
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEGFR and PI3K/m-TOR inhibitors: design, microwave assisted synthesis and anticancer activity of thiazole–coumarin hybrids†
Rasha Z. Batrana,
Eman Y. Ahmed *a,
Hanem M. Awadb,
Korany A. Alic and
Nehad A. Abdel Latif*a
*a,
Hanem M. Awadb,
Korany A. Alic and
Nehad A. Abdel Latif*a
aChemistry of Natural Compounds Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Dokki, Cairo, 12622, Egypt. E-mail: eyam_ha@yahoo.com; nehad_km@yahoo.com
bTanning Materials and Leather Technology Department, National Research Centre, Dokki, Cairo, 12622, Egypt
cApplied Organic Chemistry Department, Advanced Materials and Nanotechnology Group, National Research Centre, Dokki, Cairo, 12622, Egypt
First published on 4th October 2023
Abstract
A series of thiazoline and thiazolidinone-based 4-hydroxycoumarin derivatives were synthesized using both conventional synthesis procedures and microwave-assisted techniques. The new compounds were evaluated for their cytotoxic effect against three human cancer cell lines; MCF-7, HCT-116 and HepG2 and one normal human cell line (BJ-1). The promising anti-proliferative compounds 2a, 2b, 6a and 6b were assessed for inhibiting EGFR and PI3K/mTOR. Compound 6a showed the highest inhibition activity towards the signaling pathway. The apoptotic effect and cell cycle arrest potential of derivative 6a were examined. Moreover, the molecular docking, physicochemical properties and pharmacokinetic parameters of the promising compound were investigated, as well.
1. Introduction
Cancer is a disease of a multifactorial nature that ranks as the second leading cause of death worldwide, resulting in about 10 million deaths in 2020.1 It has been noted that a number of signaling pathways when dysregulated are subsequently involved in cancer pathogenesis. The PI3K/AKT/mTOR pathway is an intracellular signaling pathway related to a number of biological processes such as apoptosis, cell proliferation and angiogenesis. Dysregulation of this pathway is associated with a vast disturbance in cell growth, survival and metastasis which results in cancer development. Somatic mutations or gaining and losing of key genes are among the factors affecting this pathway in a number of different tumors such as breast, colon and liver cancers.2–4PI3K/AKT/mTOR pathway is initially activated by the binding of extracellular growth factors to epidermal growth factor receptor (EGFR), then phosphatidylinositol 3-kinase (PI3K) is activated which consequently activates the serine/threonine protein kinase B (PKB/AKT). This leads to some downstream effects as the activation of mechanistic target of rapamycin (mTOR). Agents that target the regulators in this pathway are either single PI3K, AKT or mTOR inhibitors or dual inhibitors of PI3K and mTOR. In theory, inhibiting EGFR and PI3K/mTOR would probably lead to the strongest inhibition of the whole PI3K/Akt/mTOR pathway.5,6
Coumarin as a privileged heterocyclic ring that displays multifarious biological activities has been utilized to design useful antitumor compounds.7–12 A closer look at the coumarin chemical structure and physicochemical properties reveals that the 2H-chromen-2-one ring is planar and lipophilic, which grants the scaffold the ability to interact with a variety of biological counterparts, whereas, the lactone group of the coumarin enable the molecule to form strong polar bonds and to acylate different protein targets.13 On the other hand, the thiazole ring is a versatile moiety that demonstrated many desirable biological activities, dabrafenib is an example of a thiazole drug that underwent clinical trials for its selectivity towards tyrosine kinase inhibition.14,15 Moreover, thiazoles are reported as potent anticancer agents that are capable of targeting certain kinase pathways, e.g. compound I, Fig. 1.16
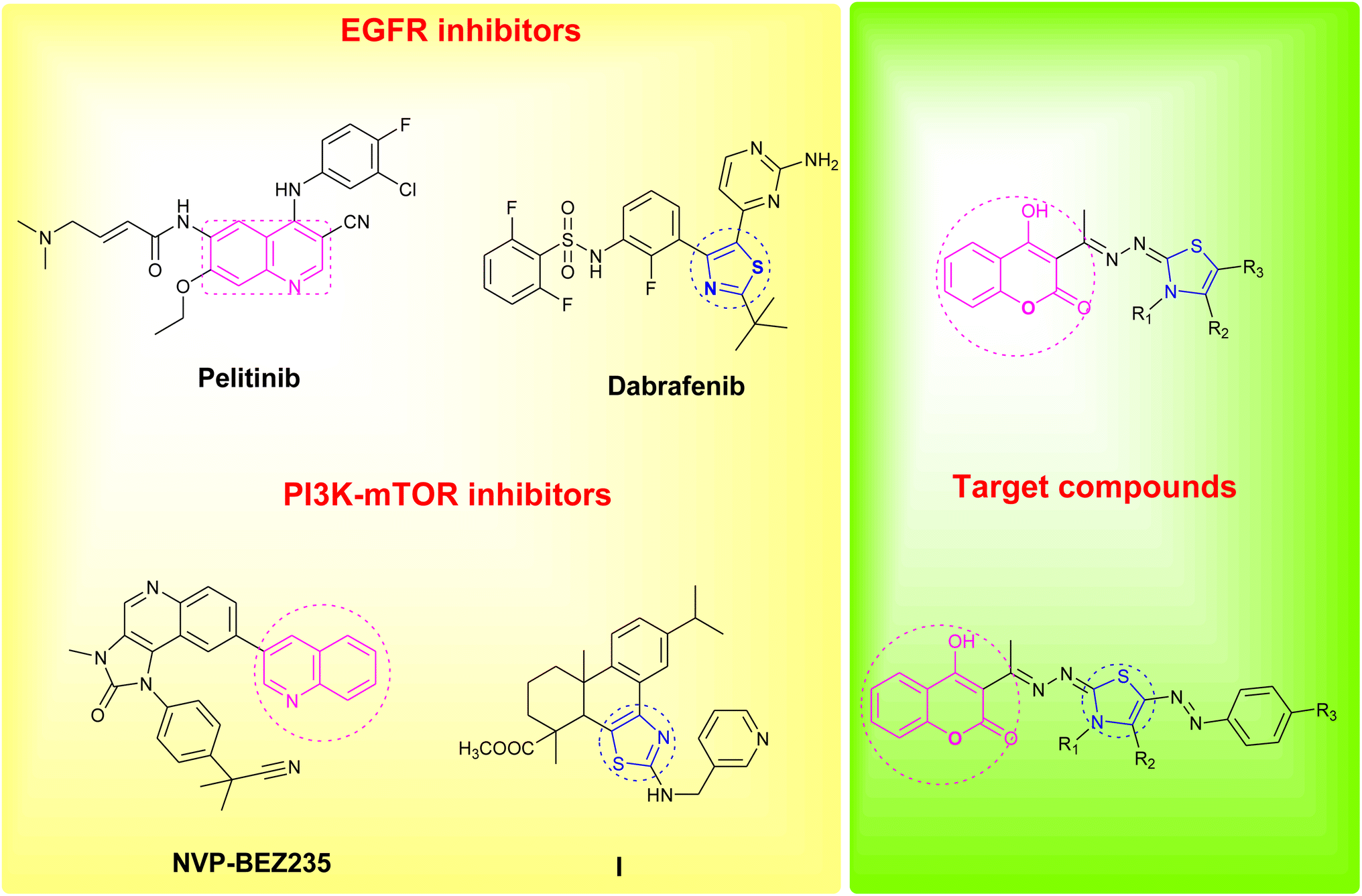 | ||
| Fig. 1 Reported EGFR and PI3K/mTOR inhibitors and design of the newly synthesized coumarin derivatives. | ||
In an attempt to pursue new antitumor agents, considering the aforementioned information, and as a continuation to our research work in cancer field,17–26 the target compounds were rationally developed by using one of the FDA approved drugs that inhibits EGFR (pelitinib) and another that is developed by Novartis and involved in PI3K/AKT/mTOR signaling pathway (NVP-BEZ235) as models, replacing their biologically active quinoline ring with its bioisostere coumarin scaffold, and hybridizing the scaffold with the bioactive thiazole moiety, Fig. 1.
2. Results and discussion
2.1. Chemistry
The target thiazolines and thiazolidinones were prepared from the intermediate thiosemicarbazones 2a–c through the reaction with different α-haloketones as outlined in Scheme 1. Thus, cyclocondensation of thiosemicarbazones 2a–c with chloroacetone and/or 3-chloroacetylacetone in ethanol using a catalytic amount of anhydrous sodium acetate yielded the corresponding 4-methylthiazolines 3a–c and/or 5-acetyl-4-methylthiazolines 4a–c, respectively. While the reaction of 2a–c with phenacyl bromide afforded the 4-phenylthiazoline congeners 5a–c. Similarly, the 4-methylthiazole-5-carboxylates 6a–c were obtained via the reaction with ethyl-2-chloroacetoacetate, Scheme 1.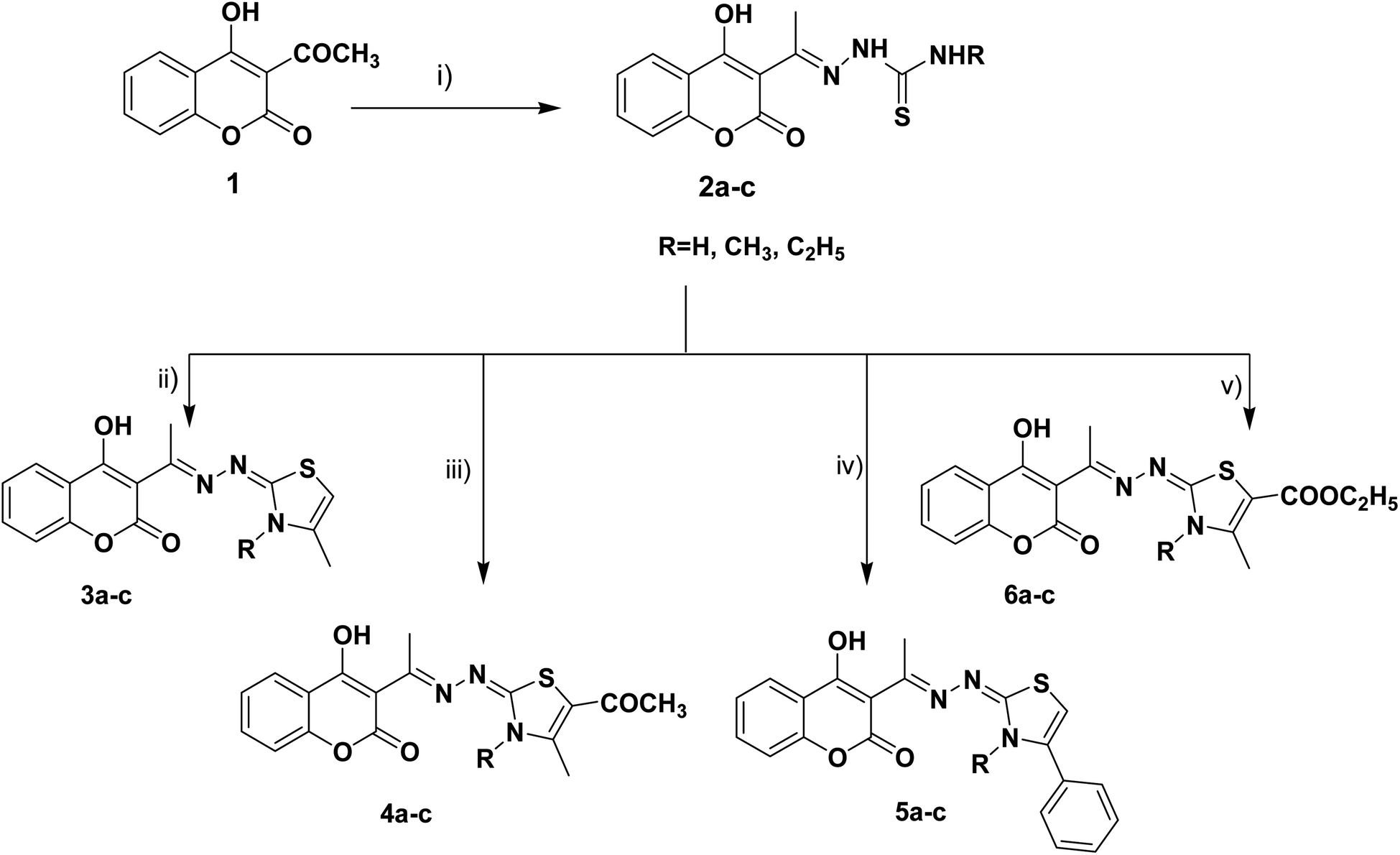 | ||
| Scheme 1 Reagents and conditions: (i) NH2NHCSNHR, EtOH, AcOH, reflux, (ii) CH3COCH2Cl, CH3COONa, EtOH, reflux, (iii) CH3COCH(Cl)COCH3, CH3COONa, EtOH, reflux, (iv) PhCOCH2Br, CH3COONa, EtOH, reflux, (v) CH3COCH(Cl)COOC2H5, CH3COONa, EtOH, reflux. | ||
Furthermore, the heterocyclization of the intermediate thiosemicarbazones 2a–c with ethyl bromoacetate and/or ethyl-2-bromopropionate under the same reaction conditions gave the corresponding thiazolidinones 7a–c and 5-methylthiazolidinones 8a–c respectively, Scheme 2.
