelec, S. Damyanova, K. Arishtirova, J. L. G. Fierro and L. Petrov, Structural and Surface Features of PtNi Catalysts for Reforming of Methane with CO2, Appl. Catal., A, 2007, 323, 188–201, DOI:10.1016/j.apcata.2007.02.017.
- V. M. Gonzalez-delacruz, F. Ternero, R. Pere, A. Caballero and J. P. Holgado, Study of Nanostructured Ni/CeO2 Catalysts Prepared by Combustion Synthesis in Dry Reforming of Methane, Appl. Catal., A, 2010, 384, 1–9, DOI:10.1016/j.apcata.2010.05.027.
- H. Ren, Q. Hao, S. Ding, Y. Zhao, M. Zhu, S. Tian, Q. Ma, W. Song, Z. Miao and Z. Liu, A High-Performance NiSiO2 Prepared by the Complexed- Impregnation Method with Citric Acid for Carbon Dioxide Reforming of Methane, Ind. Eng. Chem. Res., 2018, 57, 16257–16263, DOI:10.1021/acs.iecr.8b03897.
- J. Niu, S. E. Liland, J. Yang, K. R. Rout, J. Ran and D. Chen, Effect of Oxide Additives on the Hydrotalcite Derived Ni Catalysts for CO2 Reforming of Methane, Chem. Eng. J., 2019, 377, 1–12, DOI:10.1016/j.cej.2018.08.149.
- M. García-Diéguez, M. C. Herrera, I. S. Pieta, M. A. Larrubia and L. J. Alemany, NiBa Catalysts for CO2-Reforming of Methane, Catal. Commun., 2010, 11(14), 1133–1136, DOI:10.1016/j.catcom.2010.06.008.
- J. Niu, Y. Wang, S. E. Liland, S. K. Regli, J. Yang, K. R. Rout, J. Luo, M. Rønning, J. Ran and D. Chen, Unraveling Enhanced Activity, Selectivity, and Coke Resistance of Pt-Ni Bimetallic Clusters in Dry Reforming, ACS Catal., 2021, 11, 2398–2411, DOI:10.1021/acscatal.0c04429.
- L. M. Aparicio, Transient Isotopic Studies and Microkinetic Modeling of Methane Reforming over Nickel Catalysts, J. Catal., 1997, 165(2), 262–274, DOI:10.1006/JCAT.1997.1468.
- C. Chen, X. Wang, L. Zhang, X. Zou, W. Ding and X. Lu, Synthesis of Mesoporous
Ni–La2O3/SiO2 by Ploy(Ethylene Glycol)-Assisted Sol-Gel Route as Highly Efficient Catalysts for Dry Reforming of Methane with a H2/CO Ratio of Unity, Catal. Commun., 2017, 94, 38–41, DOI:10.1016/j.catcom.2017.02.018.
- L. Zhang, X. Wang, C. Chen, X. Zou, X. Shang, W. Ding and X. Lu, Investigation of Mesoporous NiAl2O4/MO: X (M = La, Ce, Ca, Mg)-γ-Al2O3 Nanocomposites for Dry Reforming of Methane, RSC Adv., 2017, 7, 33143–33154, 10.1039/c7ra04497f.
- A. Jawad, F. Rezaei and A. A. Rownaghi, Highly Efficient Pt/Mo-Fe/Ni-Based Al2O3-CeO2 Catalysts for Dry Reforming of Methane, Catal. Today, 2020, 350, 80–90, DOI:10.1016/j.cattod.2019.06.004.
- M. Zakrzewski, O. Shtyka, R. Ciesielski, A. Kedziora, W. Maniukiewicz, N. Arcab and T. Maniecki, Effect of Ruthenium and Cerium Oxide (IV) Promotors on the Removal of Carbon Deposit Formed during the Mixed Methane Reforming Process, Materials, 2021, 14, 1–22, DOI:10.3390/ma14247581.
- N. A. K. Aramouni, J. Zeaiter, W. Kwapinski, J. J. Leahy and M. N. Ahmad, Eclectic Trimetallic Ni–Co–Ru Catalyst for the Dry Reforming of Methane, Int. J. Hydrogen Energy, 2020, 45, 17153–17163, DOI:10.1016/j.ijhydene.2020.04.261.
- H. Wu, G. Pantaleo, V. La Parola, A. M. Venezia, X. Collard, C. Aprile and L. F. Liotta, Bi- and Trimetallic Ni Catalysts over Al2O3and Al2O3-MOx(M=Ce or Mg) Oxides for Methane Dry Reforming: Au and Pt Additive Effects, Appl. Catal., B, 2014, 156–157, 350–361, DOI:10.1016/j.apcatb.2014.03.018.
- X. Du, D. Zhang, L. Shi, R. Gao and J. Zhang, Morphology Dependence of Catalytic Properties of Ni/CeO 2 Nanostructures for Carbon Dioxide Reforming of Methane, J. Phys. Chem. C, 2012, 116, 10009–10016, DOI:10.1021/jp300543r.
- C. Yang, L. Hongrui, Z. Jianping, S. Liyi and Z. Dengsong, Promotional Effects of Rare Earth Elements (Sc, Y, Ce, and Pr) on NiMgAl Catalysts for Dry Reforming of Methane, RSC Adv., 2016, 6, 112215–112225, 10.1039/C6RA19139H.
- M.-S. Fan, A. Z. Abdullah and S. Bhatia, Catalytic Technology for Carbon Dioxide Reforming of Methane to Synthesis Gas, ChemCatChem, 2009, 1, 192–208, DOI:10.1002/cctc.200900025.
- J. Nakamura, A. Keita, S. Koichi and U. Toshio, Role of Support in Reforming of CH4 with CO2 over Rh Catalysts, Catal. Lett., 1994, 25, 265–270, DOI:10.1007/BF00816306.
