
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyethyleneimine-assisted formation of Ag–SiO2 hybrid microspheres for H2O2 sensing and SERS applications†
Swati Mehtaab,
Jitendra Bahadur *ab,
Debasis Senab,
Divya Nechiyilc,
H. Bhattdb,
Naveen Kumare and
Jyoti Prakashbc
*ab,
Debasis Senab,
Divya Nechiyilc,
H. Bhattdb,
Naveen Kumare and
Jyoti Prakashbc
aSolid State Physics Division, Bhabha Atomic Research Centre, Mumbai, 400085, India. E-mail: jbahadur@barc.gov.in
bHomi Bhabha National Institute, Mumbai, 400094, India
cMaterials Group, Bhabha Atomic Research Centre, Mumbai, 400085, India
dHigh Pressure and Synchrotron Radiation Physics Division, Bhabha Atomic Research Centre, Mumbai, 400085, India
eAtomic and Molecular Physics Division, Bhabha Atomic Research Centre, Mumbai, 400085, India
First published on 4th October 2023
Abstract
Herein, we report a simple, cost-effective, and eco-friendly approach for producing polyethyleneimine (PEI)-assisted silver nanoparticle-supported silica microspheres through evaporation-induced assembly (EIA). The silica–PEI microspheres obtained through EIA consisted of highly trapped PEI molecules owing to their electrosorption onto oppositely charged silica colloids. The trapped PEI molecules in the microspheres played a crucial role in linking silver ions to form silver ion–PEI complexes, which were then reduced to form silver nanoparticles. Further, the complex interactions between PEI and silica colloids led to enhanced porosity in the microspheres, enabling the efficient adsorption of Ag ions. The characterization of the Ag–SiO2 microspheres was carried out using various techniques, including field-emission scanning electron microscopy (FESEM), energy dispersive X-ray (EDX) spectroscopy, X-ray diffraction (XRD), small-angle X-ray scattering (SAXS), and Fourier transform infrared (FTIR) spectroscopy, which confirmed the successful formation of Ag nanoparticles on microspheres, and a plausible formation mechanism is elucidated. The Ag–SiO2 microspheres exhibited good sensing properties for hydrogen peroxide (H2O2), with an estimated limit of detection of 1.08 mM and a sensitivity of 0.033 μA mM−1 mm−2. The microspheres were also used as a surface-enhanced Raman scattering (SERS) substrate, which demonstrated high sensitivity in detecting rhodamine 6G down to a concentration of 2 × 10−6 M. The present approach elucidates a promising alternative to conventional methods that face challenges, such as scalability issues, complex and cumbersome synthesis procedures, and the use of strong reducing agents. With the potential for industrial-level scalability, this method offers a viable strategy for producing Ag–SiO2 microspheres with possible applications in biomedical and sensing technologies.
1. Introduction
In recent years, several studies have focused on the development of supported metal nanoparticles onto solid/porous substrates.1–3 This is motivated by the widespread use of metal nanoparticles in various fields, such as catalysis, sensing, electronics, photonics, and others.4–9 H2O2 is a widely known oxidant and plays a pivotal role in the fields of chemical engineering, food, and pharmaceuticals to mention a few.10–12 However, the excessive concentration of H2O2 is dangerous to human health and can cause aging, neurological disorders, and tumors in biological systems.13,14 Therefore, there is a need to develop an accurate and sensitive H2O2 sensor with the potential for industrial-scale integration. Electrochemical methods using supported metal nanoparticles represent a simple, fast, and economical candidate for hydrogen peroxide (H2O2) sensing.15,16 Further, the detection of various types of analytes, such as dyes in industrial effluents, plays an important role in environment remediation. In this regard, metal nanoparticles, particularly Ag nanoparticles, have been at the forefront owing to their unique plasmonic properties in the detection of various industrial analytes, including pesticides.17 Silver nanoparticles are commonly used in surface-enhanced Raman scattering (SERS) applications because of their strong plasmonic properties, which result in a significant enhancement of the Raman signal and used for the detection of trace amounts of analytes.4 One advantage of using supported silver nanoparticles as SERS substrates is their high stability and reproducibility, which is important for the reliable and consistent detection of analytes. The deployment of Ag nanoparticles-based SERS detection for industrial analytes needs a scalable, environmentally friendly, and cost-effective approach to fabricate the supported metal nanoparticles.Various methods have been employed to synthesize stable metal nanoparticles to date, including seed-mediated growth, electroless plating,18,19 layer-by-layer deposition,20 and hydrothermal treatment.21 However, these methods have their respective challenges for their practical application, such as the impurity of the seed used in the seeding method and the time-consuming processes involved in layer-by-layer deposition. One effective approach that can overcome these challenges is the use of mesoporous silica, such as MCM-41 and SBA-15, as a support. In this method, mesoporous silica is initially treated with noble metal ions, which are then reduced to metal nanoparticles.22–24 However, this is a two-step process. Alternatively, metal–silica hybrids can be synthesized by either adsorbing the already prepared nanoparticles onto the support25–27 or by adsorbing the metal ions on the support and subsequently reducing them by using reductants, such as sodium borohydride, hydrazine, or hydroquinone.28–31 It is important to note that the use of these strong reducing agents will generate toxic by-products, which poses challenges for large-scale production and will have a negative impact on the environment. Furthermore, some studies have reported the synthesis of colloid–metal composites by reducing metal salts using polymers in a dispersion containing polymers and colloids.32,33 However, this method requires complex and cumbersome operational processes as the reductant and support are not connected prior to the formation of the colloid–metal composites, which makes the process energy-intensive. Therefore, the major concerns in current methods include scalability issues, inaccessibility of the metal nanoparticles inside the pore channels, usage of strong reducing agents, and the complexity of the synthesis procedure. To address these issues, a facile, one-step, eco-friendly, and straightforward strategy with the usage of green reductants is essential.