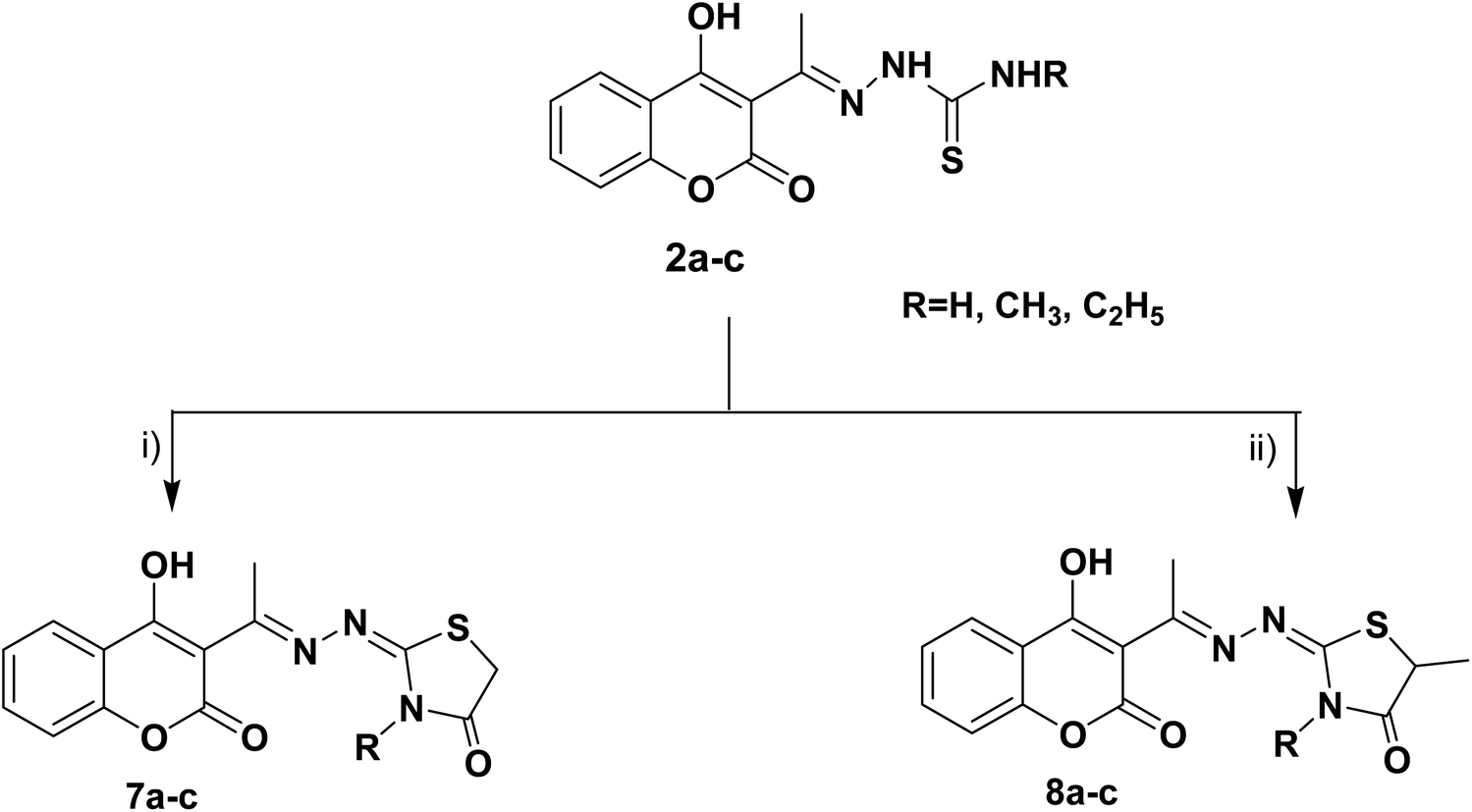 | ||
| Scheme 2 Reagents and conditions: (i) CH3CH2OCOCH2Br, CH3COONa, EtOH, reflux, (ii) CH3CH (Br) COOC2H5, CH3COONa, EtOH, reflux. | ||
Furthermore, the target compounds 9a–f were prepared via reacting the thiosemicarbazones 2a–c with hydrazonoyl chlorides in dioxane in alkaline medium, Scheme 3. The chemical structures of the synthesized target compounds were confirmed using elemental and spectral analyses (Experimental section).
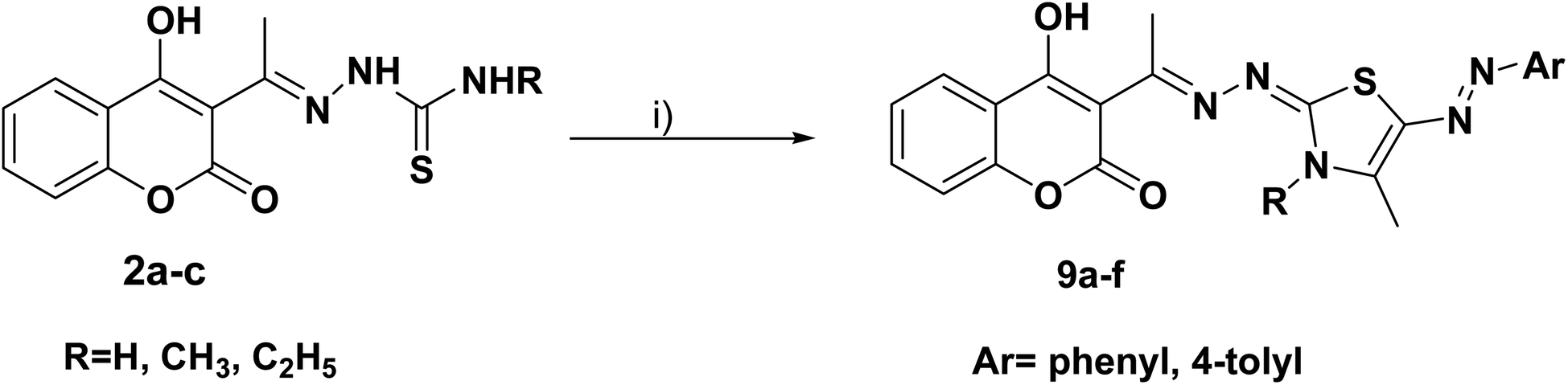 | ||
Scheme 3 Reagents and conditions: (i) CH3COCH(Cl)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–Ar, dioxane, N(C2H5)3. N–Ar, dioxane, N(C2H5)3. | ||
Synthesis was carried out using conventional synthesis procedures and reactions were modified to improve the yield and purity of products via microwave-assisted techniques. In order to study the effect of different reaction conditions on the products, microwave-assisted synthesis for the derivatives 2a–c, 3a, 4c, 5b, 5c, 6a, 7a, 7b, 8c and 9b was also carried out using the same reactants with the same molar ratios by condensation of the key intermediates 2a–c and the corresponding α-haloketones in ethanol under heating and pressure conditions, Table 1.
| Compound no. | Microwave assisted methods | Conventional method | |||||
|---|---|---|---|---|---|---|---|
| Irradiation power (W) | Reaction temperature (°C) | Pressure (Pa) | Time (min) | Yield (%) | Time (h) | Yield (%) | |
| 2a | 500 | 125 | 15 | 20 | 93.9 | 5 | 79.1 |
| 2b | 500 | 125 | 15 | 20 | 92.7 | 6 | 89.1 |
| 2c | 500 | 125 | 15 | 20 | 93.1 | 7 | 88.8 |
| 3a | 500 | 150 | 15 | 15 | 93.5 | 6 | 85.2 |
| 4c | 500 | 125 | 15 | 20 | 89.1 | 8 | 80.3 |
| 5b | 500 | 150 | 15 | 15 | 91.6 | 7 | 85.4 |
| 5c | 500 | 150 | 15 | 15 | 93.7 | 10 | 77.2 |
| 6a | 500 | 125 | 15 | 20 | 89.8 | 9 | 81.5 |
| 7a | 500 | 150 | 15 | 15 | 92.2 | 7 | 78.2 |
| 7b | 500 | 150 | 15 | 15 | 90.1 | 8 | 82.6 |
| 8c | 500 | 125 | 15 | 20 | 88.2 | 8 | 80.8 |
| 9b | 500 | 150 | 15 | 15 | 91.3 | 10 | 78.8 |
Based on the results outlined in Table 1, it is clear that the microwave irradiation approach provided products with excellent yields (88.2–93.9%) in a short time (15–20 minutes) compared to the classical synthetic methods. This can be explained by the fact that microwave heating increases the collision of reactants with each other and the high pressure conditions make the distances between the molecules of the reactants closer.
2.2. Biology
In case of MCF-7 human breast cancer cells, five compounds (6a, 8c, 2a, 6b and 2b, respectively) have significantly more potent cytotoxic activities than the standard doxorubicin (IC50 = 6.4–13.6 μM vs. 16.7 μM for doxorubicin), three compounds 9f, 6c and 2c showed remarkable cytotoxic activity (IC50 = 17.8, 20.7 and 22.7 μM, respectively); the rest of the compounds displayed lower cytotoxic activity against MCF-7 relative to the reference drug, Table 2. When the target compounds were examined against HCT-116 human colorectal carcinoma cells, it was shown that five compounds 6b, 2b, 7a, 5c and 7c have significantly more cytotoxic activities than doxorubicin (IC50 = 5.9–10.8 μM vs. 21.8 μM for doxorubicin); three compounds 2a, 2c and 6a have comparable potent cytotoxic activities to doxorubicin (IC50 = 17.8, 17.8 and 20.4 μM, respectively) and compounds 4b, 6c and 3b were slightly less active relative to the standard drug (IC50 = 24.3, 24.7 and 27.2 μM, respectively). As for the activity of the synthesized compounds on HepG2 human liver cancer cells, it was found that twelve compounds 6b, 8a, 3a, 6c, 3b, 2c, 5a, 2b, 4a, 4b, 6a, and 9d showed significant more potent cytotoxic activities than the reference drug (IC50 = 41.4–57.2 μM vs. 63.2 μM for doxorubicin), three compounds 9c, 2a, and 4c (IC50 = 60.9, 61.4 and 62.5 μM, respectively) have equipotent cytotoxic activities to doxorubicin and five compounds 9b, 7b, 5b, 8b and 3c (IC50 = 64.1–68.2 μM) have reasonable cytotoxic activity against HepG2 relative to that of doxorubicin.
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R | R1 | R2 | Ar | IC50 (μM) ± SD | ||
| MCF-7 | HCT-116 | HepG-2 | |||||
| 2a | H | — | — | — | 8.1 ± 1.1 | 17.8 ± 2.1 | 61.4 ± 5.1 |
| 2b | CH3 | — | — | — | 13.6 ± 1.5 | 7.2 ± 0.6 | 48.1 ± 3.9 |
| 2c | C2H5 | — | — | — | 22.7 ± 2.6 | 17.8 ± 1.8 | 46.4 ± 3.6 |
| 3a | H | CH3 | H | — | 34.5 ± 3.6 | 32.6 ± 3.1 | 42.5 ± 4.1 |
| 3b | CH3 | CH3 | H | — | 34.9 ± 3.1 | 27.2 ± 2.9 | 46.1 ± 3.7 |
| 3c | C2H5 | CH3 | H | — | 36.5 ± 4.2 | 36.3 ± 3.5 | 68.2 ± 5.6 |
| 4a | H | CH3 | COCH3 | — | 25.6 ± 3.1 | 31.4 ± 3.1 | 48.8 ± 3.1 |
| 4b | CH3 | CH3 | COCH3 | — | 32.7 ± 3.5 | 24.3 ± 2.7 | 57.1 ± 4.3 |
| 4c | C2H5 | CH3 | COCH3 | — | 33.7 ± 3.2 | 38.9 ± 3.2 | 62.5 ± 3.9 |
| 5a | H | Ph | H | — | 34.8 ± 4.6 | 31.8 ± 3.2 | 47.6 ± 4.1 |
| 5b | CH3 | Ph | H | — | 34.6 ± 2.9 | 42.4 ± 4.1 | 66.4 ± 5.6 |
| 5c | C2H5 | Ph | H | — | 25.9 ± 2.3 | 9.9 ± 0.9 | 86.9 ± 5.9 |
| 6a | H | CH3 | COOC2H5 | — | 6.4 ± 0.5 | 20.4 ± 1.9 | 57.2 ± 4.3 |
| 6b | CH3 | CH3 | COOC2H5 | — | 11.2 ± 1.1 | 5.9 ± 0.7 | 41.4 ± 2.9 |
| 6c | C2H5 | CH3 | COOC2H5 | — | 20.7 ± 2.1 | 24.7 ± 1.6 | 45.1 ± 4.1 |
| 7a | H | H | — | — | 26.3 ± 4.1 | 9.5 ± 1.1 | 97.6 ± 8.2 |
| 7b | CH3 | H | — | — | 32.8 ± 3.5 | 41.2 ± 3.9 | 65.7 ± 5.1 |
| 7c | C2H5 | H | — | — | 24.2 ± 3.7 | 10.8 ± 1.1 | 93.8 ± 6.1 |
| 8a | H | CH3 | — | — | 27.4 ± 1.9 | 38.3 ± 4.2 | 42.4 ± 3.5 |
| 8b | CH3 | CH3 | — | — | 26.1 ± 3.5 | 44.8 ± 4.5 | 67.2 ± 4.9 |
| 8c | C2H5 | CH3 | — | — | 7.4 ± 0.9 | 44.9 ± 4.3 | 80.2 ± 5.9 |
| 9a | H | — | — | Ph | 33.4 ± 2.8 | 33.9 ± 2.8 | 71.2 ± 5.1 |
| 9b | CH3 | — | — | Ph | 32.3 ± 3.5 | 37.9 ± 2.5 | 64.1 ± 5.5 |
| 9c | C2H5 | — | — | Ph | 32.8 ± 2.8 | 47.2 ± 4.5 | 60.9 ± 4.8 |
| 9d | H | — | — | 4-CH3Ph | 26.3 ± 2.5 | 45.6 ± 4.2 | 57.2 ± 4.6 |
| 9e | CH3 | — | — | 4-CH3Ph | 23.3 ± 1.9 | 55.2 ± 5.1 | 74.4 ± 5.9 |
| 9f | C2H5 | — | — | 4-CH3Ph | 17.8 ± 1.1 | 40.1 ± 4.7 | 82.1 ± 6.1 |
| Doxorubicin | — | — | — | — | 16.7 ± 1.5 | 21.8 ± 2.9 | 63.2 ± 5.8 |
From the above results, one can conclude that the six compounds 2a–c and 6a–c are non-selectively active on all the three human cancer types. Two compounds (3b and 4b) are active on both human colon cancer HCT-116 and liver cancer cells HepG2 but less active on human breast cancer type. Five compounds (3a, 4a, 5a, 8a, and 9d) are selectively active on only human liver cancer type but less active on both the human breast MCF-7 and human colon HCT-116 cancer types. Two compounds (8c, 9f) are selectively active on only human breast cancer type MCF-7 but not active on both human colon HCT-116 and liver HepG2 cancer types. Three compounds (5c, 7a, and 7c) are selectively active on only human colon cancer type HCT-116 but not active on the human liver HepG2 and much less active against breast MCF-7 cancer types.