Footnote |
| † Electronic supplementary information (ESI) available: Include the stability test is performed for M–Ni-based catalyst at varied temperatures (550, 650, and 700 °C) at feed containing CH4/CO2 = 50/50 for 10 h. See DOI: https://doi.org/10.1039/d3ra04809h |
|
| This journal is © The Royal Society of Chemistry 2023 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab
ab
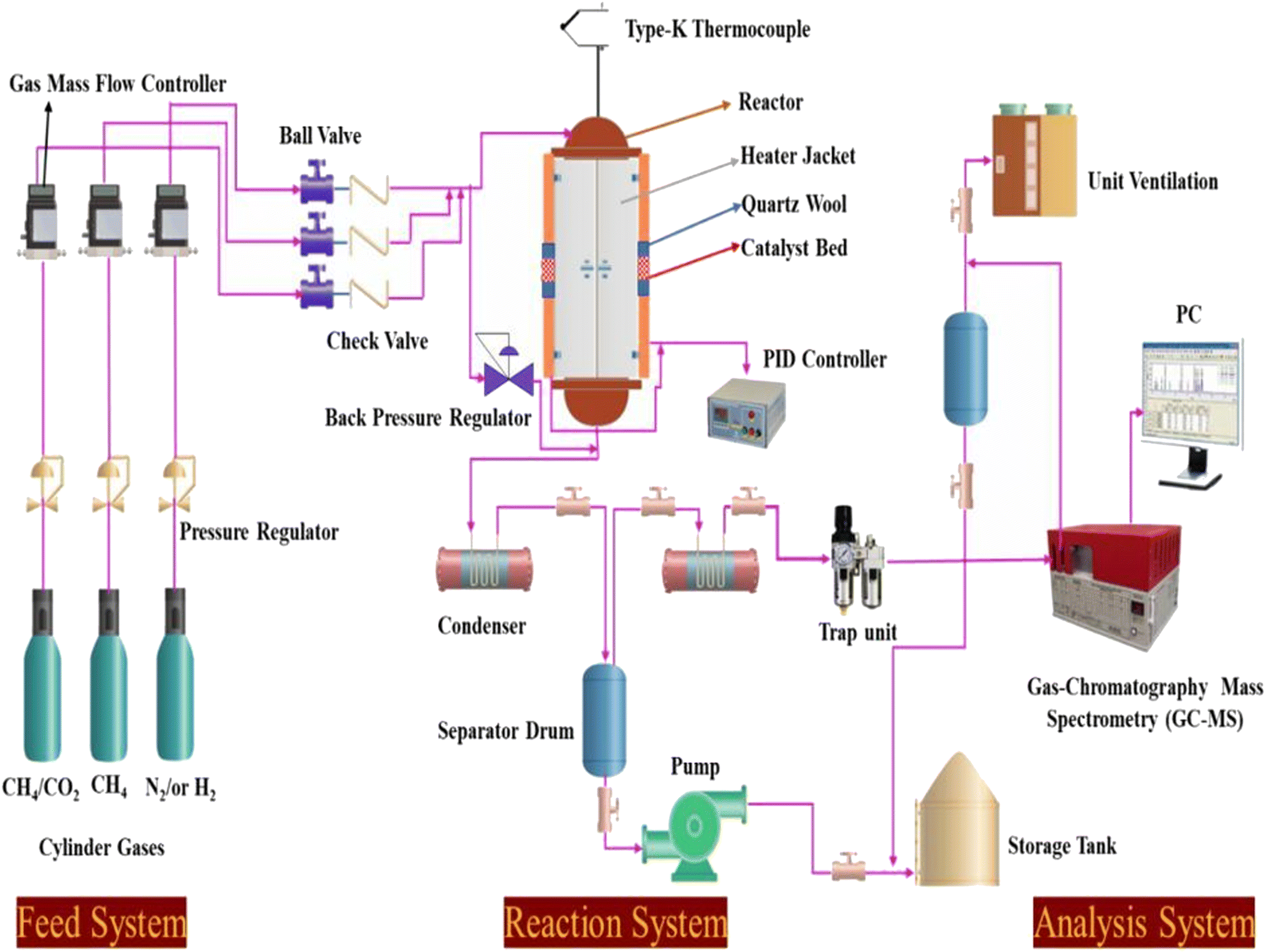
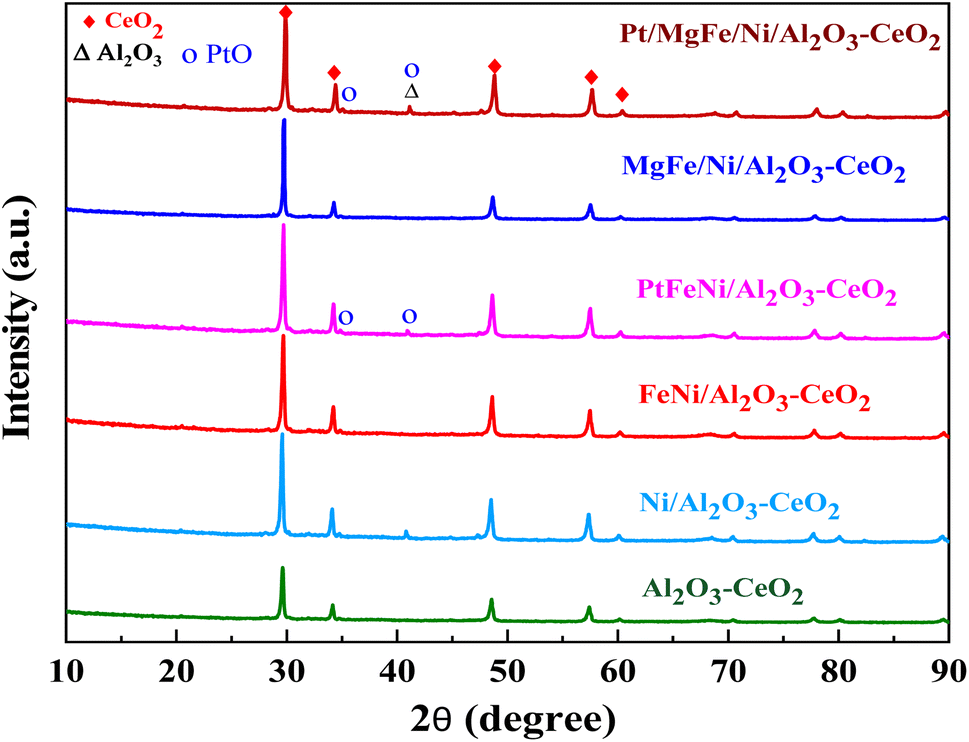
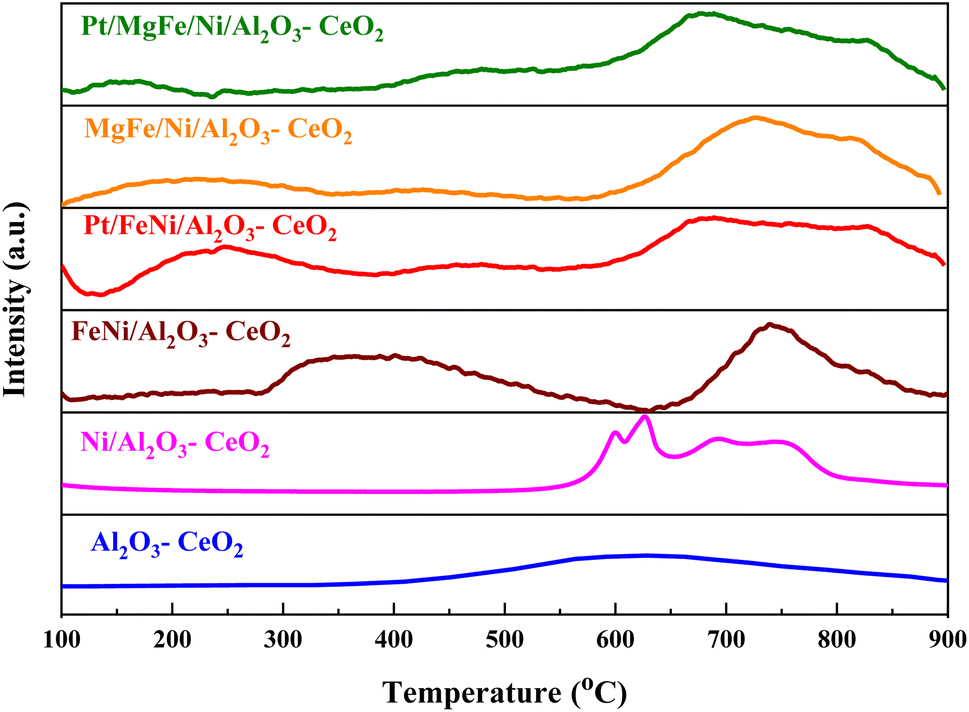
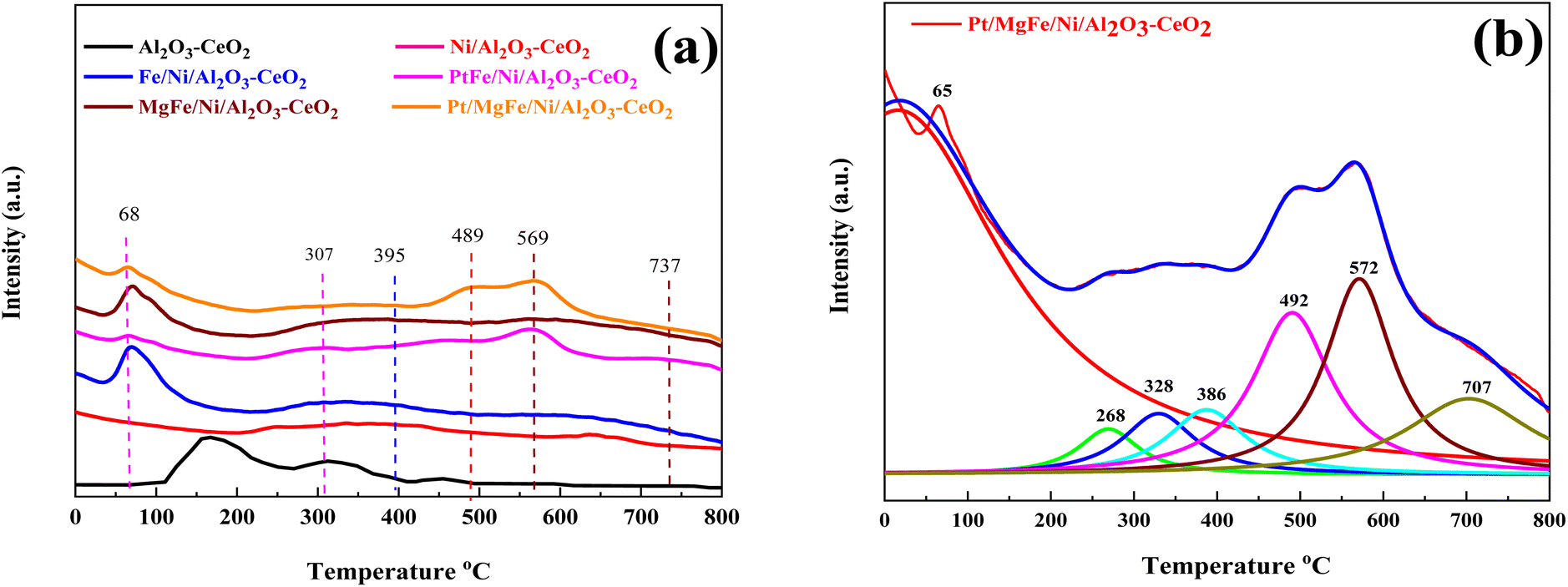
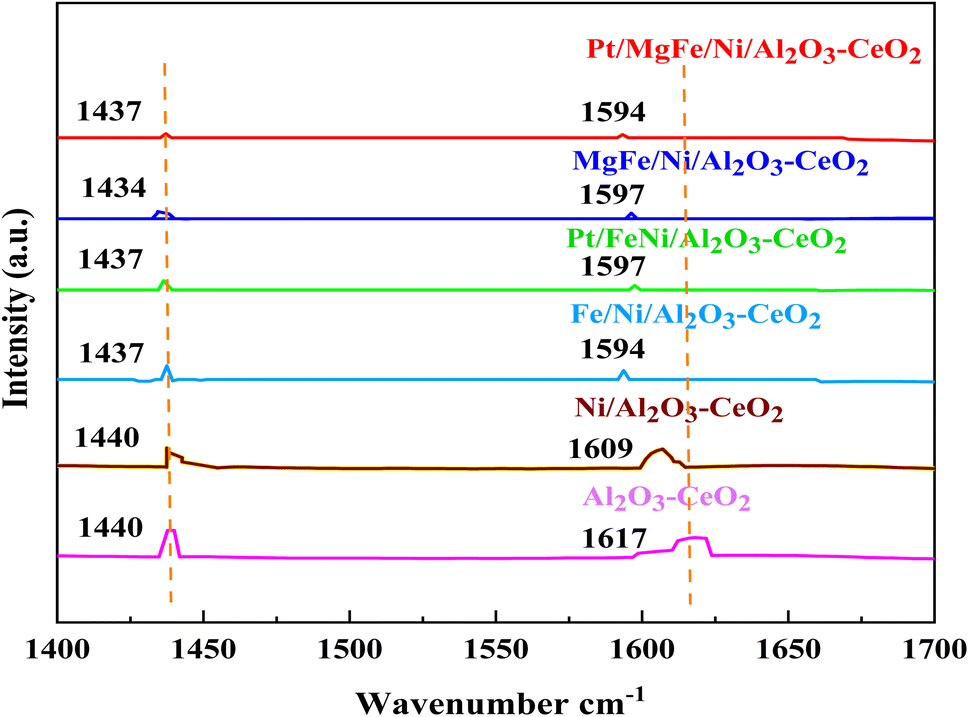
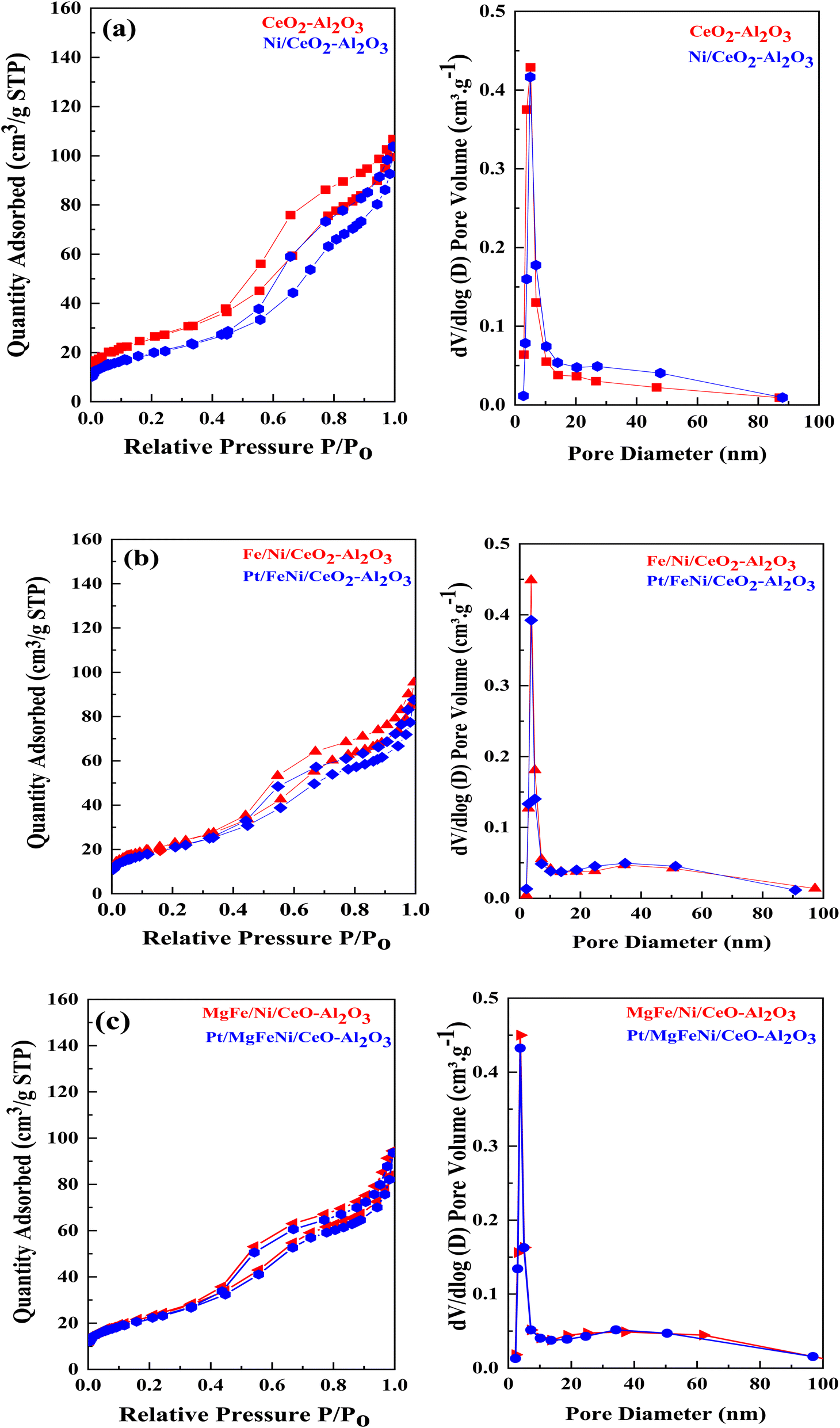
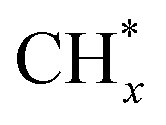 species, which
led to C* (
species, which
led to C* ( , step (5), plausible catalytic mechanism) and (ii) lower energy barriers for the oxidation of
, step (5), plausible catalytic mechanism) and (ii) lower energy barriers for the oxidation of  species (
species (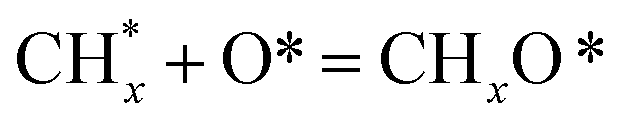 , step (17), plausible catalytic mechanism, where CHxO* is an intermediate for CO*) and C* species (C* + O* = CO* + *). As a result, the carbon concentration on the surface of Pt/M–Ni was lowered. Pt/M–Ni also weakens hydrogen dissociation, lowering the RWGS and increasing H2 generation. To better understand the influence of metal promoters on the activities of the M–Ni/Al2O3-CeO2 catalysts for the DRM, the turnover frequencies (TOFs), which reflect the intrinsic activity of the active sites, were calculated based on the Ni crystallites from XRD and the initial CH4 conversions (XCH4) at 550–700 °C, and the results are listed in Table 