In order to achieve functional composite materials, the utilization of ubiquitous natural processes, such as biomineralization and biomimetic self-assembly, would be desirable as they represent environmentally benign approaches.34–38 For instance, peptide biomimetic self-assembly is a powerful approach for the design of novel materials with tailored properties and functions, and has potential applications in various fields, including nanotechnology, biomedicine, and materials science.37,38 Peptides are molecules made up of two or more amino acids that are joined together by peptide bonds formed between the carboxyl group of one amino acid and the amine group of another amino acid.39 In biomineralization processes, the presence of the amine groups in proteins and peptides leads to interactions of the metal ions with the organic constituents,38 which could be used for the synthesis of metal nanoparticles via biogenic synthesis.40 Here, peptides act as reducing agents, stabilizers, as well as capping agents during the synthesis of metal nanoparticles. However, peptide and protein-based synthesis poses various challenges, such as cost-effectiveness and scalability. Therefore, an alternative approach is required where the peptides and proteins can be replaced with benign and less expensive molecules without compromising the advantages of the biomimetic characteristics. Due to the presence of amine groups in both polyethyleneimine (PEI) and peptides, they possess some similarities in terms of their chemical structure and biological activity, and hence, they represent a natural choice to perform the biomimetic synthesis of metal nanoparticles. Besides acting as reducing agents, amine groups are also frequently used as stabilizing agents in order to prevent agglomeration during the synthesis of metal nanoparticles, as amine groups can form complexes with metal ions.41
The realization of supported Ag nanoparticles through an amine-mediated biomimetic approach requires an effective simplistic methodology to immobilize the PEI in the silica matrix. The cooperative self-assembly of silica nanoparticles and amine molecules via evaporation-induced assembly (EIA) is one plausible method to realize amine-loaded silica microspheres. The tuning of the interaction between silica colloids and PEI molecules can dictate the nature of its binding in the silica microspheres.42 The assembly of colloids in droplet drying is one well-established method to achieve nanostructured materials with the desired morphology.42–46 The drying of micrometer-sized droplets containing silica colloids and PEI results in micrometer-sized mesoporous amine–silica microspheres in a single step. Thus, the silica–PEI microspheres, obtained through EIA, act as a support for the silver nanoparticles, which not only makes the synthesis procedure simple but also economical and environment friendly, and thus holds promise for the synthesis of metal–silica microspheres.
The present work discusses a facile, one-step approach for EIA to immobilize PEI in the inorganic matrix of embedded silica nanoparticles to obtain silica–PEI microspheres. The silica–PEI microspheres were used as a support to grow silver nanoparticles. The Ag–SiO2 microspheres were characterized using various techniques, including field-emission scanning electron microscopy (FESEM), X-ray diffraction (XRD), Fourier-transform infrared (FTIR) spectroscopy and, small-angle X-ray scattering (SAXS). The Ag–SiO2 microspheres were subsequently utilized for the electrochemical detection of hydrogen peroxide and as a SERS substrate.
2. Experimental section
2.1 Chemical reagents
Branched PEI with a molecular weight of 800 Da and 40 wt% charged stabilized colloidal silica dispersion were purchased from Sigma Aldrich. Silver nitrate (AgNO3), ethanol, Nafion, hydrogen peroxide (H2O2), and PBS buffer were also utilized in the experiments. Millipore milli Q water was utilized for the experiments.2.2 Preparation of silica–PEI microspheres
A colloidal dispersion consisting of silica nanoparticles (2 wt%) and different concentrations of PEI were spray-dried using a spray-dryer (LU 228, Lab Ultima, India) to obtain the silica–PEI microspheres. Pressure for the atomization was set to 2.0 kg cm−2 with the inlet temperature of the drying chamber set at ∼170 °C. The dispersion feeding rate was set to 2 mL min−1 and the aspiration rate was kept at 50 Nm3 h−1. The loading of PEI in the dried samples was varied at 5, 11, 20, and 33 wt% and the samples were designated as Si-p5, Si-p11, Si-p20, and Si-p33, respectively.2.3Preparation of Ag–SiO2 microspheres
A 1 M solution of silver nitrate was prepared. Next, 0.5 g of silica–PEI microspheres powder was added in to 2 mL of the 1 M AgNO3 solution. The soaked powder samples were then kept in the dark overnight. After the silver nanoparticles had formed, the samples were centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to separate the Ag–SiO2 microspheres.
000 rpm to separate the Ag–SiO2 microspheres.
2.4 Characterization of the microspheres
The morphology of the microspheres before and after silver loading was analyzed using FESEM while the Ag content was determined by EDX measurements. SAXS experiments were carried out on the microspheres using the SWAXS beamline (BL-18)45 at Indus-2 synchrotron, RRCAT, India. The scattering profiles were recorded as a function of Q (= 4πsinθ/λ, where 2θ is the scattering angle and λ is the X-ray wavelength). The experimental Q-range was between 0.07–2 nm−1. The XRD data were recorded using a CuKα-based X-ray diffractometer in a θ–θ geometry with a 0.02° step size with 2θ in the range of 10°–80°. FTIR measurements were carried out using a Bruker Vertex 80 V FT-IR spectrometer. KBr was used to disperse the samples and form pellets for the measurements.
2.5 Electrochemical detection of H2O2