For the activity against HCT cancerous cells, compounds 2a–c, 4b, 5c, 6a–c, 7a and 7c showed significant activity when compared to doxorubicin (IC50 = 5.9–24.7 μM). It was noticed that the 3,4-dimethylthiazole-5-carrboxylate 6b and methyl thiosemicarbazone 2b were the most potent anticancer agents with IC50 = 5.9 and 7.2 μM, respectively compared to doxorubicin (IC50 = 21.8 μM). The thiazolidinone 7a and its N-ethyl congener 7c as well as the N-ethyl derivative of 4-phenylthiazoline 5c showed high equipotent activity with IC50 values of 9.5, 10.8 and 9.9 μM, respectively. The thiosemicarbazone 2a and the ethylthiosemicarbazone 2c showed significant equipotent effect (IC50 = 17.8 μM). The thiazole-5-carrboxylate 6a and its N-ethyl congener 6c displayed remarkable anti-HCT effectiveness (IC50 = 20.4 and 24.7 μM, respectively). Moreover, the N-methyl derivatives of 4-methylthiazoline 3b (IC50 = 27.2 μM) and 5-acetyl-4-methylthiazoline 4b (IC50 = 24.3 μM) showed considerable potency higher than their N-unsubstituted and N-ethyl congeners.
On the other hand, the 3,4-dimethylthiazole-5-carrboxylate 6b, 5-methylthiazolidinone 8a and 4-methylthiazoline 3a are the most active derivatives against human liver cancer cell line HepG2 (IC50 = 41.4, 42.4 and 42.5 μM, respectively, vs. 63.2 μM for doxorubicin). Compound 6b was more potent than the N-desmethyl derivative 6a (IC50 = 57.2 μM) and showed equipotent cytotoxicity to its N-ethyl congener 6c (IC50 = 45.1 μM). Within the thiazolidinone series, compound 8a was the most active (IC50 = 42.4 μM) being more potent than its N-alkylated derivatives 8b, c (IC50 = 67.2 and 80.2 μM, respectively) and its desmethyl congener 7a (IC50 = 97.66 μM, respectively). The N-methyl derivatives of thiazolidinone 7b and 5-methylthiazolidinone 8b showed comparable potency to the reference doxorubicin (IC50 = 65.7 and 67.2 μM, respectively, vs. 63.2 μM for doxorubicin). The 5-acetyl-4-methylthiazoline 4a (IC50 = 48.8 μM) and 4-phenyl thiazoline 5a (IC50 = 47.6 μM) displayed higher cytotoxic activity against HepG2 cells than their N-methyl and N-ethyl congeners. Moreover, the methyl and ethyl thiosemicarbazones 2b, c showed remarkable anticancer effect than the unsubstituted thiosemicarbazone 2a (IC50 = 48.1 and 46.4 μM, respectively, vs. 61.4 μM for 2a). Within the arylazo thiazolylidene series, the 4-tolylazo compound 9d and the phenylazo derivatives 9c and 9b were the most active with comparable effect to the reference drug (IC50 = 57.2, 60.9 and 64.1 μM, respectively) (Fig. 2).