5. For all catalysts, the TOF was improved by increasing the reaction temperature, and also by secondary and tertiary metal doping, consistent with the variation in CH4 conversions. These observations are in agreement with the conversion and TPR results, which revealed the best catalytic activity and the highest reducibility for Ni-based catalyst-doped metal oxides. This could be ascribed to the high interaction and dispersion of metal dopants on the catalyst surface in accordance with XRD results (Table 1). The M–Ni tri/bimetallic surface modified with Pt had a lower binding energy for H* than the monometallic Ni-based Al2O3-CeO3 catalyst. This demonstrates that on Pt-modified M–Ni tri/bimetallic catalysts, the surface oxidation step leading to CO production (potentially CH* + O*) became the kinetically significant phase. The electrical modification by generating the Pt-modified M–Ni tri/bimetallic surface can be attributed to the catalyst composition's significant activation energy dependence.62 The trend of shifting the CO stretching frequency as a result of the charge transfer between Pt and Ni fits well with the tendency of variations in the methane activation energy. Additionally, the methane activation energy tends to be similar to the O* adsorption energy, but the CO2 activation energy tends to be similar to the C* adsorption energy with catalyst composition. This emphasizes the relationship between the heat of C* or O* adsorption and the apparent activation energy. The activation energy of CO2 increases as the C* adsorption energy increases, while the activation energy of CH4 increases