An electrochemical workstation was used for the electrochemical measurements. First, 5 mg of the Ag–Si-p20 sample was mixed in 1 mL of ethanol and 10 μL Nafion was added to it. The dispersion was sonicated for about 15 min and 3 μL of the prepared dispersion was cast on a glassy carbon electrode (GCE) twice and allowed to dry for 1 h. A three-electrode cell, with GCE as the working electrode, Ag|AgCl as the reference electrode, and platinum as the counter electrode was used. Cyclic voltammetry (CV) experiments were performed at room temperature. Prior to the experiments, sodium phosphate buffer (0.1 mol L−1) was purged with N2 for 10 min. The desired aliquots of H2O2 were successively added to the cell for the CV measurements with the variation in the H2O2 concentration.2.6 Surface-enhanced Raman scattering (SERS)
A 15 μL drop of R6G aqueous solution (2 × 10−2 M) was pipetted gently on to a glass slide and dried in ambient atmosphere for normal Raman scattering (NRS) measurements. A surface-enhanced Raman scattering (SERS) active substrate was prepared by drop-casting an aqueous dispersion of Ag–SiO2 microspheres onto a glass slide. Next, 15 μL of R6G aqueous solution, in the concentration range of 2 × 10−3 M to 2 × 10−6 M, was dropped on to the Ag–SiO2 substrate for SERS investigations. A 532 nm wavelength from a DPSS laser (OXXIUS-LC-532) was utilized to record the NRS and SERS spectra. The Raman-scattered light was detected using a CCD (ANDOR)-based monochromator (ANDOR-SR-750C) together with an edge filter. The NRS and SERS spectra were collected at 1 mW laser power, and a total of 10 scans of 4 seconds acquisition time were used to record the data. A schematic showing the instrumentation used in the Raman scattering measurements is provided in the ESI (Fig. S8†).3. Results and discussion
The FESEM micrographs in Fig. 1a, d, g and j show the morphology of the silica–PEI microspheres for different PEI loadings obtained through spray-drying. The morphologies of the Ag–SiO2 microspheres obtained for the various PEI loadings are shown in Fig. 1b, e, h and k. The deposition of silica nanoparticles on the silica microspheres was evident from the micrographs. The magnified images of the Ag nanoparticles on the silica microspheres with varying PEI loadings are depicted in Fig. 1c, f, i and l. Lower-magnification FESEM micrographs of the silica–PEI microspheres with and without silver nanoparticles are depicted in Fig. S1 in the ESI.† The level of coverage of Ag nanoparticles on the microspheres increased with the increasing loading of PEI. A lower loading of PEI resulted in a lower coverage of Ag nanoparticles on the surface, as seen in Fig. 1b, whereas a higher loading resulted in a higher coverage, as observed in Fig. 1e, h and k. These observations indicated a direct correlation between the degree of coverage of Ag nanoparticles and the loading of PEI in the silica–PEI microspheres. The higher-resolution FESEM micrographs shown in Fig. 1c, f, i and l were utilized to determine the size and degree of coverage of Ag nanoparticles anchored onto the surface of microspheres using ImageJ software.47 The estimated size distribution, as shown in Fig. 2a–c, was fitted using a normal distribution, and yielded an average size of 34 ± 6 nm for the Ag nanoparticles. In the case of Ag–Si-p33, it was difficult to estimate the size of the Ag nanoparticles due to the modifications in the surface morphology of the microspheres. Fig. 2d shows there was a quantitative variation in the percentage of degree of coverage with the increase in loading of PEI. | ||
| Fig. 1 FESEM micrographs of silica and Ag–SiO2 microspheres as a function of PEI loading: (a) Si-p5; (b, c) Si-p5–Ag; (d) Si-p11; (e, f) Si-p11–Ag; (g) Si-p20; (h, i) Si-p20–Ag; (j) Si-p33; (k, l) Si-p33–Ag. | ||
 | ||
| Fig. 2 (a–c) Size distribution of the Ag nanoparticles anchored on the surface of silica–PEI microspheres as a function of PEI loading. (d) Quantitative variation in the degree of coverage of Ag nanoparticles with the increase in PEI loading. | ||
For confirmation of the presence of Ag nanoparticles on the silica–PEI microspheres, elemental mapping of the Ag–SiO2 microspheres was performed using energy dispersive X-ray measurements.
Fig. 3(a)–(d) present the EDX maps of the Ag–SiO2 microspheres obtained for different PEI loadings. The EDX mapping illustrated the elemental distribution of Si, O, and Ag. The discreteness of the Ag nanoparticles on the surface of microspheres and an increase in the coverage of Ag nanoparticles for higher PEI loading were evident. To exhibit the discreteness of the silver nanoparticles, EDX maps of individual Ag–silica microspheres are shown in Fig. S2 in the ESI.† The content of Ag nanoparticles in the microspheres was calculated by EDX analysis (Fig. S3 in the ESI†). Fig. 2e shows that the amount of silver nanoparticles on the surface of the microspheres increased with the increase in the PEI loading. XRD measurements were carried out on the Ag–SiO2 microspheres to determine the phase of the Ag nanoparticles.
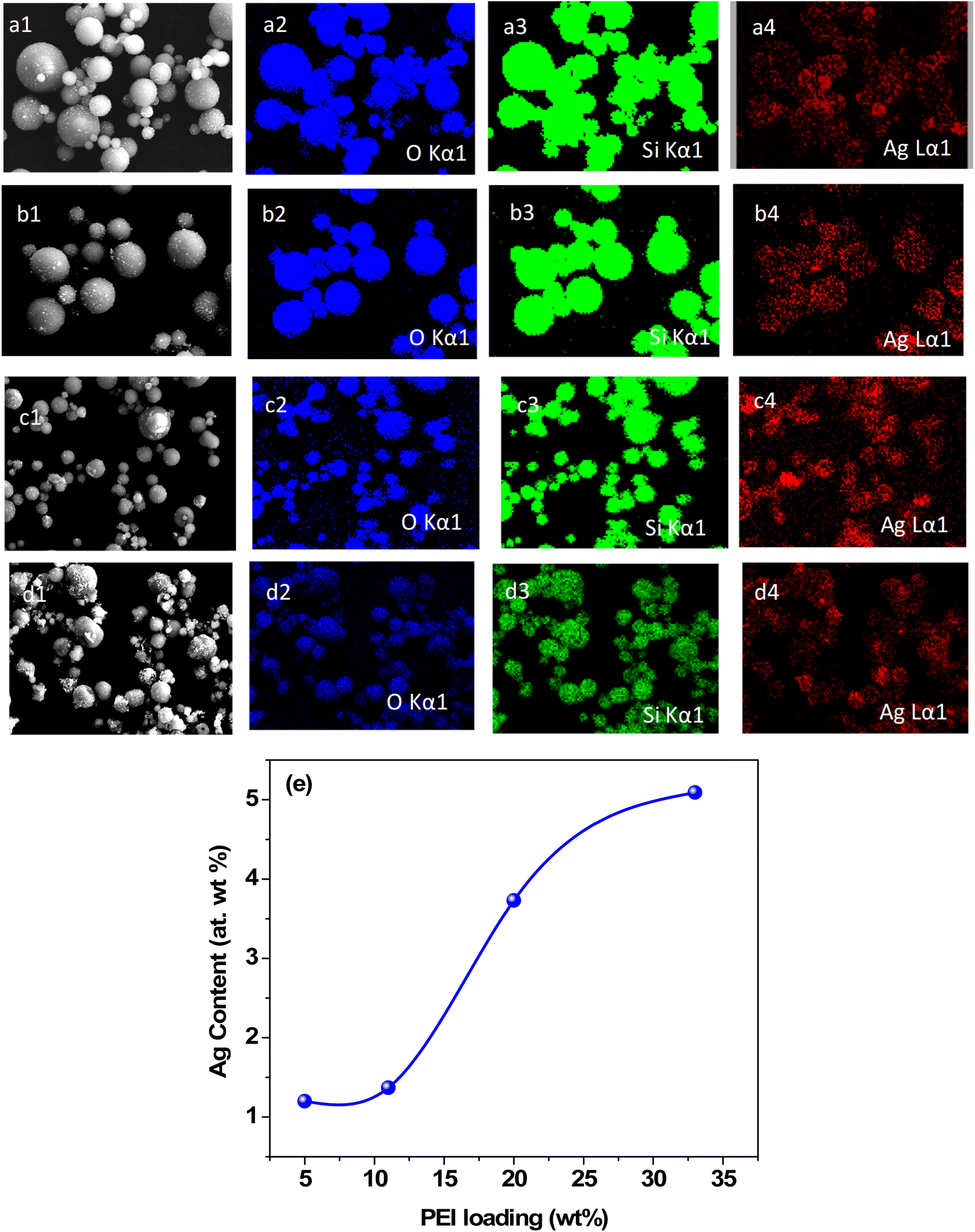 | ||
| Fig. 3 Elemental mapping of the Ag–SiO2 microspheres obtained for PEI loadings of: (a) 5 wt%, (b) 11 wt%, (c) 20 wt%, (d) 33 wt%. (e) Quantitative variation of the silver content in the Ag–SiO2 microspheres obtained for different PEI loadings. | ||
Fig. 4 illustrates the XRD patterns of the Ag–SiO2 microspheres for different silver loadings. The XRD profiles showed peaks at 38°, 44.3°, 64.3°, and 77.5°, corresponding to the (111), (200), (220), and (311) crystalline planes of Ag (JCPDS No. 04-0783),48,49 respectively. These peaks confirmed the cubic phase of the metallic Ag nanoparticles, while the broad peak observed in the range from 15°–30° was attributed to the amorphous silica nanoparticles. An additional weak peak was observed at 32.2°, corresponding to (111) plane of the face-centered cubic (FCC) phase of Ag2O (JCPDS No. 01-076-1393),50 indicating a very small presence of Ag2O, which could be attributed to slight surface oxidation of the metallic Ag nanoparticles. To estimate the crystallite size of the Ag nanoparticle from the XRD peaks, the Debye–Scherrer formula,51 as shown below, was used.