 | ||
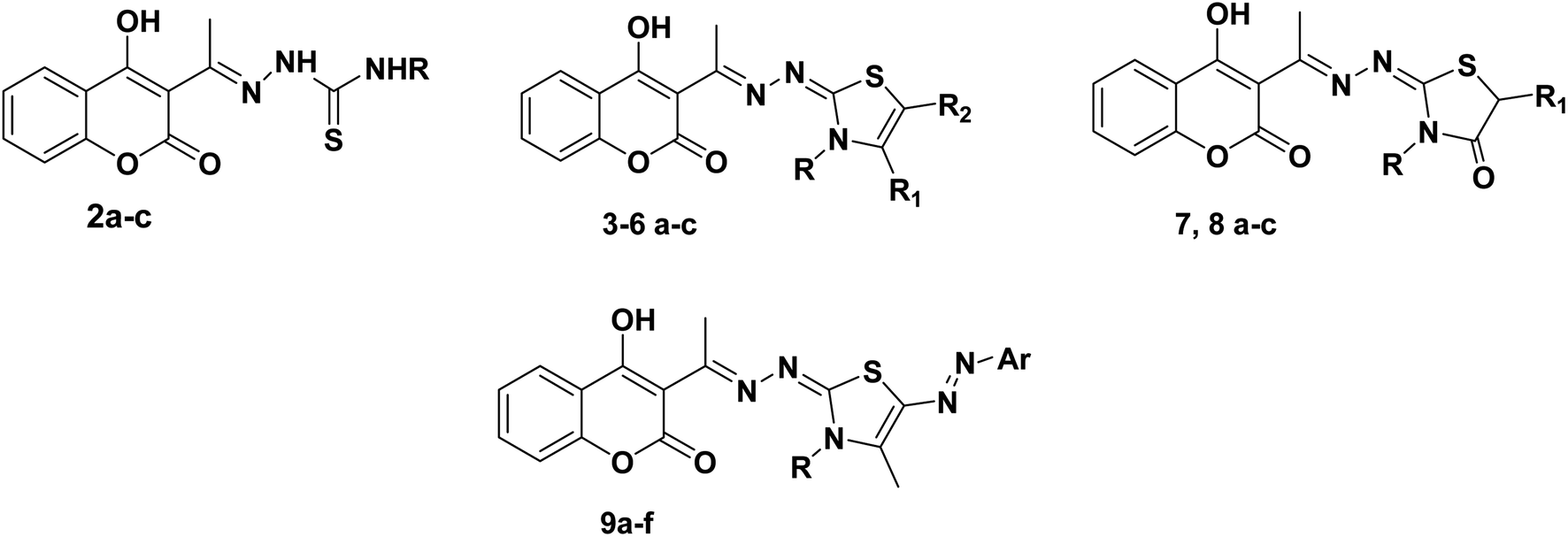
| Fig. 2 Structure activity relationship: showing the important bioactive moieties of the most potent compounds 6a–c and comparing compounds 6a–c with compounds 2a–c, 3a–c, 4a–c, 7a–c, 8a–c and 9a–c. | ||
| Compounds | IC50 (μM) | ||
|---|---|---|---|
| EGFR | m-TOR | PI3K | |
| 2a | 0.311 ± 0.02 | 1.388 ± 0.08 | 0.174 ± 0.01 |
| 2b | 2.275 ± 0.14 | 2.957 ± 0.18 | 1.331 ± 0.074 |
| 6a | 0.184 ± 0.01 | 0.719 ± 0.04 | 0.131 ± 0.007 |
| 6b | 0.563 ± 0.03 | 1.449 ± 0.08 | 0.361 ± 0.02 |
| Erlotinib | 0.088 ± 0.005 | — | — |
| Rapamycin | — | 0.396 ± 0.02 | — |
| LY294002 | — | — | 0.038 ± 0.002 |
 | ||
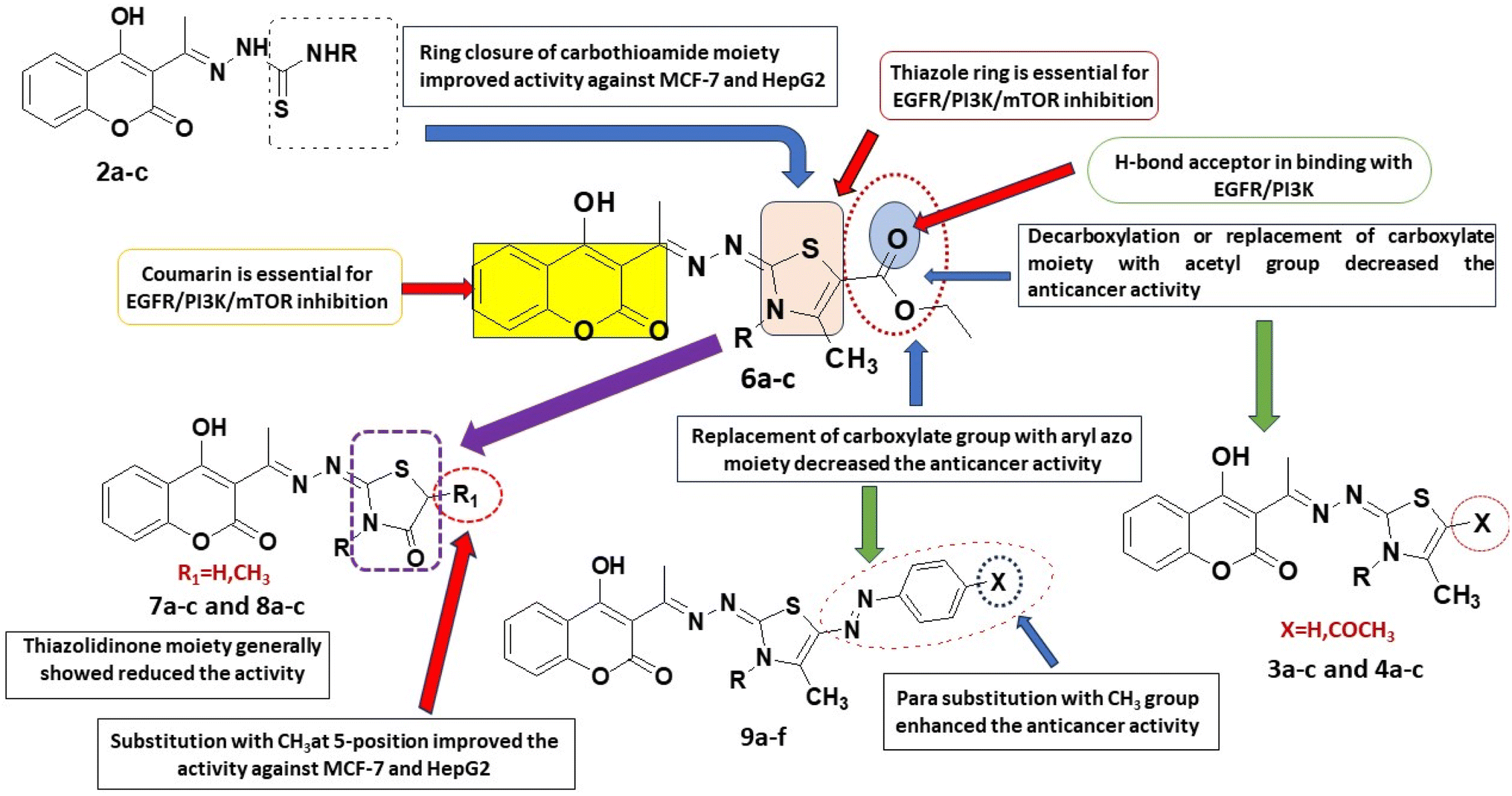
| Fig. 3 Cell cycle analysis of MCF-7 after incubation with compound 6a for 48 h. | ||
 | ||
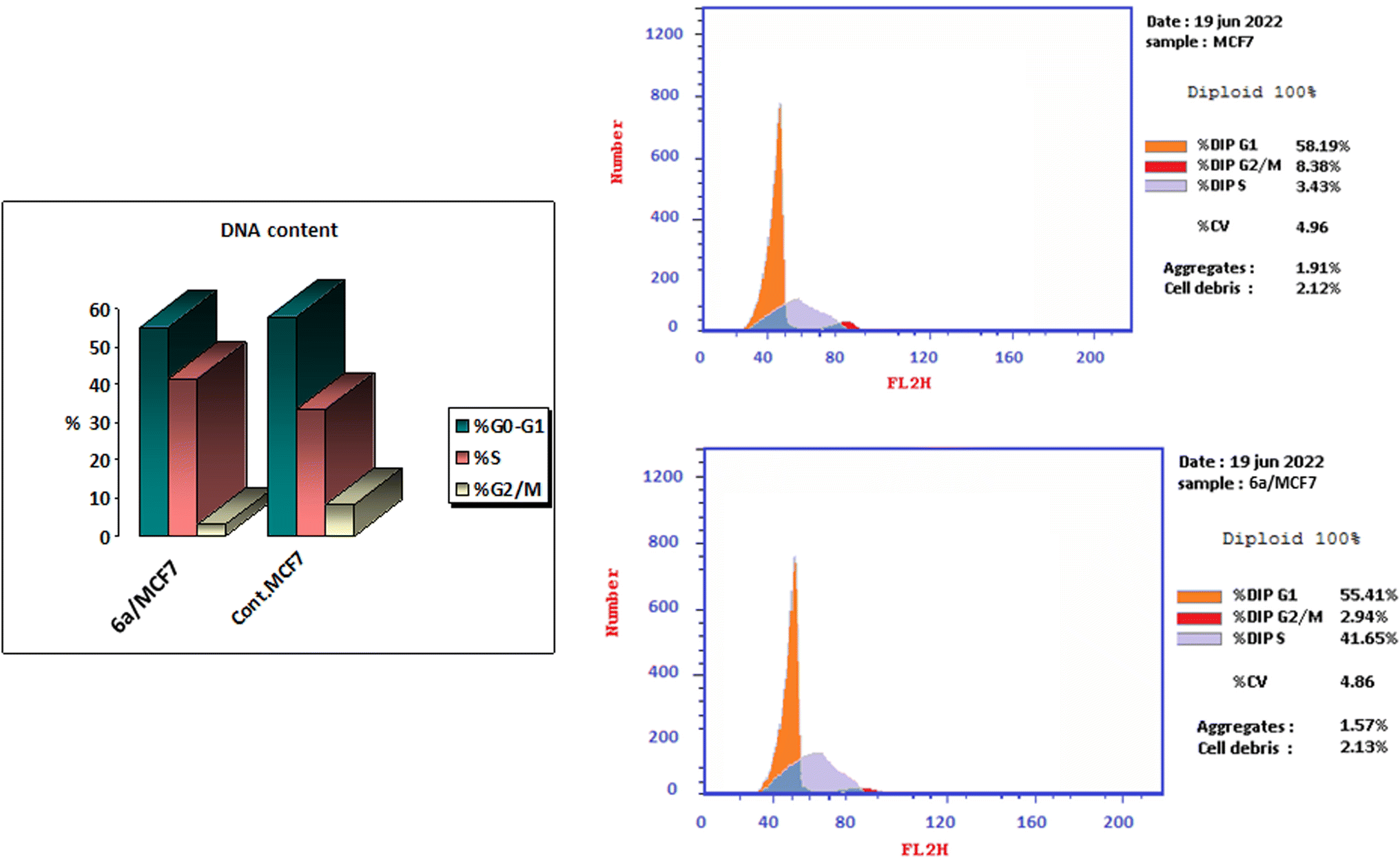
| Fig. 4 Induction of apoptosis by compound 6a. Cells were exposed to compound 6a for 48 hours and analyzed by annexin V/PI staining. | ||
2.3. Molecular docking
In a trial to explore the mode of action of the synthesized derivatives, molecular docking study was performed for the most active compound 6a. We considered the metabolism of 6a containing carboxylate group as such; we carried out docking study for its carboxylic acid metabolite as well. Docking setup was first validated by self-docking of the co-crystallized ligand, erlotinib, in the vicinity of the binding site of EGFR showing a binding score of −10.760 kcal mol−1 and RMSD value of 1.241 Å, Fig. 5.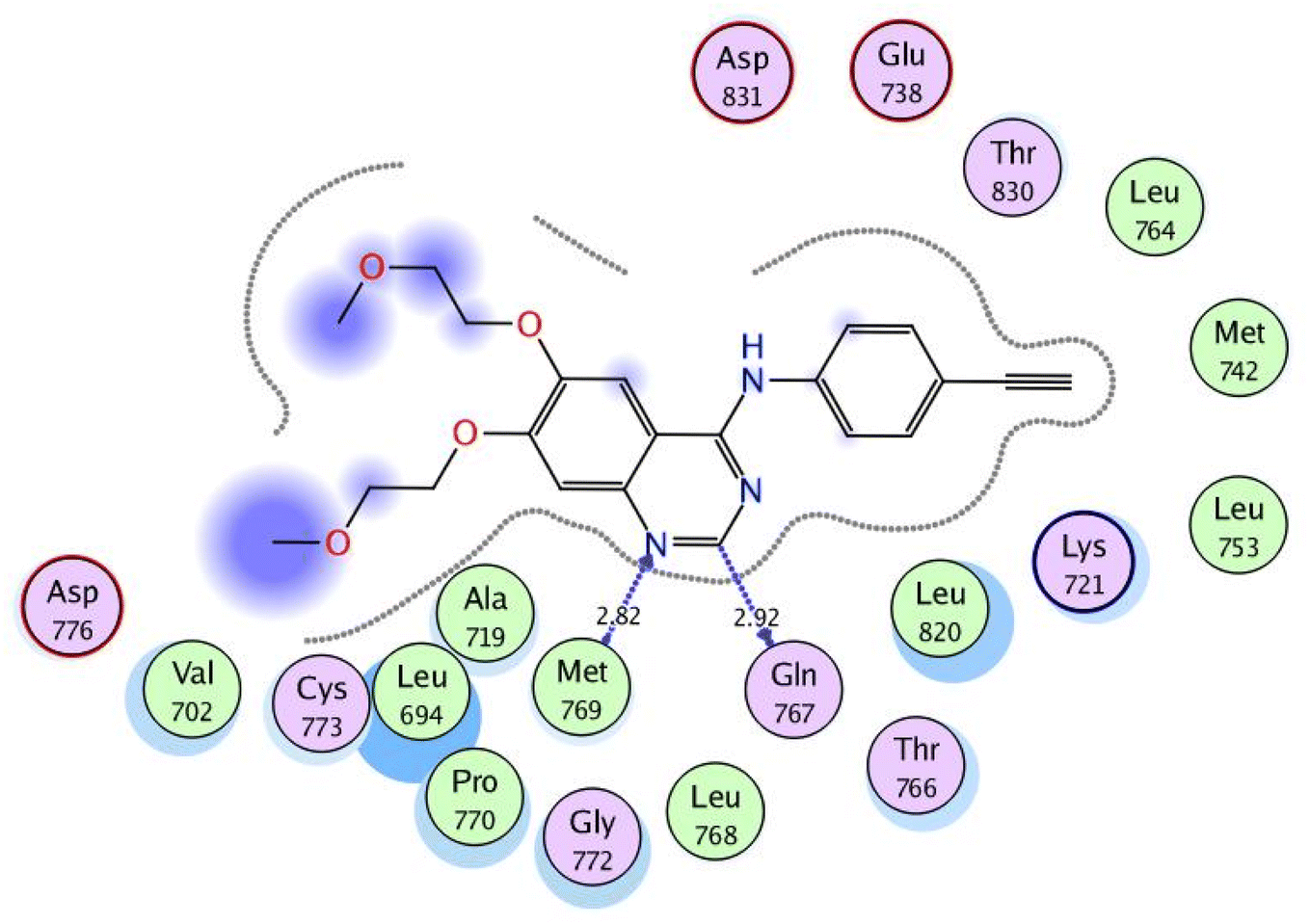 | ||
| Fig. 5 2D interaction diagram showing the co-crystallized ligand docking pose interactions with the key amino acids in EGFR binding site (PDB: 1M17). | ||
Self-docking of the co-crystallized ligand, 6-(1H-pyrazolo[3,4-b]pyridin-5-yl)-4-pyridin-4-ylquinoline, in the vicinity of the binding site of PI3K demonstrated a binding score of −7.511 kcal mol−1 and RMSD value of 0.564 Å, Fig. 6.
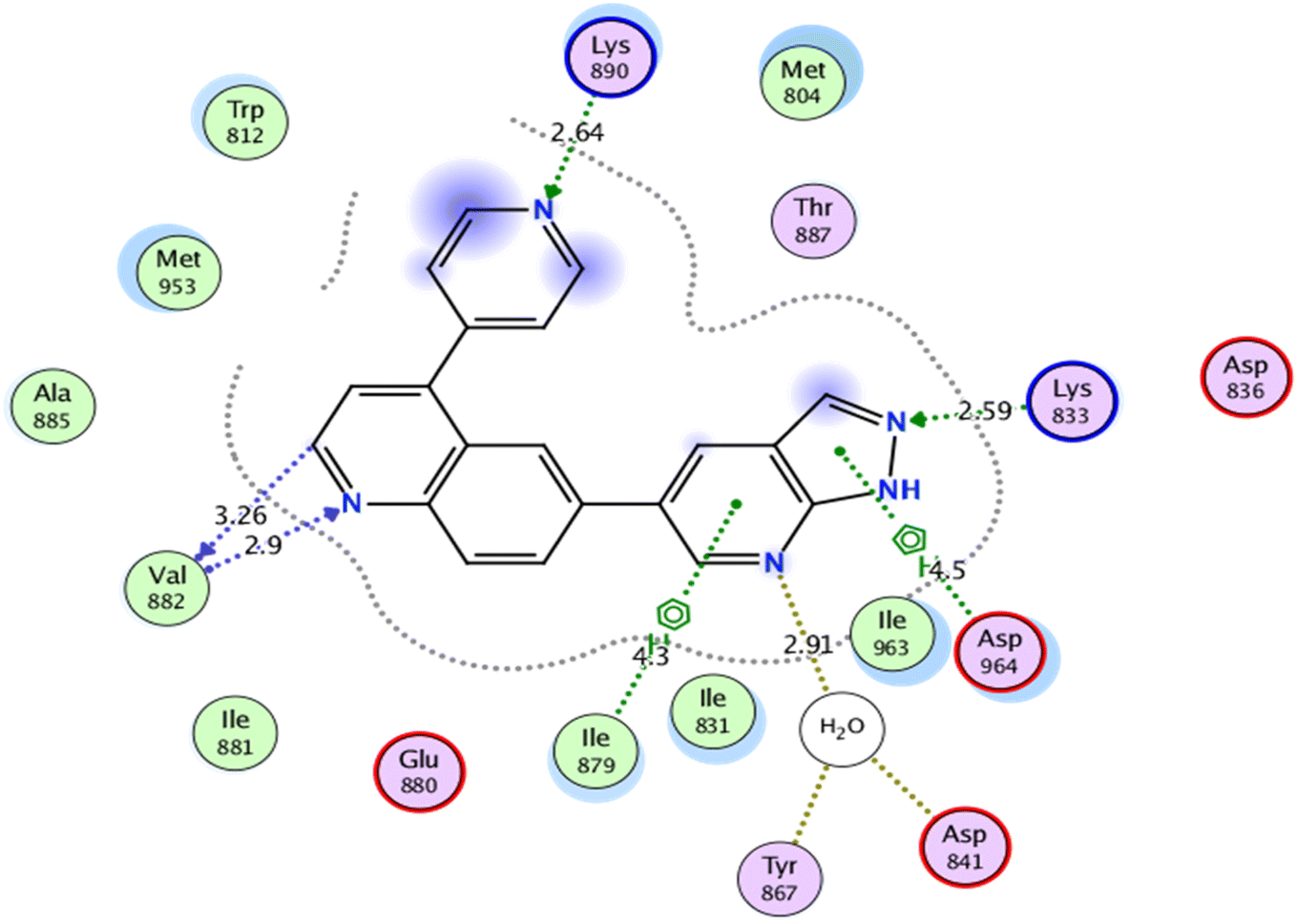 | ||
| Fig. 6 2D interaction diagram showing the co-crystallized ligand docking pose interactions with the key amino acids in PI3K binding site (PDB: 3L54). | ||
As for, the self-docking of the co-crystallized ligand, 3-(4-morpholin-4-ylpyrido[3′,2′:4,5]furo[3,2-d]pyrimidin-2-yl)phenol, in the vicinity of the binding site of mTOR, a binding score of −7.8611 kcal mol−1 and RMSD value of 0.526 Å were recorded, Fig. 7.