as the O* adsorption energy increases. This agrees with earlier literature and emphasizes the relationship between the heat of C* or O* adsorption and the apparent activation energy. The activation energy of CO2 increases as the C* adsorption energy increases, while the activation energy of CH4 increases as the O* adsorption energy increases. This agrees with previous literature.63 The results show that a minimal addition of Pt could significantly reduce the energy required to activate CO2. On the surface of catalysts, this will encourage the dissociation of CO2 into CO and O. The oxidation of surface carbon and CHx, which affects the product's H2/CO ratio and the rate of coke generation, is another process that is greatly aided by the presence of O*. Tri/bimetallic Pt-modified Ni-based Al2O3-CeO3 catalyst clusters improve catalytic activity, lower the energy barrier for CO2 activation, and encourage the creation of surface O* via CO2 adsorptive dissociation while lowering the activation energy for CH4 dissociation. Surface oxygen density may be increased by the tri/bimetallic Pt-modified Ni-based Al2O3-CeO3 catalysts. This is consistent with the findings of CO2-TPD (Fig. 3). Enhancing the catalyst's resistance to the generation of coke and extending its useful life during the reaction process is advantageous. According to H2-TPR investigations (Fig. 2), the presence of Pt in the Ni particles causes easily reducible NiO particles, thereby decreasing the reduction temperature because of the strong contact between Ni and Pt. As a result, the Ni–Pt matrix (alloy phase) generated with CO2 