 | (1) |
 | ||
| Fig. 4 X-Ray diffraction patterns of the Ag–SiO2 microspheres with different Ag loadings. | ||
The average crystallite size of the Ag nanoparticle was estimated to be approximately 10 nm based on the FWHM obtained from fitting the Lorentzian peak corresponding to the reflection (111) (Fig. S3 in the ESI†). The average size of the nanoparticles, estimated from the FESEM micrographs was larger than crystallite size due to the polycrystalline nature of the Ag nanoparticles. The UV-vis spectrum for Si-p(20)–Ag dispersed in water (Fig. S5 in the ESI†) showed a peak at 450 nm, indicating the collective absorption behavior of the Ag nanoparticles.
The SAXS profiles of pristine silica–PEI microspheres were compared with the Ag–SiO2 microspheres corresponding to loadings of 5 wt% and 33 wt% (Fig. 5). A comparison of the SAXS profiles for the other PEI loadings is shown in Fig. S6 of the ESI.† It could be observed that there was an enhancement in the intensity of the SAXS profile in the low-Q regime for the Ag–SiO2 microspheres compared to the PEI-loaded microspheres. This enhancement in the intensity was due to the scattering contribution from the Ag nanoparticles.52 The significant deviation in the SAXS profiles of Si-p(33)–Ag and Si-p(33) was attributed to the large scattering contribution from the silver nanoparticles due to higher yield of Ag nanoparticles, as also evident from the FESEM and EDX measurements.
 | ||
| Fig. 5 Comparison of the SAXS profiles of the silica–PEI microspheres and Ag–SiO2 microspheres at 5 wt% and (b) 33 wt% PEI loadings. Hollow circles (black) represent the SAXS data of pristine silica–PEI microspheres, while the hollow triangles (red) represent the SAXS profiles of the Ag–SiO2 microspheres. | ||
The SAXS profiles of Ag–SiO2 microspheres for different silver loadings are shown in Fig. 6a. As the size of the silica nanoparticles was much smaller than that of the microspheres, the total scattering intensity could be as approximated to:
| Itotal(Q) ∼ Isilica(Q) + Ims(Q) | (2) |
 | (3) |
 | (4) |
 | ||
| Fig. 6 (a) SAXS profiles of Ag–SiO2 microspheres. Solid lines represent the fitting of the data. SAXS profiles have been shifted vertically for clarity. (b) Variation in the volume fraction of the silica nanoparticles in the Ag–SiO2 microspheres obtained for different PEI loadings. Volume fraction of silica nanoparticles was obtained from the model fitting of the SAXS profiles. | ||
P(Q, r) represents the form factor of sphere of radius r, and is given by:
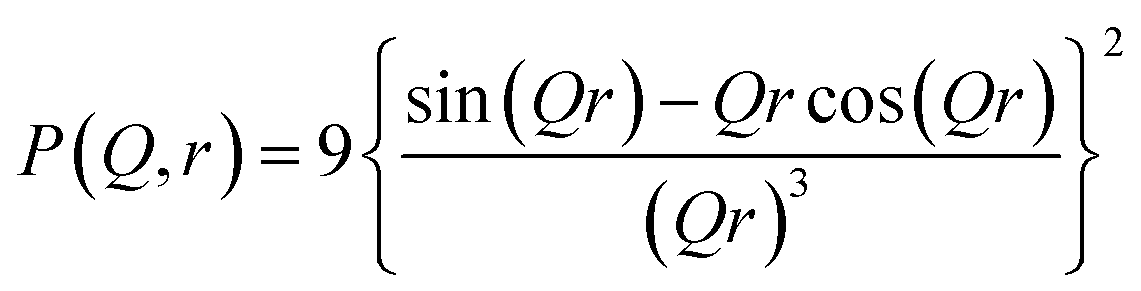 | (5) |
Ims(Q) is the combined scattering intensity from the silver nanoparticles and microspheres. Due to the large size of the Ag nanoparticles and microspheres, the scattering intensity Ims(Q) contains surface scattering contributions only. Therefore, the scattering intensity Ims(Q) can be represented as,
 | (6) |
The variation of the silica volume fraction, ϕ, at different silver loadings is depicted in Fig. 6b. The fitting of the SAXS profiles of the silica–PEI microspheres before silver loading gave identical results and the volume fraction varied in a non-monotonic fashion with the increase in PEI loading. This confirms that the embedding of the silica nanoparticles remained intact during the growth of the silver nanoparticles. This non-monotonic behavior in silica–PEI microspheres could be attributed to the complex interactions involved between the silica nanoparticles and PEI.56 Since, there was a correlation between the volume fraction and porosity of the silica–PEI microspheres, therefore the porosity of the microspheres increased at moderate PEI loadings. The increased porosity help the adsorption of Ag ions in the microspheres during the formation of silver nanoparticles.