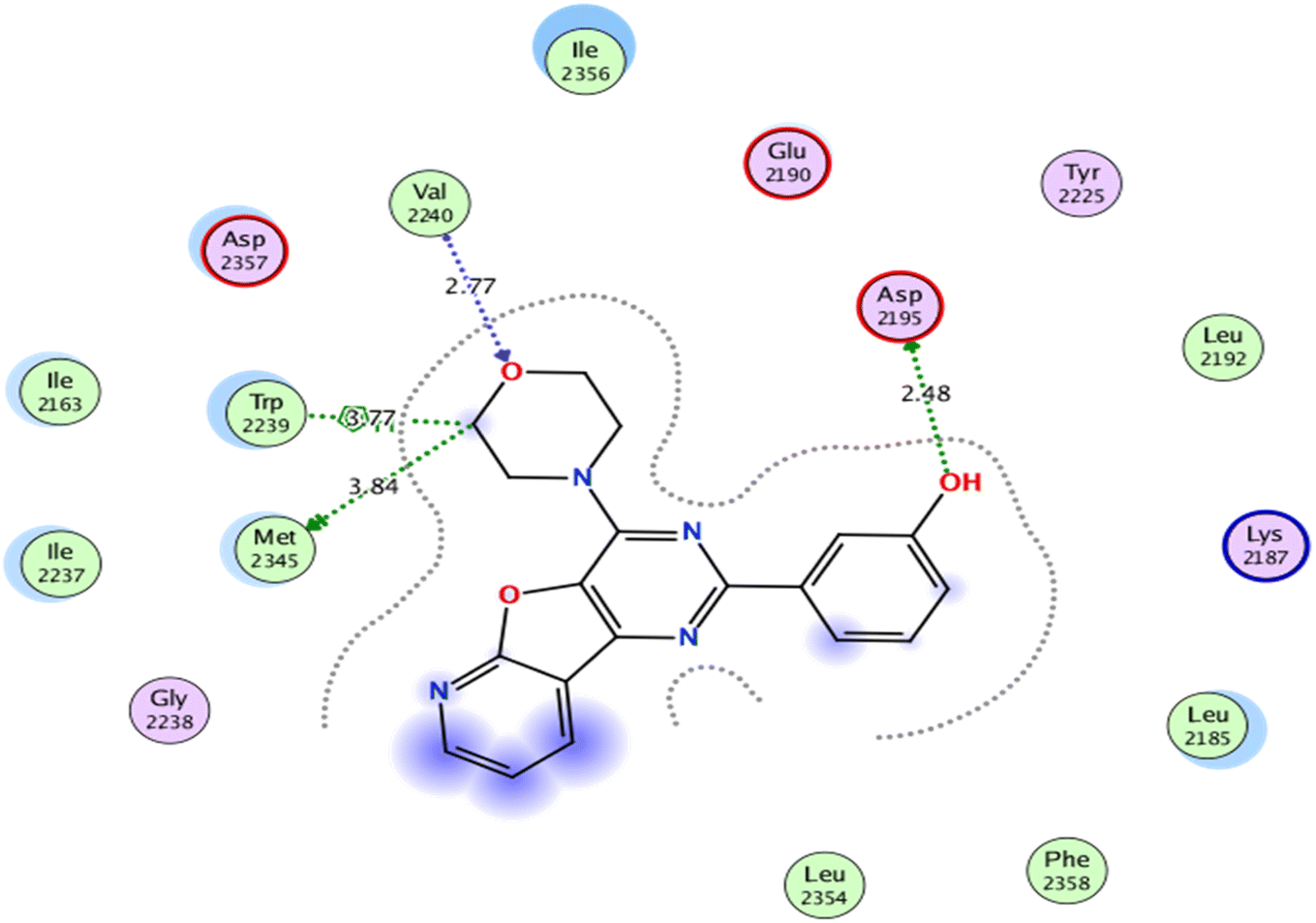 | ||
| Fig. 7 2D interaction diagram showing the co-crystallized ligand docking pose interactions with the key amino acids in mTOR binding site (PDB: 4JT6). | ||
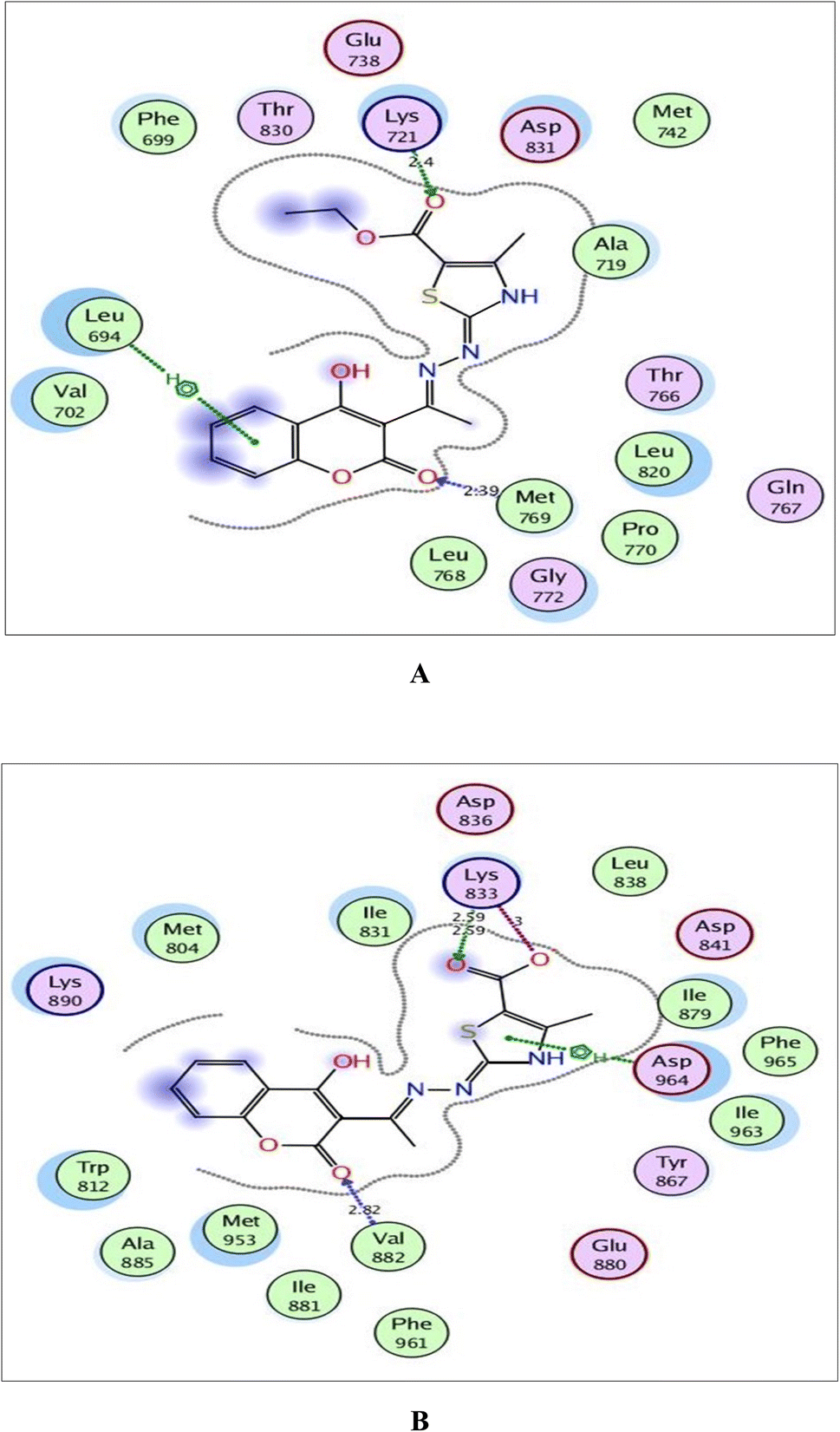
The validated setup was then used in predicting the ligand–receptor interactions at the binding site for the compound of interest 6a and its metabolite. The tested compounds displayed good binding scores and several interactions within the active sites of the target proteins. The results are summarized in Tables 4, 5 and Fig. 8–10.
| Target protein | Docking score (kcal mol−1) | Amino acids | Interacting groups | Type of interaction | Bond length (Å) |
|---|---|---|---|---|---|
| EGFR | −9.265 | Leu694 | Coumarin | Arene–H | |
| Lys721 | O(CO)CO2C2H5 |
H-bond acceptor | 2.40 | ||
| Met769 | O(CO)coumarin |
H-bond acceptor | 2.39 | ||
| PI3K | −7.320 | Lys833 | O(CO)CO2C2H5 |
H-bond acceptor | 2.83 |
| Asp836 | (CH2)CO2C2H5 | H-bond donor | 2.90 | ||
| Val882 | O(CO)coumarin |
H-bond acceptor | 2.66 | ||
| Ile963 | Thiazole | Arene–H | |||
| mTOR | −6.855 | Trp2239 | Coumarin | Arene–arene | |
| Trp2239 | Coumarin | Arene–arene | |||
| Val2240 | O(CO)coumarin |
H-bond acceptor | 2.42 | ||
| Asp2357 | Thiazole | Arene–H |
| Target protein | Docking score (kcal mol−1) | Amino acids | Interacting groups | Type of interaction | Bond length (Å) |
|---|---|---|---|---|---|
| EGFR | −9.638 | Lys721 | −O(COO)− | H-bond acceptor | 2.44 |
| Leu768 | O(CO)coumarin |
H-bond acceptor | 2.98 | ||
| Met769 | O(CO)coumarin |
H-bond acceptor | 2.52 | ||
| Thr830 | O(CO)(COO−) |
H-bond acceptor | 2.73 | ||
| Asp831 | O(CO)(COO−) |
H-bond acceptor | 2.58 | ||
| PI3K | −7.720 | Lys833 | −O(COO)− | Ionic | 3.00 |
| Lys833 | O(CO)(COO−) |
Ionic | 2.59 | ||
| Lys833 | O(CO)(COO−) |
H-bond donor | 2.59 | ||
| Val882 | O(CO)coumarin |
H-bond acceptor | 2.82 | ||
| Asp964 | Thiazole | Arene–H | |||
| mTOR | −6.751 | Lys2187 | O(CO)(COO−) |
H-bond acceptor | 3.15 |
| Glu2190 | S(thiazole) | H-bond donor | 4.15 | ||
| Trp2239 | Coumarin | Arene–arene | |||
| Val2240 | O(CO)coumarin |
H-bond acceptor | 3.28 |
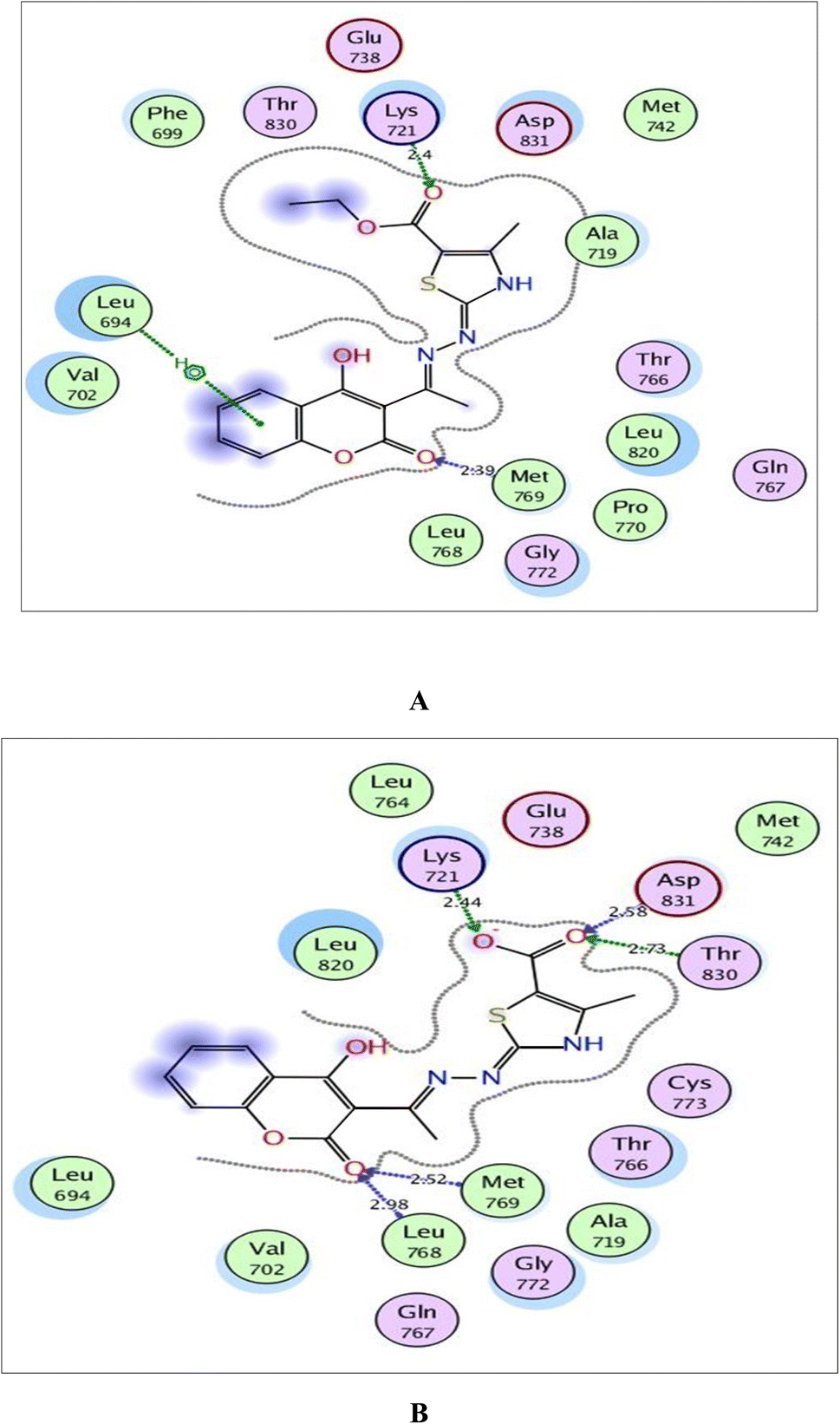 | ||
| Fig. 8 (A) and (B) Illustrated 2D diagrams of compound 6a and its metabolite, respectively showing their interactions with the EGFR binding site. | ||
 | ||
| Fig. 9 (A) and (B) Illustrated 2D diagrams of compound 6a and its metabolite, respectively showing their interactions with the PI3K binding site. | ||
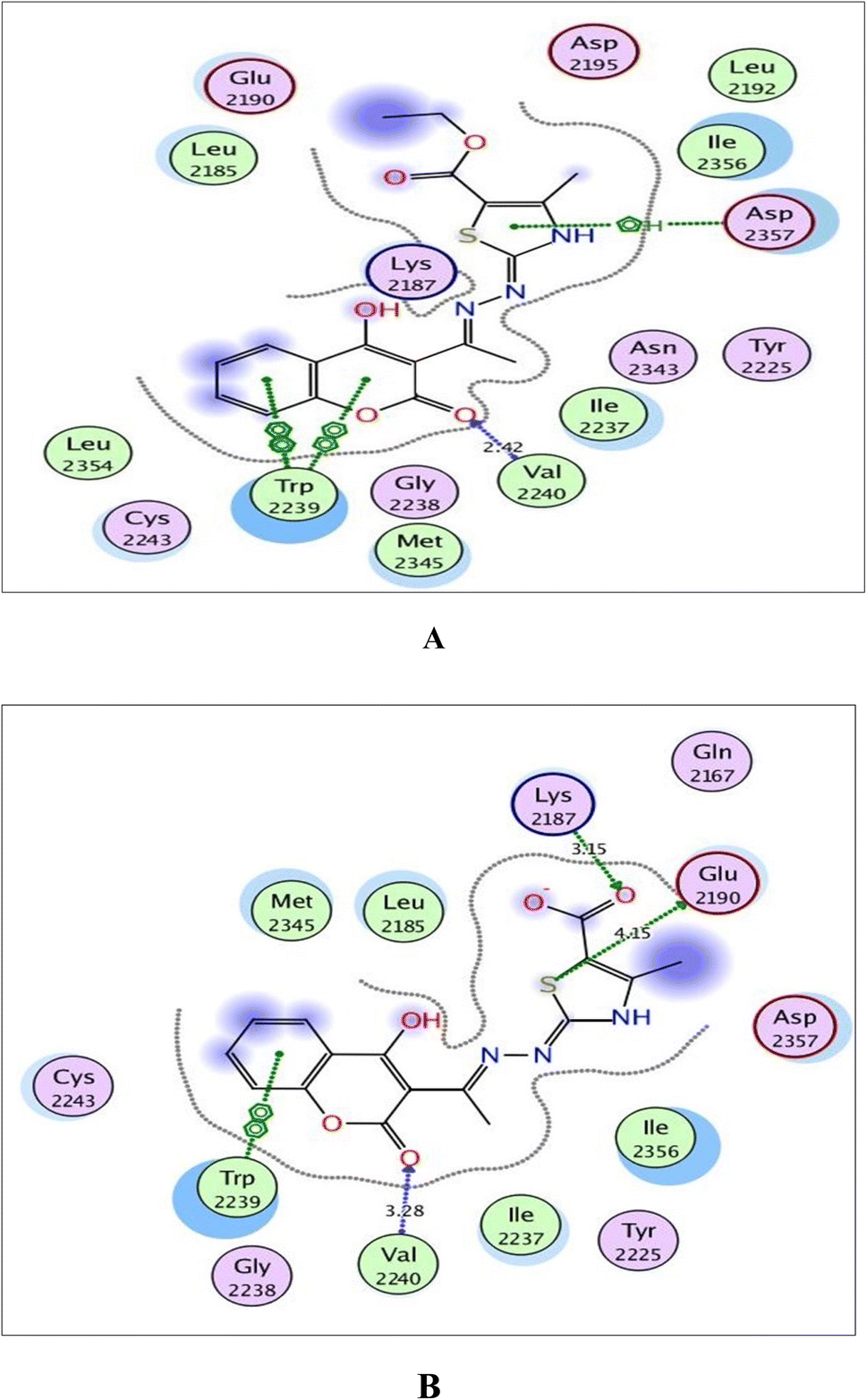 | ||
| Fig. 10 (A) and (B) Illustrated 2D diagrams of compound 6a and its metabolite, respectively showing their interactions with the mTOR binding site. | ||
Regarding the three screened receptors, it was noted that the coumarin scaffold in both 6a and its metabolite was well fitted through H-bond acceptors between the carbonyl oxygen and the key amino acids Met769 in EGFR, Val882 in PI3K and Val2240 in mTOR, resembling the original ligands (distance: 2.39 & 2.52 Å for EGFR, 2.66 & 2.82 Å for PI3K and 2.42 & 3.28 Å for mTOR, respectively).