dissociation on the metal surface is easier to remove from the surface and subsurface oxygen. Similarly, the Ni-based Al2O3-CeO3 catalyst's reducibility increased and the temperature at which the NiO species were reduced dropped after the addition of metal oxides (M = Mg and Fe). This is consistent with the findings of H2-TPR (Fig. 2). The estimated TOF numbers are consistent with the literature findings that demonstrated higher methane conversion and syngas yield at higher TOF numbers.64 In general, the coke generated by methane decomposition (step (6), plausible catalytic mechanism) and the CO disproportionation reaction (step (19), plausible catalytic mechanism) in the methane dry reforming reaction may cover the metal active sites and cause the catalyst to deactivate quickly. A 10 hours long-term durability test at 700 °C was carried out to further evaluate the potential utilization of M–Ni/Al2O3-CeO2 catalysts. The small decrease in catalytic activity seen in Fig. S1† and Table 7 could be attributed to a change in catalyst surface during the reaction process. The M–Ni/Al2O3-CeO2 catalysts exhibited strong carbon resistance over a 10 hours procedure. Fig. S1† and Table 6 indicate that the M–Ni/Al2O3-CeO2 catalysts maintained their CH4 conversion, CO2 conversion, and H2/CO ratio in the presence of minimal carbon deposition. This is due to the great dispersion of Ni on the support, which prevented Ni active site agglomeration and sintering and the strong metal-support contact, which kept the Ni nanoparticle size stable during high-temperature reactions. This is a well-established H2-TPR profile.