To study the entrapment and thermal stability of PEI, FTIR measurements of the silica–PEI microspheres, with a PEI loading of 33 wt%, were conducted at varying temperatures (Fig. 7a). There were no significant changes observed in the functional groups related to the PEI with the increase in temperature, implying a high stability of the PEI against thermally induced degradation up to 160 °C. In general, free PEI undergoes thermal degradation when the temperature is increased beyond 100 °C. However, the strong interaction between the silica nanoparticles and PEI led to a very strong trapping of PEI in the microspheres. To investigate the functional groups in the silica microspheres after Ag nanoparticle loading, FTIR analysis was performed for the Si-p(20)–Ag microspheres at room temperature. For reference, the FTIR analysis was conducted for the pure silica as well as Si-p(20), as depicted in Fig. 7b. The bands at 794 and 1084 cm−1 were due to the bending and stretching of the silanol groups, respectively. The incorporation of PEI resulted in an emergence of bands at 2839 and 2941 cm−1, corresponding to the stretching vibrations of –CH groups. Moreover, the bands at 1480 and 1572 cm−1 represented the vibrations of –CN and –NH groups,57,58 respectively, indicating the successful incorporation of PEI in the microspheres. Nevertheless, in the Ag–SiO2 microspheres, two new bands emerged at 1385 and 1632 cm−1, which indicated the stretching vibrations of –O–C–O– and carboxyl groups,58,59 respectively, which were absent in the pure silica and silica–PEI microspheres.
 | ||
| Fig. 7 (a) FTIR spectra of Si-p(33) microspheres as a function of temperature. (b) Comparison of the FTIR spectra for pure silica (black), Si-p(20) (red), and Si-p(20)–Ag (green). Data have been shifted vertically for clarity. Stars indicates the emergence of new peaks in the Ag-loaded microspheres. | ||
At this juncture, it is imperative to elaborate the processes involved in the formation of Ag nanoparticles on porous silica–PEI microspheres obtained from EIA.54,60 Silver ions are adsorbed on the silica–PEI microspheres upon the addition of an aqueous solution of silver nitrate, owing to the porous nature of the microspheres, as illustrated in Fig. 8. PEI facilitates the binding of Ag ions to form PEI–silver ion complexes.41,61,62 PEI plays a crucial role in reducing silver ions to silver nanoparticles63 akin to the peptide groups in a biomimetic process. The numerous amine groups present in PEI donate an electron from the nitrogen of the amine group to Ag+ ions, thereby leading to the formation of Ag0 nanoparticles. The trapped PEI has an additional advantage of anchoring the same amount of reduced silver ions on the surface of the microspheres as the number of amine groups in PEI molecules and maintaining the morphology of the microspheres intact, as the presence of excess silver ions ensures the complete oxidation of the adsorbed PEI. The reduction reaction of amines with silver ions oxidizes the amine groups to form carboxyl groups59 which was clearly visible in the FTIR measurements. By immobilizing PEI in the microspheres, the synthesis of Ag nanoparticles is a one-step process with PEI as a complexing agent for the ions, reducing agent, and anchoring for the nanoparticles on the surface of the microspheres. The above-discussed formation mechanism of Ag–SiO2 microspheres is illustrated in Fig. 8 in detail.
 | ||
| Fig. 8 Schematic diagram showing the formation mechanism of Ag–SiO2 microspheres. Silica–PEI microspheres adsorb the silver ions dispersed in water to form PEI–silver-ion complexes, which get further reduced to Ag nanoparticles on the microspheres, resulting in the formation of Ag–SiO2 microspheres. | ||
The Ag–SiO2 microspheres were employed in the electrochemical detection of H2O2, as depicted in Fig. 9a. The cyclic voltammetry (CV) responses of pure Si-p(20) and Si-p(20)–Ag at a PEI loading of 20% on a glassy carbon electrode with and without H2O2 in 0.1 M PBS are presented in Fig. 9b. To serve as a reference, the CV response of the bare glassy carbon electrode (GCE) was also recorded. The bare glassy carbon electrode did not exhibit a reduction peak in the presence of H2O2. A slight reduction peak was observed for the Si-p(20)-modified GCE, while a significant reduction peak at −0.4 V was observed for the Si-p(20)–Ag-modified GCE, which increased significantly in the presence of H2O2. The response current in the potential range of −0.6 V to −0.4 V and a reduction peak at −0.4 V could be attributed to the reduction of H2O2 due to the silver nanoparticles.64 The electroreduction mechanism of H2O2 on the Ag–SiO2 electrode is given by eqn (7) and (8).65,66
| H2O2 → ½O2 + H2O | (7) |
| O2 + 2e− + 2H+ → H2O2 | (8) |
 | ||
| Fig. 9 (a) Schematic diagram for the electrochemical reduction setup. (b) Cyclic voltammetry response of the bare GCE in the absence (black) and presence (red), Si-p(20) in the absence (wine) and presence (green), and Si-p(20)–Ag in the absence (cyan) and presence (blue) of H2O2. (c) CVs of Si-p(20)–Ag with the variation in H2O2 concentration. | ||
In order to study the reduction response of the silver nanoparticles, CV analysis was performed on the Si-p(20)–Ag microspheres, with the concentration of H2O2 increasing from 0 to 30 mM, as shown in Fig. 9c. The observed increase in the reduction current magnitude with the increasing H2O2 concentration (Fig. S7 in the ESI†), obtained from the peak positions in Fig. 9c, indicated the favorable reduction response of the Ag nanoparticles.22 The estimated linear detection range was found to be from 6 mM to 30 mM (Fig. S6 in the ESI†). The limit of detection was estimated using the signal-to-noise ratio method, with a signal-to-noise ratio of 3. The slope was calculated through linear fitting, as shown in Fig. S6 in the ESI.† The limit of detection was estimated to be 1.08 mM, and the sensitivity of the sensor was calculated to be 0.033 μA mM−1 mm−2, for the GCE electrode with a diameter of 3 mm. The lower sensitivity and good limit of detection for the Ag–SiO2 microspheres implied a good catalytic ability for H2O2 reduction.67,68
Further, the selectivity of the electrochemical sensor for the detection of H2O2 was studied by carrying out a series of CV measurements in the presence of varying interfering agents, such as glucose, ethanol, and ascorbic acid, and are depicted in Fig. S8a in the ESI.† The combination of H2O2 with these interfering agents did not lead to any observable change in the potential range of −0.4 V to −0.6 V. Also, no observable change was seen at the potential peak at −0.4 V. The significant change in the current response compared to the blank was due to the reduction of H2O2 in the presence of the Ag nanoparticles. This observation clearly indicated the negligible influence of these compounds on the electrochemical detection of H2O2. These findings demonstrate the good selectivity exhibited by the Ag–SiO2 microspheres-based electrochemical sensor.