By focusing upon EGFR in Fig. 8, the derivative 6a afforded arene–H interaction between coumarin moiety and Leu694. Also, there is one H-bond between carbonyl oxygen of ethylcarboxylate fragment and Lys721 (distance: 2.40 Å respectively). The metabolized form conserved its fitting through additional H-bonding between carbonyl oxygen of coumarin and the backbone of Leu768 (distance: 2.98 Å). Also, the carboxylate ion formed three H-bonds with Lys721, Thr830 and Asp831 (distance: 2.44, 2.73 and 2.58 Å, respectively).
Within the active site of PI3K, the thiazole moiety displayed arene–H interaction with Ile963 in 6a and with Asp964 in the metabolite. The ethylcarboxylate fragment in 6a revealed two H-bonds with the side chains of Lys833 and Asp836 (distance: 2.83 and 2.90 Å, respectively) while carboxylate ion in the metabolite of 6a showed two ionic bonds and one H-bond with Lys833 (distance: 3.00, 2.59 and 2.59 Å, respectively) (Fig. 9).
Concerning Fig. 10, the coumarin scaffold established arene–arene interactions, two in 6a and one in its metabolite with Trp2239 within the binding site of mTOR. Also, the thiazole ring shared fixation through arene–H interaction with Asp2357 in 6a and hydrogen bonding between sulfur atom and the side chain of Glu2190 in the metabolite form. Furthermore, the carboxylate ion in the metabolite of 6a exhibited H-bond acceptor with Lys2187 (distance: 3.15 Å).
Finally, it could be concluded that the derivative 6a and its metabolite occupied the same active pockets of EGFR, PI3K and mTOR, as the native ligands. The existence of coumarin and thiazole scaffolds in the structure of 6a and its metabolite promoted binding through different hydrophobic and hydrophilic interactions. Moreover, the presence of ethylcarboxylate fragment in 6a or the carboxylate ion in its metabolite potentiated fixation within the screened receptors.
2.4. In silico physicochemical properties, pharmacokinetics parameters and medicinal chemistry friendliness study
Compound 6a showed moderate solubility profile, with no blood–brain barrier permeation, as well as zero violations of Lipinski's rule and no pain alerts, expressing the main properties of a promising drug-like lead compound, Table 6.| Compd | #Rotatable bonds | #H-bond acceptors | #H-bond donors | MR | TPSA | WLOGP |
|---|---|---|---|---|---|---|
| 6a | 5 | 7 | 2 | 102.19 | 145.49 | 2.70 |
| SOL class | BBB permeant | Lipinski violations | Bioavailability score | PAINS alerts | Synthetic accessibility | |
| Moderately soluble | No | 0 | 0.55 | 0 | 3.79 |
3. Conclusion
In an attempt to search for new antitumor agents, a series of thiazole based 4-hydroxycoumarin derivatives was designed and synthesized. The primary cytotoxic MTT bioassay against MCF-7, HCT-116 and HepG2 showed that the 4-methylthiazole-5-carboxylates 6a–c and the thiosemicarbazones 2a–c were the most potent derivatives against MCF-7 and HCT cells and exhibited pronounced potency against HepG2 with compounds 2a, 2b, 6a and 6b being the most potent against the three cell lines. Moreover, compound 6a exhibited the best inhibition of EGFR and PI3K/mTOR. Molecular docking of 6a and its metabolite showed the importance of coumarin and thiazole moieties for binding interactions within the active sites of EGFR and PI3K/mTOR. Finally, the in silico study of the physicochemical properties and pharmacokinetics parameters of 6a showed that the new derivative is a promising lead compound.4. Experimental
4.1. Chemistry
All melting points were measured using Electrothermal IA 9000 apparatus and they were uncorrected. NMR spectra were assessed by Bruker 400 MHz spectrometer using TMS as the internal standard. The reactions under microwave irradiation were applied using the STARTSYNTH microwave synthesis lab station (Milestone Srl-Italy). The reactions were applied under 500 W and a pressure of 15 Pa. The reactions were followed by TLC (silica gel, aluminum sheets 60 F254, Merck) using benzene–ethylacetate (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) as eluent. The purity of the synthesized derivatives was determined by elemental analysis and was found to be more than 95%. Compounds 1 and 2a, b, 5a, b and 7a, b were previously prepared.10
:3 v/v) as eluent. The purity of the synthesized derivatives was determined by elemental analysis and was found to be more than 95%. Compounds 1 and 2a, b, 5a, b and 7a, b were previously prepared.10
4.1.1.1. 4-Methyl-1-(1-(4-hydroxy-2-oxo-2H-chromen-3-yl)ethylidene)thiosemicarbazide (2b). Yellow powder; yield (conventional: 85.2%, microwave: 93.5%); m.p. 210–2 °C; anal. calcd for C13H13N3O3S (291.33): C, 53.60; H, 4.50; N, 14.42; S, 11.01. Found: C, 53.69; H, 4.60; N, 14.52; S, 11.08. 1H NMR (DMSO-d6, δ, ppm): 2.61 (3H, s, CH3), 2.87, 2.88 (3H, d,
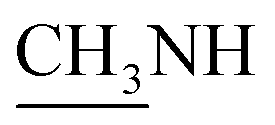 ), 7.30–8.00 (4H, m, Ar–H), 8.41 (1H, brs, NH, D2O exchangeable), 10.46 (1H, s, NH, D2O exchangeable), 15.23 (1H, s, OH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 17.75, 31.61, 96.93, 116.80, 124.46, 126.07, 134.83, 153.47, 154.67, 165.84, 183.20.
), 7.30–8.00 (4H, m, Ar–H), 8.41 (1H, brs, NH, D2O exchangeable), 10.46 (1H, s, NH, D2O exchangeable), 15.23 (1H, s, OH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 17.75, 31.61, 96.93, 116.80, 124.46, 126.07, 134.83, 153.47, 154.67, 165.84, 183.20.
4.1.2.1. 3-{1-[(4-Methyl-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (3a). Buff powder, yield (conventional: 85.2%, microwave: 93.5%); m.p. > 300 °C. Anal. calcd for C15H13N3O3S (315.35): C, 57.13; H, 4.16; N, 13.33; S, 10.17. Found: C, 57.19; H, 4.22; N, 13.40; S, 10.25. 1H NMR (DMSO-d6, δ, ppm): 2.27 (3H, s, CH3), 2.29 (3H, s, CH3), 6.63 (1H, s, CH–thiazole), 6.64–8.23 (5H, m, Ar–H and NH), 13.04 (1H, s, OH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 17.44, 17.61, 100.87, 106.97, 116.70, 117.73, 122.50, 132.75, 133.17, 147.10, 154.16, 156.18, 160.91, 163.12.
4.1.2.2. 3-{1-[(3,4-Dimethyl-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (3b). Buff powder; yield (82.6%); m.p. 241–2 °C. Anal. calcd for C16H15N3O3S (329.37): C, 58.34; H, 4.59; N, 12.76; S, 9.74. Found: C, 58.41; H, 4.66; N, 12.86; S, 9.82. 1H NMR (DMSO-d6, δ, ppm): 2.20 (3H, s, CH3), 2.79 (3H, s, CH3), 3.39 (3H, s, NCH3), 6.28 (1H, s, CH–thiazole), 7.27–7.99 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 14.21, 17.23, 31.84, 95.18, 95.31, 116.61, 119.93, 124.12, 125.63, 133.99, 138.12, 153.34, 161.66, 163.65, 164.33, 177.99.
4.1.2.3. 3-{1-[(3-Ethyl-4-methyl-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (3c). Yellow powder; yield (87.3%); m.p. 213–4 °C. Anal. calcd for C17H17N3O3S (343.4): C, 59.46; H, 4.99; N, 12.24; S, 9.34. Found: C, 59.55; H, 5.09; N, 12.34; S, 9.42. 1H NMR (DMSO-d6, δ, ppm): 1.25–1.28 (3H, t,
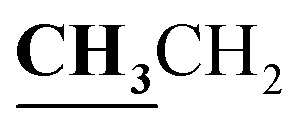 , J = 7.12 Hz), 2.23 (3H, s, CH3), 2.79 (3H, s, CH3), 3.89–3.94 (2H, q,
, J = 7.12 Hz), 2.23 (3H, s, CH3), 2.79 (3H, s, CH3), 3.89–3.94 (2H, q, 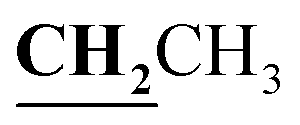 , J = 7.08 Hz), 6.27 (1H, s, CH–thiazole), 7.28–7.99 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 13.24, 13.95, 17.21, 35.04, 95.32, 95.55, 116.63, 119.93, 124.13, 125.65, 134.02, 137.57, 153.35, 161.65, 163.00, 164.43, 178.01.
, J = 7.08 Hz), 6.27 (1H, s, CH–thiazole), 7.28–7.99 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 13.24, 13.95, 17.21, 35.04, 95.32, 95.55, 116.63, 119.93, 124.13, 125.65, 134.02, 137.57, 153.35, 161.65, 163.00, 164.43, 178.01.
4.1.3.1. 3-{1-[(5-Acetyl-4-methyl-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (4a). Buff powder; yield (86.1%); m.p. > 300 °C. Anal. calcd for C17H15N3O4S (357.38): C, 57.13; H, 4.23; N, 11.76; S, 8.97. Found: C, 57.22; H, 4.29; N, 11.83; S, 9.07. 1H NMR (DMSO-d6, δ, ppm): 2.31 (3H, s, CH3), 2.46 (3H, s, CH3), 2.59 (3H, s, COCH3), 6.79–8.15 (5H, m, Ar–H and NH), 12.79 (1H, s, OH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 17.34, 18.75, 30.58, 100.27, 107.01, 116.70, 117.86, 122.43, 124.81, 132.54, 133.37, 155.49, 156.16, 160.70, 163.63, 191.10.