, step (17), plausible catalytic mechanism, where CHxO* is an intermediate for CO*) and C* species (C* + O* = CO* + *). As a result, the carbon concentration on the surface of Pt/M–Ni was lowered. Pt/M–Ni also weakens hydrogen dissociation, lowering the RWGS and increasing H2 generation. To better understand the influence of metal promoters on the activities of the M–Ni/Al2O3-CeO2 catalysts for the DRM, the turnover frequencies (TOFs), which reflect the intrinsic activity of the active sites, were calculated based on the Ni crystallites from XRD and the initial CH4 conversions (XCH4) at 550–700 °C, and the results are listed in Table 5. For all catalysts, the TOF was improved by increasing the reaction temperature, and also by secondary and tertiary metal doping, consistent with the variation in CH4 conversions. These observations are in agreement with the conversion and TPR results, which revealed the best catalytic activity and the highest reducibility for Ni-based catalyst-doped metal oxides. This could be ascribed to the high interaction and dispersion of metal dopants on the catalyst surface in accordance with XRD results (Table 1). The M–Ni tri/bimetallic surface modified with Pt had a lower binding energy for H* than the monometallic Ni-based Al2O3-CeO3 catalyst. This demonstrates that on Pt-modified M–Ni tri/bimetallic catalysts, the surface oxidation step leading to CO production (potentially CH* + O*) became the kinetically significant phase. The electrical modification by generating the Pt-modified M–Ni tri/bimetallic surface can be attributed to the catalyst composition's significant activation energy dependence.62 The trend of shifting the CO stretching frequency as a result of the charge transfer between Pt and Ni fits well with the tendency of variations in the methane activation energy. Additionally, the methane activation energy tends to be similar to the O* adsorption energy, but the CO2 activation energy tends to be similar to the C* adsorption energy with catalyst composition. This emphasizes the relationship between the heat of C* or O* adsorption and the apparent activation energy. The activation energy of CO2 increases as the C* adsorption energy increases, while the activation energy of CH4 increases as the O* adsorption energy increases. This agrees with earlier literature and emphasizes the relationship between the heat of C* or O* adsorption and the apparent activation energy. The activation energy of CO2 increases as the C* adsorption energy increases, while the activation energy of CH4 increases as the O* adsorption energy increases. This agrees with previous literature.63 The results show that a minimal addition of Pt could significantly reduce the energy required to activate CO2. On the surface of catalysts, this will encourage the dissociation of CO2 into CO and O. The oxidation of surface carbon and CHx, which affects the product's H2/CO ratio and the rate of coke generation, is another process that is greatly aided by the presence of O*. Tri/bimetallic Pt-modified Ni-based Al2O3-CeO3 catalyst clusters improve