Further, stability measurements for the Ag–SiO2 microspheres-based electrochemical sensor were conducted. By employing the analogous procedure of the earlier measurements, a GCE was fabricated utilizing the Ag–SiO2 microspheres. CV measurements with varying numbers of CV scans were carried out for the electrochemical detection of 10 mM H2O2, and the current corresponding to the reduction peak potential of −0.4 V was recorded after each 20 CV scans and is shown in Fig. S8b in the ESI.† By comparing the current responses from all the cycles, it was distinctly discernible that the sensor retained 95% of their initial current responsiveness. This observation conclusively validated that the Ag–SiO2 hybrid microspheres could function as a durable sensor, capable of sustained performance when employed for H2O2 electrochemical detection.
The Ag–SiO2 microspheres were utilized for the SERS-based detection of the model dye molecule rhodamine 6G (R6G). A schematic showing the geometry of the NRS and SERS measurements is depicted in Fig. 10a. The NRS spectrum and concentration-dependent SERS spectra of R6G in the presence of Ag–SiO2 microspheres are shown in Fig. 10b. Raman bands with an insignificant Raman signal of R6G were observed for NRS along with a large fluorescence background. However, in the presence of Ag–SiO2 microspheres, the SERS spectra of R6G recorded at a lower laser power (1 mW) could be evidently seen even at concentration down to 2 × 10−6 M, demonstrating the high sensitivity of the Ag–SiO2 microspheres as a SERS substrate. From the SERS spectra of R6G, a few strong, medium, and weak Raman peaks were observed. The typical Raman peaks of R6G at 609 cm−1 (in-plane ring bending), 768 cm−1 (out-of-plane C–H bending, in-plane ring bending), 1181 cm−1 (in-plane C–H ring bending, in-plane C–H bending, in-plane N–H bending), 1307 cm−1 (aromatic breathing, CH2 wagging), 1359 cm−1 (ring stretching, in-plane C–H bending), 1504 cm−1 (ring stretching, C–N stretching, in-plane C–H bending, in-plane N–H bending), 1570 cm−1 (ring stretching, in-plane N–H bending), and 1648 cm−1 (ring stretching, in-plane C–H bending) were observed.69
 | ||
| Fig. 10 (a) Schematic diagram showing the geometry of the NRS and SERS measurements of R6G. In the NRS measurements, R6G was drop-cast on a glass slide, whereas for the SERS measurements, the Ag–SiO2 microspheres were used as an active substrate. (b) Curve 1. NRS spectrum of R6G (2 × 10−2 M) recorded at 25 mW laser power. Curves 2–5. Concentration-dependent SERS spectra of R6G adsorbed onto Ag–SiO2 microspheres recorded at 1 mW laser power. (c) The log–log plot of the intensity of the Raman peak at 1648 cm−1 and the concentration of R6G. | ||
The concentration-dependent SERS spectra of R6G showed good signals down to the concentration of 2 × 10−6 M. Fig. 10c depicts the correlation between the intensity of the 1648 cm−1 Raman peak and the concentration of R6G. With the increase in R6G concentration from 2 × 10−6 M, the SERS signal reached a maximum at 2 × 10−4 M, and then a decrease in the intensities was observed at 2 × 10−3 M. The SERS signal is known to achieve a maximum at monolayer coverage, while the signal decreases with the formation of a multilayer.70–72 This suggests that at 2 × 10−4 M, there was a monolayer formation of R6G molecules on the Ag–SiO2 microspheres. At higher concentrations (2 × 10−3 M), the SERS signal arose from the multilayer present on the surface of the Ag–SiO2 microspheres.
To quantify the SERS enhancement using this substrate, the analytical enhancement factor (AEF) was estimated using the formula AEF = (ISERS/INRS) × (CNRS/CSERS), where CNRS and CSERS are the molar concentrations of the analyte molecule used for NRS and SERS, respectively;72,73 and ISERS and INRS are the intensities of the NRS and SERS bands. AEF was estimated using the ISERS (2 × 10−6 M) and INRS (2 × 10−2 M) of the Raman band observed at 1648 cm−1 (ring stretching, in-plane C–H bending), which was found to be ∼5 × 104.
Previously, Junfan et al.74 were able to show an AEF of ∼105 for R6G using annealed Ag nano-islands with SiO2 microsphere arrays, which corroborated well with our reported AEF. However, a double monolayer Ag@SiO2 and open nanocavity assistant soft SERS sample Ag@PDMS were able to show enhancement factors of ∼1013 and 1012, respectively.75,76 Although these materials offer significant advantages in terms of AEF, there are certain limitations to the methods used for synthesizing nanocavity-assisted soft SERS samples, as the technique used is a liquid–liquid transfer method. In the liquid–liquid transfer method, a rigid substrate is used and the fabrication method involves multiple steps, which makes it complex and cumbersome compared to our synthesis methodology, which is a one-step, cost-effective approach.
Further, a summary of the comparison of the performances of the distinct H2O2 electrochemical sensor as well as SERS substrates reported in the literature with the present study is shown in Tables S1 and S2, respectively, in the ESI.† The SERS performance of the Ag–SiO2 microspheres is acceptable for use in the detection of organic analytes in the micromolar concentration range. The detection limit for the H2O2 sensor in the present study was in the millimolar range, which is low compared to the other reported works. However, the simplistic and scalable approach presented for achieving Ag–SiO2 microspheres has the potential to be integrated at the industrial scale for H2O2 detection where the detection limit requirement is low. The primary objective of the present study was to establish a synthesis methodology that is ecologically effective, economically viable, and scalable. In the present study, PEI-loaded microspheres were utilized for the purpose of reducing Ag ions, which subsequently yielded Ag nanoparticles that were anchored on microspheres. The presence of microspheres offers additional advantages of stability and immobilization of the metal nanoparticles. Further, the electrochemical sensing of H2O2 as well as SERS sensing using the Ag–SiO2 microspheres were carried out as a feasibility study, where the microspheres-supported Ag nanoparticles offered higher accessibility to the analyte molecules and the SERS substrate could be recovered after use, which is not possible for silver nanoparticles in the dispersion form. However, there is tremendous scope for improving the parameters related to SERS and H2O2 sensing by changing the size of the Ag nanoparticles, the coverage, and the substrate geometry to mention a few, and these will be explored in our future work.