4.1.3.2. 3-{1-[(5-Acetyl-3,4-dimethyl-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (4b). Buff powder; yield (89.6%); m.p. 250–1 °C. Anal. calcd for C18H17N3O4S (371.41): C, 58.21; H, 4.61; N, 11.31; S, 8.63. Found: C, 58.30; H, 4.69; N, 11.38; S, 8.71. 1H NMR (DMSO-d6, δ, ppm): 2.36 (3H, s, CH3), 2.47 (3H, s, CH3), 2.72 (3H, s, COCH3), 3.37 (3H, s, NCH3), 7.23–7.96 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 14.26, 18.97, 30.08, 32.48, 79.40, 112.74, 113.77, 116.68, 124.60, 125.80, 134.39, 150.72, 158.63, 159.49, 165.50, 167.05, 169.97, 196.78.
4.1.3.3. 3-{1-[(5-Acetyl-3-ethyl-4-methyl-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (4c). Buff powder; yield (conventional: 85.2%, microwave: 93.5%); m.p. 277–8 °C. Anal. calcd for C19H19N3O4S (385.44): C, 59.21; H, 4.97; N, 10.90; S, 8.32. Found: C, 59.32; H, 5.09; N, 10.99; S, 8.43. 1H NMR (DMSO-d6, δ, ppm): 1.26–1.30 (3H, t,
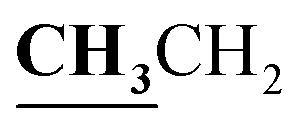 , J = 7.04 Hz), 2.46 (3H, s, CH3), 2.59 (3H, s, CH3), 2.78 (3H, s, COCH3), 3.99–4.05 (2H, q,
, J = 7.04 Hz), 2.46 (3H, s, CH3), 2.59 (3H, s, CH3), 2.78 (3H, s, COCH3), 3.99–4.05 (2H, q, 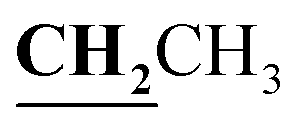 , J = 7.00 Hz), 7.27–8.03 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 13.04, 13.81, 17.36, 29.88, 40.84, 95.90, 97.96, 114.82, 116.60, 124.25, 125.83, 130.10, 134.39, 153.47, 156.74, 161.38, 164.81, 165.67, 195.92.
, J = 7.00 Hz), 7.27–8.03 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 13.04, 13.81, 17.36, 29.88, 40.84, 95.90, 97.96, 114.82, 116.60, 124.25, 125.83, 130.10, 134.39, 153.47, 156.74, 161.38, 164.81, 165.67, 195.92.
4.1.4.1. 3-{1-[(3-Methyl-4-phenyl-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (5b). Yellow powder; yield (conventional: 85.2%, microwave: 93.5%); m.p. 206–7 °C. Anal. calcd for C21H17N3O3S (391.44): C, 64.43; H, 4.38; N, 10.73; S, 8.19. Found: C, 64.51; H, 4.42; N, 10.79; S, 8.26. 1H NMR (DMSO-d6, δ, ppm): 2.82 (3H, s, CH3), 3.34 (3H, s, NCH3), 6.62 (1H, s, CH–thiazole), 7.29–8.02 (10H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 17.28, 34.03, 95.48, 98.54, 116.62, 119.81, 124.13, 125.66, 129.38, 130.12, 130.27, 134.05, 141.93, 153.35, 161.60, 163.42, 164.95, 178.01.
4.1.5.1. 4-Methyl-2-{[1-(4-hydroxy-2-oxo-2H-chromen-3-yl)-ethylidene]-hydrazono}-2,3-dihydro-thiazole-5-carboxylic acid ethyl ester (6a). Yellow powder; yield (conventional: 85.2%, microwave: 93.5%); m.p. 289–90 °C. Anal. calcd for C18H17N3O5S (387.41): C, 55.80; H, 4.42; N, 10.85; S, 8.28. Found: C, 55.92; H, 4.49; N, 10.93; S, 8.36. 1H NMR (DMSO-d6, δ, ppm): 1.26–1.29 (3H, t,
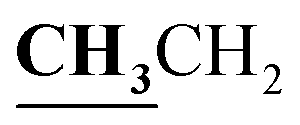 , J = 7.00 Hz), 2.39 (3H, s, CH3), 2.72 (3H, s, CH3), 4.18–4.24 (2H, q,
, J = 7.00 Hz), 2.39 (3H, s, CH3), 2.72 (3H, s, CH3), 4.18–4.24 (2H, q, 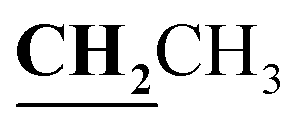 , J = 7.0 Hz), 6.80–8.00 (5H, m, Ar–H and NH), 12.52 (1H, s, OH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 14.64, 16.09, 17.42, 60.92, 85.93, 101.74, 116.75, 118.18, 124.60, 132.14, 133.60, 156.00, 156.56, 160.02, 162.79.
, J = 7.0 Hz), 6.80–8.00 (5H, m, Ar–H and NH), 12.52 (1H, s, OH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 14.64, 16.09, 17.42, 60.92, 85.93, 101.74, 116.75, 118.18, 124.60, 132.14, 133.60, 156.00, 156.56, 160.02, 162.79.
4.1.5.2. 3,4-Dimethyl-2-{[1-(4-hydroxy-2-oxo-2H-chromen-3-yl)-ethylidene]-hydrazono}-2,3-dihydro-thiazole-5-carboxylic acid ethyl ester (6b). Yellow powder; yield (87.6%); m.p. 294–5 °C. Anal. calcd for C19H19N3O5S (401.44): C, 56.85; H, 4.77; N, 10.47; S, 7.99. Found: C, 57.00; H, 4.85; N, 10.53; S, 8.08. 1H NMR (DMSO-d6, δ, ppm): 1.28–1.31 (3H, t,
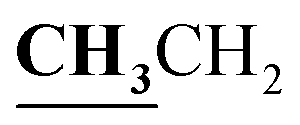 , J = 7.08 Hz), 2.58 (3H, s, CH3), 2.80 (3H, s, CH3), 3.46 (3H, s, NCH3), 4.23–4.28 (2H, q,
, J = 7.08 Hz), 2.58 (3H, s, CH3), 2.80 (3H, s, CH3), 3.46 (3H, s, NCH3), 4.23–4.28 (2H, q, 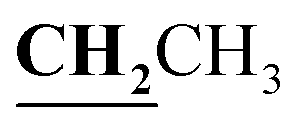 , J = 7.12 Hz), 7.27–8.04 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 13.30, 14.71, 17.37, 32.35, 61.48, 95.78, 100.67, 116.63, 119.43, 124.10, 125.80, 134.35, 149.59, 153.34, 160.18, 161.07, 161.41, 166.83.
, J = 7.12 Hz), 7.27–8.04 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 13.30, 14.71, 17.37, 32.35, 61.48, 95.78, 100.67, 116.63, 119.43, 124.10, 125.80, 134.35, 149.59, 153.34, 160.18, 161.07, 161.41, 166.83.
4.1.5.3. 3-Ethyl-4-methyl-2-{[1-(4-hydroxy-2-oxo-2H-chromen-3-yl)-ethylidene]-hydrazono}-2,3-dihydro-thiazole-5-carboxylic acid ethyl ester (6c). Orange powder; yield (88.4%); m.p. 205–6 °C. Anal. calcd for C20H21N3O5S (415.46): C, 57.82; H, 5.09; N, 10.11; S, 7.72. Found: C, 57.91; H, 5.14; N, 10.18; S, 7.80. 1H NMR (DMSO-d6, δ, ppm): 1.29–1.31 (6H, m,
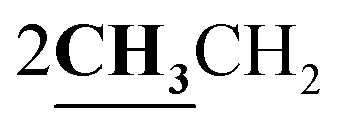 ), 2.60 (3H, s, CH3), 2.78 (3H, s, CH3), 3.97–4.02 (2H, q,
), 2.60 (3H, s, CH3), 2.78 (3H, s, CH3), 3.97–4.02 (2H, q, 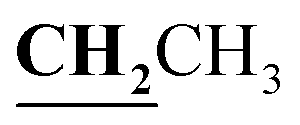 , J = 6.72 Hz), 4.24–4.28 (2H, q,
, J = 6.72 Hz), 4.24–4.28 (2H, q, 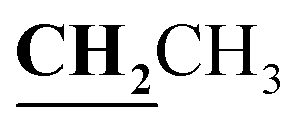 , J = 7.08 Hz), 7.28–8.03 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 12.89, 14.71, 17.29, 39.34, 61.48, 95.76, 100.98, 116.61, 119.41, 124.18, 125.78, 134.30, 148.82, 153.33, 160.18, 161.07, 161.41, 166.83, 178.15.
, J = 7.08 Hz), 7.28–8.03 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 12.89, 14.71, 17.29, 39.34, 61.48, 95.76, 100.98, 116.61, 119.41, 124.18, 125.78, 134.30, 148.82, 153.33, 160.18, 161.07, 161.41, 166.83, 178.15.
4.1.6.1. 3-Methyl-2-{[1-(4-hydroxy-2-oxo-2H-chromen-3-yl)-ethylidene]-hydrazono}-thiazolidin-4-one (7b). Yellow powder; yield (conventional: 85.2%, microwave: 93.5%); m.p. > 300 °C. Anal. calcd for C15H13N3O4S (331.35): C, 54.37; H, 3.95; N, 12.68; S, 9.68. Found: C, 54.43; H, 4.02; N, 12.77; S, 9.75. 1H NMR (DMSO-d6, δ, ppm): 2.82 (3H, s, CH3), 3.11 (3H, s, NCH3), 4.23 (2H, s, CH2–thiazolidinone), 7.31–8.00 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 14.47, 29.60, 32.71, 99.15, 117.61, 122.27, 123.77, 126.76, 138.74, 149.72, 157.37, 160.44, 162.36, 163.69, 172.24.
4.1.7.1. 5-Methyl-2-{[1-(4-hydroxy-2-oxo-2H-chromen-3-yl)-ethylidene]-hydrazono}-thiazolidin-4-one (8a). Yellow powder; yield (87.2%); m.p. 292–3 °C. Anal. calcd for C15H13N3O4S (331.35): C, 54.37; H, 3.95; N, 12.68; S, 9.68. Found: C, 54.45; H, 4.05; N, 12.77; S, 9.74. 1H NMR (DMSO-d6, δ, ppm): 1.60–1.62 (3H, d, CH3–thiazolidinone, J = 7.28 Hz), 2.77 (3H, s, CH3), 4.51–4.56 (1H, q, CH–thiazolidinone, J = 7.20 Hz), 7.31–8.01 (5H, m, Ar–H and NH), 12.34 (1H, s, OH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 18.91, 19.01, 45.75, 96.15, 116.81, 119.61, 124.43, 125.86, 134.91, 149.55, 153.55, 163.69, 164.86, 176.50.
4.1.7.2. 3,5-Dimethyl-2-{[1-(4-hydroxy-2-oxo-2H-chromen-3-yl)-ethylidene]-hydrazono}-thiazolidin-4-one (8b). Yellow powder; yield (87.9%); m.p. 241–2 °C. Anal. calcd for C16H15N3O4S (345.37): C, 55.64; H, 4.38; N, 12.17; S, 9.28. Found: C, 55.72; H, 4.48; N, 12.26; S, 9.37. 1H NMR (DMSO-d6, δ, ppm): 1.62–1.64 (3H, d, CH3–thiazolidinone, J = 7.20 Hz), 2.81 (3H, s, CH3), 3.17 (3H, s, NCH3), 4.51–4.57 (1H, q, CH–thiazolidinone, J = 7.12), 7.30–7.99 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 17.56, 18.88, 29.99, 44.44, 96.15, 116.82, 119.27, 124.48, 125.87, 134.90, 153.53, 158.91, 161.89, 165.50, 166.70, 174.76.
4.1.7.3. 3-Ethyl-5-methyl-2-{[1-(4-hydroxy-2-oxo-2H-chromen-3-yl)-ethylidene]-hydrazono}-thiazolidin-4-one (8c). Yellow powder; yield (conventional: 85.2%, microwave: 93.5%); m.p. 185–6 °C. Anal. calcd for C17H17N3O4S (359.4): C, 56.81; H, 4.77; N, 11.69; S, 8.92. Found: C, 56.89; H, 4.85; N, 11.74; S, 9.01. 1H NMR (DMSO-d6, δ, ppm): 1.19–1.23 (3H, t,
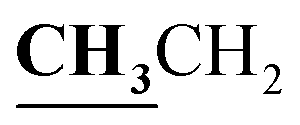 , J = 7.12 Hz), 1.62–1.64 (3H, d, CH3–thiazolidinone, J = 7.24 Hz), 2.81 (3H, s, CH3), 3.74–3.80 (2H, q,
, J = 7.12 Hz), 1.62–1.64 (3H, d, CH3–thiazolidinone, J = 7.24 Hz), 2.81 (3H, s, CH3), 3.74–3.80 (2H, q, 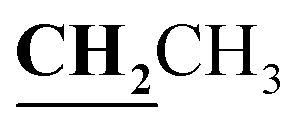 , J = 7.04 Hz), 4.53–4.59 (1H, q, CH–thiazolidinone, J = 7.16 Hz), 7.31–7.99 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 12.50, 17.47, 18.92, 38.58, 44.38, 96.14, 116.78, 119.27, 124.38, 125.84, 134.81, 153.52, 157.93, 161.29, 170.57, 174.40, 179.01.
, J = 7.04 Hz), 4.53–4.59 (1H, q, CH–thiazolidinone, J = 7.16 Hz), 7.31–7.99 (5H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 12.50, 17.47, 18.92, 38.58, 44.38, 96.14, 116.78, 119.27, 124.38, 125.84, 134.81, 153.52, 157.93, 161.29, 170.57, 174.40, 179.01.