catalytic activity, lower the energy barrier for CO2 activation, and encourage the creation of surface O* via CO2 adsorptive dissociation while lowering the activation energy for CH4 dissociation. Surface oxygen density may be increased by the tri/bimetallic Pt-modified Ni-based Al2O3-CeO3 catalysts. This is consistent with the findings of CO2-TPD (Fig. 3). Enhancing the catalyst's resistance to the generation of coke and extending its useful life during the reaction process is advantageous. According to H2-TPR investigations (Fig. 2), the presence of Pt in the Ni particles causes easily reducible NiO particles, thereby decreasing the reduction temperature because of the strong contact between Ni and Pt. As a result, the Ni–Pt matrix (alloy phase) generated with CO2 dissociation on the metal surface is easier to remove from the surface and subsurface oxygen. Similarly, the Ni-based Al2O3-CeO3 catalyst's reducibility increased and the temperature at which the NiO species were reduced dropped after the addition of metal oxides (M = Mg and Fe). This is consistent with the findings of H2-TPR (Fig. 2). The estimated TOF numbers are consistent with the literature findings that demonstrated higher methane conversion and syngas yield at higher TOF numbers.64 In general, the coke generated by methane decomposition (step (6), plausible catalytic mechanism) and the CO disproportionation reaction (step (19), plausible catalytic mechanism) in the methane dry reforming reaction may cover the metal active sites and cause the catalyst to deactivate quickly. A 10 hours long-term durability test at 700 °C was carried out to further evaluate the potential utilization of M–Ni/Al2O3-CeO2 catalysts. The small decrease in catalytic activity seen in Fig. S1† and Table 7 could be attributed to a change in catalyst surface during the reaction process. The M–Ni/Al2O3-CeO2 catalysts exhibited strong carbon resistance over a 10 hours procedure. Fig. S1† and Table 6 indicate that the M–Ni/Al2O3-CeO2 catalysts maintained their CH4 conversion, CO2 conversion, and H2/CO ratio in the presence of minimal carbon deposition. This is due to the great dispersion of Ni on the support, which prevented Ni active site agglomeration and sintering and the strong metal-support contact, which kept the Ni nanoparticle size stable during high-temperature reactions. This is a well-established H2-TPR profile.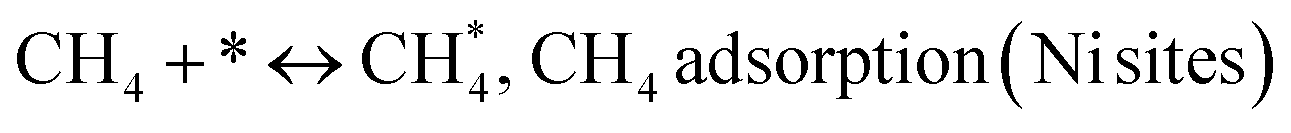
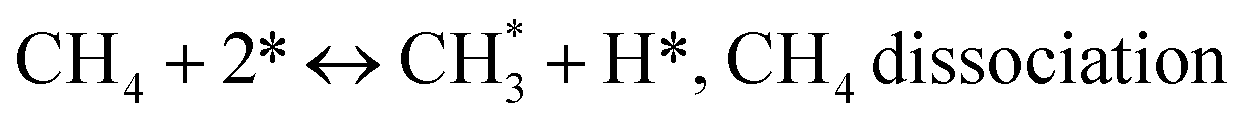
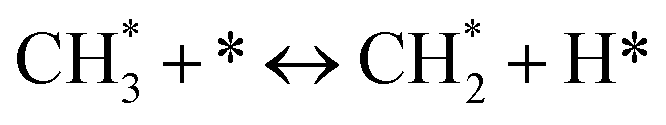
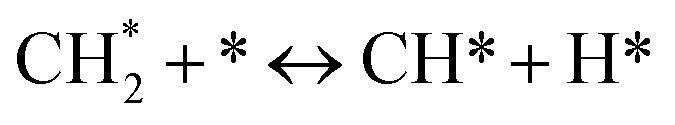
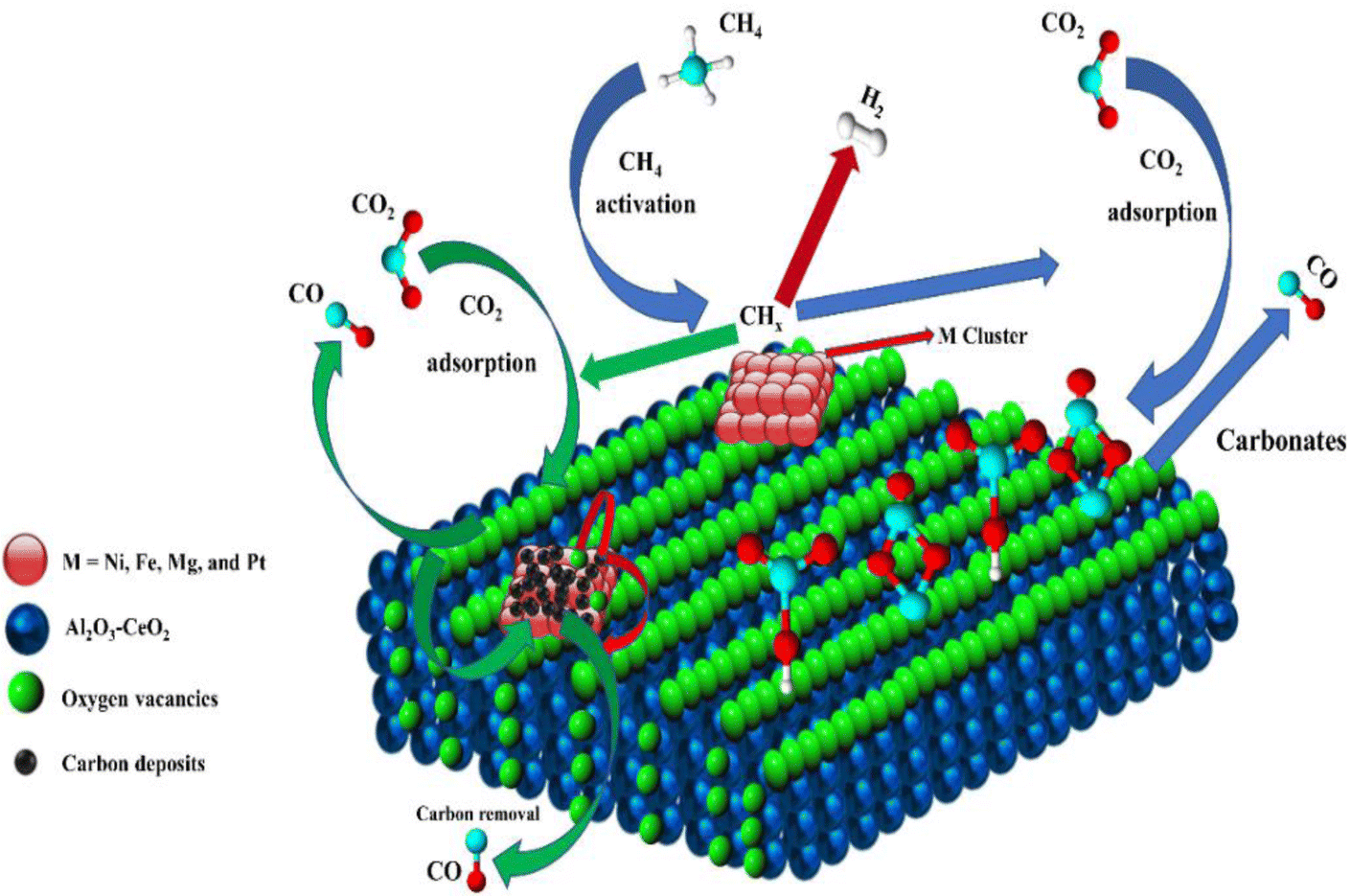
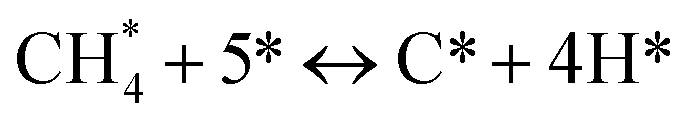

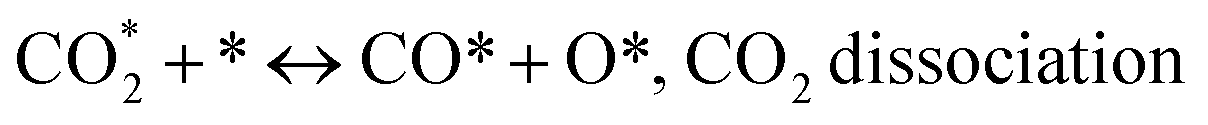
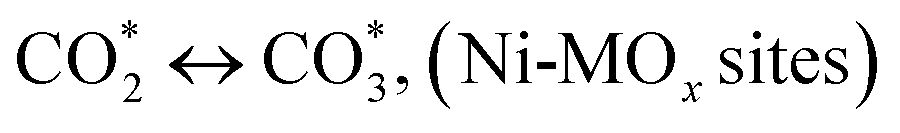
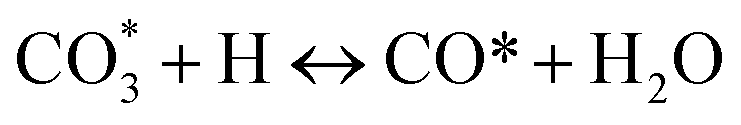
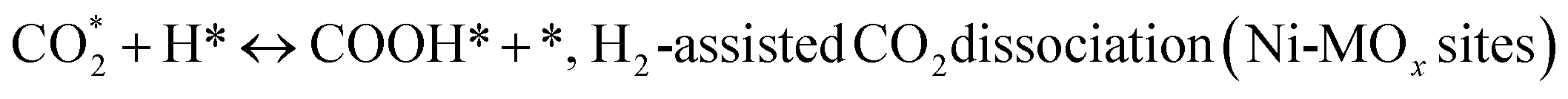
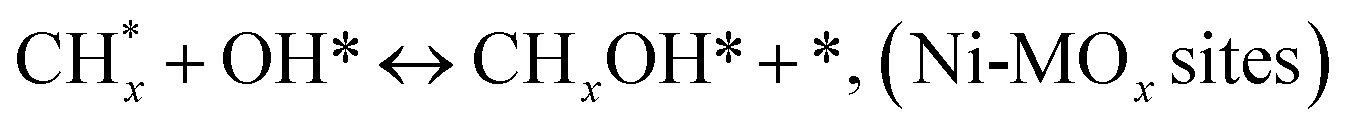
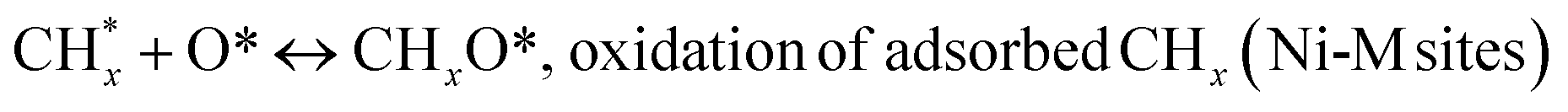
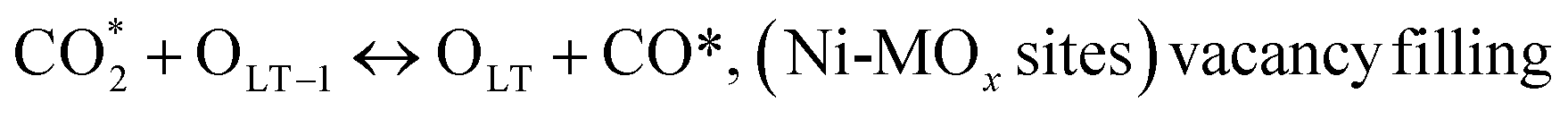
 species to give CHxO* (step (17)) and then decompose into CO* and H* (step (18)), eliminating carbon deposition and preventing catalyst deactivation. Both HCO* decomposition and