5. Conclusions
An eco-friendly, green, facile one-step method was employed to obtain Ag–SiO2 microspheres, which were then utilized for the electrochemical detection of H2O2, as well as used as a SERS substrate for the detection of R6G dye. Evaporation-induced assembly was utilized to obtain porous silica microspheres with the simultaneous immobilization of PEI. The trapped PEI offered the unique advantage of complexing silver ions onto the microspheres with an in situ reduction of silver ions to silver nanoparticles akin to in biomimetic processes. Not only did PEI act as a reductant, but it also anchored Ag nanoparticles onto the surface of the microspheres. Elemental mapping of the silver–silica composites using EDX measurements indicated an increase in the degree of coverage of Ag nanoparticles on the surface of microspheres with the increase in PEI loading. This observation clearly indicated there was a direct correlation between the amount of Ag nanoparticles formed and the loading of PEI in the microspheres. X-Ray diffraction revealed the FCC structure of the Ag nanoparticles with an estimated crystallite size of ∼10 nm. The FTIR results for the silica–PEI microspheres up to 160 °C displayed the high thermal stability of PEI and indicated their strong entrapment. The emergence of new bands corresponding to carboxyl groups in the FTIR spectra of silver-silica microspheres revealed the reduction mechanism of Ag ions to Ag nanoparticles. The Ag–SiO2 microspheres were utilized for the electrochemical detection of H2O2. A reduction peak was observed for Ag–SiO2 microspheres, which increased with the increase in the concentration of H2O2, suggesting a favorable and good sensing capability with a limit of detection of ∼1.08 mM, and the sensitivity of the sensor was estimated to be 0.033 μA mM−1 mm−2. The normal Raman scattering (NRS) of rhodamine 6G (R6G) as well as SERS measurements of R6G in the presence of Ag–SiO2 microspheres were carried out. In NRS, no Raman bands of R6G were observed, whereas in the SERS measurements carried out at 1 mW laser power, a typical Raman band at 1648 cm−1 could be clearly observed at a concentration down to 2 × 10−6 M. The green, biomimetic, and facile synthesis procedure employed in this study for the formation of Ag nanoparticles on the surface of microspheres is a scalable approach and therefore has a promising integration capability at the industrial scale in the field of sensors, as well as use as a SERS substrate for the detection of analytes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Mr Ashwani Kumar, SSPD, BARC for his help in SAXS measurements. Mr Jayakrishnan V. B. and Dr P. U. Sastry, SSPD, BARC are acknowledged for their help in XRD measurements.References
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18–45 CrossRef CAS PubMed.
- M. J. Ndolomingo, N. Bingwa and R. Meijboom, J. Mater. Sci., 2020, 55, 6195–6241 CrossRef CAS.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- Z. Deng, M. Chen and L. Wu, J. Phys. Chem. C, 2007, 111, 11692–11698 CrossRef CAS.
- C. Graf and A. van Blaaderen, Langmuir, 2002, 18, 524–534 CrossRef CAS.
- Z.-J. Jiang, C.-Y. Liu and L.-W. Sun, J. Phys. Chem. B, 2005, 109, 1730–1735 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, in Semiconductor and Metal Nanocrystals, CRC Press, 2003, pp. 415–444 Search PubMed.
- W. Wang and S. A. Asher, J. Am. Chem. Soc., 2001, 123, 12528–12535 CrossRef CAS PubMed.
- J. Zhang, J. Liu, S. Wang, P. Zhan, Z. Wang and N. Ming, Adv. Funct. Mater., 2004, 14, 1089–1096 CrossRef CAS.
- N. Lu, T. Zhang, X. Yan, Y. Gu, H. Liu, Z. Xu, H. Xu, X. Li, Z. Zhang and M. Yang, Nanoscale, 2018, 10, 14923–14930 RSC.
- P. Salazar, V. Rico and A. R. González-Elipe, Electrochim. Acta, 2017, 235, 534–542 CrossRef CAS.
- Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. r. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang and D. Bernsmeier, ACS Catal., 2018, 8, 2844–2856 CrossRef CAS.
- Q. Chen, T. Lin, J. Huang, Y. Chen, L. Guo and F. Fu, Anal. Methods, 2018, 10, 504–507 RSC.
- S. Kubendhiran, B. Thirumalraj, S.-M. Chen and C. Karuppiah, J. Colloid Interface Sci., 2018, 509, 153–162 CrossRef CAS PubMed.
- W. Lu, Y. Luo, G. Chang and X. J. B. Sun, Bioelectronics, 2011, 26, 4791–4797 CrossRef CAS PubMed.
- A. E. Vilian, S.-M. Chen and B.-S. Lou, Biosens. Bioelectron., 2014, 61, 639–647 CrossRef PubMed.
- I. Ivanišević, S. Milardović and P. Kassal, Food Technol. Biotechnol., 2021, 59, 216–237 Search PubMed.
- J. Jackson and N. Halas, J. Phys. Chem. B, 2001, 105, 2743–2746 CrossRef CAS.
- S. Oldenburg, R. Averitt, S. Westcott and N. Halas, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS.
- F. Caruso, R. A. Caruso and H. Mohwald, Science, 1998, 282, 1111–1114 CrossRef CAS PubMed.
- J. Zou, Y. Xu, B. Hou, D. Wu and Y. Sun, China Particuol., 2007, 5, 206–212 CrossRef CAS.
- J. Han, P. Fang, W. Jiang, L. Li and R. Guo, Langmuir, 2012, 28, 4768–4775 CrossRef CAS PubMed.
- J. Zheng, H. Lin, Y.-n. Wang, X. Zheng, X. Duan and Y. Yuan, J. Catal., 2013, 297, 110–118 CrossRef CAS.
- J. Zheng, H. Lin, X. Zheng, X. Duan and Y. Yuan, Catal. Commun., 2013, 40, 129–133 CrossRef CAS.
- C. Graf, S. Dembski, A. Hofmann and E. Rühl, Langmuir, 2006, 22, 5604–5610 CrossRef CAS PubMed.
- S.-E. Park, M.-Y. Park, P.-K. Han and S.-W. Lee, Bull. Korean Chem. Soc., 2006, 27, 1341–1345 CrossRef CAS.
- S. L. Westcott, S. J. Oldenburg, T. R. Lee and N. J. Halas, Langmuir, 1998, 14, 5396–5401 CrossRef CAS.
- V. Demchenko, S. Riabov, S. Kobylinskyi, L. Goncharenko, N. Rybalchenko, A. Kruk, O. Moskalenko and M. Shut, Sci. Rep., 2020, 10, 7126 CrossRef CAS PubMed.
- T. James, J. Am. Chem. Soc., 1939, 61, 648–652 CrossRef CAS.
- U. Nickel, A. zu Castell, K. Pöppl and S. Schneider, Langmuir, 2000, 16, 9087–9091 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2003, 107, 7479–7485 CrossRef.
- P. Barnickel and A. Wokaun, Mol. Phys., 1989, 67, 1355–1372 CrossRef CAS.
- A. Mayer, W. Grebner and R. Wannemacher, J. Phys. Chem. B, 2000, 104, 7278–7285 CrossRef CAS.
- C. Li and D. L. Kaplan, Curr. Opin. Solid State Mater. Sci., 2003, 7, 265–271 CrossRef CAS.