4.1.8.1. 3-{1-[(4-Methyl-5-phenylazo-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (9a). Reddish brown powder; yield (78.7%); m.p. 103–5 °C. Anal. calcd for C21H17N5O3S (419.46): C, 60.13; H, 4.09; N, 16.70; S, 7.64. Found: C, 60.07; H, 4.01; N, 16.59; S, 7.54. 1H NMR (DMSO-d6, δ, ppm): 2.45 (3H, s, CH3), 2.66 (3H, s, CH3), 7.26–8.02 (11H, m, Ar–H, OH and NH). 13C NMR (DMSO-d6, δ, ppm): 9.00, 12.18, 115.34, 117.26, 119.07, 125.01, 125.26, 125.69, 129.64, 129.85, 130.07, 130.18, 131.07, 137.15, 144.98, 158.20, 159.76, 161.00, 178.38.
4.1.8.2. 3-{1-[(3,4-Dimethyl-5-phenylazo-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (9b). Reddish brown powder; yield (conventional: 85.2%, microwave: 93.5%); m.p. 169–70 °C. Anal. calcd for C22H19N5O3S (433.48): C, 60.96; H, 4.42; N, 16.16; S, 7.40. Found: C, 60.85; H, 4.35; N, 16.08; S, 7.31. 1H NMR (DMSO-d6, δ, ppm): 2.62 (3H, s, CH3), 2.69 (3H, s, CH3), 3.57 (3H, s, CH3), 7.19–7.96 (10H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 17.81, 17.93, 30.16, 116.55, 116.74, 117.27, 123.01, 123.82, 124.05, 125.23, 125.47, 125.75, 126.07, 129.75, 131.85, 137.03, 153.35, 154.31, 154.58, 178.35.
4.1.8.3. 3-{1-[(3-Ethyl-4-methyl-5-phenylazo-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (9c). Reddish brown powder; yield (78.8%); m.p. 126–127 °C. Anal. calcd for C23H21N5O3S (447.51): C, 61.73; H, 4.73; N, 15.65; S, 7.17. Found: C, 61.80; H, 4.81; N, 15.74; S, 7.26. 1H NMR (DMSO-d6, δ, ppm): 1.36–139 (3H, t, CH3), 2.69 (3H, s, CH3), 2.79 (3H, s, CH3), 4.09–4.14 (2H, q, CH2), 7.26–8.02 (10H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 12.54, 17.26, 17.84, 30.05, 116.57, 117.30, 119.29, 122.26, 123.22, 124.13, 124.32, 125.22, 125.68, 129.65, 129.74, 130.04, 137.12, 152.13, 153.29, 161.21, 168.67, 176.89.
4.1.8.4. 3-{1-[(4-Methyl-5-(4-tolyl)azo-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (9d). Reddish brown powder; yield (76.5%); m.p. 145–6 °C. Anal. calcd for C22H19N5O3S (433.48): C, 60.96; H, 4.42; N, 16.16; S, 7.40. Found: C, 60.88; H, 4.31; N, 16.06; S, 7.29. 1H NMR (DMSO-d6, δ, ppm): 2.39 (3H, s, CH3), 2.60 (3H, s, CH3), 2.68 (3H, s, CH3), 7.17–7.84 (10H, m, Ar–H, OH and NH). 13C NMR (DMSO-d6, δ, ppm): 15.71, 17.86, 21.10, 115.36, 116.78, 117.30, 118.43, 122.49, 123.44, 123.56, 125.78, 125.71, 130.06, 130.42, 131.88, 133.77, 141.04, 150.34, 159.55.
4.1.8.5. 3-{1-[(3,4-Dimethyl-5-(4-tolyl)azo-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (9e). Reddish brown powder; yield (74.3%); m.p. 152–3 °C. Anal. calcd for C23H21N5O3S (447.51): C, 61.73; H, 4.73; N, 15.65; S, 7.17. Found: C, 61.62; H, 4.64; N, 15.55; S, 7.09. 1H NMR (DMSO-d6, δ, ppm): 2.35 (3H, s, CH3), 2.63 (3H, s, CH3), 2.74 (3H, s, CH3), 3.49 (3H, s, CH3), 7.21–7.97 (9H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 9.59, 17.87, 21.11, 25.23, 115.34, 116.92, 117.26, 118.42, 122.30, 123.44, 123.59, 124.66, 126.00, 130.10, 131.55, 135.76, 153.42, 157.46, 161.33, 164.19, 166.71, 168.70.
4.1.8.6. 3-{1-[(3-Ethyl-4-methyl-5-(4-tolyl)azo-3H-thiazol-2-ylidene)-hydrazono]-ethyl}-4-hydroxy-2H-chromen-2-one (9f). Reddish brown powder; yield (77.1%); m.p. 159–60 °C. Anal. calcd for C24H23N5O3S (461.54): C, 62.46; H, 5.02; N, 15.17. Found: C, 62.34; H, 5.09; N, 15.06. 1H NMR (DMSO-d6, δ, ppm): 1.35–1.39 (3H, t, CH3), 2.39 (3H, s, CH3), 2.59 (3H, s, CH3), 2.66 (3H, s, CH3), 4.10–4.14 (2H, q, CH2), 7.26–8.02 (9H, m, Ar–H and OH). 13C NMR (DMSO-d6, δ, ppm): 7.44, 11.72, 13.77, 25.45, 34.05, 115.35, 116.78, 117.51, 118.51, 121.59, 123.44, 123.69, 125.73, 127.14, 127.66, 128.00, 130.12, 137.19, 153.35, 154.39, 159.50, 162.91, 164.10, 167.06.
4.2. Biology
4.3. Molecular docking
All the molecular modeling studies were carried out using Molecular Operating Environment (MOE-Dock, 2014.0901) software. All minimizations were performed with MOE until an RMSD gradient of 0.1 kcal mol−1 Å−1 with MMFF94x force field and the partial charges were automatically calculated.The X-ray crystallographic structures of EGFR complexed with erlotinib (PDB ID: 1M17), PI3K complexed with 6-(1H-pyrazolo[3,4-b]pyridin-5-yl)-4-pyridin-4-ylquinoline (PDB ID: 3L54) and mTOR complexed with 3-(4-morpholin-4-ylpyrido[3′,2′:4,5]furo[3,2-d]pyrimidin-2-yl)phenol (PDB ID: 4JT6) were downloaded from the protein data bank. The proteins were prepared for docking by removal of all ligands and water molecules that are not involved in the binding. One conserved water molecule involved in the co-crystalized ligand binding to EGFR binding site was kept. It is worth to mention that four chains make up the mTOR PDB file, and two of them, designated A and B, each include an original ligand whose binding modes to those two chains are noticeably similar. Therefore, only chain A was kept in order to perform molecular docking. All the proteins were then prepared for the docking study using Protonate 3D protocol in MOE with default options. The co-crystalized ligand was used to define the binding site for docking. Triangle Matcher placement method and London dG scoring function were used for docking.
4.4. In silico physicochemical properties, pharmacokinetics parameters and medicinal chemistry friendliness study
The physicochemical as well as the leadlike nature and medicinal chemistry friendliness of the promising compound 6a were predicted using the free SwissADME web tool.Conflicts of interest
The authors declared that there are no actual or potential conflicts of interest and have approved the article.Acknowledgements
This research article was funded by NRC, Cairo, Egypt (2019–2021, ethical approval number: 19354) through the project no. 12010110 entitled “Comparative study of conventional and microwave assisted synthesis of some new N-heterocycles as anticancer agents”. The authors are grateful to Dr Eman S. Nossier, Department of Pharmaceutical Medicinal Chemistry and Drug Design, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo, 11754, Egypt, for carrying out the molecular modeling study.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- Y. Xie, X. Shi, K. Sheng, G. Han, W. Li, Q. Zhao, B. Jiang, J. Feng, J. Li and Y. Gu, Mol. Med. Rep., 2019, 19, 783–791 CAS.
- K. M. Keppler-Noreuil, V. E. R. Parker, T. N. Darling and J. A. Martinez-Agosto, Am. J. Med. Genet., Part C, 2016, 172, 402–421 CrossRef CAS PubMed.
- J. E. Paes and M. D. Ringel, Endocrinol. Metab. Clin. North Am., 2008, 37, 375–387 CrossRef CAS PubMed , viii–ix..
- F. Xu, L. Na, Y. Li and L. Chen, Cell Biosci., 2020, 10, 54 CrossRef CAS PubMed.
- V. Papadimitrakopoulou, J. Thorac. Oncol., 2012, 7, 1315–1326 CrossRef CAS PubMed.
- A. F. Kassem, R. Z. Batran, E. M. H. Abbas, S. A. Elseginy, M. N. F. Shaheen and E. M. Elmahdy, Eur. J. Med. Chem., 2019, 168, 447–460 CrossRef CAS PubMed.
- N. A. Abdel Latif, R. Z. Batran, S. F. Mohamed, M. A. Khedr, M. I. Kobeasy, S. A. F. Al-Shehri and H. M. Awad, Mini-Rev. Med. Chem., 2018, 18, 1572–1587 CrossRef CAS PubMed.
- R. Z. Batran, D. H. Dawood, S. A. El-Seginy, T. J. Maher, K. S. Gugnani and A. N. Rondon-Ortiz, Bioorg. Chem., 2017, 75, 274–290 CrossRef CAS PubMed.
- R. Z. Batran, M. A. Khedr, N. A. Abdel Latif, A. A. Abd El Aty and A. N. Shehata, J. Mol. Struct., 2019, 1180, 260–271 CrossRef CAS.
- K. Venkata Sairam, B. M. Gurupadayya, R. S. Chandan, D. K. Nagesha and B. Vishwanathan, Curr. Drug Delivery, 2016, 13, 186–201 CrossRef CAS PubMed.
- D. Srikrishna, C. Godugu and P. K. Dubey, Mini-Rev. Med. Chem., 2018, 18(2), 113–141 CrossRef CAS PubMed.
- F. F. Zhang, D. E. Haslam, M. B. Terry, J. A. Knight, I. L. Andrulis, M. B. Daly, S. S. Buys and E. M. John, Cancer, 2017, 123, 2070–2079 CrossRef CAS PubMed.
- A. Puszkiel, G. Noé, A. Bellesoeur, N. Kramkimel, M.-N. Paludetto, A. Thomas-Schoemann, M. Vidal, F. Goldwasser, E. Chatelut and B. Blanchet, Clin. Pharmacokinet., 2019, 58, 451–467 CrossRef CAS PubMed.
- G. M. Keating, Drugs, 2017, 77, 85–96 CrossRef CAS PubMed.
- N.-Y. Chen, Y.-L. Xie, G.-D. Lu, F. Ye, X.-Y. Li, Y.-W. Huang, M.-L. Huang, T.-Y. Chen and C.-P. Li, Mol. Diversity, 2021, 25, 967–979 CrossRef CAS PubMed.
- R. Z. Batran, D. H. Dawood, S. A. El-Seginy, M. M. Ali, T. J. Maher, K. S. Gugnani and A. N. Rondon-Ortiz, Arch. Pharm., 2017, 350, 1700064 CrossRef PubMed.
- N. A. Abdel Latif, R. Z. Batran, M. A. Khedr and M. M. Abdalla, Bioorg. Chem., 2016, 67, 116–129 CrossRef CAS PubMed.
- T. K. Mohamed, R. Z. Batran, S. A. Elseginy, M. M. Ali and A. E. Mahmoud, Bioorg. Chem., 2019, 85, 253–273 CrossRef CAS PubMed.
- E. Y. Ahmed, W. S. Elserwy, M. F. El-Mansy, A. M. Serry, A. M. Salem, A. M. Abdou, B. A. Abdelrahman, K. H. Elsayed and M. R. Abd Elaziz, Bioorg. Med. Chem. Lett., 2021, 48, 128258 CrossRef CAS PubMed.
- O. M. Abdelhafez, K. M. Amin, H. I. Ali, M. M. Abdalla and E. Y. Ahmed, RSC Adv., 2014, 4, 11569 RSC.
- O. M. Abdelhafez, H. I. Ali, K. M. Amin, M. M. Abdalla and E. Y. Ahmed, RSC Adv., 2015, 5, 25312–25324 RSC.
- O. M. Abdelhafez, E. Y. Ahmed, N. A. Abdel Latif, R. K. Arafa, Z. Y. Abd Elmageed and H. I. Ali, Bioorg. Med. Chem., 2019, 27, 1308–1319 CrossRef CAS PubMed.
- E. Y. Ahmed, N. A. Abdel Latif, M. F. El-Mansy, W. S. Elserwy and O. M. Abdelhafez, Bioorg. Med. Chem., 2020, 28, 115328 CrossRef CAS PubMed.
- R. Z. Batran, W. A. El-Kashak, S. M. El-Daly and E. Y. Ahmed, ChemistrySelect, 2021, 6, 11012–11021 CrossRef CAS.
- R. Z. Batran, S. M. El-Daly, W. A. El-Kashak and E. Y. Ahmed, Chem. Biol. Drug Des., 2022, 99, 470–482 CrossRef CAS PubMed.
- H. A. Abuelizz, M. Marzouk, A. H. Bakheit, H. M. Awad, M. M. Soltan, A. M. Naglah and R. Al-Salahi, Molecules, 2020, 15(25), 5944 CrossRef PubMed.
- M. A. Abd-El-Maksoud, M. El-Hussieny, H. M. Awad, A.-T. H. Mossa and F. M. Soliman, Russ. J. Gen. Chem., 2020, 90, 2356–2364 CrossRef CAS.
- R. Abdel Elatif, M. Shabana, R. Mansour and H. M. Awad, Egypt. J. Chem., 2020, 63, 1713–1721 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03483f |
| This journal is © The Royal Society of Chemistry 2023 |