species to give CHxO* (step (17)) and then decompose into CO* and H* (step (18)), eliminating carbon deposition and preventing catalyst deactivation. Both HCO* decomposition and 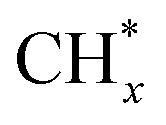 interaction with O* result in the creation of syngas. Reaction step (19) represents solid carbon, C*, primarily from the CH4 molecular dissociation, which may be converted to CO via reacting with the lattice oxygen coming either from the CeAlO3 support or from metal oxide phases (M = Pt, Mg, and Fe)-doped Ni/Al2O3-CeO3 catalysts. Reaction step (20) represents the oxidation of the reduced CeO2-Al2O3 sites (OLT-1) by reaction with CO2 molecules, and step (21) represents the reduction via
interaction with O* result in the creation of syngas. Reaction step (19) represents solid carbon, C*, primarily from the CH4 molecular dissociation, which may be converted to CO via reacting with the lattice oxygen coming either from the CeAlO3 support or from metal oxide phases (M = Pt, Mg, and Fe)-doped Ni/Al2O3-CeO3 catalysts. Reaction step (20) represents the oxidation of the reduced CeO2-Al2O3 sites (OLT-1) by reaction with CO2 molecules, and step (21) represents the reduction via 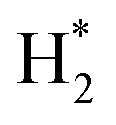 . Finally,
. Finally, 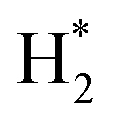 and CO* are desorbed on the catalyst to form H2(g) and CO(g) (step (22) and (23)). Fan et al.72 postulated a different reaction mechanism for catalysts with basic supports, such as CeO2, in which CO2 activation occurs on the surface of the support rather than the metal active site. In this mechanism, CO2 is adsorbed in the vicinity of the metal particles on the catalyst support, CO2 (g) = CO2 (support), resulting in the carbonate species, CO2 (support) + O2− = CO32− (support). The hydrogen is then used to reduce the carbonate (CO32− (support) + 2H = HCO2− (support) + OH−) to produce CO.73 According to the theory, CO2 is adsorbed on promoters like Ce or CeO and dissociates into CO and O (step (5)). Following this, carbon from the decomposition of CH4 is deposited on the catalyst's active sites where it reacts with the adsorbed oxygen on the promoter to generate CO (O* + C* → CO*).
and CO* are desorbed on the catalyst to form H2(g) and CO(g) (step (22) and (23)). Fan et al.72 postulated a different reaction mechanism for catalysts with basic supports, such as CeO2, in which CO2 activation occurs on the surface of the support rather than the metal active site. In this mechanism, CO2 is adsorbed in the vicinity of the metal particles on the catalyst support, CO2 (g) = CO2 (support), resulting in the carbonate species, CO2 (support) + O2− = CO32− (support). The hydrogen is then used to reduce the carbonate (CO32− (support) + 2H = HCO2− (support) + OH−) to produce CO.73 According to the theory, CO2 is adsorbed on promoters like Ce or CeO and dissociates into CO and O (step (5)). Following this, carbon from the decomposition of CH4 is deposited on the catalyst's active sites where it reacts with the adsorbed oxygen on the promoter to generate CO (O* + C* → CO*).