- S. Mann, J. Mater. Chem., 1995, 5, 935–946 RSC.
- E. Pouget, E. Dujardin, A. Cavalier, A. Moreac, C. Valéry, V. Marchi-Artzner, T. Weiss, A. Renault, M. Paternostre and F. Artzner, Nat. Mater., 2007, 6, 434–439 CrossRef CAS PubMed.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. Bernardes, E. Gazit and T. P. Knowles, Nat. Rev. Chem, 2020, 4, 615–634 CrossRef CAS.
- W. Yang, W. Guo, J. Chang and B. Zhang, J. Mater. Chem. B, 2017, 5, 401–417 RSC.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- R. Sanghi and P. Verma, Bioresour. Technol., 2009, 100, 501–504 CrossRef CAS PubMed.
- O. Krotikova, A. Ozerin, F. Radchenko and I. Novakov, Polym. Sci., 2017, 59, 288–294 CAS.
- S. Mehta, J. Bahadur, D. Sen, S. Singh and V. Polshettiwar, Soft Matter, 2022, 18, 5114–5125 RSC.
- J. Bahadur, D. Sen, S. Mazumder, S. Bhattacharya, H. Frielinghaus and G. Goerigk, Langmuir, 2011, 27, 8404–8414 CrossRef CAS PubMed.
- J. Bahadur, D. Sen, S. Mazumder, B. Paul, H. Bhatt and S. Singh, Langmuir, 2012, 28, 1914–1923 CrossRef CAS PubMed.
- A. Das, D. Sen, J. Bahadur and M. Subramanian, Colloids Surf., A, 2019, 577, 185–193 CrossRef CAS.
- D. Sen, J. S. Melo, J. Bahadur, S. Mazumder, S. Bhattacharya, S. F. D'Souza, H. Frielinghaus, G. Goerigk and R. Loidl, Soft Matter, 2011, 7, 5423–5429 RSC.
- N. Beaudoin and S. S. Beauchemin, 2002.
- C.-X. Kan, J.-J. Zhu and X.-G. Zhu, J. Phys. D: Appl. Phys., 2008, 41, 155304 CrossRef.
- C.-F. Lee, C.-L. Chang, J.-C. Yang, H.-Y. Lai and C.-H. Chen, J. Colloid Interface Sci., 2012, 369, 129–133 CrossRef CAS PubMed.
- A. A. Fayyadh and M. H. Jaduaa Alzubaidy, J. Mech. Behav. Mater., 2021, 30, 228–236 CrossRef.
- P. Debye and P. Scherrer, Phys. Z., 1916, 17, 277–283 CAS.
- P. R. Garcia, O. Prymak, V. Grasmik, K. Pappert, W. Wlysses, L. Otubo, M. Epple and C. L. Oliveira, Nanoscale Adv., 2020, 2, 225–238 RSC.
- J. S. Pedersen, Adv. Colloid Interface Sci., 1997, 70, 171–210 CrossRef CAS.
- D. Sen, J. Bahadur, S. Mazumder, G. Verma, P. Hassan, S. Bhattacharya, K. Vijai and P. Doshi, Soft Matter, 2012, 8, 1955–1963 RSC.
- R. Baxter, J. Chem. Phys., 1968, 49, 2770–2774 CrossRef CAS.
- S. Mehta, J. Bahadur, D. Sen, V. K. Aswal and J. Kohlbrecher, Phys. Chem. Chem. Phys., 2022, 24, 21740–21749 RSC.
- Z. Bacsik, N. Ahlsten, A. Ziadi, G. Zhao, A. E. Garcia-Bennett, B. Martín-Matute and N. Hedin, Langmuir, 2011, 27, 11118–11128 CrossRef CAS PubMed.
- N. Ramamurthy and S. Kannan, Rom. J. Biophys., 2007, 17, 269–276 CAS.
- C. Tian, B. Mao, E. Wang, Z. Kang, Y. Song, C. Wang and S. Li, J. Phys. Chem. C, 2007, 111, 3651–3657 CrossRef CAS.
- J. Bahadur, A. Das, J. Prakash, P. Singh, A. Khan and D. Sen, J. Appl. Phys., 2019, 126, 204301 CrossRef.
- S. S. Maw, S. Watanabe and M. T. Miyahara, Langmuir, 2020, 36, 4511–4518 CrossRef CAS PubMed.
- A. Zezin, Polym. Sci., 2019, 61, 754–764 CAS.
- X. Sun, S. Dong and E. Wang, Polymer, 2004, 45, 2181–2184 CrossRef CAS.
- S. N. Azizi, S. Ghasemi, A. Samadi-Maybodi and M. Ranjbar-Azad, Sensor. Actuator. B Chem., 2015, 216, 271–278 CrossRef CAS.
- M. Honda, T. Kodera and H. Kita, Electrochim. Acta, 1986, 31, 377–383 CrossRef CAS.
- J. Tian, H. Li, W. Lu, Y. Luo, L. Wang and X. Sun, Analyst, 2011, 136, 1806–1809 RSC.
- H. Hao, Q. Sheng and J. Zheng, Colloids Surf., A, 2017, 518, 124–129 CrossRef CAS.
- D. Yang, N. Ni, L. Cao, X. Song, Y. Alhamoud, G. Yu, J. Zhao and H. Zhou, Micromachines, 2019, 10, 268 CrossRef PubMed.
- L. Jensen and G. C. Schatz, J. Phys. Chem., 2006, 110, 5973–5977 CrossRef CAS PubMed.
- U. K. Sarkar, A. Pal, S. Chakrabarti and T. Misra, Chem. Phys. Lett., 1992, 190, 59–63 CrossRef CAS.
- P. Sanda, J. Warlaumont, J. Demuth, J. Tsang, K. Christmann and J. Bradley, Phys. Rev. Lett., 1980, 45, 1519 CrossRef CAS.
- N. Kumar, S. Thomas, R. Rao, N. Maiti and R. J. Kshirsagar, J. Raman Spectrosc., 2019, 50, 837–846 CrossRef CAS.
- E. C. Le Ru, E. Blackie, M. Meyer and P. G. Etchegoin, J. Phys. Chem. C, 2007, 111, 13794–13803 CrossRef CAS.
- C. Junfan, C. Hongxian, Z. Jie and Z. Yong, Opt. Mater. Express, 2021, 11, 2076–2088 CrossRef.
- S. HaiYang, W. Zhengkun, Z. Yong and Z. Jie, Opt. Express, 2023, 31, 16484–16494 CrossRef PubMed.
- H. Sha, Z. Wang and J. Zhang, Sensors, 2022, 22, 4595 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04095j |
| This journal is © The Royal Society of Chemistry 2023 |